
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Bioeng. Biotechnol. , 10 December 2019
Sec. Computational Genomics
Volume 7 - 2019 | https://doi.org/10.3389/fbioe.2019.00364
This article is part of the Research Topic Computational Epigenetics in Human Diseases, Cell Differentiation, and Cell Reprogramming, Volume I View all 42 articles
 Gang Liu1,2†
Gang Liu1,2† Lianlei Wang1,2†
Lianlei Wang1,2† Xinyu Wang3†Zihui Yan1,2Xinzhuang Yang2,4
Xinyu Wang3†Zihui Yan1,2Xinzhuang Yang2,4 Mao Lin1,2Sen Liu1,2Yuzhi Zuo1,2Yuchen Niu2,4Sen Zhao1,2Yanxue Zhao1,2Jianguo Zhang1,2,5Jianxiong Shen1,2,5Yipeng Wang1,2,5Guixing Qiu1,2,5Zhihong Wu2,4,5*
Mao Lin1,2Sen Liu1,2Yuzhi Zuo1,2Yuchen Niu2,4Sen Zhao1,2Yanxue Zhao1,2Jianguo Zhang1,2,5Jianxiong Shen1,2,5Yipeng Wang1,2,5Guixing Qiu1,2,5Zhihong Wu2,4,5* Nan Wu1,2,5* on behalf of the Deciphering Disorders Involving Scoliosis and Comorbidities (DISCO)
Study
Nan Wu1,2,5* on behalf of the Deciphering Disorders Involving Scoliosis and Comorbidities (DISCO)
StudyBackground: Adolescent idiopathic scoliosis (AIS) is a complex disease affecting a large number of teenagers, especially in female. This study reveals novel epigenetic perturbation to the pathogenesis of AIS.
Methods: A female monozygotic (MZ) twin pair discordant for AIS were examined for whole-exome sequencing and epigenome difference. Sets of differentially methylated regions (DMRs) were validated using MethylTarget™ method in 20 AIS female patients and 20 healthy female controls.
Results: Few exome difference but several potential DMRs were found between the MZ twins. We identified 313 hypermethylated DMRs and 397 hypomethylated DMRs, respectively. Most of them were enriched in the MAPK and PI3K-Akt signaling pathway, which may contribute to the discordance of AIS. Several DMRs related to scoliosis genes were tested, and the NDN: TSS-DMR (chr15:23932133-23932304, hg19) was confirmed in additional samples. The methylation level of this DMR was significantly higher in the AIS group than in the control group (p = 0.04).
Conclusions: We described the epigenome difference in an AIS female discordant MZ twin pair using Whole Genome Bisulfite Sequencing (WGBS). The NDN: TSS-DMR had higher methylation level in female AIS, which can help elucidate the potential etiology of AIS.
Adolescent idiopathic scoliosis (AIS) is a 3-dimensional spinal deformity affecting 1–3% of children in the world (Yawn et al., 1999). Although studies to discover the underlying mechanisms of AIS had been conducted for many years, the etiology is still unclear. Previous studies showed that genetic factors from single nucleotide variant (SNV) to copy number variants (CNV), played a pivotal role in the development of AIS (Kesling and Reinker, 1997; Li et al., 2016; Gao et al., 2017). Based on genome-wide association study (GWAS) and other similar association studies, single nucleotide polymorphisms (SNPs) of several genes such as GPR126 (Kou et al., 2013; Liu et al., 2018), PAX1 (Sharma et al., 2015; Liu et al., 2019), LBX1 (Takahashi et al., 2011; Liu et al., 2017), and BNC2 (Ogura et al., 2015), were known to increase the risk of AIS. There was also evidence that rare mutations of some genes may be responsible for AIS, such as FBN1 (Buchan et al., 2014a), AKAP2 (Li et al., 2016) and MAPK7 (Gao et al., 2017). Other studies illustrated the association between CNV and AIS (Buchan et al., 2014b). All of those genetic factors can only explain the etiology of 2–7.6% of AIS (Buchan et al., 2014a; Ikegawa, 2016).
The abnormal, or aberrant DNA methylation pattern is universally recognized as an important factor in diseases, especially complex diseases. Recent studies indicated that differential methylation of key genes or CpG site were related to AIS, such as site cg01374129 (Meng et al., 2018), COMP (Mao et al., 2018), and PITX1 (Shi et al., 2018). But there were few types of research focusing on the methylation status of AIS on the whole-genome scale and no research of monozygotic twins discordant for AIS.
Monozygotic (MZ) twins are outstanding subjects to analyze epigenetic mechanisms. Theoretically, they share almost the same genotype, but the epigenome could be different, especially for discordant MZ twins who display phenotypically discordant on disease traits. Therefore discordant MZ twins are analyzed for deciphering complex diseases, such as congenital heart disease (Lyu et al., 2018), type 1 diabetes (Elboudwarej et al., 2016), and congenital renal agenesis (Jin et al., 2014).
In this study, we enrolled a MZ female twin pair discordant for AIS from Chinese Han population. In this twin pair, one was diagnosed with AIS, but her twin sister was healthy and had no spinal deformity. Their parents had no abnormality of the skeletal system or related family history. We compared their exome variants and genome methylation difference aiming to find the contributions to the pathogenesis of AIS.
