
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Behav. Neurosci. , 08 May 2015
Sec. Individual and Social Behaviors
Volume 9 - 2015 | https://doi.org/10.3389/fnbeh.2015.00092
This article is part of the Research Topic Further Understanding of Serotonin 7 Receptors’ Neuro-Psycho-Pharmacology View all 13 articles
 Agata Antonina Rita Impellizzeri1,2,3
Agata Antonina Rita Impellizzeri1,2,3 Matteo Pappalardo4,5
Matteo Pappalardo4,5 Livia Basile4
Livia Basile4 Ornella Manfra1,2
Ornella Manfra1,2 Kjetil Wessel Andressen1,2
Kjetil Wessel Andressen1,2 Kurt Allen Krobert1,2
Kurt Allen Krobert1,2 Angela Messina3,6
Angela Messina3,6 Finn Olav Levy1,2*†
Finn Olav Levy1,2*† Salvatore Guccione4*†
Salvatore Guccione4*†The human 5-HT7 receptor is expressed in both the central nervous system and peripheral tissues and is a potential drug target in behavioral and psychiatric disorders. We examined molecular determinants of ligand binding and G protein activation by the human 5-HT7(a) receptor. The role of several key residues in the 7th transmembrane domain (TMD) and helix 8 were elucidated combining in silico and experimental mutagenesis. Several single and two double point mutations of the 5-HT7(a) wild type receptor were made (W7.33V, E7.35T, E7.35R, E7.35D, E7.35A, R7.36V, Y7.43A, Y7.43F, Y7.43T, R8.52D, D8.53K; E7.35T-R7.36V, R8.52D-D8.53K), and their effects upon ligand binding were assessed by radioligand binding using a potent agonist (5-CT) and a potent antagonist (SB269970). In addition, the ability of the mutated 5-HT7(a) receptors to activate G protein after 5-HT-stimulation was determined through activation of adenylyl cyclase. In silico investigation on mutated receptors substantiated the predicted importance of TM7 and showed critical roles of residues E7.35, W7.33, R7.36 and Y7.43 in agonist and antagonist binding and conformational changes of receptor structure affecting adenylyl cyclase activation. Experimental data showed that mutants E7.35T and E7.35R were incapable of ligand binding and adenylyl cyclase activation, consistent with a requirement for a negatively charged residue at this position. The mutant R8.52D was unable to activate adenylyl cyclase, despite unaffected ligand binding, consistent with the R8.52 residue playing an important role in the receptor-G protein interface. The mutants Y7.43A and Y7.43T displayed reduced agonist binding and AC agonist potency, not seen in Y7.43F, consistent with a requirement for an aromatic residue at this position. Knowledge of the molecular interactions important in h5-HT7 receptor ligand binding and G protein activation will aid the design of selective h5-HT7 receptor ligands for potential pharmacological use.
The 5-HT7 receptor is a seven-transmembrane spanning receptor, coupled primarily to the stimulatory G protein (Gs). It is found in multiple organ systems, such as the cardiovascular system, CNS and digestive tract. In the central nervous system it has been proposed to play a role in the action of antipsychotics and antidepressants and it seems to be involved in regulating circadian rhythms and thermoregulation, learning and memory, as well as rapid eye movement (REM) sleep via the modulation of suprachiasmatic nucleus neurons (Gellynck et al., 2013). 5-HT7 receptor agonists have been suggested in treatment of dysfunctional memory in age-related decline and Alzheimer’s disease (Meneses, 2014), as well as treatment of pain, migraine, schizophrenia, anxiety and cognitive disturbances (Gellynck et al., 2013; Gasbarri and Pompili, 2014). A possible involvement in regulation of mood suggests that 5-HT7 is a potential target for the treatment of depression (Gellynck et al., 2013). In the periphery the 5-HT7 receptor is found primarily in the smooth muscle cells of blood vessels (Ullmer et al., 1995), and in the gastrointestinal tract, where it mediates relaxation of the ileum and stomach (Prins et al., 1999), and was recently shown to be important in inflammation (Guseva et al., 2014).
Three human 5-HT7 receptor splice variants (h5-HT7(a), h5-HT7(b), h5-HT7(d)) have been identified that are structurally identical except in their predicted intracellular carboxyl terminal (C-terminal) tail. They have indistinguishable pharmacological properties and similar abilities to stimulate adenylyl cyclase, indicating that the C-terminal tail does not influence ligand binding or G-protein coupling (Krobert et al., 2001). To facilitate development of selective drugs targeting the h5-HT7 receptor, it is necessary to understand the molecular interactions involved in ligand binding to the receptor. Several studies have focused on the molecular interaction of endogenous serotonin with different 5-HT receptor subtypes. Molecular requirements for serotonin binding to its receptor include electrostatic interaction between the receptor and the amino group of the ligand, one hydrogen bond between donor-acceptor site of the receptor and the hydroxyl group of serotonin and finally van der Waals interactions.
Mutagenesis studies of serotonin receptors (Ho et al., 1992; Wang et al., 1993; Boess et al., 1998; Mialet et al., 2000) suggested that the serotonin amino group makes an electrostatic interaction with the carboxylate of the highly conserved aspartate D3.32. This interaction is usually found in biogenic amine receptors, generally involved in binding of agonists/antagonists and in receptor activation. And there is evidence that serotonin hydroxyl groups interact with a donor-acceptor hydrogen bond residue present in transmembrane domain (TMD) V. These interactions seem relevant to only some serotonin receptor subtypes including 5-HT1A, 5-HT2, 5-HT6 and 5-HT4 (Mialet et al., 2000). The role of D3.32 was not relevant for antagonist interactions at the 5-HT1A receptor and the role of the hydrogen bond donor residue of the TMDV was not unambiguously identified in either 5-HT1A or 5-HT2B receptors (Wang et al., 2013). These anomalies indicate that the role of individual residues in molecular receptor-ligand interactions vary among different serotonin receptor subtypes.
To determine the critical molecular interactions that mediate ligand binding in the h5-HT7 receptor we used an experimental and in silico site-directed mutagenesis approach. Using homology modeling of the β2-adrenergic receptor (pdb: 2RH1; Cherezov et al., 2007), a 3D model of the h5-HT7 receptor was built and stabilized by large scale simulation in membrane bilayers. We focused on several specific amino acids, located in the 7th TMD, on the basis of preliminary modeling considerations, as discussed in the results section. In order to verify the predicted interactions, several h5-HT7(a) receptor mutants were generated, expressed and assessed for ligand binding as well as agonist and antagonist effects on adenylyl cyclase activity. Our results indicated that residues E7.35, R7.36 and Y7.43 are critical for ligand binding, while residue R8.52 plays a key role in G protein activation.
A preliminary model comparison between the β2-adrenergic and h5-HT7(a) receptors was performed and the results showed a strong sequence homology, especially at the level of specific transmembrane helices. Docking of 5-HT into the orthosteric binding cavity of our 5-HT7 model revealed a critical role of residues W7.33, E7.35, Y7.43, R8.53 and D8.54, where some were similar to that previously reported for the 5-HT7 receptor based on ligand docking (Kołaczkowski et al., 2006), and for 5-HT1B and 5-HT2B receptors based on crystallization (Wang et al., 2013). To understand how specific amino acids were involved in the binding process, determination of the 5-HT7 single point mutations was based on the chemical-physical characteristics of the specific amino acid with the objective to change the amino acid charges. In addition, point mutations were made to remove the aromatic groups to determine their importance for the interaction with the specific ligands. By site-directed mutagenesis we generated several clones expressing h5-HT7(a) receptors with single or double mutated amino acids.
Molecular dynamics (MD) simulations and docking studies were carried out using an Intel Core i7 processor, 16 GB RAM, operating under Linux/Ubuntu 10.04. The homology model of the h5-HT7 receptor was kindly supplied by Prof. Ingebrigt Sylte’s group (unpublished data) and built from the crystal structure of the β2-adrenergic receptor (pdb: 2RH1; Cherezov et al., 2007) by SwissModel server.1 Conformations of residues that differ between the 5-HT7 and β2-adrenergic receptors have been optimized using the RefineModel macro of ICM (Abagyan and Totrov, 1994).
