 Esteban Martínez-García
Esteban Martínez-García Tomás Aparicio
Tomás Aparicio Víctor de Lorenzo
Víctor de Lorenzo Pablo I. Nikel
Pablo I. Nikel- Systems and Synthetic Biology Program, Centro Nacional de Biotecnología (CNB-CSIC), Madrid, Spain
Re-programming microorganisms to modify their existing functions and/or to bestow bacteria with entirely new-to-Nature tasks have largely relied so far on specialized molecular biology tools. Such endeavors are not only relevant in the burgeoning metabolic engineering arena but also instrumental to explore the functioning of complex regulatory networks from a fundamental point of view. À la carte modification of bacterial genomes thus calls for novel tools to make genetic manipulations easier. We propose the use of a series of new broad-host-range mini-Tn5-vectors, termed pBAMDs, for the delivery of gene(s) into the chromosome of Gram-negative bacteria and for generating saturated mutagenesis libraries in gene function studies. These delivery vectors endow the user with the possibility of easy cloning and subsequent insertion of functional cargoes with three different antibiotic-resistance markers (kanamycin, streptomycin, and gentamicin). After validating the pBAMD vectors in the environmental bacterium Pseudomonas putida KT2440, their use was also illustrated by inserting the entire poly(3-hydroxybutyrate) (PHB) synthesis pathway from Cupriavidus necator in the chromosome of a phosphotransacetylase mutant of Escherichia coli. PHB is a completely biodegradable polyester with a number of industrial applications that make it attractive as a potential replacement of oil-based plastics. The non-selective nature of chromosomal insertions of the biosynthetic genes was evidenced by a large landscape of PHB synthesis levels in independent clones. One clone was selected and further characterized as a microbial cell factory for PHB accumulation, and it achieved polymer accumulation levels comparable to those of a plasmid-bearing recombinant. Taken together, our results demonstrate that the new mini-Tn5-vectors can be used to confer interesting phenotypes in Gram-negative bacteria that would be very difficult to engineer through direct manipulation of the structural genes.
Introduction
Over the last few years, a number of Gram-negative bacteria have become increasingly attractive chassis for a number of synthetic biology and metabolic engineering purposes. One conspicuous case involves the environmental bacterium Pseudomonas putida as a robust host for strong oxidative bioreactions, together with its GRAS (generally recognized as safe) status and its inherent ability to grow on a wide range of substrates (Nikel et al., 2014a). This situation calls for the expansion of the available tools for re-wiring its extant genetic features to further extend its metabolic potential – or even introducing new-to-Nature functions.
One frequently used molecular biology resource for analyses and manipulations of bacterial genomes is the Tn5 transposon. Historically, a number of plasmid vectors based on both wild-type and minimized versions of Tn5 (i.e. mini-transposons) had allowed the user to introduce stable insertions of foreign DNA into the chromosome of virtually any Gram-negative bacteria (de Lorenzo et al., 1990, 1998; Herrero et al., 1990; de Lorenzo and Timmis, 1994; Martínez-García et al., 2011; Martínez-García and de Lorenzo, 2012; Nikel and de Lorenzo, 2013a). Such Tn5-derived elements present clear advantages over the use of their plasmid-based counterparts for the introduction and expression of heterologous genes into several bacterial species. These features include (but are not limited to) (i) the maintenance of the corresponding transgenes without antibiotic selective pressure, (ii) the long-term stability of the constructs and the re-usability of the functional parts, and, furthermore, (iii) Tn5 vectors admit cloning and chrosomosomal delivery of considerably long DNA fragments. Finally, as the transposase gene (tnpA) is lost following each transposition event (Berg, 1989; Reznikoff, 2006, 2008), one added value of mini-Tn5-vectors is the possibility to use them recursively in the same host, provided that they bear different selection markers. Moreover, as the TnpA transposase tends to act in cis (Phadnis et al., 1986), it promotes the insertion of DNA sequences borne by the plasmid, irrespective of previous insertions in a given target chromosome. These features allow for the integration of more than one DNA cargo into the same genome.
In this study, we report a series of synthetic, modular broad-host-range mini-Tn5-vectors for the delivery of gene(s) into the chromosome of a diversity of Gram-negative bacteria and to construct saturated mutagenesis libraries for gene function studies. These vectors were termed pBAMDs, and they enable the possibility of easy cloning and subsequent chromosomal insertion of functional cargoes with three different and interchangeable antibiotic-resistance markers (kanamycin, streptomycin, and gentamicin). Moreover, the functional parts of the new vectors can be easily swapped by digestion with the appropriate restriction enzymes, allowing for the shuffling of each element as needed. Potential applications of the new tools are illustrated in two different genetic contexts. In one case, a systematic validation of the Tn5 vectors was carried out in P. putida KT2440, demonstrating the potential of the new insertional plasmids to be used in a sequential fashion for constructing (and deconstructing) complex phenotypes. In a second case, one of the pBAMD plasmids was used to insert a gene cluster from Cupriavidus necator, encoding all the biochemical functions needed for the formation of poly(3-hydroxybutyrate) (PHB), into the chromosome of Escherichia coli, thereby resulting in a new microbial cell factory tailored for biopolymer synthesis.
Materials and Methods
Bacterial Strains, Plasmids, and Growth Conditions
The bacterial strains and plasmids used in this study are described in Table 1. Bacteria were routinely grown batchwise in LB medium (10 g l−1 tryptone, 5 g l−1 yeast extract, and 5 g l−1 NaCl) with rotary agitation (170 rpm). P. putida was grown at 30°C while E. coli cells were grown at 37°C. Selection of P. putida transconjugants was performed by spotting the cells onto M9 minimal medium agar plates (Sambrook et al., 1989) added with 0.2% (w/v) sodium citrate as the sole carbon source (to counterselect E. coli cells). PHB accumulation was assessed in selected E. coli transconjugants cultured in M9 minimal medium containing 30 g l−1 glucose as the sole carbon source. Aerobic culture conditions in experiments aimed at polymer synthesis were achieved essentially as described by Nikel et al. (2010a), by using a 1:10 culture medium-to-flask volume ratio. Antibiotics were added at the following final concentrations whenever needed: ampicillin, 150 μg ml−1 for E. coli or 500 μg ml−1 for P. putida, chloramphenicol, 30 μg ml−1; kanamycin, 50 μg ml−1, streptomycin, 80 μg ml−1; and gentamicin, 10 μg ml−1. All solid media also contained 15 g l−1 agar. Growth was estimated spectrophotometrically by measuring the optical density at 600 nm (OD600) of the cultures (appropriately diluted in 9 g l−1 NaCl whenever needed) in a Ultrospec 3000 pro UV/Visible spectrophotometer (GE Healthcare Bio-Sciences Corp., Piscataway, NJ, USA). When culturing E. coli strains that accumulate PHB, for which OD600 readings are no longer useful to estimate the cell dry weight (CDW), cells from 15 ml aliquots were washed, concentrated, and the CDW determined after drying the samples at 80°C to constant weight as previously indicated by Nikel et al. (2008a,b).
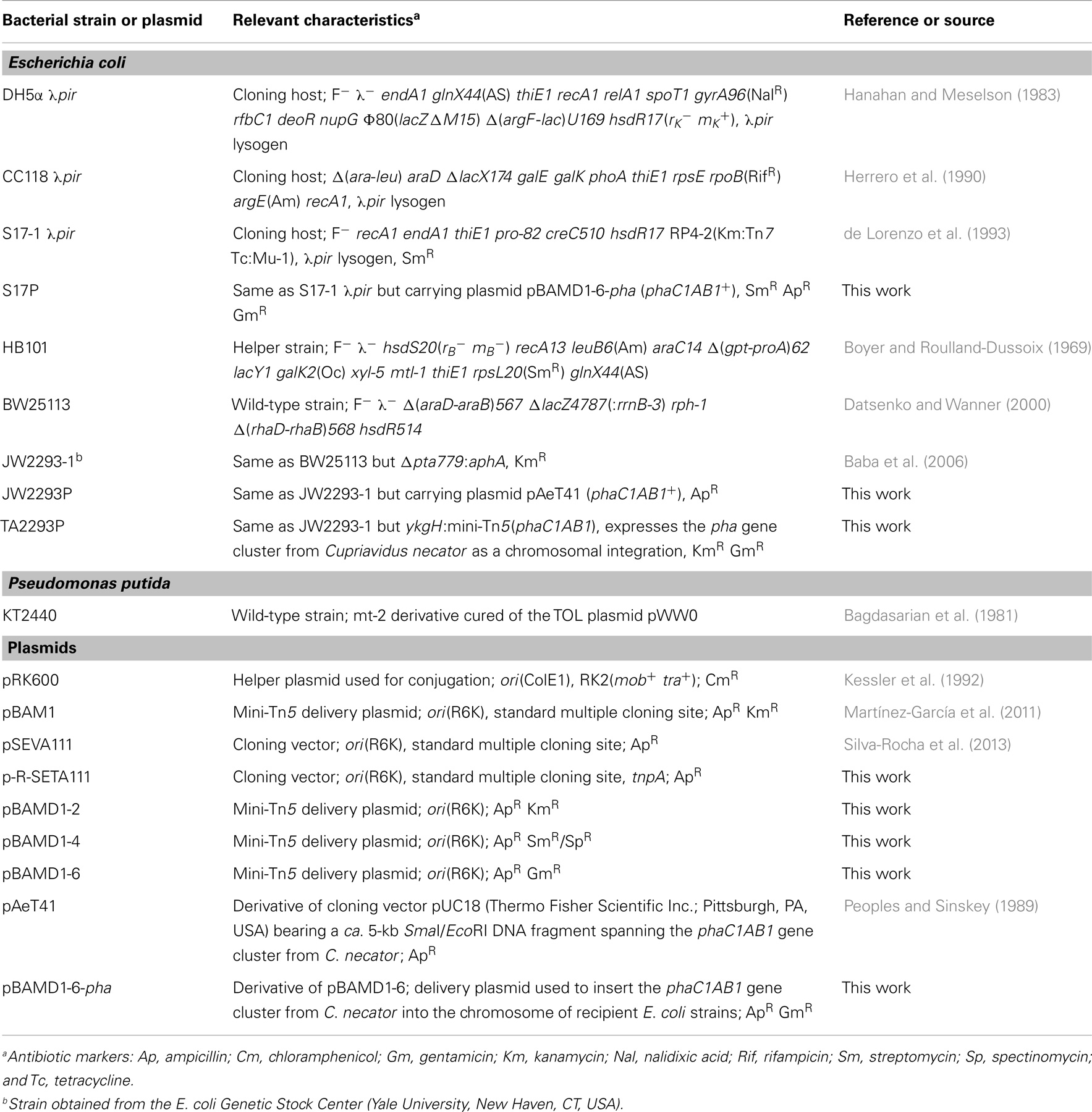
Table 1. Bacterial strains and plasmids used in this work.