Monozygotic twin discordant for AIS was recruited from Peking Union Medical College Hospital (PUMCH). There was no consanguinity between the parents. The parents and monozygotic twin of the patient were healthy without abnormalities of the skeletal system. Twenty sporadic AIS female patients and 20 healthy female controls were enrolled as a replication cohort. Genomic DNA was extracted from their peripheral blood using DNeasy Blood & Tissue Kits (QIAGEN, Eastwin Scientific, Inc., Beijing, China) according to the manufacturer's instructions. This study was approved by the Ethical Review Board of Peking Union Medical College Hospital. Written informed consent was obtained from all the participants or their parents.
WES was performed on DNA extracted from the peripheral blood. In brief, libraries were prepared from DNA samples and subjected to whole-exome capture using the VCRome SeqCap EZ Chice HGSC 96 Reactions capture reagent (Roche), followed by sequencing on an Illumina HiSeq 4000 platform with 150-bp pair-end reads mode. Whole-exome sequencing for all individuals in this family was performed and each generated fully reads with a mean depth of 107 (Table S1).
The variant-calling and annotation were performed by the in-house developed Peking Union Medical college hospital Pipeline (PUMP), same as in our previous studies (Wang et al., 2018a,b). In brief, single-nucleotide variants and internal duplications and/or deletions (aka indels) were called using the HaplotypeCaller of the Genome Analysis Toolkit (v3.4.0). Annotation of the de novo, compound heterozygotes, and recessively inherited variants was calculated with Gemini (v0.19.1) for in silico subtraction of parental variants from the proband's variants, with accounting for read number information extracted from BAM files. Computational prediction tools Gerp++ (Davydov et al., 2010), CADD (Kircher et al., 2014), SIFT (Vaser et al., 2016), Polyphen-2 (Adzhubei et al., 2010), and MutationTaster (Schwarz et al., 2014) were used to predict the conservation and pathogenicity of candidate variants. All variants were compared against Deciphering Disorders Involving Scoliosis & Comorbidities (DISCO) study in-house database and publicly available databases including the 1000 Genomes Project (http://www.internationalgenome.org/), the Exome variant server, NHLBI GO Exome Sequencing Project (ESP) (http://evs.gs.washington.edu/EVS/), and the Exome Aggregation Consortium (ExAC) (http://exac.broadinstitute.org/).
After data annotation, we first identified de novo mutations in the AIS patient taking her parents as a reference and then comparison was made between the MZ twins. For the mutations between the MZ twins including SNVs and short insertions/deletions (InDels), we classified them into four types (Figure S1). In the Equal type, both of the twins had the same mutations comparing to the Reference Genome. In the different type1, only the case had the mutation. In the different type2, the mutations only presented in the Control. In the different type3, both of the twins had different mutation compared to the Reference Genome. We hypothesized that the different type1 and type3 were potentially pathogenic.
A total amount of 5.2 micrograms genomic DNA spiked with 26 ng lambda DNA were fragmented by sonication to 200–300 bp with Covaris S220, followed by end repair and adenylation. Cytosine-methylated barcodes were ligated to sonicated DNA as per manufacturer's instructions. Then these DNA fragments were treated twice with bisulfite using EZ DNA Methylation-GoldTM Kit (Zymo Research). And the resulting single-strand DNA fragments were PCR amplificated using KAPA HiFi HotStart Uracil + ReadyMix (2X).
Library concentration was quantified by Qubit® 2.0 Fluorometer (Life Technologies, CA, USA) and quantitative PCR, and the insert size was checked on Agilent Bioanalyzer 2100 system.
The clustering of the index-coded samples was performed on a cBot Cluster Generation System using TruSeq PE Cluster Kit v3-cBot-HS (Illumia) according to the manufacturer's instructions. After cluster generation, the library preparations were sequenced on Illumina Hiseq 2500 platform and 100 bp single-end reads were generated. Image analysis and base calling were performed with the standard Illumina pipeline, and finally, 100 bp paired-end reads were generated.
Raw WGBS reads were mapped to the human reference genome (hg19, NCBI build 37.1) using BSMAP v2.90 (default parameters). Adaptor, low-quality and duplicated reads were automatically trimmed by BSMAP v2.90 with default thresholds. Single base methylation ratio was measured as the proportion of methylated Cs in all mapped reads from both strands. Only CpG sites with covered reads ≥4 reads were considered for further analysis.
Differentially methylated regions (DMRs) between the twin pair were identified using MethPipe 3.4.3 with significant differentially CpGs ≥ 5 and a minimal number of 10 CpGs that the DMR spans. DMRs were classified to “hypermethylation group” and “hypomethylation group” based on methylation level difference and these regions were ranked by absolute methylation difference between scoliosis and normal sample in the MZ twin (Figure S2). Microarray probes were designed for DMRs from the twin pair based on physical location, the methylation state of each interval was quantified by the mean of methylation beta values. Pearson correlation score was used to estimate the reliability of WGBS data.
