 Zhibin Lv
Zhibin Lv Jun Zhang
Jun Zhang Hui Ding
Hui Ding Quan Zou
Quan Zou- 1Institute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China, Chengdu, China
- 2Rehabilitation Department, Heilongjiang Province Land Reclamation Headquarters General Hospital, Harbin, China
- 3Center for Informational Biology, University of Electronic Science and Technology of China, Chengdu, China
One of the ubiquitous chemical modifications in RNA, pseudouridine modification is crucial for various cellular biological and physiological processes. To gain more insight into the functional mechanisms involved, it is of fundamental importance to precisely identify pseudouridine sites in RNA. Several useful machine learning approaches have become available recently, with the increasing progress of next-generation sequencing technology; however, existing methods cannot predict sites with high accuracy. Thus, a more accurate predictor is required. In this study, a random forest-based predictor named RF-PseU is proposed for prediction of pseudouridylation sites. To optimize feature representation and obtain a better model, the light gradient boosting machine algorithm and incremental feature selection strategy were used to select the optimum feature space vector for training the random forest model RF-PseU. Compared with previous state-of-the-art predictors, the results on the same benchmark data sets of three species demonstrate that RF-PseU performs better overall. The integrated average leave-one-out cross-validation and independent testing accuracy scores were 71.4% and 74.7%, respectively, representing increments of 3.63% and 4.77% versus the best existing predictor. Moreover, the final RF-PseU model for prediction was built on leave-one-out cross-validation and provides a reliable and robust tool for identifying pseudouridine sites. A web server with a user-friendly interface is accessible at http://148.70.81.170:10228/rfpseu.
Introduction
More than 150 types of chemical modification have been identified in cellular RNA, including adenosine methylation, cytosine modification, isomerization of uridine, and ribose modification (Boccaletto et al., 2018). These modifications have critical roles in cellular biological and physiological processes (Song and Yi, 2017). For instance, one of the most prevalent RNA modifications in eukaryotes, N6-methyladenosine (m6A), affects RNA stability (Wang et al., 2014), RNA-protein interaction (Liu et al., 2015b), RNA splicing and translation (Meyer and Jaffrey, 2014), the circadian clock (Fustin et al., 2013), immune response (Winkler et al., 2019), etc. Another widespread RNA modification is 5-methylcytosine (m5C), which has functions including preservation of the secondary structure of tRNA (Motorin and Helm, 2010), control of amino-acylation (Helm, 2006), codon identification and metabolic stability (Agris, 2008; Li et al., 2017). The pseudouridine modification is another common post-transcriptional modification in various living organisms (Zaringhalam and Papavasiliou, 2016). In 1951, pseudouridine was first identified, and experiments in 1960 revealed that it was abundant in tRNA and rRNA (Cohn, 1960). Pseudouridine results from an isomerization of uridine by breaking the glycosidic bond with 180° base rotation (Karijolich et al., 2015). This modification has been shown to have vital roles, for instance, in stabilizing RNA and in the stress response (Zhao and He, 2015; Cheng et al., 2019a; Wang et al., 2019b).
Although RNA pseudouridylation was discovered decades ago, the first transcriptome-wide RNA pseudouridylation map was not published until 2014, following the rapid development of next-generation sequencing technology (Goodwin et al., 2016). Carlile et al. (2014) developed the PseudoU-seq technology, which they used to identify more than 200 pseudouridylation sites in the regulated mRNA of yeast and human cells; in the same year, Schwartz et al. (2014) performed transcriptome-wide mapping using a similar protocol, finding more than 300 dynamic-regulated pseudouridine sites in non-coding RNA and mRNA. Li et al. (2015a) presented a chemical labeling method (CeU-Seq) that they used to pull down more than 2000 pseudouridine sites in human mRNA. Other RNA pseudouridylation sequencing protocols were also developed (Carlile et al., 2015).
As an alternative to costly and labor-intensive laboratory experiments, robust, swift, and inexpensive computational methods for RNA chemical modification prediction have emerged recently, owing to the increasing amount of data generated in this post-genomics era (Libbrecht and Noble, 2015). A large number of m6A (Chen et al., 2015, 2018a,b, 2019a; Zhou et al., 2016; Zhao et al., 2019; Zou et al., 2019) and m5C (Feng et al., 2016; Qiu et al., 2017; Li et al., 2018; Sabooh et al., 2018; Zhang et al., 2018; Yin et al., 2019) site predictors based on traditional machine learning and emerging deep learning algorithms have been proposed. However, few computational tools have been developed to predict pseudouridine sites. Li et al. (2015b) used a support vector machine (SVM) classifier to design a web server called PPUS for the identification of pseudouridine sites in Saccharomyces cerevisiae and Homo sapiens. Chen et al. (2016) constructed another SVM-based web server for pseudouridine site prediction, using the frequency composition of the nucleotides and pseudo K-tuple nucleotide composition (PseKNC) for feature representation. He et al. (2018) presented another model, PseUI, to identify pseudouridine sites in RNA sequences from three species (H. sapiens, S. cerevisiae, and M. musculus); this was an SVM-based model incorporating multiple feature-extraction technologies. Tahir et al. (2019) used convolutional neural networks to design a new predictor, iPseU-CNN; and Liu et al. (2019b) developed the eXtreme gradient boosting (XGboost) method for RNA pseudouridine site prediction (XG-PseU). Cross-validation scores for RNA pseudouridine site identification in the abovementioned three species showed the best accuracy for iPseU-CNN (66.9%) in H. sapiens, whereas XG-PseU and iPseU-CNN had the best accuracy (68.2%) in S. cerevisiae, and XG-PseU was the most accurate (72.0%) in M. musculus. According to independent testing scores, iPseU-CNN outperformed the other models, with 69.0% accuracy in H. sapiens and 73.6% accuracy in S. cerevisiae. Although the iPseU-CNN predictor had a high average cross-validation accuracy (68.9%) and independent testing accuracy (71.3%) scores, there was still room for improvement in comparison with some high-performing m6A site predictors (Chen et al., 2019a; Zou et al., 2019).
In this work, a model is developed based on the random forest algorithm, RF-PseU, for pseudouridine site recognition. The modeling overview is shown in Figure 1. RF-PseU incorporates multiple sequence feature representation technologies, and the light gradient boosting machine (LGBM) algorithm is employed to remove redundant features and rank the remaining features. Evaluation with leave-one-out (LOO) cross-validation demonstrated the robustness of the model. The average cross-validation accuracy (71.3% for 10-Fold and 71.4% for LOO) of RF-PseU was improved by 3.48–10.3% compared with existing state-of-the-art predictors, and the average independent testing accuracy (74.7%) showed a 4.8–19% increase. A user-friendly web server was also implemented, which can be accessed at http://148.70.81.170:10228/rfpseu. RF-PseU is expected to be a useful supplement to the existing tools for pseudouridine site identification.
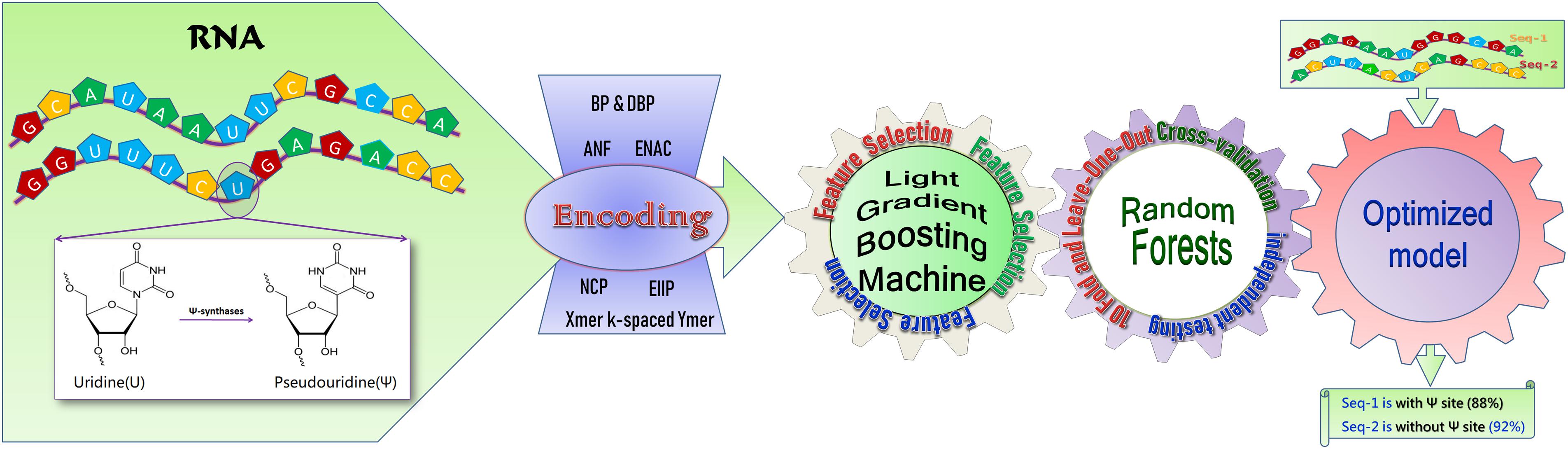
Figure 1. A schematic diagram of RF-PseU. RNA sequences with or without pseudouridine sites were encoded via seven RNA coding technologies; following removal of redundant features by light gradient boosting machine feature selection, the random forest model was trained on smaller but more relevant feature vector spaces, and was evaluated through cross-validation and independent testing to obtain an optimized model for prediction.
