 Xinyi Xu
Xinyi Xu Huichang Zhong
Huichang Zhong Weifeng Liu
Weifeng Liu Yong Tao
Yong Tao
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Bioeng. Biotechnol. , 28 April 2020
Sec. Synthetic Biology
Volume 8 - 2020 | https://doi.org/10.3389/fbioe.2020.00145
Genetic manipulations including chromosome engineering are essential techniques used to restructure cell metabolism. Lambda/Red (λ/Red)-mediated recombination is the most commonly applied approach for chromosomal modulation in Escherichia coli. However, the efficiency of this method is significantly hampered by the laborious removal of the selectable markers. To overcome the problem, the integration helper plasmid was constructed, pSBC1a-CtR, which contains Red recombinase, Cre recombinase, and exogenous orthogonal aminoacyl-transfer RNA (tRNA) synthetase/tRNA pairs, allows an unnatural amino acid (UAA) to be genetically encoded at the defined site of the antibiotic resistance gene-encoded protein. When UAAs are not in the culture medium, there was no expression in the antibiotic resistance gene-encoded protein. Accordingly, the next procedure of antibiotic gene excising is not needed. To verify this method, poxB gene was knocked out successfully. Furthermore, sequential deletion of three target genes (galR, ptsG, and pgi) was able to generate neurosporene-producing strain marked by high growth rate. Thus, the site-specific incorporation UAA mutagenesis system were used to control and expand the use of conditional selectable marker, and the technique is used to facilitate a rapid continuous genome editing in Escherichia coli.
Synthetic biology methods have greatly facilitated the optimization of processes in metabolic engineering. New promoters, ribosome binding sites, and gene circuits were designed using synthetic technologies and assembled into metabolic pathways to improve the metabolic flux and gene regulation efficiency. By these ways, the production of medicinal chemicals such as artemisinin (Chang et al., 2007) and taxol (Li et al., 2019) has been successfully optimized. Continuous genomic modification is an important tool for the optimization of processes in metabolic engineering. The λ/Red-based system was developed to mediate chromosomal homologous recombination, and resistance gene markers were used for genetic selection (Wang et al., 2016). The three plasmids of pKD4, pKD46, and pCP20 are widely used in λ/Red-mediated recombination. The pKD46 vectors include three genes: γ, β, and exo, whose products make chromosomal recombination. The plasmid pKD4 (or pKD3, pKD13) was template vector. The PCR products were generated using pairs of primers that included homology extension sequences of targeted gene and priming sequence of plasmid pKD4 (or pKD3, pKD13). The plasmid pCP20 has a temperature-sensitive replicon, antibiotic resistance marker gene, and flipase recombination enzyme (FLP) gene. FLP acts on the directly repeated FLP recognition target (FRT) sites flanking the resistance gene and makes FRT-flanked resistance gene to be eliminated. After one round of homologous recombination experiment, the vector pCP20 was electroporated into the host cells to eliminate the resistance gene, and the cells get ready to go to the next round of experiment when the pCP20 vector is lost from the cell under 42°C (Datsenko and Wanner, 2000). Although the efficiency of elimination of the antibiotic resistance gene is high (more than 80% cells lost the antibiotic resistance gene in genome), more than 80–100 nt scar, which included an FRT site, would be left behind the disrupted genes. After more than five times of chromosomal recombination experiments, the scars as mentioned above present in the host chromosome can influence expression of surrounding genes and caused the failure to achieve continuous knockout or integration experiments (Madyagol et al., 2011). To avoid this shortage, two rounds of recombination were designed and carried out by Zhang et al. (2007). In the first recombination, some of the target genes were replaced by a DNA cassette containing a chloramphenicol resistance gene (CmR) and a left sucrase gene (sacB). In the second recombination, the CmR–sacB cassette was eliminated by the selection for resistance to sucrose. However, this method also has shortcomings. The efficiency of the first homologous recombination was low, which may be due to the fact that sacB gene also had certain toxicity to Escherichia coli (E. coli) in the absence of sucrose. During the first homologous recombination, most of them were inactivated mutations of sacB, which led to a false positive when adding sucrose to screen the recombinant cells to eliminate sacB. Beyond that, the experimental procedure was laborious and time consuming, as the vector was electroporated into the host cells and eliminated from the cell under 42°C. Song and Lee developed an integration plasmid that harbors both λ/Red recombinase and cre gene under the control of two independent promoters (Song and Lee, 2013). However, in this system, the template plasmid does not contain multiple cloning sites, and the conventional p15A replication initiation site is used, which is not conducive to gene integration operations. So this method has not improved the positive rate. Researchers wanted to develop a plasmid strategy that mediates rapid one-step inactivation of genes. CRISPR-Cas9 system is an efficient genomic editing technique that is be developed (Cho et al., 2017). Unfortunately, due to the lack of homologous repair system in E. coli, the application of CRISPR-Cas9 is hampered and can be used in combination with the λ/Red recombination system. During these procedures, although selectable markers are not used, longer homologous arms and higher integration efficiency are required to ensure genomic integration. Furthermore, to recognize plasmids during each round of genomic modification, the protospacer adjacent motif site should be redesigned, which complicates the process.
The genetic encoding of unnatural amino acids (UAAs) are among the most extensively explored artificial systems. Incorporating UAAs into protein using evolved orthogonal aminoacyl-transfer RNA (tRNA) synthetase/tRNA pairs could confer the protein novel chemical and physical properties. Currently, UAAs have been wildly used in diverse bio-research fields (Young and Schultz, 2018), including exploration of protein structures, protein posttranslational modifications, and photo-cross-linking, and other specific therapeutic applications. For example, the abiotic cofactor nicotinamide flucytosine dinucleotide can be used to improve the efficiency of biological redox reactions (Ji et al., 2011). Thus, the unnatural biological molecules could provide an independent orthogonal system that does not interfere with the complex natural network in the living cell. This advantage indicates a potential application of the unnatural compounds in metabolic engineering.
In this study, we propose that UAAs can serve as a switch to turn on the specifically designed cell conditions in metabolic engineering. We generated a conditional selectable marker (CSM) by introducing the amber codon UAG into the sequence of selectable marker genes. The resistance function of these genes is eliminated in normal condition, whereas it is regained in a 3-iodo-L-tyrosine plate when corresponding orthogonal aminoacyl-tRNA synthetase/tRNA UAG pairs are introduced. This CSM system expands the power of genomic modification via reuse of the selectable marker. As CSM can be obtained for different resistance genes, the selectable marker list is significantly expanded, and continuous genomic modification can be carried out in multirounds without curing of resistance marker genes. Furthermore, all resistance marker genes can be removed in one go using Cre/lox recombination. In addition, different incompatible variants of lox site, including flank variant lox66/71 and spacing variants lox511, lox2272, m3, and m5, can provide stable curing of selectable markers at each genomic modification site. Considering that CSM can be further expanded by other UAA systems, this work will provide a rapid genomic modification strategy that can be efficiently used in metabolic engineering.