The homology model features a disulfide bridge between TMH3 and extracellular loop 2 (ECL2) which are in accordance with the template structure. All the wild type and mutants considered (W7.33V, E7.35T, E7.35R, E7.35D, E7.35A, R7.36V, Y7.43A, R8.52D, D8.53K and R8.52D-D8.53K) were embedded in a bilayer of POPC (palmitoyl-oleyl-phosphatidyl choline; 100 × 100 Å) and solvated with pre-equilibrated water molecules in the three-dimensional space (box of water) of 130 × 130 × 108 Å. The counterions (Na+ and Cl−) were placed in the proximity of the regions of the protein surface to mimic an ionic strength of 0.15 mM. Thirty-five nanoseconds of MD simulations were carried out for all the considered systems using the NAMD2 software, version 2.9 (Phillips et al., 2005) with the CHARMM27 force field where all atoms are explicitly represented and water is characterized by the TIP3P model with a dielectric constant of 1 (ε) (Jorgensen et al., 1983). All systems were energy-minimized (conjugate gradient) then gradually heated up to 300 K with a 2-fs time step and equilibrated with a 300 K thermal bath for 400 ps. The velocities were reassigned every 2 ps to achieve complete stability (Berendsen et al., 1984). Production runs were performed at 300 K. The SHAKE algorithm with a tolerance of 1 × 10−8 Å was used to fix the length of the covalent hydrogen bonds (Ryckaert et al., 1977). Non-covalent interactions were calculated at each step. To avoid edge effects and treat long-range electrostatic interactions, periodic boundary conditions and the particle-mesh-Ewald algorithm with a grid size of 130 × 130 × 108 Å (Essmann et al., 1995) respectively, were applied to all of the simulation steps. Non-bonded short-range interactions were treated by a cutoff value of 10 Å. After minimizing the protein, the helices were first equilibrated in water with constraints on the cytosolic side. Next, MD of the helices with added loops and disulphide bonds was performed. Finally, MD of the unconstrained domain embedded in a bilayer POPC was carried out.
The ten mutants of 5-HT7 were constructed based upon the optimized model obtained as described above, adopting the mutate plugin of VMD 1.91. The mutants studied were W7.33V, E7.35A, E7.35D, E7.35R, E7.35T, R7.36V, E7.35T-R7.36V, Y7.43A, Y7.43T, Y7.43F, R8.52D, D8.53K, and R8.52D-D8.53K. 30 ns of MDs calculation for each mutant was performed, using computational resources granted from the supercomputing HPC-CINECA and UK-NSC. The predicted trajectories were analyzed using the VMD 1.91 software (Humphrey et al., 1996). The average structures as extracted from the last xyz atomic coordinates (50 ps for each MD simulation) were used as input file for docking.
Semiflexible docking was carried out by the molecular docking algorithm MolDock Optimizer and the scoring function MolDock [GRID] as implemented in the Molegro Virtual Docker software, version 6 (Thomsen and Christensen, 2006). Only torsion angles in the side chains were modifed during the minimization; all other properties, including bond lengths and backbone atom positions, were held fixed, and a new receptor conformation was generated for each pose after each docking calculation. The 10 runs for each molecule were carried out with a population size of 50, maximum iteration of 2000, scaling factor of 0.50. 5-carboxamidotryptamine (5-CT) and SB269970 structures were built and minimized by the software SYBYL-X 1.3.2
All mutant h5-HT7(a) sequences used in this work were obtained by site-directed mutagenesis of the coding region of the human h5-HT7(a) sequence cloned in pcDNA3.1 vector (Krobert et al., 2001). We used the QuikChange® Site-Directed Mutagenesis kit containing a PfuTurbo DNA polymerase (Agilent). All mutated sequences were confirmed by DNA sequencing. Using this strategy we generated a set of h5-HT7(a) clones carrying a single mutation at the following positions: W7.33V, E7.35T, E7.35R, E7.35D, E7.35A, R7.36V, Y7.43A, Y7.43F, Y7.43T, R8.52D, D8.53K. We also generated h5-HT7(a) clones with the double mutations E7.35T-R7.36V and R8.52D-D8.53K.
The human 5-HT7(a) receptor mutants obtained were expressed in QBI-HEK293 cells which were grown in Dulbecco’s modified Eagle’s medium (DMEM; GIBCO) with 10% fetal bovine serum (BioWhittaker) and penicillin (100 U/ml) and streptomycin (100 μg/ml). Cells were transiently transfected with wild-type and mutated plasmid DNA using LIPOFECTAMINE™—LTX reagent (Invitrogen™) and 7.5 μg DNA per 150 mm dish and a serum-free medium (ULTRAculture, BioWhittaker; because serum contains high concentration of serotonin) supplemented with L-glutamine (2 mM), penicillin (10000 U/ml) and streptomycin (10000 U/ml).
Membrane preparations from transiently transfected QBI-HEK293 cells containing the mutated receptors were prepared 48 h after transfection as described previously (Krobert et al., 2001).
The receptor expression level of QBI-HEK293 cells expressing mutated h5-HT7(a) was determined by radioligand binding. Binding assays were performed in 96-well, round-bottom microtiter plates with total reaction volume of 50 μl, containing the indicated concentration of ligand ([3H]5-CT (serotonin agonist) or [3H]SB269970 (serotonin antagonist)) as previously described (Krobert et al., 2001). Specific binding was defined as the difference between total binding and non-specific binding (obtained in the presence of 10 μM 5-HT). G-protein-coupled receptors may exist in different conformations, such as low and high affinity states. In the presence of excess GTP most receptors will exist in the low affinity state. To avoid bias due to unpredictable ratios between the two affinity states, GTP (100 μM) was included in the assay mix.
Adenylyl cyclase activity was measured by determining conversion of [α-32P]ATP to [32P]cAMP in membrane preparations as previously described (Krobert et al., 2001). Briefly, adenylyl cyclase activities were measured on 10 μl aliquots in a final volume of 50 μl in the presence of 0.1 mM [α-32P]ATP, 4 mM MgCl2, 20 μM GTP, 1 mM EDTA, 1 mM [3H]cAMP, 1 mM 3-isobutyl-1-methylxanthine (IBMX; Sigma), a nucleoside triphosphate regenerating system and additives (FSK (100 μM), 5-CT, 5-HT and SB269970). The samples were incubated for 20 min at 32°C. Cyclic AMP formed was quantified by the double column chromatography system on Dowex 50 cation exchanger and on neutral aluminium oxide (Alumina) columns. The recoveries of each sample through the columns are monitored by adding [3H]cAMP to the assay mix, thus eliminating individual differences between the columns.
Binding and adenylyl cyclase data were analyzed by non-linear regression using Microsoft Excel 2007 with the Solver add-in, as described (Krobert et al., 2001).
Lysates of membrane preparations were separated by SDS-PAGE and electroblotted as described (Norum et al., 2005). The membranes were incubated overnight at 4°C with a 1:200 dilution (v/v) of rabbit anti-5-HT7R (Oncogene Research Products, Boston, MA) in PBS containing 5% (w/v) non-fat dry milk and 0.05% (v/v) Tween 20. Thereafter, the blots were incubated with a 1:5000 dilution (v/v) of HRP-linked anti-rabbit IgG (Amersham ECL™-HRP Linked Secondary Antibodies, GE Healthcare). The immobilized HRP-conjugated secondary antibodies were visualized with the LumiGLO Chemiluminescent Substrate (KPL, Inc.) and visualized with a UCP Sensicam (UVP Inc., CA, USA).
MD simulations of the mutants of 5-HT7 were performed in order to obtain a reliable structure for the subsequent docking calculation. After about 30 ns of equilibration root mean square deviation (RMSD) profiles were analyzed for all mutants to verify the backbone stability and perform docking analysis. All RMSD curves are steady along the whole equilibration phase and no noteworthy oscillations were observed, demonstrating that all structures were stabilized and equilibrated by 35 ns of MD. Figure 1 shows the trend of RMSD for backbone atoms of the mutated receptors E7.35T, E7.35R, R8.52D and D8.53K. All analyzed models showed a high structural rigidity; for all models the RMSD was about 0.165 Å.
