
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
TECHNOLOGY AND CODE article
Front. Big Data , 18 January 2022
Sec. Data Mining and Management
Volume 4 - 2021 | https://doi.org/10.3389/fdata.2021.727216
This article is part of the Research Topic Towards Exascale Solutions for Big Data Computing View all 5 articles
 Jinxiang Chen1†
Jinxiang Chen1† Fuyi Li2,3,4†Miao Wang1Junlong Li1Tatiana T. Marquez-Lago5,6André Leier5,6Jerico Revote2Shuqin Li1
Fuyi Li2,3,4†Miao Wang1Junlong Li1Tatiana T. Marquez-Lago5,6André Leier5,6Jerico Revote2Shuqin Li1 Quanzhong Liu1*
Quanzhong Liu1* Jiangning Song2,3*
Jiangning Song2,3*Background: Simple Sequence Repeats (SSRs) are short tandem repeats of nucleotide sequences. It has been shown that SSRs are associated with human diseases and are of medical relevance. Accordingly, a variety of computational methods have been proposed to mine SSRs from genomes. Conventional methods rely on a high-quality complete genome to identify SSRs. However, the sequenced genome often misses several highly repetitive regions. Moreover, many non-model species have no entire genomes. With the recent advances of next-generation sequencing (NGS) techniques, large-scale sequence reads for any species can be rapidly generated using NGS. In this context, a number of methods have been proposed to identify thousands of SSR loci within large amounts of reads for non-model species. While the most commonly used NGS platforms (e.g., Illumina platform) on the market generally provide short paired-end reads, merging overlapping paired-end reads has become a common way prior to the identification of SSR loci. This has posed a big data analysis challenge for traditional stand-alone tools to merge short read pairs and identify SSRs from large-scale data.
Results: In this study, we present a new Hadoop-based software program, termed BigFiRSt, to address this problem using cutting-edge big data technology. BigFiRSt consists of two major modules, BigFLASH and BigPERF, implemented based on two state-of-the-art stand-alone tools, FLASH and PERF, respectively. BigFLASH and BigPERF address the problem of merging short read pairs and mining SSRs in the big data manner, respectively. Comprehensive benchmarking experiments show that BigFiRSt can dramatically reduce the execution times of fast read pairs merging and SSRs mining from very large-scale DNA sequence data.
Conclusions: The excellent performance of BigFiRSt mainly resorts to the Big Data Hadoop technology to merge read pairs and mine SSRs in parallel and distributed computing on clusters. We anticipate BigFiRSt will be a valuable tool in the coming biological Big Data era.
Simple Sequence Repeats (SSRs), also known as short tandem repeats (STRs) or microsatellites (Fan and Chu, 2007; Madesis et al., 2013), are highly mutable nucleotide sequences (Vargas Jentzsch et al., 2013). Previous studies have shown that copy number alterations in tandem repeat DNA are associated with at least 31 different human diseases (Mitsuhashi et al., 2019). As a particular type of tandem repeats, SSRs are also related to many diseases such as colon cancer (Velasco et al., 2019) and humans' neurodegenerative disease (Cao et al., 2014), human triplet-repeat expansion diseases (Caskey et al., 1992; Mitas, 1997). Furthermore, as one of the most popular molecular markers (Guang et al., 2019), SSRs have been widely applied in numerous scientific researches including ecological investigation (Selkoe and Toonen, 2010), human population (Willems et al., 2014), genome evolution (Cavagnaro et al., 2010), plant genetics (Zalapa et al., 2012) and forensic analysis (de Knijff, 2018), and have several biomedical applications (Girgis and Sheetlin, 2013). Notably, repeats in the genome are species-specific (Girgis, 2015), SSRs are likely to be unknown for new genomes. Therefore, SSRs identification in new genomes is fundamentally important for understanding microsatellite evolution mechanisms (Ellegren, 2004).
Conventional experimental methods for SSR identification, such as labeled probes, are often labor-intensive and -expensive (Fernandez-Silva and Toonen, 2013). Computational SSR identification methods provide a valuable and alternative strategy for large-scale experimental design efficiently. Given the importance and value of computational methods for SSR identification, there has been encouraging progress in the development of computational methods and tools for SSR identification. Lim et al. (2013) provided a review of these methods developed before 2013. Various methods/tools have been developed in recent years. These tools are broadly classified into four categories: (i) graphical interface-based methods including GMATo (Wang et al., 2013) and GMATA (Wang and Wang, 2016), (ii) web interface-based methods including ProGeRF (Lopes et al., 2015), QDD (Meglécz et al., 2014), MISA-web (Beier et al., 2017), (iii) database-based methods including SSRome (Mokhtar and Atia, 2018) and MSDB (Avvaru et al., 2017a), and (iv) stand-alone-based methods including SA-SSR (Pickett et al., 2016), Kmer-SSR (Pickett et al., 2017), PERF (Avvaru et al., 2017b), Dot2dot (Genovese et al., 2019) and Look4TRs (Velasco et al., 2019). Most existing methods are generally designed to identify SSRs for species with the entire genome sequence available. These methods rely heavily on a high-quality assembled genome (Guo et al., 2018). However, many non-model species have no entire genomes. Fortunately, new NGS technologies can produce large numbers of genomics data for any species, and this has made it possible to identify SSRs from the newly assembled genome (Andersen and Mills, 2014). However, it is the biggest challenge to assemble a genome using short reads (Magoc and Salzberg, 2011). Moreover, it presents a significant obstacle to aligning reads within the repeat regions to the reference genome (Nashta-ali et al., 2017). As a result, the assembled genome often misses highly repetitive regions (Chu et al., 2016); even good-quality human reference genomes often contain missing bases in repeat regions (Chu et al., 2016). Thus, it is difficult to assemble a high-quality genome (Gnerre et al., 2011; Pickett et al., 2016). In scenarios where the target patterns are very sparse in the genomes, such as clustered repeats like CRISPR region, it is basically wasteful to find the repetitive sequences by assembling all sequencing reads into the genomes (Chen et al., 2019). To address this, in recent years various methods (Castoe et al., 2012; Gymrek et al., 2012; Miller et al., 2013; Fungtammasan et al., 2015; Tang and Nzabarushimana, 2017) have been proposed to identify SSRs from raw sequence data generated by NGS. After identifying SSRs in reads, the non-repetitive flanking sequence of SSR-containing reads can be used to map to the reference for increasing the alignment specificity (Gymrek et al., 2012). Furthermore, analyses of SSRs based on NGS have been used in a range of applications, including forensic analysis (Van Neste et al., 2014; Børsting and Morling, 2015; Parson et al., 2016; van der Gaag et al., 2016; Hoogenboom et al., 2017; de Knijff, 2018; Ganschow et al., 2018), SSRs genotyping (Bornman et al., 2012; Kistler et al., 2017; Budiš et al., 2018), stutter analysis (Vilsen et al., 2018), population genetic (Wirtz et al., 2016) and SSR Markers in Plants (Taheri et al., 2018).
Generally, a typical SSR locus is represented in the repeat modules surrounded by both flanking regions (Budiš et al., 2018). An example of SSR allele is “ACGATGATCGATAGATAGATAGATAGATAGATAGATAGATAGTCAGAGCACC”, which means that the sequences “ACGATGATC” and “GTCAGAGCACC” represent the upstream and downstream region around the motif GATA with eight repeats, respectively. Certain NGS technologies such as Roche 454 could provide reads that fully contain SSRs along with suitable flanking sequences (Perry and Rowe, 2011). In recent years, emerging NGS technologies such as PacBio and Nanopore can produce long reads (Mardis, 2017). However, the most commonly used NGS platforms (e.g., Illumina) on the market often provide short paired-end reads (Escalona et al., 2016; Wang, 2016). In most cases, short reads do not contain full SSR allele regions (Budiš et al., 2018). Thus, constructing longer reads by merging paired-end reads has been used as a common strategy prior to identifying SSR motifs (van der Gaag et al., 2016; Hoogenboom et al., 2017; Ganschow et al., 2018). Several paired-end read merging algorithms have been proposed in recent years, which include FLASH (Magoc and Salzberg, 2011), leeHom (Renaud et al., 2014), PEAR (Zhang et al., 2013), BBMerge (Bushnell et al., 2017) and Konnector (Vandervalk et al., 2014), OverlapPER (Oliveira et al., 2018), Cope (Liu et al., 2012), and XORRO (Dickson and Gloor, 2013). There also exist approaches and tools such as SSRs-pipeline (Miller et al., 2013) and RAD-seq-Assembly-Microsatellite (Xue et al., 2017), which integrate paired-end reads merging and SSRs mining into a single pipeline.
These computational methods and tools have been used for merging paired-end reads and identifying novel SSRs. Other analysis tools such as iLearn (Chen et al., 2020, 2021), BioSeq-Analysis (Liu, 2019; Liu et al., 2019) and BioSeq-BLM (Li et al., 2021) were recently developed to handle with the avalanche of biological sequences. However, with the continued development of NGS technologies, there is a strong need to develop new paired-end read merging and SSRs mining methods that better meet the “Big Data” analysis (Wordsworth et al., 2018). As NGS technology can often generate hundreds of gigabytes (GB) sequence data in compressed FASTQ format in every single run (Wang, 2016), it is becoming more and more difficult and time-consuming to use these stand-alone methods and tools to merge paired-end reads and identify SSR loci from such large-scale datasets. To the best of our knowledge, there are currently no methods and tools to date that are developed based on Big Data techniques for merging paired-end reads and mining SSRs. Thus, it would be highly desirable and valuable to significantly enhance the performance of paired-end reads merging and SSRs mining tools by combining the cutting-edge Big Data techniques. In this way, the computational SSRs mining approaches could keep up with the pace of data explosion and efficiently deal with the growth of such large-scale data.
Traditional parallel computing technique based on message passing interface (Gropp et al., 1999) is more effective for moderately sized data and computational-intensive problem (Kang et al., 2015). It is not the best choice to deal with the vast amount of data (Samadi et al., 2018). Hadoop (White, 2009) and Spark (Zaharia et al., 2010) have become two standard big data technologies to handle huge the size of data (Samadi et al., 2018). They have been widely used in the bioinformatics area to deal with the rapid growth and accumulation of biomedical Big Data. Table 1 summarizes the bioinformatics tools developed based on Big Data technologies for handling large-scale sequence data. These methods are involved in many different tasks, including alignment and mapping, sequence analysis, genome analysis, sequence assembly, error correction, duplicate DNA reads and clustering analysis. However, there is no bioinformatics method based on Big Data techniques for merging paired-end reads and mining SSRs from large-scale NGS sequence data, highlighting the critical needs and value of developing and deploying such strategies to bridge the knowledge gap.