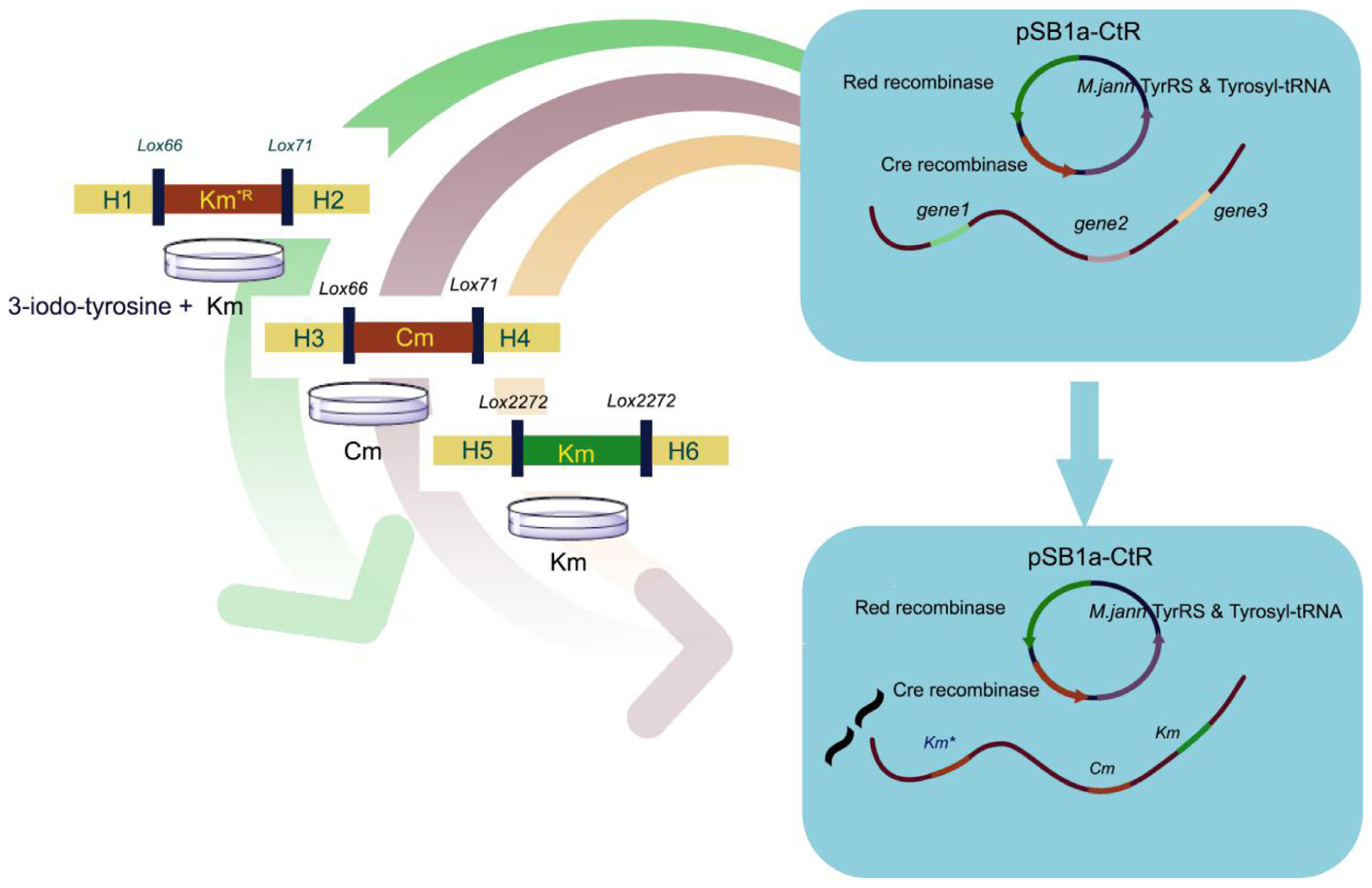
GRAPHICAL ABSTRACT Simultaneous triple gene deletion experiments. In the first deletion procedure, the selective marker KmR gene 160th site codon was mutated to the amber stop codon. Cells were spread onto agar with UAA to select the KmR transformants. After confirmation by PCR, the cells were processed to knockout genes in experiments with different antibiotic-resistant genes and loxP site without iterative plasmid transformation or curing steps.
Strains and plasmids used in this study are listed in Table 1. E. coli BW25113 was used as a host strain in gene knockout experiments, whereas the E. coli strain DH5α (TransGen Biotech Co., China) was used in all cloning experiments. For the construction of plasmids and strains, E. coli cells were grown in Luria–Bertani (LB) medium or on medium plate with 1.5% agar at appropriate temperature. Bioconversion medium was prepared by adding 4% (w/v) glucose to the Minimum Essential medium. The appropriate antibiotics were added in the following concentrations: 50 or 100 μg/ml of kanamycin, 100 μg/ml of ampicillin, and 25 μg/ml chloramphenicol. A total of 1 mM isopropyl-β-D-thiogalactoside (IPTG, Sigma-Aldrich) and 0.2% L-arabinose (Sigma-Aldrich) were used for induction. The transformed bacterial cells were grown at 30°C in LB medium plate supplemented with 1 mM 3-iodo-L-tyrosine (Sigma-Aldrich). All flask cultures were grown in ZYM-5052 autoinducing medium supplemented with glucose and with appropriate antibiotics (Studier, 2005). For the cultivation of an engineered E. coli strain for neurosporene production, a preculture was prepared by inoculating a single colony, which was stored at −80°C until use, in a 25-ml test tube containing 5 ml LB medium and cultivated at 37°C and 220 rpm in a shaking incubator after thawing. Cells (3 ml) that had grown for 12 h were transferred to a 300-ml baffled flask containing 100 ml ZYM-5052 autoinducing medium and were incubated for further 12 h. Following this, 10 ml 40% (wt/vol) of glucose was added to the baffled flask and incubated for 24 h.
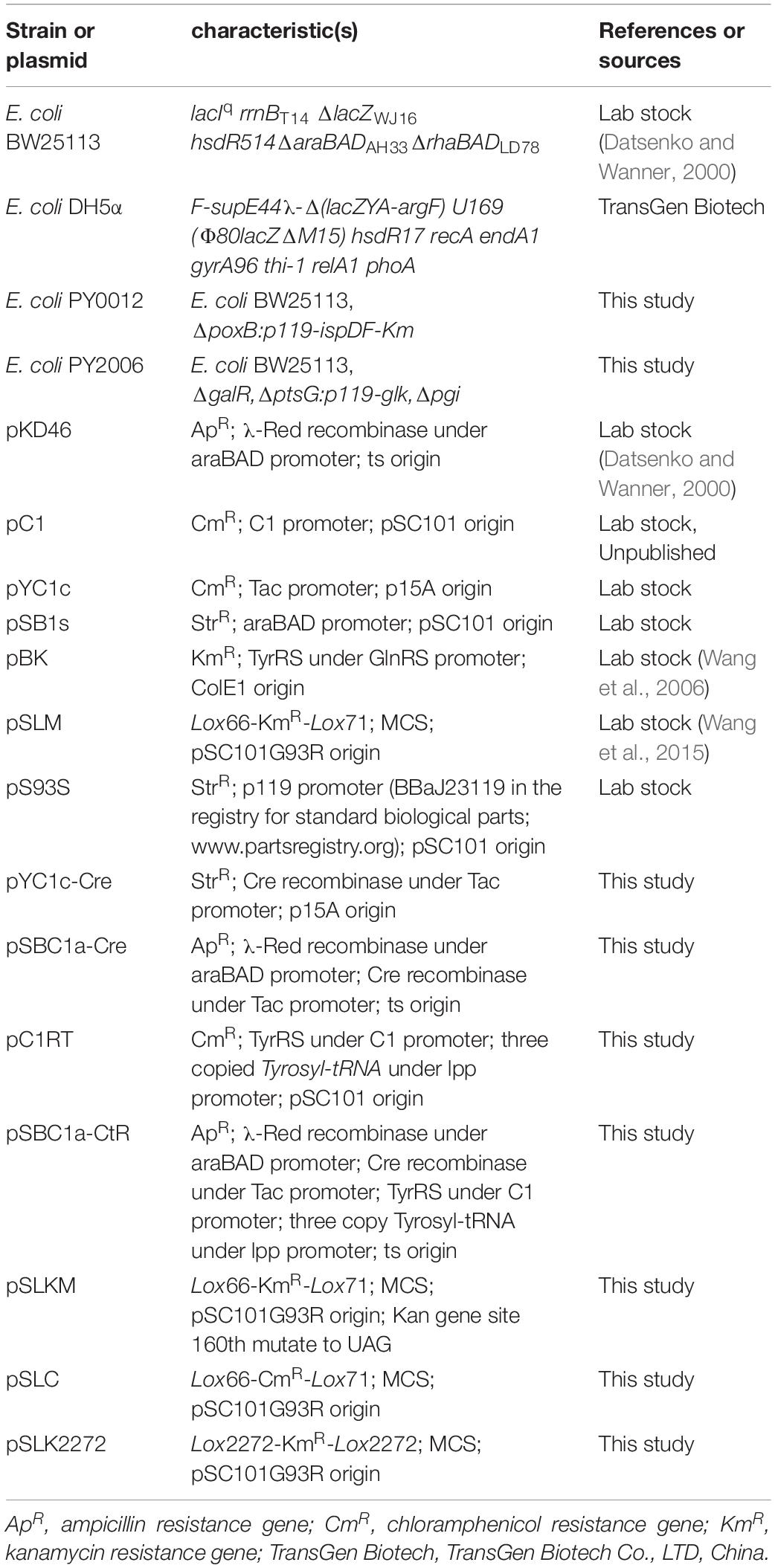
Table 1. Bacterial strains and plasmids used in this study.