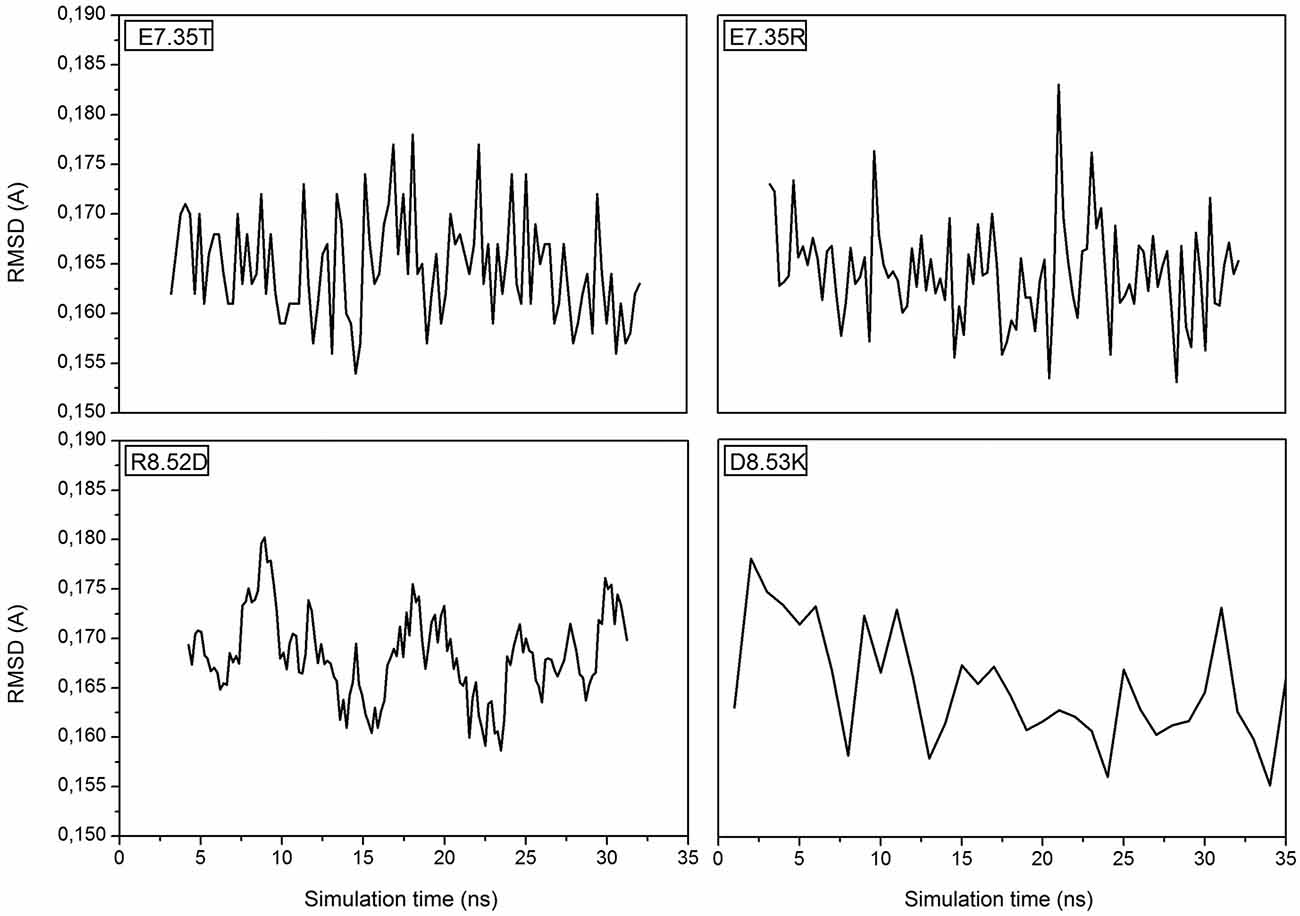
Figure 1. Root Mean Square Deviation (RMSD) is referring to the backbone of the four mutants E7.35T, E7.35R, R8.52D and D8.53K.
In addition, in Figure 2, the secondary structures of the models were plotted over time, in order to verify that the predicted secondary structure does not change significantly. The backbone of the seven trans-membrane domains seems to be stable in helix conformation during the simulation. On the other hand, residues of the extracellular loops appear to be more flexible, and they do not have a stable conformation. At the end of 30 ns of equilibration, our model consists of seven transmembrane helices (Figure 2) and a large cavity defined by the helices III, V, VI and VII, in accordance with Wang et al. (1993). Comparison of our 5-HT7 model with the X-ray structure of 5-HT1B (4IAQ) (Wang et al., 2013) and 5-HT2B (4IB4) (Wacker et al., 2013) receptors, confirms that the main folding pattern of our model appears to be consistent with experimental data, as reported in panel A of Figure 3. While the three structures here compared have slightly different primary sequences (e.g., T6.46 in 5-HT7, M6.46 in 5-HT2B and A6.46 in 5-HT1B), the same region with different amino acids has the same spatial orientation of the side chains, as evidenced in Figure 3B. Particularly, glutamic acid is conserved in all the models, along with its orientation. Moreover, in our model a formation of a hydrogen bond between T7.35 and R6.58 is observed, as discussed later in the discussion section, which should play a role in ligand binding. All the data indicate that our models appear good candidates for further study.
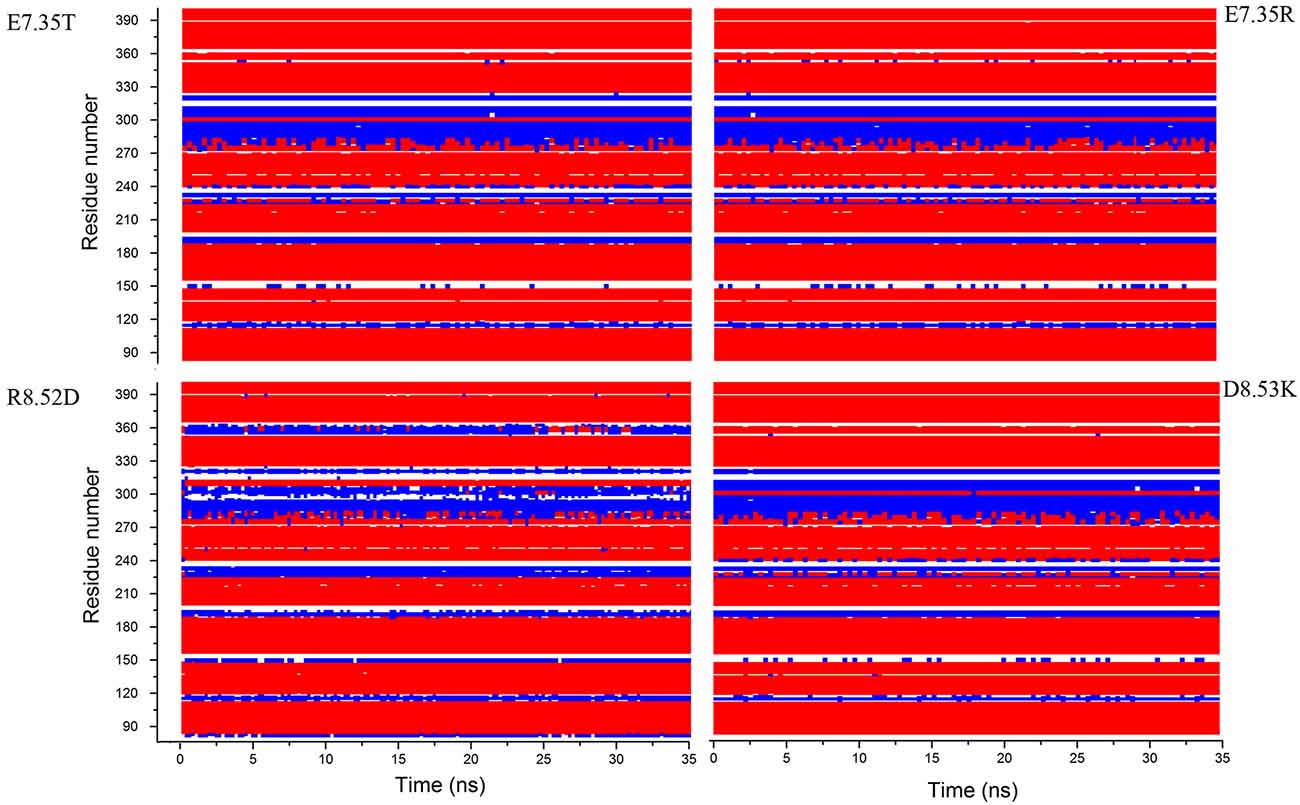
Figure 2. Secondary structure for the mutants E7.35T, E7.35R, R8.52D and D8.53K calculated over the molecular dynamic (MD) trajectory. Red represents helix conformation, blue represents turn conformation, white represents random coil conformation.