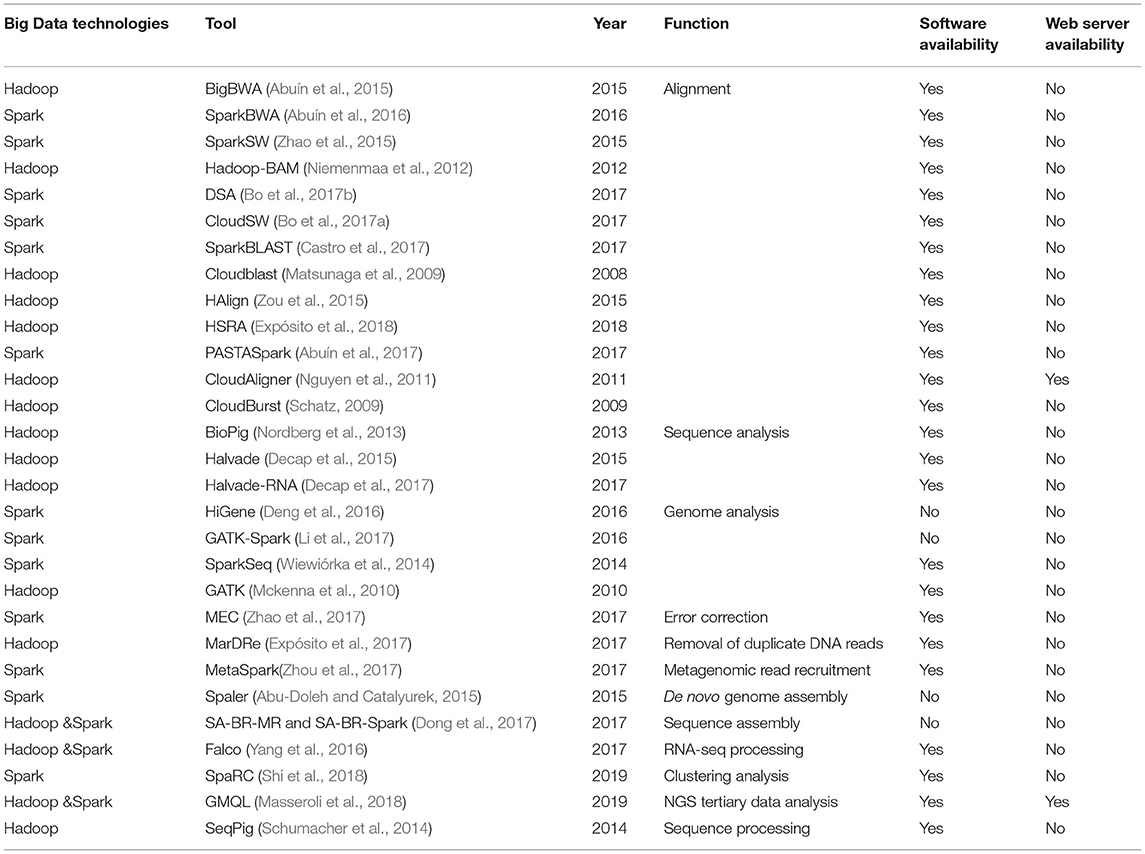
Table 1. Bioinformatics tools developed based on Big Data technologies for handling large-scale sequence datasets.
Both Hadoop and Spark can deal with the above problems. Generally, Spark has better performances for iterative algorithms than Hadoop (Samadi et al., 2018). However, Spark is in-memory computing, and it becomes slower than Hadoop when the cluster has not enough memory. Thus, Hadoop is a better choice for the system without sufficient memory (Samadi et al., 2018). For merging paired-end reads and mining SSRs, we do not need to perform operations over the same data recursively and only need to choose Hadoop to address these two problems. In this work, we propose BigFiRSt (Big data-based Flash and peRf algorithm for mining Ssrs), a novel Hadoop-based program suite and is specifically designed to integrate paired-end reads merging and SSRs search into an effective computational pipeline. There are two fundamental modules in BigFiRSt: BigFLASH and BigPERF. They represent two implementations of the well-known stand-alone algorithms FLASH (Magoc and Salzberg, 2011) and PERF (Avvaru et al., 2017b) based on Hadoop techniques. Due to the advantages of the Hadoop big data technology, BigFLASH and BigPERF have significantly improved the computational efficiency compared with the baseline FLASH and PERF, respectively. Moreover, BigFiRSt allows users to apply BigFLASH and BigPERF separately and provides a pipeline functionality to enable users to run them consecutively. It allows the program to take short read pairs as the input and return the mined SSRs. These outputs can be used for genotyping analysis and other custom analyses (Budiš et al., 2018) to better suit users' specific needs.
Intuitively, it is more convenient for biologists to process and analyse large-scale sequences by a user-friendly web interface. However, in practice, it remains a challenging problem for users to upload large scale datasets from their local machines to the online web interface (Zou et al., 2014). To facilitate users to merge read pairs and subsequently identify SSRs in relatively small datasets, we provide a publicly available web interface of BigFiRSt, which is available at http://bigdata.biocie.cn/BigFiRSt/. There is no other such web interface integrating these two processes currently available in the research community to the best of our knowledge. On the other hand, for handling massive datasets and facilitating the data process using local computers, we also provide the source codes of BigFiRSt for download https://github.com/JinxiangChenHome/BigFiRSt such that users can configure and execute the BigFiRSt program on a cluster supported by the Hadoop.
BigFiRSt was developed based on the Big Data Hadoop technology (White, 2009). Hadoop has been regarded as a milestone of big data processing (Petrillo et al., 2019). It is an open-source framework that can be installed on a Linux cluster for distributed processing of large-scale data sets using the MapReduce model (Dean and Ghemawat, 2008). MapReduce is a computation mode that allows users to specify a map and a reduce operation for parallelising the extensive computation. Generally, a Hadoop MapReduce job requires three core modules, namely, Hadoop Distributed File System (HDFS) (Shvachko et al., 2010), Hadoop MapReduce and Yet Another Resource Negotiator (YARN) (Vavilapalli et al., 2013). The input large-scale data sets are split into independent blocks and stored in HDFS across all Hadoop cluster computing nodes. Independent data blocks are processed by map tasks in a completely parallel manner. Reduce tasks fetch the corresponding partitioned data from the output of map tasks. YARN is responsible for resource management of the cluster and job scheduling/monitoring. Altogether, HDFS and YARN are able to provide the fault tolerance and data locality of Hadoop clusters (Taylor, 2010; Alnasir and Shanahan, 2018).
The overall framework of the BigFiRSt methodology is illustrated in Figure 1. BigFiRSt contains two modules: BigFLASH (Figure 1A) and BigPERF (Figure 1B). BigFLASH is used to merge short read pairs and output long consensus reads, while BigPERF extracts SSRs from large-scale reads. These modules can be further integrated into a pipeline that takes the output of BigFLASH as the input to BigPERF. The red line in Figure 1 highlights the pipeline that connects BigFLASH with BigPERF.
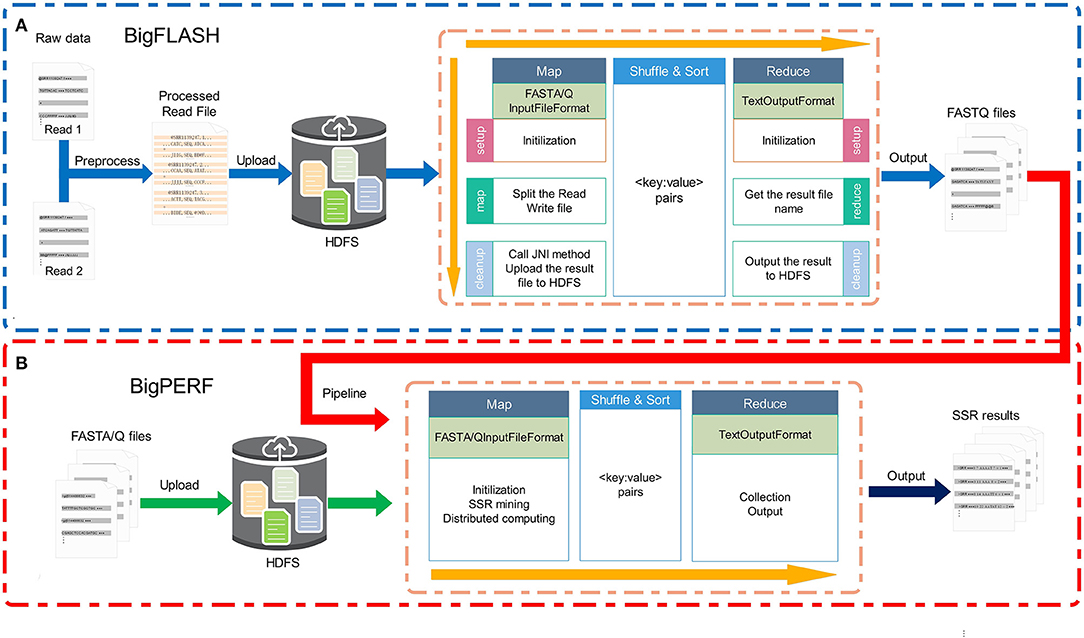
Figure 1. The overall framework of the BigFiRSt methodology. BigFiRSt contains two modules. (A) BigFLASH is used to merge short read pairs. (B) BigPERF is used to mine SSRs contained in reads.
The Hadoop MapReduce module provides the Mapper interface with the Map method and the Reducer interface with the reduce method, respectively. A Hadoop application generally implements these two interfaces to create the map and reduce tasks. The number of map tasks depends on the number of InputSplits, which is a logical split of input files. InputSplits are created from data blocks, which exist physically on disk across Datanodes of clusters. In BigFiRSt, the size of InputSplit is the same as the block size, by default. The Hadoop MapReduce framework creates one map task to process each InputSplit in a completely parallel manner. Each InputSplit is generated by the InputFormat. In the Hadoop framework, FileInputFormat is the base class of all file-based InputFormat. The default InputFormat is TextInputFormat (a subclass of FileInputFormat), which breaks file into lines. The text of each line as value is processed by the map task. For BigFiRSt, the input data with the FASTQ format denotes a read for every four lines. The first line is the sequence title/identifier, which starts with a character “@.” The second line represents the nucleotide sequence of this read. The nucleotides in the sequence are usually presented in the upper case. The third line starts with “+” and contains a full repeat of the title line (the first line). The fourth line denotes the quality string of the sequence. Its length was equal to the sequence string (the second line). Hadoop cannot directly handle sequences with FASTQ format. We used a subclass of FileInputFormat written in (Ferraro Petrillo et al., 2017) to convert each InputSplit to a format that Hadoop can handle.
