 Michael Colla
Michael Colla Steffi Weidt
Steffi Weidt Erich Seifritz
Erich Seifritz Golo Kronenberg
Golo Kronenberg- Department of Psychiatry, Psychotherapy and Psychosomatics, Hospital of Psychiatry, University of Zurich, Zurich, Switzerland
The serendipitous discovery of ketamine’s antidepressant effects represents one of the major landmarks in neuropsychopharmacological research of the last 50 years. Ketamine provides an exciting challenge to traditional concepts of antidepressant drug therapy, producing rapid antidepressant effects seemingly without targeting monoaminergic pathways in the conventional way. In consequence, the advent of ketamine has spawned a plethora of neurobiological research into its putative mechanisms. Here, we provide a brief overview of current theories of antidepressant drug action including monoaminergic signaling, disinhibition of glutamatergic neurotransmission, neurotrophic and neuroplastic effects, and how these might relate to ketamine. Given that research into ketamine has not yet yielded new therapies beyond ketamine itself, current knowledge gaps and limitations of available studies are also discussed.
Introduction
Ketamine, synthesized in 1962 by the research team of Calvin Stevens, was the culmination of Parke-Davis’s drive to find a short-acting intravenous anesthetic with favorable cardiovascular and respiratory characteristics (Mion, 2017). Ketamine replaced its congener phencyclidine (PCP), which, after a brief period of use as an anesthetic agent under the brand name Sernyl ®, had to be abandoned owing to high rates of postoperative dysphoria and hallucinations (Meyer et al., 1959). The distinctive state produced by ketamine — characterized by analgesia, catalepsy, and amnesia, while maintaining respiratory reflexes and hemodynamic stability — was first described by Corssen and Domino (1965), who dubbed it “dissociative anesthesia.” Its wide therapeutic index makes ketamine an excellent agent for use in emergency medical practice, battlefield pain management, and, more generally, in resource-stripped settings such as the developing world. Ketamine remains on the most recent WHO Model List of Essential Medicines as an injectable general anesthetic (World Health Organization [WHO], 2019).
Ketamine burst on the scene of antidepressant psychopharmacology in 2000 with the publication of its first double-blind placebo-controlled trial in major depression. This pilot investigation of seven patients found significant improvements in mood within 72 h of a single subanesthetic dose of intravenous racemic ketamine hydrochloride (Berman et al., 2000). A number of follow-up studies have confirmed the fast-onset antidepressant effects of ketamine infusions (Zarate et al., 2006a; aan het Rot et al., 2010; Murrough et al., 2013a,b). Moreover, adjunctive intravenous ketamine has emerged as a powerful new treatment option for patients suffering from treatment-resistant depression (TRD; Diazgranados et al., 2010; Fava et al., 2020). In the interim, variant forms of ketamine therapy including treatment with the S-enantiomer (i.e., S-ketamine) and administration via the nasal (Popova et al., 2019) and oral route (Domany et al., 2019) have also been demonstrated to confer rapid antidepressant benefit. Table 1 summarizes key studies of ketamine in depression.
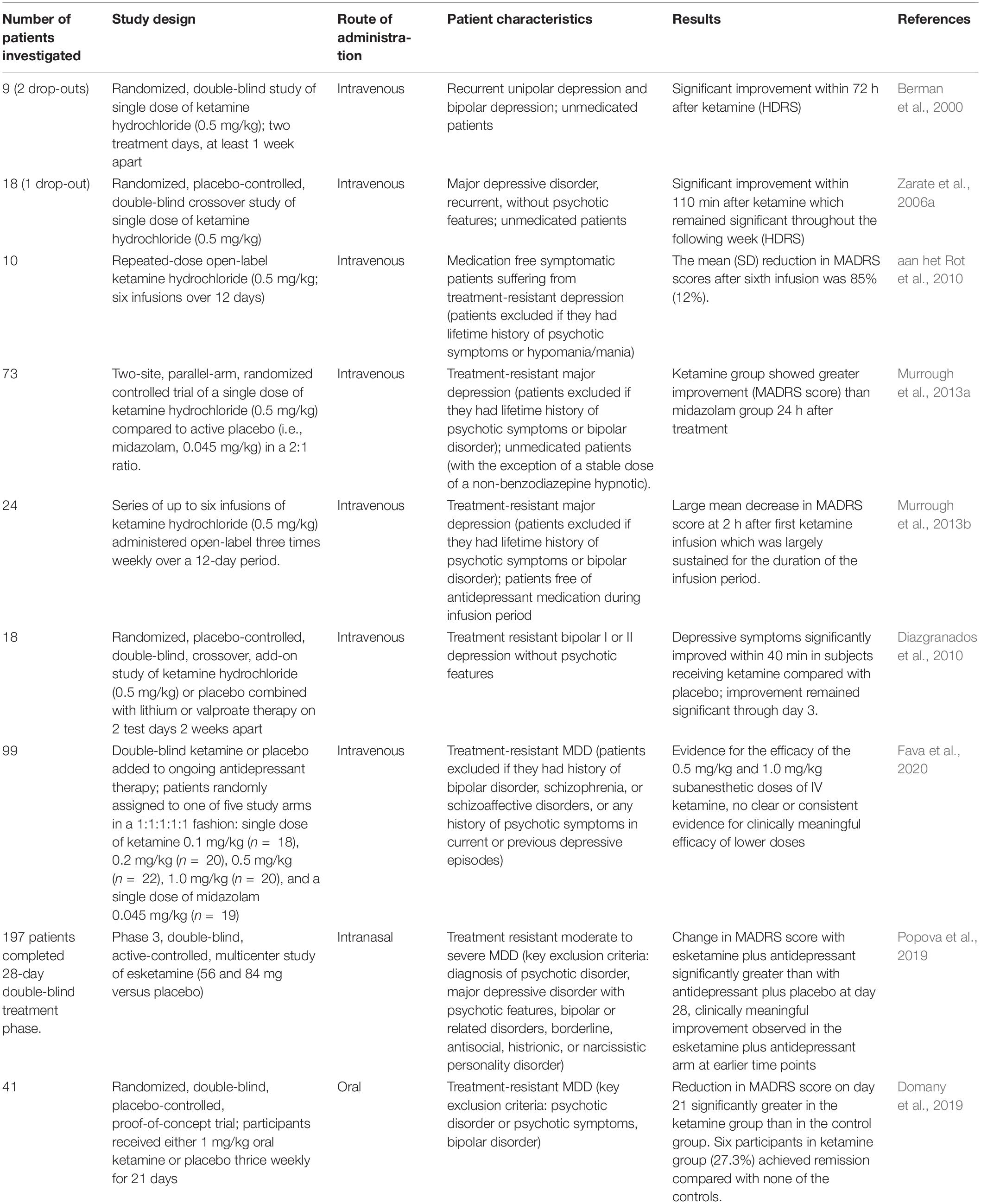
Table 1. Overview of key studies of ketamine in depression.
From a neuroscience perspective, the uncanny rapidity of ketamine’s antidepressant action (often within a few hours) sets it apart from conventional antidepressants, providing a new window on the neurobiology of the antidepressant response with exciting possibilities for translational and, maybe even more interesting, reverse translational research. The purpose of this mini-review is, therefore, to provide an overview of current thinking on ketamine’s putative mechanisms of action within the context of antidepressant drug discovery and development.
