 Xun Yu Choong
Xun Yu Choong Justin L. Tosh
Justin L. Tosh Laura J. Pulford
Laura J. Pulford Elizabeth M. C. Fisher
Elizabeth M. C. Fisher- 1Department of Neurodegenerative Disease, Institute of Neurology, University College London, London, UK
- 2The LonDownS Consortium, London, UK
Down syndrome (DS) is a common genetic condition caused by the presence of three copies of chromosome 21 (trisomy 21). This greatly increases the risk of Alzheimer disease (AD), but although virtually all people with DS have AD neuropathology by 40 years of age, not all develop dementia. To dissect the genetic contribution of trisomy 21 to DS phenotypes including those relevant to AD, a range of DS mouse models has been generated which are trisomic for chromosome segments syntenic to human chromosome 21. Here, we consider key characteristics of human AD in DS (AD-DS), and our current state of knowledge on related phenotypes in AD and DS mouse models. We go on to review important features needed in future models of AD-DS, to understand this type of dementia and so highlight pathogenic mechanisms relevant to all populations at risk of AD.
Introduction: AD-DS, the Most Common Genetic Form of AD
Down syndrome (DS) is a complex, heterogeneous disorder caused by the presence of an extra copy of human chromosome 21. Trisomy 21 is a common condition, with an incidence of 1 in 750 live births (Parker et al., 2010). Prevalence in many countries is growing due to increasing maternal age, the greatest risk factor for DS (Loane et al., 2013), together with rises in DS life expectancy (Yang et al., 2002; Bittles and Glasson, 2004). In Northern Europe, for example, the number of people aged over 40 years with DS is approximately double what it was in 1990, and in the UK this age group accounts for a third of the estimated 40,000 people with DS (Wu and Morris, 2013).
The clinical presentation of DS varies extensively and includes features present in all individuals, such as cognitive deficits, and those seen in only some people, such as heart defects (Zigman, 2013; Jensen and Bulova, 2014). Alzheimer disease (AD) pathology is found in the brains of virtually all people with DS by 40 years of age (Wisniewski et al., 1985; Mann and Esiri, 1989), and trisomy 21 causes an increased risk of dementia such that approximately one third of the DS population has AD (“AD-DS”) by the age of 60, with an estimated lifetime prevalence of 90% for all people with DS (Prasher and Krishnan, 1993; Holland et al., 1998; Coppus et al., 2006; Margallo-Lana et al., 2007; McCarron et al., 2014). However, while AD-DS is one of the largest contributors to morbidity and mortality in DS (Coppus et al., 2008), not all individuals develop dementia, even by 70 years of age (Krinsky-McHale et al., 2008; Ghezzo et al., 2014). Thus, the DS population has the most common genetic form of early-onset AD, caused by trisomy 21. Studying AD-DS allows investigation of the initial pathogenic events leading to AD and the development of dementia, relevant to both people with DS and to the general population.
One approach to dissecting human disease is through studying mouse models, and a large number of transgenic strains have been generated to understand specific aspects of AD pathology, most of which have human gene mutations that give rise to rare early-onset familial Alzheimer disease (FAD; Braidy et al., 2012; Webster et al., 2014). In the last decade, chromosome engineering techniques have enabled the generation of an array of DS mouse models that will allow us to dissect the genetic contribution of chromosome 21 (Hsa21), or regions of the mouse genome syntenic to Hsa21, to DS phenotypes. These models recapitulate a wide range of DS features, including neurobiological, behavioral and aging-related aspects (Zhang et al., 2012b; Ruparelia et al., 2013). Thus, in the study of AD-DS, mouse models of DS offer an increasingly important approach to understanding pathogenic mechanisms, so informing us about pathways and networks relevant to all populations at risk of dementia.
Here, we present an overview of clinical features of AD-DS, compared to other genetic forms of AD, to highlight human phenotypes that may be assessed in mechanistic studies of mouse models. We then give examples of data from DS mouse models compared to transgenic mice modeling aspects of AD pathology, to illustrate informative findings from both types of model. We also offer examples of potentially helpful data for investigating AD-DS from the outcomes of overexpressing single genes from Hsa21. Finally, we consider the important features for mouse models to enhance our understanding of AD-DS, and therefore the pathogenetic mechanisms relevant to all AD. For brevity, citations may not necessarily be the original papers, but useful reviews or later references.
Genetic Forms of AD, Including AD-DS
The APP gene lies on Hsa21 and encodes the amyloid precursor protein that is at the heart of the amyloid cascade hypothesis of Alzheimer disease (Glenner and Wong, 1984; Hardy and Higgins, 1992; Hardy and Selkoe, 2002). This hypothesis was generated partly from the observation that extracellular plaques in brains of people with AD are composed of Aβ peptides that are products of APP metabolism. The hypothesis suggests that abnormal APP metabolism initiates AD pathogenesis by triggering a set of events that result in Aβ aggregation, particularly of the Aβ42 peptide, in these extracellular plaques. This leads to the formation of intracellular neurofibrillary tangles, primarily composed of the protein tau, and eventually loss of synapses and neurons. The relationship between the histopathological features of AD and dementia is not yet clear (Castellani and Perry, 2014).
The amyloid cascade hypothesis is currently the most widely-accepted paradigm guiding investigations of AD pathogenesis, and is supported at least in part by the rare cases of FAD caused by different mutations in APP, and in the presenilin genes PSEN1 and PSEN2 that affect APP processing. APP mutations may, for example, result in an increase in total Aβ production, or a relative increase in Aβ species associated with pathogenicity (Ryan and Rossor, 2010).
Importantly for understanding AD-DS, the link between APP and AD also extends to gene dose: in rare forms of FAD, duplication of the wildtype APP locus alone (“Dup-APP”) is sufficient to cause highly penetrant early-onset AD (Rovelet-Lecrux et al., 2006; Sleegers et al., 2006). Dup-APP cases demonstrate that the three doses of APP arising from trisomy 21 are likely to be causative for AD-DS. Conversely, although very rare, partial trisomy 21 excluding APP (i.e., with two “doses” of APP) does not appear to lead to AD (Prasher et al., 1998; Korbel et al., 2009).
While people with DS and Dup-APP are at high risk of dementia, presumably in both cases because of APP triplication, there are some intriguing differences in their AD-related clinical features (Wiseman et al., 2015). Examining the effects of different APP genotypes may therefore provide insights into the modulation of APP pathogenesis. Table 1 shows key examples of phenotypes in AD-DS and how these compare with Dup-APP, FAD due to other APP mutations (primarily point mutations) and late-onset sporadic AD (SAD). Mutations in PSEN1 and PSEN2, which do not map to Hsa21, are not included.
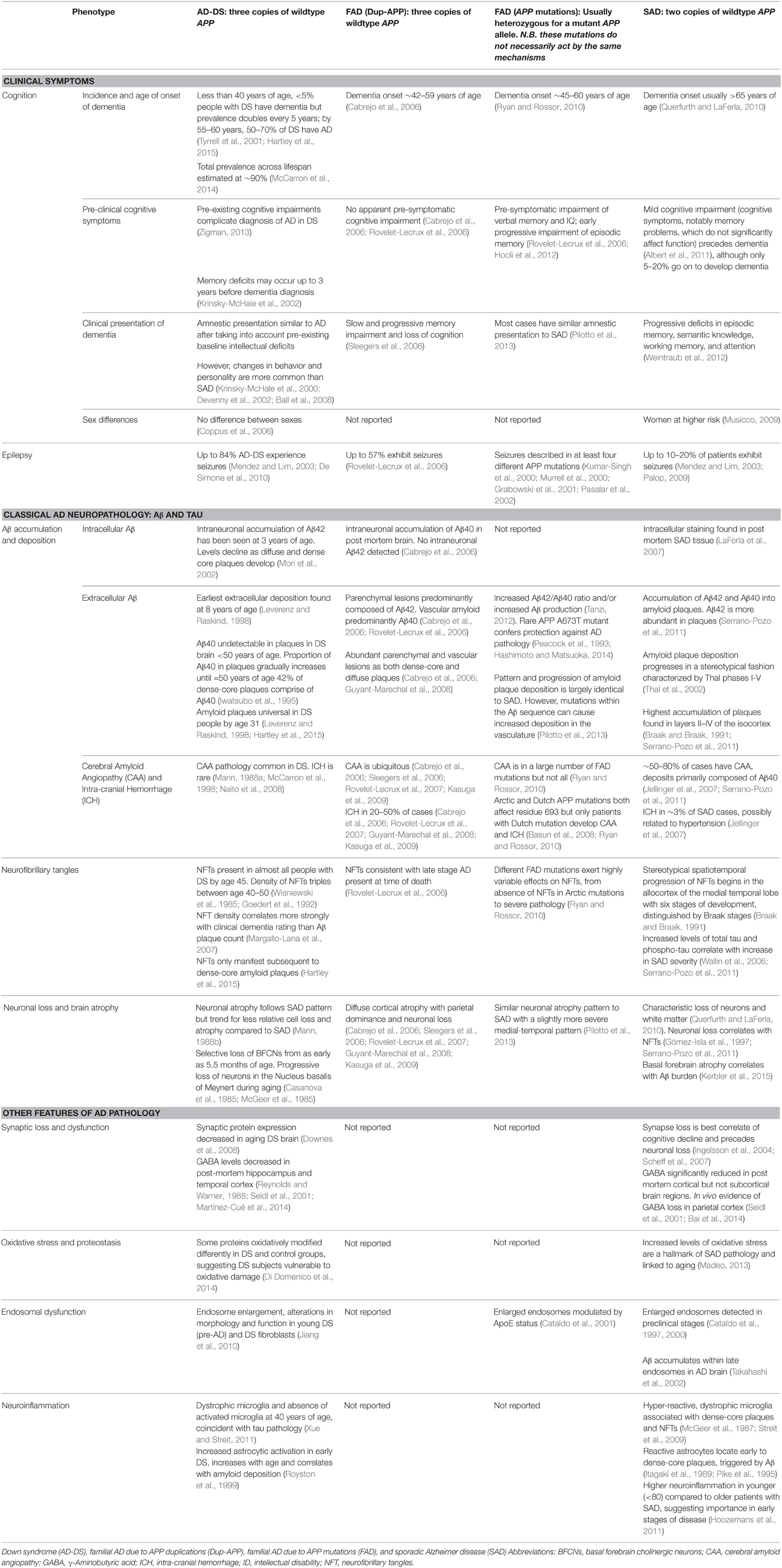
Table 1. Comparison of phenotypes from different genetic forms of human Alzheimer disease.
However, a difficulty in analysing phenotypes is the considerable heterogeneity in clinical presentation within each APP genotype, even within families with the same mutation. For example, there is a wide variety of non-cognitive symptoms and behavioral changes across all four AD genotypes, including personality changes (Nelson et al., 2001; Ball et al., 2008), hallucinations (Sleegers et al., 2006; Basun et al., 2008; Guyant-Marechal et al., 2008), paranoia (Sleegers et al., 2006; Pilotto et al., 2013), and delusions (Burns et al., 1990), some of which are associated with cognitive decline (Adams and Oliver, 2010). Another important issue in diagnosing AD in AD-DS is that dementia is an additional cognitive deficit acquired on top of the baseline cognitive impairment found in people with DS: distinguishing between cognitive deficits due to intellectual disability, and decline at early stages of AD, is therefore an important challenge. However, diagnosis of dementia by experienced clinicians has been shown to be accurate in DS, and even more reliable than recent operational dementia criteria (Sheehan et al., 2015). Further, a few clinical features stand out in AD-DS—a striking example, albeit one of unknown relevance to AD, is seizure susceptibility in adulthood, which appears heightened by APP duplication, as both AD-DS (84%) and Dup-APP (57%) have significantly higher rates of seizures than SAD (10–20%). This may indicate specific pathways that are progressively disrupted by APP duplication, resulting in damaging electrical activity in the brain.
Dup-APP and FAD caused by APP mutations are relatively rare, and much information about these conditions remains to be gathered, for example, on synaptic dysfunction, oxidative stress and neuroinflammation. In contrast, AD-DS arises in a population with a well-defined genetic basis and a sizeable prevalence, which means it is of great value for investigating AD pathogenesis for everyone at risk of dementia.
Modeling DS, Including AD-DS, in Mice
Human chromosome 21 has synteny with the mouse genome, such that its ortholog genes are found in three blocks with conserved order and gene orientation on mouse chromosomes 10 (Mmu10), Mmu16, and Mmu17 (Hattori et al., 2000; Dierssen et al., 2009); the mouse App gene lies on Mmu16 (Figure 1). Mice with precisely-defined trisomies (or monosomies) have been generated, now usually by chromosome engineering (Brault et al., 2006; Tybulewicz and Fisher, 2006), to provide a set of models that are segmentally trisomic for regions orthologous to Hsa21 (Davisson et al., 1993; Sago et al., 1998; Olson et al., 2004; Li et al., 2007; Herault et al., 2009; Pereira et al., 2009; Yu et al., 2010a; Liu et al., 2011, 2014; Brault et al., 2015).
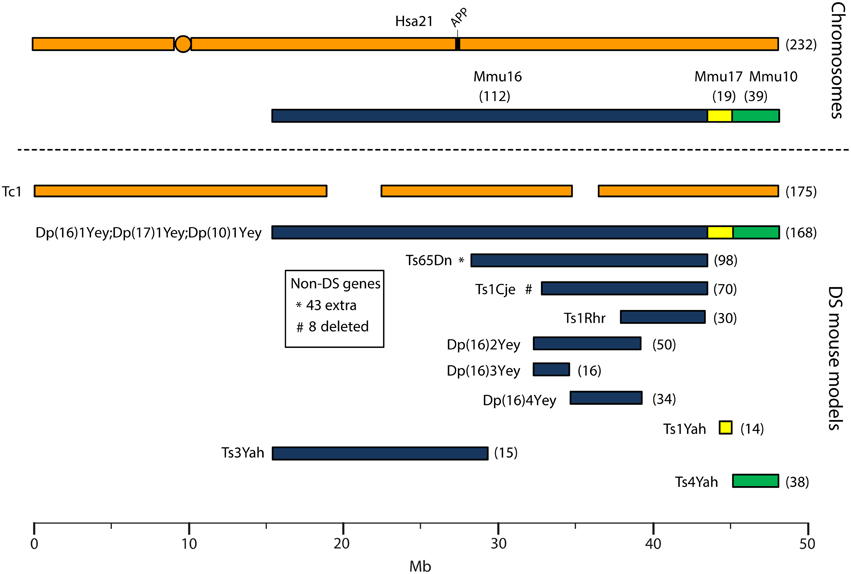
Figure 1. Human chromosome 21 (Hsa21), orthologous mouse chromosomes (Mmu), and key mouse models of Down syndrome. Diagram representing Hsa21 and its alignment with syntenic regions on Mmus 16, 17, and 10. The orange circle represents the human centromere and mouse models are color-coded and aligned according to the chromosomal segment for which they are trisomic. Numbers in brackets represent the number of protein-coding Hsa21 orthologous genes within each region or mouse model, according to Ensembl release 79 and the breakpoints published in papers referenced here. The Tc1 mouse is the only model which carries Hsa21, though genomic rearrangements and deletions (indicated by breaks in the chromosome) mean the mouse is functionally trisomic for only ~75% of Hsa21 genes (Gribble et al., 2013). All other mouse models carry duplications of mouse orthologues. The Dp1(16)Yey;Dp1(17)Yey;Dp1(10)Yey (or Ts1Yey;Ts3Yey;Ts2Yey) mouse was generated by crossing together three partial trisomy models (Yu et al., 2010a) and spans the entirety of the Hsa21-syntenic regions. The Ts65Dn mouse (Davisson et al., 1993) contains a freely segregating segment of Mmu16, however it is also trisomic for 43 extra protein-coding genes on the centromeric section of Mmu17 that are not relevant to DS (indicated by an asterisk (*) and accompanying text box; Duchon et al., 2011; Reinholdt et al., 2011). The Ts1Cje mouse (Sago et al., 1998) also contains a monosomy of eight protein-coding genes on Mmu12, irrelevant to the DS phenotype (indicated by “#” and accompanying text box. Gene numbers are based on Ensembl release 79, compared to the original seven monosomic genes detailed in Duchon et al., 2011). Other mice are Ts1Rhr or Dp1(16)Rhr mice (Olson et al., 2004); Ts1Yah mice (Pereira et al., 2009); Ts3Yah (previously published as Ts2Yah; Brault et al., 2015); and Ts4Yah mice (previously published as Ts3Yah mice; Herault et al., 2009). Other useful examples of mouse models include the Ts43H model (not shown) which is partially trisomic for Mmu17 including some genes with ortholog on Hsa21 (Vacík et al., 2005). The scale is in megabase pairs (Mb).