Materials and Methods
Data Sets
Given that there were small differences between the benchmark data sets used in the studies of Chen et al. (2018a) and Liu et al. (2019b), data sets obtained from Chen et al. (2018a) were used to train and test our models. The training data sets included data for three species. That is, H. sapiens training dataset with 495 psedouridine-sites-containing sequences and 495 non-psedouridine-sites-containing; S. cerevisiae training dataset contains 314 psedouridine-sites-sequences and 314 non-psedouridine-sites-sequences; M. musculus training dataset consists of 944 sequences, half of which is positive samples. Whereas the independent testing data sets covered only two species, H. sapiens and S. cerevisiae, both of which contain 100 positive samples and 100 negative samples. For the H. sapiens and M. musculus data sets, the window size was 21, i.e. the positive samples were psedouridine site centroid sequences of 21 base pairs each, whereas those for the S. cerevisiae samples window site was 31, with psedouridine site centroid sequences of 31 base pairs. Negative samples, in which no psedouridine sites were detected, consisted of 21 base pairs for H. sapiens and M. musculus, and 31 base pairs for S. cerevisiae. The benchmark data sets can be downloaded from http://lin-group.cn/server/iRNAPseu/data.
Feature Representation
Several widely used and convenient bio-sequence feature representation tools have been developed (Mrozek et al., 2013; Liu et al., 2015a, 2019c; Yu et al., 2015, 2019; Cheng and Hu, 2018; Hu et al., 2019; Muhammod et al., 2019). The two main tools used in this work were iLearn (Hu et al., 2019) and PyFeat (Muhammod et al., 2019).
Nucleotide Binary Profiles
Binary profiles encode the four bases (ACGU) as (1,0,0,0), (0,1,0,0), (0,0,1,0), and (0,0,0,1), whereas dibinary profiles encode the 16 dinucleotides (AA, AC, AG, AU, CA, CC, CG, CU, GA, GC, GG, GU, UA, UC, UG, and UU) as (0,0,0,0), (0,0,0,1), (0,0,1,0), (0,0,1,1),…, (1,1,1,1).
Accumulated Nucleotide Frequency
Suppose si is a base (ACGU) at the ith position of a RNA sequence. Then we can determine the si density di of the ith prefix subsequence of a RNA sequence as follows:
where L is the sequence length and q is one of the four nucleotides (ACGU).
Nucleotide Chemical Properties
The four RNA nucleotides (ACGU) are different from each other in terms of chemical structure and chemical bonds. On the basis of these differences, AGCU can be categorized into three different classes (Table 1) and encoded using a three-dimensional coordinate, i.e. A is denoted by (1,1,1), C by (0,1,0), G by (1,0,0), and U by (0,0,1).

Table 1. ACGU categories based on chemical properties.
Electron-Ion Interaction Pseudopotentials (EIIP)
Nair and Sreenadhan (2006) used the EIIP values of A, G, C, and T (A: 0.1260, G: 0.0806, C: 0.1340, T: 0.1335) to directly represent the nucleotides in a DNA sequence. Here, iLearn was used to encode each nucleotide in the RNA sequences into EIIP feature vectors.
Enhanced Nucleic Acid Composition
The nucleotide composition was calculated for a fixed-length window of the RNA sequence, allowing the fixed window (length = 5) to continuously slide from the 5′ to the 3′ terminus. RNA sequences were then encoded into feature vectors of equal length.
Xmer k-Spaced Ymer Composition Frequency
This method is used to count the composition of a subsequence of X and Y consecutive nucleotides with intervals k, e.g. AGU@AU, A@CU, GU@@@A, where @ indicates a one-interval space, @@ a two-interval space, and so on. Generally, using Xmer k-spaced Ymer to encode an RNA sequence will generate a 4X × 4Y feature vector. In this study, X, Y, and k were set to 1, 2, or 3; and eight XYK combinations (except for 3mer-kspaced-3mer) were used for encoding. The PyFeat tool developed by Rafsanjani et al. (Muhammod et al., 2019) was used to convert RNA sequences into vectors.
Feature Selection
Feature selection is an effective way to remove redundant information and prevent over-fitting in machine learning modeling (Tang et al., 2017; Xu et al., 2018a; Cheng et al., 2019a; Liu, 2019; Sun et al., 2019; Yu et al., 2019). Several feature selection technologies, including ANOVA (Lv et al., 2019b) and MRMD (Zou et al., 2016), have been developed and are widely used for DNA, RNA, and protein identification (Xu et al., 2018b). In this work, an LGBM (Ke et al., 2017)1 wrapper was used to select appropriate feature spaces for model training. In this process, raw training data were fed into the LGBM model and their features were ranked by importance value as calculated with the LGBM algorithm. Features with importance values greater than the average were selected to compose the feature space for modeling.
Model Evaluation Metrics and Methods
The proposed models were evaluated by five commonly used metrics, accuracy (ACC), sensitivity (Sn), specificity (Sp), Matthew correlation coefficient (MCC), and integral area under the receiver operating characteristic curve (auROC). These metrics were calculated using the following equations, where TP, TN, FP, and FN stand for true positive, true negative, false positive, and false negative, respectively (Cheng et al., 2016, 2019b,c; Wei et al., 2017d, e; Liu et al., 2019a). For the ROC curve, 1-specificity was plotted on the horizontal axis, and sensitivity on the vertical axis.
LOO, K-Fold cross-validation, and independent testing are the most widely used methods for predictor evaluation (Mrozek et al., 2015; Cao and Cheng, 2016; Chen et al., 2017, 2018a, 2019b; Pan et al., 2017; He et al., 2018, 2019; Jiang et al., 2018; Xiong et al., 2018; Yu et al., 2018; Zhang et al., 2018; Ding et al., 2019; Feng et al., 2019; Kong and Zhang, 2019; Li and Liu, 2019; Lv et al., 2019a; Manavalan et al., 2019; Shan et al., 2019; Wang et al., 2019a; Wei et al., 2019a, b; Xu et al., 2019; Yu and Dai, 2019). That is the general machine learning evaluation methods (training, validation and testing) are used for optimized model evaluation. To test the efficiency of the classification, LOO cross-validation was performed for a data set containing n items, of which n-1 items were used for training and the remaining one was used for validation. This procedure was repeated until every sequence in the training data set had been used once as a validation testing sample. LOO cross-validation is robust but time-consuming for a large data set. To compare the performance of the model with that of existing predictors, 10-Fold cross-validation was also used. The training data set was stochastically divided into 10 subsets, with one subset for validation and the remaining nine for training. This process was repeated 10 times and the average results were used to evaluate the model. Finally, independent testing was performed to obtain a data set that was completely distinct from the training data set for evaluation of the trained model.
Algorithm
The random forest method is a bagging-type ensemble learning algorithm (Cheng et al., 2018a, b). By combining multiple weak classifiers, the final results can be voted or averaged to obtain an overall model with higher accuracy, better general performance, and resistance to overfitting. This algorithm has been extensively used in bioinformatics and other areas, and has been confirmed to be an effective modeling technique in various domains (Ding et al., 2016a,b; Mrozek et al., 2016; Qiu et al., 2016; Wang et al., 2017; Wei et al., 2017a,b,c; Yu et al., 2017a; Zheng et al., 2017; Tang et al., 2018, 2019a; Xue et al., 2018; Degenhardt et al., 2019; Xu et al., 2019). In this study, the scikit-learn toolkit, available at https://scikit-learn.org, was used to establish the models.