BigFLASH implements the FLASH (Magoc and Salzberg, 2011) algorithm based on the Hadoop technology. FLASH has been extensively used for pre-processing large-scale NGS sequence data and facilitating the downstream analysis. Generally, it works by first merging read pairs into a consensus read preceding the analysis of SSR profiles based on NGS (van der Gaag et al., 2016; Hoogenboom et al., 2017; Ganschow et al., 2018). If cases where read pairs end within an SSR sequence, then the SSR sequence can be truncated after the read pairs are merged by FLASH. Accordingly, we used a Modified Version (1.2.11) of FLASH (2015) to implement BigFLASH.
The original FLASH algorithm was written in C programming language. However, Hadoop was programmed using the Java language and as such, it provides many useful Java APIs for Hadoop based application development. In general, Hadoop-based applications are implemented in Java in order to enable better interactions with Hadoop. Therefore, in BigFLASH, we used the Java Native Interface (JNI) (Liang, 1999) to integrate Java programming codes with the FLASH C code and effectively enable such interactions. This renders rewriting the source codes of FLASH unnecessary and ensures that no further modification of the original algorithm is required. We only used the FLASH source codes to build an additional shared library file named “libflash.so.” BigFLASH is able to parse the input parameters and then pass them on to the main method of FLASH by loading “libflash.so.”
The BigFLASH process comprises of three major steps, which are illustrated in Figure 1A. The detailed workflow of BigFLASH is shown in Figure 2A. As can be seen, the first step is data pre-processing. The read pairs are stored in two separate FASTQ files. Considering that there is no API available in Hadoop for handling read pairs storing in two separate FASTQ files, we compiled a Python script (downloadable from the BigFiRSt web site) that can conveniently convert the two input FASTQ files into one single FASTQ file. The pseudo-code of the Python script is provided in the Supplementary Material. At the second step, the pre-processed data is uploaded to the HDFS, where large-scale data files are divided into fixed-size blocks. The third step is the MapReduce phase. BigFLASH applies the FASTAInputFileFormat/FASTQInputFileFormat function of FASTdoop (Ferraro Petrillo et al., 2017) to convert each data block to the Hadoop-acceptable format. Each block is processed by a Mapper. Each Mapper calls FLASH to merge the read pairs located in processed blocks, and all Mappers are executed in parallel. Lastly, the Reduce phase generates files of the merged reads by collecting the output of each Mapper. The key feature of BigFLASH is its Mappers, whose detailed procedures of implementation are described as follows: First, BigFLASH overrides the “setup” method from parent Class Mapper to parse the input parameters. The “setup” method will be invoked automatically, to initialize input parameters required by FLASH. Second, the “map” method from parent Class Mapper is overridden, to parse each InputSplits into two FASTQ files, which will be handled by FLASH. And finally, BigFLASH overrides the method “cleanup,” which passes the input parameters to the main method of FLASH by calling the declared native method.
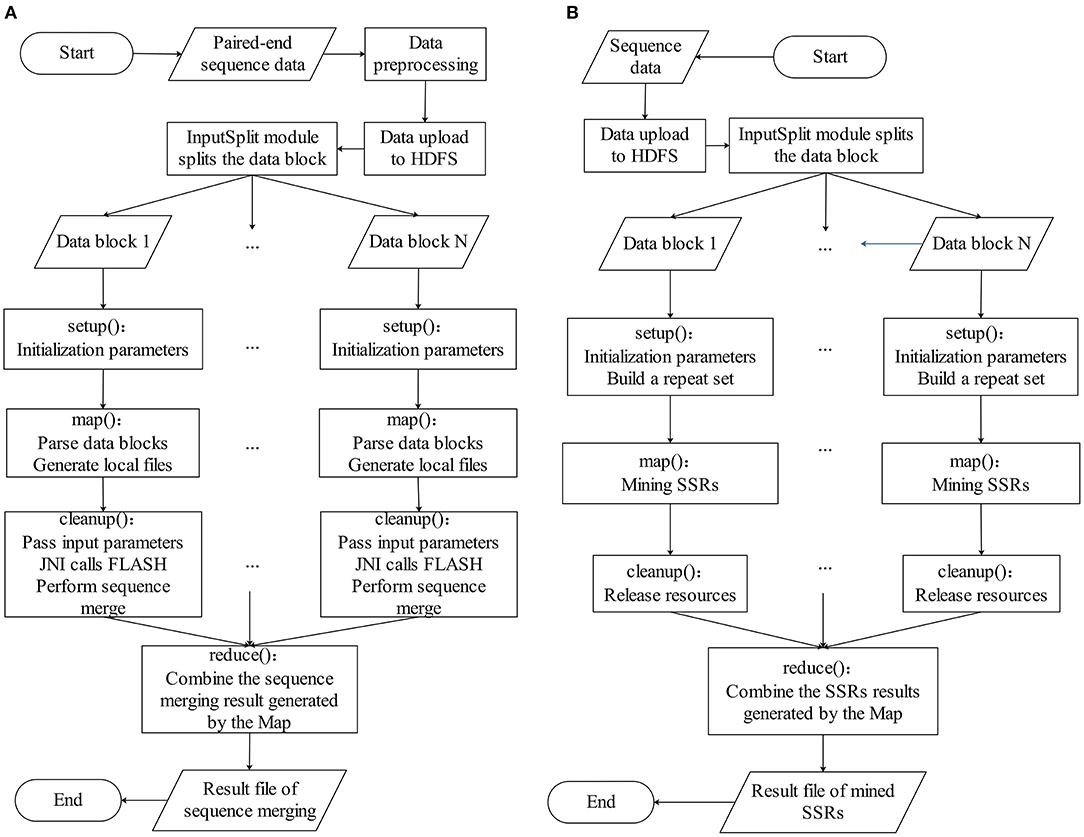
Figure 2. The detailed workflow of (A) BigFLASH and (B) BigPERF.
As aforementioned, the original PERF program was written in Python (Avvaru et al., 2017b) and in this study, we rewrote it in Java to develop and implement BigPERF. The overall framework of BigPERF is shown in Figure 1B. The detailed workflow of BigFLASH is shown in Figure 2B. There exist three steps involved in the development of BigPERF. The first step is to upload the user input files in the FASTA/FASTQ format to HDFS. Similar to the second step in BigFLASH, the input data files are divided into fixed-size blocks and the FASTAInputFileFormat/FASTQInputFileFormat function is used to convert each block to a Hadoop-acceptable format. BigPERF first overrides the “setup” method from the parent Class Mapper to parse the input parameters and build a repeat set, which is then used for lookup during repeat identification. Then, the map method from the parent Class Mapper is overridden to mine all SSRs by extending the substrings appearing in the repeat set in a completely parallel manner. At this phase, each mapper generates a result file. The third step is the Reduce phase, where BigPERF collects the results from the map phase to generate the final results.
In the Reduce phase, BigFLASH and BigPERF only collect results from the output result files generated by each mapper. A complete Reduce task contains three primary phases: shuffle, sort and reduce. The Hadoop framework sorts the outputs of the mappers by keys simultaneously, and the shuffle phase fetches the relevant partitioned output of all mappers. Finally, the reduce phase calls the reduce method for each <key, (list of values)> pair in the grouped inputs.
Users can use the method “setNumReduceTasks” to set the number of reduce-tasks. A combined result output would be generated when the number of Reducers is set 1. The number of the result files depends on the predefined reducer number. Users are allowed to set the number of reduce-tasks to zero if no reduction is desirable. If the number of reduce-tasks is set to zero, the output of all the mappers is the result.
In BigFLASH and BigPERF, users have the option to select to use the “reduce” phase. In cases where such option is selected, according to the Hadoop MapReduce tutorial, the right number for “reduces” seems to be 0.95 or 1.75 multiplied by (<no. of nodes> * <no. of maximum containers per node>). The detailed information refers to the Hadoop tutorial on the official website.
We have implemented and deployed an online web server of BigFiRSt in order to facilitate users to merge read pairs and/or mine SSRs in small-scale datasets (up to 30 MB). The web server of BigFiRSt is freely accessible at http://bigdata.biocie.cn/BigFiRSt/. The two algorithms FLASH and PERF, as well as the computational pipeline have also been made available at this web server.
The BigFiRSt web server is managed by Tomcat 7 and hosted on a Linux server, equipped with a 1-core CPU, 40 GB hard disk and 2 GB memory. Using the web server, users can upload files, select desired parameters and obtain the result files.
Users can merge paired-end reads by the FLASH algorithm via the web interface of BigFiRSt. This module works as follows: First, users need to upload both FASTQ format data files that respectively store the forward and reverse reads. Alternatively, users can also input the sequences in the FASTQ format in the text area. Second, users can update default parameter values of the FLASH algorithm using the web interface. Thirdly, users click to submit the job. Alternatively, users can also provide their email addresses in order to receive a notification Email after the submitted job is finished. Finally, when the submitted job is completed successfully, users can view the job details and download the generated results. In this case, users should have received such notification email and can check to review the job details by clicking a hyperlink in the email.
This module uses the PERF algorithm to mine SSRs from DNA sequences in FASTA format. Similar to using FLASH, users need to first upload sequence data and update default parameter values, and then submit this job. After the submitted job is completed, the user can view the detailed results of mined SSRs in a table. Moreover, users can input a preferred SSR and retrieve all reads containing this SSR from the result table. In addition, users can also export the mined results in the CSV/Excel format for the follow-up analysis in local computers.
The function of this module is to integrate FLASH with PERF into a pipeline. The pipeline first calls FLASH to merge read pairs of the input data, and then calls PERF to mine SSRs from the output of FLASH. Users only need to upload two FASTQ files containing pair-end reads or paste the data to this module, then update the default parameter value and submit the job, and finally obtain the result.
The web interface of BigFiRSt also provides other auxiliary functions including source code download, search for submitted jobs, view of all submitted jobs, and contact information, etc.
The experimental environment of BigFiRSt includes HDFS (version 2.7.3), YARN (version 2.7.3), MapReduce2 (version 2.7.3), Java (version 1.8), and Python (version 2.7.3).
We evaluated the performance of BigFiRSt using a five-node Hadoop cluster on the Research Cloud server of Monash University. The structure of this five-node Hadoop cluster with detailed hardware configurations is illustrated in Figure 3. One node of this cluster is a master node while the other four are computing nodes.