Nucleic Acid Manipulations and General Cloning Techniques
DNA manipulations followed routine laboratory techniques as described by Sambrook et al. (1989) and Martínez-García and de Lorenzo (2012). Plasmid DNA was obtained using the QIAprep Spin™ Miniprep kit (Qiagen, Inc., Valencia, CA, USA). Restriction enzymes were obtained from New England Biolabs Inc. (Ipswich, MA, USA), and T4 DNA ligase was purchased from Roche Applied Science Co. (Indianapolis, IN, USA). Plasmid p-R-SETA111 was constructed using isothermal assembly essentially as detailed by Gibson et al. (2009) but using a home-made mixture of enzymes. Colony PCR was performed using a single colony from a fresh LB agar plate and transferred directly into the reaction tube. PCR reactions were purified either with the NucleoSpin™ Gel and PCR clean-up kit (Macherey-Nagel GmbH & Co. KG, Düren, Germany) or the ExoSAP-IT™ PCR product clean-up kit (USB, Affymetrix Ltd., Santa Clara, CA, USA). Oligonucleotides were purchased from Sigma-Aldrich Co. (St. Louis, MO, USA). The oligonucleotides used in this work for specific DNA constructions are indicated in Table S1 in the Supplementary Material; the oligonucleotides used to identify the location of the chromosomal insertions are indicated in Table 2 (see also next section). The three different mini-Tn5 modules were chemically synthesized de novo by GeneCust Europe S.A. (Dudelange, Luxembourg). DNA sequencing was carried out by Secugen SL (Madrid, Spain).

Table 2. Oligonucleotides used in this work to identify and sequence the chromosomal locus of mini-Tn5 insertions.
Three separate DNA segments, carrying the transposon module along with the corresponding antibiotic-resistance determinant, were obtained as follows. In the first step, we used pBAM1 (Martínez-García et al., 2011) as the template to amplify the bla gene using primers Ap-AsiSI-F and Ap-MluI-R (Table S1 in the Supplementary Material), thereby substituting the SwaI and PshAI restriction sites by target recognition sites for AsiSI and MluI, respectively, while maintaining the transcriptional control of bla through the native P3 promoter (Brosius et al., 1982). The backbone of plasmid pSEVA111 (Silva-Rocha et al., 2013) was amplified with the SEVA111-F and SEVA111-R oligonucleotide pair to obtain the second DNA fragment. Finally, the tnpA gene from pBAM1 was obtained using the oligonucleotides tnpA-SanDI-F and tnpA-AsiSI-R that add the corresponding SanDI and AsiSI restriction sites to the amplified fragment. These fragments were joined together by isothermal assembly, giving rise to plasmid p-R-SETA111 (Figure S1 in Supplementary Material).
Tripartite conjugative matings were set using E. coli CC118 λpir (carrying pBAMD1-x, where x stands for any of the three antibiotic markers; see below) as the donor strain, the mating-helper strain E. coli HB101 (carrying pRK600), and P. putida KT2440 as the recipient strain. Conjugative matings were performed as described elsewhere (Martínez-García et al., 2011; Martínez-García and de Lorenzo, 2012). Briefly, the OD600 from overnight cultures grown in LB medium with the appropriate antibiotics was adjusted to 1, then the cells were washed twice with 10 mM MgSO4 to remove antibiotics from the culture medium, and each bacterial suspension was added to a 10 ml tube containing 5 ml of 10 mM MgSO4 to obtain a final OD600 of ca. 0.03. Biparental matings were done by following a similar procedure, but using E. coli S17-1 λpir as the donor strain.
The mixture was concentrated by filtration and the cells were laid onto a filter disk (0.45 μm pore-size, 23 mm diameter, EMD Millipore Corp., Billerica, MA, USA). The filter was placed onto the surface of an LB agar plate and incubated at 30°C for 6 h. Finally, the biomass from the filter was suspended in 5 ml of 10 mM MgSO4 and different dilutions were plated onto a suitable selective media.
Localization of the mini-Tn5 Transposon Insertion Sites by Arbitrary PCR
In order to specifically map the landing sites of the mini-transposon within the chromosome, a set of oligonucleotides was designed that specifically hybridizes in each of the mini-Tn5 elements (Table 2). Transconjugants were streaked onto M9 minimal medium agar plates with 0.2% (w/v) sodium citrate as the sole carbon source and supplemented with the appropriate antibiotics to obtain isolated colonies, which were re-streaked again in the same culture medium to obtain isolated clones. These colonies were used as the template for arbitrary PCR. The identification of the transposon insertion site could be independently obtained using either the oligonucleotides that are close to the ME-O end or with the ones designed for the ME-I end. However, when these plasmids carry any heterologous DNA in the multiple cloning site (MCS), the use of the oligonucleotides that are close to the ME-O end is the preferable option, since the ones for the ME-I end are located downstream the MCS and therefore would amplify through it. In the later case, and depending on the length of the DNA cloned in the MCS, the corresponding amplicon may not provide long enough a sequence to ascertain the insertion site of the mini-transposon module.
The conditions of the first round of arbitrary PCR were as follows: 5 min at 95°C (initial denaturation); six cycles of 30 s at 95°C, 30 s at 30°C, and 90 s at 72°C; and 30 cycles of 30 s at 95°C, 30 s at 45°C, and 90 s at 72°C (Das et al., 2005). The ARB6 oligonucleotide was used together with the external oligonucleotides within the mini-transposon (indicated with the “Ext” acronym in Table 2). Then, we used 1 μl of the first PCR round as the template for the second round of arbitrary PCR by applying the following conditions: 1 min at 95°C (initial denaturation); 30 cycles of 30 s at 95°C, 30 s at 52°C, and 90 s at 72°C; followed by an extra extension of 4 min at 72°C (Das et al., 2005). For the second round of arbitrary PCR, the ARB2 oligonucleotide was used together with the internal primers within the mini-transposon (indicated with the “Int” acronym in Table 2). Finally, the PCR amplification product obtained in the second round was directly purified and sent for sequencing with the corresponding internal oligonucleotide.
DNA sequences were thoroughly inspected visually for any error and analyzed using the Pseudomonas Genome Database (Winsor et al., 2011), and BlastN (Altschul et al., 1990, 1997) was subsequently employed to map the precise transposon insertion point. To ascertain the conservation level of the 9-bp target sequence of Tn5 transposase, we resorted to the web-based application WebLogo 3.4 (Crooks et al., 2004).
Analytical Procedures
For a coarse estimation of the PHB content in E. coli transconjugants, we resorted to a fluorimetric assay based on Nile red staining (Spiekermann et al., 1999). Cells were grown overnight in LB medium with the proper antibiotic. The cultures were diluted to an OD600 of 0.1 in fresh LB medium containing 30 g l−1 glucose and 200 μl aliquots were placed in a 96 well microtiter plate (Costar™ black plates with clear bottom; Thermo Fisher Scientific Inc.). After growing the cells for 24 h at 37°C, 0.002 volume of a Nile red stock solution, freshly prepared by dissolving the dye (purchased from Sigma-Aldrich Co.) to 1 mg ml−1 in dimethyl sulfoxide, were added to each well. The microtiter plates were incubated at 37°C in the dark for 30 min, and the fluorescence at 585 nm was measured in a SpectraMax M2e plate reader (Molecular Devices, LLC., Sunnyvale, CA, USA) in cells before and after staining with Nile red. The raw fluorescence readings were normalized to the biomass in each well by dividing the values by the OD600 of the corresponding culture.
The quantification of PHB content in E. coli via fluorescence-activated cell sorting (FACS) was conducted by following a slight modification of the protocol described by Tyo et al. (2006). In brief, cultures to be analyzed were promptly cooled to 4°C by placing them in an ice bath for 15 min. Cells were harvested by centrifugation (5 min, 5,000 × g, 4°C), resuspended to an OD600 of 0.4 in cold TES buffer [10 mM Tris·HCl (pH = 7.5), 2.5 mM EDTA, and 10% (w/v) sucrose], and further incubated on ice for 15 min. Bacteria were recovered by centrifugation as explained above, and finally resuspended in the same volume of cold 1 mM MgCl2. A 1 ml aliquot of this suspension was added with 3 μl of an 1 mg ml−1 Nile red solution and incubated in the dark at 4°C for 30 min. Cells were analyzed by FACS immediately after the staining procedure. FACS was carried out in a MACSQuant™ VYB cytometer (Miltenyi Biotec GmbH, Bergisch Gladbach, Germany). Cells were excited with an Ar laser (488 nm, diode-pumped solid state), and the Nile red fluorescence at 585 nm was detected with a 614/50 nm band-pass filter. FACS analysis was done on at least 50,000 cells and the results were analyzed with the built-in MACSQuantify™ software 2.5 (Miltenyi Biotec). The geometric mean of fluorescence in each sample was correlated to the PHB content (expressed as a percentage) through a calibration curve as described previously (Tyo et al., 2006).
