 Gus R. McFarlane*
Gus R. McFarlane* Katie L. Robinson
Katie L. Robinson Kim Whitaker
Kim Whitaker John WebsterLauren DrysdaleLillian Brancalion
John WebsterLauren DrysdaleLillian Brancalion Ashlea Webster
Ashlea Webster Brendon O’Rourke
Brendon O’Rourke Daniel R. Bogema
Daniel R. Bogema- NSW Department of Primary Industries, Elizabeth Macarthur Agricultural Institute, Menangle, NSW, Australia
Varroa destructor is a major pest of European honey bees (Apis mellifera), causing significant economic and welfare impacts. Australia remained the last continent free from V. destructor. In June 2022, a detection of V. destructor was reported in sentinel colonies at the Port of Newcastle, Australia. Rapid and accurate identification of the species was critical for timely response and management. In this case study, two Nanopore DNA sequencing methods, PCR amplicon sequencing and Cas9-targeted sequencing, were used to rapidly diagnose the species and mitochondrial haplotype of Varroa mites in parasitized colonies. Nanopore PCR amplicon sequencing provided molecular identification of the species and halogroup determination within 24 hours based on a 458 bp amplicon of the mitochondrial Cytochrome c oxidase subunit I (COXI) gene. We also developed and applied a Cas9-targeted Nanopore sequencing technique that used eight guide RNAs to enrich for 5240 bp of the mitochondrial genome. This method delivered richer data for identification within the same timeframe. Our results underscore the efficacy of Nanopore amplicon sequencing and represent the first reported application of Cas9-targeted Nanopore sequencing within a biosecurity framework. These findings enhance the repertoire of diagnostic tools available for biosecurity applications.
Introduction
Varroa destructor is an ectoparasite of European honey bee (Apis mellifera) and stands as the most severe pest for honey bees worldwide (Figure 1A). The mite inflicts severe damage to colonies and has a profound impact on beekeeping and pollination activities around the globe (Traynor et al., 2020). Until recently, Australia remained the last Varroa-free continent, excluding Antarctica (Roberts et al., 2017; Traynor et al., 2020). In June 2022, biosecurity surveillance in the Australian state of New South Wales (NSW) detected Varroa mites in government-managed sentinel colonies at the Port of Newcastle. Given the existence of four Varroa species that parasitise honey bees, rapid genetic identification of the species was the immediate goal for the NSW Government’s Department of Primary Industries (NSW DPI) (Roberts et al., 2015). In this case study, we highlight an early component of a broad response effort: our application of amplicon and Cas9-targeted Nanopore sequencing to rapidly and accurately identify the Varroa species and mitochondrial haplotype at the onset of the outbreak.

Figure 1 Varroa destructor in Australia. (A) Varroa destructor parasitising a bee pupa. (B) In June 2022, a detection at the Port of Newcastle was reported, and several other parasitized colonies were later found in the that month in the surrounding regions. (C) Within this study, thirteen colonies were analysed from five different locations. All locations are from a 20km radius around the Port of Newcastle. Exact locations have been anonymised. Ten colonies were analysed by Nanopore amplicon sequencing of the COXI gene, and three were analysed by Cas9-targeted Nanopore sequencing of the Varroa destructor mitochondrial genome.
Species identification for Varroa mites is typically based on PCR amplification of 458 bp of the mitochondrial Cytochrome c oxidase subunit I (COXI) gene, followed by Sanger sequencing of the resultant PCR amplicon (Dietemann et al., 2013; Lin et al., 2018). This conventional method requires at least 48 hours to yield results, thus delaying responsive actions. In contrast, Nanopore sequencing facilitates rapid, real-time, and portable DNA sequencing (Parker et al., 2017; Wang et al., 2021). This technology has gained widespread acceptance in multiple disciplines, including epidemiology and forensic science, and is emerging as a powerful biosecurity diagnostic tool (Yang et al., 2014; Hall et al., 2020; Abeynayake et al., 2021). At the outbreak’s onset, we rapidly tested the initial sample from the Port of Newcastle, followed by nine subsequent parasitized colonies from surrounding areas, using PCR amplification of COXI and Nanopore amplicon sequencing (Figures 1B, C). This approach enabled us to determine species identification and mitochondrial haplotype.