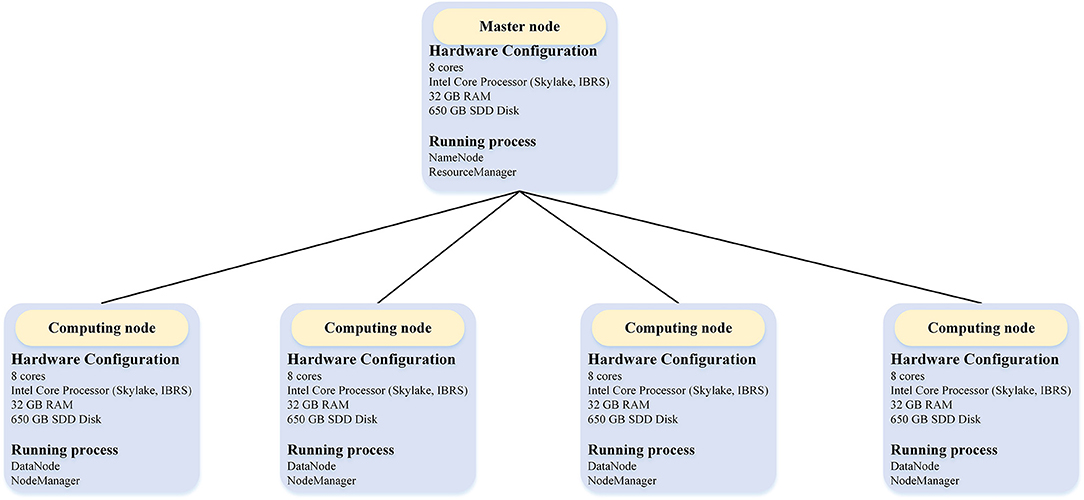
Figure 3. The overall architecture of the Hadoop cluster in the experiment.
The master node is used for launching and managing the computational tasks, while four computing nodes are responsible for the Map/Reduce tasks. Table 2 provides the detailed information of the configuration of each machine used in the experiment. Each node had eight cores and 32 GB RAM memory. We configured the Hadoop “yarn-site.xml” file to allocate 4 GB memory for each of the eight cores for each node. Among these, seven cores were allocated to computational tasks and one core to the operating system. Accordingly, each node can run up to seven Map/Reduce tasks at the same time. That is, a total of 28 Map/Reduce tasks are allocated for four computing nodes. This arrangement also means that when we performed a 32-core experiment in a Hadoop cluster, we needed to set the master node as a compute node as well (i.e., using 4/8 cores of the master node for computing). The block size was set to 128 M by the Hadoop configure file.
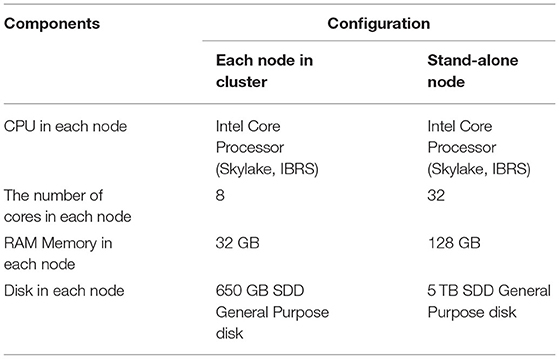
Table 2. Configurations for each machine used in the experiment.
For the performance comparison of BigFLASH, we conducted a comparative experiment in which the numbers of FLASH threads and Hadoop cluster cores were set as the same. Therefore, we added a 32-core stand-alone machine with the same hardware configurations as any of the machines in the cluster for experimentation (refer to Table 2 for more detail).
We employed three experimental datasets from The 1000 Genomes Project Consortium (2010) to examine the performance of BigFiRSt. A statistical summary of the three datasets used is provided in Table 3.
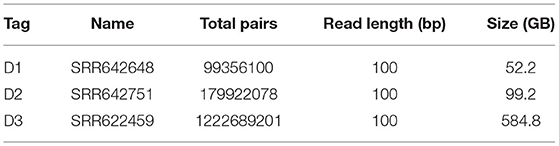
Table 3. Main characteristics of the input datasets for read pairs merging.
The main characteristics of the datasets used for the read pair merging phase is shown in Table 3. We compared the execution time between BigFLASH and FLASH for this process and obtained the performance results by averaging the execution time over the five experiments for each method. We ran FLASH five times on a 32-core stand-alone machine with the same hardware configurations as any of the machines in the cluster. All parameters used in the test were set as the default. The experimental results are shown in Table 4.
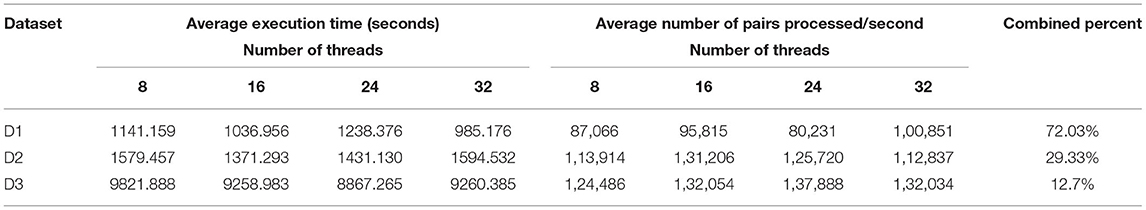
Table 4. Experimental results for merging read pairs by the original FLASH algorithm.
Here the number of reduce-tasks was set to zero when running the BigFLASH. The average execution time for merging read pairs is shown in Table 5. We controlled the total number of cores in the cluster by modifying the mapred-site.xml and yarn-site.xml configuration files in Hadoop. The experimental results show that, for any one of the experimental datasets and as the number of cores in the cluster increases, the shorter the cluster execution time, in a roughly similar scale (as the number of splits in the dataset in Hadoop is greater than the number of cluster cores). As shown in Tables 4, 5, when employed 8 CPU cores cluster for BigFLASH, speedup ratios reach 2.630, 1.670, 1.832 on D1, D2, and D3, respectively. In comparison, when applied 32 CPU cores cluster, speedup ratios are improved to 5.950, 5.623, and 5.432 on D1, D2, and D3, respectively. In general, more CPU cores achieved more speedup ratios.
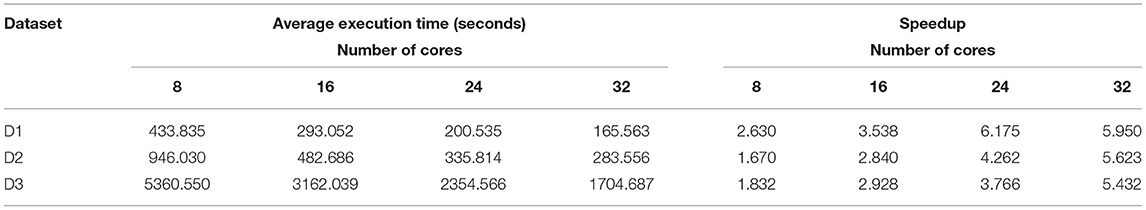
Table 5. Average execution time for merging read pairs by BigFLASH in the cluster.
On the other hand, we can also measure the performance of BigFLASH in terms of the number of read pairs processed per second in the Map phase. The sum of the execution time of all map tasks for each experiment is shown in Table 6. As can be seen, when using more cores in the cluster, the total running time of all Mappers would be more than those of all Mappers when less cores are used in the cluster. When more Mappers in each node would be running at the same time, each Mapper would be cost more times. However, more Mappers could be running at the same time when more cores are available in the cluster, thus it would cost less time to finish the map tasks (refer to Table 5).
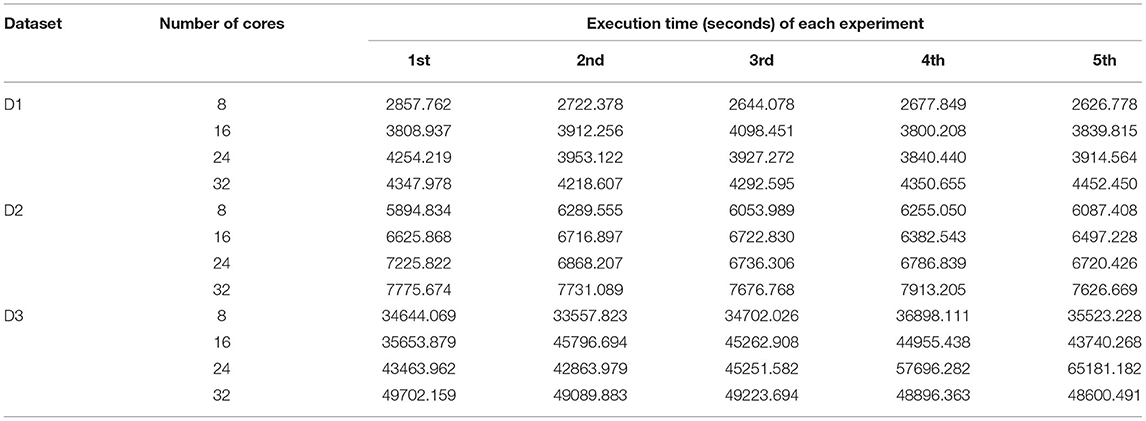
Table 6. Execution time of all map tasks of BigFLASH in five experiments.
Table 7 shows the average number of read pairs processed per second, which can be calculated as follows:
where avePairsPerSec denotes the average number of read pairs processed per second, totalReadsNum is the total number of read pairs of the processed dataset, aveExecutionTime means the average execution time shown, while numOfCores denotes the number of cluster cores. numOfCores − 1 indicates that YARN's ApplicationMaster process occupied a single core for resource management and task monitoring and did not participate in calculations. From Table 7 we can find that BigFLASH can handle more pairs each second than FLASH and achieve the highest speed up rate 7.16. Taken together, we conclude that the performance of BigFLASH was considerably better than the original serial algorithm, greatly shortening the execution time of the original program and reducing the user waiting time.
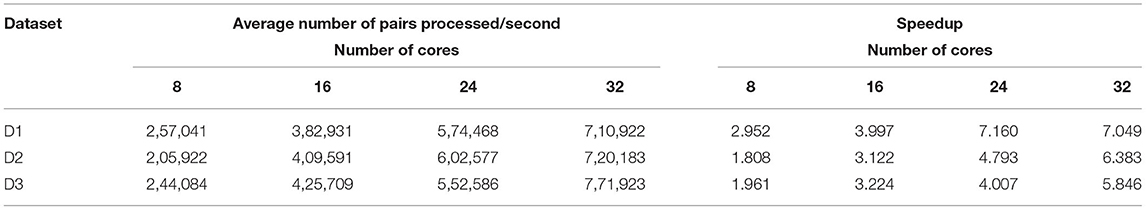
Table 7. Amount of data processed in the Map phase of BigFLASH.
The three datasets used for BigPERF and PERF performance evaluations in terms of SSRs mining are shown in Table 8. Note that these datasets were derived from the merged results of BigFLASH for the three datasets in Table 3.
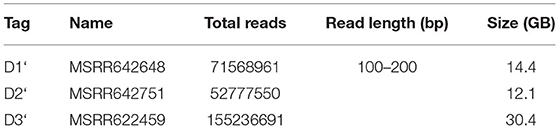
Table 8. Input datasets for mining SSRs.
We compared the execution time between PERF and BigPERF for the SSRs mining process. The resultant execution time on each dataset was obtained by averaging the time of the five randomized experiments. We randomly used one node of the cluster to run the original PERF algorithm five times on each dataset. The parameters used in the comparison experiments and the averaged running time (seconds) are shown in Table 9.