The candidate variants including the six different mutation sites within the active center of the sequence of kanamycin resistance gene (aminoglycoside kinase APH 3′IIa, KmR) were designed and obtained. The mutations were obtained using the PCR method. With the addition of 1 mM of 3-iodo-L-tyrosine, the growth state of the strains that harbor plasmids containing KmR mutants were detected by means of kanamycin plate coating. Accordingly, we improved the expression level of tyrosyl-tRNA synthetase (TyrRS) using promoter replacement by strong constitutive PC1 promoter and placed three copies of tRNAtyr (CUA) to get pC1TR.
Gene cloning procedures, including PCR amplification, restriction, and ligation, were carried out according to the established protocols. All oligonucleotides are listed in the supporting information (Supplementary Table S1). E. coli cells were transformed by the electrotransformation method. The Gibson method was used for assembling multiple overlapping DNA fragments into a plasmid in single step without any restriction enzyme sites.
To improve the efficiency of continuously conducted gene modification, we optimized the plasmids used in the developed UAA conditional selectable marker (UCM) system. First, an integrated plasmid pKD was obtained using pKD46 as template. The TryRS gene and triple tRNAs were inserted into pKD46. The plasmid pC1RT that contains artificial aminoacyl-tRNA synthetase/tRNA pair was constructed. The pYC1c plasmids were double digested by KpnI and EcoRI and then ligated to the cre gene, which was synthesized using the vector pJL519 (Patel et al., 2010) and was amplified by PCR (Cre-kpnI-F and Cre-EcoRI-R). The final vector was named pYC1c-Cre. The vector pC1 was digested with NcoI and XhoI and assembled with the TyrRS gene, which were amplified from pBK-Ityr plasmid by PCR (Wang et al., 2006). The TyrRS gene and three copies of tyrosyl-tRNA genes were cloned into a module plasmid pC1RT using the Gibson assembly method (Davidson et al., 2002). Vector pKD46 was digested to linear segment, and cre genes under PTac promoter were amplified from the plasmid pYC1C-Cre. The TyrRS gene under C1 promoter and three copies of tRNA under lpp promoter were amplified from plasmid pC1RT by PCR. The three aforementioned DNA segments were integrated using pSBC1a-CtR plasmid by the Gibson method.
Three types of template plasmids containing antibiotic resistance genes flanked by the mutated loxP sites were used in PCR amplification. The first plasmid, pSLKM, contains the loxLE-Km-gene-loxRE region constructed in our previous study (Wang et al., 2015) and contains mutation KmR. The second plasmid, pSLC, had the same backbone as pSLM, but contained CmR, a different antibiotic gene. To construct the plasmid pSLC, the PCR products containing CmR gene that flanked NotI and NheI restriction sites from pSC1c (our lab stocked) were ligated into vector plasmid pSLM digested by restriction endonuclease NotI and NheI. The third plasmid, pSLK2272, was the same as pSLM, but contained lox2272 sites flanking the KmR gene. To construct pSLK2272, the KmR gene was amplified from the plasmid pKD4 using the primers lox2272Km-F and lox2272Km-R. The pSLM plasmids were digested into linear sequence by restriction endonuclease NotI and NheI. The PCR products included the lox2272-KmR-lox2272 gene and were ligated into linear pSLM fragments using the Gibson method. The above DNA sequence was confirmed by genetic sequencing.
To validate our method, the pSLKM plasmid was used as a template vector for the amplification of the linear gene and knockout of DNA fragments. This pSLKM vector was digested into the linear gene by restriction endonucleases and ligated to recombination gene. Each linear knockout fragment was amplified by overlapping two PCR procedures. The first PCR was performed using the primers F1 and R1 for each gene, which would be knocked out. F1 sequence has 70-bp homologous arm sequences of the target gene. R2 has internal homologous sequence of the according antibiotic resistance gene. The second PCR was performed using primers (F2 and R1). F2 sequence has internal homologous sequence of the according antibiotic resistance gene. R1 has 70-bp homologous arm sequences of the target gene. The two PCR products that contained 20-bp overlapping gene sequences were used as templates to perform the final PCR using the primers (F1 and R1) to create full-length PCR product. The PCR product was named poxB-PCR, which included 70-bp homologous arm sequences of poxB, loxLE/RE site, and KmR mutation gene with an amber codon UAG substituted for the 160th site.
For simultaneous three-gene knockout experiments, the pSLKM, pSLC, and pSLK2272 plasmids were used as template vectors to amplify the linear PCR product. DNA amplification was the same as the two overlapping PCR procedures described above. All the primers used in this study are listed in Supplementary Table S1. The first PCR product was named galR-PCR and had 50-bp homologous arm sequences of GalR, loxLE/RE site, and KmR with an amber codon substituted for 160th site in the mutation gene. The second PCR product amplified from the pLSC-glk vector, in which the glk gene with p119 promoter were inserted to pLSC by the Gibson method, was named ptsG-PCR and had 50-bp homologous arm sequences complementary to the downstream and upstream regions of the ptsG gene, respectively (the primers were glk-F1 and glk-R1). The third PCR product was named pgi-PCR and was constructed using the pSLK2272 vector and the primers pgi-F1 and pgi-R1 with 50-bp homologous arm sequences complementary to the downstream and upstream regions of the pgi gene, respectively. PCR products were gel purified, digested with DpnI, repurified, and suspended in an elution buffer (0.5 mM Tris, pH 8.0). Transformants carrying pSBC1a-CtR plasmid were grown in LB medium with ampicillin and L-arabinose and harvested at an OD600 nm of about∼0.5 by centrifugation at 1122 × g and 4°C. Cell pellets were washed twice with 10% (w/v) glycerol and suspended in 100 μl of the same glycerol solution.