Genomic position distribution of DMRs was performed using ChIPseeker (v1.14.0) Bioconductor package. Every DMR starting from 3 kb downstream to 3 kb upstream of the TSS was assigned to the corresponding RefSeq gene. Functional enrichment analysis of DMRs associated genes at KEGG pathways was performed using clusterProfiler (v3.6.0) Bioconductor package.
We searched public database including HPO, OMIM, Clinvar, and Pubmed using scoliosis as key term, 940 associated genes were collected as a scoliosis related gene set (Table S2). For these genes, an enrichment P-value was calculated based on hypergeometric testing:F
N represents the total gene counts of RefSeq database, M means the total number of scoliosis-related genes, k is the number of genes that have mutations identified from WES data, and x is the number of intersected genes between these two gene sets.
For the 940 scoliosis related genes, an enrichment P-value was also calculated based on hypergeometric testing above. Here N is total RefSeq genes (27967), M is the number of scoliosis-related genes, k is the number of DMRs associated genes, and x is the number of intersected genes between the two gene sets.
DNA methylation level was analysis by MethylTargetTM (Genesky Biotechnologies Inc., Shanghai, China), an NGS-based multiple Targeted CpG methylation analysis method. Specifically, the genomic regions of interest were analyzed and transformed into bisulfite-converted sequences by geneCpG software. PCR primer sets were designed with the Methylation Primer software from bisulfate converted DNA.
Genomic DNA (400 ng) was subjected to sodium bisulfite treatment using EZ DNA Methylation™-GOLD Kit (Zymo Research) according to the manufacturer's protocols. Multiplex PCR was performed with optimized primer sets combination. A 20 μl PCR reaction mixture was prepared for each reaction and included 1x reaction buffer (Takara), 3 mM Mg2+, 0.2 mM dNTP, 0.1 μM of each primer, 1U HotStarTaq polymerase (Takara) and 2 μl template DNA. The cycling program was 95°C for 2 min; 11 cycles of 94°C for 20 s, 63°C for 40s with a decreasing temperature step of 0.5°C per cycle, 72°C for 1 min; then followed by 24 cycles of 94°C for 20 s, 65°C for 30 s, 72°C for 1 min; 72°C for 2 min.
PCR amplicons were diluted and amplified using indexed primers. Specifically, a 20 μl mixture was prepared for each reaction and included 1x reaction buffer (NEB Q5TM), 0.3 mM dNTP, 0.3 μM of F primer, 0.3 μM of index primer, 1 U Q5TM DNA polymerase (NEB) and 1 μL diluted template. The cycling program was 98°C for 30 s; 11 cycles of 98°C for 10 s, 65°C for 30 s, 72°C for 30 s; 72°C for 5 min. PCR amplicons (170bp-270bp) were separated by agarose electrophoresis and purified using QIAquick Gel Extraction kit (QIAGEN).
Libraries from different samples were quantified and pooled together, followed by sequencing on the Illumina MiSeq platform according to manufacturer's protocols. Sequencing was performed with a 2 × 150 bp paired-end mode.
Quality control of sequencing reads was performed by FastQC. Filtered reads were mapped to genome by Blast. After reads recalibration with USEARCH, methylation and haplotype were analyzed using Perl script. Statistics were performed by U-test and ANOVA.
The monozygotic twins were 15-year-old girls, one of them (OS029) was diagnosed with AIS at 12. The patient had two apexes of scoliosis, T3-T11 with Cobb angle of about 27 degrees and T12-L3 with Cobb angle of about 26 degrees. The spine images of the healthy sister (OS030) and parents were normal (Figures 1A,B). In the replication cohort, the mean age of the AIS patients was 15 years old, and the mean age of the controls was 28 years old. All of the participants were Chinese females.

Figure 1. Clinical and WES findings of the AIS discordant monozygotic twin. (A) Pedigree of the patient. (B) Clinical characteristics and imaging of the proband and her healthy twin sister*. (C) Enrichment of WES mutation genes with scoliosis related genes. *Written informed consent was obtained from their parents for the publication of this image.
After strict filtering and genomic annotation, we failed to identify pathogenic variants responsible for the discordant phenotype in the MZ twin. Finally, only three de novo variants were identified, one missense SNV in the exon of PASD1 (c.425C>T), one missense SNV in the exon of SLC44A4 (c.1281G>C) and one synonymous SNV in the exon of AGTR2 (c.1080C>A), but all the de novo exome variants occur in both MZ twins (Table S3). Then we compared all the exome variants between the MZ twins. There were 30,700 same mutations, 32 Type 1 mutations and 1,830 Type 3 mutations. There were no Type 2 mutations (Table 1). Altogether there were 1,862 potential pathogenic mutations located in 1,381 genes (Table S4), and 81 were overlapped with scoliosis related genes (Figure 1C). By comparing with randomized gene list, these potential pathogenic genes undergoing mutations were significantly enriched in scoliosis genes (P = 4.45 × 10−07). Since both of the parents had no spinal deformity, these potential pathogenic mutations may only increase the susceptibility of scoliosis without causing scoliosis directly.