Monoamine Mechanisms
The short history of the development of antidepressant drugs is riddled with accidental yet transformative discoveries. At the risk of recounting well-known facts, here is a summary of the milestones: Iproniazid, initially developed and marketed by Hoffmann La-Roche as an antibiotic to treat tuberculosis (Marsilid ®), was serendipitously identified as possessing antidepressant characteristics (Loomer et al., 1957; deVerteuil and Lehmann, 1958). A connection was quickly made with iproniazid’s strong inhibitory effect on monoamine oxidase (MAO), paving the way for the targeted development of other, more refined, and ultimately safer MAO inhibitors, which are still widely prescribed today (e.g., Stefanis et al., 1982). The antidepressant activity of imipramine, the first tricyclic antidepressant, was recognized almost coevally with the discovery of iproniazid’s antidepressant properties (Kuhn, 1957). Inhibition of the reuptake of biogenic amines was swiftly identified as the primary molecular mechanism of tricyclics (Axelrod et al., 1961; Carlsson et al., 1966, 1968; Fuxe and Ungerstedt, 1968). The observation that blood-pressure lowering drug reserpine may precipitate depression (Harris, 1957) provided further support for a link between brain levels of biogenic amines and mood states. Taken together, and in historical perspective, this “monoamine hypothesis” of depression has proven incredibly useful in the development of newer classes of antidepressants (such as selective serotonin reuptake inhibitors, noradrenaline reuptake inhibitors, dual reuptake inhibitors, etc.) that are usually superior to the older compounds with comparable efficacy yet fewer side effects and a greater therapeutic index. Nevertheless, the clinical limitations of monoamine-based agents, in particular relatively high rates of non-response and even resistance to treatment, have long led to calls to focus more research on alternative mechanisms (Berton and Nestler, 2006).
An obvious conceptual problem with the monoamine hypothesis lies in the fact that changes in neurotransmitter concentrations (along with the onset of typical side effects) occur within a few hours while conventional antidepressants typically require several days to weeks to take effect. Neurobiological research into the mechanisms underpinning the antidepressant response has therefore pivoted to longer-term adaptive changes downstream of the acute effects on biogenic amines.
Ketamine’s principal pharmacological action is as an N-methyl-D-aspartate (NMDA) receptor antagonist. However, in a manner somewhat reminiscent of clozapine, ketamine is a “dirty” drug. Multiple off-target effects including on monoamine systems need to be considered. In vitro, ketamine displays affinity to dopamine D2 and serotonin 5-HT2 receptors in the same range as its affinity for the NMDA receptor (Kapur and Seeman, 2002). It has also been reported that ketamine inhibits monoamine transporters in cultured cells (Nishimura et al., 1998) and blocks the uptake of [3H]-dopamine into rat striatal synaptosomes (Keita et al., 1996).
Repeated ketamine injections increase the firing rate of norepinephrine neurons in the locus coeruleus and of dopaminergic neurons in the ventral tegmental area in rats (Iro et al., 2021). Microdialysis studies have demonstrated increased serotonin release by ketamine in the rodent prefrontal cortex (Ago et al., 2019; López-Gil et al., 2019). Several groups have found that serotonin depletion abrogates the antidepressive-like effects of ketamine in the forced swim test (Gigliucci et al., 2013; Fukumoto et al., 2015; du Jardin et al., 2016; Pham et al., 2017). Even so, measurable occupancy of the serotonin transporter in vivo was not detectable by positron emission tomography in twelve healthy human subjects after infusion of an antidepressant dose of ketamine (Spies et al., 2018).
While the available literature indicates that ketamine leads to increased dopamine levels in frontal cortex, striatum, and nucleus accumbens in rodents, the picture is less clear for the primate and human brain, given methodological issues and the scant available literature (Kokkinou et al., 2018). From a clinical perspective, the fact that haloperidol is able to ameliorate ketamine-induced psychosis argues for a role of dopaminergic pathways in ketamine’s psychotropic effects (Giannini et al., 2000).
Ketamine and the Glutamatergic System
Racemic ketamine acts as a non-competitive NMDA receptor antagonist (Figure 1A). It is believed that the dissociative and psychotomimetic effects of PCP and ketamine relate directly to the affinity of these molecules to the NMDA receptor. Based on displacement binding studies with [3H]-MK801 as the marker ligand, S-ketamine exhibits an approximately three- to fourfold higher affinity to the NMDA receptor than R-ketamine (Moaddel et al., 2013). The pharmacokinetic profiles of racemic ketamine and its two enantiomers do not differ significantly in humans (White et al., 1985). Serum ketamine concentrations at the point of regaining consciousness and orientation during the course of experimental anesthesia of human volunteers indicate an S:R ketamine isomer potency ratio of 4:1. Similarly, S-ketamine has an approximately three- to fivefold greater ability to impair psychomotor function than R-ketamine (White et al., 1985). The available literature, though scant, seems to suggests that, in humans, subanesthetic doses of R-ketamine lack the dissociative potential of racemic ketamine (Vollenweider et al., 1997; Leal et al., 2021).