Support vector machine (Cortes and Vapnik, 1995) is a generalized linear classifier that classifies data based on supervised learning; its decision boundary is the maximum-margin hyperplane required to solve the learning sample. SVM has been widely used in a variety of fields (Xiong et al., 2012; Ding et al., 2017; Yu et al., 2017b; Fu et al., 2018; Fang et al., 2019; Lai et al., 2019; Meng et al., 2019; Shen et al., 2019; Tang et al., 2019b; Zhang et al., 2019; Zhu et al., 2019). Here, it was used for modeling comparisons. SVM was also implemented via the scikit-learn toolkit, using the Gaussian radial basis functions, with the critical hyper-parameters (C and γ) of SVM optimized in a range from 10–6 to 106 with exponent step 100.5.
Results and Discussion
Optimization With Different Feature Spaces
To determine optimal feature spaces, we first used the LGBM algorithm to sort the features from maximum to minimum according to their importance value. All the features with importance value greater than the average were kept. Second, we used an incremental feature selection strategy; as shown in Figure 2A, the 10-Fold cross-validation and independent testing accuracy varied as features were added. Initially, the accuracy increased rapidly for each species. As shown in Figure 2 (A1) and Figure 2 (A2), when the feature dimensions for H. sapiens and S. cerevisiae reached 257 and 397, the model achieved maximum independent testing accuracies of 75.0 and 77.0%, respectively. Owing to the lack of independent test data sets for M. musculus, Figure 2 (A3) shows only the cross-validation accuracy curve, with its peak value (74.8%) at a feature dimension of 161. The optimal feature space dimensions selected for each species were 257, 397, and 161, respectively. These values were used for further experiments and optimization.
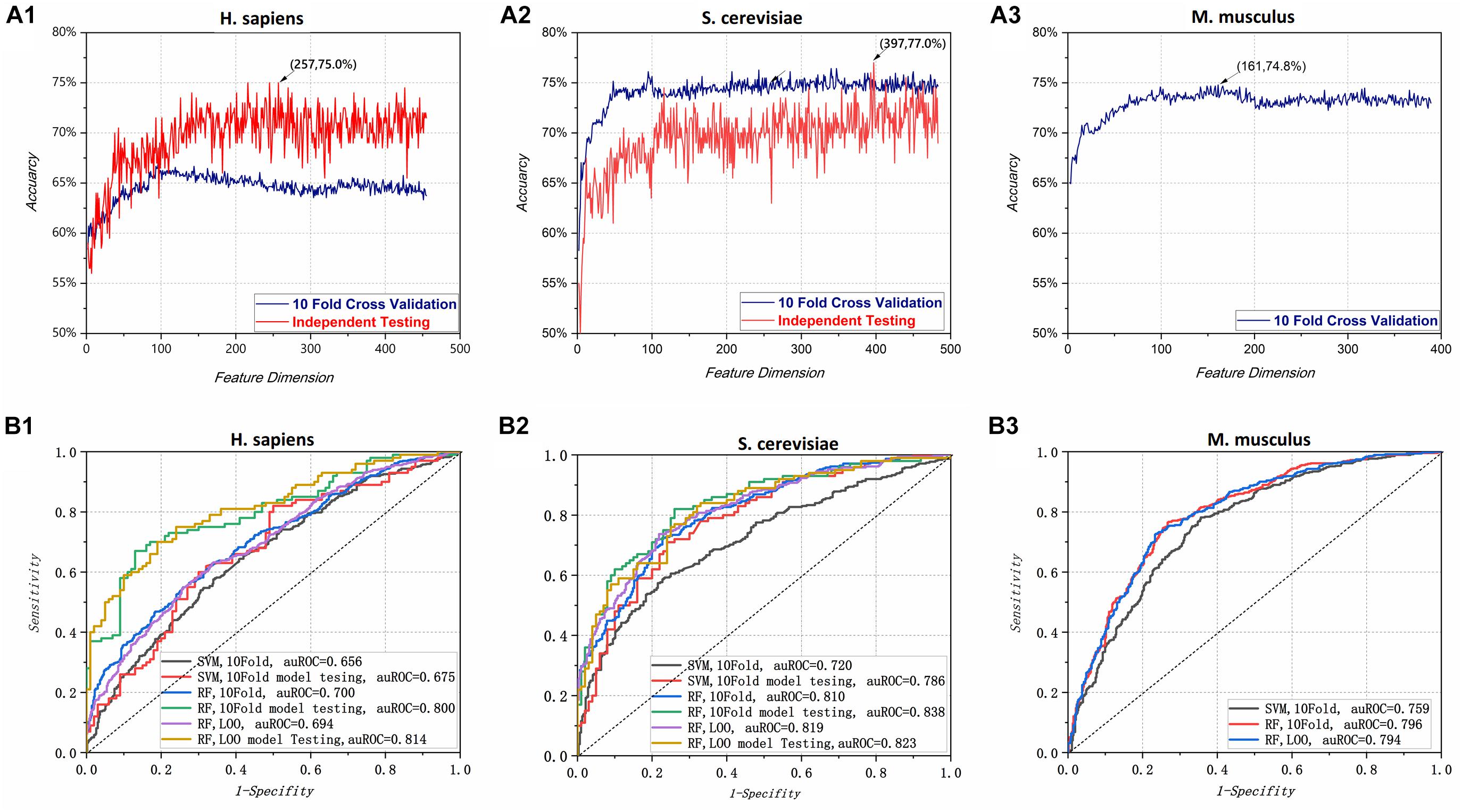
Figure 2. (A) Accuracy of the random forest predictor varied with feature dimension for all three species: (A1) H. sapiens; (A2) S. cerevisiae; (A3) M. musculus. The best independent accuracies for H. sapiens and S. cerevisiae were 75.0% with 257 features and 77.0% with 397 features, respectively, and the best 10-Fold cross-validated accuracy for M. musculus was 74.8% with 161 features. (B) Receiver operating characteristic curve (ROC) and area under the ROC curve (auROC) for different species under various conditions. (B1) is for H. sapiens, (B2) is for S. cerevisiae and (B3) is M. musculus. A support vector machine (SVM) was used for comparison with the random forest (RF) model. 10-Fold (10-Fold) model testing and leave-one-out (LOO) model testing indicate the model with the best 10-Fold and LOO cross-validation scores in independent testing. In cross-validation (10-Fold and LOO) and testing process, the training datasets have divided into training part and validation part. That is, they have used the general machine learning evaluation methods (training, validation and testing) for model optimization. In the figure, the 10-fold cross-validation and LOO cross-validation metric values are obtained from the validation part of training part, while the independent testing metric values are obtained from the independent testing datasets.
Comparison With SVM Predictors
Given that PPUS (Li et al., 2015b), iRNA-PseU (Chen et al., 2016), and PseUI (He et al., 2018) were all based on SVM, an optimized SVM model for pseudouridine site identification with the same feature spaces as the RF model was constructed to determine the effects of the SVM and RF on prediction performance. The performances of the two models are shown in Table 2. Overall, the models based on RF showed markedly better performance than those based on SVM. For instance, in terms of 10-Fold cross-validation accuracy, the RF models for H. sapiens, S. cerevisiae, and M. musculus outperformed the corresponding SVM models by 3.71%, 10.8%, and 5.80%, respectively. The independent testing accuracy scores showed an even greater contrast. For example, the RF model had 75.0% accuracy for H. sapiens, exactly 1.17 times that of the SVM model. The ROC curve and auROC value shown in Figure 2B also demonstrate that the optimized RF models performed better than the optimized SVM models for the same feature spaces. Thus, non-SVM models such as XG-PseU (Liu et al., 2019b), iPseU-CNN (Tahir et al., 2019), and our RF-PseU model might be more suitable for distinguishing pseudouridine sites from non- pseudouridine sites.
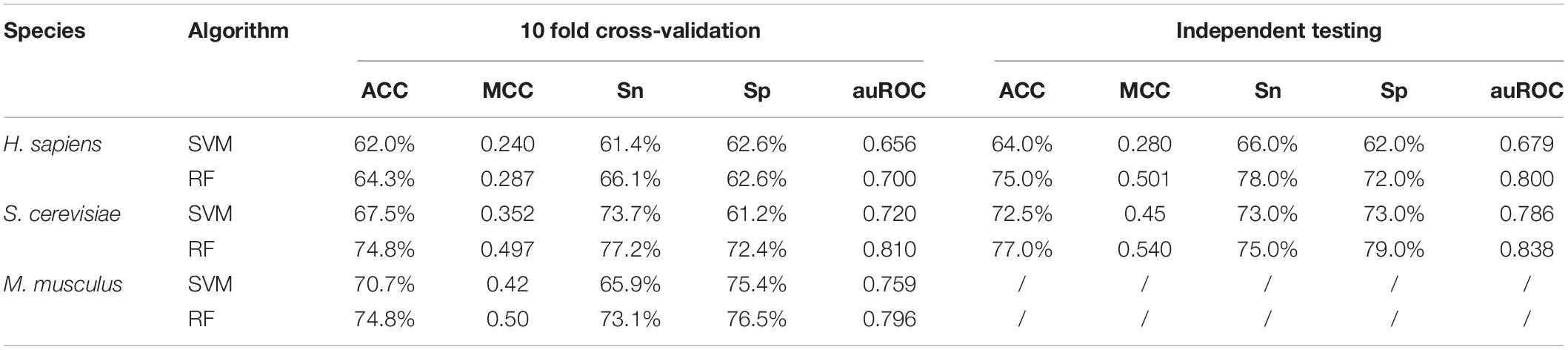
Table 2. Cross-validation and independent testing scores of two different classifiers for three species.