For the gene knockout experiment, the procedure was as follows. First, approximately 1–2 μg of PCR products was added to the 200 μl E. coli BW25113 competent cells containing the pSBC1a-CtR plasmid. Electroporation was then performed using Gene pulser II (Bio-Rad) and 2-mm electroporation gap cuvettes (Bio-Rad). The electroporated cells expressing λ-Red recombinase were transferred with the linear gene knockout DNA fragment to trigger a recombination event. Transferred cells were incubated at 30°C, and the positive colonies were selected on LB agar plates containing 1 mM 3-iodo-tyrosine and 50 μg/ml kanamycin. Positive recombinants were verified by colony PCR using two confirming primers. The primer F1 is a copy of the sequence to the outside left of the target gene. The primer R1 is a copy of the sequence to the outside right of the target gene. The primer Km-TAA-F is a copy of the reverse sequence originated from UAA codon of the KmR gene. The primer Cm-TAA-F is a copy of the reverse sequence originated from UAA codon of the CmR gene. The confirmed colonies were streaked and grown on LB agar plates containing 1 mM IPTG and ampicillin to excise the KmR and CmR genes. Control colonies were always tested side by side. All the primers used in this study are listed in Supplementary Table S1.
To test our method, simultaneous triple gene deletion experiments were conducted using three selected target genes galR, ptsG, and pgi. In some experiments, glk gene with p119 promoter was used instead of gene ptsG. The selection and transfection procedures were the same as described above. Cells that contained the galR-PCR product were incubated for 1 h at 30°C. The cells were then spread for 30 min on agar with 3-iodo-Tyr to select KmR and ampicillin resistance gene (ApR) transformants at 30°C. Recombinants were confirmed by colony-testing PCR with primers as noted above using the forward primer Km-TAA-F and the reverse primers galR-F. The confirmed single colony with knocked out galR was selected. After the first step, the ptsG-PCR products were added to the 200-μl confirmed cells harboring the pSBC1a-CtR plasmid. The cells had been incubated at 30°C for 1 h, and then half was spread onto agar to select CmR and ApR transformants at 30°C. Recombinants were confirmed by colony-testing PCR using the forward Cm-TAA-F primer and the reverse PTSG-RC primer. The final confirmed colonies were spread and grown on LB agar plates containing kanamycin and ampicillin. The fragment of KmR gene was amplified from plasmids pSLK2272 using primers pgi-F1 and pgi-R1, then was electrotransfected into cells harboring pSBC1a-CtR. The transformants were spread on plate containing kanamycin and ampicillin to screen for positive colonies. In the final step, 1 mM IPTG was added to the culture medium, and the cells were induced by the cre recombinase expression to excise the three resistance genes simultaneously. After all sequential gene deletion experiments had been completed, pSBC1a-CtR was eliminated at 42°C.
To confirm the efficiency of this method for the sequential deletion of multiple genes, three genes were sequentially deleted to develop a neurosporene-producing E. coli strain as a proof-of-concept example. This strain was developed by the deletion of the galR, ptsG, and pgi gene; meanwhile, glk genes were increased in E. coli. During the cultivation, cell turbidity was monitored spectrophotometrically at 600 nm with a UV-1800 spectrophotometer (SHIMADZU, Japan). Neurosporene was extracted from cell pellets with acetone under agitation conditions, and its concentration was determined using absorption spectrum at 470 nm using a UV–visible (UV–vis) spectrophotometer. Neurosporene content was calculated by normalization of neurosporene production to dry cell weight (Farasat et al., 2014).
CSM was created by insertion of amber stop codon UAG to replace the sense codons within the sequence of KmR. We mutated the six genes of different amino acid sites to UAG within the active center of KmR. We compared the impact of different mutations on the loss of kanamycin-resistance phenotype. Strains harboring pC1RT vector and either of the pSLM plasmids that contained the mutated KmR genes did not grow on kanamycin plates (Supplementary Figure S1, left). When 1 mM of 3-iodo-L-tyrosine was added, all of these strains grew (Supplementary Figure S1, right). We selected the mutation with the KmR gene of GLU160 to UAG.
The evolution from Methanococcus jannaschii TyrRS that charges a modified M. jannaschii tRNAtyr(CUA) with 3-iodo-L-tyrosine was expressed under PC1 constitutive promoter site, whereas orthogonal tRNA gene was expressed under its native PLpp promoter. Accordingly, we improved the expression level of placed three copies of tRNAtyr (CUA). As expected, the BW25113 cells (contained three plasmids: pLSM with KmR 160th site mutation, pC1, pKD46) grew well on the plates containing kanamycin, chloramphenicol, ampicillin, and 3-iodotyrosine (Supplementary Figure S2).
The knockout system mainly includes two helper plasmids. As one of the two helper plasmids, the integration helper plasmid pSBC1a-CtR was constructed and was composed of arabinose-inducible Red recombinase gene, Cre recombinase gene, and exogenous orthogonal aminoacyl-tRNA synthetase/tRNA genes. The cells containing the plasmid pSBC1a-CtR was able to express the active antibiotic resistance gene-encoded protein that was with an UAA at the defined site (Figure 1A and Supplementary Table S3). To eliminate the background expression of cre gene under the control of PTac promoter, all manipulations were performed on plates supplemented with 1% glucose.