Generating many models with different partial trisomies creates a mapping panel in which individual phenotypes may be assessed in several strains, and so assigned to specific trisomic chromosomal region(s). As all DS phenotypes presumably arise from abnormal gene dosage, candidate genes that when present in three copies give rise to all or part of the phenotype, can be chosen from the trisomic critical region. Individual candidate genes can then be studied, for example, in overexpression or knockout models, to assess the effects of different copy numbers of the gene. Figure 1 is an overview of DS mouse models and the chromosomal segments for which they are trisomic. Table 2 details the gene content for each DS mouse model shown, including protein-coding and non-protein-coding genes relevant to human trisomy 21.
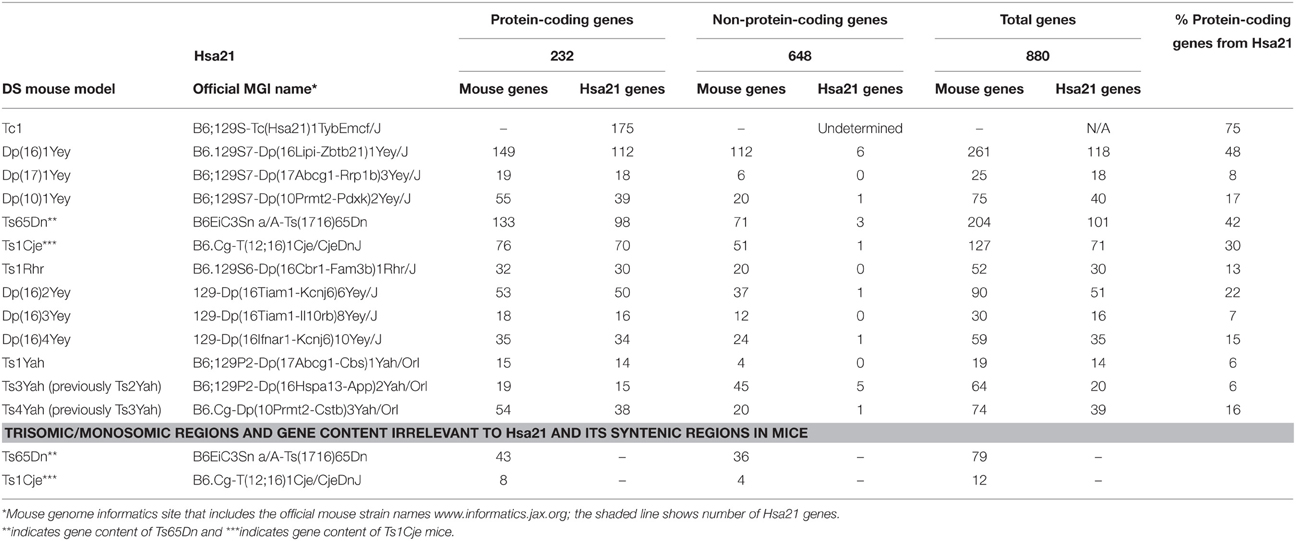
Table 2. Trisomic region and triplicated gene content in Down syndrome mouse models shown in Figure 1 compared with Hsa21 (Ensembl release 79).
The most complete mouse model to date, Dp(10)1Yey/+;Dp(16)1Yey/+;Dp(17)1Yey/+, is trisomic for all Hsa21 syntenic regions and was generated by crossing three DS mouse models, each carrying duplications of the respective Hsa21 orthologous regions on Mmu10, Mmu16 and Mmu17 (Li et al., 2007; Yu et al., 2010a,b; Figure 1). However, the vast majority of studies relating to AD-DS have been performed on the Ts65Dn mouse, as this has been an extremely important “standard model” of DS for many years, prior to the development of newer strains by chromosome engineering (Davisson et al., 1993; Reeves et al., 1995; Table 2). The Ts65Dn mouse carries a Robertsonian translocation resulting in trisomy of ~42% of the protein-coding genes orthologous to Hsa21, but it also has 79 additional genes (including long non-coding sequences) from Mmu17 that are outside the Hsa21 region of synteny, and these need to be taken into account when analysing phenotypes (Duchon et al., 2011; Reinholdt et al., 2011). These extra triplicated genes that do not relate to DS happen to include non-Hsa21 genes, such as SYNJ2 and TIAM2 that have Hsa21/Mmu16 paralogues (SYNJ1, TIAM1), which may complicate phenotype-genotype correlations (Duchon et al., 2011). Other triplicated genes in Ts65Dn irrelevant to DS include several genes encoding dynein light chains that may influence endosomal trafficking, and so potentially affect neuronal phenotypes (Hartley et al., 2015).
A different type of mouse model of DS is the “humanized” transchromosomic “Tc1” mouse that carries a freely-segregating Hsa21 (O'Doherty et al., 2005), which is functionally trisomic for ~75% of Hsa21 protein-coding genes (Gribble et al., 2013). However, this extra chromosome is rearranged, and lost stochastically at different rates in different mouse tissues—thus, Tc1 mice are mosaic for the human chromosome. With respect to AD research, the APP gene is not functionally trisomic in Tc1 mice because of a rearrangement that has occurred by chance, so this animal expresses just the two endogenous copies of mouse App (Sheppard et al., 2012).
While many DS mouse models have been published, there is no single complete model, and the usefulness of these strains lies in their comparative and complementary use in studying genotype-phenotype relationships, including AD-related phenotypes (Table 3). These studies enable us to map critical dosage-sensitive genes because each locus is likely expressed at trisomic levels, mimicking human DS transcription. We can also study the interactions of Hsa21 dosage-sensitive genes with the rest of the genome (Hsa21 and non-Hsa21), as well as effects exerted by aneuploidy per se.
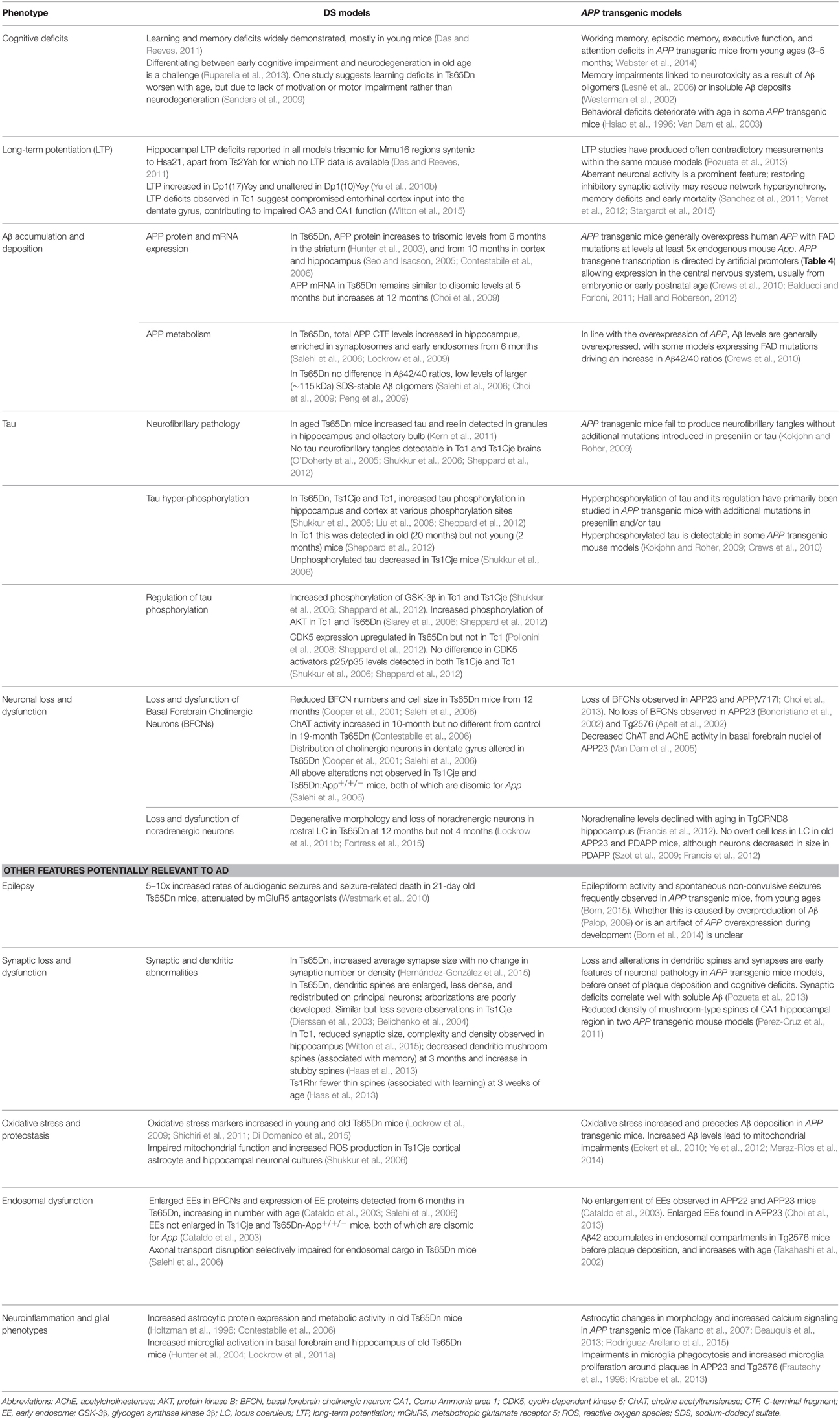
Table 3. Examples of AD phenotypes studied in DS mouse models, and related findings in APP transgenic strains described in Table 4.
Modeling Amyloid Deposition in Mice
In contrast to the segmental duplication of tens of endogenous wildtype genes in DS mouse strains, AD models are primarily transgenic lines that overexpress one or more of the human mutant genes that cause FAD. These transgenes usually insert at random sites in the genome and may be driven by artificial promoters (see examples in Table 4), which vary in terms of their spatial and temporal expression patterns, and result in expression at often 5–10 fold compared to endogenous mouse orthologue (Balducci and Forloni, 2011; Hall and Roberson, 2012). Overexpressing wildtype human APP or mouse App does not result in amyloid deposition (Elder et al., 2010); hence the need to use known AD-causative mutant sequences in transgenic mice.
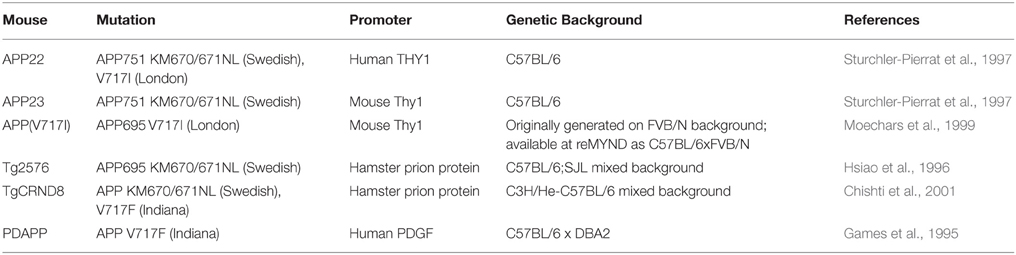
Table 4. Human APP overexpressing transgenic mice referred to in this review (information obtained from Alzforum.org).
In general, while mutant APP transgenic mice develop robust amyloid deposition, synaptotoxic features and memory impairments, none of them reproduces tau-containing neurofibrillary tangles, the hallmark pathology of AD which most closely correlates with dementia (Hall and Roberson, 2012). The combined overexpression of mutant APP and mutant human tau is required to reproduce both amyloid and tau pathology, although these tau mutations in humans do not alone cause AD but another form of neurodegeneration, frontotemporal dementia. Mutant APP transgenics may be best considered models of APP/Aβ pathology (amyloid deposition) rather than full AD.
Studying AD-DS Phenotypes in Mice
In Table 3, we summarize examples of findings that may be informative for AD-DS from different DS (mainly Ts65Dn) mice and examples of AD models (Table 4). With respect to AD, a wide range of mutant APP transgenic strains are available in the literature, so we have chosen a few well-known examples [APP22, APP23, APP (V717I), PDAPP, Tg2576, TgCRND8] to illustrate some potential phenotypes of interest. We note that the expression of wildtype mouse APP, and wildtype or mutant human APP protein in these different models can influence amyloid pathology (Kokjohn and Roher, 2009). For example, because of amino acid differences between the two species, mouse APP may be processed with little BACE1 cleavage and so may yield three times less Aβ than wildtype human APP (De Strooper et al., 1995). In addition, the genetic background of AD mouse strains affects a range of APP/Aβ phenotypes, including plaque deposition, APP metabolism, survival, and seizure rates (Carlson et al., 1997; Lehman et al., 2003; Krezowski et al., 2004; Lassalle et al., 2008; Rustay et al., 2010; Jackson et al., 2015). Similarly, phenotypes observed in DS mice may be influenced by genetic background (O'Doherty et al., 2005; Galante et al., 2009; Costa et al., 2010; Deitz and Roper, 2011; Haydar and Reeves, 2012). We consider only APP transgenic models of AD, as the other genes used in such models (PSEN1, PSEN2, and MAPT) are not encoded on Hsa21, and therefore are not directly relevant to AD-DS.
In studying mouse phenotypes to understand AD-DS, we are presented with two key issues. Firstly, we need to test longitudinally DS models to look for changes in older mice that are not apparent early on, and so may indicate aging or neurodegenerative processes rather than neurodevelopmental deficits. Secondly, we need to separate normal aging processes in DS from those connected specifically to AD-DS. The thoughtful use of the increasing range of different mouse models is enabling us to dissect these issues to further our understanding of AD-DS.
A study that has addressed both (1) neurodegenerative vs. neurodevelopmental and (2) normal aging vs. AD phenotypes has been performed in the Ts65Dn mouse. This study concerned the neurodegenerative phenotype loss of basal forebrain cholinergic neurons (BFCNs), and was carried out through an experimental design involving optimal crossing of different mouse models and assessment of the genetically-distinct progeny (Salehi et al., 2006). Firstly, Salehi and colleagues quantified the known loss of BFCNs in Ts65Dn mice, and showed this loss to be progressive, thus an aging or an AD-related phenotype in this DS mouse model. The authors then compared BFCN loss in Ts65Dn and Ts1Cje DS mouse models (Figure 1), and were able to map a dosage-sensitive critical region that had to contain a candidate gene for this phenotype: Ts65Dn mice lose BFCNs but Ts1Cje mice turned out to have no loss compared to wildtype mice. Therefore, the dosage-sensitive gene(s), that when present in three copies is responsible for BFCN loss, must map within the region of trisomy present in Ts65Dn but not in Ts1Cje. A key candidate in this region was the App gene. By crossing Ts65Dn mice to heterozygous App knockout mice, the authors generated cohorts of progeny that carried the trisomic region with either two or three copies of wildtype App. Assessing BFCN loss in these cohorts led to the conclusion that the phenotype arises mainly from having three copies of App and, further, that it is associated with impairments in nerve growth factor retrograde transport, linked to early endosomes, which are enlarged (Salehi et al., 2006).
Given the role of APP triplication in this phenotype, there is likely a strong link to AD and AD-DS. In people with early AD pathology or mild cognitive impairment, neurofibrillary pathology has been detected in BFCNs (Mesulam et al., 2004; Grudzien et al., 2007), while their loss has been observed in patients with SAD (and other neurodegenerative disorders; Zarow et al., 2003). Interestingly, enlarged early endosomes have been detected in cortical tissues from cognitively intact individuals with mild AD pathology, and in young individuals with DS (under 12 years old), suggesting that endosome enlargement is an early feature in AD pathogenesis (Cataldo et al., 2000).
DS Models in the Study of Candidate Genes Influencing AD
As illustrated in Table 1, while people with DS have three copies of APP and develop early AD neuropathology, their clinical presentation is variable, suggesting that other genetic and environmental factors influence pathogenesis. In addition to APP, many genes on Hsa21 have been studied in the context of neurodegeneration and/or AD, and it is conceivable that a three-copy dose of any of these genes could contribute to disease and dysfunction.