Comparison With Previous Predictors
The performance of RF-PseU was also compared with that of state-of-the-art predictors including iRNA-PseU (Chen et al., 2016), PseUI (He et al., 2018), iPseU-CNN (Tahir et al., 2019), and XG-PseU (Liu et al., 2019b). First, we compared the evaluation scores for the three species. Table 3 compares the cross-validation and independent testing scores for the state-of-the-art pseudouridine sites predictors with those of RF-PseU. In terms of cross-validation scores, the LOO accuracy values for S. cerevisiae and M. musculus were 75.4% and 74.5%, respectively, representing increments of approximately 10.5% and 3.47% over the values for the existing predictor (XG-PseU) with the best cross-validation score. However, the LOO accuracy of RF-PseU for H. sapiens, at 64.0%, showed a decrease of 4.0% compared with the best H. sapiens pseudouridine site predictor, PseU-CNN. In terms of independent testing, as shown in Table 3, RF-PseU scored higher than the existing predictors in all aspects. For comprehensive comparison, the average scores for different species were calculated. The results, shown in Table 4, demonstrate that RF-PseU performed better overall than the other four predictors. The cross-validation accuracy scores of RF-PseU were 3.48% higher than those of the best existing predictor, iPseU-CNN; in terms of independent testing scores, RF-PseU showed a marked improvement of 4.7–10.6% compared with iPseU-CNN. The overall performance of RF-PseU was also significantly better than those of the other predictors, indicating that RF-PseU can discriminate true pseudouridine sites from non-pseudouridine sites more precisely than the existing predictors.
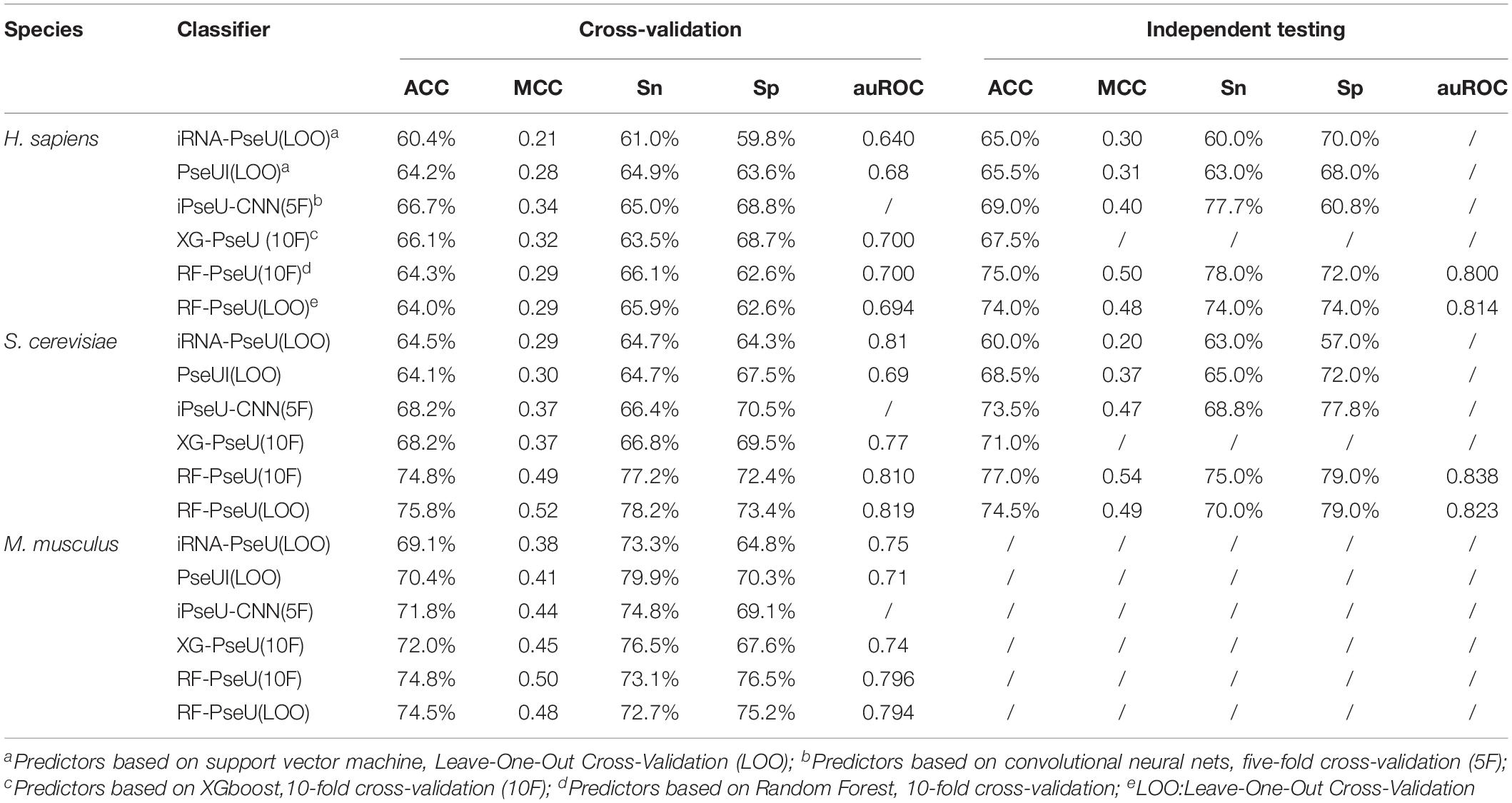
Table 3. Comparison of cross-validation and independent testing scores of existing state-of-the-art pseudouridine site predictors and RF-PseU.

Table 4. Comparison of average accuracies for state-of-the-art predictors.
Web Server Implementation
For convenience, a webserver with an easy-to-use interface was developed (see screenshot in Figure 3), which can be accessed freely at http://148.70.81.170:10228/rfpseu. A step-by-step user guide is given here. First, users select a species from the drop-down box and paste or type the query RNA sequences in FASTA format into the textbox. Second, after clicking the submit button, the query results will be shown in a table on the same page after a wait. Note that once a query task has been submitted, the submit button will be disabled. Third, the user can click the clear button to empty the input text box and enable the submit button, and return to step one to enter a new query task.