Cell-free extracts were obtained from cells harvested by centrifugation from an appropriate culture volume at 4,000 × g at 4°C for 10 min and processed as described previously (Nikel and de Lorenzo, 2013b; Nikel et al., 2014b). The total protein concentration in cell extracts was assessed by means of the Bradford method (Bradford, 1976) using a commercially available kit from BioRad Laboratories, Inc. (Hercules, CA, USA), with crystalline bovine serum albumin as the standard for determinations. In vitro quantification of the specific 3-ketoacyl-coenzyme A (CoA) thiolase activity in the thiolysis direction was conducted according to the protocols developed by Palmer et al. (1991) and Slater et al. (1998), with some modifications. The assay mixture (1 ml) contained 65 mM Tris–HCl (pH = 7.5), 50 mM MgCl2, 62.5 μM CoA, and 65 μM 3-acetoacetyl-CoA (Sigma-Aldrich Co.). Solutions of both CoA and 3-acetoacetyl-CoA were freshly prepared just prior to the assay. The assay was initiated upon the prompt addition of the cell-free extract, and the disappearance of 3-acetoacetyl-CoA was measured with time at 304 nm (using an extinction coefficient for 3-acetoacetyl-CoA ε304 = 16.9 × 103 M−1 cm−1). One enzyme unit was defined as the amount of enzyme catalyzing the conversion of 1 μmol of substrate to product per min at 25°C.
Residual glucose and acetate concentrations in culture supernatants were determined in selected samples using adequate enzymatic kits (R-Biopharm AG, Darmstadt, Germany), essentially as per the manufacturer’s instructions. In either case, control mock assays were made by spiking M9 minimal medium with different amounts of the metabolite under examination. Metabolite yields and kinetic culture parameters were analytically calculated from the raw growth data as described elsewhere (Nikel et al., 2008a, 2010a, 2014b; Nikel and de Lorenzo, 2013a,b).
Statistical Analysis
The reported experiments were independently repeated at least twice (as indicated in the corresponding figure legend), and the mean value of the corresponding parameter ± SD is presented. When appropriate, data were statistically treated with an unpaired Student’s t test, and 95% confidence intervals for each parameter were calculated to demonstrate a statistically significant difference in means among the experimental samples. For the flow cytometry experiments, the geometric mean values (from which the PHB content is derived) were analyzed via the Mann–Whitney U test.
Nucleotide Sequence Accession Numbers
The sequences of the pBAMD vectors were deposited in the GenBank database with the following GenBank accession numbers: KM403113 (pBAMD1-2), KM403114 (pBAMD1-4), and KM403115 (pBAMD1-6).
Results and Discussion
Rationale, Design, and General Characteristics of the mini-Tn5-Based Vectors pBAMDs
Vector pBAM1 (born again mini-transposon) is a synthetic and modular plasmid with a number of features to facilitate the genome editing of Gram-negative bacteria (Martínez-García et al., 2011). We decided to further extend the range of such applications by constructing a new set of pBAM1-derivative plasmids, which are compatible with the rules set in the Standard European Vector Architecture (SEVA) format (Silva-Rocha et al., 2013). We decided to accomplish this challenge by constructing a standardized version of the mini-transposon delivery plasmid that take full advantage of all the benefits of the pBAM1 plasmid together with the functional elements available within the SEVA collection. The starting idea was to design a plasmid series in which the mini-transposon module, the antibiotic-resistance marker, and the tnpA could be easily interchanged at the user’s will. In doing so, several changes were needed to re-structure the constituents of pBAM1, giving rise to three plasmids that were collectively termed pBAMD (i.e., pBAM derivative) vectors. These insertion plasmids share all the advantages and several structural features of their pBAM1 predecessor. In brief, these features include (i) the narrow host-range origin of replication of plasmid R6K [ori(R6K)], dependent on the Π protein (encoded by the pir gene of plasmid R6K); (ii) an origin of transfer, oriT, that allows for the conjugative transfer of the plasmid from a host strain to a new bacterial recipient through RK2-mediated mobilization; (iii) the bla-encoded β-lactamase marker that confers resistance to ampicillin as a selective marker of the backbone vector; and [iv] a modified, hyper-active transposase encoded by tnpA just outside (but adjacent to) a DNA segment that is flanked by the terminal sequences of Tn5 (i.e., the mini-transposon module itself). The Tn5-transposition system is an optimal source of biological parts because of its genetic promiscuity, and it can be considered to operate as a virtually orthogonal part, since it displays an autonomous behavior with respect to the host metabolic and regulatory traits.
We first constructed an intermediary plasmid, named p-R-SETA111, that was later used as the backbone in which the different mini-Tn5 antibiotic modules were implanted to obtain the pBAMD1-x vectors. A mixture of three separate DNA fragments were isothermally assembled (Gibson et al., 2009) to construct the p-R-SETA111 intermediary plasmid (Figure S1 in Supplementary Material). The sequence of p-R-SETA111 was thoroughly checked after assembling with the set of oligonucleotides described in Table S1 in the Supplementary Material. This intermediate vector bears an R6K origin of replication that depends on the Π protein supplied in trans (Kolter et al., 1978) for replication. This situation calls for the use of E. coli pir+ strains to propagate these plasmids (Miller and Mekalanos, 1988; Herrero et al., 1990), such as E. coli DH5α λpir, CC118 λpir, or S17-1 λpir (Table 1). Another feature of plasmid p-R-SETA111 is the presence of a minimized origin of transfer (oriT) from the promiscuous conjugative plasmid RP4 (Lyras and Rood, 1998; Silva-Rocha et al., 2013). Another trait of this intermediary vector shared with the SEVA plasmids is that the cargo module is flanked by the strong T1 and T0 transcriptional terminators (Silva-Rocha et al., 2013), which isolate transcriptionally any DNA sequence cloned in the MCS of p-R-SETA111. Importantly, the modular design of this plasmid allows for the convenient exchange of the tnpA by restriction of p-R-SETA111 with the rare cutters SanDI (5′-GG/GWCCC-3′, W = A or T; Simcox et al., 1995) and AsiSI (5′-GCGAT/CGC-3′). Likewise, the antibiotic marker of the plasmid backbone (bla in this particular case), can be easily exchanged by enzymatic restriction with AsiSI and MluI (5′-A/CGCGT-3′).
Three different mini-Tn5 modules were designed as the cargo segments to be implanted into the p-R-SETA111 plasmid. These elements were devised as cargoes for the SEVA plasmid collection (Silva-Rocha et al., 2013; Durante-Rodríguez et al., 2014), and so they were bracketed by PacI and SpeI restriction sites. The cargo modules have a similar structural design, only differing in the antibiotic marker placed within the mini-transposon (see below). The mini-Tn5 modules are flanked by the two mosaic end (ME) sequences. ME elements are optimized 19-bp DNA sequences recognized by the Tn5 TnpA transposase to promote the specific transposition of any DNA segment bracketed by these elements (Zhou et al., 1998). Even though they are identical in sequence, and with the aim to facilitate the orientation of each functional element within the plasmid, we termed them as either ME-I (the one just after the PacI recognition site) or ME-O (the one close to the SpeI recognition site).
The next relevant feature, placed right after the ME-I element, is a 10-bp buffer DNA sequence that is immediately followed by a MCS (spanning recognition sites for AvrII, SfiI, NotI, EcoRI, SacI, SmaI, BamHI, XbaI, SalI, PstI, SphI, HindIII, and NotI, in the 5′ → 3′direction). This DNA stretch has the same restriction sites as those present in cargo 1 in the SEVA database. The synthetic T500 terminator (Yarnell and Roberts, 1999) was placed after the MCS in order to avoid any transcriptional read-through that any heterologous DNA cloned within the cargo can leak into the following component of the plasmid. The next functional element of the mini-transposon is the antibiotic selection marker that allows for the proper selection of transconjugants. For this set of plasmids, we have used resistances to kanamycin (aphA, encoding an aminoglycoside 3′-phosphotransferase; Martínez-García et al., 2011), streptomycin/spectinomycin (aadA, encoding a streptomycin 3′(9)-O-nucleotidyl transferase; Fling et al., 1985), and gentamicin (aacC1, encoding a gentamicin 3′-N-acetyltransferase; Kovach et al., 1995). These antibiotic-resistance cassettes are coded as 2, 4, and 6 in the SEVA database. Since the antibiotic selection cassettes are flanked by SwaI and PshAI recognition sites, the user has the ability to further expand the plasmid collection by exchanging the cognate resistance genes with any of the two other markers present in the SEVA collection, i.e., cat (encoding a chloramphenicol O-acetyltransferase) or tetA (encoding a tetracycline efflux protein). The rho-independent transcriptional terminator from the gene 32 encoded in the phage T4 genome (Gorski et al., 1985; Miller et al., 2003; Martínez-García et al., 2011) was placed immediately downstream to the antibiotic-resistance gene to prevent any possible read-through from the corresponding promoter elements. Furthermore, the motif 5′-GGGACCC-3′ was changed to 5′-CGGACCC-3′ to eliminate a SanDI restriction target within the existing terminator sequence.
The last step in the construction of the pBAMD1-x vectors was the insertion of the mini-transposon modules themselves into the p-R-SETA111 backbone. The sequences of the three mini-Tn5 modules were edited in silico to follow the SEVA rules and synthesized de novo. The mini-transposon modules were restricted using PacI and SpeI and inserted into p-R-SETA111 previously digested with the same enzymes. The correctness of this last cloning step was verified by DNA sequencing with the SEVA oligonucleotides PS1 and PS2 (Table S1 in the Supplementary Material), that flank the cargo module. The resulting delivery plasmids were termed pBAMD1-2 (KmR), pBAMD1-4 (SmR/SpR), and pBAMD1-6 (GmR) (Figure 1A). Note that the first digit numeric nomenclature stems for the fact that all the pBAMD1-x vectors are derivatives of the pSEVA111 vector, while the second number identify the antibiotic-resistance marker. All the vectors share the same MCS (Figure 1B), also compatible with the rest of SEVA vectors already available.