Table 9. Running information of PERF original algorithm.
Here the number of reduce-tasks was set to zero when running the BigPERF. The execution time results are shown in Table 10. The sum of the execution time of all map tasks for each experiment is shown in Table 11, and the amounts of data processed by BigPERF in the Map phase are shown in Table 12, respectively. Similar to the experimental results for merging read pairs, the experimental results of SSR mining also exhibited consistent results. That is, the Hadoop-based algorithms (i.e., BigFLASH and BigPERF) are much more efficient compared with their original counterparts. Remarkably, we found that the performance improvement of BigPERF was extremely pronounced. For example, in terms of the execution time, BigPERF was at least 21 times faster (in the case of the D2‘ data set and the 8-core cluster) and at most 68 times (in the case of the D3‘ data set and the 32-core cluster) faster than that of PERF. In terms of the number of reads processed per second in the map phase, BigPERF runs at least 22 times faster (in the case of the D2‘ data set and the 8-core cluster) and at most 76 times faster (in the case of the D3‘ data set and the 32-core cluster) than PERF.
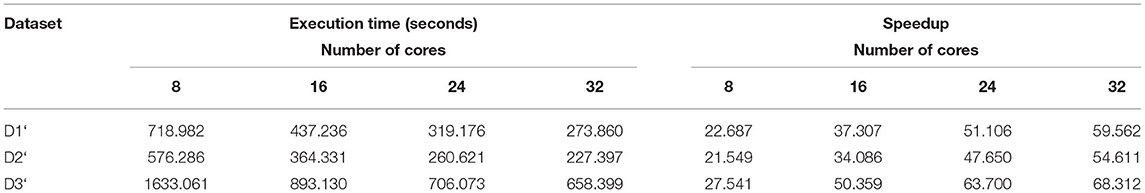
Table 10. Execution time of BigPERF for searching SSRs.
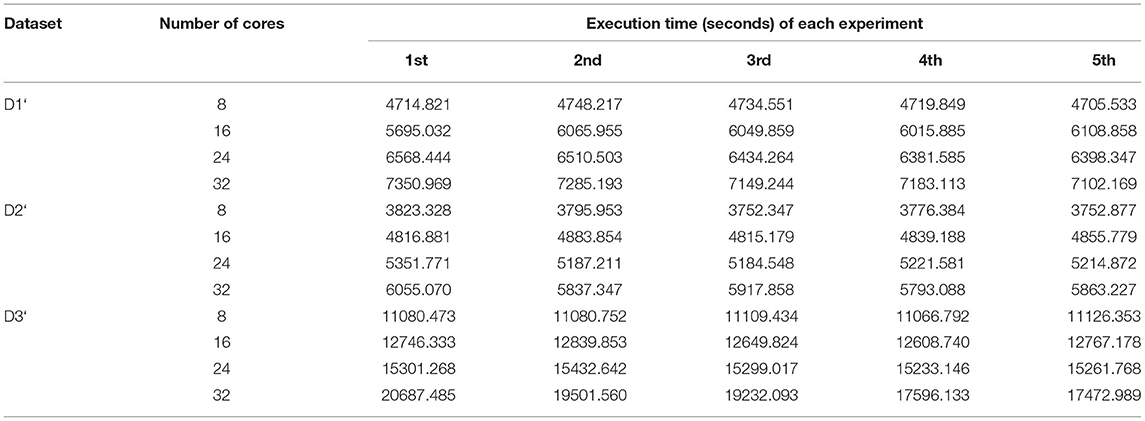
Table 11. Execution times of all map tasks of BigPERF in five experiments.
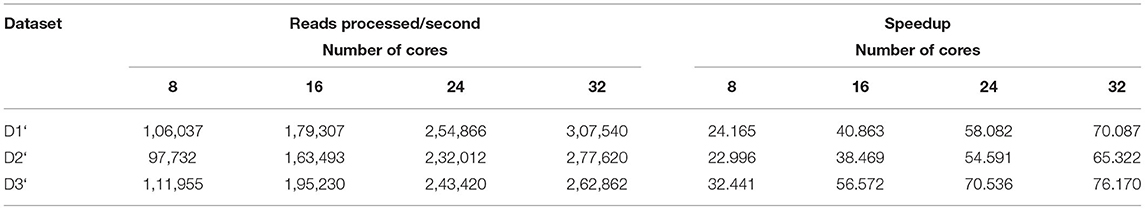
Table 12. Amount of data processed by BigPERF in the Map phase.
SSRs-pipeline (Miller et al., 2013) is a stand-alone tool that integrates read pairs merging and SSRs mining into a single pipeline. SSRs-pipeline first uses FLASH as a pre-processing algorithm of merging short read pairs from Illumina high-throughput DNA sequencing data and then employs a regular expression-based method to mine SSRs from merged read sequences. FLASH has been extensively used for pre-processing large-scale NGS sequence data and facilitating the downstream analysis (van der Gaag et al., 2016; Hoogenboom et al., 2017; Ganschow et al., 2018). Comprehensive experiments in Avvaru et al. (2017b) have shown that PERF is an extremely fast algorithm for mining SSRs. Moreover, PERF does not need to construct an extra complicated data structure for each read sequence. Thus, in this work, we selected FLASH and PERF to implement BigFiRSt using Big Data Technology. Obviously, there are many other well-known methods for mining SSRs and merging read pairs (review in the introduction section). The idea proposed in this paper can also be applied to implement other methods based on Big Data technologies.
Currently, no published parallel methods for merging read pairs and mining SSRs are available. Thus, we only compared the performance of BigFLASH with FLASH, and that of BigPERF with PERF. Figure 4 illustrates the runtime performance comparison results between BigFLASH and FLASH. Although the original FLASH algorithm was a multithreaded algorithm with up to five threads, the execution time by FLASH was not apparently reduced as the number of the used cores increased. The reason is that only up to five threads could be used in FLASH. Thus, a supercomputer or cluster cannot further improve the performance of FLASH by simply adding more cores or more nodes. Compared with FLASH, BigFLASH significantly reduced the execution time for merging read pairs. For instance, FLASH had a running time of more than 2.46 h on the D3 dataset, which was more than 580 GB large. In contrast, BigFLASH (with 32 cores) only consumed 0.47 h to process the entire dataset. In addition, we can also see that the execution time was gradually reduced as the number of used cores increased, for each experimental dataset (across D1 to D3). Please refer to read pair merging for more detailed discussions.
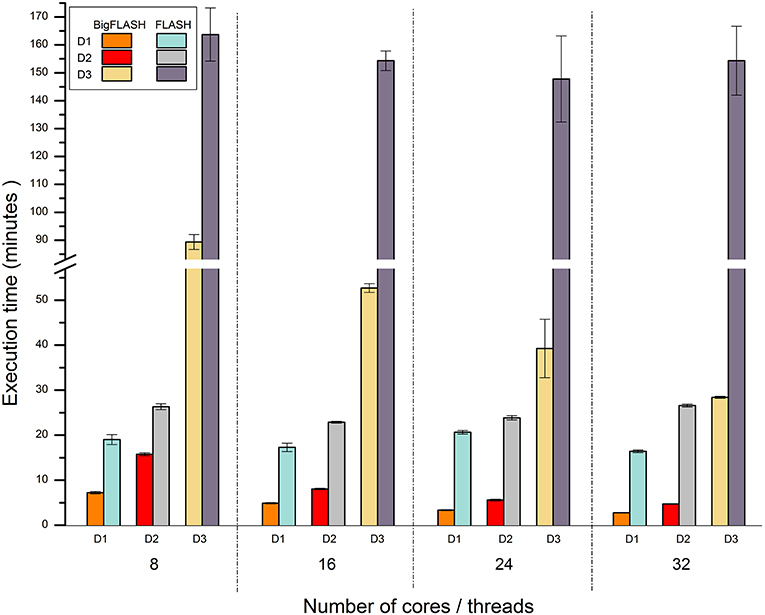
Figure 4. Runtime performance comparison between BigFLASH and FLASH for merging read pairs.
Figure 5 shows the performance comparison results between BigPERF and PERF. PERF required more than 12.49 h to process the D3' dataset using one node of our cluster, while it only took BigPERF (with 32 cores) 0.18 h to process the dataset on the same cluster. Similar to BigFLASH, the execution time of BigPERF could be gradually reduced for each experimental dataset with the increasing number of cores added. Please refer to SSR mining for more detailed discussions on this aspect.
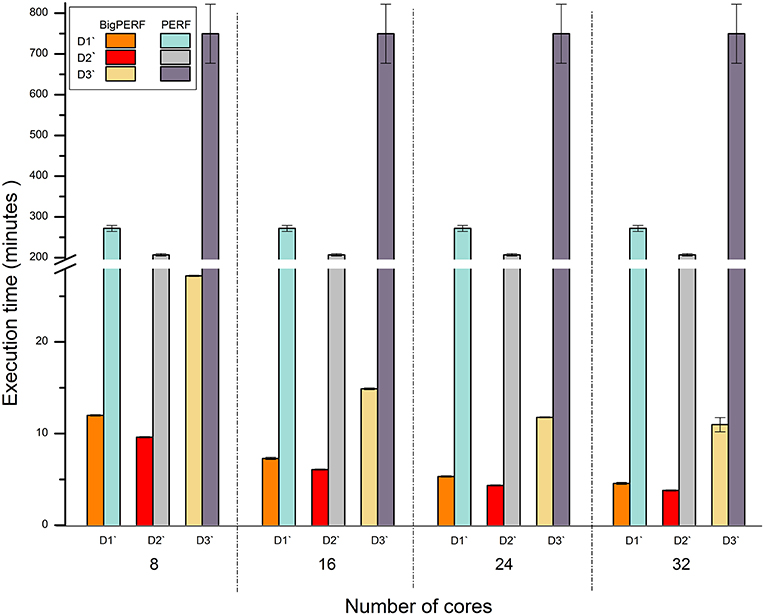
Figure 5. Runtime performance comparison between BigPERF and PERF for mining SSRs.
Despite BigFiRSt improves the performance of the computational efficiency of read pair merging and SSR mining, it has the following limitations. It is great challenges for biologists to deploy a big data-based running environment. Although some commercial cloud-based big data platforms are available to run big data technique-based software, it remains a challenging problem for users to upload large scale datasets from their local machines to cloud platform. In addition, many large-scale datasets generated by NGS are costly, even some datasets may be private. Once datasets are uploaded to cloud platform, these datasets would be divulged. BigFiRSt have the above limitations. Thus, it is very interesting work to address the above issues for handling large scale sequences generated by NGS.