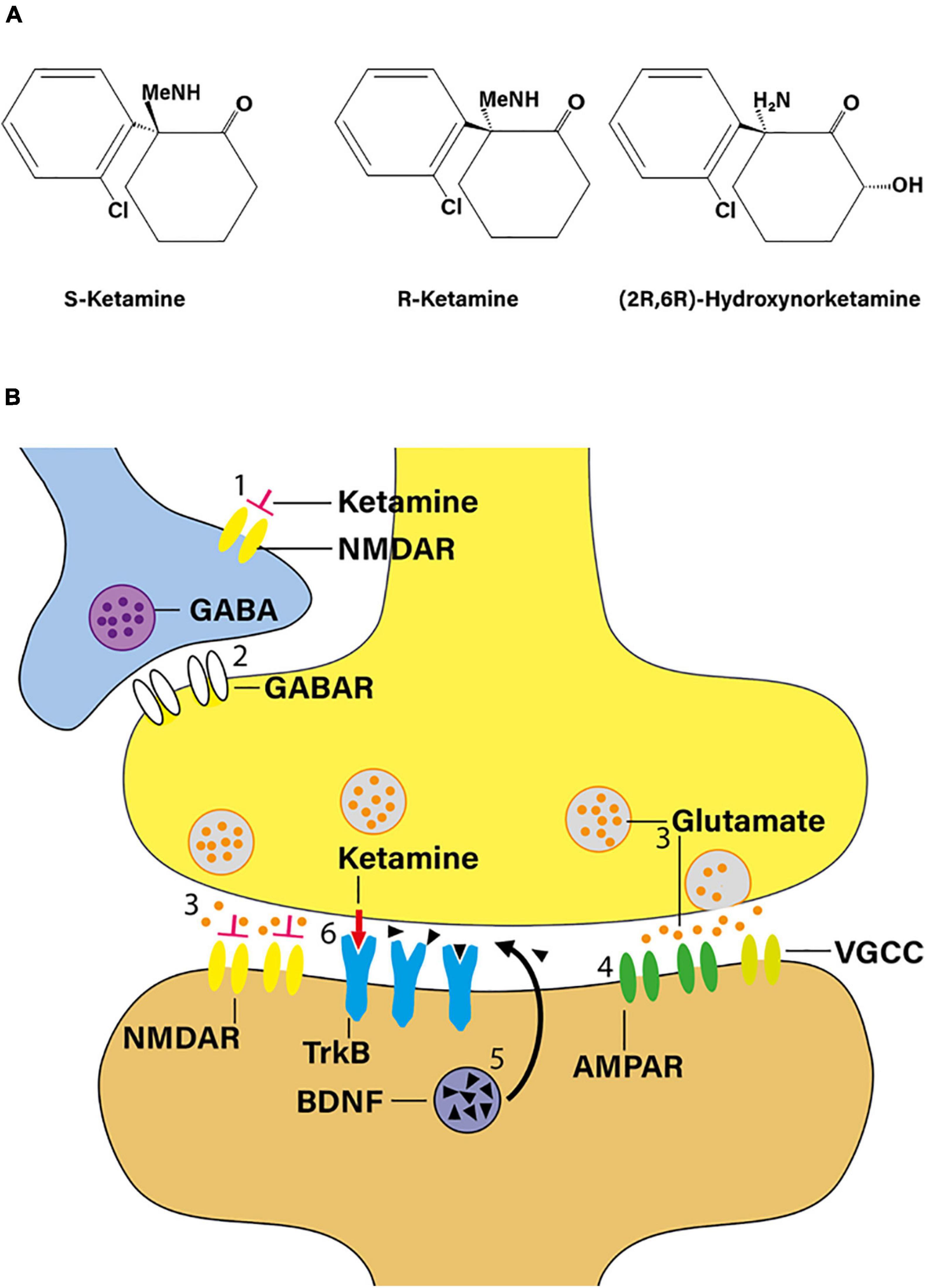
Figure 1. Ketamine as a novel antidepressant. (A) Structural formula of S-ketamine, R-ketamine, and R-ketamine metabolite 2R,6R-hydroxynorketamine (adapted from Zanos et al., 2016). Me, methyl moiety. (B) According to the “disinhibition hypothesis” of ketamine action, NMDA receptor blockade by ketamine may increase glutamatergic outflow. When administered in a subanesthetic dose, ketamine blocks NMDA receptors on γ-aminobutyric acid (GABA) interneurons (1), thereby reducing GABA release (2) on principal neurons, and, in turn, increasing presynaptic release of glutamate (3). Preferential activation of postsynaptic α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptors (AMPAR) in mood-regulating synapses (4) is believed to play a critical role in mediating ketamine’s rapid antidepressant response, triggering downstream changes such as inducing BDNF signaling (5). In addition, ketamine may also interact with the TrkB receptor directly (red arrow; 6). TrkB, tropomyosin receptor kinase B; VGCC, voltage-gated calcium channel (adapted from Shinohara et al., 2020).
It is tempting to speculate that the dissociative and the antidepressant effects of ketamine might be separable. In the context of double-blind placebo-controlled drug testing, this is a complex issue because questions around functional unblinding due to ketamine’s dissociative effects and the potential use of active comparators have to be considered (Ballard and Zarate, 2020). At least so far, the bulk of the available clinical evidence seems to favor an association between racemic ketamine’s hallucinogenic/dissociative and antidepressant effects (Mathai et al., 2020).
While clinical research into a possible role for R-ketamine in depression is still in its infancy, sufficient data has already accrued to recommend the use of S-ketamine. Intravenous S-ketamine has been shown to produce rapid onset of robust antidepressant effects in patients with TRD after a 40-min infusion (Singh et al., 2016). Further, there is meta-analytical evidence for the adjunctive intranasal use of S-ketamine in TRD and in depressed patients with acute suicidality (Papakostas et al., 2020). Moreover, a recent randomized double-blind head-to-head comparison of intravenous S-ketamine (0.25 mg/kg) and racemic ketamine (0.5 mg/kg) as adjunctive therapy in TRD confirmed non-inferiority of S-ketamine (Correia-Melo et al., 2020).
N-methyl-D-aspartate receptor blockade may augment glutamatergic outflow, e.g., in the prefrontal cortex. Indeed, one plausible mechanism of this seemingly paradoxical effect is that ketamine, when administered in a subanesthetic dose, blocks NMDA receptors on γ-aminobutyric acid interneurons, thereby increasing presynaptic release of glutamate (Moghaddam et al., 1997; Pothula et al., 2020). According to this “disinhibition hypothesis” (Figure 1B), downstream activation of α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid (AMPA) receptors in mood-regulating synapses is believed to play a crucial role in mediating ketamine’s rapid antidepressant response as evidenced by the fact that pre-treatment with NBQX, an AMPA receptor antagonist, attenuates the behavioral effects of ketamine in experimental mice and rats (Maeng et al., 2008; Koike and Chaki, 2014). It thus appears that, ultimately, ketamine produces increased glutamatergic throughput of AMPA receptors, as compared to NMDA receptors, triggering rapid downstream changes on the molecular, structural, and network levels (Figure 1B; Jourdi et al., 2009; Li et al., 2010; Autry et al., 2011).
While clinical research has, so far, focused primarily on the S-ketamine stereoisomer, it has been hypothesized, based on behavioral studies in experimental mice, that R-ketamine should show the greater antidepressant potency (Yang et al., 2015; Zanos et al., 2016). To our knowledge, there is currently only one small pilot trial that has investigated the effects of R-ketamine in major depression (Leal et al., 2021). That open-label study of seven patients reported a significant decrease in Montgomery-Åsberg Depression Rating Scale scores within 24 h of a single intravenous infusion of R-ketamine (0.5 mg/kg).
There is extensive metabolism of ketamine stereoisomers via cytochrome P450 enzymes producing a broad array of catabolites including norketamine, hydroxyketamines, dehydronorketamine, and the hydroxynorketamines (Kharasch and Labroo, 1992; Desta et al., 2012). In particular, potent antidepressant properties have been ascribed to the (2R,6R)-hydroxynorketamine [(2R,6R)-HNK] metabolite (Figure 1A), which is exclusively derived from R-ketamine (Zanos et al., 2016). Mechanistically, (2R,6R)-HNK acts through AMPA receptor-mediated mechanisms, with the AMPA receptor antagonist NBQX reversing its antidepressant-like effects (Zanos et al., 2016). Moreover, (2R,6R)-HNK recapitulates key downstream events observed in the rodent brain in response to ketamine such as increased neurotrophic signaling and rapid dendritic and synaptic plasticity (Autry et al., 2011; Zanos et al., 2016).