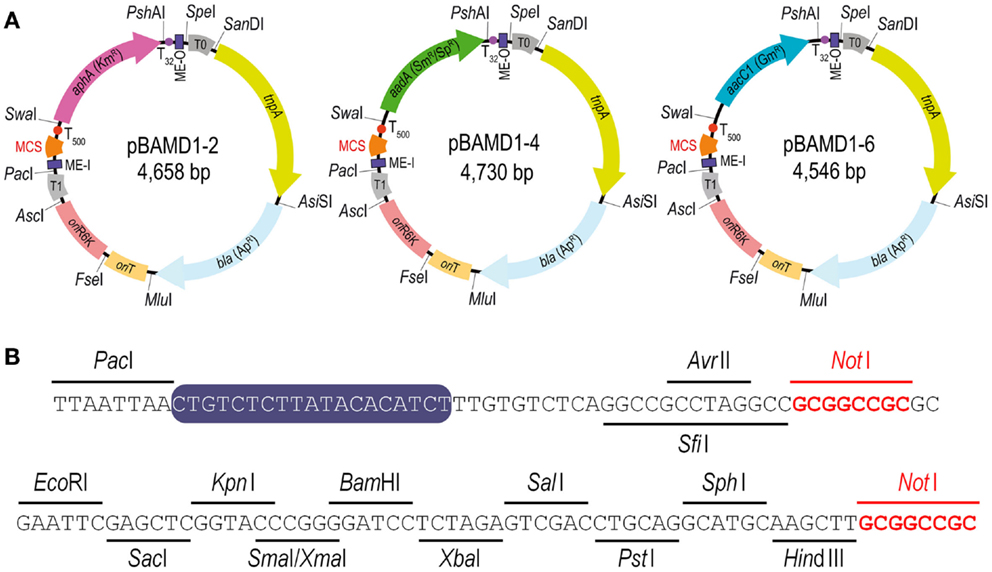
Figure 1. Functional features of the pBAMD1-x delivery vectors. (A) Schematic representation of the pBAMD1-x plasmid series. The functional elements of each plasmid include the relevant restriction sites used for assembling the vectors, the antibiotic-resistance markers (Ap, ampicillin; Km, kanamycin; Sm, streptomycin; Sp, spectinomycin; and Gm, gentamicin), the hyper-active TnpA transposase encoded by tnpA, a conditional origin of replication [ori(R6K)] dependent of the π protein, an origin of transfer (oriT), two mosaic elements (termed ME-I and ME-O), the transcriptional terminators T0 and T1 located just outside the transposon module, and a multiple cloning site (MCS) compatible with any plasmid belonging to the Standard European Vector Architecture (SEVA) initiative (Silva-Rocha et al., 2013; Durante-Rodríguez et al., 2014). Note that the antibiotic-resistance gene determines the full name of each plasmid. (B) Restriction enzymes targets within the multiple cloning site of the pBAMD1-x delivery vectors (x = 2, KmR; x = 4, SmR/SpR; and x = 6, GmR). The MCS starts with the unique AvrII/SfiI recognition sites, and two NotI recognition sites (highlighted in red) were included in the MCS sequence to enable the consecutive assembly of different cargos from the SEVA collection. The sequence of ME-I is indicated by a purple box.
Functional Validation of the pBAMD1-x Vectors in P. putida KT2440
To evaluate the functionality of the new set of Tn5 plasmids, the frequency of transposition into the environmental bacterium P. putida KT2440 was firstly tested in 6 h triparental mating assays. In order to estimate the frequency of transposition events, the number of antibiotic-resistant colonies was assessed after 24 h of incubation at 30°C, and normalized to the total of 1.5 × 108 recipient cells used in each experiment. The average frequency of transposition obtained at 6 h was (3.8 ± 2.9) × 10−4 transconjugants (ranging from a minimum of 2.5 × 10−5 to a maximum of 1.0 × 10−3 transconjugant cells). These figures were independent of the antibiotic marker used (either pBAMD1-2, pBAMD1-4, or pBAMD1-6), and no spontaneous antibiotic-resistant clones (i.e., KmR, SmR, or GmR, respectively) were detected under such growth conditions (data not shown).
The next step was to differentiate between bona fide transposition events and non-specific plasmid integration events in the P. putida genome. To do so, transconjugants cells were re-streaked onto LB medium plates containing 500 μg ml−1 ampicillin and incubated for 24 h to check for possible growth – which would indicate that the corresponding pBAMD vector had integrated into the target chromosome instead of transposing the MEs-flanked DNA cargo. Besides this simple test, the user could also perform colony PCR amplifications to confirm that the transconjugants do not have the pBAMD plasmid backbone by using any of the following two SEVA oligonucleotides combination. If the plasmid is present, the PS5-PS4 oligonucleotide pair will produce a 225-bp amplicon within the oriT region, and the PS5-PS6 oligonucleotide pair will generate a 665-bp amplicon including the oriT segment and the R6K origin of replication (Silva-Rocha et al., 2013). By conducting these two assays, we noticed that 4.2 ± 2.4% of the potential transconjugants obtained with the pBAMD vectors resulted from plasmid co-integration events (a percentage very similar to that reported for other Tn5-based plasmid systems; de Lorenzo et al., 1990). Therefore, it is highly recommended to confirm the nature of the antibiotic-resistant clones obtained after each round of insertions.
We also studied whether the pBAMD mini-transposon delivery plasmids can be used serially to generate double and even triple transconjugant mutants by taking advantage of the three different antibiotic resistance markers (Figure 2A). A first round of transposition was performed with the three individual pBAMD plasmids using P. putida KT2440 as the recipient strain. Transconjugants were re-streaked onto M9 minimal medium plates containing 0.2% (w/v) citrate and supplemented with the appropriate antibiotics to obtain isolated colonies. These colonies were re-streaked again in the same culture medium to obtain pure clones. We randomly picked 22 clones obtained with each pBAMD1-x plasmid and characterized the corresponding transposon insertion site. In 12 out of 22 transconjugant clones, the ME-O related oligonucleotides (Table 2) were used, while in the remaining 10 transconjugant clones, the sequence was obtained by using the ME-I related oligonucleotides for the arbitrary PCR amplification. One KmR clone after the first round of transposition with pBAMD1-2 was selected and used as the recipient strain for the pBAMD1-4 and pBAMD1-6 mini-transposon plasmids. After this second insertion round, we selected four transconjugants obtained with each system, and mapped the landing point of the mini-transposon using only the ME-O related oligonucleotides. Finally, we selected one P. putida KT2440 KmR and GmR and one P. putida KT2440 KmR and SmR to perform the third round of transposition with either pBAMD1-4 or pBAMD1-6, respectively. In the last step, we have chosen one mutant per plasmid system and characterized again the insertion site of the three mini-transposons using the ME-O oligonucleotides. The precise site of transposon insertion could be ascertained in 30 transconjugants out of 32 independent clones (Table 3). Specifically, in one of the cases in which it was not possible to identify the localization of the mini-Tn5 element, the transposon insertion could have happened in either PP2612 or PP3616, since both genes share a 97% sequence identity. In the other case, the transposon insertion site could not be precisely mapped due to the presence of a large number of internal repeats within the lapA gene (PP0168).
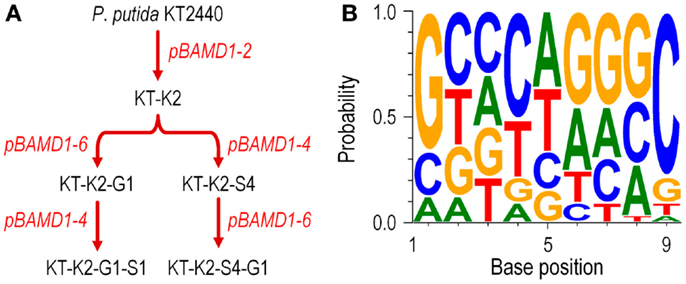
Figure 2. Functional characterization of the pBAMD1-x delivery vectors in P. putida KT2440. (A) Sequential insertion of different mini-Tn5 modules from pBAMD1-x plasmids carrying all the three possible antibiotic-resistance determinants. The flowchart shows the procedure followed for the combinatorial integrations, starting from the wild-type strain KT2440. The names given to the intermediate strains reflect the order in which each antibiotic was delivered into the recipient bacteria (K, kanamycin; S, streptomycin/spectinomycin; and G, gentamicin). The exact chromosomal localization of the insertions in these strains is given in Table 3. Plasmids used in each round of integration are indicated in red. (B) Assessment of the possible sequence preference in the target DNA during the insertion process of the pBAMD1-x delivery vectors in P. putida KT2440. The WebLogo 3.4 software was used to identify the DNA signature (if any) in which the transposon lands in the chromosome of recipient bacteria. The software was fed with the 9-bp DNA sequence targeted by mini-Tn5 in independent trials (Table 3). Note the slight preference for G/C pairs at both ends of the target DNA motif (i.e., in positions 1 and 9).