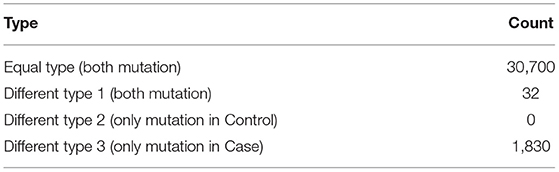
Table 1. Different mutations in the MZ twin discordant for AIS.
The global methylation status between the MZ twins were very similar, only a small percentage of promoter existed difference (348/27967, 1.24%, Table S5). We compared whole-genome DNA methylation level between the MZ twins, and found that both the hyper- and hypo- DMRs were differently distributed (Figure 2A). Finally, we identified 313 hypermethylated DMRs and 397 hypomethylated DMRs (Table S5). The average hypermethylation difference was 38% and the average hypomethylation difference was 31% between the two samples. We found that more DMRs locate in the promoter regions (Figure 2B) and most of them located in CGI (CpG island) (Figure 2C). Significant methylation level difference between the twins may influence the function of DMR-related genes and contribute to the etiology of this disease. Interestingly, these DMRs overlapped with 25 allele-specific methylated regions, suggesting that they were related to abnormal genomic imprinting (Figure S3) (de Sa Machado Araujo et al., 2018).
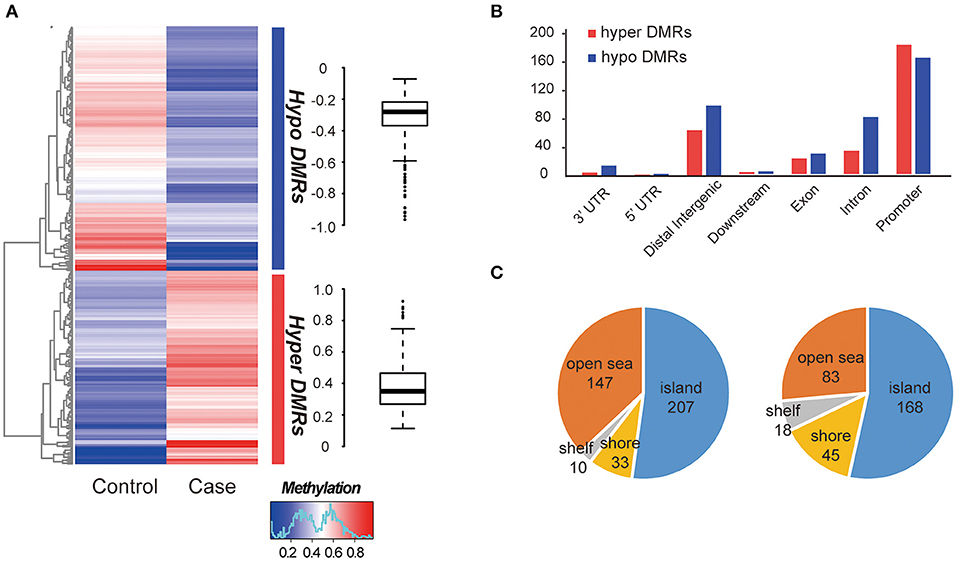
Figure 2. Comparison of DMRs between two samples. (A) Hierarchical clustering of DNA methylation profiles of AIS twins. The distribution of the methylation difference was shown in the box plot at the right side. (B) Characteristics of different functional element DNA methylation profiles of AIS twins. (C) CpG island distributions of hypomethylated DMRs (left) and hypermethylated DMRs (right).
We enriched the genes associated with these DMRs and then subjected these genes to KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway analysis. It revealed that DMR associated genes were enriched in several pathways. Three pathways with the most significant association were the MAPK signaling pathway, PI3K-Akt signaling pathway and Rap1 signaling pathway (Figure 3A), indicating that these pathways could be associated with AIS.

Figure 3. Annotations of DMR related genes between two samples. (A) Scatterplot of DMR related genes enrichment with KEGG pathway. (B) Enrichment of DMR related genes with scoliosis related genes.
To explore the relationship between these DMRs and scoliosis, we enriched the DMR related genes and found they were significantly overlapped with scoliosis related genes. In hypermethylated DMR associated genes group, 37 genes were overlapped with these scoliosis genes. In hypomethylated DMR associated genes group, 30 genes were overlapped with scoliosis related genes (Figure 3B). After comparing to randomized gene list, the DMR associated genes were significantly overlapped with scoliosis genes (p = 9.11 × 10−12 and 1.42 × 10−05, respectively), indicating that the methylation of scoliosis-related genes may be associated with the risk of AIS development.
For further identification of specific pathogenic epigenetic variants, we selected 14 DMRs from the literature for validation based on the related gene's function in the replication cohort (Table 2). All of the 14 genes were related to scoliosis and manifested with high methylation difference in the MZ twins. We found the methylation level of NDN: TSS-DMR (chr15:23932133-23932304, hg19) was significantly differentiated in the replication cohort. The mean methylation level in the AIS group was significantly higher than the mean methylation level in the control group (3.78 × 10−1 and 3.64 × 10−1, p = 0.04). This DMR locates at the promoter region of gene NDN. The different methylation of this gene may influence gene expression and be associated with the phenotype of AIS.