Neurotrophic Signaling, Neuroplasticity, and Stress
Profound structural changes such as neuronal atrophy, loss of synapses, and a decrease in hippocampal neurogenesis reflect the deleterious effects of stress, stress hormones, and major depression on the brain (Duman et al., 2016). Brain-derived neurotrophic factor (BDNF)/tropomyosin receptor kinase B (TrkB) signaling is of crucial importance to neuronal plasticity, morphogenesis, and survival (Huang and Reichardt, 2001). Numerous pre-clinical studies have connected stress and an excess of corticosteroids with reduced BDNF signaling in depression-related brain areas (Smith et al., 1995; Schaaf et al., 2000; Vollmayr et al., 2000; Oh et al., 2019). Conversely, bilateral infusion of BDNF into the hippocampal dentate gyrus has been shown to produce antidepressant-like effects in behavioral models of depression (Shirayama et al., 2002). Antidepressant interventions such as electroconvulsive therapy (Nibuya et al., 1995), physical activity (Sleiman et al., 2016), and conventional antidepressant pharmacotherapy (Conti et al., 2002) have all been linked with a rise in brain BDNF levels. Likewise, ketamine administration has been shown to raise BDNF mRNA and protein levels in hippocampus (Choi et al., 2017). BDNF signaling seems to be central to ketamine’s distinct antidepressant activity because ketamine fails to produce rapid antidepressant-like effects in either BDNF or TrkB conditional knockout mice (Autry et al., 2011). Quite unexpectedly, some very recent research has demonstrated an exciting new mode of action of several antidepressants, including ketamine, beyond increasing BDNF concentrations, namely, to directly bind to TrkB (Casarotto et al., 2021). Antidepressant binding to TrkB could then facilitate BDNF action and the attendant cellular as well as structural plasticity (Casarotto et al., 2021). An important intracellular signaling pathway activated in response to ketamine is the mammalian target of rapamycin pathway. Activation of this pathway promotes rapid synaptic plasticicity with increased synaptic signaling proteins and increased number and function of synapses (Li et al., 2010). In this context, and given that the anti-dementia drug memantine, which shares with ketamine the property of non-competitive NMDA antagonism, is widely prescribed in Alzheimer’s disease, it may be worthwhile to assess the effects of ketamine in patients with dementia (Smalheiser, 2019).
So far, few studies have investigated the effects of ketamine on hippocampal neurogenesis (Deyama and Duman, 2020). It has been reported that ketamine increases cell proliferation in the hippocampal dentate gyrus of rats showing a depressive-like phenotype (Michaëlsson et al., 2019). However, since neurogenesis is a multi-step process that unfolds over several weeks (Kempermann et al., 2004), it is unlikely that an overall increase in neurogenesis explains ketamine’s rapid antidepressant effects. Still, increased recruitment of adult-born neurons into hippocampal circuitry (i.e., an acceleration in the final stages of neurogenesis) in response to ketamine is an obvious possibility, especially considering the importance of these immature cells for shaping memory processes (Anacker and Hen, 2017).
Open Questions and Outlook
An honest appraisal of where the field stands today must acknowledge the fact that, so far, “decades of ‘murinization”’ have contributed relatively little to antidepressant development (Holsboer, 2014). From the sole perspective of drug discovery, the poor predictability of antidepressant efficacy based on behavioral assays in rodents is probably chief among today’s challenges. On the other hand, it should be noted that the concept of NMDA antagonism in the treatment of depression was developed against a rich backdrop of experimental research (reviewed in Skolnick et al., 1996), demonstrating, among other things, that chronic administration of desipramine inhibits glutamatergic neurotransmission at NMDA receptors (Mjellem et al., 1993), and that both conventional antidepressants and electroconvulsive therapy alter the ligand-binding properties of the NMDA receptor complex (Paul et al., 1993, 1994). Given the short nature of this mini-review, the considerable body of preclinical evidence demonstrating ketamine’s antidepressant activity in rodent models of depression has been largely passed over. For a detailed overview of this subject, the reader is referred to Polis et al. (2019) and Rincón-Cortés and Grace (2020).
How will the field evolve in the future? As a logical next step, the R-ketamine enantiomer is currently in the early stages of clinical development. Moreover, certain ketamine metabolites may hold promise as possessing equal antidepressant efficacy to the racemic parent molecule, possibly with fewer side effects, especially (2R,6R)-HNK. From a broader view, however, the prospect of discovering other molecules, not directly related to ketamine itself but tapping into the same neurobiological mechanisms, remains uncertain, at least for the time being. So far, the principle of NMDA antagonism has, unfortunately, not translated into tangible new drugs. Also, side-effects beyond psychotic symptoms have to be considered. Merck & Co’s dizocilpine (commonly referred to as MK-801 in the lab), a strong NMDA receptor antagonist, was shown to produce acute pathomorphological lesions in specific populations of neurons when administered acutely to adult rats in comparatively low doses (Olney et al., 1989). MK-801 is no longer in active clinical development for this reason. Similar evidence of neurotoxicity (the eponymous “Olney’s” lesions) has also been observed in experimental rodents after ketamine and PCP (reviewed in Ellison, 1995) and, more worryingly, in human ketamine addicts (Wang et al., 2013). Given these findings, it will be important, from a safety standpoint, to monitor the long-term effects of NMDA antagonist therapy (including with ketamine and S-ketamine) on brain structure and patients’ cognitive trajectories.
Moving beyond neurotoxicity, which may represent a class effect, investigations of NMDA receptor antagonists other than ketamine in depression have, so far, failed to produce clinically relevant outcomes. Memantine proved ineffective as an antidepressant in two double-blind placebo-controlled trials (Zarate et al., 2006b; Smith et al., 2013). Similarly, rislenemdaz (also known as MK-0657), an NR2B subunit-specific NMDA receptor antagonist, failed to produce antidepressant effects in TRD, either when used as a monotherapy or in conjunction with other antidepressants (Ibrahim et al., 2012; Henter et al., 2021). Lanicemine, an NMDA blocker with low rates of associated psychotomimetic effects, does not come near to replicating ketamine’s antidepressant effects (Zarate et al., 2013; Sanacora et al., 2017). More recently, three phase-III clinical trials of rapastinel, an NMDA receptor modulator with glycine-site partial agonist features, also failed to demonstrate antidepressant effects (Henter et al., 2021). This outcome is sobering, given that pre-clinical research had demonstrated antidepressant-like effects of rapastinel in mice and rats (Burgdorf et al., 2013; Yang et al., 2016).
The possibility of still other modes of action should also not be overlooked. It has long been known that ketamine possesses certain anti-inflammatory properties, which may especially benefit patients undergoing major surgery or septic patients requiring sedation (Kawasaki et al., 1999; Welters et al., 2011). Intriguingly, lipopolysaccharide-induced sickness behavior in mice can be blocked by ketamine (Walker et al., 2013). Moreover, the antidepressant effects of the two ketamine enantiomers in the chronic social defeat stress model of depression have been linked with restoration of gut microbiota in mice (Yang et al., 2017).
Opioid effects have also been implicated in ketamine’s clinical profile. Both S- and R-ketamine bind to and activate mu and kappa opioid receptors (Bonaventura et al., 2021). Further, it has recently been reported that naltrexone blocks the antidepressant effects of ketamine in depressed patients (Williams et al., 2018).
In the aggregate, ketamine represents the first major breakthrough in antidepressant development in the last half-century. As described above, it engages novel mechanisms beyond monoaminergic neurotransmission, resulting in a much faster onset of action than conventional monoamine-based therapeutics. Although much remains to be elucidated, the advent of ketamine signals exciting new opportunities to extend and refine our knowledge of the neurobiological mechanisms underlying the antidepressant response. Given the accruing evidence of ketamine’s therapeutic effects in TRD, it seems that the time has arrived to assign a central position to ketamine as an augmentation in the treatment algorithms for TRD patients.