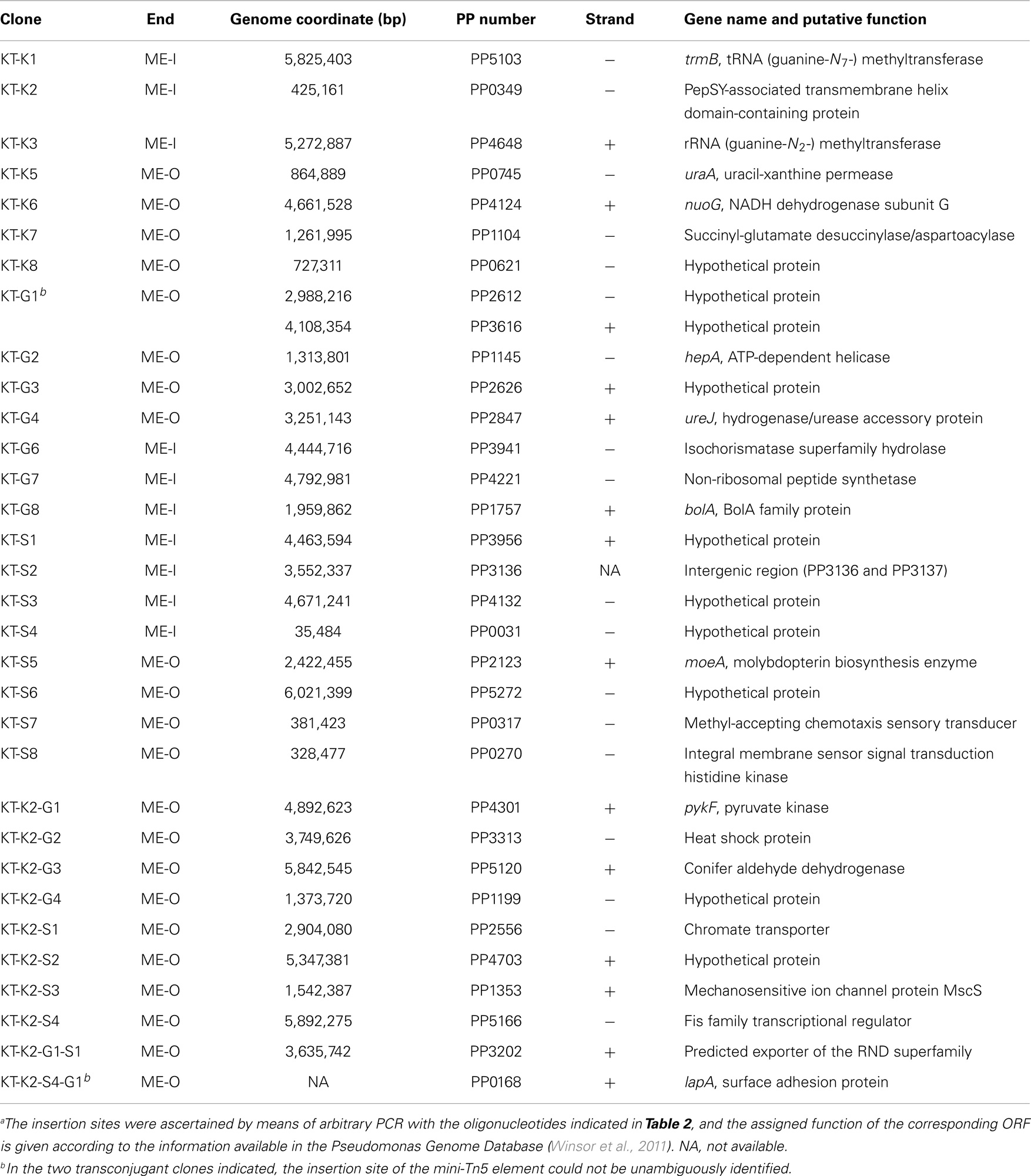
Table 3. Insertion sites of the mini-transposon born by the different pBAMD1-x vectors in the genome of P. putida KT2440a.
We then used the insertion sequence data of each mapped clone in Table 3 to detect any sequence preference for the integration of the mini-Tn5 cassettes within the genome of P. putida KT2440. The web-based application WebLogo 3.4 (Crooks et al., 2004) was fed with the 9-bp landing sequence targeted by the mini-transposon. The program was set to show the probability of having a defined base at any specific position within the 9-bp motif, and the G + C percentage of the genome was adjusted to the value of P. putida KT2440 (G + C = 61.5%; Nelson et al., 2002). The results shown in Figure 2B reveal that there is no DNA sequence bias for the integration site of the mini-transposon, in a similar fashion as observed for plasmid pBAM1 (Martínez-García et al., 2011). However, a relatively minor preference for G/C pairs at both ends of the target DNA motif could be observed, as detected for other systems based on Tn5 (Lodge et al., 1988). These experiments confirm that the three mini-transposons borne by the pBAMD1-x vectors could be used serially to generate a second or even a third round of insertion mutagenesis procedure, or also to stably integrate multiple genetic devices into the genome of a single microbial strain.
Design and Construction of a Microbial Cell Factory for Poly(3-hydroxybutyrate) Synthesis
Engineering an stable PHB+ phenotype in E. coli
Poly(3-hydroxybutyrate) is an isotactic polyester composed by 3-hydroxybutyrate units (Anderson and Dawes, 1990). The PHB synthesis pathway in C. necator (formerly known as Ralstonia eutropha) comprises three enzymes (Figure 3A) (Steinbüchel and Hein, 2001). PhaA, a 3-ketoacyl-CoA thiolase, condenses two acetyl-CoA moities, yielding 3-acetoacetyl-CoA. This intermediate is the substrate for PhaB, a NADPH-dependent 3-acetoacetyl-CoA reductase (encoded by phaB1). In the final step of this biosynthetic pathway, (R)-(–)-3-hydroxybutyryl-CoA is polymerized to PHB by PhaC, a poly(3-hydroxyalkanoate) synthase (encoded by phaC1). The very idea of a thermoplastic and biocompatible material, which is also readily biodegraded by a number of bacteria has become very attractive in an era of increasing environmental concern (Keshavarz and Roy, 2010). A number of different recombinant E. coli strains designed for polymer accumulation have been constructed thus far (Li et al., 2007; Chen et al., 2013; Ruiz et al., 2013), outsourcing the pha genes from several bacteria (Verlinden et al., 2007). However, most of the PHB production systems available thus far suffer from a number of drawbacks (Wang et al., 2014). Among them, the transcriptional regulation of the pha genes is of particular importance. In natural producer bacteria, PHB accumulation is triggered by an imbalance in the availability of critical nutrients (e.g., the N or S source; Anderson and Dawes, 1990). In recombinant E. coli, however, the constitutive expression of the pha genes leads to a growth-dependent accumulation of PHB, which normally results in metabolic burden in the producing cells (Wang and Lee, 1997). Controlling the rate of polymer accumulation in recombinant E. coli is thus of paramount importance for the design of efficient microbial cell factories. Besides this feature, the segregational stability of plasmids in E. coli recombinants could also be an issue in prolonged fermentation processes aimed at biopolymer production (Nikel et al., 2010b; Ruiz et al., 2013).
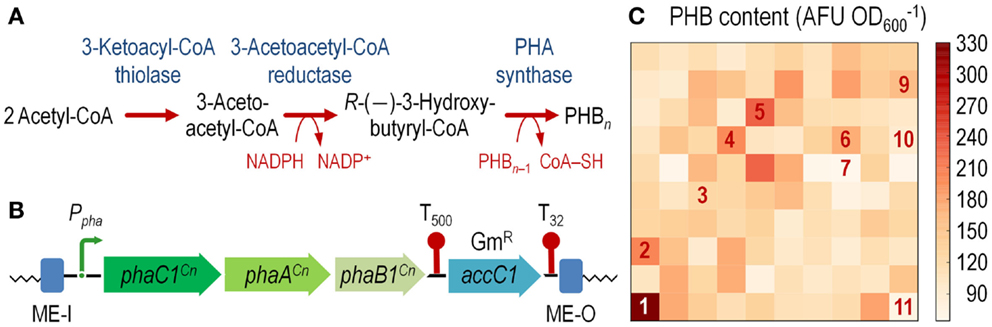
Figure 3. Construction of an E. coli cell factory expressing the phaC1AB1 gene cluster from C. necator as a chromosomal insertion. (A) Poly(3-hydroxybutyrate) (PHB) biosynthesis pathway. Three enzymes are necessary for de novo synthesis of PHB in C. necator: a 3-ketoacyl-coenzyme A (CoA) thiolase (PhaA), a NADPH-dependent 3-acetoacetyl-CoA reductase (PhaB1), and a PHB synthase (PhaC1). PhaA and PhaB1 catalyze the condensation of two molecules of acetyl-CoA to 3-acetoacetyl-CoA and the reduction of acetoacetyl-CoA to R-(–)-3-hydroxybutyryl-CoA, respectively. PhaC1 polymerizes these monomers to PHB, whereas one CoA-SH molecule per monomer is released. The resulting PHB polymer is stored as water-insoluble granules in the cytoplasm of the cells. (B) Organization of the functional elements borne by plasmid pBAM1-6-pha and transferred into the chromosome of the recipient E. coli strain. The transcriptional terminators included in the plasmid backbone, which flank the gentamicin resistance (GmR) determinant (accC1), are depicted as T500 and T32. Note that the elements in this outline are not drawn to scale. (C) Exploring the landscape of PHB synthesis in E. coli transconjugants. The phaC1AB1 gene cluster from C. necator was randomly integrated into the chromosome of E. coli JW2293-1 (Δpta), and 24-h cultures of individual colonies were analyzed for PHB accumulation by fluorimetry after staining the cells with Nile red (see Materials and Methods for details). Several colonies, identified by numbers in the heat-map, were kept and further analyzed to establish the precise site of mini-Tn5(phaC1AB1) insertion (Table S2 in the Supplementary Material). AFU, arbitrary fluorescence units.
To overcome this state of affairs, we decided to explore the landscape of potentially useful transcription levels in E. coli by randomly integrating the pha genes from C. necator into the chromosome. To this end, we used vector pBAMD1-6 as the backbone to clone a 5.3-kb DNA fragment spanning the phaC1AB1 genes. Plasmid pAeT41 (Peoples and Sinskey, 1989) was digested with EcoRI and SmaI to liberate the aforementioned DNA segment, and inserted into the corresponding restriction sites of pBAMD1-6 to generate plasmid pBAMD1-6-pha (Table 1). The functional parts of the DNA element to be transferred into E. coli are shown in Figure 3B.
As acetyl-CoA is the precursor metabolite for PHB formation, competing pathways that use this intermediate are expected to drain building blocks of the biopolymer synthesis. In E. coli, the acetate formation pathway, comprising Pta and AckA (phosphotransacetylase and acetate kinase), uses acetyl-CoA as the starting metabolite (Neidhardt et al., 1990). This pathway, which diverts a considerable amount of carbon from the central metabolism (Wolfe, 2005), is active under both oxic and anoxic conditions (Clark, 1989). For this reason, the phaBAC gene cluster was delivered into a Δpta recipient strain (Table 1), in which the acetate formation is expected to be low. Plasmid pBAMD1-6-pha was first transferred to E. coli S17-1 λpir to perform a biparental mating. E. coli S17-1 λpir or SM10 λpir are the preferred donor strains when mobilizing RP4-based plasmid to E. coli recipient strains since they bear the functional tra and mob elements integrated in the genome, thus avoiding the inadvertent transfer of the mating-helper plasmid (pRK600) alongside the mini-transposon delivery system. Alternatively, E. coli strain MFD pir+ (Ferrières et al., 2010), devoid of the Mu element present in the strains detailed above, could be used for transposon insertions.