Single gene overexpressing transgenics do not model DS, or AD-DS, but may provide some insights if carefully considered. For example, seizures and neuronal network abnormalities remain challenging areas to investigate but important phenotypes to be explored in DS, AD-DS, and APP overexpression models of AD (i.e., which are single gene transgenic models). In SAD, seizures have been associated with early cognitive decline (Vossel et al., 2013), while the incidence of seizures in AD-DS is high and is associated with increased risk of dementia (for example, McCarron et al., 2014). To date, seizure phenotypes and epileptiform activity have been characterized across numerous APP transgenic mice (Born, 2015), but it is unclear whether these phenotypes are primarily driven by amyloid overproduction (Mucke and Selkoe, 2012) or are an effect of unphysiological APP overexpression during development (Born et al., 2014). Antiepileptic drugs, such as levetiracetam, which improve seizures in DS (Sangani et al., 2010) and in AD (Cumbo and Ligori, 2010), also ameliorate synaptic and memory dysfunctions in APP transgenic mice by suppressing neuronal network dysfunction (Sanchez et al., 2012; Devi and Ohno, 2013).
So, while single gene transgenic models do not model human trisomy 21 or AD because they usually express the gene by many-fold, from ectopic promoters, they offer insights into some of the functional consequences of overexpression, albeit at non-trisomic levels. Table 5 presents a list of Hsa21 gene candidates, in chromosomal order, that have been investigated for overexpression-related phenotypes linked with AD across different mouse, fruitfly, and cellular models. We also compare, where data are available, how related changes in these genes have been explored in humans with AD and/or DS. Making optimal use of mouse genetics, some of the single-gene-overexpressing mouse transgenics have been crossed with AD models, to look for changes in phenotypes that may be informative. For example, crossing an S100β overexpression model with the Tg2576 APP transgenic mouse generates double mutant progeny with exacerbated cerebral amyloidosis and reactive gliosis. This suggests that increased expression of S100β could contribute to AD pathogenesis possibly by promoting amyloidogenic APP processing (Mori et al., 2010).
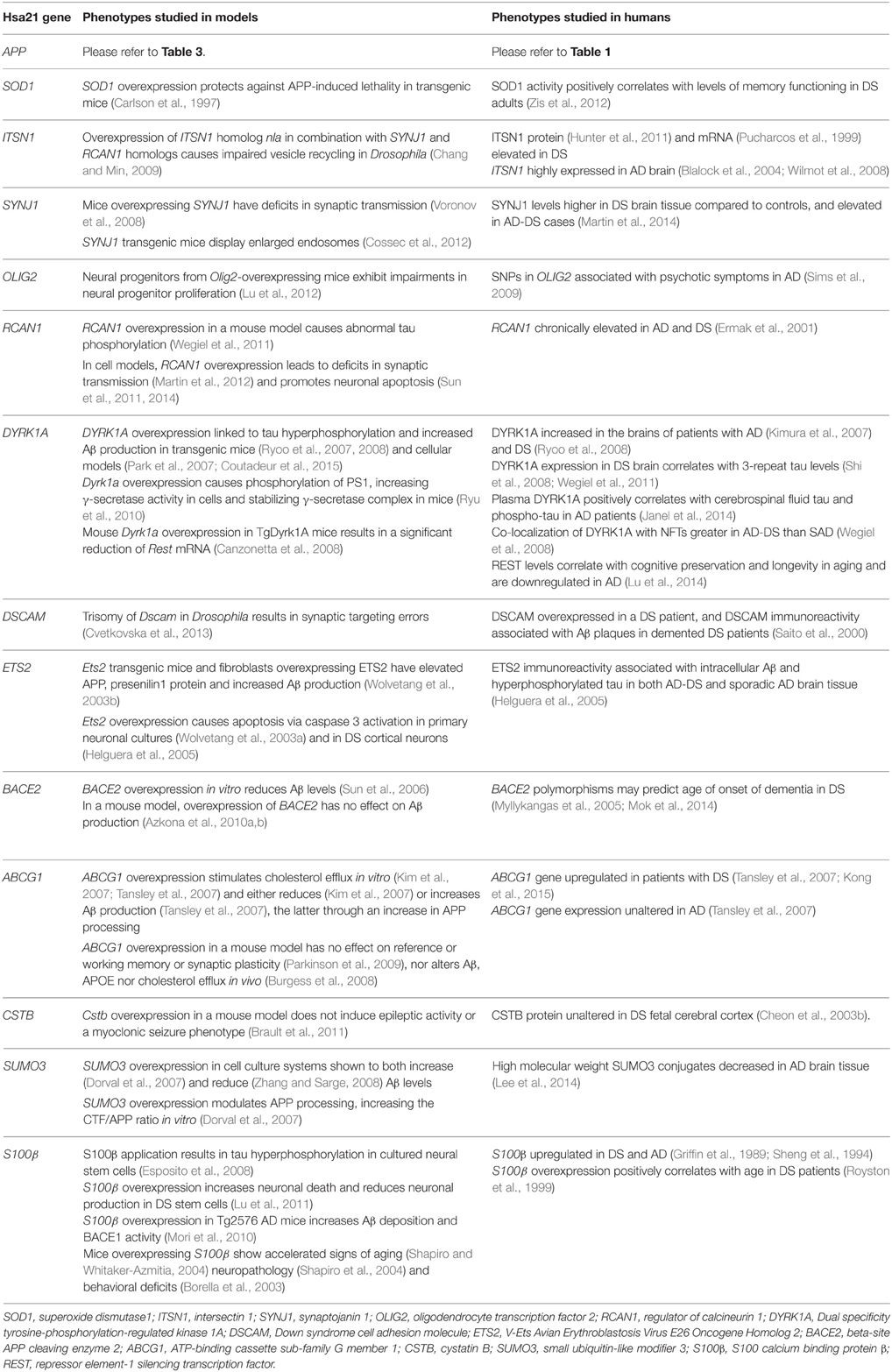
Table 5. Single gene overexpression models from Hsa21, with relevance to AD phenotypes. Genes are listed in order from centromere to Hsa21q telomere.
Other key Hsa21 gene candidates DYRK1A and RCAN1 have been linked to AD pathogenesis through their effects on tau. The toxic neurofibrillary tangles (NFTs) that accumulate in AD are formed of hyperphosphorylated tau protein. Overexpression of DYRK1A in transgenic mice resulted in tau hyperphosphorylation (Ryoo et al., 2007, 2008), and DYRK1A has been shown to co-localize with NFTs more frequently in AD-DS brain compared to SAD (Wegiel et al., 2008). Similarly, overexpression of RCAN1 in a mouse model resulted in abnormal tau hyperphosphorylation (Wegiel et al., 2011). This suggests that the increased expression of DYRK1A and RCAN1 in DS could promote the formation of NFTs, a hallmark feature of AD pathology.
Triplication of Hsa21 genes in DS does not necessarily lead to a 1.5-fold increase (compared to euploid individuals) in their RNA or protein expression. For example, a study in DS fetal cortical tissue revealed multiple Hsa21 proteins in fact expressed at similar or lower levels than in disomic controls (Cheon et al., 2003a,b,c,d). Assessments at transcriptomic and proteomic levels, together with meta-analysis across these studies, provide useful resources for understanding patterns of alteration in gene expression (for example, see Vilardell et al., 2011). As a few of the studies in Table 5 have demonstrated, it is important to verify the effect of trisomy on candidate gene expression, in relevant tissues and contexts, before further characterization of any potential downstream effects of trisomy.
Prospects for Research
Individuals with DS manifest the most common genetic form of AD, and this undoubtedly largely arises from expressing three copies of APP (Ness et al., 2012; Hartley et al., 2015). Therefore, studying and modeling this population will assist in understanding the contribution of APP to AD pathogenesis, and evaluating the amyloid cascade hypothesis. However, the variation in clinical presentation of AD-DS shows that many other genetic and environmental factors contribute, almost certainly including protective factors. The thoughtful use of models will thus provide insight into these factors.
To study mouse models of AD-DS, it is critical to dissect neurodevelopmental from neurodegenerative effects (Bothwell and Giniger, 2000; Contestabile et al., 2010). To be of interest for AD-DS, such phenotypes should differ from normal aging in the mouse strain of interest, although this can be difficult to determine, particularly as DS has been characterized as a syndrome of accelerated aging in both clinical (Lott, 2012; Zigman, 2013) and epigenetic terms (Horvath et al., 2015), and because aging remains the clearest non-genetic risk factor for all forms of AD (Fratiglioni, 1996; Bush and Beail, 2004). The longitudinal study of cognitive decline in DS mice poses similar challenges to those in people with DS, and tests need to distinguish between dysfunction due to dementia, as opposed to aging or baseline learning deficits. For example, variations of a learning procedure involving incremental repeated acquisition tasks suggest that declining performances by Ts65Dn mice with age may be due to motor impairments and/or decreased motivation, rather than neurodegenerative-related effects (Sanders et al., 2009). To improve behavioral testing in mouse models of AD-DS, a potential avenue to explore capitalizes on the association of dementia with deficits in episodic memory. The development of tests based on, for example, visuo-spatial data, should therefore highlight age-dependent, dementia-related deficits in mouse models, because they rely on the encoding and binding of information spontaneously, and do not challenge other cognitive domains (Iordanova et al., 2009).
As well as the hypothesis-driven study of AD-DS phenotypes, one of the greatest strengths of working with mouse models is our ability to undertake unbiased hypothesis-generating research, by mapping phenotypes to genomic critical regions using the range of strains now available. These include chromosome-engineered panels of partially trisomic mice (Figure 1) as well as single gene knockout animals, such as the App+∕− heterozygous mice, which may be crossed to partially trisomic strains, to generate progeny with altered single gene copy numbers on different trisomic region backgrounds. The cohorts of progeny from these crosses provide ideal groups for testing the contributions of single Hsa21 genes to AD-DS.
Mouse genome engineering continues to offer new models and approaches for teasing apart AD-DS relevant phenotypes, and new strains are being published regularly to help refine experimental strategies. For example, the recent genomically humanized NLF mouse (Saito et al., 2014), which has human amino acid residues at key sites within APP that affect its processing, may yield new insights into the biology of both AD and AD-DS, partly through expressing mutant APP at physiological levels. The strategic breeding of new APP models with DS segmental trisomies will contribute to determining which phenotypes are downstream of an amyloid cascade. Furthermore, independent study of partial trisomies without three copies of App may help tease out effects of other factors, for example oxidative stress, cholesterol metabolism or immune system dysfunction, in the development of dementia (Wiseman et al., 2015).
DS mouse models also give us the flexibility to investigate the effects of potentially dosage-sensitive non-coding regions. For example, microRNAs (miRs)—short (20–23 nucleotide) RNAs that downregulate the transcription of target genes—have increasingly been investigated in AD pathogenesis due to their differential regulation in molecular pathways associated with AD (Veerappan et al., 2013). Hsa21 encodes 29 miRs (MirBase release 21, Griffiths-Jones, 2004), and their potential overexpression in trisomy may contribute to genetic dysregulation relevant to AD-DS. Overexpression of the Hsa21-encoded miR-155 in DS has been reported to increase Aβ production via the downregulation of sorting nexin 27, a membrane-trafficking component found in early endosomes, that modulates γ-secretase activity (Wang et al., 2013, 2014).
Hsa21 also encodes genes involved in post-translational histone modification, including DYRK1A, ETS2, HMGN1, BRWD1, and RUNX1 (Dekker et al., 2014), which may be investigated for their potential roles leading to the aberrant histone modifications observed in AD (Zhang et al., 2012a; Narayan et al., 2015). Histone methylation (specifically H3K4me3) has been shown to correlate highly with genome-wide domains of dysregulated gene expression in DS, which are highly conserved between humans and Ts65Dn mice (Letourneau et al., 2014). DS mouse models therefore model epigenetic structures in humans and may be used to study the effects of its dysregulation in AD-DS.
Finally, mouse model research must be undertaken in parallel with other rapid advances in the AD-DS field. The advent of human induced pluripotent stem (iPS) cells (Hunsberger et al., 2015) for DS provides for the first time a trisomic human in vitro model that recapitulates hallmarks of some AD pathology (Shi et al., 2012; Chang et al., 2015; Moore et al., 2015; Murray et al., 2015). The further development of this technology (Hunsberger et al., 2015) will prove valuable to phenotyping and drug target discovery, alongside in vivo research and in vitro primary cultures from DS mice. An increasing call is being made for partnerships to build up large cohorts of, and biobanks from, people with DS for the systematic longitudinal study of AD-DS progression (Hartley et al., 2015). In-depth phenotypic studies across development with infants and adults with DS are already underway (Wiseman et al., 2015). These will allow greater power to identify biomarkers for the prediction of AD in this large, genetically well-defined population, for example, through plasma (Dekker et al., 2015; Schupf et al., 2015), cerebrospinal fluid (Portelius et al., 2014a,b), and neuroimaging studies (Beacher et al., 2009; Landt et al., 2011; Powell et al., 2014; Sabbagh et al., 2015). Biomarker studies are also being performed in AD models, including at very early phases of Aβ deposition (Maia et al., 2015). Extending these studies to mouse models of DS and AD-DS will contribute to elucidating the genotype-phenotype relationships that ultimately lead to dementia.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank members of the London Down Syndrome Consortium (LonDownS) for their constructive feedback to this review, in particular Annette Karmiloff-Smith, Carla Startin, Andre Strydom, Victor Tybulewicz, Frances Wiseman, as well as Veronique Brault, Mark Good, Eva Lana-Elola and Sheona Watson-Scales for their generous comments and advice. XYC is funded by the Brain Research Trust, JT is funded by the Alzheimer's Society, LP is funded by Alzheimer's Research UK, EF is funded by the Wellcome Trust.
References
Adams, D., and Oliver, C. (2010). The relationship between acquired impairments of executive function and behaviour change in adults with Down syndrome. J. Intellect. Disabil. Res. 54, 393–405. doi: 10.1111/j.1365-2788.2010.01271.x
Albert, M. S., DeKosky, S. T., Dickson, D., Dubois, B., Feldman, H. H., Fox, N. C., et al. (2011). The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimer's Dementia 7, 270–279. doi: 10.1016/j.jalz.2011.03.008
Apelt, J., Kumar, A., and Schliebs, R. (2002). Impairment of cholinergic neurotransmission in adult and aged transgenic Tg2576 mouse brain expressing the Swedish mutation of human beta-amyloid precursor protein. Brain Res. 953, 17–30. doi: 10.1016/S0006-8993(02)03262-6
Azkona, G., Amador-Arjona, A., Obradors-Tarragó, C., Varea, E., Arqué, G., Pinacho, R., et al. (2010a). Characterization of a mouse model overexpressing beta-site APP-cleaving enzyme 2 reveals a new role for BACE2. Genes Brain Behav. 9, 160–172. doi: 10.1111/j.1601-183X.2009.00538.x
Azkona, G., Levannon, D., Groner, Y., and Dierssen, M. (2010b). In vivo effects of APP are not exacerbated by BACE2 co-overexpression: behavioural characterization of a double transgenic mouse model. Amino Acids 39, 1571–1580. doi: 10.1007/s00726-010-0662-8
Bai, X., Edden, R. A. E., Gao, F., Wang, G., Wu, L., Zhao, B., et al. (2014). Decreased γ-aminobutyric acid levels in the parietal region of patients with Alzheimer's disease. Magn. Reson. Imaging 41, 1326–1331. doi: 10.1002/jmri.24665
Balducci, C., and Forloni, G. (2011). APP transgenic mice: their use and limitations. NeuroMol. Med. 13, 117–137. doi: 10.1007/s12017-010-8141-7
Ball, S. L., Holland, A. J., Treppner, P., Watson, P. C., and Huppert, F. A. (2008). Executive dysfunction and its association with personality and behaviour changes in the development of Alzheimer's disease in adults with Down syndrome and mild to moderate learning disabilities. Br. J. Clin. Psychol. 47, 1–29. doi: 10.1348/014466507X230967
Basun, H., Bogdanovic, N., Ingelsson, M., Almkvist, O., Näslund, J., Axelman, K., et al. (2008). Clinical and neuropathological features of the arctic APP gene mutation causing early-onset Alzheimer disease. Arch. Neurol. 65, 499–505. doi: 10.1001/archneur.65.4.499
Beacher, F., Daly, E., Simmons, A., Prasher, V., Morris, R., Robinson, C., et al. (2009). Alzheimer's disease and Down's syndrome: an in vivo MRI study. Psychol. Med. 39, 675–684. doi: 10.1017/S0033291708004054
Beauquis, J., Pavía, P., Pomilio, C., Vinuesa, A., Podlutskaya, N., Galvan, V., et al. (2013). Environmental enrichment prevents astroglial pathological changes in the hippocampus of APP transgenic mice, model of Alzheimer's disease. Exp. Neurol. 239, 28–37. doi: 10.1016/j.expneurol.2012.09.009
Belichenko, P. V., Masliah, E., Kleschevnikov, A. M., Villar, A. J., Epstein, C. J., Salehi, A., et al. (2004). Synaptic structural abnormalities in the Ts65Dn mouse model of Down Syndrome. J. Comp. Neurol. 480, 281–298. doi: 10.1002/cne.20337
Bittles, A. H., and Glasson, E. J. (2004). Clinical, social, and ethical implications of changing life expectancy in Down syndrome. Dev. Med. Child Neurol. 46, 282–286. doi: 10.1111/j.1469-8749.2004.tb00483.x
Blalock, E. M., Geddes, J. W., Chen, K. C., Porter, N. M., Markesbery, W. R., and Landfield, P. W. (2004). Incipient Alzheimer's disease: microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc. Natl. Acad. Sci. U.S.A. 101, 2173–2178. doi: 10.1073/pnas.0308512100
Boncristiano, S., Calhoun, M. E., Kelly, P. H., Pfeifer, M., Bondolfi, L., Stalder, M., et al. (2002). Cholinergic changes in the APP23 transgenic mouse model of cerebral amyloidosis. J. Neurosci. 22, 3234–3243.