Researchers have identified at least six mitochondrial haplotypes of V. destructor based on the 458 bp COXI amplicon, each with a unique set of characteristics. Varroa destructor mitochondrial haplotypes, referred to herein as haplogroups, are named after the first country they are identified in and include Sri Lanka, Nepal, China, Vietnam, Japan/Thailand and Korea haplogroups (Li et al., 2019). Only two of them, however, the Korea and Japan/Thailand haplogroup, have been found to infest European honey bees (Traynor et al., 2020). The Korean haplogroup is the most widespread and problematic. Not only is this haplogroup found in numerous regions across the globe, but it also presents formidable challenges to control measures. These challenges are multifaceted; they include a heightened tolerance to commonly used miticides, remarkable adaptability to a range of climatic conditions, and an aggressive reproductive behaviour that enhances its capability to colonise new habitats rapidly (Bahreini et al., 2020; Giliba et al., 2020; Traynor et al., 2020). Understanding these haplogroups and their respective traits assists in formulating a targeted biosecurity control effort.
Sequencing of the 458 bp COXI amplicon yields valuable data for species identification and haplogroup determination. Varroa destructor are, however, notorious for their low levels of genetic diversity within invasion populations, making the small COXI amplicon uninformative for outbreak tracing. The limited genetic diversity within the species is largely attributed to genetic bottleneck events, haplodiploid sex determination, and the mite’s reproductive behaviour, known as arrhenotokous parthenogenesis (Rosenkranz et al., 2010; Dynes et al., 2017). This specialised form of reproduction allows females to produce male offspring from unfertilised eggs and female offspring from fertilised eggs. During the reproductive cycle, an adult female mite enters a bee brood cell and lays several eggs. Generally, a single male is born first and fertilises his subsequent sisters within the same cell (Rosenkranz et al., 2010). This reproductive strategy commonly leads to considerable inbreeding, which, in turn, limits genetic diversity (Traynor et al., 2020). As a result, the utility of the 458 bp COXI amplicon that is routinely used for species identification and haplogroup determination is not informative for detailed epidemiological investigation.
To enhance NSW DPI’s genetic tracing capabilities for incoming V. destructor samples and other potential exotic insect incursions, we sought to develop a rapid DNA sequencing method that provides more genetic data. To achieve this, we turned to Cas9-targeted Nanopore sequencing, which has proven successful in enriching for DNA targets in biomedical research but has yet to be applied in a biosecurity context (Gilpatrick et al., 2020; McDonald et al., 2021; Rubben et al., 2022). We developed a Cas9-targeting method capable of enriching for 5420 bp of the V. destructor mitochondrial genome.
The Cas9-targeted library preparation incorporates a two-step process to facilitate targeted sequencing. DNA termini undergo dephosphorylation in the initial phase, rendering them unresponsive to adapter ligation. In the subsequent phase, Cas9 nuclease is used to cleave the targeted DNA fragments at both ends of the specified region of interest. These newly cleaved DNA segments generate phosphorylated termini, which are conducive to adapter ligation, thus facilitating preferential sequencing of these fragments (Quan et al., 2019; Gilpatrick et al., 2020).
Our Cas9-targeted approach utilised a set of eight guide RNAs (gRNAs), each engineered to direct the Cas9 nuclease to a unique 20 nucleotide (nt) sequence within the V. destructor mitochondrial genome. This 20 nt sequence, known as the CRISPR protospacer, must be immediately followed at the 3’ end by a protospacer-adjacent motif (PAM) sequence of 5’-NGG for the Cas9-mediated double-stranded DNA break to occur at the target site. The cleavage typically happens 3 bp upstream of the PAM site, and is most often described as a blunt-end double-stranded DNA break (Jinek et al., 2012; McFarlane et al., 2023). We hypothesised that any residual Cas9 enzyme remaining bound to the DNA post-cleavage could be removed through a combined treatment of Proteinase K and heating at 80°C to denature the enzyme.
We applied this Cas9-targeted Nanopore sequencing technique to V. destructor samples obtained from three parasitized colonies located in geographically distinct areas. Both Nanopore amplicon sequencing of COXI and Cas9-targeted mitochondrial sequencing proved effective for species identification, haplogroup determination, and the creation of informative phylogenetic trees. The methodologies applied in this case study enrich the biosecurity diagnostic toolkit and hold particular significance for situations where a rapid outcome is critical to containing and mitigating biosecurity risks.
