


94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
GENERAL COMMENTARY article
Front. Behav. Neurosci. , 18 August 2011
Sec. Learning and Memory
volume 5 - 2011 | https://doi.org/10.3389/fnbeh.2011.00051
A commentary on:
On the temporary nature of disruption of fear-potentiated startle following PKMζ inhibition in the amygdale
by Nader, K. (2011). Front. Behav. Neurosci. 5:29. doi: 10.3389/fnbeh.2011.00029
Recently we published data showing that under some circumstances the effect of protein kinase M zeta (PKMζ) inhibition in the amygdala on the expression of fear-potentiated startle (FPS) is a function of when the drug was applied relative to when testing occurred (Parsons and Davis, 2011). We showed in three separate experiments that if memory is tested anywhere from 10 to 15 days after infusion of the PKMζ inhibitor zeta-pseudosubstrate inhibitory peptide (ZIP) there was no evidence of memory impairment. However, there was a reliable impairment if testing took place a few hours or a few days after infusion of ZIP. A commentary was published by Nader (2011) critiquing several aspects of our findings and the conclusions drawn. Here, we respond to the criticism.
The medial and cortical nuclei of the amygdala receive dense olfactory input from both the olfactory bulb and piriform cortex, but there is also olfactory input to lateral/basolateral amygdala from the piriform and perirhinal cortices (reviewed in Otto et al., 2000). Several studies have argued that it is the BLA, and not the medial/cortical amygdala, that is the critical site of amygdala plasticity underlying odor-signaled fear learning. First, lesions of the BLA block olfactory fear conditioning (Cousens and Otto, 1998), as do lesions of the perirhinal cortex (Herzog and Otto, 1998) which provide the olfactory input into the BLA. Work from our lab found similar results in that an NMDA antagonist applied to the BLA blocked the acquisition of olfactory fear learning, but had no effect when infused into the medial nuclei (Walker et al., 2005). These results are consistent with prior studies showing that NMDA antagonists delivered to the BLA block the acquisition of fear memory whether the shock is signaled by a light (Miserendino et al., 1990) or noise (Campeau et al., 1992). Finally, olfactory fear conditioning has been shown to activate the mRNA for BDNF in the BLA, but not the medial nuclei (Rattiner et al., 2004) which is also the case when a visual cue is used to signal shock (Rattiner et al., 2004). Thus, just as is the case when visual or auditory cues are used to signal shock, olfactory fear conditioning depends on plasticity within the BLA. Therefore, the results of our experiments studying the role of PKMζ on olfactory fear memory are very unlikely to be unique to the modality of the conditioned stimulus.
The issue of whether we used a dose of ZIP (10 nmol/μL, 0.5 μl/side) that was inadequate to permanently disrupt fear memory is one that we think will ultimately need to be answered with data rather than speculation. Nonetheless, it is an important issue to consider. Dr. Nader states that “The dose used by the authors was four times lower that used in 9 of 10 studies on the topic.” We are puzzled as to where the “9 of 10 studies” claim comes from. At the time our manuscript was accepted into publication only one paper (Migues et al., 2010) had used a dose four times higher than what we used. There were two other papers employing amygdala infusions of ZIP, one of those used the same dose we did (Kwapis et al., 2009) and the other (Serrano et al., 2008) used the same concentration, but twice the volume (i.e., twice the dose). We have no reason to believe that the concentration of ZIP we used was inadequate because it has been used successfully to disrupt memory in many other tasks (e.g., Pastalkova et al., 2006) and we replicated the work in several studies using this dose when testing occurred 2 h or a few days after infusion of ZIP (Serrano et al., 2008; Kwapis et al., 2009).
We believe the same argument applies to the potential impact of the volume of infusion. We infused 0.5 μL/hemisphere of ZIP, which we would estimate is the volume used in a vast majority of published works employing microinfusions into the rat amygdala. Other studies have used higher volumes of ZIP when infusions are made into a larger area like the dorsal hippocampus (e.g., Pastalkova et al., 2006). To contend that our dose was inadequate by contrasting it to experiments where the same concentration of ZIP is being infused into a large structure is not an appropriate comparison. Finally, it should also be pointed out that we directly replicated (our Figure 3B) the findings of the previous paper (Migues et al., 2010) that ZIP interfered with memory recall when tested for a second time at a 10-day interval, using one-fourth of the dose. If our dose was inadequate, then why does ZIP block memory expression during the 10-day test under some circumstances?