Author Contributions
GK drafted the manuscript with substantive input from all authors. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors are conducting a study of oral ketamine in TRD funded by Ketabon GmbH. ES serves on advisory boards of Janssen.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
aan het Rot, M., Collins, K. A., Murrough, J. W., Perez, A. M., Reich, D. L., Charney, D. S., et al. (2010). Safety and efficacy of repeated-dose intravenous ketamine for treatment-resistant depression. Biol. Psychiatry 67, 139–145. doi: 10.1016/j.biopsych.2009.08.038
Ago, Y., Tanabe, W., Higuchi, M., Tsukada, S., Tanaka, T., Yamaguchi, T., et al. (2019). (R)-Ketamine induces a greater increase in prefrontal 5-HT Release than (S)-Ketamine and ketamine metabolites via an AMPA Receptor-Independent Mechanism. Int. J. Neuropsychopharmacol. 22, 665–674. doi: 10.1093/ijnp/pyz041
Anacker, C., and Hen, R. (2017). Adult hippocampal neurogenesis and cognitive flexibility - linking memory and mood. Nat. Rev. Neurosci. 18, 335–346. doi: 10.1038/nrn.2017.45
Autry, A. E., Adachi, M., Nosyreva, E., Na, E. S., Los, M. F., Cheng, P. F., et al. (2011). NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 475, 91–95. doi: 10.1038/nature10130
Axelrod, J., Whitby, L. G., and Hertting, G. (1961). Effect of psychotropic drugs on the uptake of H3-norepinephrine by tissues. Science 133, 383–384. doi: 10.1126/science.133.3450.383
Ballard, E. D., and Zarate, C. A. Jr. (2020). The role of dissociation in ketamine’s antidepressant effects. Nat. Commun. 11:6431. doi: 10.1038/s41467-020-20190-4
Berman, R. M., Cappiello, A., Anand, A., Oren, D. A., Heninger, G. R., Charney, D. S., et al. (2000). Antidepressant effects of ketamine in depressed patients. Biol. Psychiatry 47, 351–354. doi: 10.1016/s0006-3223(99)00230-9
Berton, O., and Nestler, E. (2006). New approaches to antidepressant drug discovery: beyond monoamines. Nat. Rev. Neurosci. 7, 137–151. doi: 10.1038/nrn1846
Bonaventura, J., Lam, S., Carlton, M., Boehm, M. A., Gomez, J. L., Solís, O., et al. (2021). Pharmacological and behavioral divergence of ketamine enantiomers: implications for abuse liability. Mol. Psychiatry [Epub Online ahead of print]. doi: 10.1038/s41380-021-01093-2
Burgdorf, J., Zhang, X. L., Nicholson, K. L., Balster, R. L., Leander, J. D., Stanton, P. K., et al. (2013). GLYX-13, a NMDA receptor glycine-site functional partial agonist, induces antidepressant-like effects without ketamine-like side effects. Neuropsychopharmacology 38, 729–742. doi: 10.1038/npp.2012.246
Carlsson, A., Fuxe, K., Hamberger, B., and Lindqvist, M. (1966). Biochemical and histochemical studies on the effects of imipramine-like drugs and (+)-amphetamine on central and peripheral catecholamine neurons. Acta Physiol. Scand. 67, 481–497. doi: 10.1111/j.1748-1716.1966.tb03334.x
Carlsson, A., Fuxe, K., and Ungerstedt, U. (1968). The effect of imipramine on central 5-hydroxytryptamine neurons. J. Pharm. Pharmacol. 20, 150–151. doi: 10.1111/j.2042-7158.1968.tb09706.x
Casarotto, P. C., Girych, M., Fred, S. M., Kovaleva, V., Moliner, R., Enkavi, G., et al. (2021). Antidepressant drugs act by directly binding to TRKB neurotrophin receptors. Cell 184, 1299–1313. doi: 10.1016/j.cell.2021.01.034
Choi, M., Lee, S. H., Park, M. H., Kim, Y. S., and Son, H. (2017). Ketamine induces brain-derived neurotrophic factor expression via phosphorylation of histone deacetylase 5 in rats. Biochem. Biophys. Res. Commun. 489, 420–425. doi: 10.1016/j.bbrc.2017.05.157
Conti, A. C., Cryan, J. F., Dalvi, A., Lucki, I., and Blendy, J. A. (2002). cAMP response element-binding protein is essential for the upregulation of brain-derived neurotrophic factor transcription, but not the behavioral or endocrine responses to antidepressant drugs. J. Neurosci. 22, 3262–3268. doi: 10.1523/jneurosci.22-08-03262.2002
Correia-Melo, F. S., Leal, G. C., Vieira, F., Jesus-Nunes, A. P., Mello, R. P., Magnavita, G., et al. (2020). Efficacy and safety of adjunctive therapy using esketamine or racemic ketamine for adult treatment-resistant depression: a randomized, double-blind, non-inferiority study. J. Affect Disord. 264, 527–534. doi: 10.1016/j.jad.2019.11.086
Corssen, G., and Domino, E. F. (1965). Dissociative Anesthesia: Further Pharmacologic Studies and First Clinical Experience with the Phencyclidine Derivative CI-581. Anesth Analg. 45, 29–40. doi: 10.1213/00000539-196601000-00007
Desta, Z., Moaddel, R., Ogburn, E. T., Xu, C., Ramamoorthy, A., Venkata, S. L., et al. (2012). Stereoselective and regiospecific hydroxylation of ketamine and norketamine. Xenobiotica 42, 1076–1087. doi: 10.3109/00498254.2012.685777
deVerteuil, R. L., and Lehmann, H. E. (1958). Therapeutic Trial of Iproniazid (Marsilid) in depressed and apathetic patients. Can. Med. Assoc. J. 78, 131–133.