There are two different types of de novo methods of SSRs identification, which mine SSRs from the entire genome and read sequences (Guo et al., 2018), respectively. The former heavily relies on high-quality entire genomes. It is practically very difficult to obtain a sufficiently good reference genome. Even for the human reference genome, several repeats may still be missing (Chu et al., 2016). In this scenario, it is beneficial for the latter to directly mine SSRs from large-scale sequencing reads generated by NGS techniques. While sequence reads generated by NGS are representative big data, conventional stand-alone methods often suffer from computational bottlenecks.
Thus, in this work, we have developed a program suite termed BigFiRSt based on the Big Data Hadoop technology to address the critical need of efficiently mining SSRs from large-scale NGS sequence datasets. For long enough reads produced by third-generation sequencing (e.g., Nanopore, PacBio), we need only use BigPERF (one module of BigFiRSt) to search SSRs contained in reads. For the short length of paired-end reads generated by second-generation sequencing (e.g., Illumina, SOLiD, IonTorrent), we can use the pipeline of BigFiRSt to first merge overlapping read pairs and then mine SSRs contained in merged read sequences. Alternatively, we used BigFLASH (another module of BigFiRSt) as pre-processing to merge read pairs into consensus sequences for other downstream analyses. Extensive benchmarking tests have shown that BigFiRSt has significantly improved the computational efficiency when merging read pairs and mining SSRs from the large-scale datasets. In the future era of big data, especially given the development of new sequencing techniques and rapid generation of sequence data, we anticipate that BigFiRSt will prove to be a valuable tool.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
JS and QL conceived the initial idea and designed the methodology. JC and FL implemented the experiments and processed the results. All authors drafted, revised, and approved the final manuscript.
QL was supported by the grant from National Natural Science Foundation of China (61972322), the Provincial Natural Science Foundation of Shaanxi Province (2021JM-110). JS was supported by grants from the National Health and Medical Research Council of Australia (NHMRC) (APP490989, APP1127948, and APP1144652), the Australian Research Council (ARC) (LP110200333 and DP120104460), and the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (R01 AI111965).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We acknowledge the reviewers' constructive comments, which have greatly helped to improve the scientific quality of this study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fdata.2021.727216/full#supplementary-material
SSRs, Simple Sequence Repeats; NGS, next-generation sequencing; STRs, short tandem repeats; BigFiRSt, Big data-based Flash and peRf algorithm for mining Ssrs; HDFS, Hadoop Distributed File System; YARN, Yet Another Resource Negotiator.
Abu-Doleh, A., and Catalyurek, U. V. (2015). “Spaler: spark and GraphX based de novo genome assembler,” in IEEE International Conference on Big Data (Santa Clara. CA). doi: 10.1109/BigData.2015.7363853
Abuín, J. M., Pena, T. F., and Pichel, J. C. (2017). PASTASpark: multiple sequence alignment meets Big Data. Bioinformatics 33, 2948–2950. doi: 10.1093/bioinformatics/btx354
Abuín, J. M., Pichel, J. C., Pena, T. F., and Amigo, J. (2015). BigBWA: approaching the Burrows–Wheeler aligner to big data technologies. Bioinformatics 31, 4003–4005. doi: 10.1093/bioinformatics/btv506
Abuín, J. M., Pichel, J. C., Pena, T. F., and Amigo, J. (2016). SparkBWA: speeding up the alignment of high-throughput DNA sequencing data. PLoS ONE 11:e0155461. doi: 10.1371/journal.pone.0155461
Alnasir, J. J., and Shanahan, H. P. (2018). The application of Hadoop in structural bioinformatics. Brief. Bioinform. 21, 96–105. doi: 10.1093/bib/bby106
Andersen, J. C., and Mills, N. J. (2014). iMSAT: a novel approach to the development of microsatellite loci using barcoded Illumina libraries. BMC Genomics 15:858. doi: 10.1186/1471-2164-15-858
Avvaru, A. K., Saxena, S., Sowpati, D. T., and Mishra, R. K. (2017a). MSDB: a comprehensive database of simple sequence repeats. Genome Biol. Evol. 9:1797. doi: 10.1093/gbe/evx132
Avvaru, A. K., Sowpati, D. T., and Mishra, R. K. (2017b). PERF: an exhaustive algorithm for ultra-fast and efficient identification of microsatellites from large DNA sequences. Bioinformatics 34, 943–948. doi: 10.1093/bioinformatics/btx721
Beier, S., Thiel, T., Münch, T., Scholz, U., and Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585. doi: 10.1093/bioinformatics/btx198
Bo, X., Li, C., Hang, Z., Wang, J., Wang, Q., Zhou, J., et al. (2017b). “DSA: Scalable Distributed Sequence Alignment System Using SIMD Instructions,” in IEEE/ACM International Symposium on Cluster (Madrid).
Bo, X., Li, C., Hang, Z., Wang, J., and Zhou, X. (2017a). “Efficient distributed Smith-Waterman Algorithm based on apache spark,” in IEEE International Conference on Cloud Computing (Honololu, HI).
Bornman, D. M., Hester, M. E., Schuetter, J. M., Kasoji, M. D., Minard-Smith, A., Barden, C. A., et al. (2012). Short-read, high-throughput sequencing technology for STR genotyping. Biotech. Rapid Dispatches 2012, 1–6. doi: 10.2144/000113857
Børsting, C., and Morling, N. (2015). Next generation sequencing and its applications in forensic genetics. Forensic Sci. Int. 18, 78–89. doi: 10.1016/j.fsigen.2015.02.002
Budiš, J., Kucharík, M., Duriš, F., Gazdarica, J., Zrubcová, M., Ficek, A., et al. (2018). Dante: genotyping of known complex and expanded short tandem repeats. Bioinformatics 35, 1310–1317 doi: 10.1093/bioinformatics/bty791
Bushnell, B., Rood, J., and Singer, E. (2017). BBMerge–accurate paired shotgun read merging via overlap. PLoS ONE 12:e0185056. doi: 10.1371/journal.pone.0185056
Cao, M. D., Balasubramanian, S., and Bodén, M. (2014). Sequencing technologies and tools for short tandem repeat variation detection. Brief. Bioinform. 16, 193–204. doi: 10.1093/bib/bbu001
Caskey, C. T., Pizzuti, A., Fu, Y. H., Fenwick, R. G., and Nelson, D. L. (1992). Triplet repeat mutations in human-disease. Science 256, 784–789. doi: 10.1126/science.256.5058.784
Castoe, T. A., Poole, A. W., de Koning, A. J., Jones, K. L., Tomback, D. F., Oyler-McCance, S. J., et al. (2012). Rapid microsatellite identification from Illumina paired-end genomic sequencing in two birds and a snake. PLoS ONE 7:e30953. doi: 10.1371/journal.pone.0030953
Castro, M. R. D., Tostes, C. D. S., Dávila, A. M. R., Senger, H., and Silva, F. A. B. D. (2017). SparkBLAST: scalable BLAST processing using in-memory operations. BMC Bioinformatics 18:318. doi: 10.1186/s12859-017-1723-8
Cavagnaro, P. F., Senalik, D. A., Yang, L., Simon, P. W., Harkins, T. T., Kodira, C. D., et al. (2010). Genome-wide characterization of simple sequence repeats in cucumber (Cucumis sativus L.). BMC Genomics 11:569. doi: 10.1186/1471-2164-11-569
Chen, S., Chen, Y., Sun, F., Waterman, M. S., and Zhang, X. (2019). A new statistic for efficient detection of repetitive sequences. Bioinformatics (Oxford, England) 35, 4596–4606. doi: 10.1093/bioinformatics/btz262
Chen, Z., Zhao, P., Li, C., Li, F., Xiang, D., Chen, Y.-Z., et al. (2021). iLearnPlus: a comprehensive and automated machine-learning platform for nucleic acid and protein sequence analysis, prediction and visualization. Nucleic Acids Res. 49:e60. doi: 10.1093/nar/gkab122
Chen, Z., Zhao, P., Li, F., Marquez-Lago, T. T., Leier, A., Revote, J., et al. (2020). iLearn: an integrated platform and meta-learner for feature engineering, machine-learning analysis and modeling of DNA, RNA and protein sequence data. Brief. Bioinform. 21, 1047–1057. doi: 10.1093/bib/bbz041
Chu, C., Nielsen, R., and Wu, Y. (2016). REPdenovo: inferring de novo repeat motifs from short sequence reads. PLoS ONE 11:e0150719. doi: 10.1371/journal.pone.0150719
de Knijff, P. (2018). From next generation sequencing to now generation sequencing in forensics. Forensic Sci. Int. 38, 175–180. doi: 10.1016/j.fsigen.2018.10.017
Dean, J., and Ghemawat, S. (2008). MapReduce: simplified data processing on large clusters. Commun. ACM 51, 107–113. doi: 10.1145/1327452.1327492
Decap, D., Reumers, J., Herzeel, C., Costanza, P., and Fostier, J. (2015). Halvade: scalable sequence analysis with MapReduce. Bioinformatics 31, 2482–2488. doi: 10.1093/bioinformatics/btv179
Decap, D., Reumers, J., Herzeel, C., Costanza, P., and Fostier, J. (2017). Halvade-RNA: parallel variant calling from transcriptomic data using MapReduce. PLoS ONE 12:e0174575. doi: 10.1371/journal.pone.0174575
Deng, L., Huang, G., Zhuang, Y., Wei, J., and Yan, Y. (2016). “HiGene: a high-performance platform for genomic data analysis,” in IEEE International Conference on Bioinformatics & Biomedicine (Shenzhen).
Dickson, R. J., and Gloor, G. B. (2013). XORRO: Rapid Paired-End Read Overlapper. arxiv [preprint].arxiv:1304.4620.