Borella, A., Sumangali, R., Ko, J., and Whitaker-Azmitia, P. M. (2003). Characterization of social behaviors and oxytocinergic neurons in the S-100 beta overexpressing mouse model of Down Syndrome. Behav. Brain Res. 141, 229–236. doi: 10.1016/S0166-4328(02)00373-X
Born, H. A., Kim, J. Y., Savjani, R. R., Das, P., Dabaghian, Y. A., Guo, Q., et al. (2014). Genetic suppression of transgenic APP rescues Hypersynchronous network activity in a mouse model of Alzeimer's disease. J. Neurosci. 34, 3826–3840. doi: 10.1523/jneurosci.5171-13.2014
Born, H. A. (2015). Seizures in Alzheimer's disease. Neuroscience 286C, 251–263. doi: 10.1016/j.neuroscience.2014.11.051
Bothwell, M., and Giniger, E. (2000). Alzheimer's Disease. Cell 102, 271–273. doi: 10.1016/S0092-8674(00)00032-5
Braak, H., and Braak, E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259. doi: 10.1007/BF00308809
Braidy, N., Muñoz, P., Palacios, A. G., Castellano-Gonzalez, G., Inestrosa, N. C., Chung, R. S., et al. (2012). Recent rodent models for Alzheimer's disease: clinical implications and basic research. J. Neural Transm. 119, 173–195. doi: 10.1007/s00702-011-0731-5
Brault, V., Duchon, A., Romestaing, C., Sahun, I., Pothion, S., Karout, M., et al. (2015). Opposite phenotypes of muscle strength and locomotor function in mouse models of partial trisomy and monosomy 21 for the proximal Hspa13-App region. PLoS Genet. 11:e1005062. doi: 10.1371/journal.pgen.1005062
Brault, V., Martin, B., Costet, N., Bizot, J. C., and Hérault, Y. (2011). Characterization of PTZ-induced seizure susceptibility in a down syndrome mouse model that overexpresses CSTB. PLoS ONE 6:e27845. doi: 10.1371/journal.pone.0027845
Brault, V., Pereira, P., Duchon, A., and Hérault, Y. (2006). Modeling chromosomes in mouse to explore the function of genes, genomic disorders, and chromosomal organization. PLoS Genet. 2:e86. doi: 10.1371/journal.pgen.0020086
Burgess, B. L., Parkinson, P. F., Racke, M. M., Hirsch-Reinshagen, V., Fan, J., Wong, C., et al. (2008). ABCG1 influences the brain cholesterol biosynthetic pathway but does not affect amyloid precursor protein or apolipoprotein E metabolism in vivo. J. Lipid Res. 49, 1254–1267. doi: 10.1194/jlr.M700481-JLR200
Burns, A., Jacoby, R., and Levy, R. (1990). Psychiatric phenomena in Alzheimer's disease. I Disorders of thought content. Br. J. Psychiatry 157, 72–76. doi: 10.1192/bjp.157.1.72
Bush, A., and Beail, N. (2004). Risk factors for dementia in people with down syndrome: issues in assessment and diagnosis. Am. J. Ment. Retard. 109, 83–97. doi: 10.1352/0895-8017(2004)109<83:RFFDIP>2.0.CO;2
Cabrejo, L., Guyant-Maréchal, L., Laquerrière, A., Vercelletto, M., De la Fournière, F., Thomas-Antérion, C., et al. (2006). Phenotype associated with APP duplication in five families. Brain 129, 2966–2976. doi: 10.1093/brain/awl237
Canzonetta, C., Mulligan, C., Deutsch, S., Ruf, S., O'Doherty, A., Lyle, R., et al. (2008). DYRK1A-dosage imbalance perturbs NRSF/REST levels, deregulating pluripotency and embryonic stem cell fate in Down syndrome. Am. J. Hum. Genet. 83, 388–400. doi: 10.1016/j.ajhg.2008.08.012
Carlson, G. A., Borchelt, D. R., Dake, A., Turner, S., Danielson, V., Coffin, J. D., et al. (1997). Genetic modification of the phenotypes produced by amyloid precursor protein overexpression in transgenic mice. Hum. Mol. Genet. 6, 1951–1959. doi: 10.1093/hmg/6.11.1951
Casanova, M. F., Walker, L. C., Whitehouse, P. J., and Price, D. L. (1985). Abnormalities of the nucleus basalis in Down's syndrome. Ann. Neurol. 18, 310–313. doi: 10.1002/ana.410180306
Castellani, R. J., and Perry, G. (2014). The complexities of the pathology-pathogenesis relationship in Alzheimer disease. Biochem. Pharmacol. 88, 671–676. doi: 10.1016/j.bcp.2014.01.009
Cataldo, A., Rebeck, G. W., Ghetri, B., Hulette, C., Lippa, C., Van Broeckhoven, C., et al. (2001). Endocytic disturbances distinguish among subtypes of Alzheimer's disease and related disorders. Ann. Neurol. 50, 661–665. doi: 10.1002/ana.1254
Cataldo, A. M., Barnett, J. L., Pieroni, C., and Nixon, R. A. (1997). Increased Neuronal Endocytosis and Protease Delivery to Early Endosomes in Sporadic Alzheimer's Disease: neuropathologic Evidence for a Mechanism of Increased beta -Amyloidogenesis. J. Neurosci. 17, 6142–6151.
Cataldo, A. M., Petanceska, S., Peterhoff, C. M., Terio, N. B., Epstein, C. J., Villar, A., et al. (2003). App Gene dosage modulates endosomal abnormalities of Alzheimer's Disease in a segmental Trisomy 16 mouse model of down syndrome. J. Neurosci. 23, 6788–6792.
Cataldo, A. M., Peterhoff, C. M., Troncoso, J. C., Gomez-Isla, T., Hyman, B. T., and Nixon, R. A. (2000). Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer's disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am. J. Pathol. 157, 277–286. doi: 10.1016/S0002-9440(10)64538-5
Chang, C.-Y., Chen, S.-M., Lu, H.-E., Lai, S.-M., Lai, P.-S., Shen, P.-W., et al. (2015). N-butylidenephthalide attenuates Alzheimer's Disease-like Cytopathy in down syndrome induced pluripotent stem cell-derived neurons. Sci. Rep. 5, 8744. doi: 10.1038/srep08744
Chang, K. T., and Min, K. T. (2009). Upregulation of three Drosophila homologs of human chromosome 21 genes alters synaptic function: implications for Down syndrome. Proc. Natl. Acad. Sci. U.S.A. 106, 17117–17122. doi: 10.1073/pnas.0904397106
Cheon, M. S., Bajo, M., Kim, S. H., Claudio, J. O., Stewart, A. K., Patterson, D., et al. (2003a). Protein levels of genes encoded on chromosome 21 in fetal Down syndrome brain: challenging the gene dosage effect hypothesis (Part II). Amino Acids 24, 119–125. doi: 10.1007/s00726-002-0337-1
Cheon, M. S., Kim, S. H., Ovod, V., Kopitar Jerala, N., Morgan, J. I., Hatefi, Y., et al. (2003b). Protein levels of genes encoded on chromosome 21 in fetal Down syndrome brain: challenging the gene dosage effect hypothesis (Part III). Amino Acids 24, 127–134. doi: 10.1007/s00726-002-0340-6
Cheon, M. S., Kim, S. H., Yaspo, M.-L., Blasi, F., Aoki, Y., Melen, K., et al. (2003c). Protein levels of genes encoded on chromosome 21 in fetal Down syndrome brain: challenging the gene dosage effect hypothesis (Part I). Amino Acids 24, 111–117. doi: 10.1007/s00726-002-0336-2
Cheon, M. S., Shim, K. S., Kim, S. H., Hara, A., and Lubec, G. (2003d). Protein levels of genes encoded on chromosome 21 in fetal Down syndrome brain: challenging the gene dosage effect hypothesis (Part IV). Amino Acids 25, 41–47. doi: 10.1007/s00726-003-0009-9
Chishti, M. A., Yang, D. S., Janus, C., Phinney, A. L., Horne, P., Pearson, J., et al. (2001). Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J. Biol. Chem. 276, 21562–21570. doi: 10.1074/jbc.M100710200
Choi, J. H. K., Berger, J. D., Mazzella, M. J., Morales-Corraliza, J., Cataldo, A. M., Nixon, R. A., et al. (2009). Age-dependent dysregulation of brain amyloid precursor protein in the Ts65Dn Down syndrome mouse model. J. Neurochem. 110, 1818–1827. doi: 10.1111/j.1471-4159.2009.06277.x
Choi, J. H. K., Kaur, G., Mazzella, M. J., Morales-Corraliza, J., Levy, E., and Mathews, P. M. (2013). Early endosomal abnormalities and cholinergic neuron degeneration in Amyloid-β protein precursor transgenic mice. J. Alzheimers Dis. 34, 691–700. doi: 10.3233/JAD-122143
Contestabile, A., Benfenati, F., and Gasparini, L. (2010). Communication breaks-Down: from neurodevelopment defects to cognitive disabilities in Down syndrome. Prog. Neurobiol. 91, 1–22. doi: 10.1016/j.pneurobio.2010.01.003
Contestabile, A., Fila, T., Bartesaghi, R., Contestabile, A., and Ciani, E. (2006). Choline acetyltransferase activity at different ages in brain of Ts65Dn mice, an animal model for Down's syndrome and related neurodegenerative diseases. J. Neurochem. 97, 515–526. doi: 10.1111/j.1471-4159.2006.03769.x
Cooper, J. D., Salehi, A., Delcroix, J. D., Howe, C. L., Belichenko, P. V., Chua-Couzens, J., et al. (2001). Failed retrograde transport of NGF in a mouse model of Down's syndrome: reversal of cholinergic neurodegenerative phenotypes following NGF infusion. Proc. Natl. Acad. Sci. U.S.A. 98, 10439–10444. doi: 10.1073/pnas.181219298
Coppus, A., Evenhuis, H., Verberne, G.-J., Visser, F., van Gool, P., Eikelenboom, P., et al. (2006). Dementia and mortality in persons with Down's syndrome. J. Intellect. Disabil. Res. 50, 768–777. doi: 10.1111/j.1365-2788.2006.00842.x
Coppus, A. M. W., Evenhuis, H. M., Verberne, G.-J., Visser, F. E., Oostra, B. A., Eikelenboom, P., et al. (2008). Survival in elderly persons with Down syndrome. J. Am. Geriatr. Soc. 56, 2311–2316. doi: 10.1111/j.1532-5415.2008.01999.x
Cossec, J. C., Lavaur, J., Berman, D. E., Rivals, I., Hoischen, A., Stora, S., et al. (2012). Trisomy for synaptojanin1 in down syndrome is functionally linked to the enlargement of early endosomes. Hum. Mol. Genet. 21, 3156–3172. doi: 10.1093/hmg/dds142
Costa, A. C. S., Stasko, M. R., Schmidt, C., and Davisson, M. T. (2010). Behavioral validation of the Ts65Dn mouse model for Down syndrome of a genetic background free of the retinal degeneration mutation Pde6brd1. Behav. Brain Res. 206, 52–62. doi: 10.1016/j.bbr.2009.08.034
Coutadeur, S., Benyamine, H., Delalonde, L., de Oliveira, C., Leblond, B., Foucourt, A., et al. (2015). A novel DYRK1A (Dual specificity tyrosine phosphorylation-regulated kinase 1A) inhibitor for the treatment of Alzheimer's disease: effect on Tau and amyloid pathologies in vitro. J. Neurochem. 133, 440–451. doi: 10.1111/jnc.13018
Crews, L., Rockenstein, E., and Masliah, E. (2010). APP transgenic modeling of Alzheimer's disease: mechanisms of neurodegeneration and aberrant neurogenesis. Brain Struct. Funct. 214, 111–126. doi: 10.1007/s00429-009-0232-6
Cumbo, E., and Ligori, L. D. (2010). Levetiracetam, lamotrigine, and phenobarbital in patients with epileptic seizures and Alzheimer's disease. Epilepsy Behav. 17, 461–466. doi: 10.1016/j.yebeh.2010.01.015
Cvetkovska, V., Hibbert, A. D., Emran, F., and Chen, B. E. (2013). Overexpression of Down syndrome cell adhesion molecule impairs precise synaptic targeting. Nat. Neurosci. 16, 677–682. doi: 10.1038/nn.3396
Das, I., and Reeves, R. H. (2011). The use of mouse models to understand and improve cognitive deficits in Down syndrome. Dis. Model. Mech. 4, 596–606. doi: 10.1242/dmm.007716
Davisson, M. T., Schmidt, C., Reeves, R. H., Irving, N. G., Akeson, E. C., Harris, B. S., et al. (1993). Segmental trisomy as a mouse model for Down syndrome. Prog. Clin. Biol. Res. 384, 117–133.
De Simone, R., Puig, X. S., Gelisse, P., Crespel, A., Genton, P., and Gélisse, P. (2010). Senile myoclonic epilepsy: delineation of a common condition associated with Alzheimer's disease in Down syndrome. Seizure 19, 383–389. doi: 10.1016/j.seizure.2010.04.008
De Strooper, B., Simons, M., Multhaup, G., Van Leuven, F., Beyreuther, K., and Dotti, C. G. (1995). Production of intracellular amyloid-containing fragments in hippocampal neurons expressing human amyloid precursor protein and protection against amyloidogenesis by subtle amino acid substitutions in the rodent sequence. EMBO J. 14, 4932–4938.