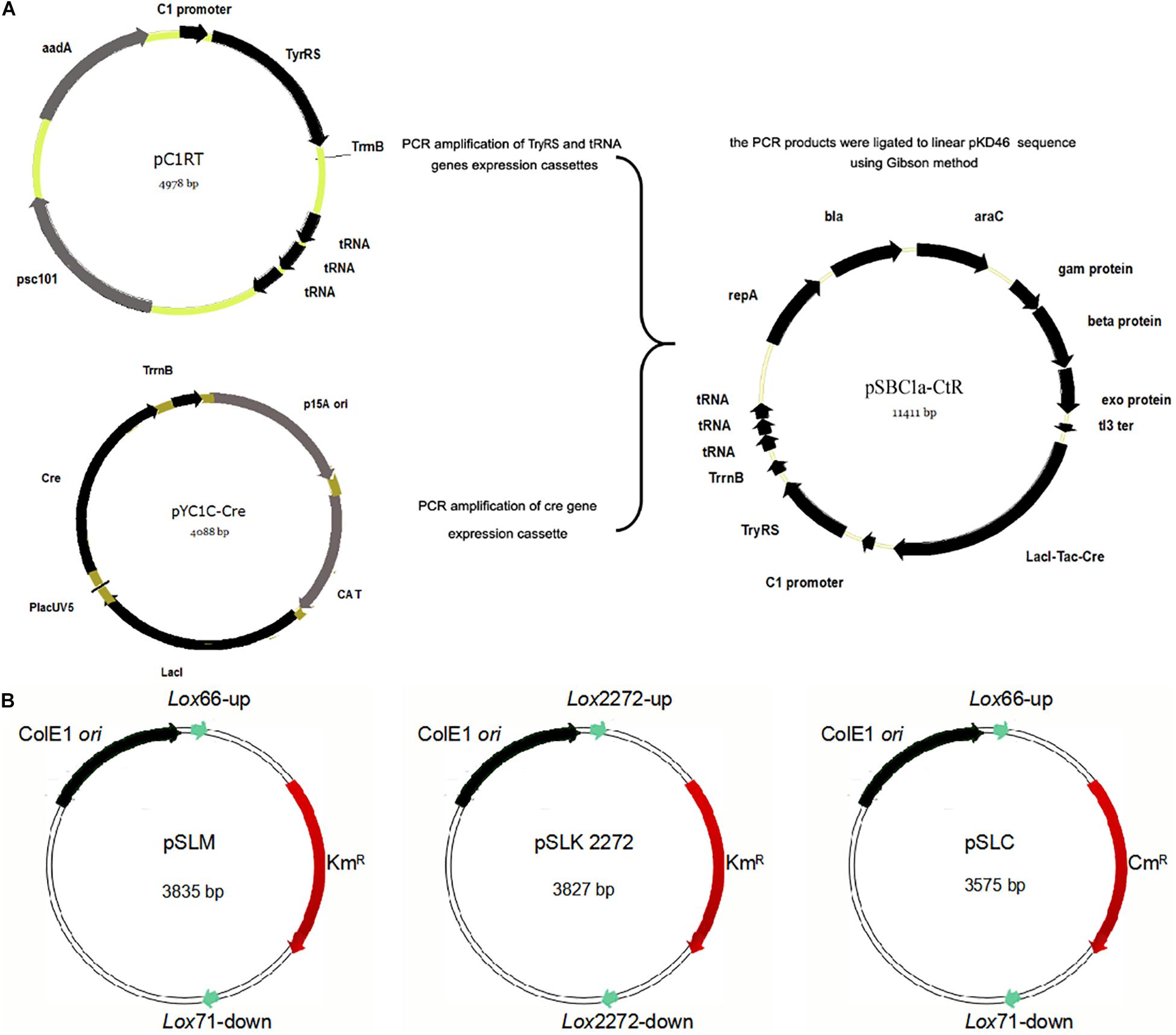
Figure 1. Construction of the integration helper pSBC1a-CtR and three template plasmids. (A) The IPTG-inducible Red recombinase gene under PTac promoter was amplified from pYC1c-Cre and the TyrRS gene by PCR. Three copies of transfer RNA (tRNA) were amplified from plasmid pC1RT by PCR. Vector for construction of the pSBC1a-CtR plasmid by overlap PCR method was used to create the three copies of Tyrosyl-tRNA PCR product. TyrRS gene PCR products and three copies of Tyrosyl-tRNA PCR products were constructed using the vector named pC1RT via Gibson method. pKD46 was digested to linear segment and ligated to the PCR products mentioned above by Gibson method. The final vector was named pSBC1a-CtR. (B) Map of template plasmids pSLKM, pSLC, and pSLK2272 is shown.
To obtain a selectable marker-free genetic background for further rounds of modifications, it is necessary to remove the selectable markers used in each previous round. In the developed UCM system of genomic modification, we have introduced different lox variants including lox2272/lox2272 and lox66/71 that were flanked to the selectable marker genes. The plasmid pSLKM (or pSLC, pSLC2272) (Figure 1B) was used as the template vector. The template vector has two functional areas. First, the Lox66/Lox71 or Lox2272/Lox2272 sequences were the action sites of FLP. The FRT-flanked resistance genes were lost under action of FLP (Cre recombinase). Second, the multicloning sites (MCSs) were on one of the two sides of the resistance marker gene. The sequence of the MCS and FRT site sequence is shown in Supplementary Table S2. Subsequently, multiresistance markers were excised simultaneously when the Cre recombinase was expressed on an LB agar plate containing ampicillin and IPTG. The excision of four resistance markers was confirmed using colony-based PCR.
To illustrate the application of UCM system for the modification of chromosomal genes in E. coli, the mutant KmR was used as selectable marker for the deletion of poxB gene. Replacing the poxB gene with the ispDF gene can result in redirection of central carbon flux and lead to various metabolic responses. The fragment of KmR gene was amplified from plasmids using primers, then was electrotransfected into cells harboring pKD46 and pC1RT. The process of the recombination is presented in Figure 2 (left). The transformants were spread on the plate containing kanamycin and 1 mM 3-iodo-L-tyrosine to screen for the positive colonies. Altogether, 21 colonies were picked out, and gene expression was confirmed using PCR method with primers RC and Km-TAA-F, as shown in Figure 2 (left). All colonies were tested positively. The five colonies were chosen randomly and further verified by PCR and sequenced to prove the correct expression. The rate of selected positive clone was 83% (Figure 3A). This result indicates that the CSM can be used for chromosomal modification.
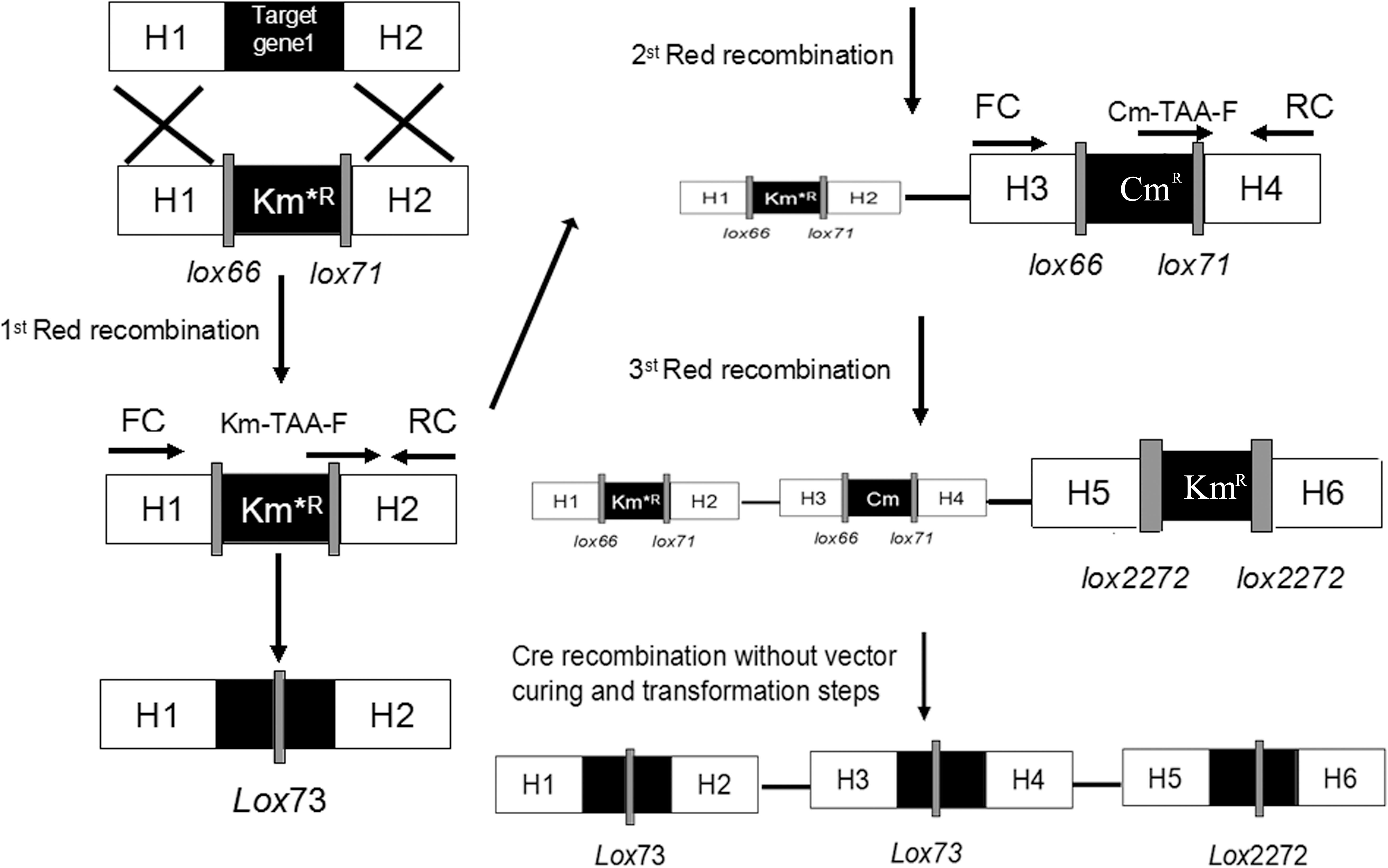
Figure 2. Schemes for gene knockout and excision of markers. Procedures are shown for the single-gene deletion (left) and simultaneous tripe-gene deletion (right) using the pSBC1a-CtR plasmids.