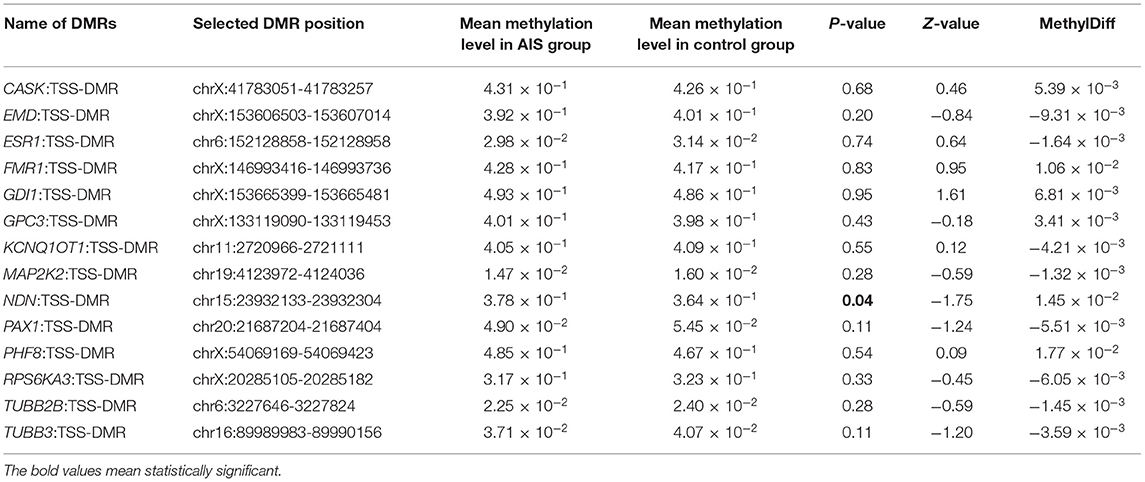
Table 2. Selected epigenome DNAm difference region in the replicated cohort.
This study represented the first combined analysis of whole exome and whole epigenome of MZ twins discordant for AIS. According to previous studies, genetic variations can predispose to AIS (Buchan et al., 2014a; Gao et al., 2017). Therefore, we first performed WES to detect genetic variations. We found three de novo SNVs, but the healthy twin sister also had these SNVs, indicating that pathogenesis of AIS may not be on account of these variants. Epigenetic variation is widely acknowledged to be involved in the pathogenesis of various diseases (Jin et al., 2014; Lyu et al., 2018). In this study, we compared whole-genome DNA methylation between the discordant twins using WGBS. Potential pathological DMRs were identified. The DMR related genes were enriched predominantly in the MAPK signaling pathway, and other 12 signaling pathways (i.e., PI3K-Akt and Rap1 signaling pathways etc.). The MAPK and PI3K-Akt signaling pathways have been reported to play an important role in osteoblast differentiation and skeletogenesis (Ge et al., 2007; Zhang et al., 2014; Iezaki et al., 2018). Previous studies also indicated that Rap1 signaling pathway is critical for myogenic differentiation and osteoclast function (Pizon et al., 1996; Zou et al., 2013). Abnormalities of these pathways could give rise to disorders manifested with scoliosis (Tiffin et al., 2013; Martinez-Lopez et al., 2017; Tsai et al., 2018). The previous study also showed that MAPK signaling pathway and PI3K-Akt signaling pathway were downregulated in bone marrow mesenchymal stem cells (BM-MSCs) of AIS, which may contribute to the AIS initiation and development (Zhuang et al., 2016). We hypothesized that hypermethylation of these DMR related genes could regulate MAPK, PI3K-Akt, and Rap1 signaling pathways, which may influence the initiation of AIS. This regulation mechanism may contribute to the discordance of AIS in this twin pair.
We also found the DMR associated genes were significantly overlapped with scoliosis related genes, which indicated that the methylation of scoliosis-related genes may play a pivotal role in the development of AIS. To explore specific epigenetic variants, we compared the specific methylation difference of several DMRs in the replication cohort. We found the NDN: TSS-DMR (chr15:23932133-23932304, hg19) was significantly differentiated in the replication cohort. The AIS group had higher methylation level of this DMR than the control group. This DMR locates in the promoter area of NDN and has 146 base pairs distance to TSS. NDN locates at 15q11.2 and is an intronless gene located in the Prader-Willi syndrome (PWS) deletion region. Previous studies hypothesized that lack of its coding protein Necdin during development may contribute to PWS (Jay et al., 1997; Miller et al., 2009). PWS is a rare disease associated with a variety of musculoskeletal abnormalities, about 43.4% of patients were afflicted with scoliosis (Odent et al., 2008). Besides, NDN is a well-known maternally imprinted gene and is expressed exclusively from the paternal allele, and its methylation are persistent markers of gene regulation (Lau et al., 2004). Previous studies had proved that imprinted DMRs (iDMRs) could be perturbed in kinds of diseases (de Sa Machado Araujo et al., 2018). In our study, we hypothesized that higher methylation could decrease the expression of NDN, which predisposed the patient to AIS.