Deitz, S. L., and Roper, R. J. (2011). Trisomic and allelic differences influence phenotypic variability during development of Down syndrome mice. Genetics 189, 1487–1495. doi: 10.1534/genetics.111.131391
Dekker, A. D., Coppus, A. M. W., Vermeiren, Y., Aerts, T., van Duijn, C. M., Kremer, B. P., et al. (2015). Serum MHPG strongly predicts conversion to Alzheimer's disease in behaviorally characterized subjects with Down syndrome. J. Alzheimers Dis. 43, 871–891. doi: 10.3233/JAD-140783
Dekker, A. D., De Deyn, P. P., and Rots, M. G. (2014). Epigenetics: the neglected key to minimize learning and memory deficits in Down syndrome. Neurosci. Biobehav. Rev. 45, 72–84. doi: 10.1016/j.neubiorev.2014.05.004
Devenny, D. A., Zimmerli, E. J., Kittler, P., and Krinsky-McHale, S. J. (2002). Cued recall in early-stage dementia in adults with Down's syndrome. J. Intellect. Disabil. Res. 46, 472–483. doi: 10.1046/j.1365-2788.2002.00417.x
Devi, L., and Ohno, M. (2013). Effects of levetiracetam, an antiepileptic drug, on memory impairments associated with aging and Alzheimer's disease in mice. Neurobiol. Learn. Mem. 102, 7–11. doi: 10.1016/j.nlm.2013.02.001
Di Domenico, F., Pupo, G., Mancuso, C., Barone, E., Paolini, F., Arena, A., et al. (2015). Bach1 overexpression in Down syndrome correlates with the alteration of the HO-1/BVR-a system: insights for transition to Alzheimer's disease. J. Alzheimers Dis. 44, 1107–1120. doi: 10.3233/JAD-141254
Di Domenico, F., Pupo, G., Tramutola, A., Giorgi, A., Schininà, M. E., Coccia, R., et al. (2014). Redox proteomics analysis of HNE-modified proteins in Down syndrome brain: clues for understanding the development of Alzheimer disease. Free Radic. Biol. Med. 71, 270–280. doi: 10.1016/j.freeradbiomed.2014.03.027
Dierssen, M., Benavides-Piccione, R., Martínez-Cué, C., Estivill, X., Flórez, J., Elston, G. N., et al. (2003). Alterations of neocortical pyramidal cell phenotype in the Ts65Dn mouse model of Down syndrome: effects of environmental enrichment. Cereb. Cortex 13, 758–764. doi: 10.1093/cercor/13.7.758
Dierssen, M., Herault, Y., and Estivill, X. (2009). Aneuploidy: from a physiological mechanism of variance to Down syndrome. Physiol. Rev. 89, 887–920. doi: 10.1152/physrev.00032.2007
Dorval, V., Mazzella, M. J., Mathews, P. M., Hay, R. T., and Fraser, P. E. (2007). Modulation of Abeta generation by small ubiquitin-like modifiers does not require conjugation to target proteins. Biochem. J. 404, 309–316. doi: 10.1042/bj20061451
Downes, E. C., Robson, J., Grailly, E., Abdel-All, Z., Xuereb, J., Brayne, C., et al. (2008). Loss of synaptophysin and synaptosomal-associated protein 25-kDa (SNAP-25) in elderly Down syndrome individuals. Neuropathol. Appl. Neurobiol. 34, 12–22. doi: 10.1111/j.1365-2990.2007.00899.x
Duchon, A., Raveau, M., Chevalier, C., Nalesso, V., Sharp, A. J., and Herault, Y. (2011). Identification of the translocation breakpoints in the Ts65Dn and Ts1Cje mouse lines: relevance for modeling Down syndrome. Mamm. Genome 22, 674–684. doi: 10.1007/s00335-011-9356-0
Eckert, A., Schulz, K. L., Rhein, V., and Götz, J. (2010). Convergence of amyloid-β and tau pathologies on mitochondria in vivo. Mol. Neurobiol. 41, 107–114. doi: 10.1007/s12035-010-8109-5
Elder, G. A., Gama Sosa, M. A., and De Gasperi, R. (2010). Transgenic mouse models of Alzheimer's disease. Mt. Sinai J. Med. 77, 69–81. doi: 10.1002/msj.20159
Ermak, G., Morgan, T. E., and Davies, K. J. (2001). Chronic overexpression of the calcineurin inhibitory gene DSCR1 (Adapt78) is associated with Alzheimer's disease. J. Biol. Chem. 276, 38787–38794. doi: 10.1074/jbc.M102829200
Esposito, G., Scuderi, C., Lu, J., Savani, C., De Filippis, D., Iuvone, T., et al. (2008). S100B induces tau protein hyperphosphorylation via Dickopff-1 up-regulation and disrupts the Wnt pathway in human neural stem cells. J. Cell. Mol. Med. 12, 914–927. doi: 10.1111/j.1582-4934.2008.00159.x
Fortress, A. M., Hamlett, E. D., Vazey, E. M., Aston-Jones, G., Cass, W. A., Boger, H. A., et al. (2015). Designer receptors enhance memory in a mouse model of down syndrome. J. Neurosci. 35, 1343–1353. doi: 10.1523/JNEUROSCI.2658-14.2015
Francis, B. M., Yang, J., Hajderi, E., Brown, M. E., Michalski, B., McLaurin, J., et al. (2012). Reduced Tissue Levels of Noradrenaline Are Associated with Behavioral Phenotypes of the TgCRND8 Mouse Model of Alzheimer's Disease. Neuropsychopharmacology 37, 1934–1944. doi: 10.1038/npp.2012.40
Fratiglioni, L. (1996). Epidemiology of Alzheimer's disease and current possibilities for prevention. Acta Neurol. Scand. Suppl. 165, 33–40. doi: 10.1111/j.1600-0404.1996.tb05870.x
Frautschy, S. A., Yang, F., Irrizarry, M., Hyman, B., Saido, T. C., Hsiao, K., et al. (1998). Microglial response to amyloid plaques in APPsw transgenic mice. Am. J. Pathol. 152, 307–317.
Galante, M., Jani, H., Vanes, L., Daniel, H., Fisher, E. M. C., Tybulewicz, V. L. J., et al. (2009). Impairments in motor coordination without major changes in cerebellar plasticity in the Tc1 mouse model of Down syndrome. Hum. Mol. Genet. 18, 1449–1463. doi: 10.1093/hmg/ddp055
Games, D., Adams, D., Alessandrini, R., Barbour, R., Berthelette, P., Blackwell, C., et al. (1995). Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature 373, 523–527. doi: 10.1038/373523a0
Ghezzo, A., Salvioli, S., Solimando, M. C., Palmieri, A., Chiostergi, C., Scurti, M., et al. (2014). Age-related changes of adaptive and neuropsychological features in persons with Down Syndrome. PLoS ONE 9:e113111. doi: 10.1371/journal.pone.0113111
Glenner, G. G., and Wong, C. W. (1984). Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 120, 885–890.
Goedert, M., Spillantini, M. G., Cairns, N. J., and Crowther, R. A. (1992). Tau proteins of Alzheimer paired helical filaments: abnormal phosphorylation of all six brain isoforms. Neuron 8, 159–168. doi: 10.1016/0896-6273(92)90117-V
Gómez-Isla, T., Hollister, R., West, H., Mui, S., Growdon, J. H., Petersen, R. C., et al. (1997). Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann. Neurol. 41, 17–24. doi: 10.1002/ana.410410106
Grabowski, T. J., Cho, H. S., Vonsattel, J. P., Rebeck, G. W., and Greenberg, S. M. (2001). Novel amyloid precursor protein mutation in an Iowa family with dementia and severe cerebral amyloid angiopathy. Ann. Neurol. 49, 697–705. doi: 10.1002/ana.1009
Gribble, S. M., Wiseman, F. K., Clayton, S., Prigmore, E., Langley, E., Yang, F., et al. (2013). Massively parallel sequencing reveals the complex structure of an irradiated human chromosome on a mouse background in the Tc1 model of Down syndrome. PLoS ONE 8:e60482. doi: 10.1371/journal.pone.0060482
Griffin, W. S., Stanley, L. C., Ling, C., White, L., MacLeod, V., Perrot, L. J., et al. (1989). Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 86, 7611–7615. doi: 10.1073/pnas.86.19.7611
Griffiths-Jones, S. (2004). The microRNA Registry. Nucleic Acids Res. 32, D109–D111. doi: 10.1093/nar/gkh023
Grudzien, A., Shaw, P., Weintraub, S., Bigio, E., Mash, D. C., and Mesulam, M. M. (2007). Locus coeruleus neurofibrillary degeneration in aging, mild cognitive impairment and early Alzheimer's disease. Neurobiol. Aging 28, 327–335. doi: 10.1016/j.neurobiolaging.2006.02.007
Guyant-Marechal, I., Berger, E., Laquerrière, A., Rovelet-Lecrux, A., Viennet, G., Frebourg, T., et al. (2008). Intrafamilial diversity of phenotype associated with app duplication. Neurology 71, 1925–1926. doi: 10.1212/01.wnl.0000339400.64213.56
Haas, M. A., Bell, D., Slender, A., Lana-Elola, E., Watson-Scales, S., Fisher, E. M. C., et al. (2013). Alterations to dendritic spine morphology, but not dendrite patterning, of cortical projection neurons in Tc1 and Ts1Rhr mouse models of Down syndrome. PLoS ONE 8:e78561. doi: 10.1371/journal.pone.0078561
Hall, A. M., and Roberson, E. D. (2012). Mouse models of Alzheimer's disease. Brain Res. Bull. 88, 3–12. doi: 10.1016/j.brainresbull.2011.11.017
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356. doi: 10.1126/science.1072994
Hardy, J. A., and Higgins, G. A. (1992). Alzheimer's disease: the amyloid cascade hypothesis. Science 256, 184–185. doi: 10.1126/science.1566067
Hartley, D., Blumenthal, T., Carrillo, M., DiPaolo, G., Esralew, L., Gardiner, K., et al. (2015). Down syndrome and Alzheimer's disease: common pathways, common goals. Alzheimers Dementia 11, 700–709. doi: 10.1016/j.jalz.2014.10.007
Hashimoto, Y., and Matsuoka, M. (2014). A mutation protective against Alzheimer's disease renders amyloid β precursor protein incapable of mediating neurotoxicity. J. Neurochem. 130, 291–300. doi: 10.1111/jnc.12717
Hattori, M., Fujiyama, A., Taylor, T. D., Watanabe, H., Yada, T., Park, H. S., et al. (2000). The DNA sequence of human chromosome 21. Nature 405, 311–319. doi: 10.1038/35012518
Haydar, T. F., and Reeves, R. H. (2012). Trisomy 21 and early brain development. Trends Neurosci. 35, 81–91. doi: 10.1016/j.tins.2011.11.001
Helguera, P., Pelsman, A., Pigino, G., Wolvetang, E., Head, E., and Busciglio, J. (2005). ets-2 promotes the activation of a mitochondrial death pathway in Down's syndrome neurons. J. Neurosci. 25, 2295–2303. doi: 10.1523/jneurosci.5107-04.2005
Herault, Y., Lopes, P. P., Magnol, L., Sahun, I., Duchon, A., Prandini, P., et al. (2009). “Tackling the complexity of the genotype–phenotype relationship in the Down syndrome with the mouse aneuploidy zoo: a resource of new models to study aneuploidies involving human chromosome 21,” in The American Society of Human Genetics 59th Annual Meeting. (Honolulu HI).
Hernández-González, S., Ballestín, R., López-Hidalgo, R., Gilabert-Juan, J., Blasco-Ibáñez, J. M., Crespo, C., et al. (2015). Altered distribution of hippocampal interneurons in the murine Down Syndrome Model Ts65Dn. Neurochem. Res. 40, 151–164. doi: 10.1007/s11064-014-1479-8
Holland, A. J., Hon, J., Huppert, F. A., Stevens, F., and Watson, P. (1998). Population-based study of the prevalence and presentation of dementia in adults with Down's syndrome. Br. J. Psychiatry 172, 493–498.
Holtzman, D. M., Santucci, D., Kilbridge, J., Chua-Couzens, J., Fontana, D. J., Daniels, S. E., et al. (1996). Developmental abnormalities and age-related neurodegeneration in a mouse model of Down syndrome. Proc. Natl. Acad. Sci. U.S.A. 93, 13333–13338. doi: 10.1073/pnas.93.23.13333
Hooli, B. V., Mohapatra, G., Mattheisen, M., Parrado, A. R., Roehr, J. T., Shen, Y., et al. (2012). Role of common and rare APP DNA sequence variants in Alzheimer disease. Neurology 78, 1250–1257. doi: 10.1212/WNL.0b013e3182515972
Hoozemans, J. J. M., Rozemuller, A. J. M., van Haastert, E. S., Eikelenboom, P., and van Gool, W. A. (2011). Neuroinflammation in Alzheimer's disease wanes with age. J. Neuroinflammation 8:171. doi: 10.1186/1742-2094-8-171
Horvath, S., Garagnani, P., Bacalini, M. G., Pirazzini, C., Salvioli, S., Gentilini, D., et al. (2015). Accelerated epigenetic aging in Down syndrome. Aging Cell 14, 491–495. doi: 10.1111/acel.12325
Hsiao, K., Chapman, P., Nilsen, S., Eckman, C., Harigaya, Y., Younkin, S., et al. (1996). Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274, 99–102.
Hunsberger, J., Efthymiou, A. G., Malik, N., Behl, M., Mead, I. L., Zeng, X., et al. (2015). Induced pluripotent stem cell models to enable in vitro models for screening in the CNS. Stem Cells Dev. 24, 1852–1864. doi: 10.1089/scd.2014.0531
Hunter, C. L., Bimonte-Nelson, H. A., Nelson, M., Eckman, C. B., and Granholm, A.-C. (2004). Behavioral and neurobiological markers of Alzheimer's disease in Ts65Dn mice: effects of estrogen. Neurobiol. Aging 25, 873–884. doi: 10.1016/j.neurobiolaging.2003.10.010
Hunter, C. L., Isacson, O., Nelson, M., Bimonte-Nelson, H., Seo, H., Lin, L., et al. (2003). Regional alterations in amyloid precursor protein and nerve growth factor across age in a mouse model of Down's syndrome. Neurosci. Res. 45, 437–445. doi: 10.1016/S0168-0102(03)00005-1
Hunter, M. P., Nelson, M., Kurzer, M., Wang, X., Kryscio, R. J., Head, E., et al. (2011). Intersectin 1 contributes to phenotypes in vivo: implications for Down's syndrome. Neuroreport 22, 767–772. doi: 10.1097/WNR.0b013e32834ae348
Ingelsson, M., Fukumoto, H., Newell, K. L., Growdon, J. H., Hedley-Whyte, E. T., Frosch, M. P., et al. (2004). Early Abeta accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology 62, 925–931. doi: 10.1212/01.WNL.0000115115.98960.37
Iordanova, M. D., Burnett, D. J., Aggleton, J. P., Good, M., and Honey, R. C. (2009). The role of the hippocampus in mnemonic integration and retrieval: complementary evidence from lesion and inactivation studies. Eur. J. Neurosci. 30, 2177–2189. doi: 10.1111/j.1460-9568.2009.07010.x
Itagaki, S., McGeer, P. L., Akiyama, H., Zhu, S., and Selkoe, D. (1989). Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J. Neuroimmunol. 24, 173–182. doi: 10.1016/0165-5728(89)90115-X
Iwatsubo, T., Mann, D. M., Odaka, A., Suzuki, N., and Ihara, Y. (1995). Amyloid beta protein (A beta) deposition: a beta 42(43) precedes A beta 40 in Down syndrome. Ann. Neurol. 37, 294–299. doi: 10.1002/ana.410370305
Jackson, H. M., Onos, K. D., Pepper, K. W., Graham, L. C., Akeson, E. C., Byers, C., et al. (2015). DBA/2J genetic background exacerbates spontaneous lethal seizures but lessens amyloid deposition in a mouse model of Alzheimer's disease. PLoS ONE 10:e0125897. doi: 10.1371/journal.pone.0125897
Janel, N., Sarazin, M., Corlier, F., Corne, H., de Souza, L. C., Hamelin, L., et al. (2014). Plasma DYRK1A as a novel risk factor for Alzheimer's disease. Transl. Psychiatry 4:e425. doi: 10.1038/tp.2014.61
Jellinger, K. A., Lauda, F., and Attems, J. (2007). Sporadic cerebral amyloid angiopathy is not a frequent cause of spontaneous brain hemorrhage. Eur. J. Neurol. 14, 923–928. doi: 10.1111/j.1468-1331.2007.01880.x
Jensen, K. M., and Bulova, P. D. (2014). Managing the care of adults with Down's syndrome. BMJ 349:g5596. doi: 10.1136/bmj.g5596