In his commentary, Dr. Nader selects out our Figure 3B and makes the conclusion that “the vast majority of observed changes in behavior are due to reduced performance of the control groups (forgetting), as opposed to increased performance by the ZIP treated groups.” Performance during the 10-day test was lower, yet the ZIP treated rats tested only at 10 days still showed over twice as much potentiation compared the animals tested 1 day after infusion (48 vs. 19%). We would also like to point to our Figure 2B where the levels of potentiation in the control group tested at 15 days differed very little from the 2-day group and the same pattern of data was observed, with ZIP rats tested at 15 days showing nearly twice as much fear as those tested at 2 days (46 vs. 26%). In fact, an ANOVA on these data using test interval and drug as between-subjects factors found there was no effect of test time but a significant main effect of drug (F = 5.27, p < 0.05) and, most germane to our argument, a significant interaction (F = 6.17, p < 0.05). This indicates that the effect of ZIP on memory was quite large during the test shortly after infusion, but when memory was tested at later time points there was absolutely no difference.
Another factor important to consider is that the percent potentiation value is a relative measure. In our paper, the percent FPS values reported were computed by comparing the amplitude of startle in the presence of the odor vs. to the startle stimuli that occur between each odor presentation. However, what we often see when testing occurs at long intervals is that startle levels on the startle alone trials increase once the conditioned stimulus comes on (e.g., Walker and Davis, 2002), and consequently those percentages are lower. An alternative way to assess the results is to compute the percent potentiation relative to the startle stimuli presented during the test session just before the odor is first presented (what we call “leaders”). We have done this for the three experiments where we tested long-term and those data are depicted in Figure 1. The ZIP groups are all showing greater than 100% potentiation, except for those rats tested 1 or 2 days later. And, once again we find the critical significant interaction when computed in this way when we consider the 15 and 10-day results (F = 9.67, p < 0.01), showing again the effect of ZIP on memory depended on the time after infusion memory was measured. Thus, we think the data indicates very little weakening of the memory over time.
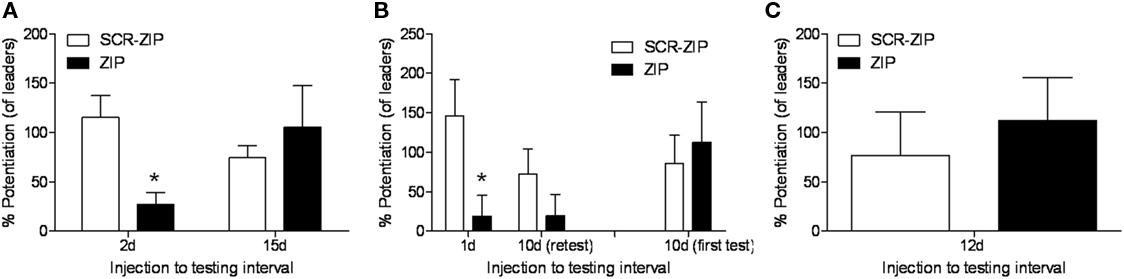
Figure 1. Percent fear-potentiated startle values relative to the baseline startle stimuli (“leaders”) for the three experiments (Parsons and Davis, 2011) where memory was tested long after infusion of ZIP into the BLA. ZIP disrupted the expression of memory when tested 2 (A) or 1 days (B) after infusion, but not at 10 (B), 12 (C), or 15 days (A).
Dr. Nader states that in our Figure S2 that the “SE of their control group is so large that it is unclear if any impairment could be found significantly different statistically.” It is certainly the case that the SE is higher in this experiment, and it is possible that for this single experiment it would be difficult show a significant disruption in memory. However, we would argue that the group means in this experiment and the two other similar ones (our Figures 2b, and 3b), paint a very clear picture. In no case was there ever a hint of impairment in ZIP treated rats, so it made little sense to increase the power and lower the SE by doubling or tripling the number of subjects in these experiments.
Dr. Nader asserts that “the impairment induced by ZIP could be partial erasure of the memory” and that “increases in performance might be mediated by incubation of that memory (increases in responding over time), new learning adding onto the residual memory.” While these are important considerations, we believe they are unlikely to be a factor in our study because there is little evidence of incubation over time in our experiments and a reminder shock does not lead to any recovery in ZIP treated rats which were impaired 2 days after infusion (our supplementary Figure 1). If some residual trace existed in ZIP treated rats, the reminder shock should have lead to recovery in those animals (Squire, 2006).
The experiments in our paper were not designed to provide a definitive answer as to the nature of the performance deficit after PKMζ inhibition. Instead they were designed to simply test if those deficits are observed long after infusion of ZIP into the amygdala. Too many of the prior published experiments (e.g., Serrano et al., 2008) infused ZIP, tested memory a few hours and/or a few days later, and concluded that the drug had erased memory. Some of the published studies tested longer after ZIP infusion, but in some cases the deficits were weaker and ZIP was applied to a different brain area (Shema et al., 2007; their Figure 1A), alternative behavioral metrics were used (Pastalkova et al., 2006; their Figure 3C), or the animals were tested repeatedly (Migues et al., 2010; their Figure 1). Thus, we felt that the evidence that PKMζ supported the permanent storage of memory in the amygdala was weak and incomplete, and thus necessitated further testing. It could very well be that this kinase may be involved in memory maintenance in other brain areas (e.g., insular cortex), in fact the data from studies of taste aversion are compelling (e.g., Shema et al., 2007, 2011).