Materials and methods
DNA extraction
Samples from parasitized colonies from within a 20 km radius around the Port of Newcastle were collected after destroying colonies using petrol vapour. Bees with Varroa mites were preserved in 100% ethanol. In the laboratory, the mites were detached and air-dried to evaporate any residual ethanol. One mite per colony was utilised for Nanopore amplicon sequencing, and four per colony for Cas9-targeted sequencing. DNA was extracted using a DNeasy Blood & Tissue Kit (Qiagen), in accordance with the manufacturer’s guidelines for tissue samples.
PCR amplification of the mitochondrial COXI gene
A 458 bp region of the mitochondrial COXI gene was amplified using primers CoxF and CoxRa (Figure 2) (Anderson and Fuchs, 1998). The PCR reaction mixture contained 1 ul of gDNA, 1 x ImmunoBuffer, 1 mM dNTPs, 1.5 mM MgCl2, 0.4 µM each primer and 5U Immolase DNA polymerase (Meridian Bioscience) in a total volume of 50 µl. Cycle conditions were 95˚C for 10 min; 35 cycles of 95˚C for 30 sec, 45˚C for 30 sec, 72˚C for 30 sec; 72˚C for 5 min. PCR amplicons were purified using AMPure XP magnetic beads (Beckman Coulter) and 1.8X volume and eluted in 22.5 ul of Milli-Q water.
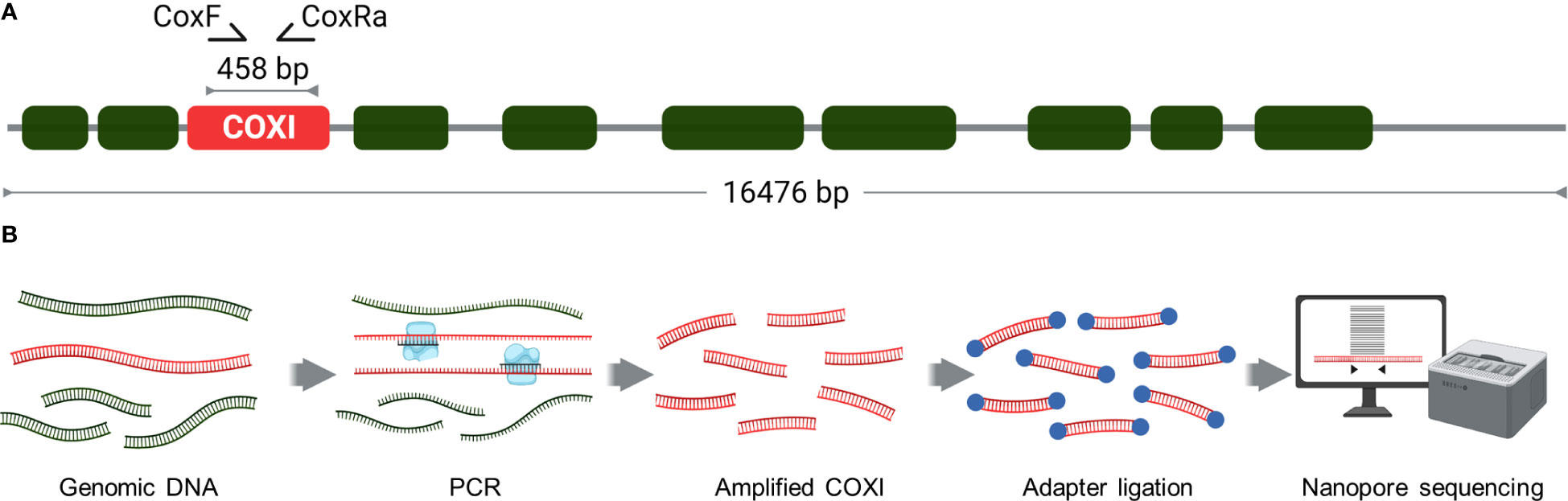
Figure 2 Nanopore amplicon sequencing of Varroa destructor mitochondrial COXI gene. (A) COXI gene was amplified from the mitochondrial genome using CoxF and CoxRa primers to generate a 458 bp amplicon. (B) Schematic of the workflow for Nanopore amplicon sequencing of the Varroa destructor COXI gene. The process involved PCR and amplicon purification, adapter ligation and barcoding before subsequent sequencing and data analysis. This can be completed within 24 hours, inclusive of DNA extraction.