After integration of the mini-Tn5:phaC1AB1 device into E. coli JW2293-1, individual GmR colonies were purified and separately grown in microtiter plates as explained in the Section “Materials and Methods” to explore PHB accumulation in 100 independent transconjugants after 24 h of incubation. Figure 3C shows the level of PHB accumulation in the transconjugants as compared to that of E. coli JW2293-1 carrying the pAeT41 plasmid, in which the expression of the phaC1AB1 gene cluster is driven by the native promoter. All the transconjugants tested accumulated the polymer from mono-copy chromosomal insertions of the PHB biosynthesis pathway to some extent, ranging from 3% up to 78% of the accumulation levels observed in E. coli JW2293P (which carries plasmid pAeT41). Table S2 in the Supplementary Material shows the insertion locus for some selected phaC1AB1+ transconjugants, clearly illustrating the non-selective nature of the chromosomal incorporation of Tn5 (and consequently, the wide range of PHB accumulation levels), as it was already observed in P. putida transconjugants. We selected one of the transconjugants, termed E. coli TA2293P, that accumulated high polymer levels (clone 1 in Figure 3C and Table S2 in Supplementary Material), and the insertion site of the transposon was determined to be ykgH, an open reading frame encoding a predicted inner membrane protein (Table S2 in the Supplementary Material). The physiology of PHB accumulation of this strain was studied as detailed below.
Physiological and biochemical characterization of E. coli TA2293P as a microbial cell factory for PHB synthesis
The growth parameters of several E. coli strains were determined to explore their potential as biopolymer cell factories (Table 4). We decided to compare the performance of strains in which the pha genes are expressed either in a multi-copy plasmid or as a mono-copy insertion in the bacterial chromosome side-by-side. Interestingly, the elimination of Pta resulted in a reduction of the specific growth rate, probably by an imbalance in the acetyl-CoA pool that was partially restored by the heterologous expression of the phaC1AB1 gene cluster. This positive effect was more evident in the strain in which the genes were inserted into the chromosome (the specific growth rate attained ca. 85% of that in the wild-type strain), thus suggesting that the adequate expression level of the PHB biosynthesis genes is important to recover a homeostatic acetyl-CoA balance. This kinetic pattern was also mirrored in the final biomass density of the cultures. In fact, among the strains tested, E. coli TA2293P attained the highest cell density. This result highlights the advantage of integrating the pha genes in the chromosome, as such approach not only avoids the metabolic burden usually associated with heterologous gene expression from plasmids and other extra-chromosomal elements but it also allows for the selection of an integrant strain exhibiting the appropriate level of transcription of the corresponding genes. Moreover, the selection of ykgH as a target was not at all obvious, indicating, again, the value of random insertion of the genes of interest in the bacterial chromosome.
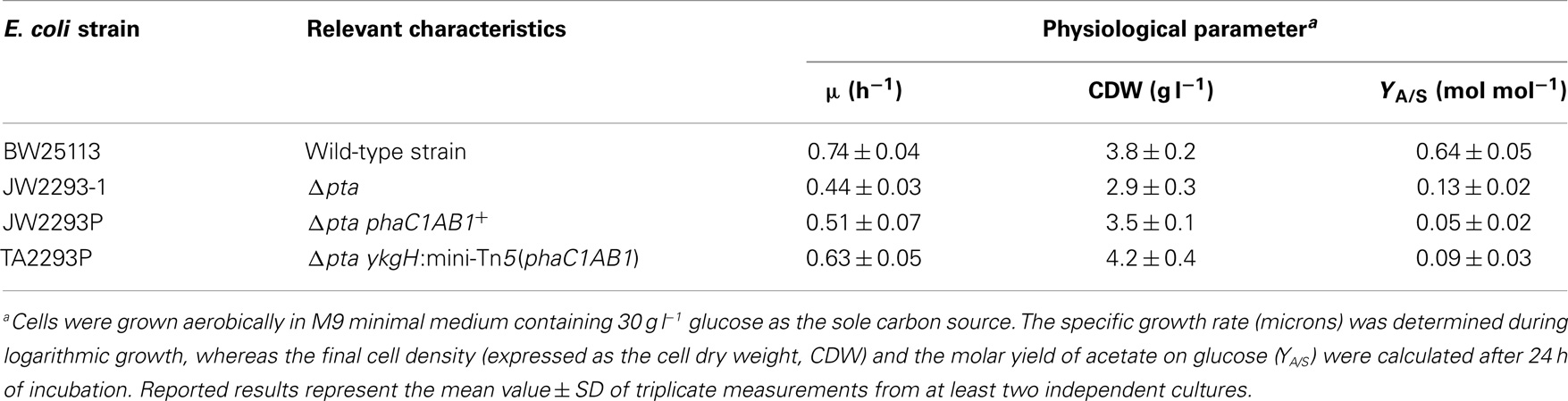
Table 4. Physiological characterization of wild-type and mutant E. coli strains as microbial cell factories for PHB biosynthesis in shaken-flask cultures.
Since we selected a pta mutant of E. coli as the recipient strain, in which acetate formation is expected to be impaired, by-product formation was also explored in these cultures as a measure of the carbon flow from glucose to PHB (Table 4). In cultures of the wild-type strain, up to 60% of the total carbon source was converted into acetate, pinpointing this metabolite as the key by-product of hexose catabolism in E. coli (and therefore, as the main side pathway competing for acetyl-CoA). The pta mutant still produced some acetate (probably through the action of pyruvate oxidase, PoxB); however, the molar conversion of glucose into acetate reached only ca. 20% of that observed in the wild-type strain. Acetate formation in both E. coli strains carrying PhaC1AB1 was comparable, and much lower than the other two strains. In all, these results bear witness of (i) the suitability of a pta mutant, deficient in acetate formation, as the starting point to construct a PHB cell factory, and (ii) the effect of the PHB biosynthetic pathway in using acetyl-CoA as the precursor metabolite. Once the coarse physiological characterization of these strains was completed, the next relevant question was how they perform as PHB producers.
Since PhaA is the first committed enzymatic step of PHB formation from acetyl-CoA, the in vitro activity of this enzyme was assayed as a proxy of the activity of the whole biosynthetic pathway in shaken-flask cultures using glucose as the sole carbon source (Figure 4A). Note that there was some degree of thiolase activity in both E. coli BW25113 and JW2293-1, probably represented by FadA (an enzyme normally involved in the degradation of fatty acids via the β-oxidation cycle). However, the specific PhaA activity was six and fourfold higher in the strains carrying the phaC1AB1 gene cluster (E. coli JW2293P and TA2293P, respectively) as compared to that of E. coli JW2293-1. As expected, the highest enzymatic activity corresponded to the strain carrying the pha genes in a multi-copy plasmid. Nevertheless, E. coli TA2293P had an activity level ca. 60% of the strain bearing pAeT41, indicating that the appropriate insertion of the gene cluster could result in thiolase activities similar to those of a typical recombinant PHB producer. Yet, how do these activities translate into PHB accumulation?
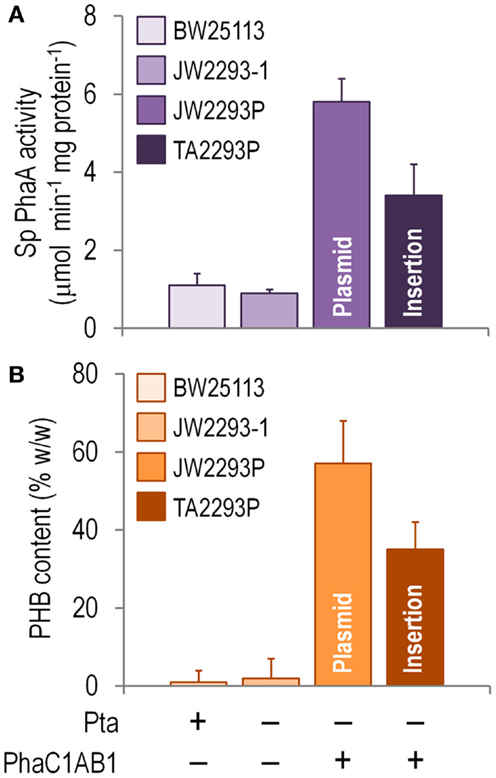
Figure 4. Biochemical characterization of E. coli TA2293P as a microbial cell factory for PHB synthesis. (A) In vitro determination of the specific (Sp) 3-ketoacyl-coenzyme A thiolase (PhaA) activity. Cells were harvested after growing them for 24 h in M9 minimal medium added with 30 g l−1 glucose as the sole carbon source, and the activity of PhaA was determined in the cell-free extract as detailed in the Section “Materials and Methods.” (B) Poly(3-hydroxybutyrate) (PHB) accumulation. The PHB content (expressed as a percentage of the cell dry weight) was assessed by flow cytometry after growing the cells for 24 h in M9 minimal medium added with 30 g l−1 glucose as the sole carbon source. In all cases, each bar represents the mean value of the corresponding enzymatic activity ± SD of triplicate measurements from at least two independent experiments. The strains used to explore these biochemical traits were E. coli BW25113 (wild-type strain), E. coli JW2293-1 (Δpta), E. coli JW2293P (Δpta, carrying the phaC1AB1 gene cluster in a multi-copy plasmid), and E. coli TA2293P [Δpta, ykgH:mini-Tn5(phaC1AB1)]. See Table 1 for further details about the genotype of each E. coli strain. The relevant features of each strain are indicated at the bottom of the figure.
Figure 4B shows that E. coli TA2293P accumulated PHB up to 62% of the level observed in the same strain but expressing the pha genes in a plasmid (i.e., E. coli JW2293P). As previously observed in other traits during the physiological characterization, the later strain had the highest PHB accumulation level among all the strains tested. That E. coli TA2293P accumulates such a high amount of PHB is a somewhat surprising (and welcome) result considering the difference in copy number between the two phaC1AB1+ strains under comparison. Note that a possible effect of the absence (or an altered expression level) of YkgH on the properties of E. coli TA2293P cannot be completely ruled out. Interestingly, when this strain was persistently cultured in LB medium without any selective pressure, its phenotypic traits, particularly regarding polymer accumulation, remained unchanged. By contrast, the segregational stability of pAeT41 was assessed in cultures of E. coli JW2293P, and, after growing the cells in LB medium and sub-culturing them daily seven times without any antibiotic, <25% of the cells were resistant to ampicillin.