Jiang, Y., Mullaney, K. A., Peterhoff, C. M., Che, S., Schmidt, S. D., Boyer-Boiteau, A., et al. (2010). Alzheimer's-related endosome dysfunction in Down syndrome is Abeta-independent but requires APP and is reversed by BACE-1 inhibition. Proc. Natl. Acad. Sci. U.S.A. 107, 1630–1635. doi: 10.1073/pnas.0908953107
Kasuga, K., Shimohata, T., Nishimura, A., Shiga, A., Mizuguchi, T., Tokunaga, J., et al. (2009). Identification of independent APP locus duplication in Japanese patients with early-onset Alzheimer disease. J. Neurol. Neurosurg. Psychiatry 80, 1050–1052. doi: 10.1136/jnnp.2008.161703
Kerbler, G. M., Fripp, J., Rowe, C. C., Villemagne, V. L., Salvado, O., Rose, S., et al. (2015). Basal forebrain atrophy correlates with amyloid β burden in Alzheimer's disease. NeuroImage Clin. 7, 105–113. doi: 10.1016/j.nicl.2014.11.015
Kern, D. S., Maclean, K. N., Jiang, H., Synder, E. Y., Sladek, J. R., and Bjugstad, K. B. (2011). Neural stem cells reduce hippocampal tau and reelin accumulation in aged Ts65Dn Down syndrome mice. Cell Transplant. 20, 371–379. doi: 10.3727/096368910X528085
Kim, W. S., Rahmanto, A. S., Kamili, A., Rye, K. A., Guillemin, G. J., Gelissen, I. C., et al. (2007). Role of ABCG1 and ABCA1 in regulation of neuronal cholesterol efflux to apolipoprotein E discs and suppression of amyloid-beta peptide generation. J. Biol. Chem. 282, 2851–2861. doi: 10.1074/jbc.M607831200
Kimura, R., Kamino, K., Yamamoto, M., Nuripa, A., Kida, T., Kazui, H., et al. (2007). The DYRK1A gene, encoded in chromosome 21 Down syndrome critical region, bridges between beta-amyloid production and tau phosphorylation in Alzheimer disease. Hum Mol Genet. 16, 15–23. doi: 10.1093/hmg/ddl437
Kokjohn, T. A., and Roher, A. E. (2009). Amyloid precursor protein transgenic mouse models and Alzheimer's disease: understanding the paradigms, limitations, and contributions. Alzheimers Dementia 5, 340–347. doi: 10.1016/j.jalz.2009.03.002
Kong, X. D., Liu, N., Xu, X. J., Zhao, Z. H., and Jiang, M. (2015). Screening of human chromosome 21 genes in the dorsolateral prefrontal cortex of individuals with Down syndrome. Mol. Med. Rep. 11, 1235–1239. doi: 10.3892/mmr.2014.2855
Korbel, J. O., Tirosh-Wagner, T., Urban, A. E., Chen, X.-N., Kasowski, M., Dai, L., et al. (2009). The genetic architecture of Down syndrome phenotypes revealed by high-resolution analysis of human segmental trisomies. Proc. Natl. Acad. Sci. U.S.A. 106, 12031–12036. doi: 10.1073/pnas.0813248106
Krabbe, G., Halle, A., Matyash, V., Rinnenthal, J. L., Eom, G. D., Bernhardt, U., et al. (2013). Functional impairment of microglia coincides with Beta-amyloid deposition in mice with Alzheimer-like pathology. PLoS ONE 8:e60921. doi: 10.1371/journal.pone.0060921
Krezowski, J., Knudson, D., Ebeling, C., Pitstick, R., Giri, R. K., Schenk, D., et al. (2004). Identification of loci determining susceptibility to the lethal effects of amyloid precursor protein transgene overexpression. Hum. Mol. Genet. 13, 1989–1997. doi: 10.1093/hmg/ddh210
Krinsky-McHale, S. J., Devenny, D. A., Gu, H., Jenkins, E. C., Kittler, P., Murty, V. V., et al. (2008). Successful aging in a 70-year-old man with down syndrome: a case study. Intellect. Dev. Disabil. 46, 215–228. doi: 10.1352/2008.46:215-228
Krinsky-McHale, S. J., Devenny, D. A., and Silverman, W. P. (2002). Changes in explicit memory associated with early dementia in adults with Down's syndrome. J. Intellect. Disabil. Res. 46, 198–208. doi: 10.1046/j.1365-2788.2002.00365.x
Krinsky-McHale, S. J., Devenny, D. A., Sersen, G., and Silverman, W. P. (2000). Sequence of cognitive decline in dementia in adults with Down's syndrome. J. Intellect. Disabil. Res. 44, 654–665. doi: 10.1111/j.1365-2788.2000.00305.x
Kumar-Singh, S., De Jonghe, C., Cruts, M., Kleinert, R., Wang, R., Mercken, M., et al. (2000). Nonfibrillar diffuse amyloid deposition due to a gamma(42)-secretase site mutation points to an essential role for N-truncated A beta(42) in Alzheimer's disease. Hum. Mol. Genet. 9, 2589–2598. doi: 10.1093/hmg/9.18.2589
LaFerla, F. M., Green, K. N., and Oddo, S. (2007). Intracellular amyloid-beta in Alzheimer's disease. Nat. Rev. Neurosci. 8, 499–509. doi: 10.1038/nrn2168
Landt, J., D'Abrera, J. C., Holland, A. J., Aigbirhio, F. I., Fryer, T. D., Canales, R., et al. (2011). Using positron emission tomography and Carbon 11-labeled Pittsburgh Compound B to image brain Fibrillar β-amyloid in adults with down syndrome: safety, acceptability, and feasibility. Arch. Neurol. 68, 890–896. doi: 10.1001/archneurol.2011.36
Lassalle, J. M., Halley, H., Daumas, S., Verret, L., and Francés, B. (2008). Effects of the genetic background on cognitive performances of TG2576 mice. Behav. Brain Res. 191, 104–110. doi: 10.1016/j.bbr.2008.03.017
Lee, L., Dale, E., Staniszewski, A., Zhang, H., Saeed, F., Sakurai, M., et al. (2014). Regulation of synaptic plasticity and cognition by SUMO in normal physiology and Alzheimer's disease. Sci. Rep. 4:7190. doi: 10.1038/srep07190
Lehman, E. J. H., Kulnane, L. S., Gao, Y., Petriello, M. C., Pimpis, K. M., Younkin, L., et al. (2003). Genetic background regulates beta-amyloid precursor protein processing and beta-amyloid deposition in the mouse. Hum. Mol. Genet. 12, 2949–2956. doi: 10.1093/hmg/ddg322
Lesné, S., Koh, M. T., Kotilinek, L., Kayed, R., Glabe, C. G., Yang, A., et al. (2006). A specific amyloid-beta protein assembly in the brain impairs memory. Nature 440, 352–357. doi: 10.1038/nature04533
Letourneau, A., Santoni, F. A., Bonilla, X., Sailani, M. R., Gonzalez, D., Kind, J., et al. (2014). Domains of genome-wide gene expression dysregulation in Down's syndrome. Nature 508, 345–350. doi: 10.1038/nature13200
Leverenz, J. B., and Raskind, M. A. (1998). Early amyloid deposition in the medial temporal lobe of young Down syndrome patients: a regional quantitative analysis. Exp. Neurol. 150, 296–304. doi: 10.1006/exnr.1997.6777
Li, Z., Yu, T., Morishima, M., Pao, A., LaDuca, J., Conroy, J., et al. (2007). Duplication of the entire 22.9 Mb human chromosome 21 syntenic region on mouse chromosome 16 causes cardiovascular and gastrointestinal abnormalities. Hum. Mol. Genet. 16, 1359–1366. doi: 10.1093/hmg/ddm086
Liu, C., Belichenko, P. V., Zhang, L., Fu, D., Kleschevnikov, A. M., Baldini, A., et al. (2011). Mouse models for Down syndrome-associated developmental cognitive disabilities. Dev. Neurosci. 33, 404–413. doi: 10.1159/000329422
Liu, C., Morishima, M., Jiang, X., Yu, T., Meng, K., Ray, D., et al. (2014). Engineered chromosome-based genetic mapping establishes a 3.7 Mb critical genomic region for Down syndrome-associated heart defects in mice. Hum. Genet. 133, 743–753. doi: 10.1007/s00439-013-1407-z
Liu, F., Liang, Z., Wegiel, J., Hwang, Y.-W., Iqbal, K., Grundke-Iqbal, I., et al. (2008). Overexpression of Dyrk1A contributes to neurofibrillary degeneration in Down syndrome. FASEB J. 22, 3224–3233. doi: 10.1096/fj.07-104539
Loane, M., Morris, J. K., Addor, M.-C., Arriola, L., Budd, J., Doray, B., et al. (2013). Twenty-year trends in the prevalence of Down syndrome and other trisomies in Europe: impact of maternal age and prenatal screening. Eur. J. Hum. Genet. 21, 27–33. doi: 10.1038/ejhg.2012.94
Lockrow, J., Boger, H., Bimonte-Nelson, H., and Granholm, A. C. (2011a). Effects of long-term memantine on memory and neuropathology in Ts65Dn mice, a model for Down syndrome. Behav. Brain Res. 221, 610–622. doi: 10.1016/j.bbr.2010.03.036
Lockrow, J., Boger, H., Gerhardt, G., Aston-Jones, G., Bachman, D., and Granholm, A. C. (2011b). A noradrenergic lesion exacerbates neurodegeneration in a down syndrome mouse model. J. Alzheimers Dis. 23, 471–489. doi: 10.3233/JAD-2010-101218
Lockrow, J., Prakasam, A., Huang, P., Bimonte-Nelson, H., Sambamurti, K., and Granholm, A. C. (2009). Cholinergic degeneration and memory loss delayed by vitamin E in a Down syndrome mouse model. Exp. Neurol. 216, 278–289. doi: 10.1016/j.expneurol.2008.11.021
Lott, I. T. (2012). Neurological phenotypes for Down syndrome across the life span. Prog. Brain Res. 197, 101–121. doi: 10.1016/B978-0-444-54299-1.00006-6
Lu, J., Esposito, G., Scuderi, C., Steardo, L., Delli-Bovi, L. C., Hecht, J. L., et al. (2011). S100B and APP promote a gliocentric shift and impaired neurogenesis in down syndrome neural progenitors. PLoS ONE 6:e22126. doi: 10.1371/journal.pone.0022126
Lu, J., Lian, G., Zhou, H., Esposito, G., Steardo, L., Delli-Bovi, L. C., et al. (2012). OLIG2 over-expression impairs proliferation of human Down syndrome neural progenitors. Hum. Mol. Genet. 21, 2330–2340. doi: 10.1093/hmg/dds052
Lu, T., Aron, L., Zullo, J., Pan, Y., Kim, H., Chen, Y., et al. (2014). REST and stress resistance in ageing and Alzheimer disease. Nature 507, 448–454. doi: 10.1038/nature13163
Madeo, J. (2013). The Role of Oxidative Stress in Alzheimer's Disease. J. Alzheimers Dis. Park. 03:116. doi: 10.4172/2161-0460.1000116
Maia, L. F., Kaeser, S. A., Reichwald, J., Lambert, M., Obermüller, U., Schelle, J., et al. (2015). Increased CSF Aβ during the very early phase of cerebral Aβ deposition in mouse models. EMBO Mol. Med. 7, 895–903. doi: 10.15252/emmm.201505026
Mann, D. M., and Esiri, M. M. (1989). The pattern of acquisition of plaques and tangles in the brains of patients under 50 years of age with Down's syndrome. J. Neurol. Sci. 89, 169–179.
Mann, D. M. (1988a). The pathological association between Down syndrome and Alzheimer disease. Mech. Ageing Dev. 43, 99–136. doi: 10.1016/0047-6374(88)90041-3
Mann, D. M. (1988b). Alzheimer's disease and Down's syndrome. Histopathology 13, 125–137. doi: 10.1111/j.1365-2559.1988.tb02018.x
Margallo-Lana, M. L., Moore, P. B., Kay, D. W. K., Perry, R. H., Reid, B. E., Berney, T. P., et al. (2007). Fifteen-year follow-up of 92 hospitalized adults with Down's syndrome: incidence of cognitive decline, its relationship to age and neuropathology. J. Intellect. Disabil. Res. 51, 463–477. doi: 10.1111/j.1365-2788.2006.00902.x
Martin, K. R., Corlett, A., Dubach, D., Mustafa, T., Coleman, H. A., Parkington, H. C., et al. (2012). Over-expression of RCAN1 causes Down syndrome-like hippocampal deficits that alter learning and memory. Hum. Mol. Genet. 21, 3025–3041. doi: 10.1093/hmg/dds134
Martin, S. B., Dowling, A. L., Lianekhammy, J., Lott, I. T., Doran, E., Murphy, M. P., et al. (2014). Synaptophysin and Synaptojanin-1 in Down Syndrome are Differentially Affected by Alzheimer's Disease. J. Alzheimers Dis. 42, 767–775. doi: 10.3233/jad-140795
Martínez-Cué, C., Delatour, B., and Potier, M.-C. (2014). Treating enhanced GABAergic inhibition in Down syndrome: use of GABA α5-selective inverse agonists. Neurosci. Biobehav. Rev. 46, 218–227. doi: 10.1016/j.neubiorev.2013.12.008
McCarron, M., McCallion, P., Reilly, E., and Mulryan, N. (2014). A prospective 14-year longitudinal follow-up of dementia in persons with Down syndrome. J. Intellect. Disabil. Res. 58, 61–70. doi: 10.1111/jir.12074
McCarron, M. O., Nicoll, J. A., and Graham, D. I. (1998). A quartet of Down's syndrome, Alzheimer's disease, cerebral amyloid angiopathy, and cerebral haemorrhage: interacting genetic risk factors. J. Neurol. Neurosurg. Psychiatry 65, 405–406. doi: 10.1136/jnnp.65.3.405
McGeer, E. G., Norman, M., Boyes, B., O'Kusky, J., Suzuki, J., and McGeer, P. L. (1985). Acetylcholine and aromatic amine systems in postmortem brain of an infant with Down's syndrome. Exp. Neurol. 87, 557–570. doi: 10.1016/0014-4886(85)90184-0
McGeer, P. L., Itagaki, S., Tago, H., and McGeer, E. G. (1987). Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci. Lett. 79, 195–200. doi: 10.1016/0304-3940(87)90696-3
Mendez, M., and Lim, G. (2003). Seizures in elderly patients with dementia: epidemiology and management. Drugs Aging 20, 791–803. doi: 10.2165/00002512-200320110-00001
Meraz-Ríos, M. A., Franco-Bocanegra, D., Toral Rios, D., and Campos-Peña, V. (2014). Early onset Alzheimer's disease and oxidative stress. Oxid. Med. Cell. Longev. 2014, 375968. doi: 10.1155/2014/375968
Mesulam, M., Shaw, P., Mash, D., and Weintraub, S. (2004). Cholinergic nucleus basalis tauopathy emerges early in the aging-MCI-AD continuum. Ann. Neurol. 55, 815–828. doi: 10.1002/ana.20100
Moechars, D., Dewachter, I., Lorent, K., Reversé, D., Baekelandt, V., Naidu, A., et al. (1999). Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J. Biol. Chem. 274, 6483–6492.
Mok, K. Y., Jones, E. L., Hanney, M., Harold, D., Sims, R., Williams, J., et al. (2014). Polymorphisms in BACE2 may affect the age of onset Alzheimer's dementia in Down syndrome. Neurobiol. Aging 35, e1–e5. doi: 10.1016/j.neurobiolaging.2013.12.022
Moore, S., Evans, L. D. B., Andersson, T., Portelius, E., Smith, J., Dias, T. B., et al. (2015). APP Metabolism Regulates Tau Proteostasis in Human Cerebral Cortex Neurons. Cell Rep. 11, 689–696. doi: 10.1016/j.celrep.2015.03.068
Mori, C., Spooner, E. T., Wisniewsk, K. E., Wisniewski, T. M., Yamaguch, H., Saido, T. C., et al. (2002). Intraneuronal Abeta42 accumulation in Down syndrome brain. Amyloid 9, 88–102.
Mori, T., Koyama, N., Arendash, G. W., Horikoshi-Sakuraba, Y., Tan, J., and Town, T. (2010). Overexpression of human S100B exacerbates cerebral amyloidosis and gliosis in the Tg2576 mouse model of Alzheimer's disease. Glia 58, 300–314. doi: 10.1002/glia.20924
Mucke, L., and Selkoe, D. J. (2012). Neurotoxicity of amyloid β-protein: synaptic and network dysfunction. Cold Spring Harb. Perspect. Med. 2, a006338. doi: 10.1101/cshperspect.a006338
Murray, A., Letourneau, A., Canzonetta, C., Stathaki, E., Gimelli, S., Sloan-Bena, F., et al. (2015). Brief report: isogenic induced pluripotent stem cell lines from an adult with mosaic down syndrome model accelerated neuronal ageing and neurodegeneration. Stem Cells 33, 2077–2084. doi: 10.1002/stem.1968
Murrell, J. R., Hake, A. M., Quaid, K. A., Farlow, M. R., and Ghetti, B. (2000). Early-onset Alzheimer disease caused by a new mutation (V717L) in the amyloid precursor protein gene. Arch. Neurol. 57, 885–887. doi: 10.1001/archneur.57.6.885
Musicco, M. (2009). Gender differences in the occurrence of Alzheimer's disease. Funct. Neurol. 24, 89–92.