Cas9-targeting of the mitochondrial genome
CRISPR RNAs (crRNAs) were designed using Geneious Prime’s CRISPR sites tool (Dotmatics) to fragment the V. destructor mitochondrial reference genome (NC_004454) into eight fragments, ranging in size from 1601 to 2490 bp fragments (Figure 3A; Supplementary 1 for crRNA sequences.). The guide RNAs (gRNAs) were assembled as duplexes from custom synthetic crRNAs (Integrated DNA Technologies; IDT) and universal trans-activating crRNAs (tracrRNAs; IDT), following the manufacturer’s guidelines to generate a 10 μM solution. Ribonucleoprotein (RNP) was formed by combining 10 pmol of gRNA duplex with 10 pmol of HiFi Cas9 Nuclease V3 (IDT) in 1X CutSmart Buffer (New England Biolabs, NEB) to produce a final volume of 30 μl at 333 nM.
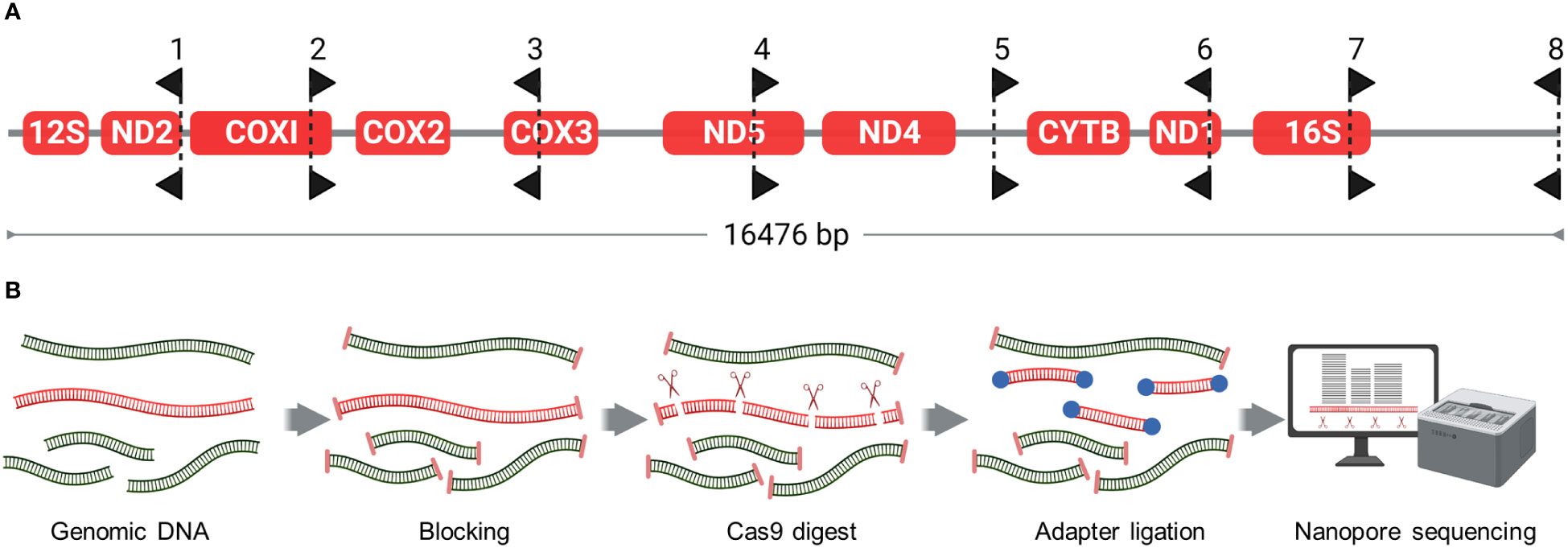
Figure 3 Cas9-targeted Nanopore sequencing (A) Placement and directionality of Cas9 gRNAs. gRNAs were designed to fragment the Varroa destructor mitochondrial genome into eight fragments. Black arrow-shaped markers point away from Protospacer Adjacent Motif (PAM) sequence, such that the PAM sequence of each gRNA resides on the flat side of each marker. (B) Schematic of the Cas9-Targeted Nanopore Sequencing workflow. The process involves dephosphorylation of DNA ends to block their reactivity, followed by Cas9-target cleavage to generate new phosphorylated ends. These phosphorylated ends undergo barcoding and adapter ligation, before subsequent sequencing and data analysis. This can be completed within 24 hours, inclusive of DNA extraction.