Conclusion
In our current study, we presented a set of new mini-Tn5-derived vectors that can be used to engineer the genome of Gram-negative bacteria. While the worth of these tools has been exposed in two model bacteria, P. putida and E. coli, the inherent promiscuity of Tn5 ensures its functioning in a number of different microbial hosts. Of particular importance is the possibility of sequentially using the three pBAMD1-x vectors, thereby enabling the user to accumulate insertions in the same genetic background in a combinatorial fashion. This is a particularly interesting feature for the construction of complex phenotypes, such as biopolymer formation, which depend on more than one enzyme. Although the expression of the biosynthetic genes in a multi-copy plasmid is in principle enough to bestow the desired phenotype on the recipient bacterium, fine-tuned expression levels (together with the appearance of emergent phenotypic properties in the host, brought about by the insertion process itself) can be easily achieved by randomly integrating the structural genes into the chromosome. In this way, and since the genetic context of each integration will surely result in different regulatory patterns at the transcriptional level, the user could choose among a library of insertions those clones that meet any desired criterion (in our case, PHB accumulation). Moreover, as the delivery plasmids described in this study can be used in a sequential manner, other polymer-associated enzymes, such as phasins, can also be incorporated in the same strain to enhance further polymer production.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are indebted to Prof. A. Sinskey (Massachusetts Institute of Technology) for sharing research materials and to A. Goñi (CNB-CSIC) for his help in image processing. I. Benedetti (CNB-CSIC) is gratefully acknowledged for her help in FACS measurements. This study was supported by the BIO Program of the Spanish Ministry of Economy and Competitiveness, the ST-FLOW and ARISYS Contracts of the EU, the ERANET-IB Program, and the PROMT Project of the CAM to VDL. PIN is a researcher from the Consejo Nacional de Investigaciones Científicas y Técnicas (Argentina) and holds a Marie Curie Actions Program grant from the EC (ALLEGRO, UE-FP7-PEOPLE-2011-IIF-300508). The authors declare no conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/Journal/10.3389/fbioe.2014.00046/abstract
References
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi:10.1093/nar/25.17.3389
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Anderson, A. J., and Dawes, E. A. (1990). Occurrence, metabolism, metabolic role, and industrial uses of bacterial polyhydroxyalkanoates. Microbiol. Rev. 54, 450–472.
Baba, T., Ara, T., Hasegawa, M., Takai, Y., Okumura, Y., Baba, M., et al. (2006). Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2, 2006.0008. doi:10.1038/msb4100050
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bagdasarian, M., Lurz, R., Rückert, B., Franklin, F. C., Bagdasarian, M. M., Frey, J., et al. (1981). Specific-purpose plasmid cloning vectors. II. Broad host range, high copy number, RSF1010-derived vectors, and a host-vector system for gene cloning in Pseudomonas. Gene 16, 237–247. doi:10.1016/0378-1119(81)90080-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Berg, D. E. (1989). “Transposon Tn5,” in Mobile DNA, eds D. E. Berg and M. M. Howe (Washington, DC: American Society for Microbiology Press), 185–210.
Boyer, H. W., and Roulland-Dussoix, D. (1969). A complementation analysis of the restriction and modification of DNA in Escherichia coli. J. Mol. Biol. 41, 459–472. doi:10.1016/0022-2836(69)90288-5
Bradford, M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254. doi:10.1016/0003-2697(76)90527-3
Brosius, J., Cate, R. L., and Perlmutter, A. P. (1982). Precise location of two promoters for the β-lactamase gene of pBR322. S1 mapping of ribonucleic acid isolated from Escherichia coli or synthesized in vitro. J. Biol. Chem. 257, 9205–9210.
Chen, X., Zhou, L., Tian, K., Kumar, A., Singh, S., Prior, B. A., et al. (2013). Metabolic engineering of Escherichia coli: a sustainable industrial platform for bio-based chemical production. Biotechnol. Adv. 31, 1200–1223. doi:10.1016/j.biotechadv.2013.02.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Clark, D. P. (1989). The fermentation pathways of Escherichia coli. FEMS Microbiol. Rev. 5, 223–234. doi:10.1111/j.1574-6968.1989.tb03398.x
Crooks, G. E., Hon, G., Chandonia, J. M., and Brenner, S. E. (2004). WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190. doi:10.1101/gr.849004
Das, S., Noe, J. C., Paik, S., and Kitten, T. (2005). An improved arbitrary primed PCR method for rapid characterization of transposon insertion sites. J. Microbiol. Methods 63, 89–94. doi:10.1016/j.mimet.2005.02.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Datsenko, K. A., and Wanner, B. L. (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645. doi:10.1073/pnas.120163297
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
de Lorenzo, V., Cases, I., Herrero, M., and Timmis, K. N. (1993). Early and late responses of TOL promoters to pathway inducers: identification of postexponential promoters in Pseudomonas putida with lacZ-tet bicistronic reporters. J. Bacteriol. 175, 6902–6907.
de Lorenzo, V., Herrero, M., Jakubzik, U., and Timmis, K. N. (1990). Mini-Tn5 transposon derivatives for insertion mutagenesis, promoter probing, and chromosomal insertion of cloned DNA in Gram-negative Eubacteria. J. Bacteriol. 172, 6568–6572.
de Lorenzo, V., Herrero, M., Sánchez, J. M., and Timmis, K. N. (1998). Mini-transposons in microbial ecology and environmental biotechnology. FEMS Microbiol. Ecol. 27, 211–224. doi:10.1111/j.1574-6941.1998.tb00538.x
de Lorenzo, V., and Timmis, K. N. (1994). Analysis and construction of stable phenotypes in Gram-negative bacteria with Tn5- and Tn10-derived minitransposons. Meth. Enzymol. 235, 386–405. doi:10.1016/0076-6879(94)35157-0
Durante-Rodríguez, G., de Lorenzo, V., and Martínez-García, E. (2014). The Standard European Vector Architecture (SEVA) plasmid toolkit. Methods Mol. Biol. 1149, 469–478. doi:10.1007/S978-1-4939-0473-0_36
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ferrières, L., Hémery, G., Nham, T., Guérout, A. M., Mazel, D., Beloin, C., et al. (2010). Silent mischief: bacteriophage Mu insertions contaminate products of Escherichia coli random mutagenesis performed using suicidal transposon delivery plasmids mobilized by broad-host-range RP4 conjugative machinery. J. Bacteriol. 192, 6418–6427. doi:10.1128/JB.00621-10
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fling, M. E., Kopf, J., and Richards, C. (1985). Nucleotide sequence of the transposon Tn7 gene encoding an aminoglycoside-modifying enzyme, 3”(9)-O-nucleotidyltransferase. Nucleic Acids Res. 13, 7095–7106. doi:10.1093/nar/13.19.7095
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gibson, D. G., Young, L., Chuang, R. Y., Venter, J. C., Hutchison, C. A., and Smith, H. O. (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345. doi:10.1038/nmeth.1318
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gorski, K., Roch, J. M., Prentki, P., and Krisch, H. M. (1985). The stability of bacteriophage T4 gene 32 mRNA: a 5′ leader sequence that can stabilize mRNA transcripts. Cell 43, 461–469. doi:10.1016/0092-8674(85)90176-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hanahan, D., and Meselson, M. (1983). Plasmid screening at high colony density. Meth. Enzymol. 100, 333–342. doi:10.1016/0076-6879(83)00066-X
Herrero, M., de Lorenzo, V., and Timmis, K. N. (1990). Transposon vectors containing non-antibiotic resistance selection markers for cloning and stable chromosomal insertion of foreign genes in Gram-negative bacteria. J. Bacteriol. 172, 6557–6567.
Keshavarz, T., and Roy, I. (2010). Polyhydroxyalkanoates: bioplastics with a green agenda. Curr. Opin. Microbiol. 13, 321–326. doi:10.1016/j.mib.2010.02.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kessler, B., de Lorenzo, V., and Timmis, K. N. (1992). A general system to integrate lacZ fusions into the chromosomes of Gram-negative Eubacteria: regulation of the Pm promoter of the TOL plasmid studied with all controlling elements in monocopy. Mol. Gen. Genet. 233, 293–301. doi:10.1007/BF00587591
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kolter, R., Inuzuka, M., and Helinski, D. R. (1978). trans-Complementation-dependent replication of a low molecular weight origin fragment from plasmid R6K. Cell 15, 1199–1208. doi:10.1016/0092-8674(78)90046-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kovach, M. E., Elzer, P. H., Hill, D. S., Robertson, G. T., Farris, M. A., Roop, R. M., et al. (1995). Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166, 175–176. doi:10.1016/0378-1119(95)00584-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, R., Zhang, H., and Qi, Q. (2007). The production of polyhydroxyalkanoates in recombinant Escherichia coli. Bioresour. Technol. 98, 2313–2320. doi:10.1016/j.biortech.2006.09.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lodge, J. K., Weston-Hafer, K., and Berg, D. E. (1988). Transposon Tn5 target specificity: preference for insertion at G/C pairs. Genetics 120, 645–650.
Lyras, D., and Rood, J. I. (1998). Conjugative transfer of RP4-oriT shuttle vectors from Escherichia coli to Clostridium perfringens. Plasmid 39, 160–164. doi:10.1006/plas.1997.1325
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Martínez-García, E., Calles, B., Arévalo-Rodríguez, M., and de Lorenzo, V. (2011). pBAM1: an all-synthetic genetic tool for analysis and construction of complex bacterial phenotypes. BMC Microbiol. 11:38. doi:10.1186/1471-2180-11-38
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Martínez-García, E., and de Lorenzo, V. (2012). Transposon-based and plasmid-based genetic tools for editing genomes of Gram-negative bacteria. Methods Mol. Biol. 813, 267–283. doi:10.1007/978-1-61779-412-4_16
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Miller, E. S., Kutter, E., Mosig, G., Arisaka, F., Kunisawa, T., and Rüger, W. (2003). Bacteriophage T4 genome. Microbiol. Mol. Biol. Rev. 67, 86–156. doi:10.1128/MMBR.67.1.86-156.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Miller, V. L., and Mekalanos, J. J. (1988). A novel suicide vector and its use in construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR. J. Bacteriol. 170, 2575–2583.