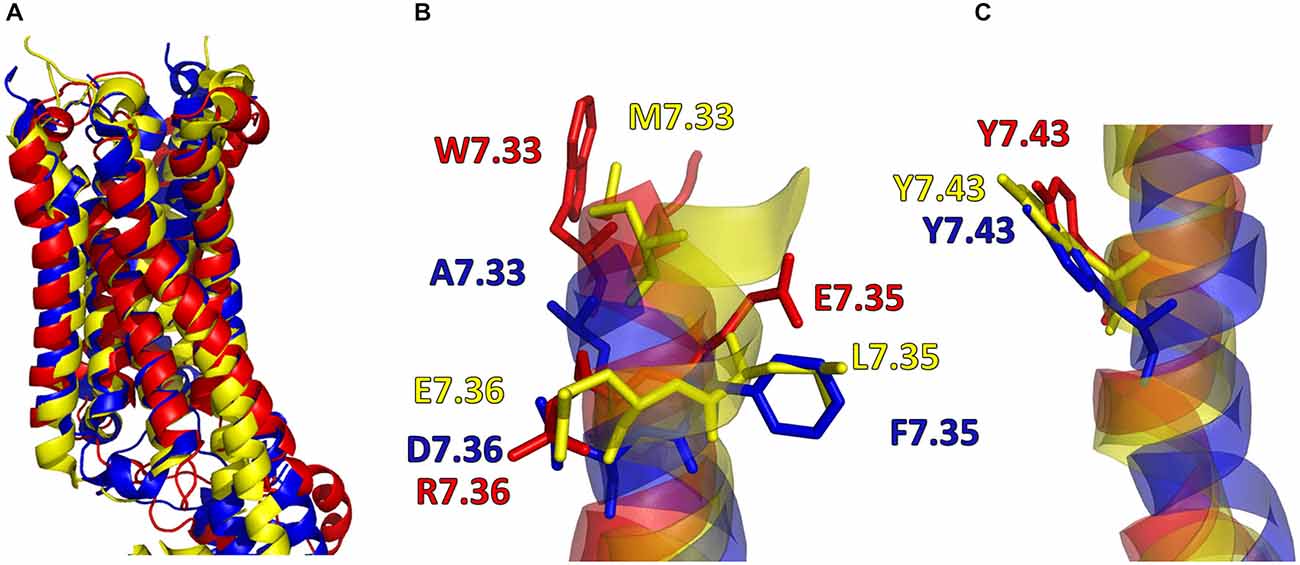
Figure 3. (A) Superimposition of 5-HT7 after 30 ns of equilibration (red) with 5-HT1B (blue) (Wang et al., 2013) and 5-HT2B (yellow) (Wacker et al., 2013). (B,C) Main chain of the three receptors represented as stick and aminoacid residues as lines.
Docking results have been summarized in Table 1. All the mutants are predicted to bind both the agonist 5-CT and the antagonist SB269970. Tables 2, 3 list the favorable and unfavorable interactions of the two reference molecules with the wild type and mutant receptors. Overall, the antagonist (SB269970) forms a higher number of positive interactions and no unfavorable steric interactions that allow the formation of more stable complex (more negative docking score) than the agonist. Mutation of E7.35 to Asp, Thr and Arg were predicted to produce better binding compared to the wild type protein, while mutation of E7.35 to Ala was predicted to maintain high binding capacity. On the contrary, mutation of R8.52 to Asp was predicted to produce weaker binding. Only mutant D8.53K was predicted to show slightly lower binding. In this case the increased steric hindrance of the mutations plays a key role in binding properties.
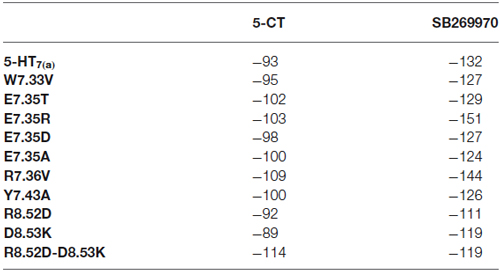
Table 1. MolDock score (a.u.) from docking analysis of wild type and mutant receptors with 5-CT and SB269970.

Table 2. List of favorable and unfavorable interactions after docking between SB269970 and the target molecules.

Table 3. List of favorable and unfavorable interactions after docking between 5-CT and the target molecules.
The mutant receptors were examined by comparing the binding properties and ability to activate adenylyl cyclase (cyclic AMP production), using transiently transfected QBI-HEK293 cells. The affinities of the agonist (5-CT) and the antagonist (SB269970) determined from saturation binding experiments with up to 3 nM of [3H]5-CT and up to 2.5 nM [3H]SB269970 varied from being not modified, reduced or, in some mutants, no specific binding was detected. The receptor density of all the analyzed mutants was only slightly reduced in a subset of mutants, so receptor density is unlikely to account for changes in affinity (shown in Table 4). In addition, the ability to activate AC was either unmodified or reduced in the mutated receptors compared to wild-type (Table 5). Most importantly, the mutagenesis studies revealed critical roles of residues E7.35, R7.36 and Y7.43 in ligand binding and R8.52 in activation of AC.
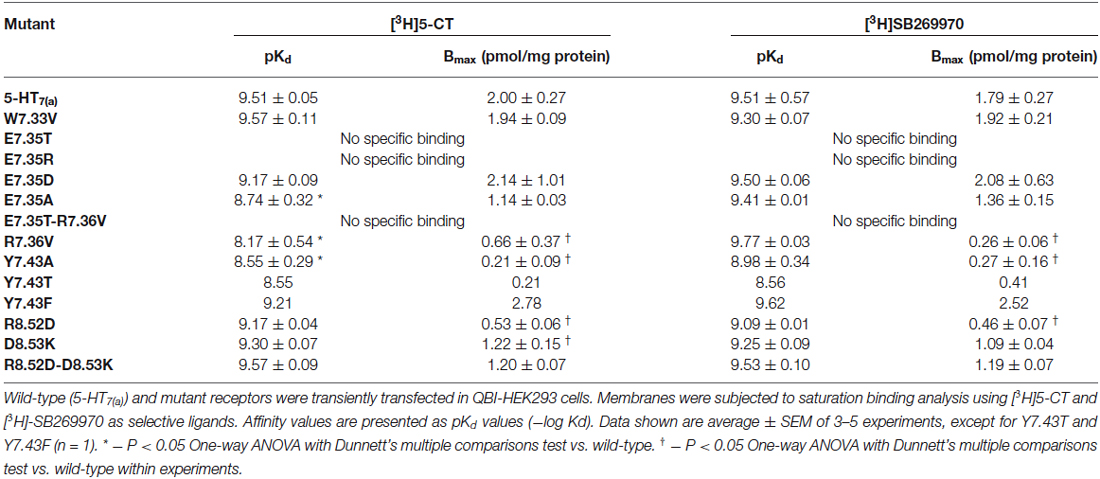
Table 4. Binding properties of wild-type and mutant 5-HT7(a) receptors expressed in QBI-HEK293 cells.