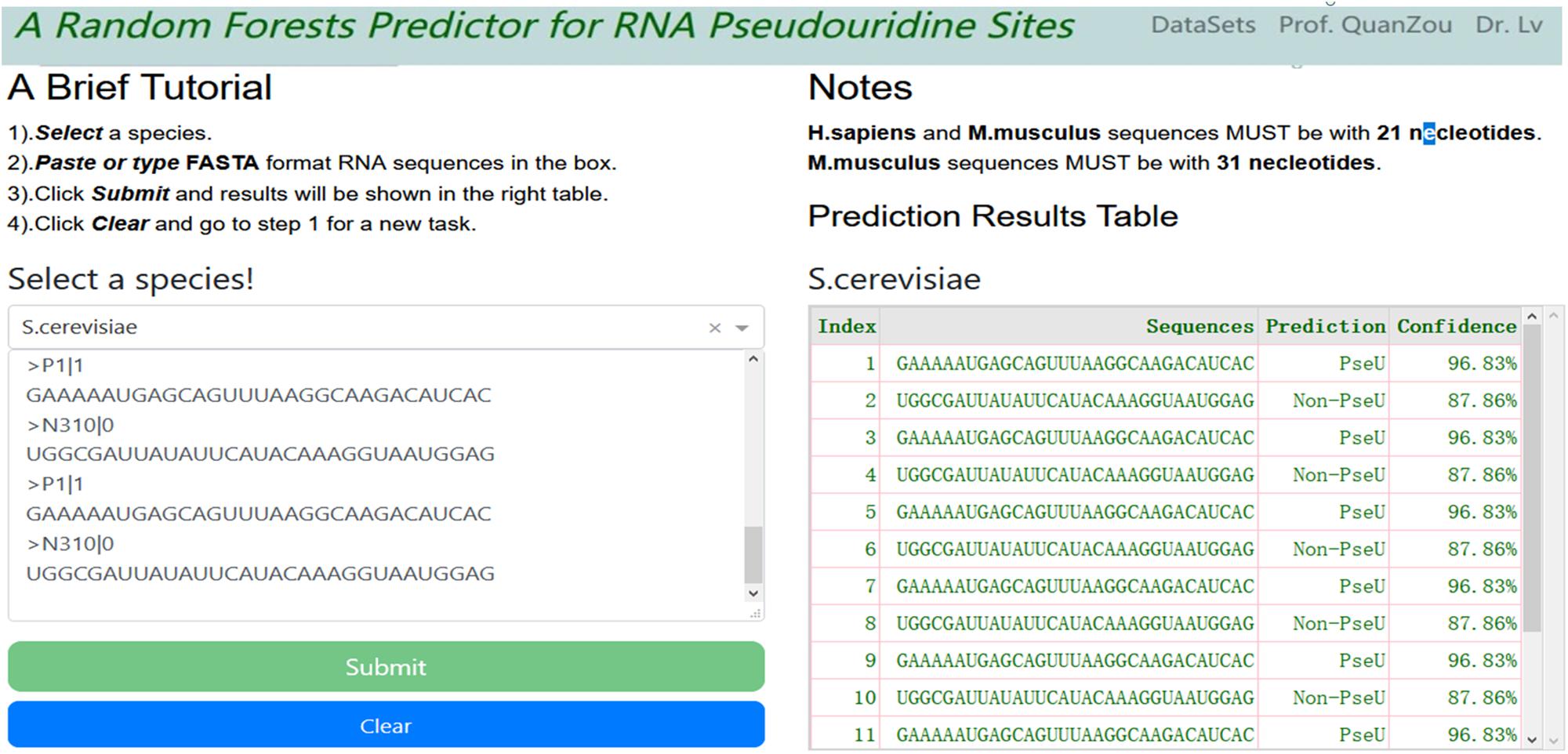
Figure 3. A screenshot of RF-PseU web server interface. The web server allows users to type or paste FASTA format text into the textbox and click submit button; the results are displayed in the right-hand table.
Conclusion
In this study, a new model, named RF-PseU, for predicting RNA pseudouridine sites in multiple species is presented. For given feature spaces, the random forest algorithm was shown to be more efficient than SVM models for discriminating pseudouridine sites from non-pseudouridine sites. In terms of average cross-validation and independent testing accuracy scores, RF-PseU showed improvements of 3.6–10% and 4.8–21%, respectively, compared with state-of-the-art predictors. Moreover, a web server with a user-friendly interface is available. It is anticipated that RF-PseU will be a useful tool for RNA pseudouridine site analysis. However, the model requires further development via combination with other technologies before it is suitable for use as a classifier for RNA pseudouridine sites. Future work will explore emerging methods such as Gene2Vec (Zou et al., 2019), m6Acomet (Wu et al., 2019), and iterative feature representation (Wei et al., 2019b) to improve the model’s performance.
Data Availability Statement
Publicly available datasets were analyzed in this study. This data can be found here: http://lin-group.cn/server/iRNAPseu/data.
Author Contributions
ZL and JZ were responsible for experiments and manuscripts preparation. HD participated in discussions. QZ worked as supervisor for all procedures.
Funding
The work was supported by the National Natural Science Foundation of China (Nos. 61922020, 61771331, and 91935302), and the Scientific Research Foundation in Shenzhen (JCYJ20170818100431895 and JCYJ20180306172207178).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
References
Agris, P. F. (2008). Bringing order to translation: the contributions of transfer RNA anticodon-domain modifications. Embo Rep. 9, 629–635. doi: 10.1038/embor.2008.104
Boccaletto, P., Machnicka, M. A., Purta, E., Piatkowski, P., Baginski, B., and Wirecki, T. K. (2018). MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res. 46, D303–D307. doi: 10.1093/nar/gkx1030
Cao, R., and Cheng, J. (2016). Protein single-model quality assessment by feature-based probability density functions. Sci. Rep. 6:23990. doi: 10.1038/srep23990
Carlile, T. M., Rojas-Duran, M. F., and Gilbert, W. V. (2015). Pseudo-seq: genome-wide detection of pseudouridine modifications in RNA. Methods Enzymol. 560, 219–245. doi: 10.1016/bs.mie.2015.03.011
Carlile, T. M., Rojas-Duran, M. F., Zinshteyn, B., Shin, H., Bartoli, K. M., and Gilbert, W. V. (2014). Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature 515, 143–146. doi: 10.1038/nature13802
Chen, K. Q., Wei, Z., Zhang, Q., Wu, X. Y., Rong, R., Lu, Z. L., et al. (2019a). WHISTLE: a high-accuracy map of the human N-6-methyladenosine (m(6)A) epitranscriptome predicted using a machine learning approach. Nucleic Acids Res. 47:e41. doi: 10.1093/nar/gkz074
Chen, W., Ding, H., Zhou, X., Lin, H., and Chou, K. C. (2018a). iRNA(m6A)-PseDNC: identifying N-6-methyladenosine sites using pseudo dinucleotide composition. Anal. Biochem. 561–562, 59–65. doi: 10.1016/j.ab.2018.09.002
Chen, W., Feng, P. M., Ding, H., Lin, H., and Chou, K. C. (2015). iRNA-Methyl: identifying N-6-methyladenosine sites using pseudo nucleotide composition. Anal. Biochem. 490, 26–33. doi: 10.1016/j.ab.2015.08.021
Chen, W., Feng, P. M., Yang, H., Ding, H., Lin, H., and Chou, K. C. (2018b). iRNA-3typeA: identifying three types of modification at RNA’s adenosine sites. Mol. Ther. Nucleic Acids 11, 468–474. doi: 10.1016/j.omtn.2018.03.012
Chen, W., Lv, H., Nie, F., and Lin, H. (2019b). i6mA-Pred: identifying DNA N6-methyladenine sites in the rice genome. Bioinformatics (Oxf. Engl.) 35, 2796–2800. doi: 10.1093/bioinformatics/btz015
Chen, W., Tang, H., Ye, J., Lin, H., and Chou, K. C. (2016). iRNA-PseU: identifying RNA pseudouridine sites. Mol. Ther. Nucleic Acids 5:e332. doi: 10.1038/mtna.2016.37
Chen, W., Yang, H., Feng, P., Ding, H., and Lin, H. (2017). iDNA4mC: identifying DNA N-4-methylcytosine sites based on nucleotide chemical properties. Bioinformatics 33, 3518–3523. doi: 10.1093/bioinformatics/btx479
Cheng, L., and Hu, Y. (2018). Human disease system biology. Curr. Gene Ther. 18, 255–256. doi: 10.2174/1566523218666181101143116
Cheng, L., Hu, Y., Sun, J., Zhou, M., and Jiang, Q. (2018a). DincRNA: a comprehensive web-based bioinformatics toolkit for exploring disease associations and ncRNA function. Bioinformatics 34, 1953–1956. doi: 10.1093/bioinformatics/bty002
Cheng, L., Jiang, Y., Ju, H., Sun, J., Peng, J., Zhou, M., et al. (2018b). InfAcrOnt: calculating cross-ontology term similarities using information flow by a random walk. BMC Genomics 19(Suppl. 1):919. doi: 10.1186/s12864-017-4338-6
Cheng, L., Qi, C., Zhuang, H., Fu, T., and Zhang, X. (2019a). gutMDisorder: a comprehensive database for dysbiosis of the gut microbiota in disorders and interventions. Nucleic Acids Res. 48(Suppl. 1):gkz843. doi: 10.1093/nar/gkz843
Cheng, L., Sun, J., Xu, W. Y., Dong, L. X., Hu, Y., and Zhou, M. (2016). OAHG: an integrated resource for annotating human genes with multi-level ontologies. Sci. Rep. 6, 1–9. doi: 10.1038/srep34820
Cheng, L., Yang, H., Zhao, H., Pei, X., Shi, H., Sun, J., et al. (2019b). MetSigDis: a manually curated resource for the metabolic signatures of diseases. Brief. Bioinform. 20, 203–209. doi: 10.1093/bib/bbx103
Cheng, L., Zhuang, H., Ju, H., Yang, S., Han, J., Tan, R., et al. (2019c). Exposing the causal effect of body mass index on the risk of type 2 diabetes mellitus: a mendelian randomization study. Front. Genet. 10:94. doi: 10.3389/fgene.2019.00094
Cohn, W. E. (1960). Pseudouridine, a carbon-carbon linked ribonucleoside in ribonucleic acids: isolation, structure, and chemical characteristics. J. Biol. Chem. 235, 1488–1498. doi: 10.1002/jbmte.390020410
Cortes, C., and Vapnik, V. (1995). Support-vector networks. Mach. Learn. 20, 273–297. doi: 10.1007/BF00994018
Degenhardt, F., Seifert, S., and Szymczak, S. (2019). Evaluation of variable selection methods for random forests and omics data sets. Brief. Bioinform. 20, 492–503. doi: 10.1093/bib/bbx124
Ding, Y., Tang, J., and Guo, F. (2016a). Identification of protein–protein interactions via a novel matrix-based sequence representation model with amino acid contact information. Int. J. Mol. Sci. 17:1623. doi: 10.3390/ijms17101623
Ding, Y., Tang, J., and Guo, F. (2016b). Predicting protein-protein interactions via multivariate mutual information of protein sequences. BMC Bioinformatics 17:398. doi: 10.1186/s12859-016-1253-9