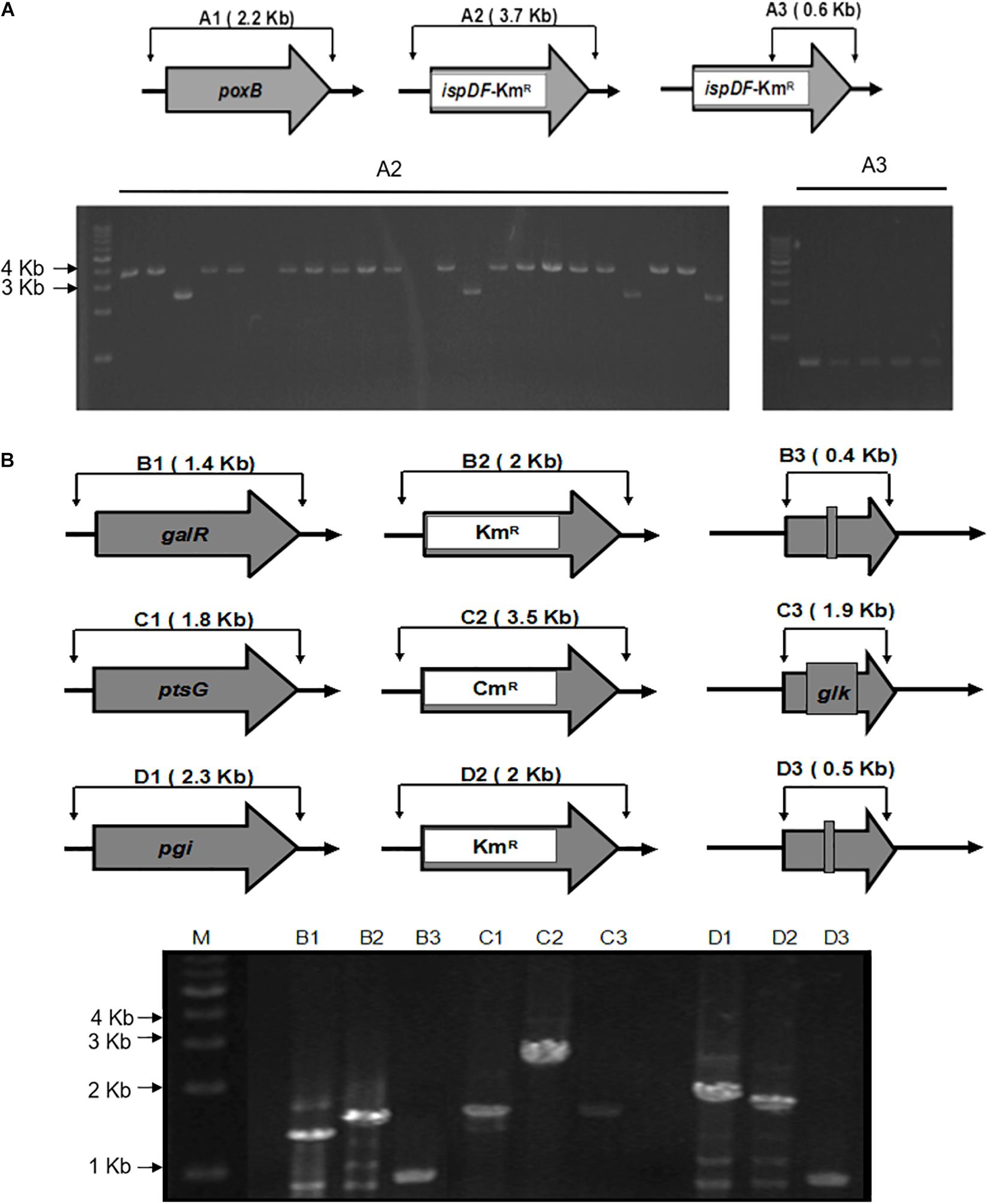
Figure 3. Schemes for gene knockout and excision of markers. (A) The gel image of PCR results is shown for the single knockout of the poxB genes and replacing with ispDF gene. A1, wild type; A2 and A3, mutants with a resistance marker and ispDF. A1 and A2 knockout mutants were confirmed using the FC and RC primers for each gene. A3 mutants were confirmed with resistance markers using the RC primers for each gene and the Km-TAA-F primer. (B) The gel image of the PCR-amplified chromosomal DNA fragments. The gel image of PCR results during the triple knockout of the galR, ptsG, and pgi genes is shown. B, galR; C, ptsG; D, pgi; 1, wild type; 2, mutant with a resistance marker; 3, knockout mutant; M, marker. Routine colony PCR was performed to check the recombination events. Wild-type and knockout mutants were confirmed using the FC and RC primers for each gene.
As shown in Figure 2, the fragments that contain UAG mutational kanamycin gene flanked two homology regions at galR gene site were amplified for the first round of deletion. The plasmid pSLC was used as a template for PCR amplification. Targeted fragments were then transferred into the cells. The transformants were spread on the plate containing kanamycin and 1 mM 3-iodo-L-tyrosine to screen for positive colonies. Chosen positive strains were subjected to the next round of deletion. Then, the PCR product that replaced the ptsG with glk was transformed to the cells without removing the mutated KmR and galR gene loci. The transformants were screened on chloramphenicol plates. The chosen positive strains were subjected to the next round of deletion. The obtained double-genes deletion strain that retains pSBC1a-CtR was subjected to further rounds of gene deletion. In the third round of experiments, deletion of pgi can result in the redirection of central carbon flux and lead to various metabolic responses. The fragment of KmR gene was amplified from plasmids using primers, then was electrotransfected into cells containing pSBC1a-CtR from the last deletion experiment. The transformants were spread on the plate containing kanamycin to screen for positive colonies. About 60% of those colonies were identified as positive using the colony PCR method. These results confirm that continuous genomic modifications can be successfully carried out using the UCM system without the step of curing selectable marker (Figure 3).
The gal operon repressor, GalR regulator, was deleted to increase the galactose content in the cell. Furthermore, glucose internalization by the phosphotransferase system (PTS) represents a drawback for the production of isoprene because the section of phosphoenol pyruvate was consumed during glucose phosphorylation in E. coli. Thus, the yield of carotenoids can be increased if ptsG gene is deleted. Besides, previous research demonstrated that Glk levels enabled a PTS– strain to sustain high growth rates. Thus, the yield of isoprene would be increased in the cell with deleted ptsG but increased Glk. In addition, pgi-knockout strain demonstrated that the flux through the glyoxylate shunt was increased, while lycopene production was decreased in comparison with the parent strain. Double deletion of galR and ptsG genes can block PTS-mediated glucose uptake, leaving non-PTS glucose transport unaffected.