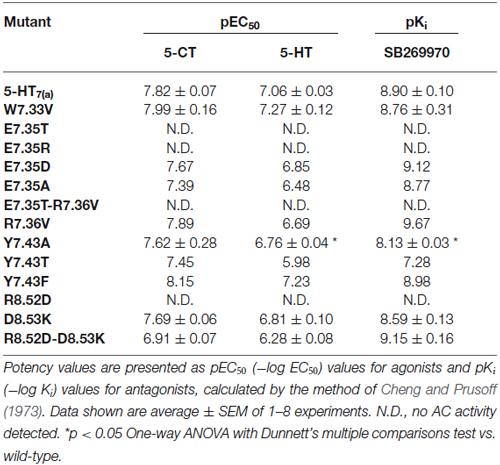
Table 5. Activation of adenylyl cyclase (AC) by the agonists 5-CT and 5-HT and inhibition of 10 μM 5-HT-stimulated AC by the antagonist SB269970.
Initially, E7.35 was mutated to Thr to remove the negative charge in this part of the receptor and this new receptor showed a complete lack of ability to bind either agonist or antagonist. To understand which kind of interaction is critical in this part of the receptor for the ligand-receptor-interaction, other mutations were performed at E7.35: (1) to Asp (E7.35D) to understand the importance of amino acid charge; (2) to Ala (E7.35A) to test if the length of the side chain is important for ligand binding; and (3) to Arg (E7.35R) to determine if there are electrostatic interactions involved. These new mutants (E7.35D, E7.35A, E7.35R) were analyzed as above in the binding assays. While the E7.35R mutant receptor was completely unable to bind either agonist or antagonist, the E7.35A mutant displayed reduced binding affinity for agonist, but not for antagonist, and the E7.35D mutation did not change the ability of the receptor to bind any ligands (Table 4). Mutation of the neighboring residue, R7.36V, resulted in decreased binding affinity for agonist only and a reduced receptor expression. The double mutant E7.35T-R7.36V was unable to bind any ligands.
The mutation R8.52D, where the positive charge was replaced by a negative one, did not alter the affinity for 5-CT or SB269970 (but the receptor expression was significantly reduced), neither did the mutation D8.53K nor the double mutant R8.52D-D8.53K. Another TMH7 mutation evaluated was Y7.43A, where the aromatic group of residue Y7.43 was replaced with the non-polar Ala residue. This mutation caused a significant reduction in agonist binding affinity and a possible reduction in antagonist binding affinity, in addition to a reduction in receptor expression. To determine if the aromatic group or OH-group was involved in the changes elicited by mutation Y7.43A, mutant receptors Y7.43F and Y7.43T were constructed. Preliminary data indicate that Y7.43T showed a reduction in ligand potency and receptor expression, whereas Y7.43F was without effect upon ligand binding.
We next examined the effect of the different mutations within TMH7 of the h5-HT7(a) receptor on the transductional response (G protein activation) by measuring their ability to activate adenylyl cyclase (AC). As shown in Table 5 and Figure 4, the W7.33V, E7.35D, R7.36V, Y7.43A, and D8.53K mutated receptors stimulated AC activity with the same potency as the wild-type receptor. A possible higher affinity for the antagonist SB269970 of the R7.36V mutant receptor measured in AC assays (Figure 4; Table 5) was not supported by binding data (Table 4) and therefore not investigated further. Particularly interesting was the finding that the R8.52D mutant which displayed unaltered binding affinity for both agonist and antagonist, was not able to activate AC constitutively (no effect of SB269970), or by either of the agonists 5-CT or 5-HT (Table 5). In contrast, the Y7.43A mutant displayed a lower binding affinity for 5-CT, a lower potency on AC for 5-HT and SB269970, but no change in potency to activate AC for 5-CT (Table 5). In line with the lack of binding, the mutants E7.35T and E7.35R showed no ability to activate AC, whereas the conservative mutation E7.35D showed no change in AC activation and the E7.35A mutant activated AC with reduced potency.
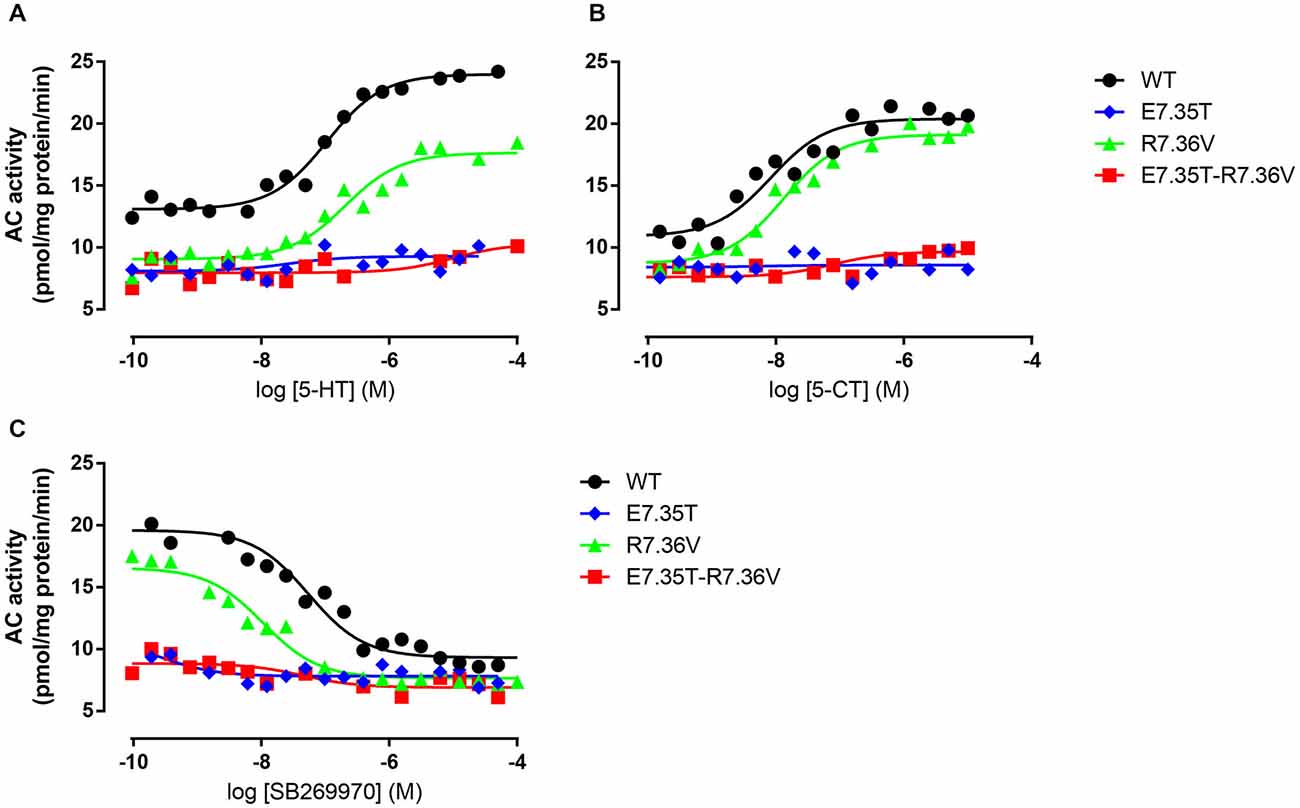
Figure 4. Ability of mutant receptors to activate adenylyl cyclase. Adenylyl cyclase (AC) activity in response to increasing concentrations of 5-HT (A) or 5-CT (B) in membranes from QBI-HEK293 cells transiently expressing the wild-type (WT) or indicated mutant receptors. In (C) the ability of increasing concentrations of SB269970 to antagonize 10 μM 5-HT was determined. AC activity was measured as described under Materials and Methods, and the data shown are representative of those obtained from 1–3 independent experiments.