There are several limitations in our study. First, since WES only covered most exons of the human reference genome, we did not assess the rest of the genome and other types of variants, such as karyotype or structural variants. Therefore, we cannot exclude the possibility that genome variants contribute to the etiology of this MZ twin pair discordant for AIS. Second, since all of the AIS patients were teenagers, the age of the control group is not matched with the case group in the replication cohort. The unmatched age may cause bias of the implications. Third, the sample size of the MZ twins and the replication cohort was relatively small. Larger samples are needed for the replication.
In conclusion, we described the genome methylation difference in an AIS discordant MZ twin pair using WGBS and selected DMR methylation difference in a replication cohort. We found that the NDN: TSS-DMR had higher methylation level in AIS, which may elucidate the potential etiology of AIS.
The datasets generated for this study can be found in the http://discostudy.org/data/DISCO2019-1.html.
The studies involving human participants were reviewed and approved by Ethical Review Board of Peking Union Medical College Hospital. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
GL, LW, and NW performed the research, analyzed, and interpreted the data. GL drafted the manuscript. XW, XY, ML, ZY, SL, YZu, YN, SZ, and YZh helped sample collection. JZ and JS performed phenotyping of patients. XW, ZY, and XY helped analysis and interpretation of NGS data. YN provided technique support. GQ, YW, ZW, JZ, and JS offered professional discussions and instructions. ZW and NW conceived and designed the study, revised the manuscript, and provided the final approval of the manuscript.
This research was funded in part by the National Natural Science Foundation of China (81822030 to NW, 81902271 to GL, 81772299 and 81930068 to ZW, 81772301 and 81972132 to GQ, 81672123 to JZ, 81572097 and 81871746 to YW), Beijing Natural Science Foundation (7172175 to NW, 7191007 to ZW, and 7184232 to SL), CAMS Initiative Fund for Medical Sciences (2016-I2M-3-003 to GQ and NW, 2016-I2M-2-006 and 2017- I2M-2-001 to ZW), the Central Level Public Interest Program for Scientific Research Institute (2018RC31003 to NW), the National Key Research and Development Program of China (No. 2018YFC0910506 to NW and ZW), and the International Program Associate grant from RIKEN of Japan (190038 to ML).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We give our great thanks to Dr. Shugang Li, Hong Zhao, and Yu Zhao for the patient enrollment and clinical evaluation; and to the patients for their active participation.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbioe.2019.00364/full#supplementary-material
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P., et al. (2010). A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249. doi: 10.1038/nmeth0410-248
Buchan, J. G., Alvarado, D. M., Haller, G., Aferol, H., Miller, N. H., Dobbs, M. B., et al. (2014b). Are copy number variants associated with adolescent idiopathic scoliosis? Clin. Orthop. Relat. Res. 472, 3216–3225. doi: 10.1007/s11999-014-3766-8
Buchan, J. G., Alvarado, D. M., Haller, G. E., Cruchaga, C., Harms, M. B., Zhang, T., et al. (2014a). Rare variants in FBN1 and FBN2 are associated with severe adolescent idiopathic scoliosis. Hum. Mol. Genet. 23, 5271–5282. doi: 10.1093/hmg/ddu224
Davydov, E. V., Goode, D. L., Sirota, M., Cooper, G. M., Sidow, A., and Batzoglou, S. (2010). Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput. Biol. 6:e1001025. doi: 10.1371/journal.pcbi.1001025
de Sa Machado Araujo, G., da Silva Francisco Junior, R., Dos Santos Ferreira, C., Mozer Rodrigues, P. T., Terra Machado, D., Louvain de Souza, T., et al. (2018). Maternal 5(m)CpG imprints at the PARD6G-AS1 and GCSAML differentially methylated regions are decoupled from parent-of-origin expression effects in multiple human tissues. Front. Genet. 9:36. doi: 10.3389/fgene.2018.00036
Elboudwarej, E., Cole, M., Briggs, F. B., Fouts, A., Fain, P. R., Quach, H., et al. (2016). Hypomethylation within gene promoter regions and type 1 diabetes in discordant monozygotic twins. J. Autoimmun. 68, 23–29. doi: 10.1016/j.jaut.2015.12.003
Gao, W., Chen, C., Zhou, T., Yang, S., Gao, B., Zhou, H., et al. (2017). Rare coding variants in MAPK7 predispose to adolescent idiopathic scoliosis. Hum. Mutat. 38, 1500–1510. doi: 10.1002/humu.23296
Ge, C., Xiao, G., Jiang, D., and Franceschi, R. T. (2007). Critical role of the extracellular signal-regulated kinase-MAPK pathway in osteoblast differentiation and skeletal development. J. Cell Biol. 176, 709–718. doi: 10.1083/jcb.200610046