Myllykangas, L., Wavrant-De Vrieze, F., Polvikoski, T., Notkola, I. L., Sulkava, R., Niinisto, L., et al. (2005). Chromosome 21 BACE2 haplotype associates with Alzheimer's disease: a two-stage study. J. Neurol. Sci. 236, 17–24. doi: 10.1016/j.jns.2005.04.008
Naito, K.-S., Sekijima, Y., and Ikeda, S.-I. (2008). Cerebral amyloid angiopathy-related hemorrhage in a middle-aged patient with Down's syndrome. Amyloid 15, 275–277. doi: 10.1080/13506120802524981
Narayan, P. J., Lill, C., Faull, R., Curtis, M. A., and Dragunow, M. (2015). Increased acetyl and total histone levels in post-mortem Alzheimer's disease brain. Neurobiol. Dis. 74, 281–294. doi: 10.1016/j.nbd.2014.11.023
Nelson, L. D., Orme, D., Osann, K., and Lott, I. T. (2001). Neurological changes and emotional functioning in adults with Down Syndrome. J. Intellect. Disabil. Res. 45, 450–456. doi: 10.1046/j.1365-2788.2001.00379.x
Ness, S., Rafii, M., Aisen, P., Krams, M., Silverman, W., and Manji, H. (2012). Down's syndrome and Alzheimer's disease: towards secondary prevention. Nat. Rev. Drug Discov. 11, 655–656. doi: 10.1038/nrd3822
O'Doherty, A., Ruf, S., Mulligan, C., Hildreth, V., Errington, M. L., Cooke, S., et al. (2005). An aneuploid mouse strain carrying human chromosome 21 with Down syndrome phenotypes. Science 309, 2033–2037. doi: 10.1126/science.1114535
Olson, L. E., Richtsmeier, J. T., Leszl, J., and Reeves, R. H. (2004). A chromosome 21 critical region does not cause specific Down syndrome phenotypes. Science 306, 687–690. doi: 10.1126/science.1098992
Palop, J. J. (2009). Epilepsy and Cognitive Impairments in Alzheimer Disease. Arch. Neurol. 66, 435. doi: 10.1001/archneurol.2009.15
Park, J., Yang, E. J., Yoon, J. H., and Chung, K. C. (2007). Dyrk1A overexpression in immortalized hippocampal cells produces the neuropathological features of Down syndrome. Mol. Cell Neurosci. 36, 270–279. doi: 10.1016/j.mcn.2007.07.007
Parker, S. E., Mai, C. T., Canfield, M. A., Rickard, R., Wang, Y., Meyer, R. E., et al. (2010). Updated National Birth Prevalence estimates for selected birth defects in the United States, 2004-2006. Birth Defects Res. A. Clin. Mol. Teratol. 88, 1008–1016. doi: 10.1002/bdra.20735
Parkinson, P. F., Kannangara, T. S., Eadie, B. D., Burgess, B. L., Wellington, C. L., and Christie, B. R. (2009). Cognition, learning behaviour and hippocampal synaptic plasticity are not disrupted in mice over-expressing the cholesterol transporter ABCG1. Lipids Heal. Dis. 8:5. doi: 10.1186/1476-511x-8-5
Pasalar, P., Najmabadi, H., Noorian, A. R., Moghimi, B., Jannati, A., Soltanzadeh, A., et al. (2002). An Iranian family with Alzheimer's disease caused by a novel APP mutation (Thr714Ala). Neurology 58, 1574–1575. doi: 10.1212/WNL.58.10.1574
Peacock, M. L., Warren, J. T., Roses, A. D., and Fink, J. K. (1993). Novel polymorphism in the A4 region of the amyloid precursor protein gene in a patient without Alzheimer's disease. Neurology 43, 1254–1256. doi: 10.1212/WNL.43.6.1254
Peng, S., Garzon, D. J., Marchese, M., Klein, W., Ginsberg, S. D., Francis, B. M., et al. (2009). Decreased brain-derived neurotrophic factor depends on amyloid aggregation state in transgenic mouse models of Alzheimer's disease. J. Neurosci. 29, 9321–9329. doi: 10.1523/JNEUROSCI.4736-08.2009
Pereira, P. L., Magnol, L., Sahún, I., Brault, V., Duchon, A., Prandini, P., et al. (2009). A new mouse model for the trisomy of the Abcg1-U2af1 region reveals the complexity of the combinatorial genetic code of down syndrome. Hum. Mol. Genet. 18, 4756–4769. doi: 10.1093/hmg/ddp438
Perez-Cruz, C., Nolte, M. W., van Gaalen, M. M., Rustay, N. R., Termont, A., Tanghe, A., et al. (2011). Reduced spine density in specific regions of CA1 pyramidal neurons in two transgenic mouse models of Alzheimer's disease. J. Neurosci. 31, 3926–3934. doi: 10.1523/JNEUROSCI.6142-10.2011
Pike, C. J., Cummings, B. J., and Cotman, C. W. (1995). Early association of reactive astrocytes with senile plaques in Alzheimer's disease. Exp. Neurol. 132, 172–179. doi: 10.1016/0014-4886(95)90022-5
Pilotto, A., Padovani, A., and Borroni, B. (2013). Clinical, biological, and imaging features of monogenic Alzheimer's Disease. Biomed. Res. Int. 2013:689591. doi: 10.1155/2013/689591
Pollonini, G., Gao, V., Rabe, A., Palminiello, S., Albertini, G., and Alberini, C. M. (2008). Abnormal expression of synaptic proteins and neurotrophin-3 in the Down syndrome mouse model Ts65Dn. Neuroscience 156, 99–106. doi: 10.1016/j.neuroscience.2008.07.025
Portelius, E., Hölttä, M., Soininen, H., Bjerke, M., Zetterberg, H., Westerlund, A., et al. (2014a). Altered cerebrospinal fluid levels of amyloid β and amyloid precursor-like protein 1 peptides in Down's syndrome. NeuroMol. Med. 16, 510–516. doi: 10.1007/s12017-014-8302-1
Portelius, E., Soininen, H., Andreasson, U., Zetterberg, H., Persson, R., Karlsson, G., et al. (2014b). Exploring Alzheimer molecular pathology in Down's syndrome cerebrospinal fluid. Neurodegener. Dis. 14, 98–106. doi: 10.1159/000358800
Powell, D., Caban-Holt, A., Jicha, G., Robertson, W., Davis, R., Gold, B. T., et al. (2014). Frontal white matter integrity in adults with Down syndrome with and without dementia. Neurobiol. Aging 35, 1562–1569. doi: 10.1016/j.neurobiolaging.2014.01.137
Pozueta, J., Lefort, R., and Shelanski, M. L. (2013). Synaptic changes in Alzheimer's disease and its models. Neuroscience 251, 51–65. doi: 10.1016/j.neuroscience.2012.05.050
Prasher, V. P., Farrer, M. J., Kessling, A. M., Fisher, E. M., West, R. J., Barber, P. C., et al. (1998). Molecular mapping of Alzheimer-type dementia in Down's syndrome. Ann. Neurol. 43, 380–383. doi: 10.1002/ana.410430316
Prasher, V. P., and Krishnan, V. H. R. (1993). Age of onset and duration of dementia in people with down syndrome: integration of 98 reported cases in the literature. Int. J. Geriatr. Psychiatry 8, 915–922. doi: 10.1002/gps.930081105
Pucharcos, C., Fuentes, J. J., Casas, C., de la Luna, S., Alcantara, S., Arbones, M. L., et al. (1999). Alu-splice cloning of human Intersectin (ITSN), a putative multivalent binding protein expressed in proliferating and differentiating neurons and overexpressed in Down syndrome. Eur. J. Hum. Genet. 7, 704–712. doi: 10.1038/sj.ejhg.5200356
Querfurth, H. W., and LaFerla, F. M. (2010). Alzheimer's disease. N. Engl. J. Med. 362, 329–344. doi: 10.1056/NEJMra0909142
Reeves, R. H., Irving, N. G., Moran, T. H., Wohn, A., Kitt, C., Sisodia, S. S., et al. (1995). A mouse model for Down syndrome exhibits learning and behaviour deficits. Nat. Genet. 11, 177–184. doi: 10.1038/ng1095-177
Reinholdt, L. G., Ding, Y., Gilbert, G. J., Gilbert, G. T., Czechanski, A., Solzak, J. P., et al. (2011). Molecular characterization of the translocation breakpoints in the Down syndrome mouse model Ts65Dn. Mamm. Genome 22, 685–691. doi: 10.1007/s00335-011-9357-z
Reynolds, G. P., and Warner, C. E. J. (1988). Amino acid neurotransmitter deficits in adult Down's syndrome brain tissue. Neurosci. Lett. 94, 224–227. doi: 10.1016/0304-3940(88)90299-6
Rodríguez-Arellano, J. J., Parpura, V., Zorec, R., and Verkhratsky, A. (2015). Astrocytes in physiological aging and Alzheimer's disease. Neuroscience. doi: 10.1016/j.neuroscience.2015.01.007. [Epub ahead of print].
Rovelet-Lecrux, A., Frebourg, T., Tuominen, H., Majamaa, K., Campion, D., and Remes, A. M. (2007). APP locus duplication in a Finnish family with dementia and intracerebral haemorrhage. J. Neurol. Neurosurg. Psychiatry 78, 1158–1159. doi: 10.1136/jnnp.2006.113514
Rovelet-Lecrux, A., Hannequin, D., Raux, G., Le Meur, N., Laquerrière, A., Vital, A., et al. (2006). APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat. Genet. 38, 24–26. doi: 10.1038/ng1718
Royston, M. C., McKenzie, J. E., Gentleman, S. M., Sheng, J. G., Mann, D. M., Griffin, W. S., et al. (1999). Overexpression of S100beta in Down's syndrome: correlation with patient age and with beta-amyloid deposition. Neuropathol. Appl. Neurobiol. 25, 387–393. doi: 10.1046/j.1365-2990.1999.00196.x
Ruparelia, A., Pearn, M. L., and Mobley, W. C. (2013). Aging and intellectual disability: insights from mouse models of Down syndrome. Dev. Disabil. Res. Rev. 18, 43–50. doi: 10.1002/ddrr.1127
Rustay, N. R., Cronin, E. A., Curzon, P., Markosyan, S., Bitner, R. S., Ellis, T. A., et al. (2010). Mice expressing the Swedish APP mutation on a 129 genetic background demonstrate consistent behavioral deficits and pathological markers of Alzheimer's disease. Brain Res. 1311, 136–147. doi: 10.1016/j.brainres.2009.11.040
Ryan, N. S., and Rossor, M. N. (2010). Correlating familial Alzheimer's disease gene mutations with clinical phenotype. Biomark. Med. 4, 99–112. doi: 10.2217/bmm.09.92
Ryoo, S. R., Cho, H. J., Lee, H. W., Jeong, H. K., Radnaabazar, C., Kim, Y. S., et al. (2008). Dual-specificity tyrosine(Y)-phosphorylation regulated kinase 1A-mediated phosphorylation of amyloid precursor protein: evidence for a functional link between Down syndrome and Alzheimer's disease. J. Neurochem. 104, 1333–1344. doi: 10.1111/j.1471-4159.2007.05075.x
Ryoo, S. R., Jeong, H. K., Radnaabazar, C., Yoo, J. J., Cho, H. J., Lee, H. W., et al. (2007). DYRK1A-mediated hyperphosphorylation of Tau. A functional link between Down syndrome and Alzheimer disease. J. Biol. Chem. 282, 34850–34857. doi: 10.1074/jbc.M707358200
Ryu, Y. S., Park, S. Y., Jung, M. S., Yoon, S. H., Kwen, M. Y., Lee, S. Y., et al. (2010). Dyrk1A-mediated phosphorylation of Presenilin 1: a functional link between Down syndrome and Alzheimer's disease. J. Neurochem. 115, 574–584. doi: 10.1111/j.1471-4159.2010.06769.x
Sabbagh, M. N., Chen, K., Rogers, J., Fleisher, A. S., Liebsack, C., Bandy, D., et al. (2015). Florbetapir PET, FDG PET, and MRI in Down syndrome individuals with and without Alzheimer's dementia. Alzheimers Dementia 11, 994–1004. doi: 10.1016/j.jalz.2015.01.006
Sago, H., Carlson, E. J., Smith, D. J., Kilbridge, J., Rubin, E. M., Mobley, W. C., et al. (1998). Ts1Cje, a partial trisomy 16 mouse model for Down syndrome, exhibits learning and behavioral abnormalities. Proc. Natl. Acad. Sci. U.S.A. 95, 6256–6261. doi: 10.1073/pnas.95.11.6256
Saito, T., Matsuba, Y., Mihira, N., Takano, J., Nilsson, P., Itohara, S., et al. (2014). Single App knock-in mouse models of Alzheimer's disease. Nat. Neurosci. 17, 661–663. doi: 10.1038/nn.3697
Saito, Y., Oka, A., Mizuguchi, M., Motonaga, K., Mori, Y., Becker, L. E., et al. (2000). The developmental and aging changes of Down's syndrome cell adhesion molecule expression in normal and Down's syndrome brains. Acta Neuropathol. 100, 654–664. doi: 10.1007/s004010000230
Salehi, A., Delcroix, J.-D., Belichenko, P. V., Zhan, K., Wu, C., Valletta, J. S., et al. (2006). Increased App expression in a mouse model of Down's syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron 51, 29–42. doi: 10.1016/j.neuron.2006.05.022
Sanchez, M. M., Moghadam, S., Naik, P., Martin, K. J., and Salehi, A. (2011). Hippocampal network alterations in Alzheimer's disease and Down syndrome: from structure to therapy. J. Alzheimers Dis. 26(Suppl 3), 29–47. doi: 10.3233/JAD-2011-0050
Sanchez, P. E., Zhu, L., Verret, L., Vossel, K. A., Orr, A. G., Cirrito, J. R., et al. (2012). Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer's disease model. Proc. Natl. Acad. Sci. U.S.A. 109, E2895–E2903. doi: 10.1073/pnas.1121081109
Sanders, N. C., Williams, D. K., and Wenger, G. R. (2009). Does the learning deficit observed under an incremental repeated acquisition schedule of reinforcement in Ts65Dn mice, a model for Down syndrome, change as they age? Behav. Brain Res. 203, 137–142. doi: 10.1016/j.bbr.2009.04.031
Sangani, M., Shahid, A., Amina, S., and Koubeissi, M. (2010). Improvement of myoclonic epilepsy in Down syndrome treated with levetiracetam. Epileptic Disord. 12, 151–154. doi: 10.1684/epd.2010.0306
Scheff, S. W., Price, D. A., Schmitt, F. A., DeKosky, S. T., and Mufson, E. J. (2007). Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology 68, 1501–1508. doi: 10.1212/01.wnl.0000260698.46517.8f
Schupf, N., Lee, A., Park, N., Dang, L.-H., Pang, D., Yale, A., et al. (2015). Candidate genes for Alzheimer's disease are associated with individual differences in plasma levels of beta amyloid peptides in adults with Down syndrome. Neurobiol. Aging 36, 2907.e1–2907.e10. doi: 10.1016/j.neurobiolaging.2015.06.020
Seidl, R., Cairns, N., Singewald, N., Kaehler, S. T., and Lubec, G. (2001). Differences between GABA levels in Alzheimer's disease and Down syndrome with Alzheimer-like neuropathology. Naunyn. Schmiedebergs. Arch. Pharmacol. 363, 139–145. doi: 10.1007/s002100000346
Seo, H., and Isacson, O. (2005). Abnormal APP, cholinergic and cognitive function in Ts65Dn Down's model mice. Exp. Neurol. 193, 469–480. doi: 10.1016/j.expneurol.2004.11.017
Serrano-Pozo, A., Frosch, M. P., Masliah, E., and Hyman, B. T. (2011). Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 1:a006189. doi: 10.1101/cshperspect.a006189
Shapiro, L. A., Marks, A., and Whitaker-Azmitia, P. M. (2004). Increased clusterin expression in old but not young adult S100B transgenic mice: evidence of neuropathological aging in a model of Down Syndrome. Brain Res. 1010, 17–21. doi: 10.1016/j.brainres.2003.12.057
Shapiro, L. A., and Whitaker-Azmitia, P. M. (2004). Expression levels of cytoskeletal proteins indicate pathological aging of S100B transgenic mice: an immunohistochemical study of MAP-2, drebrin and GAP-43. Brain Res. 1019, 39–46. doi: 10.1016/j.brainres.2004.05.100
Sheehan, R., Sinai, A., Bass, N., Blatchford, P., Bohnen, I., Bonell, S., et al. (2015). Dementia diagnostic criteria in Down syndrome. Int. J. Geriatr. Psychiatry 30, 857–863. doi: 10.1002/gps.4228
Sheng, J. G., Mrak, R. E., and Griffin, W. S. (1994). S100 beta protein expression in Alzheimer disease: potential role in the pathogenesis of neuritic plaques. J. Neurosci. Res. 39, 398–404. doi: 10.1002/jnr.490390406