To determine if the complete inability of the E7.35T and E7.35R mutants to bind the radioligands [3H]5-CT and [3H]SB269970 resulted from the absence of receptor expression, we determined if these receptors were in fact expressed by subjecting membrane preparations to SDS-PAGE and determining 5-HT7 expression using a polyclonal antibody directed against the N-terminus of the h5-HT7 receptor. In those receptors analyzed (even in mutants with reduced or absent radioligand binding), a protein band of about ~50 kDa, corresponding with the size of the h5-HT7 receptor, was detected, whereas in non-transfected control cells no band was detected (data not shown). These data indicate that the absence of ligand binding of the various mutant receptors does not result from a lack of receptor expression despite displaying an absence of radioligand binding.
GPCRs are challenging targets in drug design (Overington et al., 2006). Despite the recent surge of GPCR structural data, structures have only been determined for a minor fraction of GPCRs. Therefore, in silico tools are key to obtain structural information to integrate and rationalize the design of experimental studies of receptor-ligand interactions. Homology modeling has been successfully applied for several different GPCRs (see Sandal et al., 2013 and references therein). Combining in silico with experimental tools can lead to accurate structural characterization and accelerate drug design. In this work, the mechanism of molecular recognition between the h5-HT7 receptor and ligands was investigated through in silico molecular docking and well-established in vitro experimental approaches.
The modeling data predicted that the residue E7.35 of the wild type was relevant for ligand binding. In particular, this residue located in the TMH7 is involved in the formation of a three-member salt bridge with R7.36 and R6.58. Therefore, we focused our attention on this residue and performed single point mutagenesis in order to evaluate the effect of specific amino acids on the binding affinity of the receptor. The chosen mutants E7.35T, E7.35R, E7.35A, E7.35D were created by site-directed mutagenesis and analyzed with radioligand binding assays and the ability to activate AC.
The ability to bind both the agonist (5-CT) and antagonist (SB269970) radioligand was completely abolished in the E7.35T mutant, despite expression of the mutant receptor as determined by Western analysis. An analysis of the MD trajectory suggests that the inability to activate the receptor may result from the formation of a H bond between T7.35 and R6.58 in the E7.35T mutant. Possibly, this H bond acts as a locked gate preventing the entry of the ligands to the binding cavity (Figure 5). Likewise, no specific binding of either agonist or antagonist ligands was observed in the mutant E7.35R. The MD analysis of E7.35R suggests that the guanidine side chain of R7.35 in the E7.35R mutant moves towards the guanidine side chain of R6.58 blocking the entrance to the orthosteric pocket of the protein (Figure 6). Consistent with the lack of binding, the E7.35T and E7.35R mutant receptors were unable to activate adenylyl cyclase activity (Figure 4; Table 5). Not surprisingly, the conservatively changed mutant E7.35D receptor, maintaining the charged residue, behaved essentially as the WT receptor both regarding binding and AC activation, consistent with the important role of the electrostatic interactions with this residue. Also consistent with this was the finding that the E7.35A mutated receptor, replacing Glu with the nonpolar Ala, showed reduced binding affinity to both agonist (5-CT) and antagonist (SB269970) radioligand (Table 4), and was still able to activate AC, but with reduced potency in line with the reduced binding affinity (Table 5).
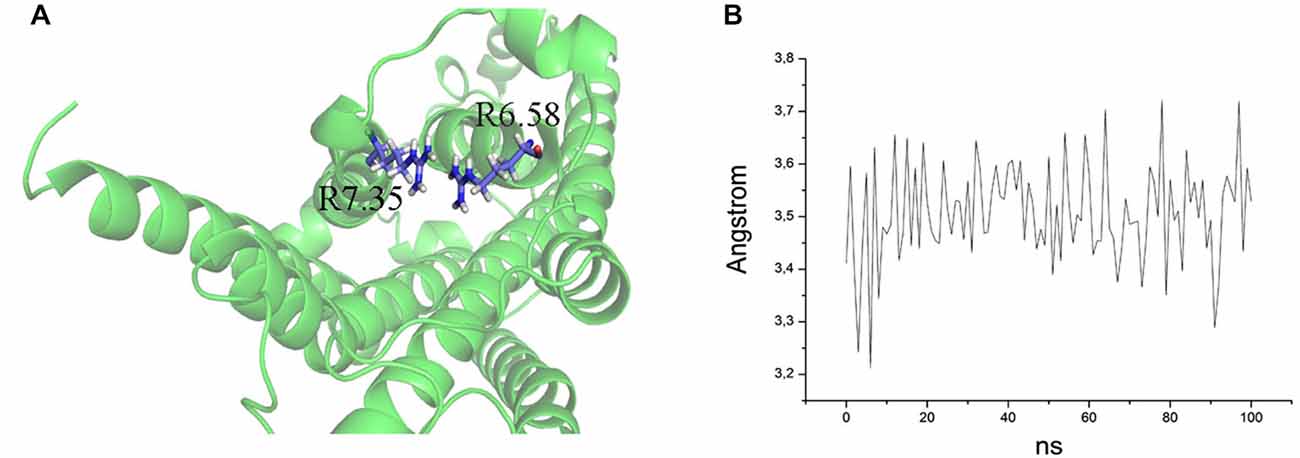
Figure 5. (A) Overlap of R7.35 and R6.58 in the mutant E7.35R. The backbone is shown in cartoon representation and the residues are indicated in stick format. (B) Distance between the two guanidine groups of R7.35 and R6.58 measured over the MD trajectory.
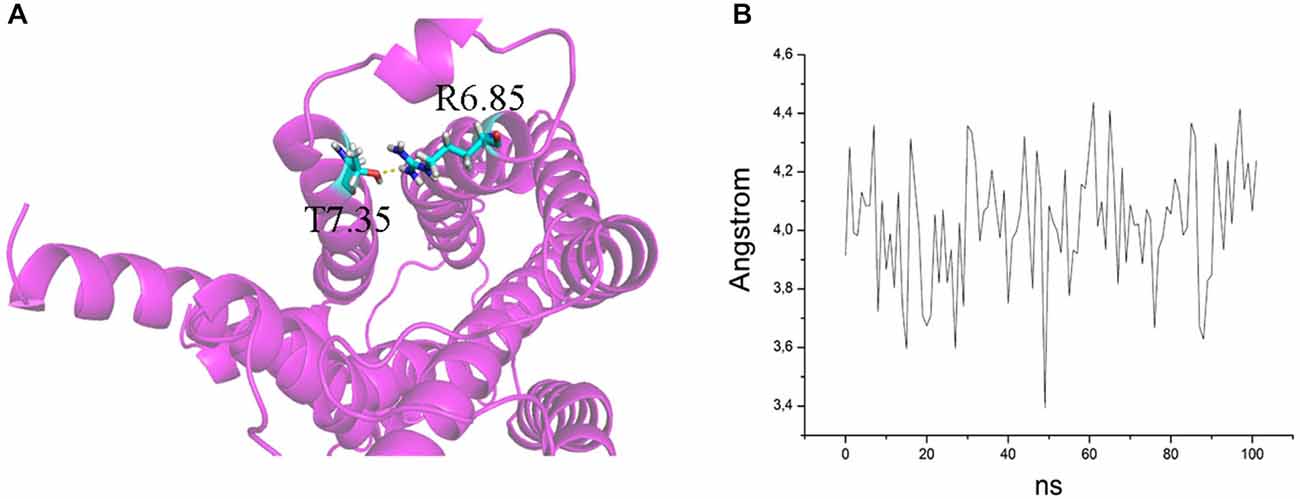
Figure 6. (A) H bond between T7.35 and R6.58 in the mutant E7.35T. The backbone is shown in cartoon representation and the residues are indicated in stick format. (B) Distance between the oxygen atom of T7.35 and hydrogen aminic atom of R6.58 measured over the MD trajectory.