Finally, Dr. Nader argues that “the authors used a task that that (sic) cannot differentiate between storage and retrieval impairment views of recovery from amnesia.” Let us assume this is the case. Then most, if not all, of the dozen or so tasks used in papers where ZIP disrupted memory after infusion into the amygdala or other brain areas must all suffer this same drawback. Yet, nearly all of these papers came to the conclusion that the ZIP-induced deficits were indicative of an impairment of memory storage. If we want to conclude that the paradigms used so far to study the effects of ZIP are inadequate, then what have we learned about the role of PKMζ in memory?
Campeau, S., Miserendino, M. J., and Davis, M. (1992). Intra-amygdala infusion of the N-methyl-D-aspartate receptor antagonist AP5 blocks acquisition but not expression of fear-potentiated startle to an auditory conditioned stimulus. Behav. Neurosci. 106, 569–574.
Cousens, G., and Otto, T. (1998). Both pre- and posttraining excitotoxic lesions of the basolateral amygdala abolish the expression of olfactory and contextual fear conditioning. Behav. Neurosci. 112, 1092–1103.
Herzog, C., and Otto, T. (1998). Contributions of anterior perirhinal cortex to olfactory and contextual fear conditioning. Neuroreport 9, 1855–1859.
Kwapis, J. L., Jarome, T. J., Lonergan, M. E., and Helmstetter, F. J. (2009). Protein kinase Mzeta maintains fear memory in the amygdala but not in the hippocampus. Behav. Neurosci. 123, 844–850.
Migues, P. V., Hardt, O., Wu, D. C., Gamache, K., Sacktor, T. C., Wang, Y. T., and Nader, K. (2010). PKMzeta maintains memories by regulating GluR2-dependent AMPA receptor trafficking. Nat. Neurosci. 13, 630–634.
Miserendino, M. J., Sananes, C. B., Melia, K. R., and Davis, M. (1990). Blocking of acquisition but not expression of conditioned fear-potentiated startle by NMDA antagonists in the amygdala. Nature 345, 716–718.
Nader, K. (2011). On the temporary nature of disruption of fear-potentiated startle following PKM zeta inhibition in the amygdale. Front. Behav. Neurosci. 5:29. doi: 10.3389/fnbeh.2011.00029
Otto, T., Cousens, G., and Herzog, C. (2000). Behavioral and neuropsychological foundations of olfactory fear conditioning. Behav. Brain Res. 110, 119–128.
Parsons, R. G., and Davis, M. (2011). Temporary disruption of fear-potentiated startle following PKMzeta inhibition in the amygdala. Nat. Neurosci. 14, 295–296.
Pastalkova, E., Serrano, P., Pinkhasova, D., Wallace, E., Fenton, A. A., and Sacktor, T. C. (2006). Storage of spatial information by the maintenance mechanism of LTP. Science 313, 1141–1144.
Rattiner, L. M., Davis, M., French, C. T., and Ressler, K. J. (2004). Brain-derived neurotrophic factor and tyrosine kinase receptor B involvement in amygdala-dependent fear conditioning. J. Neurosci. 24, 4796–4806.
Serrano, P., Friedman, E. L., Kenney, J., Taubenfeld, S. M., Zimmerman, J. M., Hanna, J., Alberini, C., Kelley, A. E., Maren, S., Rudy, J. W., Yin, J. C., Sacktor, T. C., and Fenton, A. A. (2008). PKMzeta maintains spatial, instrumental, and classically conditioned long-term memories. PLoS Biol. 6, 2698–2706. doi: 10.1371/journal.pbio.0060318
Shema, R., Haramati, S., Ron, S., Hazvi, S., Chen, A., Sacktor, T. C., and Dudai, Y. (2011). Enhancement of consolidated long-term memory by overexpression of protein kinase Mzeta in the neocortex. Science 331, 1207–1210.
Shema, R., Sacktor, T. C., and Dudai, Y. (2007). Rapid erasure of long-term memory associations in the cortex by an inhibitor of PKM zeta. Science 317, 951–953.
Squire, L. R. (2006). Lost forever or temporarily misplaced? The long debate about the nature of memory impairment. Learn. Mem. 13, 522–529.
Walker, D. L., and Davis, M. (2002). The role of amygdala glutamate receptors in fear learning, fear-potentiated startle, and extinction. Pharmacol. Biochem. Behav. 71, 379–392.
Citation: Parsons RG and Davis M (2011) Gone but not forgotten. Front. Behav. Neurosci. 5:51. doi: 10.3389/fnbeh.2011.00051
Received: 10 July 2011; Accepted: 01 August 2011;
Published online: 18 August 2011.
Copyright: © 2011 Parsons and Davis. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence:cnBhcnNvM0BlbW9yeS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.