Iezaki, T., Fukasawa, K., Horie, T., Park, G., Robinson, S., Nakaya, M., et al. (2018). The MAPK Erk5 is necessary for proper skeletogenesis through a molecular axis that involves Smurfs-Smads-Sox9. Development 145:dev164004. doi: 10.1242/dev.164004
Ikegawa, S. (2016). Genomic study of adolescent idiopathic scoliosis in Japan. Scoliosis Spinal Disord. 11:5. doi: 10.1186/s13013-016-0067-x
Jay, P., Rougeulle, C., Massacrier, A., Moncla, A., Mattei, M. G., Malzac, P., et al. (1997). The human necdin gene, NDN, is maternally imprinted and located in the Prader-Willi syndrome chromosomal region. Nat. Genet. 17, 357–361. doi: 10.1038/ng1197-357
Jin, M., Zhu, S., Hu, P., Liu, D., Li, Q., Li, Z., et al. (2014). Genomic and epigenomic analyses of monozygotic twins discordant for congenital renal agenesis. Am. J. Kidney Dis. 64, 119–122. doi: 10.1053/j.ajkd.2014.01.423
Kesling, K. L., and Reinker, K. A. (1997). Scoliosis in twins. A meta-analysis of the literature and report of six cases. Spine 22, 2009–2014; discussion 2015. doi: 10.1097/00007632-199709010-00014
Kircher, M., Witten, D. M., Jain, P., O'Roak, B. J., Cooper, G. M., and Shendure, J. (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315. doi: 10.1038/ng.2892
Kou, I., Takahashi, Y., Johnson, T. A., Takahashi, A., Guo, L., Dai, J., et al. (2013). Genetic variants in GPR126 are associated with adolescent idiopathic scoliosis. Nat. Genet. 45, 676–679. doi: 10.1038/ng.2639
Lau, J. C., Hanel, M. L., and Wevrick, R. (2004). Tissue-specific and imprinted epigenetic modifications of the human NDN gene. Nucleic Acids Res. 32, 3376–3382. doi: 10.1093/nar/gkh671
Li, W., Li, Y., Zhang, L., Guo, H., Tian, D., Li, Y., et al. (2016). AKAP2 identified as a novel gene mutated in a Chinese family with adolescent idiopathic scoliosis. J. Med. Genet. 53, 488–493. doi: 10.1136/jmedgenet-2015-103684
Liu, G., Liu, S., Li, X., Chen, J., Chen, W., Zuo, Y., et al. (2019). Genetic polymorphisms of PAX1 are functionally associated with different PUMC types of adolescent idiopathic scoliosis in a northern Chinese Han population. Gene 688, 215–220. doi: 10.1016/j.gene.2018.12.013
Liu, G., Liu, S., Lin, M., Li, X., Chen, W., Zuo, Y., et al. (2018). Genetic polymorphisms of GPR126 are functionally associated with PUMC classifications of adolescent idiopathic scoliosis in a Northern Han population. J. Cell. Mol. Med. 22, 1964–1971. doi: 10.1111/jcmm.13486
Liu, S., Wu, N., Zuo, Y., Zhou, Y., Liu, J., Liu, Z., et al. (2017). Genetic polymorphism of LBX1 is associated with adolescent idiopathic scoliosis in northern chinese han population. Spine 42, 1125–1129. doi: 10.1097/BRS.0000000000002111
Lyu, G., Zhang, C., Ling, T., Liu, R., Zong, L., Guan, Y., et al. (2018). Genome and epigenome analysis of monozygotic twins discordant for congenital heart disease. BMC Genomics 19:428. doi: 10.1186/s12864-018-4814-7
Mao, S. H., Qian, B. P., Shi, B., Zhu, Z. Z., and Qiu, Y. (2018). Quantitative evaluation of the relationship between COMP promoter methylation and the susceptibility and curve progression of adolescent idiopathic scoliosis. Eur. Spine J. 27, 272–277. doi: 10.1007/s00586-017-5309-y
Martinez-Lopez, A., Blasco-Morente, G., Perez-Lopez, I., Herrera-Garcia, J. D., Luque-Valenzuela, M., Sanchez-Cano, D., et al. (2017). CLOVES syndrome: review of a PIK3CA-related overgrowth spectrum (PROS). Clin. Genet. 91, 14–21. doi: 10.1111/cge.12832
Meng, Y., Lin, T., Liang, S., Gao, R., Jiang, H., Shao, W., et al. (2018). Value of DNA methylation in predicting curve progression in patients with adolescent idiopathic scoliosis. EBioMedicine 36, 489–496. doi: 10.1016/j.ebiom.2018.09.014
Miller, N. L., Wevrick, R., and Mellon, P. L. (2009). Necdin, a Prader-Willi syndrome candidate gene, regulates gonadotropin-releasing hormone neurons during development. Hum. Mol. Genet. 18, 248–260. doi: 10.1093/hmg/ddn344
Odent, T., Accadbled, F., Koureas, G., Cournot, M., Moine, A., Diene, G., et al. (2008). Scoliosis in patients with Prader-Willi Syndrome. Pediatrics 122, e499–e503. doi: 10.1542/peds.2007-3487
Ogura, Y., Kou, I., Miura, S., Takahashi, A., Xu, L., Takeda, K., et al. (2015). A functional SNP in BNC2 is associated with adolescent idiopathic scoliosis. Am. J. Hum. Genet. 97, 337–342. doi: 10.1016/j.ajhg.2015.06.012
Pizon, V., Cifuentes-Diaz, C., Mege, R. M., Baldacci, G., and Rieger, F. (1996). Expression and localization of RAP1 proteins during myogenic differentiation. Eur. J. Cell Biol. 69, 224–235.