The MD analysis showed that another residue was important in the interaction with the ligands: R7.36. The Arg was changed to Val (R7.36V) and the mutation reduced the affinities of agonist (5-CT), but not for antagonist (SB269970) radioligand (Table 4). But the mutant receptor displayed 5-CT-stimulated AC activity with essentially the same properties as the wild-type receptor (Table 5; Figures 4A–C). MD simulations indicated that the R7.36 is involved in a network of interactions. This residue formed an electrostatic interaction with D2.65, which is partially instable (0.8% of whole simulation), due to the formation, as said before, of a three-member salt bridge R7.36-E7.35-R6.58. Consequently, the extracellular part of TMH7 is constrained by two salt bridges to TMH2 and TMH6.
The release of this ionic lock could be a key step in receptor activation. In order to test this hypothesis, charge-neutralizing mutations of E5.67 and of D3.49 were made in the β2-adrenergic receptor (Ballesteros et al., 2001). Experimental data by Ballesteros et al. (2001), together with the high-resolution structure of rhodopsin (Palczewski et al., 2000) suggest that ionic interactions between D/E3.49, R3.50, and E6.30 may constitute a common switch governing the activation of many rhodopsin-like G-protein-coupled receptors. In addition, semi-flexible docking calculations were performed at the binding cavity defined by the residues from the 3rd, 5th, 6th, 7th helices and the ECL2, including an orthosteric pocket embedded in the 7TM core and a long binding pocket close to the extracellular site. The binding mode is in accordance with that recently reported for the crystal structure of the h5-HT1B G-protein-coupled receptor bound to ergotamine or dihydroergotamine, which are accommodated at the orthosteric pocket and an extended binding pocket close to the ECL2, respectively (Wang et al., 2013). The residues belonging to the architecture of this pocket are conserved in 5-HT receptors.
The movements of transmembrane segments (TMs) III and VI at the cytoplasmic side of the membrane play an important role in the activation of G-protein-coupled receptors. There is evidence for the existence of an ionic lock that constrains the relative mobility of the cytoplasmic ends of TM3 and TM6 in the inactive state of the β2-adrenergic receptor (Ballesteros et al., 2001). The highly conserved R3.50 at the cytoplasmic end of TM3 interacts both with the adjacent D3.49 and with E6.30 at the cytoplasmic end of TM6. Such a network of ionic interactions has now been directly supported by the high-resolution structure of the inactive state of the β2-adrenergic receptor (Dror et al., 2009) and would serve to constrain the receptor in the inactive state.
The R8.52D mutant receptor showed a different behavior: although the receptor displayed normal affinity for ligands and a significantly reduced expression (Table 4), the receptor was unable to activate adenylyl cyclase (Table 5). The MD data showed that this residue pointed toward the water during simulation; its position was at the base of helix VII and near the G protein binding domain. We examined the next residue D8.53 to better understand the importance of this region. We changed the Asp to Lys and found that the affinity of both agonist and antagonist ligands for the D8.53K mutant was similar to that observed for the wild-type receptor. A similar result was obtained in the adenylyl cyclase assays, where the D8.53K mutant receptor activated the enzyme with the same efficiency as the wild-type receptor. However, the D8.53K mutation was able to rescue the ability of the R8.52D-mutated receptor to activate AC, since both 5-CT and 5-HT were able to stimulate adenylyl cyclase activity through the double mutated R8.52D-D8.53K receptor with essentially unchanged potency (Table 5) and only a decrease in the efficacy (not shown) compared with the wild-type receptor. Thus the mutagenesis data on the R8.52 residue might be explained by the R8.52D mutation destroying the receptor-G protein interface, possibly by removing the positive charge.
To better understand which kind of interactions are important to bind the specific ligands, to activate the receptor and the downstream pathway, we analyzed the aromatic residues W7.33 and Y7.43. The W7.33 was replaced with Val and Y7.43 with Ala. The mutated receptor W7.33V didn’t change the affinities for 5-CT and SB269970 ligands and stimulated cAMP production with the same efficiency as the wild-type receptor.
The Y7.43A mutant showed a significantly reduced affinity of agonist binding, whereas the reduction in antagonist binding did not reach significance. Kołaczkowski et al. (2006) reported this amino acid to be involved in formation of hydrogen bond with ligands. The two mutants Y7.43F and Y7.43T were designed to assess if the aromatic or the OH-group is involved in the binding process. Based on preliminary data, only the mutant Y7.43T showed a reduction in the ability to bind both the ligands. No changes were observed for the mutant Y7.43F (Table 4). The data are generally in agreement with that for AC activation and antagonism (Table 5). These experimental data were explained by MD simulations, which pointed out that another highly stable salt bridge is formed between Y7.43 and D2.65 in the TMH2 of the 5-HT7 receptor.
The actions of ligands at receptors depend on the affinity for the receptor and the activation of a signaling system, termed efficacy, which is positive for agonists as a result of conformational changes. While some of the above mentioned mutations do not seem to affect ligand recognition, they may still prevent or reduce receptor activation and G protein coupling, illustrating independent contributions of these residues in the WT to stabilizing the bound ligands and/or formation of a ligand-induced active state of the receptor (Osaka et al., 1998; Strange, 2008).
Based on the above reported findings there seems to be a non-ionic lock between helix III and helix VI in the 5-HT7 receptor. However, when the salt-bridge between D3.49 and R3.50 is broken a new one is formed between R3.50 and S2.39. The active and inactive states of the 5-HT7 receptor are characterized by D3.49-R3.50 and R3.50 and S2.39 salt bridges, respectively. The mutant R8.52D in helix VIII lacks cyclase activation. This residue points toward the water during simulation; its position is at the base of helix VII and near the G protein binding domain. Thus, the mutagenesis data on this residue might be explained by destruction of the receptor-G protein interface, and we can assume that the charge is important.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by The Norwegian Council on Cardiovascular Disease, The Research Council of Norway, The Kristian Gerhard Jebsen Foundation, Anders Jahre’s Foundation for the Promotion of Science, The Family Blix Foundation, South-Eastern Norway Regional Health Authority, University of Oslo and the COST Action CM1207 (GLISTEN).3 All calculations were granted by project GPCR-4D from PRACE Distributed European Computing Initiative (DECI-9) and project HP10BKEPYB from Italian Super Computing Resource Allocation (ISCRA). Prof. Ingebrigt Sylte, Department of Medical Biology, Faculty of Health Sciences, UiT, The Arctic University of Norway, NO-9037 Tromsø, Norway is gratefully acknowledged for the homology model of the h5-HT7, the helpful discussions and suggestions.