Ding, Y., Tang, J., and Guo, F. (2017). Identification of drug-target interactions via multiple information integration. Inf. Sci. 418–419, 546–560. doi: 10.1016/j.ins.2017.08.045
Ding, Y., Tang, J., and Guo, F. (2019). Identification of drug-side effect association via multiple information integration with centered kernel alignment. Neurocomputing 325, 211–224. doi: 10.1016/j.neucom.2018.10.028
Fang, T., Zhang, Z., Sun, R., Zhu, L., He, J., Huang, B., et al. (2019). RNAm5CPred: prediction of RNA 5-methylcytosine sites based on three different kinds of nucleotide composition. Mol. Ther. Nucleic Acids 18, 739–747. doi: 10.1016/j.omtn.2019.10.008
Feng, P., Yang, H., Ding, H., Lin, H., Chen, W., and Chou, K.-C. (2019). iDNA6mA-PseKNC: identifying DNA N-6-methyladenosine sites by incorporating nucleotide physicochemical properties into PseKNC. Genomics 111, 96–102. doi: 10.1016/j.ygeno.2018.01.005
Feng, P. M., Ding, H., Chen, W., and Lin, H. (2016). Identifying RNA 5-methylcytosine sites via pseudo nucleotide compositions. Mol. Biosyst. 12, 3307–3311. doi: 10.1039/c6mb00471g
Fu, J., Tang, J., Wang, Y., Cui, X., Yang, Q., Hong, J., et al. (2018). Discovery of the consistently well-performed analysis chain for SWATH-MS Based pharmacoproteomic quantification. Front. Pharmacol. 9:681. doi: 10.3389/fphar.2018.00681
Fustin, J.-M., Doi, M., Yamaguchi, Y., Hida, H., Nishimura, S., Yoshida, M., et al. (2013). RNA-methylation-dependent RNA processing controls the speed of the circadian clock. Cell 155, 793–806. doi: 10.1016/j.cell.2013.10.026
Goodwin, S., McPherson, J. D., and McCombie, W. R. (2016). Coming of age: ten years of next-generation sequencing technologies. Nat. Rev. Genet. 17, 333–351. doi: 10.1038/nrg.2016.49
He, J. J., Fang, T., Zhang, Z. Z., Huang, B., Zhu, X. L., and Xiong, Y. (2018). PseUI: pseudouridine sites identification based on RNA sequence information. BMC Bioinformatics 19:11. doi: 10.1186/s12859-018-2321-0
He, W., Jia, C., and Zou, Q. (2019). 4mCPred: machine learning methods for DNA N-4-methylcytosine sites prediction. Bioinformatics 35, 593–601. doi: 10.1093/bioinformatics/bty668
Helm, M. (2006). Post-transcriptional nucleotide modification and alternative folding of RNA. Nucleic Acids Res. 34, 721–733. doi: 10.1093/nar/gkj471
Hu, Y., Zhao, T., Zhang, N., Zhang, Y., and Cheng, L. (2019). A review of recent advances and research on drug target identification methods. Curr. Drug Metab. 20, 209–216. doi: 10.2174/1389200219666180925091851
Jiang, L., Ding, Y., Tang, J., and Guo, F. (2018). MDA-SKF: similarity kernel fusion for accurately discovering miRNA-disease association. Front. Genet. 9:618. doi: 10.3389/fgene.2018.00618
Karijolich, J., Yi, C. Q., and Yu, Y. T. (2015). Transcriptome-wide dynamics of RNA pseudouridylation. Nat. Rev. Mol. Cell Biol. 16, 581–585. doi: 10.1038/nrm4040
Ke, G., Meng, Q., Finley, T., Wang, T., Chen, W., Ma, W., et al. (2017). “LightGBM: a highly efficient gradient boosting decision tree,” in Advances in Neural Information Processing Systems 30, eds I. Guyon, U. V. Luxburg, S. Bengio, H. Wallach, R. Fergus, and S. Vishwanathan, (Red Hook, NY: Curran Associates, Inc), 3146–3154.
Kong, L., and Zhang, L. (2019). i6mA-DNCP: computational identification of DNA N6-methyladenine sites in the rice genome using optimized dinucleotide-based features. Genes 10:828. doi: 10.3390/genes10100828
Lai, H. Y., Zhang, Z. Y., Su, Z. D., Su, W., Ding, H., Chen, W., et al. (2019). iProEP: a computational predictor for predicting promoter. Mol. Ther. Nucleic Acids 17, 337–346. doi: 10.1016/j.omtn.2019.05.028
Li, B., Tang, J., Yang, Q., Li, S., Cui, X., and Li, Y. (2017). NOREVA: normalization and evaluation of MS-based metabolomics data. Nucleic Acids Res. 45, W162–W170. doi: 10.1093/nar/gkx449
Li, C.-C., and Liu, B. (2019). MotifCNN-fold: protein fold recognition based on fold-specific features extracted by motif-based convolutional neural networks. Brief. Bioinform. bbz133. doi: 10.1093/bib/bbz133
Li, X. Y., Zhu, P., Ma, S. Q., Song, J. H., Bai, J. Y., Sun, F. F., et al. (2015a). Chemical pulldown reveals dynamic pseudouridylation of the mammalian transcriptome. Nat. Chem. Biol. 11, 592–597. doi: 10.1038/nchembio.1836
Li, Y. H., Zhang, G. G., and Cui, Q. H. (2015b). PPUS: a web server to predict PUS-specific pseudouridine sites. Bioinformatics 31, 3362–3364. doi: 10.1093/bioinformatics/btv366
Li, Y. Z., Fan, Y.-X., and Yang, H.-H. (2018). KELMPSP: pseudouridine sites identification based on kernel extreme learning machine. Chin. J. Biochem. Mol. Biol. 34, 785–793. doi: 10.13865/j.cnki.cjbmb2018.07.14
Libbrecht, M. W., and Noble, W. S. (2015). Machine learning applications in genetics and genomics. Nat. Rev. Genet. 16, 321–332. doi: 10.1038/nrg3920
Liu, B. (2019). BioSeq-Analysis: a platform for DNA, RNA, and protein sequence analysis based on machine learning approaches. Brief. Bioinform. 20, 1280–1294. doi: 10.1093/bib/bbx165
Liu, B., Li, C.-C., and Yan, K. (2019a). DeepSVM-fold: protein fold recognition by combining support vector machines and pairwise sequence similarity scores generated by deep learning networks. Brief. Bioinform. bbz098. doi: 10.1093/bib/bbz098
Liu, B., Liu, F. L., Wang, X. L., Chen, J. J., Fang, L. Y., and Chou, K. C. (2015a). Pse-in-One: a web server for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nucleic Acids Res. 43, W65–W71. doi: 10.1093/nar/gkv458
Liu, K., Chen, W., and Lin, H. (2019b). XG-PseU: an eXtreme gradient boosting based method for identifying pseudouridine sites. Mol. Genet. Genomics 295, 13–21. doi: 10.1007/s00438-019-01600-9
Liu, N., Dai, Q., Zheng, G., He, C., Parisien, M., and Pan, T. (2015b). N-6-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature 518, 560–564. doi: 10.1038/nature14234
Liu, S., Zheng, B., Sheng, Y., Kong, Q., Jiang, Y., Yang, Y., et al. (2019c). Identification of cancer dysfunctional subpathways by integrating DNA methylation, copy number variation, and gene-expression data. Front. Genet. 10:441. doi: 10.3389/fgene.2019.00441
Lv, H., Dao, F.-Y., Guan, Z.-X., Zhang, D., Tan, J.-X., and Jiu-Xin, Tan (2019a). iDNA6mA-Rice: a computational tool for detecting N6-methyladenine sites in rice. Front. Genet. 10:793. doi: 10.3389/fgene.2019.00793
Lv, Z., Jin, S., Ding, H., and Zou, Q. (2019b). A random forest sub-Golgi protein classifier optimized via dipeptide and amino acid composition features. Front. Bioeng. Biotechnol. 7:215. doi: 10.3389/fbioe.2019.00215
Manavalan, B., Basith, S., Shin, T. H., Wei, L., and Lee, G. (2019). Meta-4mCpred: a sequence-based meta-predictor for accurate DNA 4mC site prediction using effective feature representation. Mol. Ther. Nucleic Acids 16, 733–744. doi: 10.1016/j.omtn.2019.04.019