Schwarz, J. M., Cooper, D. N., Schuelke, M., and Seelow, D. (2014). MutationTaster2: mutation prediction for the deep-sequencing age. Nat. Methods 11, 361–362. doi: 10.1038/nmeth.2890
Sharma, S., Londono, D., Eckalbar, W. L., Gao, X., Zhang, D., Mauldin, K., et al. (2015). A PAX1 enhancer locus is associated with susceptibility to idiopathic scoliosis in females. Nat. Commun. 6:6452. doi: 10.1038/ncomms7452
Shi, B., Xu, L., Mao, S., Xu, L., Liu, Z., Sun, X., et al. (2018). Abnormal PITX1 gene methylation in adolescent idiopathic scoliosis: a pilot study. BMC Musculoskelet. Disord. 19:138. doi: 10.1186/s12891-018-2054-2
Takahashi, Y., Kou, I., Takahashi, A., Johnson, T. A., Kono, K., Kawakami, N., et al. (2011). A genome-wide association study identifies common variants near LBX1 associated with adolescent idiopathic scoliosis. Nat. Genet. 43, 1237–1240. doi: 10.1038/ng.974
Tiffin, H. R., Jenkins, Z. A., Gray, M. J., Cameron-Christie, S. R., Eaton, J., Aftimos, S., et al. (2013). Dysregulation of FHL1 spliceforms due to an indel mutation produces an emery-dreifuss muscular dystrophy plus phenotype. Neurogenetics 14, 113–121. doi: 10.1007/s10048-013-0359-8
Tsai, I. C., McKnight, K., McKinstry, S. U., Maynard, A. T., Tan, P. L., Golzio, C., et al. (2018). Small molecule inhibition of RAS/MAPK signaling ameliorates developmental pathologies of Kabuki Syndrome. Sci. Rep. 8:10779. doi: 10.1038/s41598-018-28709-y
Vaser, R., Adusumalli, S., Leng, S. N., Sikic, M., and Ng, P. C. (2016). SIFT missense predictions for genomes. Nat. Protoc. 11, 1–9. doi: 10.1038/nprot.2015.123
Wang, K., Zhao, S., Liu, B., Zhang, Q., Li, Y., Liu, J., et al. (2018a). Perturbations of BMP/TGF-beta and VEGF/VEGFR signalling pathways in non-syndromic sporadic brain arteriovenous malformations (BAVM). J. Med. Genet. 55, 675–684. doi: 10.1136/jmedgenet-2017-105224
Wang, K., Zhao, S., Zhang, Q., Yuan, J., Liu, J., Ding, X., et al. (2018b). Whole-exome sequencing reveals known and novel variants in a cohort of intracranial vertebral-basilar artery dissection (IVAD). J. Hum. Genet. 63, 1119–1128. doi: 10.1038/s10038-018-0496-x
Yawn, B. P., Yawn, R. A., Hodge, D., Kurland, M., Shaughnessy, W. J., Ilstrup, D., et al. (1999). A population-based study of school scoliosis screening. JAMA 282, 1427–1432. doi: 10.1001/jama.282.15.1427
Zhang, Y., Pizzute, T., and Pei, M. (2014). A review of crosstalk between MAPK and Wnt signals and its impact on cartilage regeneration. Cell Tissue Res. 358, 633–649. doi: 10.1007/s00441-014-2010-x
Zhuang, Q., Mao, W., Xu, P., Li, H., Sun, Z., Li, S., et al. (2016). Identification of differential genes expression profiles and pathways of bone marrow mesenchymal stem cells of adolescent idiopathic scoliosis patients by microarray and integrated gene network analysis. Spine 41, 840–855. doi: 10.1097/BRS.0000000000001394
Keywords: adolescent idiopathic scoliosis (AIS), monozygotic twins, whole exome sequencing (WES), DNA methylation, whole genome bisulfite sequencing (WGBS)
Citation: Liu G, Wang L, Wang X, Yan Z, Yang X, Lin M, Liu S, Zuo Y, Niu Y, Zhao S, Zhao Y, Zhang J, Shen J, Wang Y, Qiu G, Wu Z and Wu N (2019) Whole-Genome Methylation Analysis of Phenotype Discordant Monozygotic Twins Reveals Novel Epigenetic Perturbation Contributing to the Pathogenesis of Adolescent Idiopathic Scoliosis. Front. Bioeng. Biotechnol. 7:364. doi: 10.3389/fbioe.2019.00364
Received: 19 August 2019; Accepted: 12 November 2019;
Published: 10 December 2019.
Edited by:
Yongchun Zuo, Inner Mongolia University, ChinaReviewed by:
Enrique Medina-Acosta, Universidade Estadual do Norte Fluminense Darcy Ribeiro, BrazilCopyright © 2019 Liu, Wang, Wang, Yan, Yang, Lin, Liu, Zuo, Niu, Zhao, Zhao, Zhang, Shen, Wang, Qiu, Wu and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nan Wu, ZHIud3VuYW5AcHVtY2guY24=; Zhihong Wu, b3J0aG9zY2llbmNlQDEyNi5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.