To demonstrate the efficiency of the UCM method in synthetic biology, we developed a neurosporene-producing E. coli strain as a proof-of-concept example for the sequential deletion of multiple genes including galR, ptsG, and pgi. Meanwhile, the strain had increased content of glk genes. Neurosporene was produced by the triple-genes deleted strain. The cell growth curve shows that the final strain grew faster than the wild-type cells (Figure 4A). Notably, 43.24 g/L of neurosporene was produced by the final strain PY2006 (Figure 4B). It is emphasized that the sequential deletion of target genes took overall 10 days and that transformation and curing steps were not required. The antibiotic resistance gene was used repeatedly, which took about one-third of the time required during the application of the conventional methods.
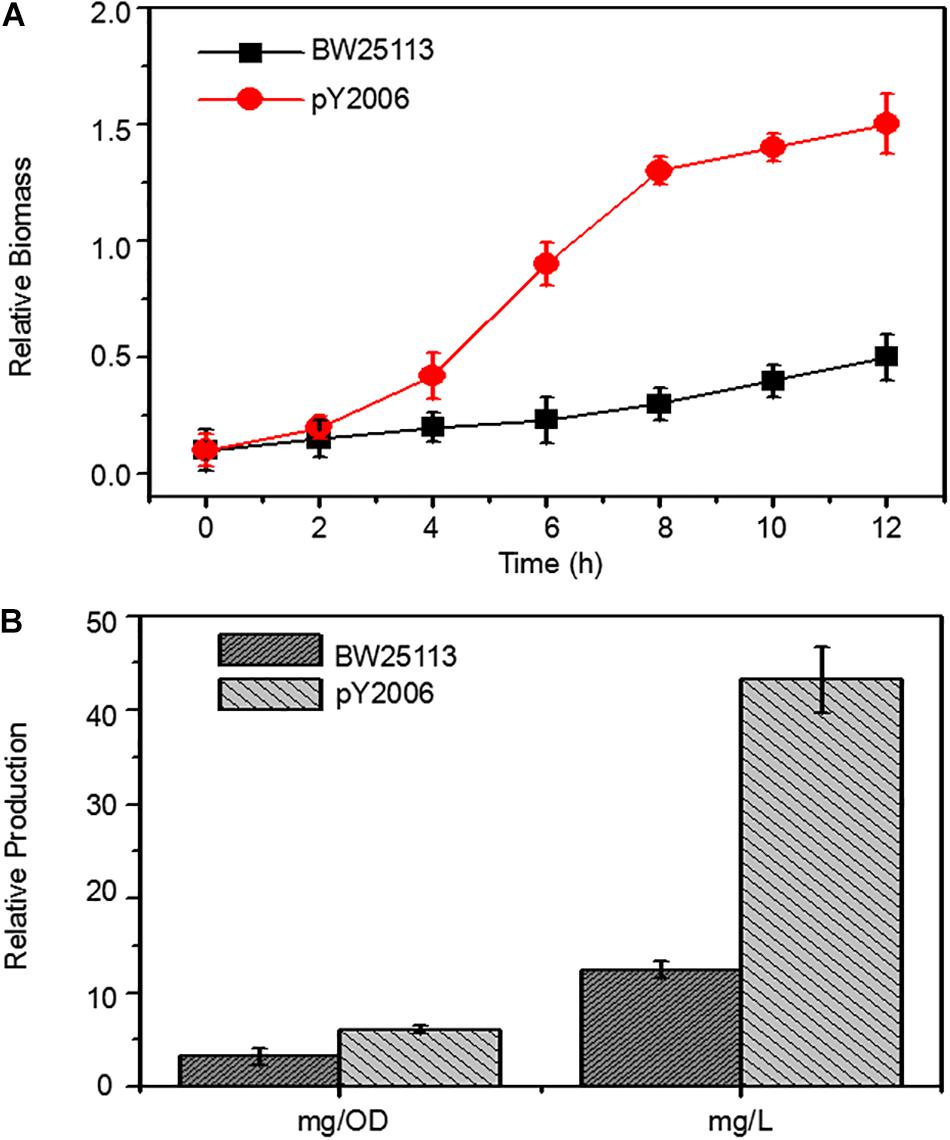
Figure 4. Metabolic engineering of E. coli to produce neurosporene. (A) The PY2006 cells (the genotypes shown in Table 1) with three knocked out genes grew faster than wild-type cell in Minimum Essential medium. (B) Comparison of neurosporene production in the engineered strains pY2006 and wild-type strain BW25113 in ZYM-5052 autoinducing medium. Data are expressed as the mean ± SD of three independent experiments.
This study describes the novel UAA mutagenesis system that utilizes a pair of exogenous M. jannaschii tyrosyl-tRNA and TyrRS construct. To confirm that the system runs effectively, three different experimental designs were used. First, to reduce the false-positive rate due to plasmid incompatibility, the vector pSBC1a-CtR and the template vector including pSLKM, pSLC, and pSLK2272 were constructed with low-copy number replication origin pSC101. When ampicillin and kanamycin were added to the plate, homologous recombination occurred. The cells lost the template vectors via the pressure of incompatibility during replication. The false-positive products of recombination were eliminated when antibiotic resistance gene was introduced as the selectable marker. Second, according to the previous study, once the expression of the TyRS was increased, overall yields of mutant proteins were also effectively increased (Hernández-Montalvo et al., 2003). C1 promoter (laboratory stocked) was used instead of glutamine promoter to increase the efficiency of the UAA mutagenesis system. The test data showed that TyrRS expression was the same as that of vector pBK under promoter gln and replication origin ColE1 in cells where the TyrRS gene was present under the C1 constitute promoter (Young et al., 2010). Third, Glu160 site of aminoglycoside-3′-O-phosphotransferase gene is an enzyme activity site. 3-Iodo-tyrosine, which is slightly different from the normal amino acid, was introduced to Glu160 of aminoglycoside-3′-O-phosphotransferase gene using amber stop codon UAG. The mutated loxP sequences (loxLE/loxRE or lox2272) were used for the gene knockout studies (Usui et al., 2012) to avoid unwanted recombination events. In our study, three template plasmids, pSLKM, pSLC, and pSLK2272, were used for the generation of linear gene knockout DNA fragments in simultaneous triple-gene knockout experiments.
The new knockout system simplifies plasmid transformation using Cre recombinase induction and expression and KmR gene elimination. When 3-iodotyrosine was added to the culture media, the mutational antibiotic KmR gene was translated to the full-length protein, and the cells were able to grow in this medium. After the first selection stage, the mutants confirmed by colony PCR were restreaked on a plate containing mutation KmR gene and pSBC1a-CtR at the second selection stage. Despite the gene knockout, DNA linear fragments with or without different antibiotic resistance gene were transferred into the cells. The plates without 3-iodotyrosine allowed the KmR mutation gene in the chromosome to be translated to truncation, and E. coli cells did not grow due to the activity of antibiotic resistance genes. For the sequential multiple gene knockout, the KmR genes were used as the selectable marker. Finally, cells were suspended in LB and were spread on a plate containing ampicillin and IPTG to induce loxP-mediated excision of the resistance marker. All colonies lost the resistance marker successfully. There were some limitations due to the limited number of resistance markers used for selection during simultaneous deletion approach. This problem could be lessened with the application of the developed gene knockout system with new plasmid pSBC1a-CtR. Gene deletions could be continued until the desired multiple-gene knockout mutants are obtained. The additional steps curing antibiotic resistance gene could be eliminated and the antibiotic label used many times without restriction of the antibiotic resistance gene.