Dong, G., Fu, X., and Pan, L., i. H. (2017). An accurate sequence assembly algorithm for livestock, plants and microorganism based on Spark. Int. J. Pattern Recogn. Artif. Intell. 31:1750024. doi: 10.1142/S0218001417500240
Ellegren, H. (2004). Microsatellites: simple sequences with complex evolution. Nat. Rev. Genet. 5, 435–445. doi: 10.1038/nrg1348
Escalona, M., Rocha, S., and Posada, D. (2016). A comparison of tools for the simulation of genomic next-generation sequencing data. Nat. Rev. Genet. 17, 459–469. doi: 10.1038/nrg.2016.57
Expósito, R. R., González-Domínguez, J., and Touriño, J. (2018). HSRA: hadoop-based spliced read aligner for RNA sequencing data. PLoS ONE 13:e0201483. doi: 10.1371/journal.pone.0201483
Expósito, R. R., Veiga, J., González-Domínguez, J., and Touriño, J. (2017). MarDRe: efficient MapReduce-based removal of duplicate DNA reads in the cloud. Bioinformatics 33, 2762–2764. doi: 10.1093/bioinformatics/btx307
Fan, H., and Chu, J. Y. (2007). A brief review of short tandem repeat mutation. Genomic Proteomics Bioinform. 5, 7–14. doi: 10.1016/S1672-0229(07)60009-6
Fernandez-Silva, I., and Toonen, R. J. (2013). Optimizing selection of microsatellite loci from 454 pyrosequencing via post-sequencing bioinformatic analyses. Methods Mol. Biol. 1006, 101–120. doi: 10.1007/978-1-62703-389-3_7
Ferraro Petrillo, U., Roscigno, G., Cattaneo, G., and Giancarlo, R. (2017). FASTdoop: a versatile and efficient library for the input of FASTA and FASTQ files for MapReduce Hadoop bioinformatics applications. Bioinformatics 33, 1575–1577. doi: 10.1093/bioinformatics/btx010
Fungtammasan, A., Ananda, G., Hile, S. E., Su, M. S. W., Sun, C., Harris, R., et al. (2015). Accurate typing of short tandem repeats from genome-wide sequencing data and its applications. Genome Res. 25, 736–749. doi: 10.1101/gr.185892.114
Ganschow, S., Silvery, J., Kalinowski, J., and Tiemann, C. (2018). toaSTR: a web application for forensic STR genotyping by massively parallel sequencing. Forensic Sci. Int. 37, 21–28. doi: 10.1016/j.fsigen.2018.07.006
Genovese, L. M., Mosca, M. M., Pellegrini, M., and Geraci, F. (2019). Dot2dot: accurate whole-genome tandem repeats discovery. Bioinformatics 35, 914–922. doi: 10.1093/bioinformatics/bty747
Girgis, H. Z. (2015). Red: an intelligent, rapid, accurate tool for detecting repeats de-novo on the genomic scale. BMC Bioinformatics 16:227. doi: 10.1186/s12859-015-0654-5
Girgis, H. Z., and Sheetlin, S. L. (2013). MsDetector: toward a standard computational tool for DNA microsatellites detection. Nucleic Acids Res. 41:e22. doi: 10.1093/nar/gks881
Gnerre, S., MacCallum, I., Przybylski, D., Ribeiro, F. J., Burton, J. N., Walker, B. J., et al. (2011). High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl. Acad. Sci. 108, 1513–1518. doi: 10.1073/pnas.1017351108
Gropp, W., Lusk, E., and Skjellum, A. (1999). Using MPI: Portable Parallel Programming with the Message-Passing Interface. Cambridge, MA: MIT Press. doi: 10.7551/mitpress/7056.001.0001
Guang, X.-M., Xia, J.-Q., Lin, J.-Q., Yu, J., Wan, Q.-H., and Fang, S.-G. (2019). IDSSR: an efficient pipeline for identifying polymorphic microsatellites from a single genome sequence. Int. J. Mol. Sci. 20:3497. doi: 10.3390/ijms20143497
Guo, R., Li, Y. R., He, S., Le, O. Y., Sun, Y., Zhu, Z. (2018). RepLong: de novo repeat identification using long read sequencing data. Bioinformatics 34, 1099–1107. doi: 10.1093/bioinformatics/btx717
Gymrek, M., Golan, D., Rosset, S., and Erlich, Y. (2012). lobSTR: a short tandem repeat profiler for personal genomes. Genome Res. 22, 1154–1162. doi: 10.1101/gr.135780.111
Hoogenboom, J., van der Gaag, K. J., de Leeuw, R. H., Sijen, T., de Knijff, P., and Laros, J. F. (2017). FDSTools: a software package for analysis of massively parallel sequencing data with the ability to recognise and correct STR stutter and other PCR or sequencing noise. Forensic Sci. Int. 27, 27–40. doi: 10.1016/j.fsigen.2016.11.007
Kang, S. J., Lee, S. Y., and Lee, K. M. (2015). Performance comparison of OpenMP, MPI, and MapReduce in practical problems. Adv. Multimedia 2015:9. doi: 10.1155/2015/575687
Kistler, L., Johnson, S. M., Irwin, M. T., Louis, E. E., Ratan, A., and Perry, G. H. (2017). A massively parallel strategy for STR marker development, capture, and genotyping. Nucleic Acids Res. 45, e142–e142. doi: 10.1093/nar/gkx574
Li, H. L., Pang, Y. H., and Liu, B. (2021). BioSeq-BLM: a platform for analyzing DNA, RNAand protein sequences based on biological language models. Nucleic Acids Res. 49:e129. doi: 10.1093/nar/gkab829
Li, X., Tan, G., Zhang, C., Xu, L., and Sun, N. (2017). “Accelerating large-scale genomic analysis with Spark,” in IEEE International Conference on Bioinformatics & Biomedicine (Shenzhen).
Liang, S. (1999). The Java Native Interface: Programmer's Guide and Specification. Boston, MA: Addison-Wesley Professional.
Lim, K. G., Kwoh, C. K., Hsu, L. Y., and Wirawan, A. (2013). Review of tandem repeat search tools: a systematic approach to evaluating algorithmic performance. Brief. Bioinform. 14, 67–81. doi: 10.1093/bib/bbs023
Liu, B. (2019). BioSeq-Analysis: a platform for DNA, RNA and protein sequence analysis based on machine learning approaches. Brief. Bioinform. 20, 1280–1294. doi: 10.1093/bib/bbx165
Liu, B., Gao, X., and Zhang, H. (2019). BioSeq-Analysis2.0: an updated platform for analyzing DNA, RNA and protein sequences at sequence level and residue level based on machine learning approaches. Nucleic Acids Res. 47:e127. doi: 10.1093/nar/gkz740
Liu, B., Yuan, J., Yiu, S. M., Li, Z., Xie, Y., Chen, Y., et al. (2012). COPE: an accurate k-mer-based pair-end reads connection tool to facilitate genome assembly. Bioinformatics 28, 2870–2874. doi: 10.1093/bioinformatics/bts563
Lopes, R. S., Moraes, W. J. L., Rodrigues, T. S., and Bartholomeu, D. C. (2015). ProGeRF: proteome and genome repeat finder utilizing a fast parallel hash function. BioMed Res. Int. 2015:394157. doi: 10.1155/2015/394157
Madesis, P., Ganopoulos, I., and Tsaftaris, A. (2013). Microsatellites: Evolution and Contribution. New York, NY: Springer. doi: 10.1007/978-1-62703-389-3_1
Magoc, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Mardis, E. R. (2017). DNA sequencing technologies: 2006-2016. Nat. Protocols 12, 213–218. doi: 10.1038/nprot.2016.182
Masseroli, M., Canakoglu, A., Pinoli, P., Kaitoua, A., Gulino, A., Horlova, O., et al. (2018). Processing of big heterogeneous genomic datasets for tertiary analysis of Next Generation Sequencing data. Bioinformatics 35, 729-736. doi: 10.1093/bioinformatics/bty688
Matsunaga, A. M., Tsugawa, M. O., and Fortes, J. A. B. (2009). “CloudBLAST: combining MapReduce and virtualization on distributed resources for bioinformatics applications,” in IEEE Fourth International Conference on Escience (Indianapolis. IN). doi: 10.1109/eScience.2008.62
Mckenna, A., Hanna, M. E., Sivachenko, A., Cibulskis, K., Kernytsky, A., Garimella, K., et al. (2010). The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
Meglécz, E., Pech, N., Gilles, A., Dubut, V., Hingamp, P., Trilles, A., et al. (2014). QDD version 3.1: a user-friendly computer program for microsatellite selection and primer design revisited: experimental validation of variables determining genotyping success rate. Mol. Ecol. Resour. 14, 1302–1313. doi: 10.1111/1755-0998.12271
Miller, M. P., Knaus, B. J., Mullins, T. D., and Haig, S. M. (2013). SSR_pipeline: a bioinformatic infrastructure for identifying microsatellites from paired-end Illumina high-throughput DNA sequencing data. J. Heredity 104, 881–885. doi: 10.1093/jhered/est056
Mitas, M. (1997). Trinucleotide repeats associated with human disease. Nucleic Acids Res. 25, 2245–2253. doi: 10.1093/nar/25.12.2245
Mitsuhashi, S., Frith, M. C., Mizuguchi, T., Miyatake, S., Toyota, T., Adachi, H., et al. (2019). Tandem-genotypes: robust detection of tandem repeat expansions from long DNA reads. Genome Biol. 20:58. doi: 10.1186/s13059-019-1667-6
Modified Version (1.2.11) of FLASH (2015). Available online at: https://github.com/Jerrythafast/FLASH-lowercase-overhang
Mokhtar, M. M., and Atia, M. A. M. (2018). SSRome: an integrated database and pipelines for exploring microsatellites in all organisms. Nucleic Acids Res. 47, D244–D252. doi: 10.1093/nar/gky998
Nashta-ali, D., Aliyari, A., Moghadam, A. A., Edrisi, M. A., Motahari, S. A., and Khalaj, B. H. (2017). Meta-aligner: long-read alignment based on genome statistics. BMC Bioinformatics 18:126. doi: 10.1186/s12859-017-1518-y
Nguyen, T., Shi, W., and Ruden, D. (2011). CloudAligner: a fast and full-featured MapReduce based tool for sequence mapping. BMC Res. Notes 4:171. doi: 10.1186/1756-0500-4-171
Niemenmaa, M., Kallio, A., Schumacher, A., Klemelä, P., Korpelainen, E., and Heljanko, K. (2012). Hadoop-BAM: directly manipulating next generation sequencing data in the cloud. Bioinformatics 28, 876–877. doi: 10.1093/bioinformatics/bts054
Nordberg, H., Bhatia, K., Wang, K., and Wang, Z. (2013). BioPig: a Hadoop-based analytic toolkit for large-scale sequence data. Bioinformatics 29, 3014–3019. doi: 10.1093/bioinformatics/btt528
Oliveira, R. R. M., Nunes, G. L., de Lima, T. G. L., Oliveira, G., and Alves, R. (2018). PIPEBAR and OverlapPER: tools for a fast and accurate DNA barcoding analysis and paired-end assembly. BMC Bioinformatics 19:297. doi: 10.1186/s12859-018-2307-y