Deyama, S., and Duman, R. S. (2020). Neurotrophic mechanisms underlying the rapid and sustained antidepressant actions of ketamine. Pharmacol. Biochem. Behav. 188:172837. doi: 10.1016/j.pbb.2019.172837
Diazgranados, N., Ibrahim, L., Brutsche, N. E., Newberg, A., Kronstein, P., Khalife, S., et al. (2010). A randomized add-on trial of an N-methyl-D-aspartate antagonist in treatment-resistant bipolar depression. Arch. Gen. Psychiatry 67, 793–802. doi: 10.1001/archgenpsychiatry.2010.90
Domany, Y., Bleich-Cohen, M., Tarrasch, R., Meidan, R., Litvak-Lazar, O., Stoppleman, N., et al. (2019). Repeated oral ketamine for out-patient treatment of resistant depression: randomised, double-blind, placebo-controlled, proof-of-concept study. Br. J. Psychiatry 214, 20–26. doi: 10.1192/bjp.2018.196
du Jardin, K. G., Liebenberg, N., Müller, H. K., Elfving, B., Sanchez, C., and Wegener, G. (2016). Differential interaction with the serotonin system by S-ketamine, vortioxetine, and fluoxetine in a genetic rat model of depression. Psychopharmacology 233, 2813–2825. doi: 10.1007/s00213-016-4327-5
Duman, R. S., Aghajanian, G. K., Sanacora, G., and Krystal, J. H. (2016). Synaptic plasticity and depression: new insights from stress and rapid-acting antidepressants. Nat. Med. 22, 238–249. doi: 10.1038/nm.4050
Ellison, G. (1995). The N-methyl-D-aspartate antagonists phencyclidine, ketamine and dizocilpine as both behavioral and anatomical models of the dementias. Brain Res. Brain Res. Rev. 20, 250–267. doi: 10.1016/0165-0173(94)00014-g
Fava, M., Freeman, M. P., Flynn, M., Judge, H., Hoeppner, B. B., Cusin, C., et al. (2020). Double-blind, placebo-controlled, dose-ranging trial of intravenous ketamine as adjunctive therapy in treatment-resistant depression (TRD). Mol. Psychiatry 25, 1592–1603. doi: 10.1038/s41380-018-0256-5
Fukumoto, K., Iijima, M., and Chaki, S. (2015). The antidepressant effects of an mGlu2/3 receptor antagonist and ketamine require AMPA receptor stimulation in the mPFC and subsequent activation of the 5-HT neurons in the DRN. Neuropsychopharmacology 41, 1046–1056. doi: 10.1038/npp.2015.233
Fuxe, K., and Ungerstedt, U. (1968). Histochemical studies on the effect of (+)-amphetamine, drugs of the imipramine group and tryptamine on central catecholamine and 5-hydroxytryptamine neurons after intraventricular injection of catecholamines and 5-hydroxytryptamine. Eur. J. Pharmacol. 4, 135–144. doi: 10.1016/0014-2999(68)90169-6
Giannini, A. J., Underwood, N. A., and Condon, M. (2000). Acute ketamine intoxication treated by haloperidol: a preliminary study. Am. J. Ther. 7, 389–391. doi: 10.1097/00045391-200007060-00008
Gigliucci, V., O’Dowd, G., Casey, S., Egan, D., Gibney, S., and Harkin, A. (2013). Ketamine elicits sustained antidepressant-like activity via a serotonin-dependent mechanism. Psychopharmacology 228, 157–166. doi: 10.1007/s00213-013-3024-x
Harris, T. H. (1957). Depression induced by Rauwolfia compounds. Am. J. Psychiat. 113:950. doi: 10.1176/ajp.113.10.950
Henter, I. D., Park, L. T., and Zarate, C. A. Jr. (2021). Novel glutamatergic modulators for the treatment of mood disorders: current status. CNS Drugs 35, 527–543. doi: 10.1007/s40263-021-00816-x
Holsboer, F. (2014). Redesigning antidepressant drug discovery. Dialogues Clin. Neurosci. 16, 5–7. doi: 10.31887/dcns.2014.16.1/fholsboer
Huang, E. J., and Reichardt, L. F. (2001). Neurotrophins: roles in neuronal development and function. Annu. Rev. Neurosci. 24, 677–736. doi: 10.1146/annurev.neuro.24.1.677
Ibrahim, L., Diaz Granados, N., Jolkovsky, L., Brutsche, N., Luckenbaugh, D. A., Herring, W. J., et al. (2012). A Randomized, placebo-controlled, crossover pilot trial of the oral selective NR2B antagonist MK-0657 in patients with treatment-resistant major depressive disorder. J. Clin. Psychopharmacol. 32, 551–557. doi: 10.1097/JCP.0b013e31825d70d6
Iro, C. M., Hamati, R., El Mansari, M., and Blier, P. (2021). Repeated but not single administration of ketamine prolongs increases of the firing activity of norepinephrine and dopamine neurons. Int. J. Neuropsychopharmacol. 24, 570–579. doi: 10.1093/ijnp/pyab010
Jourdi, H., Hsu, Y. T., Zhou, M., Qin, Q., Bi, X., and Baudry, M. (2009). Positive AMPA receptor modulation rapidly stimulates BDNF release and increases dendritic mRNA translation. J. Neurosci. 29, 8688–8697. doi: 10.1523/JNEUROSCI.6078-08.2009
Kapur, S., and Seeman, P. (2002). NMDA receptor antagonists ketamine and PCP have direct effects on the dopamine D2 and serotonin 5-HT2 receptors—implications for models of schizophrenia. Mol. Psychiatry 7, 837–844. doi: 10.1038/sj.mp.4001093
Kawasaki, T., Ogata, M., Kawasaki, C., Ogata, J., Inoue, Y., and Shigematsu, A. (1999). Ketamine suppresses proinflammatory cytokine production in human whole blood in vitro. Anesth. Analg. 89, 665–669. doi: 10.1213/00000539-199909000-00024
Keita, H., Lecharny, J. B., Henzel, D., Desmonts, J. M., and Mantz, J. (1996). Is inhibition of dopamine uptake relevant to the hypnotic action of i.v. anaesthetics? Br. J. Anaesth. 77, 254–256. doi: 10.1093/bja/77.2.254
Kempermann, G., Jessberger, S., Steiner, B., and Kronenberg, G. (2004). Milestones of neuronal development in the adult hippocampus. Trends Neurosci. 27, 447–452. doi: 10.1016/j.tins.2004.05.013
Kharasch, E. D., and Labroo, R. (1992). Metabolism of ketamine stereoisomers by human liver microsomes. Anesthesiology 77, 1201–1207. doi: 10.1097/00000542-199212000-00022
Koike, H., and Chaki, S. (2014). Requirement of AMPA receptor stimulation for the sustained antidepressant activity of ketamine and LY341495 during the forced swim test in rats. Behav. Brain Res. 271, 111–115. doi: 10.1016/j.bbr.2014.05.065
Kokkinou, M., Ashok, A. H., and Howes, O. D. (2018). The effects of ketamine on dopaminergic function: meta-analysis and review of the implications for neuropsychiatric disorders. Mol. Psychiatry 23, 59–69. doi: 10.1038/mp.2017.190
Kuhn, R. (1957). Treatment of depressive states with an iminodibenzyl derivative (G 22355). Schweiz. Med. Wochenschr. 87, 1135–1140.
Leal, G. C., Bandeira, I. D., Correia-Melo, F. S., Telles, M., Mello, R. P., Vieira, F., et al. (2021). Intravenous arketamine for treatment-resistant depression: open-label pilot study. Eur. Arch. Psychiatry Clin. Neurosci. 271, 577–582. doi: 10.1007/s00406-020-01110-5
Li, N., Lee, B., Liu, R. J., Banasr, M., Dwyer, J. M., Iwata, M., et al. (2010). mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 329, 959–964. doi: 10.1126/science.1190287
Loomer, H. P., Saunders, J. C., and Kline, N. S. (1957). A clinical and pharmacodynamic evaluation of iproniazid as a psychic energizer. Psychiatr. Res. Rep. Am. Psychiatr. Assoc. 8, 129–141.