Meng, C. L., Jin, S. S., Wang, L., Guo, F., and Zou, Q. (2019). AOPs-SVM: a sequence-based classifier of antioxidant proteins using a support vector machine. Front. Bioeng. Biotechnol. 7:10. doi: 10.3389/fbioe.2019.00224
Meyer, K. D., and Jaffrey, S. R. (2014). The dynamic epitranscriptome: N-6-methyladenosine and gene expression control. Nat. Rev. Mol. Cell Biol. 15, 313–326. doi: 10.1038/nrm3785
Motorin, Y., and Helm, M. (2010). tRNA stabilization by modified nucleotides. Biochemistry 49, 4934–4944. doi: 10.1021/bi100408z
Mrozek, D., Gosk, P., and Malysiak-Mrozek, B. (2015). Scaling Ab initio predictions of 3D protein structures in microsoft azure cloud. J. Grid Comput. 13, 561–585. doi: 10.1007/s10723-015-9353-8
Mrozek, D., Malysiak-Mrozek, B., and Siaznik, A. (2013). search GenBank: interactive orchestration and ad-hoc choreography of web services in the exploration of the biomedical resources of the national center For biotechnology information. BMC Bioinformatics 14:18. doi: 10.1186/1471-2105-14-73
Mrozek, D., Socha, B., Kozielski, S., and Malysiak-Mrozek, B. (2016). An efficient and flexible scanning of databases of protein secondary structures. J. Intell. Inf. Syst. 46, 213–233. doi: 10.1007/s10844-014-0353-0
Muhammod, R., Ahmed, S., Md Farid, D., Shatabda, S., Sharma, A., and Dehzangi, A. (2019). PyFeat: a Python-based effective feature generation tool for DNA, RNA and protein sequences. Bioinformatics (Oxf. Engl.) 35, 3831–3833. doi: 10.1093/bioinformatics/btz165
Nair, A. S., and Sreenadhan, S. P. (2006). A coding measure scheme employing electron-ion interaction pseudopotential (EIIP). Bioinformation 1, 197–202.
Pan, G., Tang, J., and Guo, F. (2017). Analysis of co-associated transcription factors via ordered adjacency differences on motif distribution. Sci. Rep. 7:43597. doi: 10.1038/srep43597
Qiu, W.-R., Jiang, S.-Y., Xu, Z.-C., Xiao, X., and Chou, K.-C. (2017). iRNAm5C-PseDNC: identifying RNA 5-methylcytosine sites by incorporating physical-chemical properties into pseudo dinucleotide composition. Oncotarget 8, 41178–41188. doi: 10.18632/oncotarget.17104
Qiu, W. R., Sun, B. Q., Xiao, X., Xu, Z. C., and Chou, K. C. (2016). iPTM-mLys: identifying multiple lysine PTM sites and their different types. Bioinformatics 32, 3116–3123. doi: 10.1093/bioinformatics/btw380
Sabooh, M. F., Iqbal, N., Khan, M., Khan, M., and Maqbool, H. F. (2018). Identifying 5-methylcytosine sites in RNA sequence using composite encoding feature into Chou’s PseKNC. J. Theor. Biol. 452, 1–9. doi: 10.1016/j.jtbi.2018.04.037
Schwartz, S., Bernstein, D. A., Mumbach, M. R., Jovanovic, M., Herbst, R. H., and León-Ricardo, B. X. (2014). Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell 159, 148–162. doi: 10.1016/j.cell.2014.08.028
Shan, X., Wang, X., Li, C. D., Chu, Y., Zhang, Y., and Xiong, Y. I. (2019). Prediction of CYP450 enzyme-substrate selectivity based on the network-based label space division method. J. Chem. Inf. Model. 59, 4577–4586. doi: 10.1021/acs.jcim.9b00749
Shen, Y., Tang, J., and Guo, F. (2019). Identification of protein subcellular localization via integrating evolutionary and physicochemical information into Chou’s general PseAAC. J. Theor. Biol. 462, 230–239. doi: 10.1016/j.jtbi.2018.11.012
Song, J. H., and Yi, C. Q. (2017). Chemical modifications to RNA: a new layer of gene expression regulation. ACS Chem. Biol. 12, 316–325. doi: 10.1021/acschembio.6b00960
Sun, W., Han, Y., Yang, S., Zhuang, H., Zhang, J., and Cheng, L. (2019). The assessment of Interleukin-18 on the risk of coronary heart disease. Med. Chem. doi: 10.2174/1573406415666191004115128 [Epub ahead of print].
Tahir, M., Tayara, H., and Chong, K. T. (2019). iPseU-CNN: identifying RNA pseudouridine sites using convolutional neural networks. Mol. Ther. Nucleic Acids 16, 463–470. doi: 10.1016/j.omtn.2019.03.010
Tang, H., Cao, R.-Z., Wang, W., Liu, T.-S., Wang, L.-M., and He, C.-M. (2017). A two-step discriminated method to identify thermophilic proteins. Int. J. Biomath. 10:1750050. doi: 10.1142/s1793524517500504
Tang, J., Fu, J., Wang, Y., Li, B., Li, Y., and Yang, Q. (2019a). ANPELA: analysis and performance assessment of the label-free quantification workflow for metaproteomic studies. Brief. Bioinform. 10:bby127. doi: 10.1093/bib/bby127
Tang, J., Fu, J., Wang, Y., Luo, Y., Yang, Q., Li, B., et al. (2019b). Simultaneous improvement in the precision, accuracy, and robustness of label-free proteome quantification by optimizing data manipulation chains. Mol. Cell. Proteomics 18, 1683–1699. doi: 10.1074/mcp.RA118.001169
Tang, W., Wan, S. X., Yang, Z., Teschendorff, A. E., and Zou, Q. (2018). Tumor origin detection with tissue-specific miRNA and DNA methylation markers. Bioinformatics 34, 398–406. doi: 10.1093/bioinformatics/btx622
Wang, P., Zhang, X., Fu, T., Li, S., Li, B., Xue, W., et al. (2017). Differentiating physicochemical properties between addictive and nonaddictive ADHD drugs revealed by molecular dynamics simulation studies. ACS Chem. Neurosci. 8, 1416–1428. doi: 10.1021/acschemneuro.7b00173
Wang, X., Lu, Z., Gomez, A., Hon, G. C., Yue, Y., and Han, D. (2014). N-6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505, 117–120. doi: 10.1038/nature12730
Wang, X., Zhu, X., Ye, M., Wang, Y., Li, C.-D., and Xiong, Y. (2019a). STS-NLSP: a network-based label space partition method for predicting the specificity of membrane transporter substrates using a hybrid feature of structural and semantic similarity. Front. Bioeng. Biotechnol. 7:306. doi: 10.3389/fbioe.2019.00306
Wang, Y., Zhang, S., Li, F., Zhou, Y., Zhang, Y., and Wang, Z. (2019b). Therapeutic target database 2020: enriched resource for facilitating research and early development of targeted therapeutics. Nucleic Acids Res. 48, D1031–D1041. doi: 10.1093/nar/gkz981
Wei, L., Luan, S., Nagai, L. A. E., Su, R., and Zou, Q. (2019a). Exploring sequence-based features for the improved prediction of DNA N4-methylcytosine sites in multiple species. Bioinformatics 35, 1326–1333. doi: 10.1093/bioinformatics/bty824
Wei, L., Su, R., Luan, S., Liao, Z., Manavalan, B., Zou, Q., et al. (2019b). Iterative feature representations improve N4-methylcytosine site prediction. Bioinformatics 35, 4930–4937. doi: 10.1093/bioinformatics/btz408
Wei, L., Xing, P., Su, R., Shi, G., Ma, Z., and Zou, Q. (2017a). CPPred-RF: a sequence-based predictor for identifying cell-penetrating peptides and their uptake efficiency. J. Proteome Res. 16, 2044–2053. doi: 10.1021/acs.jproteome.7b00019
Wei, L., Xing, P., Tang, J., and Zou, Q. (2017b). PhosPred-RF: a novel sequence-based predictor for phosphorylation sites using sequential information only. IEEE Trans. Nanobioscience 16, 240–247. doi: 10.1109/TNB.2017.2661756