Parson, W., Ballard, D., Budowle, B., Butler, J. M., Gettings, K. B., Gill, P., et al. (2016). Massively parallel sequencing of forensic STRs: considerations of the DNA commission of the International Society for Forensic Genetics (ISFG) on minimal nomenclature requirements. Forensic Sci. Int. 22, 54–63. doi: 10.1016/j.fsigen.2016.01.009
Perry, J. C., and Rowe, L. (2011). Rapid microsatellite development for water striders by next-generation sequencing. J. Hered 102, 125–129. doi: 10.1093/jhered/esq099
Petrillo, U. F., Sorella, M., Cattaneo, G., Giancarlo, R., and Rombo, S. E. (2019). Analyzing big datasets of genomic sequences: fast and scalable collection of k-mer statistics. BMC Bioinformatics 20:138. doi: 10.1186/s12859-019-2694-8
Pickett, B., Karlinsey, S., Penrod, C., Cormier, M., Ebbert, M. T., Shiozawa, D. K., et al. (2016). SA-SSR: a suffix array-based algorithm for exhaustive and efficient SSR discovery in large genetic sequences. Bioinformatics 32, 2707–2709. doi: 10.1093/bioinformatics/btw298
Pickett, B. D., Miller, J. B., and Ridge, P. G. (2017). Kmer-SSR: a fast and exhaustive SSR search algorithm. Bioinformatics 33, 3922–3928. doi: 10.1093/bioinformatics/btx538
Renaud, G., Stenzel, U., and Kelso, J. (2014). leeHom: adaptor trimming and merging for Illumina sequencing reads. Nucleic Acids Res. 42, e141–e141. doi: 10.1093/nar/gku699
Samadi, Y., Zbakh, M., and Tadonki, C. (2018). Performance comparison between Hadoop and Spark frameworks using HiBench benchmarks. Concurrency Comput. Practice Exp. 30:e4367. doi: 10.1002/cpe.4367
Schatz, M. C. (2009). CloudBurst: highly sensitive read mapping with MapReduce. Bioinformatics 25, 1363–1369. doi: 10.1093/bioinformatics/btp236
Schumacher, A., Pireddu, L., Niemenmaa, M., Kallio, A., Korpelainen, E., Zanetti, G., et al. (2014). SeqPig: simple and scalable scripting for large sequencing data sets in Hadoop. Bioinformatics 30, 119–120. doi: 10.1093/bioinformatics/btt601
Selkoe, K. A., and Toonen, R. J. (2010). Microsatellites for ecologists: a practical guide to using and evaluating microsatellite markers. Ecol. Lett. 9, 615–629. doi: 10.1111/j.1461-0248.2006.00889.x
Shi, L., Meng, X., Tseng, E., Mascagni, M., and Wang, Z. (2018). SpaRC: scalable sequence clustering using Apache Spark. Bioinformatics 35, 760–768. doi: 10.1093/bioinformatics/bty733
Shvachko, K., Kuang, H., Radia, S., and Chansler, R. (2010). “The hadoop distributed file system,” in IEEE 26th Symposium on Mass Storage Systems and Technologies (MSST) (Washington, DC), 1–10. doi: 10.1109/MSST.2010.5496972
Taheri, S., Abdullah, T. L., Yusop, M. R., Hanafi, M. M., Sahebi, M., Azizi, P., et al. (2018). Mining and development of novel SSR markers using Next Generation Sequencing (NGS) data in plants. Molecules 23:399. doi: 10.3390/molecules23020399
Tang, H., and Nzabarushimana, E. (2017). STRScan: targeted profiling of short tandem repeats in whole-genome sequencing data. BMC Bioinformatics 18:398. doi: 10.1186/s12859-017-1800-z
Taylor, R. C. (2010). An overview of the Hadoop/MapReduce/HBase framework and its current applications in bioinformatics. BMC Bioinformatics 11(Suppl 12):S1. doi: 10.1186/1471-2105-11-S12-S1
The 1000 Genomes Project Consortium (2010). A map of human genome variation from population-scale sequencing. Nature 467:1061–1073. doi: 10.1038/nature09534
van der Gaag, K. J., de Leeuw, R. H., Hoogenboom, J., Patel, J., Storts, D. R., Laros, J. F., et al. (2016). Massively parallel sequencing of short tandem repeats—population data and mixture analysis results for the PowerSeq™ system. Forensic Sci. Int. 24, 86–96. doi: 10.1016/j.fsigen.2016.05.016
Van Neste, C., Vandewoestyne, M., Van Criekinge, W., Deforce, D., and Van Nieuwerburgh, F. (2014). My-Forensic-Loci-queries (MyFLq) framework for analysis of forensic STR data generated by massive parallel sequencing. Forensic Sci. Int. 9, 1–8. doi: 10.1016/j.fsigen.2013.10.012
Vandervalk, B. P., Jackman, S. D., Raymond, A., Mohamadi, H., Yang, C., Attali, D. A., et al. (2014). “Konnector: connecting paired-end reads using a bloom filter de Bruijn graph,” in 2014 IEEE International Conference on Bioinformatics and Biomedicine (BIBM) (Belfast), 51–58. doi: 10.1109/BIBM.2014.6999126
Vargas Jentzsch, I. M., Bagshaw, A., Buschiazzo, E., Merkel, A., and Gemmell, N. J. (2013). Evolution of Microsatellite DNA. Hoboken, NJ: John Wiley & Sons, Ltd. doi: 10.1002/9780470015902.a0020847.pub2
Vavilapalli, V. K., Murthy, A. C., Douglas, C., Agarwali, S., and Konar, M. (2013). “Apache Hadoop YARN: Yet Another Resource Negotiator,” in Proceedings of the 4th Annual Symposium on Cloud Computing (Santa Clara, CA), 330–339. doi: 10.1145/2523616.2523633
Velasco, A., James, B. T., Wells, V. D., and Girgis, H. Z. (2019). Look4TRs: a de-novo tool for detecting simple tandem repeats using self-supervised hidden Markov models. Bioinformatics (Oxford, England) 36, 380−387. doi: 10.1093/bioinformatics/btz551
Vilsen, S. B., Tvedebrink, T., Eriksen, P. S., Bøsting, C., Hussing, C., Mogensen, H. S., et al. (2018). Stutter analysis of complex STR MPS data. Forensic Sci. Int. 35, 107–112. doi: 10.1016/j.fsigen.2018.04.003
Wang, X. (2016). Next-Generation Sequencing Data Analysis. Boca Raton, FL: CRC Press, Inc. doi: 10.1201/b19532
Wang, X., Lu, P., and Luo, Z. (2013). GMATo: a novel tool for the identification and analysis of microsatellites in large genomes. Bioinformation 9:541. doi: 10.6026/97320630009541
Wang, X., and Wang, L. (2016). GMATA: an integrated software package for genome-scale SSR mining, marker development and viewing. Front. Plant Sci. 7:1350. doi: 10.3389/fpls.2016.01350
White, T. (2009). Hadoop: The Definitive Guide: MapReduce for the Cloud. Sebastopol, CA: O'Reilly Media, Inc.
Wiewiórka, M. S., Antonio, M., Alicja, P., Sergio, M., Piotr, G., and Okoniewski, M. J. (2014). SparkSeq: fast, scalable and cloud-ready tool for the interactive genomic data analysis with nucleotide precision. Bioinformatics 30, 2652–2653. doi: 10.1093/bioinformatics/btu343
Willems, T., Gymrek, M., Highnam, G., Mittelman, D., Erlich, Y., and Consortium, G. P. (2014). The landscape of human STR variation. Genome Res. 24, 1894–1904. doi: 10.1101/gr.177774.114
Wirtz, S., Böhm, C., Fritz, J., Hankeln, T., and Hochkirch, A. (2016). Isolation of microsatellite loci by next-generation sequencing of the critically endangered Northern Bald ibis, Geronticus eremita. J. Heredity 107, 363–366. doi: 10.1093/jhered/esw013
Wordsworth, S., Doble, B., Payne, K., Buchanan, J., and Marshall, D. A. (2018). McCabe C, Regier DA: using “big data” in the cost-effectiveness analysis of next-generation sequencing technologies: challenges and potential solutions. Value Health 21, 1048–1053. doi: 10.1016/j.jval.2018.06.016
Xue, D. X., Li, Y. L., and Liu, J. X. (2017). A rapid and cost-effective approach for the development of polymorphic microsatellites in non-model species using paired-end RAD sequencing. Mol. Genet. Genomics 292, 1165–1174. doi: 10.1007/s00438-017-1337-x
Yang, A., Troup, M., Lin, P., and Ho, J. W. K. (2016). Falco: a quick and flexible single-cell RNA-seq processing framework on the cloud. Bioinformatics 33:btw732. doi: 10.1093/bioinformatics/btw732
Zaharia, M., Chowdhury, M., Franklin, M. J., Shenker, S., and Stoica, I. (2010). “Spark: cluster computing with working sets,” in USENIX Conference on Hot Topics in Cloud Computing (Boston, MA).
Zalapa, J. E., Hugo, C., Huayu, Z., Shawn, S., Douglas, S., Eric, Z., et al. (2012). Using next-generation sequencing approaches to isolate simple sequence repeat (SSR) loci in the plant sciences. Am. J. Bot. 99, 193–208. doi: 10.3732/ajb.1100394
Zhang, J., Kobert, K., Flouri, T., and Stamatakis, A. (2013). PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 30, 614–620. doi: 10.1093/bioinformatics/btt593
Zhao, G., Ling, C., and Sun, D. (2015). “Sparksw: scalable distributed computing system for large-scale biological sequence alignment,” in 2015 15th IEEE/ACM International Symposium on Cluster, Cloud and Grid Computing (Shenzhen), 845–852. doi: 10.1109/CCGrid.2015.55
Zhao, L., Chen, Q., Li, W., Jiang, P., Wong, L., and Li, J. (2017). MapReduce for accurate error correction of next-generation sequencing data. Bioinformatics 33, 3844–3851. doi: 10.1093/bioinformatics/btx089
Zhou, W., Li, R., Yuan, S., Liu, C. C., Yao, S., Luo, J., et al. (2017). MetaSpark: a spark-based distributed processing tool to recruit metagenomic reads to reference genomes. Bioinformatics 33:1090. doi: 10.1093/bioinformatics/btw750
Zou, Q., Hu, Q., Guo, M., and Wang, G. (2015). HAlign: fast multiple similar DNA/RNA sequence alignment based on the centre star strategy. Bioinformatics 31, 2475–2481. doi: 10.1093/bioinformatics/btv177
Keywords: next-generation sequencing, read pairs, Simple Sequence Repeats (SSR), Hadoop, big data
Citation: Chen J, Li F, Wang M, Li J, Marquez-Lago TT, Leier A, Revote J, Li S, Liu Q and Song J (2022) BigFiRSt: A Software Program Using Big Data Technique for Mining Simple Sequence Repeats From Large-Scale Sequencing Data. Front. Big Data 4:727216. doi: 10.3389/fdata.2021.727216
Received: 18 June 2021; Accepted: 13 December 2021;
Published: 18 January 2022.
Edited by:
Javier Garcia-Blas, Universidad Carlos III de Madrid, SpainReviewed by:
Ping Guo, University of Illinois at Springfield, United StatesCopyright © 2022 Chen, Li, Wang, Li, Marquez-Lago, Leier, Revote, Li, Liu and Song. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiangning Song, amlhbmduaW5nLnNvbmdAbW9uYXNoLmVkdQ==; Quanzhong Liu, bGl1cXpob25nQG53c3VhZi5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.