López-Gil, X., Jiménez-Sánchez, L., Campa, L., Castro, E., Frago, C., and Adell, A. (2019). Role of Serotonin and Noradrenaline in the Rapid Antidepressant Action of Ketamine. ACS Chem. Neurosci. 10, 3318–3326. doi: 10.1021/acschemneuro.9b00288
Maeng, S., Zarate, C. A. Jr., Du, J., Schloesser, R. J., McCammon, J., Chen, G., et al. (2008). Cellular mechanisms underlying the antidepressant effects of ketamine: role of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biol. Psychiatry 63, 349–352. doi: 10.1016/j.biopsych.2007.05.028
Mathai, D. S., Meyer, M. J., Storch, E. A., and Kosten, T. R. (2020). The relationship between subjective effects induced by a single dose of ketamine and treatment response in patients with major depressive disorder: a systematic review. J. Affect Disord. 264, 123–129. doi: 10.1016/j.jad.2019.12.023
Meyer, J. S., Greifenstein, F., and Devault, M. (1959). A new drug causing symptoms of sensory deprivation. J. Nerv. Ment. Dis. 129, 54–61. doi: 10.1097/00005053-195907000-00006
Michaëlsson, H., Andersson, M., Svensson, J., Karlsson, L., Ehn, J., Culley, G., et al. (2019). The novel antidepressant ketamine enhances dentate gyrus proliferation with no effects on synaptic plasticity or hippocampal function in depressive-like rats. Acta Physiol. (Oxf) 225:e13211. doi: 10.1111/apha.13211
Mion, G. (2017). History of anaesthesia: the ketamine story - past, present and future. Eur. J. Anaesthesiol. 34, 571–575. doi: 10.1097/EJA.0000000000000638
Mjellem, N., Lund, A., and Hole, K. (1993). Reduction of NMDA-induced behaviour after acute and chronic administration of desipramine in mice. Neuropharmacology 32, 591–595. doi: 10.1016/0028-3908(93)90055-8
Moaddel, R., Abdrakhmanova, G., Kozak, J., Jozwiak, K., Toll, L., Jimenez, L., et al. (2013). Sub-anesthetic concentrations of (R,S)-ketamine metabolites inhibit acetylcholine-evoked currents in alpha7 nicotinic acetylcholine receptors. Eur. J. Pharmacol. 698, 228–234. doi: 10.1016/j.ejphar.2012.11.023
Moghaddam, B., Adams, B., Verma, A., and Daly, D. (1997). Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J. Neurosci. 17, 2921–2927. doi: 10.1523/JNEUROSCI.17-08-02921.1997
Murrough, J. W., Iosifescu, D. V., Chang, L. C., Al Jurdi, R. K., Green, C. E., Perez, A. M., et al. (2013a). Antidepressant efficacy of ketamine in treatment-resistant major depression: a two-site randomized controlled trial. Am. J. Psychiatry 170, 1134–1142. doi: 10.1176/appi.ajp.2013.13030392
Murrough, J. W., Perez, A. M., Pillemer, S., Stern, J., Parides, M. K., aan het Rot, M., et al. (2013b). Rapid and longer-term antidepressant effects of repeated ketamine infusions in treatment-resistant major depression. Biol. Psychiatry 74, 250–256. doi: 10.1016/j.biopsych.2012.06.022
Nibuya, M., Morinobu, S., and Duman, R. S. (1995). Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J. Neurosci. 15, 7539–7547. doi: 10.1523/JNEUROSCI.15-11-07539.1995
Nishimura, M., Sato, K., Okada, T., Yoshiya, I., Schloss, P., Shimada, S., et al. (1998). Ketamine inhibits monoamine transporters expressed in human embryonic kidney 293 cells. Anesthesiology 88, 768–774. doi: 10.1097/00000542-199803000-00029
Oh, H., Piantadosi, S. C., Rocco, B. R., Lewis, D. A., Watkins, S. C., and Sibille, E. (2019). The role of dendritic brain-derived neurotrophic factor transcripts on altered inhibitory circuitry in depression. Biol. Psychiatry 85, 517–526. doi: 10.1016/j.biopsych.2018.09.026
Olney, J. W., Labruyere, J., and Price, M. T. (1989). Pathological changes induced in cerebrocortical neurons by phencyclidine and related drugs. Science 244, 1360–1362. doi: 10.1126/science.2660263
Papakostas, G. I., Salloum, N. C., Hock, R. S., Jha, M. K., Murrough, J. W., Mathew, S. J., et al. (2020). Efficacy of esketamine augmentation in major depressive disorder: a meta-analysis. J. Clin. Psychiatry 81:19r12889. doi: 10.4088/JCP.19r12889
Paul, I. A., Layer, R. T., Skolnick, P., and Nowak, G. (1993). Adaptation of the NMDA receptor in rat cortex following chronic electroconvulsive shock or imipramine. Eur. J. Pharmacol. 247, 305–311. doi: 10.1016/0922-4106(93)90199-J
Paul, I. A., Nowak, G., Layer, R. T., Popik, P., and Skolnick, P. (1994). Adaptation of the N-methyl-D-aspartate receptor complex following chronic antidepressant treatments. J. Pharmacol. Exp. Ther. 269, 95–102.
Pham, T. H., Mendez-David, I., Defaix, C., Guiard, B. P., Tritschler, L., David, D. J., et al. (2017). Ketamine treatment involves medial prefrontal cortex serotonin to induce a rapid antidepressant-like activity in BALB/cJ mice. Neuropharmacology 112, 198–209. doi: 10.1016/j.neuropharm.2016.05.010
Polis, A. J., Fitzgerald, P. J., Hale, P. J., and Watson, B. O. (2019). Rodent ketamine depression-related research: finding patterns in a literature of variability. Behav. Brain Res. 376:112153. doi: 10.1016/j.bbr.2019.112153
Popova, V., Daly, E. J., Trivedi, M., Cooper, K., Lane, R., Lim, P., et al. (2019). Efficacy and safety of flexibly dosed esketamine nasal spray combined with a newly initiated oral antidepressant in treatment-resistant depression: a randomized double-blind active-controlled study. Am. J. Psychiatry 176, 428–438. doi: 10.1176/appi.ajp.2019.19020172
Pothula, S., Kato, T., Liu, R. J., Wu, M., Gerhard, D., Shinohara, R., et al. (2020). Cell-type specific modulation of NMDA receptors triggers antidepressant actions. Mol. Psychiatry [Epub Online ahead of print]. doi: 10.1038/s41380-020-0796-3
Rincón-Cortés, M., and Grace, A. A. (2020). Antidepressant effects of ketamine on depression-related phenotypes and dopamine dysfunction in rodent models of stress. Behav. Brain Res. 379:112367. doi: 10.1016/j.bbr.2019.112367
Sanacora, G., Johnson, M. R., Khan, A., Atkinson, S. D., Riesenberg, R. R., Schronen, J. P., et al. (2017). Adjunctive Lanicemine (AZD6765) in patients with major depressive disorder and history of inadequate response to antidepressants: a randomized, placebo-controlled study. Neuropsychopharmacology 42, 844–853. doi: 10.1038/npp.2016.224
Schaaf, M. J., De Kloet, E. R., and Vreugdenhil, E. (2000). Corticosterone effects on BDNF expression in the hippocampus. Implications for memory formation. Stress 3, 201–208. doi: 10.3109/10253890009001124
Shinohara, R., Aghajanian, G. K., and Abdallah, C. G. (2020). Neurobiology of the rapid-acting antidepressant effects of ketamine: impact and opportunities. Biol. Psychiatry 90, 85–95. doi: 10.1016/j.biopsych.2020.12.006