Our Cas9-targeted library preparation was based on a methodology developed by Gilpatrick et al. (2020) (Figure 3B). Total genomic DNA from four mites (~400 ng) was resuspended in 90 μl of 1X CutSmart buffer and dephosphorylated with 6 μl of Quick CIP enzyme (NEB) for 10 min at 37°C, followed by heating for 2 min at 80°C for enzyme inactivation. After allowing the sample to return to room temperature, 28 μl of the prepared RNP was added. In the same tube, 1 μl of 10 mM dATP and 1 μl of Taq DNA polymerase (NEB) was also added for A-tailing of the DNA. The sample was then incubated at 37°C for 1 hour for Cas9 cleavage, followed by 10 min at 72°C for A-tailing. Cas9 was inactivated by adding 1 ul (800U) of Thermolabile Proteinase K (NEB) and incubated at 37°C for 15 min, followed by 10 min at 80°C for enzyme inactivation. The sample was purified using AMPure XP magnetic beads (Beckman Coulter) at 1.8X volume and eluted into 22.5ul of Milli-Q water.
Nanopore sequencing
For the initial sample (Colony 1), purified PCR amplicons of COXI were sequenced using the Oxford Nanopore Technologies (ONT) Ligation Sequencing Kit (SQK-LSK109) without barcoding to expedite a result. DNA sequencing was carried out on a GridION DNA sequencer with a MinION flow cell (R9.4.1; ONT). The sequencing was managed using MinKNOW software (v.19.2.2; ONT). Sequencing of mites collected from colonies (2-13) followed the same methodology with the addition of barcoding using the Native Barcode Expansion 1-12 library kit (EXP-NBD104; ONT) on a GridION or MinION DNA sequencer. Barcoded PCR amplicons and Cas9-targeted libraries were sequenced on separate flow cells.
Data analysis
All Nanopore sequencing data was basecalled with Guppy (v6.4.6) in high accuracy mode. COXI consensus sequences were generated from Nanopore amplicon sequencing for each of the samples taken from the colonies 1-10. Reads were filtered for quality ≥9 and at least 300 reads were then aligned in Geneious Prime using MAFFT (v1.5.0; Katoh & Standley, 2013). Consensus sequences were generated from the alignment with 95% base call threshold. The species identity of the COXI incursion sequence was identified by MegaBLAST search of the GenBank nt database (NCBI) and by alignment to V. destructor, Varroa jacobsoni and Varroa underwoodi references using MAFFT. A phylogenetic tree was constructed from 107 COXI Varroa sequences sourced from NCBI. A maximum likelihood phylogenetic tree based on the MAFFT gap-trimmed alignment was generated using RAxML (v4.0) GTR-CAT model with 500 bootstrap replicates (Stamatakis, 2014).
The reads from the Cas9-targeted Nanopore sequencing, were filtered at phred quality ≥9 and a read length >1000 nt. Reads were mapped to the V. destructor mitochondrial genome (NC_004454) using minimap2. The depth of coverage was graphed using Geneious Prime and GraphPad Prism 9 (v9.3.0). Data from an independent Illumina V. destructor whole genome sequencing project conducted by NSW DPI (unpublished) were used for comparing the mean percentage of reads mapped to the V. destructor mitochondrial and nuclear genomes. Illumina data were mapped to the V. destructor reference genome (GCF_002443255.2) using bwa-mem2 (v2.2.1; Md et al., 2019), whereas Cas9-targeted data was mapped to the same reference genome utilising minimap2 (v2.22; Li, 2018). Mapping percentages were calculated as the ratio of mapped reads to the total reads in each run and were graphed using GraphPad.