Abagyan, R., and Totrov, M. (1994). Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J. Mol. Biol. 235, 983–1002. doi: 10.1006/jmbi.1994.1052
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ballesteros, J. A., Jensen, A. D., Liapakis, G., Rasmussen, S. G., Shi, L., Gether, U., et al. (2001). Activation of the β2-adrenergic receptor involves disruption of an ionic lock between the cytoplasmic ends of transmembrane segments 3 and 6. J. Biol. Chem. 276, 29171–29177. doi: 10.1074/jbc.m103747200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ballesteros, J. A., and Weinstein, H. (1995). Integrated methods for modeling G-protein coupled receptors. Methods Neurosci. 25, 366–428. doi: 10.1016/S1043-9471(05)80049-7
Berendsen, H. J. C., Postma, J. P. L., van Gunsteren, W. F., DiNola, A. R. H. J., and Haak, J. R. (1984). Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690. doi: 10.1063/1.448118
Boess, F. G., Monsma, F. J. Jr., and Sleight, A. J. (1998). Identification of residues in transmembrane regions III and VI that contribute to the ligand binding site of the serotonin 5-HT6 receptor. J. Neurochem. 71, 2169–2177. doi: 10.1046/j.1471-4159.1998.71052169.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cheng, Y., and Prusoff, W. H. (1973). Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22, 3099–3108. doi: 10.1016/0006-2952(73)90196-2
Cherezov, V., Rosenbaum, D. M., Hanson, M. A., Rasmussen, S. G., Thian, F. S., Kobilka, T. S., et al. (2007). High-resolution crystal structure of an engineered human β2-adrenergic G protein-coupled receptor. Science 318, 1258–1265. doi: 10.1126/science.1150577
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dror, R. O., Arlow, D. H., Borhani, D. W., Jensen, M. Ø., Piana, S., and Shaw, D. E. (2009). Identification of two distinct inactive conformations of the β2-adrenergic receptor reconciles structural and biochemical observations. Proc. Natl. Acad. Sci. U S A 106, 4689–4694. doi: 10.1073/pnas.0811065106
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Essmann, U., Perera, L., Berkowitz, M. L., Darden, T., Lee, H., and Pedersen, L. G. (1995). A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593. doi: 10.1063/1.470117
Gasbarri, A., and Pompili, A. (2014). Serotonergic 5-HT7 receptors and cognition. Rev. Neurosci. 25, 311–323. doi: 10.1515/revneuro-2013-0066
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gellynck, E., Heyninck, K., Andressen, K. W., Haegeman, G., Levy, F. O., Vanhoenacker, P., et al. (2013). The serotonin 5-HT7 receptors: two decades of research. Exp. Brain Res. 230, 555–568. doi: 10.1007/s00221-013-3694-y
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Guseva, D., Holst, K., Kaune, B., Meier, M., Keubler, L., Glage, S., et al. (2014). Serotonin 5-HT7 receptor is critically involved in acute and chronic inflammation of the gastrointestinal tract. Inflamm. Bowel Dis. 20, 1516–1529. doi: 10.1097/MIB.0000000000000150
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ho, B. Y., Karschin, A., Branchek, T., Davidson, N., and Lester, H. A. (1992). The role of conserved aspartate and serine residues in ligand binding and in function of the 5-HT1A receptor: a site-directed mutation study. FEBS Lett. 312, 259–262. doi: 10.1016/0014-5793(92)80948-g
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Humphrey, W., Dalke, A., and Schulten, K. (1996). VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38. doi: 10.1016/0263-7855(96)00018-5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W., and Klein, M. T. (1983). Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935. doi: 10.1063/1.445869
Kołaczkowski, M., Nowak, M., Pawłowski, M., and Bojarski, A. J. (2006). Receptor-based pharmacophores for serotonin 5-HT7R antagonists-implications to selectivity. J. Med. Chem. 49, 6732–6741. doi: 10.1021/jm060300c
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Krobert, K. A., Bach, T., Syversveen, T., Kvingedal, A. M., and Levy, F. O. (2001). The cloned human 5-HT7 receptor splice variants: a comparative characterization of their pharmacology, function and distribution. Naunyn Schmiedebergs Arch. Pharmacol. 363, 620–632. doi: 10.1007/s002100000369
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Meneses, A. (2014). Memory formation and memory alterations: 5-HT6 and 5-HT7 receptors, novel alternative. Rev. Neurosci. 25, 325–356. doi: 10.1515/revneuro-2014-0001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mialet, J., Dahmoune, Y., Lezoualc’h, F., Berque-Bestel, I., Eftekhari, P., Hoebeke, J., et al. (2000). Exploration of the ligand binding site of the human 5-HT4 receptor by site-directed mutagenesis and molecular modeling. Br. J. Pharmacol. 130, 527–538. doi: 10.1038/sj.bjp.0703356
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Norum, J. H., Méthi, T., Mattingly, R. R., and Levy, F. O. (2005). Endogenous expression and PKA-dependent phosphorylation of the guanine nucleotide exchange factor Ras-GRF1 in HEK293 cells. FEBS J. 272, 2304–2316. doi: 10.1111/j.1742-4658.2005.04658.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Osaka, H., Sugiyama, N., and Taylor, P. (1998). Distinctions in agonist and antagonist specificity conferred by anionic residues of the nicotinic acetylcholine receptor. J. Biol. Chem. 273, 12758–12765. doi: 10.1074/jbc.273.21.12758
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Overington, J. P., Al-Lazikani, B., and Hopkins, A. L. (2006). How many drug targets are there? Nat. Rev. Drug Discov. 5, 993–996. doi: 10.1038/nrd2199
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Palczewski, K., Kumasaka, T., Hori, T., Behnke, C. A., Motoshima, H., Fox, B. A., et al. (2000). Crystal structure of rhodopsin: a G protein-coupled receptor. Science 289, 739–745. doi: 10.1126/science.289.5480.739
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Phillips, J. C., Braun, R., Wang, W., Gumbart, J., Tajkhorshid, E., Villa, E., et al. (2005). Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802. doi: 10.1002/jcc.20289
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Prins, N. H., Briejer, M. R., Van Bergen, P. J., Akkermans, L. M., and Schuurkes, J. A. (1999). Evidence for 5-HT7 receptors mediating relaxation of human colonic circular smooth muscle. Br. J. Pharmacol. 128, 849–852. doi: 10.1038/sj.bjp.0702762
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ryckaert, J. P., Ciccotti, G., and Berendsen, H. J. C. (1977). Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comp. Phys. 23, 327–341. doi: 10.1016/0021-9991(77)90098-5
Sandal, M., Duy, T. P., Cona, M., Zung, H., Carloni, P., Musiani, F., et al. (2013). GOMoDo: a GPCRs online modeling and docking webserver. PLoS One 8:e74092. doi: 10.1371/journal.pone.0074092
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Strange, P. G. (2008). Agonist binding, agonist affinity and agonist efficacy at G protein-coupled receptors. Br. J. Pharmacol. 153, 1353–1363. doi: 10.1038/sj.bjp.0707672
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Thomsen, R., and Christensen, M. H. (2006). MolDock: a new technique for high-accuracy molecular docking. J. Med. Chem. 49, 3315–3321. doi: 10.1021/jm051197e
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ullmer, C., Schmuck, K., Kalkman, H. O., and Lübbert, H. (1995). Expression of serotonin receptor mRNAs in blood vessels. FEBS Lett. 370, 215–221. doi: 10.1016/0014-5793(95)00828-w
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wacker, D., Wang, C., Katritch, V., Han, G. W., Huang, X. P., Vardy, E., et al. (2013). Structural features for functional selectivity at serotonin receptors. Science 340, 615–619. doi: 10.1126/science.1232808
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, C. D., Gallaher, T. K., and Shih, J. C. (1993). Site-directed mutagenesis of the serotonin 5-hydroxytrypamine2 receptor: identification of amino acids necessary for ligand binding and receptor activation. Mol. Pharmacol. 43, 931–940.
Wang, C., Jiang, Y., Ma, J., Wu, H., Wacker, D., Katritch, V., et al. (2013). Structural basis for molecular recognition at serotonin receptors. Science 340, 610–614. doi: 10.1126/science.1232807
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: homology modeling, mutagenesis, molecular dynamics, docking, G protein, adenylyl cyclase
Citation: Impellizzeri AAR, Pappalardo M, Basile L, Manfra O, Andressen KW, Krobert KA, Messina A, Levy FO and Guccione S (2015) Identification of essential residues for binding and activation in the human 5-HT7(a) serotonin receptor by molecular modeling and site-directed mutagenesis. Front. Behav. Neurosci. 9:92. doi: 10.3389/fnbeh.2015.00092
Received: 30 January 2015; Accepted: 31 March 2015;
Published online: 08 May 2015.
Edited by:
Walter Adriani, Istituto Superiore di Sanità, ItalyReviewed by:
Tetsuo Yamamori, National Institue for Basic Biology, JapanCopyright © 2015 Impellizzeri, Pappalardo, Basile, Manfra, Andressen, Krobert, Messina, Levy and Guccione. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Finn Olav Levy, Department of Pharmacology, Institute of Clinical Medicine, University of Oslo and Oslo University Hospital, Sognsvannsveien 20, P.O.Box 1057 Blindern, N-0316 Oslo, Norway,Zi5vLmxldnlAbWVkaXNpbi51aW8ubm8=;
Salvatore Guccione, Department of Drug Sciences, University of Catania, V.le A. Doria 6 Ed. 2, Città Universitaria, I-95125 Catania, Italy,Z3VjY2lvbmVAdW5pY3QuaXQ=
† Shared senior authors.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.