Shirayama, Y., Chen, A. C., Nakagawa, S., Russell, D. S., and Duman, R. S. (2002). Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. J. Neurosci. 22, 3251–3261. doi: 10.1523/JNEUROSCI.22-08-03251.2002
Singh, J. B., Fedgchin, M., Daly, E., Xi, L., Melman, C., De Bruecker, G., et al. (2016). Intravenous esketamine in adult treatment-resistant depression: a double-blind, double-randomization, placebo-controlled study. Biol. Psychiatry 80, 424–431. doi: 10.1016/j.biopsych.2015.10.018
Skolnick, P., Layer, R. T., Popik, P., Nowak, G., Paul, I. A., and Trullas, R. (1996). Adaptation of N-methyl-D-aspartate (NMDA) receptors following antidepressant treatment: implications for the pharmacotherapy of depression. Pharmacopsychiatry 29, 23–26. doi: 10.1055/s-2007-979537
Sleiman, S. F., Henry, J., Al-Haddad, R., El Hayek, L., Abou Haidar, E., Stringer, T., et al. (2016). Exercise promotes the expression of brain derived neurotrophic factor (BDNF) through the action of the ketone body β-hydroxybutyrate. Elife 5:e15092. doi: 10.7554/eLife.15092
Smalheiser, N. R. (2019). Ketamine: a neglected therapy for Alzheimer disease. Front. Aging Neurosci. 11:186. doi: 10.3389/fnagi.2019.00186
Smith, E. G., Deligiannidis, K. M., Ulbricht, C. M., Landolin, C. S., Patel, J. K., and Rothschild, A. J. (2013). Antidepressant augmentation using the N-methyl-D-aspartate antagonist memantine: a randomized, double-blind, placebo-controlled trial. J. Clin. Psychiatry 74, 966–973. doi: 10.4088/JCP.12m08252
Smith, M. A., Makino, S., Kvetnansky, R., and Post, R. M. (1995). Stress and glucocorticoids affect the expression of brain-derived neurotrophic factor and neurotrophin-3 mRNAs in the hippocampus. J. Neurosci. 15, 1768–1777. doi: 10.1523/JNEUROSCI.15-03-01768.1995
Spies, M., James, G. M., Berroterán-Infante, N., Ibeschitz, H., Kranz, G. S., Unterholzner, J., et al. (2018). Assessment of ketamine binding of the serotonin transporter in humans with positron emission tomography. Int. J. Neuropsychopharmacol. 21, 145–153. doi: 10.1093/ijnp/pyx085
Stefanis, C. N., Alevizos, B. H., and Papadimitriou, G. N. (1982). Antidepressant effect of Ro 11-1163, a new MAO inhibitor. Int. Pharmacopsychiatry 17, 43–48. doi: 10.1159/000468556
Vollenweider, F. X., Leenders, K. L., Oye, I., Hell, D., and Angst, J. (1997). Differential psychopathology and patterns of cerebral glucose utilisation produced by (S)- and (R)-ketamine in healthy volunteers using positron emission tomography (PET). Eur. Neuropsychopharmacol. 7, 25–38. doi: 10.1016/S0924-977X(96)00042-9
Vollmayr, B., Faust, H., Lewicka, S., and Henn, F. A. (2000). Brain-derived-neurotrophic-factor (BDNF) stress response in rats bred for learned helplessness. Mol. Psychiatry 6, 471–474. doi: 10.1038/sj.mp.4000907
Walker, A. K., Budac, D. P., Bisulco, S., Lee, A. W., Smith, R. A., Beenders, B., et al. (2013). NMDA receptor blockade by ketamine abrogates lipopolysaccharide-induced depressive-like behavior in C57BL/6J mice. Neuropsychopharmacology 38, 1609–1616. doi: 10.1038/npp.2013.71
Wang, C., Zheng, D., Xu, J., Lam, W., and Yew, D. T. (2013). Brain damages in ketamine addicts as revealed by magnetic resonance imaging. Front. Neuroanat. 7:23. doi: 10.3389/fnana.2013.00023
Welters, I. D., Feurer, M. K., Preiss, V., Müller, M., Scholz, S., Kwapisz, M., et al. (2011). Continuous S-(+)-ketamine administration during elective coronary artery bypass graft surgery attenuates pro-inflammatory cytokine response during and after cardiopulmonary bypass. Br. J. Anaesth. 106, 172–179. doi: 10.1093/bja/aeq341
White, P. F., Schüttler, J., Shafer, A., Stanski, D. R., Horai, Y., and Trevor, A. J. (1985). Comparative pharmacology of the ketamine isomers. Br. J. Anesthesiol. 57, 197–203. doi: 10.1093/bja/57.2.197
Williams, N. R., Heifets, B. D., Blasey, C., Sudheimer, K., Pannu, J., Pankow, H., et al. (2018). Attenuation of antidepressant effects of ketamine by opioid receptor antagonism. Am. J. Psychiatry 175, 1205–1215. doi: 10.1176/appi.ajp.2018.18020138
World Health Organization [WHO] (2019). Model List of Essential Medicines. Available Online at: https://www.who.int/publications/i/item/WHOMVPEMPIAU2019.06 (accessed April 1, 2021).
Yang, B., Zhang, J. C., Han, M., Yao, W., Yang, C., Ren, Q., et al. (2016). Comparison of R-ketamine and rapastinel antidepressant effects in the social defeat stress model of depression. Psychopharmacology (Berl) 233, 3647–3657. doi: 10.1007/s00213-016-4399-2
Yang, C., Qu, Y., Fujita, Y., Ren, Q., Ma, M., Dong, C., et al. (2017). Possible role of the gut microbiota-brain axis in the antidepressant effects of (R)-ketamine in a social defeat stress model. Transl. Psychiatry 7:1294. doi: 10.1038/s41398-017-0031-4
Yang, C., Shirayama, Y., Zhang, J. C., Ren, Q., Yao, W., Ma, M., et al. (2015). R-ketamine: a rapid-onset and sustained antidepressant without psychotomimetic side effects. Transl. Psychiatry 5:e632. doi: 10.1038/tp.2015.136
Zanos, P., Moaddel, R., Morris, P. J., Georgiou, P., Fischell, J., Elmer, G. I., et al. (2016). NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature 533, 481–486. doi: 10.1038/nature17998
Zarate, C. A. Jr., Mathews, D., Ibrahim, L., Chaves, J. F., Marquardt, C., Ukoh, I., et al. (2013). A randomized trial of a low-trapping nonselective N-methyl-D-aspartate channel blocker in major depression. Biol. Psychiatry 74, 257–264. doi: 10.1016/j.biopsych.2012.10.019
Zarate, C. A. Jr., Singh, J. B., Carlson, P. J., Brutsche, N. E., Ameli, R., Luckenbaugh, D. A., et al. (2006a). A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch. Gen. Psychiatry 63, 856–864. doi: 10.1001/archpsyc.63.8.856
Keywords: depression, antidepressant, treatment-resistant depression, ketamine, BDNF, neurogenesis, monoamines, glutamate
Citation: Colla M, Scheerer H, Weidt S, Seifritz E and Kronenberg G (2021) Novel Insights Into the Neurobiology of the Antidepressant Response From Ketamine Research: A Mini Review. Front. Behav. Neurosci. 15:759466. doi: 10.3389/fnbeh.2021.759466
Received: 16 August 2021; Accepted: 28 October 2021;
Published: 03 December 2021.
Edited by:
Matthew O. Parker, University of Portsmouth, United KingdomReviewed by:
Juan Francisco Rodríguez-Landa, Universidad Veracruzana, MexicoDarcy Litteljohn, Independent Researcher, Toronto, ON, Canada
Copyright © 2021 Colla, Scheerer, Weidt, Seifritz and Kronenberg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Golo Kronenberg, Z29sby5rcm9uZW5iZXJnQHB1a3poLmNo