The method based on the engineering of modular orthogonal UAA makes possible the genetic encoding of the UAAs in E. coli. Applications of genetically encoded UAAs are widespread. However, the applications were mostly used to address protein structure and function. This study introduces new tools to optimize the cell knockout/in system. Our procedure allows the sequential deletion of three genes in one week, a much shorter time period than previously required for the conventional cre recombinase mediated gene knockout method. In summary, using the UAA mutagenesis system, we developed a simple and highly efficient method of integrating DNA into chromosomal genes in E. coli. This method could be widely applied in gene manipulation.
The datasets generated for this study can be found in the https:// www.ncbi.nlm.nih.gov/nuccore/NC_000913.3?report=genbank& from=4233758&to=4235407, https://www.ncbi.nlm.nih.gov/nuc core/NC_011750.1?report=genbank&from=3405133&to=340616 4, https://www.ncbi.nlm.nih.gov/nuccore/NC_000913.3?report= genbank&from=1157869&to=1159302.
XX carried out the experiment and wrote the manuscript. HZ, WL, and YT analyzed the data and reviewed the manuscript.
This work was supported, in part, by the National Key R&D Program of China under Grant No. 2018YFC0910600, the National Natural Science Foundation of China under Grant Nos. 81627807, 81871397, and 81701853, the Natural Science Basic Research Plan in Shaanxi Province of China under Grant No. 2019JQ-519, and the Fundamental Research Funds for the Central Universities (JB181203).
HZ was employed by company Xiamen Huison Biotech Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbioe.2020.00145/full#supplementary-material
ApR, ampicillin resistance gene; CmR, chloramphenicol resistance gene; E. coli, Escherichia coli; IPTG isopropyl- β -D-thiogalactoside; KmR, kanamycin resistance gene; TyrRS, tyrosyl-tRNA synthetase; UAA, unnatural amino acid; UCM, UAA conditional selectable marker.
Chang, M. C. Y., Eachus, R. A., Trieu, W., Ro, D. K., and Keasling, J. D. (2007). Engineering Escherichia coli for production of functionalized terpenoids using plant P450s. Nat. Chem. Biol. 3, 274–277. doi: 10.1038/nchembio875
Cho, J. S., Choi, K. R., Prabowo, C. P. S., Shin, J. H., Yang, D., Jang, J., et al. (2017). CRISPR/Cas9-coupled recombineering for metabolic engineering of Corynebacterium glutamicum. Metab. Eng. 42, 157–167. doi: 10.1016/j.ymben.2017.06.010
Datsenko, K. A., and Wanner, B. L. (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645. doi: 10.1073/pnas.120163297
Davidson, R. C., Blankenship, J. R., Kraus, P. R., de, Jesus Berrios M., Hull, C. M., D’Souza, C., et al. (2002). A PCR-based strategy to generate integrative targeting alleles with large regions of homology. Microbiology 148, 2607–2615. doi: 10.1099/00221287-148-8-2607
Farasat, I., Kushwaha, M., Collens, J., Easterbrook, M., Guido, M., and Salis, H. M. (2014). Efficient search, mapping, and optimization of multi-protein genetic systems in diverse bacteria. Mol. Syst. Biol. 10, 731–749. doi: 10.15252/msb.20134955
Hernández-Montalvo, V., Martínez, A., Hernández-Chavez, G., Bolivar, F., Valle, F., and Gosset, G. (2003). Expression of galP and glk in a Escherichia coli PTS mutant restores glucose transport and increases glycolytic flux to fermentation products. Biotechnol. Bioeng 83, 687–694. doi: 10.1002/bit.10702
Ji, D., Wang, L., Hou, S., Liu, W., Wang, J., Wang, Q., et al. (2011). Creation of bioorthogonal redox systems depending on nicotinamide flucytosine dinucleotide. J. Am. Chem. Soc. 133, 20857–20862. doi: 10.1021/ja2074032
Li, J., Mutanda, I., Wang, K., Yang, L., Wang, J., and Wang, Y. (2019). Chloroplastic metabolic engineering coupled with isoprenoid pool enhancement for committed taxanes biosynthesis in Nicotiana benthamiana. Nat. Commun. 10:4850. doi: 10.1038/s41467-019-12879-y
Madyagol, M., Al-Alami, H., Levarski, Z., Drahovska, H., and Turna, J. (2011). Gene replacement techniques for Escherichia coli genome modification. Folia Microbiol. 56, 253–263. doi: 10.1007/s12223-011-0035-z
Patel, R. D., Lodge, J. K., and Baker, L. G. (2010). Going green in Cryptococcus neoformans: the recycling of a selectable drug marker. Fungal Genet. Biol. 47, 191–198. doi: 10.1016/j.fgb.2009.11.007
Song, C. W., and Lee, S. Y. (2013). Rapid one-step inactivation of single or multiple genes in Escherichia coli. Biotechnol. J. 8, 776–784. doi: 10.1002/biot.201300153
Studier, F. W. (2005). Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41, 207–234. doi: 10.1016/j.pep.2005.01.016
Usui, Y., Hirasawa, T., Furusawa, C., Shirai, T., Yamamoto, N., Mori, H., et al. (2012). Investigating the effects of perturbations to pgi and eno gene expression on central carbon metabolism in Escherichia coli using C-13 metabolic flux analysis. Microbial. Cell Fact. 11:87. doi: 10.1186/1475-2859-11-87
Wang, K., Fredens, J., Brunner, S. F., Kim, S. H., Chia, T., and Chin, J. W. (2016). Defining synonymous codon compression schemes by genome recoding. Nature 539, 59–64. doi: 10.1038/nature20124
Wang, J., Xie, J., and Schultz, P. G. (2006). A genetically encoded fluorescent amino acid. J. Am. Chem. Soc 128, 8738–8739. doi: 10.1021/ja062666k
Wang, Y., Chen, N., Xu, X., Tao, Y., Xu, Y., and Liu, W. (2015). Novel efficient strategy for λ-Red-mediated gene knock-out/in in Escherichia coli using SCLM system. Microbiology China 42, 699–711. doi: 10.13344/j.microbiol.china.140339
Young, D. D., and Schultz, P. G. (2018). Playing with the Molecules of Life. ACS Chem. Biol. 13, 854–870. doi: 10.1021/acschembio.7b00974
Young, T. S., Ahmad, I., Yin, J. A., and Schultz, P. G. (2010). An enhanced system for unnatural amino acid mutagenesis in E. coli. J. Mol. Biol. 395, 361–374. doi: 10.1016/j.jmb.2009.10.030
Keywords: unnatural amino acids, Cre-mediated recombination system, antibiotic resistance gene, neurosporene, Escherichia coli
Citation: Xu X, Zhong H, Liu W and Tao Y (2020) Extension of Genetic Marker List Using Unnatural Amino Acid System: An Efficient Genomic Modification Strategy in Escherichia coli. Front. Bioeng. Biotechnol. 8:145. doi: 10.3389/fbioe.2020.00145
Received: 06 November 2019; Accepted: 12 February 2020;
Published: 28 April 2020.
Edited by:
Pablo Ivan Nikel, Novo Nordisk Foundation Center for Biosustainability (DTU Biosustain), DenmarkReviewed by:
Jiantao Guo, University of Nebraska–Lincoln, United StatesCopyright © 2020 Xu, Zhong, Liu and Tao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weifeng Liu, bGl1d2Z2QDE2My5jb20=; Yong Tao, dGFveW9uZ0BpbS5hYy5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.