Cas9-targeted reads mapped to the V. destructor mitochondrial genome (NC_004454) were extracted and aligned using MAFFT. Consensus sequences were created in Geneious Prime with a 95% base-call threshold. For comparative genetics, four complete V. destructor mitochondrial genomes were downloaded from NCBI. Due to the limited number of assembled V. destructor mitochondrial genomes publicly available, eight additional V. destructor mitochondrial genomes were assembled from publicly accessible Illumina whole genome V. destructor sequencing data. NCBI accession numbers for the eight datasets selected for assembly are listed in Supplementary 1. Numerous other V. destructor whole-genome datasets are accessible for assembly in NCBI. Mitochondrial genome assemblies were generated using an automated script that mapped reads to the V. destructor mitochondrial reference genome (NC_004454) with bwa-mem2, extracted the mapped reads, and assembled the cleaned and filtered reads using SPAdes (v3.15.3; Bankevich et al., 2012). The assembly script and further details can be found at github.com/gustomc/auto_mito_asm.
A phylogenetic tree of the Cas9-enriched mitochondrial region was constructed using Jukes-Cantor UPGMA model based on a MAFFT alignment (Sneath and Sokal, 1973). The alignment included mitochondrial genomes from three Australian samples and twelve additional V. destructor data sets. The Jukes-Cantor UPGMA model was employed over RaxML, IQ-TREE, FastTree and other more complex models to prevent overfitting of the small and low-complexity dataset, which was observed when using more sophisticated models.
Results
COXI amplicon sequencing
Nanopore sequencing of COXI amplicons for Colony 1 yielded a consensus sequence within 24 hours of receiving the Varroa-parasitized bees in the laboratory. The samples from the subsequent nine colonies were barcoded and sequenced together on a single flow cell. A consensus sequence was generated for each case (see Supplementary 2). The COXI sequences were identical across all ten colonies. A megaBLAST search revealed a 100% identity match between the incursion sequence and the V. destructor reference sequence. In Figure 4, the incursion sequence was aligned to the COXI reference sequences of V. destructor, V. jacobsoni, and V. underwoodi, further confirming a 100% identity match to V. destructor. Given the significant threat that V. destructor poses to Australian industries, all 10 samples were subjected to additional confirmatory tests, including Sanger sequencing, which matched the Nanopore sequencing results, and morphological assessment to genus level (data not shown). Dichotomous generic keys and descriptions of Varroa and other bee mites were used to confirm the morphological identification of the specimens as female Varroa sp (Oudemans, 1904; Baker, 1999; Krantz and Walter, 2009). Morphometric measurements and examination of the idiosoma, ventral shields, palps, coxae and body setation were made using light and compound microscopes. These observations further confirmed this generic identification.

Figure 4 Varroa COXI sequence alignment using MAFFT. The samples from the 10 colonies assessed by Nanopore amplicon sequencing had identical Varroa destructor amplicon sequences. This sequence was aligned to reference sequences for Varroa destructor, Varroa underwoodi and Varroa jacobsoni. Sequence disagreements to the Australia June 2022 sequence are highlighted with black lines. Refer to Supplementary 2 for a nucleotide-level alignment.
A phylogenetic tree was constructed using an additional 107 COXI sequences to clarify the haplogroup of the detected V. destructor (Figure 5). Owing to the global ubiquity of the Korean haplogroup, it constitutes the majority of sequences available in the NCBI database, a fact clearly reflected in Figure 5. As shown in this figure, the COXI amplicon sequence, which was consistent across all ten evaluated Australian colonies, unambiguously placed the NSW V. destructor within the Korean haplogroup.