Neidhardt, F. C., Ingraham, J. L., and Schaechter, M. (1990). Physiology of the Bacterial Cell: A Molecular Approach. Sunderland, MA: Sinauer Associates.
Nelson, K. E., Weinel, C., Paulsen, I. T., Dodson, R. J., Hilbert, H., Martins dos Santos, V. A. P., et al. (2002). Complete genome sequence and comparative analysis of the metabolically versatile Pseudomonas putida KT2440. Environ. Microbiol. 4, 799–808. doi:10.1046/j.1462-2920.2002.00366.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nikel, P. I., de Almeida, A., Pettinari, M. J., and Méndez, B. S. (2008a). The legacy of HfrH: mutations in the two-component system CreBC are responsible for the unusual phenotype of an Escherichia coli arcA mutant. J. Bacteriol. 190, 3404–3407. doi:10.1128/JB.00040-08
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nikel, P. I., Pettinari, M. J., Galvagno, M. A., and Méndez, B. S. (2008b). Poly(3-hydroxybutyrate) synthesis from glycerol by a recombinant Escherichia coli arcA mutant in fed-batch microaerobic cultures. Appl. Microbiol. Biotechnol. 77, 1337–1343. doi:10.1007/s00253-007-1255-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nikel, P. I., and de Lorenzo, V. (2013a). Implantation of unmarked regulatory and metabolic modules in Gram-negative bacteria with specialised mini-transposon delivery vectors. J. Biotechnol. 163, 143–154. doi:10.1016/j.jbiotec.2012.05.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nikel, P. I., and de Lorenzo, V. (2013b). Engineering an anaerobic metabolic regime in Pseudomonas putida KT2440 for the anoxic biodegradation of 1,3-dichloroprop-1-ene. Metab. Eng. 15, 98–112. doi:10.1016/j.ymben.2012.09.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nikel, P. I., Giordano, A. M., de Almeida, A., Godoy, M. S., and Pettinari, M. J. (2010a). Elimination of d-lactate synthesis increases poly(3-hydroxybutyrate) and ethanol synthesis from glycerol and affects cofactor distribution in recombinant Escherichia coli. Appl. Environ. Microbiol. 76, 7400–7406. doi:10.1128/AEM.02067-10
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nikel, P. I., Pettinari, M. J., Galvagno, M. A., and Méndez, B. S. (2010b). Metabolic selective pressure stabilizes plasmids carrying biosynthetic genes for reduced biochemicals in Escherichia coli redox mutants. Appl. Microbiol. Biotechnol. 88, 563–573. doi:10.1007/s00253-010-2774-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nikel, P. I., Martínez-García, E., and de Lorenzo, V. (2014a). Biotechnological domestication of pseudomonads using synthetic biology. Nat. Rev. Microbiol. 12, 368–379. doi:10.1038/nrmicro3253
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nikel, P. I., Kim, J., and de Lorenzo, V. (2014b). Metabolic and regulatory rearrangements underlying glycerol metabolism in Pseudomonas putida KT2440. Environ. Microbiol. 16, 239–254. doi:10.1111/1462-2920.12224
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Palmer, M. A. J., Differding, E., Gamboni, R., Williams, S. F., Peoples, O. P., Walsh, C. T., et al. (1991). Biosynthetic thiolase from Zoogloea ramigera. Evidence for a mechanism involving Cys-378 as the active site base. J. Biol. Chem. 266, 8369–8375.
Peoples, O. P., and Sinskey, A. J. (1989). Poly-β-hydroxybutyrate (PHB) biosynthesis in Alcaligenes eutrophus H16. Identification and characterization of the PHB polymerase gene (phbC). J. Biol. Chem. 264, 15298–15303.
Phadnis, S. H., Sasakawa, C., and Berg, D. E. (1986). Localization of action of the IS50-encoded transposase protein. Genetics 112, 421–427.
Pratt, L. A., and Kolter, R. (1998). Genetic analysis of Escherichia coli biofilm formation: roles of flagella, motility, chemotaxis and type I pili. Mol. Microbiol. 30, 285–293. doi:10.1046/j.1365-2958.1998.01061.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Reznikoff, W. S. (2006). Tn5 transposition: a molecular tool for studying protein structure-function. Biochem. Soc. Trans. 34, 320–323. doi:10.1042/BST20060320
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Reznikoff, W. S. (2008). Transposon Tn5. Annu. Rev. Genet. 42, 269–286. doi:10.1146/annurev.genet.42.110807.091656
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ruiz, J. A., de Almeida, A., Godoy, M. S., Mezzina, M. P., Bidart, G. N., Méndez, B. S., et al. (2013). Escherichia coli redox mutants as microbial cell factories for the synthesis of reduced biochemicals. Comput. Struct. Biotechnol. J. 3, e201210019. doi:10.5936/csbj.201210019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sambrook, J., Maniatis, T., and Fritsch, E. F. (1989). Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
Silva-Rocha, R., Martínez-García, E., Calles, B., Chavarría, M., Arce-Rodríguez, A., de Las Heras, A., et al. (2013). The Standard European Vector Architecture (SEVA): a coherent platform for the analysis and deployment of complex prokaryotic phenotypes. Nucleic Acids Res. 41, D666–D675. doi:10.1093/nar/gks1119
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Simcox, T. G., Fabian, L., Kretz, K., Hedden, V., and Simcox, M. E. (1995). SanDI, a new type-II restriction endonuclease that recognizes 5′-GG/GWCCC-3′. Gene 155, 129–130. doi:10.1016/0378-1119(94)00711-Z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Slater, S., Houmiel, K. L., Tran, M., Mitsky, T. A., Taylor, N. B., Padgette, S. R., et al. (1998). Multiple β-ketothiolases mediate poly(β-hydroxyalkanoate) copolymer synthesis in Ralstonia eutropha. J. Bacteriol. 180, 1979–1987.
Spiekermann, P., Rehm, B. H., Kalscheuer, R., Baumeister, D., and Steinbüchel, A. (1999). A sensitive, viable-colony staining method using Nile red for direct screening of bacteria that accumulate polyhydroxyalkanoic acids and other lipid storage compounds. Arch. Microbiol. 171, 73–80. doi:10.1007/s002030050681
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Steinbüchel, A., and Hein, S. (2001). Biochemical and molecular basis of microbial synthesis of polyhydroxyalkanoates in microorganisms. Adv. Biochem. Eng. Biotechnol. 71, 81–123. doi:10.1007/3-540-40021-4_3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tyo, K. E., Zhou, H., and Stephanopoulos, G. N. (2006). High-throughput screen for poly-3-hydroxybutyrate in Escherichia coli and Synechocystis sp. strain PCC6803. Appl. Environ. Microbiol. 72, 3412–3417. doi:10.1128/AEM.72.5.3412-3417.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Verlinden, R. A., Hill, D. J., Kenward, M. A., Williams, C. D., and Radecka, I. (2007). Bacterial synthesis of biodegradable polyhydroxyalkanoates. J. Appl. Microbiol. 102, 1437–1449. doi:10.1111/j.1365-2672.2007.03335.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, F., and Lee, S. Y. (1997). Production of poly(3-hydroxybutyrate) by fed-batch culture of filamentation-suppressed recombinant Escherichia coli. Appl. Environ. Microbiol. 63, 4765–4769.
Wang, Y., Yin, J., and Chen, G. Q. (2014). Polyhydroxyalkanoates, challenges and opportunities. Curr. Opin. Biotechnol. 30, 59–65. doi:10.1016/j.copbio.2014.06.001
Winsor, G. L., Lam, D. K. W., Fleming, L., Lo, R., Whiteside, M. D., Yu, N. Y., et al. (2011). Pseudomonas genome database: improved comparative analysis and population genomics capability for Pseudomonas genomes. Nucleic Acids Res. 39, D596–D600. doi:10.1093/nar/gkq869
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wolfe, A. J. (2005). The acetate switch. Microbiol. Mol. Biol. Rev. 69, 12–50. doi:10.1128/MMBR.69.1.12-50.2005
Yarnell, W. S., and Roberts, J. W. (1999). Mechanism of intrinsic transcription termination and antitermination. Science 284, 611–615. doi:10.1126/science.284.5414.611
Zhou, M., Bhasin, A., and Reznikoff, W. S. (1998). Molecular genetic analysis of transposase-end DNA sequence recognition: cooperativity of three adjacent base-pairs in specific interaction with a mutant Tn5 transposase. J. Mol. Biol. 276, 913–925. doi:10.1006/jmbi.1997.1579
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: metabolic engineering, Pseudomonas putida, Escherichia coli, transposon mini-Tn5, central metabolism, chromosomal integration, polyhydroxyalkanoates
Citation: Martínez-García E, Aparicio T, de Lorenzo V and Nikel PI (2014) New transposon tools tailored for metabolic engineering of Gram-negative microbial cell factories. Front. Bioeng. Biotechnol. 2:46. doi: 10.3389/fbioe.2014.00046
Received: 22 August 2014; Accepted: 13 October 2014;
Published online: 28 October 2014.
Edited by:
Jean Marie François, CNRS, FranceReviewed by:
M. Kalim Akhtar, University College London, UKDaehee Lee, Korea Research Institute of Bioscience and Biotechnology, South Korea
Copyright: © 2014 Martínez-García, Aparicio, de Lorenzo and Nikel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pablo I. Nikel, Systems and Synthetic Biology Program (CNB-CSIC), C/Darwin, 3, Madrid 28049, Spain e-mail:cGFibG8ubmlrZWxAY25iLmNzaWMuZXM=