Sheppard, O., Plattner, F., Rubin, A., Slender, A., Linehan, J. M., Brandner, S., et al. (2012). Altered regulation of tau phosphorylation in a mouse model of down syndrome aging. Neurobiol. Aging 33, e31–e44. doi: 10.1016/j.neurobiolaging.2011.06.025
Shi, J., Zhang, T., Zhou, C., Chohan, M. O., Gu, X., Wegiel, J., et al. (2008). Increased dosage of Dyrk1A alters alternative splicing factor (ASF)-regulated alternative splicing of tau in Down syndrome. J. Biol. Chem. 283, 28660–28669. doi: 10.1074/jbc.M802645200
Shi, Y., Kirwan, P., Smith, J., MacLean, G., Orkin, S. H., and Livesey, F. J. (2012). A human stem cell model of early Alzheimer's disease pathology in Down syndrome. Sci. Transl. Med. 4, 124ra29. doi: 10.1126/scitranslmed.3003771
Shichiri, M., Yoshida, Y., Ishida, N., Hagihara, Y., Iwahashi, H., Tamai, H., et al. (2011). α-Tocopherol suppresses lipid peroxidation and behavioral and cognitive impairments in the Ts65Dn mouse model of Down syndrome. Free Radic. Biol. Med. 50, 1801–1811. doi: 10.1016/j.freeradbiomed.2011.03.023
Shukkur, E. A., Shimohata, A., Akagi, T., Yu, W., Yamaguchi, M., Murayama, M., et al. (2006). Mitochondrial dysfunction and tau hyperphosphorylation in Ts1Cje, a mouse model for Down syndrome. Hum. Mol. Genet. 15, 2752–2762. doi: 10.1093/hmg/ddl211
Siarey, R. J., Kline-Burgess, A., Cho, M., Balbo, A., Best, T. K., Harashima, C., et al. (2006). Altered signaling pathways underlying abnormal hippocampal synaptic plasticity in the Ts65Dn mouse model of Down syndrome. J. Neurochem. 98, 1266–1277. doi: 10.1111/j.1471-4159.2006.03971.x
Sims, R., Hollingworth, P., Moskvina, V., Dowzell, K., O'Donovan, M. C., Powell, J., et al. (2009). Evidence that variation in the oligodendrocyte lineage transcription factor 2 (OLIG2) gene is associated with psychosis in Alzheimer's disease. Neurosci. Lett. 461, 54–59. doi: 10.1016/j.neulet.2009.05.051
Sleegers, K., Brouwers, N., Gijselinck, I., Theuns, J., Goossens, D., Wauters, J., et al. (2006). APP duplication is sufficient to cause early onset Alzheimer's dementia with cerebral amyloid angiopathy. Brain 129, 2977–2983. doi: 10.1093/brain/awl203
Stargardt, A., Swaab, D. F., and Bossers, K. (2015). The storm before the quiet: neuronal hyperactivity and Aβ in the presymptomatic stages of Alzheimer's disease. Neurobiol. Aging 36, 1–11. doi: 10.1016/j.neurobiolaging.2014.08.014
Streit, W. J., Braak, H., Xue, Q.-S., and Bechmann, I. (2009). Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer's disease. Acta Neuropathol. 118, 475–485. doi: 10.1007/s00401-009-0556-6
Sturchler-Pierrat, C., Abramowski, D., Duke, M., Wiederhold, K. H., Mistl, C., Rothacher, S., et al. (1997). Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc. Natl. Acad. Sci. U.S.A. 94, 13287–13292. doi: 10.1073/pnas.94.24.13287
Sun, X., He, G., and Song, W. (2006). BACE2, as a novel APP theta-secretase, is not responsible for the pathogenesis of Alzheimer's disease in Down syndrome. FASEB J. 20, 1369–1376. doi: 10.1096/fj.05-5632com
Sun, X., Wu, Y., Chen, B., Zhang, Z., Zhou, W., Tong, Y., et al. (2011). Regulator of calcineurin 1 (RCAN1) facilitates neuronal apoptosis through caspase-3 activation. J. Biol. Chem. 286, 9049–9062. doi: 10.1074/jbc.M110.177519
Sun, X., Wu, Y., Herculano, B., and Song, W. (2014). RCAN1 Overexpression exacerbates calcium overloading-induced neuronal apoptosis. PLoS ONE 9:e95471. doi: 10.1371/journal.pone.0095471
Szot, P., Van Dam, D., White, S. S., Franklin, A., Staufenbiel, M., and De Deyn, P. P. (2009). Age-dependent changes in noradrenergic locus coeruleus system in wild-type and APP23 transgenic mice. Neurosci. Lett. 463, 93–97. doi: 10.1016/j.neulet.2009.07.055
Takahashi, R. H., Milner, T. A., Li, F., Nam, E. E., Edgar, M. A., Yamaguchi, H., et al. (2002). Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am. J. Pathol. 161, 1869–1879. doi: 10.1016/S0002-9440(10)64463-X
Takano, T., Han, X., Deane, R., Zlokovic, B., and Nedergaard, M. (2007). Two-photon imaging of astrocytic Ca2+ signaling and the microvasculature in experimental mice models of Alzheimer's disease. Ann. N. Y. Acad. Sci. 1097, 40–50. doi: 10.1196/annals.1379.004
Tansley, G. H., Burgess, B. L., Bryan, M. T., Su, Y., Hirsch-Reinshagen, V., Pearce, J., et al. (2007). The cholesterol transporter ABCG1 modulates the subcellular distribution and proteolytic processing of beta-amyloid precursor protein. J. Lipid Res. 48, 1022–1034. doi: 10.1194/jlr.M600542-JLR200
Tanzi, R. E. (2012). The genetics of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2:a006296. doi: 10.1101/cshperspect.a006296
Thal, D. R., Rüb, U., Orantes, M., and Braak, H. (2002). Phases of A β-deposition in the human brain and its relevance for the development of AD. Neurology 58, 1791–1800. doi: 10.1212/WNL.58.12.1791
Tybulewicz, V. L. J., and Fisher, E. M. C. (2006). New techniques to understand chromosome dosage: mouse models of aneuploidy. Hum. Mol. Genet. 15, R103–R109. doi: 10.1093/hmg/ddl179
Tyrrell, J., Cosgrave, M., McCarron, M., McPherson, J., Calvert, J., Kelly, A., et al. (2001). Dementia in people with Down's syndrome. Int. J. Geriatr. Psychiatry 16, 1168–1174. doi: 10.1002/gps.502
Vacík, T., Ort, M., Gregorová, S., Strnad, P., Blatny, R., Conte, N., et al. (2005). Segmental trisomy of chromosome 17: a mouse model of human aneuploidy syndromes. Proc. Natl. Acad. Sci. U.S.A. 102, 4500–4505. doi: 10.1073/pnas.0500802102
Van Dam, D., D'Hooge, R., Staufenbiel, M., Van Ginneken, C., Van Meir, F., and De Deyn, P. P. (2003). Age-dependent cognitive decline in the APP23 model precedes amyloid deposition. Eur. J. Neurosci. 17, 388–396. doi: 10.1046/j.1460-9568.2003.02444.x
Van Dam, D., Marescau, B., Engelborghs, S., Cremers, T., Mulder, J., Staufenbiel, M., et al. (2005). Analysis of cholinergic markers, biogenic amines, and amino acids in the CNS of two APP overexpression mouse models. Neurochem. Int. 46, 409–422. doi: 10.1016/j.neuint.2004.11.005
Veerappan, C. S., Sleiman, S., and Coppola, G. (2013). Epigenetics of Alzheimer's disease and frontotemporal dementia. Neurotherapeutics 10, 709–721. doi: 10.1007/s13311-013-0219-0
Verret, L., Mann, E. O., Hang, G. B., Barth, A. M. I., Cobos, I., Ho, K., et al. (2012). Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 149, 708–721. doi: 10.1016/j.cell.2012.02.046
Vilardell, M., Rasche, A., Thormann, A., Maschke-Dutz, E., Pérez-Jurado, L. A., Lehrach, H., et al. (2011). Meta-analysis of heterogeneous Down Syndrome data reveals consistent genome-wide dosage effects related to neurological processes. BMC Genomics 12:229. doi: 10.1186/1471-2164-12-229
Voronov, S. V., Frere, S. G., Giovedi, S., Pollina, E. A., Borel, C., Zhang, H., et al. (2008). Synaptojanin 1-linked phosphoinositide dyshomeostasis and cognitive deficits in mouse models of Down's syndrome. Proc. Natl. Acad. Sci. U.S.A. 105, 9415–9420. doi: 10.1073/pnas.0803756105
Vossel, K. A., Beagle, A. J., Rabinovici, G. D., Shu, H., Lee, S. E., Naasan, G., et al. (2013). Seizures and epileptiform activity in the early stages of Alzheimer disease. JAMA Neurol. 70, 1158–1166. doi: 10.1001/jamaneurol.2013.136
Wallin, A. K., Blennow, K., Andreasen, N., and Minthon, L. (2006). CSF biomarkers for Alzheimer's Disease: levels of beta-amyloid, tau, phosphorylated tau relate to clinical symptoms and survival. Dement. Geriatr. Cogn. Disord. 21, 131–138. doi: 10.1159/000090631
Wang, X., Huang, T., Zhao, Y., Zheng, Q., Thompson, R. C., Bu, G., et al. (2014). Sorting nexin 27 regulates Abeta production through modulating gamma-secretase activity. Cell Rep. 9, 1023–1033. doi: 10.1016/j.celrep.2014.09.037
Wang, X., Zhao, Y., Zhang, X., Badie, H., Zhou, Y., Mu, Y., et al. (2013). Loss of sorting nexin 27 contributes to excitatory synaptic dysfunction by modulating glutamate receptor recycling in Down's syndrome. Nat. Med. 19, 473–480. doi: 10.1038/nm.3117
Webster, S. J., Bachstetter, A. D., Nelson, P. T., Schmitt, F. A., and Van Eldik, L. J. (2014). Using mice to model Alzheimer's dementia: an overview of the clinical disease and the preclinical behavioral changes in 10 mouse models. Front. Genet. 5:88. doi: 10.3389/fgene.2014.00088
Wegiel, J., Dowjat, K., Kaczmarski, W., Kuchna, I., Nowicki, K., Frackowiak, J., et al. (2008). The role of overexpressed DYRK1A protein in the early onset of neurofibrillary degeneration in Down syndrome. Acta Neuropathol. 116, 391–407. doi: 10.1007/s00401-008-0419-6
Wegiel, J., Kaczmarski, W., Barua, M., Kuchna, I., Nowicki, K., Wang, K. C., et al. (2011). Link between DYRK1A overexpression and several-fold enhancement of neurofibrillary degeneration with 3-repeat tau protein in Down syndrome. J. Neuropathol. Exp. Neurol. 70, 36–50. doi: 10.1097/NEN.0b013e318202bfa1
Weintraub, S., Wicklund, A. H., and Salmon, D. P. (2012). The neuropsychological profile of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2, a006171. doi: 10.1101/cshperspect.a006171
Westerman, M. A., Cooper-Blacketer, D., Mariash, A., Kotilinek, L., Kawarabayashi, T., Younkin, L. H., et al. (2002). The relationship between Abeta and memory in the Tg2576 mouse model of Alzheimer's disease. J. Neurosci. 22, 1858–1867.
Westmark, C. J., Westmark, P. R., and Malter, J. S. (2010). Alzheimer's disease and Down syndrome rodent models exhibit audiogenic seizures. J. Alzheimer's Dis. 20, 1009–1013. doi: 10.3233/JAD-2010-100087
Wilmot, B., McWeeney, S. K., Nixon, R. R., Montine, T. J., Laut, J., Harrington, C. A., et al. (2008). Translational gene mapping of cognitive decline. Neurobiol. Aging 29, 524–541. doi: 10.1016/j.neurobiolaging.2006.11.008
Wiseman, F. K., Al-Janabi, T., Hardy, J., Karmiloff-Smith, A., Nizetic, D., Tybulewicz, V. L. J., et al. (2015). A genetic cause of Alzheimer disease: mechanistic insights from Down syndrome. Nat. Rev. Neurosci. 16, 564–574. doi: 10.1038/nrn3983
Wisniewski, K. E., Wisniewski, H. M., and Wen, G. Y. (1985). Occurrence of neuropathological changes and dementia of Alzheimer's disease in Down's syndrome. Ann. Neurol. 17, 278–282. doi: 10.1002/ana.410170310
Witton, J., Padmashri, R., Zinyuk, L. E., Popov, V. I., Kraev, I., Line, S. J., et al. (2015). Hippocampal circuit dysfunction in the Tc1 mouse model of Down syndrome. Nat. Neurosci. 18, 1291–1298. doi: 10.1038/nn.4072
Wolvetang, E. J., Wilson, T. J., Sanij, E., Busciglio, J., Hatzistavrou, T., Seth, A., et al. (2003a). ETS2 overexpression in transgenic models and in Down syndrome predisposes to apoptosis via the p53 pathway. Hum. Mol. Genet. 12, 247–255. doi: 10.1093/hmg/ddg015
Wolvetang, E. W., Bradfield, O. M., Tymms, M., Zavarsek, S., Hatzistavrou, T., Kola, I., et al. (2003b). The chromosome 21 transcription factor ETS2 transactivates the beta-APP promoter: implications for Down syndrome. Biochim. Biophys. Acta 1628, 105–110. doi: 10.1016/S0167-4781(03)00121-0
Wu, J., and Morris, J. K. (2013). The population prevalence of Down's syndrome in England and Wales in 2011. Eur. J. Hum. Genet. 21, 1016–1019. doi: 10.1038/ejhg.2012.294
Xue, Q.-S., and Streit, W. J. (2011). Microglial pathology in Down syndrome. Acta Neuropathol. 122, 455–466. doi: 10.1007/s00401-011-0864-5
Yang, Q., Rasmussen, S. A., and Friedman, J. M. (2002). Mortality associated with Down's syndrome in the USA from 1983 to 1997: a population-based study. Lancet 359, 1019–1025. doi: 10.1016/S0140-6736(02)08092-3
Ye, X., Tai, W., and Zhang, D. (2012). The early events of Alzheimer's disease pathology: from mitochondrial dysfunction to BDNF axonal transport deficits. Neurobiol. Aging 33, e1–e10. doi: 10.1016/j.neurobiolaging.2011.11.004
Yu, T., Liu, C., Belichenko, P., Clapcote, S. J., Li, S., Pao, A., et al. (2010b). Effects of individual segmental trisomies of human chromosome 21 syntenic regions on hippocampal long-term potentiation and cognitive behaviors in mice. Brain Res. 1366, 162–171. doi: 10.1016/j.brainres.2010.09.107
Yu, T., Li, Z., Jia, Z., Clapcote, S. J., Liu, C., Li, S., et al. (2010a). A mouse model of Down syndrome trisomic for all human chromosome 21 syntenic regions. Hum. Mol. Genet. 19, 2780–2791. doi: 10.1093/hmg/ddq179
Zarow, C., Lyness, S. A., Mortimer, J. A., and Chui, H. C. (2003). Neuronal Loss Is Greater in the Locus Coeruleus Than Nucleus Basalis and Substantia Nigra in Alzheimer and Parkinson Diseases. Arch. Neurol. 60, 337. doi: 10.1001/archneur.60.3.337
Zhang, K., Schrag, M., Crofton, A., Trivedi, R., Vinters, H., and Kirsch, W. (2012a). Targeted proteomics for quantification of histone acetylation in Alzheimer's disease. Proteomics 12, 1261–1268. doi: 10.1002/pmic.201200010
Zhang, L., Fu, D., Belichenko, P. V., Liu, C., Kleschevnikov, A. M., Pao, A., et al. (2012b). Genetic analysis of Down syndrome facilitated by mouse chromosome engineering. Bioeng. Bugs 3, 8–12. doi: 10.4161/bbug.3.1.17696
Zhang, Y. Q., and Sarge, K. D. (2008). Sumoylation of amyloid precursor protein negatively regulates Abeta aggregate levels. Biochem. Biophys. Res. Commun. 374, 673–678. doi: 10.1016/j.bbrc.2008.07.109
Zigman, W. B. (2013). Atypical aging in Down syndrome. Dev. Disabil. Res. Rev. 18, 51–67. doi: 10.1002/ddrr.1128
Keywords: Alzheimer disease, APP, Down syndrome, mouse models, trisomy 21
Citation: Choong XY, Tosh JL, Pulford LJ and Fisher EMC (2015) Dissecting Alzheimer disease in Down syndrome using mouse models. Front. Behav. Neurosci. 9:268. doi: 10.3389/fnbeh.2015.00268
Received: 22 May 2015; Accepted: 21 September 2015;
Published: 13 October 2015.
Edited by:
Roger H. Reeves, Johns Hopkins University, USAReviewed by:
Sebastian Herbert Scharf, F. Hoffmann-La Roche Ltd, SwitzerlandYann Herault, Centre National de la Recherche Scientifique, France
Alexander M. Kleschevnikov, University of California, San Diego, USA
Copyright © 2015 Choong, Tosh, Pulford and Fisher. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elizabeth M. C. Fisher, Department of Neurodegenerative Disease, UCL Institute of Neurology, Queen Square, London WC1N 3BG, UK,ZS5maXNoZXJAcHJpb24udWNsLmFjLnVr