Wei, L. Y., Tang, J. J., and Zou, Q. (2017c). Local-DPP: an improved DNA-binding protein prediction method by exploring local evolutionary information. Inf. Sci. 384, 135–144. doi: 10.1016/j.ins.2016.06.026
Wei, L. Y., Wan, S. X., Guo, J. S., and Wong, K. K. L. (2017d). A novel hierarchical selective ensemble classifier with bioinformatics application. Artif. Intell. Med. 83, 82–90. doi: 10.1016/j.artmed.2017.02.005
Wei, L. Y., Xing, P. W., Zeng, J. C., Chen, J. X., Su, R., and Guo, F. (2017e). Improved prediction of protein-protein interactions using novel negative samples, features, and an ensemble classifier. Artif. Intell. Med. 83, 67–74. doi: 10.1016/j.artmed.2017.03.001
Winkler, R., Gillis, E., Lasman, L., Safra, M., Geula, S., and Soyris, C. (2019). m(6)A modification controls the innate immune response to infection by targeting type I interferons. Nat. Immunol. 20, 173–182. doi: 10.1038/s41590-018-0275-z
Wu, X. Y., Wei, Z., Chen, K. Q., Zhang, Q., Su, J. L., Liu, H., et al. (2019). m6Acomet: large-scale functional prediction of individual m(6)A RNA methylation sites from an RNA co-methylation network. BMC Bioinformatics 20:223. doi: 10.1186/s12859-019-2840-3
Xiong, Y., Liu, J., Zhang, W., and Zeng, T. (2012). Prediction of heme binding residues from protein sequences with integrative sequence profiles. Proteome Sci. 10(Suppl. 1):S20. doi: 10.1186/1477-5956-10-S1-S20
Xiong, Y., Wang, Q., Yang, J., Zhu, X., and Wei, D. Q. (2018). PredT4SE-Stack: prediction of bacterial Type IV secreted effectors from protein sequences using a stacked ensemble method. Front. Microbiol. 9:2571. doi: 10.3389/fmicb.2018.02571
Xu, L., Liang, G., Liao, C., Chen, G.-D., and Chang, C.-C. (2018a). An efficient classifier for Alzheimer’s disease genes identification. Molecules 23:3140. doi: 10.3390/molecules23123140
Xu, L., Liang, G., Liao, C., Chen, G.-D., and Chang, C.-C. (2019). k-Skip-n-Gram-RF: a random forest based method for Alzheimer’s disease protein identification. Front. Genet. 10:33. doi: 10.3389/fgene.2019.00033
Xu, L., Liang, G., Wang, L., and Liao, C. (2018b). A novel hybrid sequence-based model for identifying anticancer peptides. Genes 9:158. doi: 10.3390/genes9030158
Xue, W., Yang, F., Wang, P., Zheng, G., Chen, Y., Yao, X., et al. (2018). What contributes to serotonin-norepinephrine reuptake inhibitors’ dual-targeting mechanism? The key role of transmembrane domain 6 in human serotonin and norepinephrine transporters revealed by molecular dynamics simulation. ACS Chem. Neurosci. 9, 1128–1140. doi: 10.1021/acschemneuro.7b00490
Yin, J., Sun, W., Li, F., Hong, J., Li, X., and Zhou, Y. (2019). VARIDT 1.0: variability of drug transporter database. Nucleic Acids Res. 48, D1042–D1050. doi: 10.1093/nar/gkz779
Yu, H., and Dai, Z. (2019). SNNRice6mA: a deep learning method for predicting DNA N6-methyladenine sites in rice genome. Front. Genet. 10:1071. doi: 10.3389/fgene.2019.01071
Yu, L., Huang, J. B., Ma, Z. X., Zhang, J., Zou, Y. P., and Gao, L. (2015). Inferring drug-disease associations based on known protein complexes. BMC Med. Genomics 8:13. doi: 10.1186/1755-8794-8-s2-s2
Yu, L., Su, R., Wang, B., Zhang, L., Zou, Y., Zhang, J., et al. (2017a). Prediction of novel drugs for hepatocellular carcinoma based on multi-source random walk. IEEE ACM Trans. Comput. Biol. Bioinform. 14, 966–977. doi: 10.1109/tcbb.2016.2550453
Yu, L., Yao, S., Gao, L., and Zha, Y. (2019). Conserved disease modules extracted from multilayer heterogeneous disease and gene networks for understanding disease mechanisms and predicting disease treatments. Front. Genet. 9:745. doi: 10.3389/fgene.2018.00745
Yu, L., Zhao, J., and Gao, L. (2017b). Drug repositioning based on triangularly balanced structure for tissue-specific diseases in incomplete interactome. Artif. Intell. Med. 77, 53–63. doi: 10.1016/j.artmed.2017.03.009
Yu, L., Zhao, J., and Gao, L. (2018). Predicting potential drugs for breast cancer based on miRNA and tissue specificity. Int. J. Biol. Sci. 14, 971–980. doi: 10.7150/ijbs.23350
Zaringhalam, M., and Papavasiliou, F. N. (2016). Pseudouridylation meets next-generation sequencing. Methods 107, 63–72. doi: 10.1016/j.ymeth.2016.03.001
Zhang, M., Li, F. Y., Marquez-Lago, T. T., Leier, A., Fan, C., Kwoh, C. K., et al. (2019). MULTiPly: a novel multi-layer predictor for discovering general and specific types of promoters. Bioinformatics 35, 2957–2965. doi: 10.1093/bioinformatics/btz016
Zhang, M., Xu, Y., Li, L., Liu, Z., Yang, X., and Yu, D.-J. (2018). Accurate RNA 5-methylcytosine site prediction based on heuristic physical-chemical properties reduction and classifier ensemble. Anal. Biochem. 550, 41–48. doi: 10.1016/j.ab.2018.03.027
Zhao, B. S., and He, C. (2015). Pseudouridine in a new era of RNA modifications. Cell Res. 25, 153–154. doi: 10.1038/cr.2014.143
Zhao, X. W., Zhang, Y., Ning, Q., Zhang, H. R., Ji, J. C., and Yin, M. H. (2019). Identifying N-6-methyladenosine sites using extreme gradient boosting system optimized by particle swarm optimizer. J. Theor. Biol. 467, 39–47. doi: 10.1016/j.jtbi.2019.01.035
Zheng, G., Xue, W., Yang, F., Zhang, Y., Chen, Y., Yao, X., et al. (2017). Revealing vilazodone’s binding mechanism underlying its partial agonism to the 5-HT1A receptor in the treatment of major depressive disorder. Phys. Chem. Chem. Phys. 19, 28885–28896. doi: 10.1039/c7cp05688e
Zhou, Y., Zeng, P., Li, Y. H., Zhang, Z. D., and Cui, Q. H. (2016). SRAMP: prediction of mammalian N-6-methyladenosine (m(6)A) sites based on sequence-derived features. Nucleic Acids Res. 44:e91. doi: 10.1093/nar/gkw104
Zhu, X., He, J., Zhao, S., Tao, W., Xiong, Y., and Bi, S. (2019). A comprehensive comparison and analysis of computational predictors for RNA N6-methyladenosine sites of Saccharomyces cerevisiae. Brief. Funct. Genomics 18, 367–376. doi: 10.1093/bfgp/elz018
Zou, Q., Xing, P. W., Wei, L. Y., and Liu, B. (2019). Gene2vec: gene subsequence embedding for prediction of mammalian N-6-methyladenosine sites from mRNA. RNA 25, 205–218. doi: 10.1261/rna.069112.118
Keywords: pseudouridine sites, light gradient boosting, random forest, machine learning, RNA
Citation: Lv Z, Zhang J, Ding H and Zou Q (2020) RF-PseU: A Random Forest Predictor for RNA Pseudouridine Sites. Front. Bioeng. Biotechnol. 8:134. doi: 10.3389/fbioe.2020.00134
Received: 17 January 2020; Accepted: 10 February 2020;
Published: 26 February 2020.
Edited by:
Yongchun Zuo, Inner Mongolia University, ChinaReviewed by:
Yi Xiong, Shanghai Jiao Tong University, ChinaHongmin Cai, South China University of Technology, China
Copyright © 2020 Lv, Zhang, Ding and Zou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Zhang, emhhbmdqdW4xMzkwMjAwM0AxNjMuY29t; Quan Zou, em91cXVhbkBuY2xhYi5uZXQ=