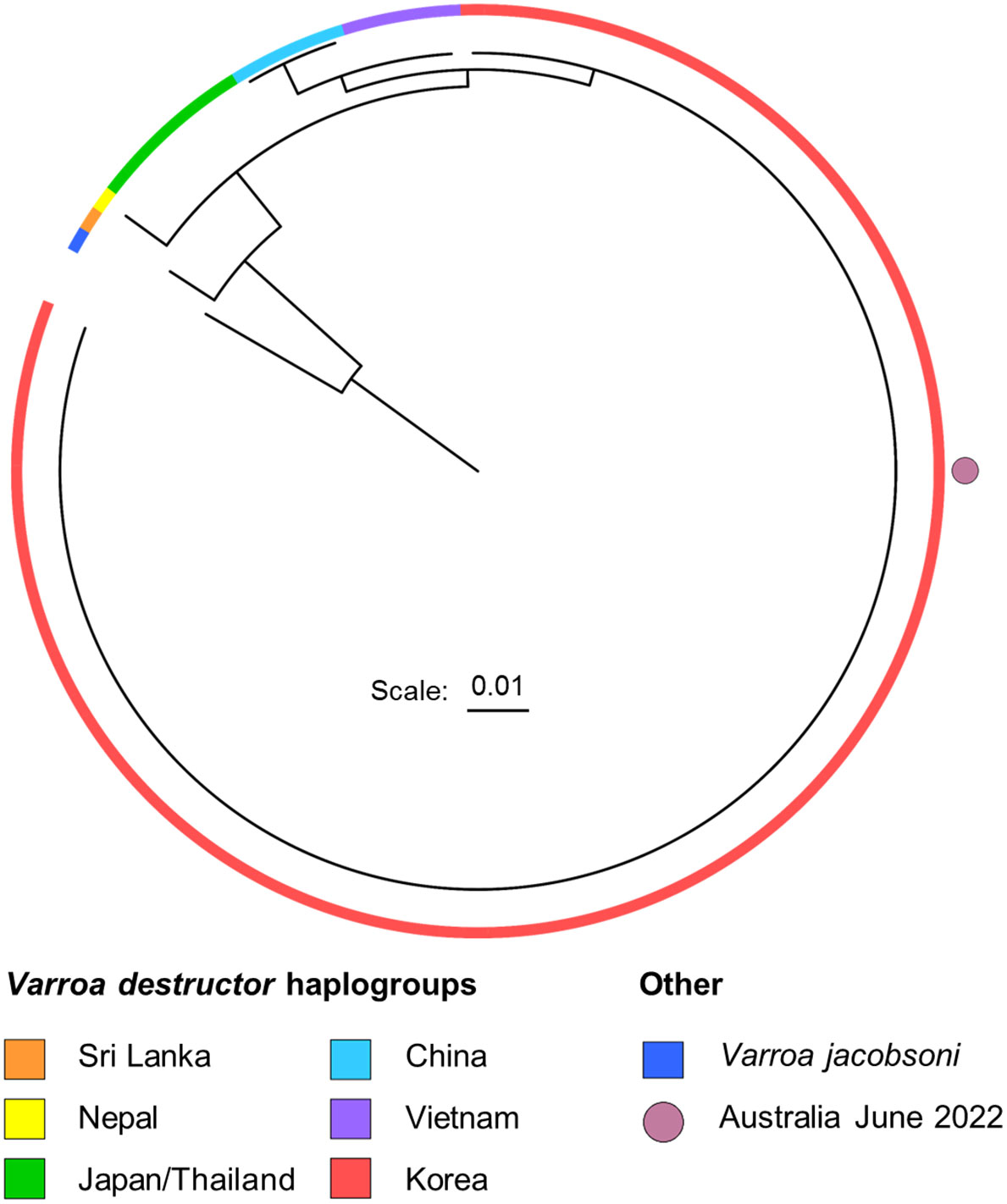
Figure 5 Maximum likelihood phylogenetic tree generated from RaxML GTR-CAT, based on 107 V. destructor COXI sequences. Varroa jacobsoni serves as an outgroup. The ten Australia June 2022 samples had identical COXI sequences and were grouped within the red Korean haplogroup. The tree leaf representing the Australia June 2022 sequence is highlighted with a magenta circle. For a phylogenetic tree with labelled leaves, see Supplementary 3.
Cas9-targeted sequencing of mitochondrial genome
In light of the genetic uniformity observed in the COXI amplicons from the initial ten Australian colonies limiting epidemiological tracing, we deployed Cas9-targeted Nanopore sequencing of the V. destructor mitochondrial genome to get higher-resolution genetic data that could enable more effective epidemiological investigation, as well as species identification and haplogroup determination. The technique was tested across colonies from three geographically distinct locations separated by approximately 20km (colonies 11, 12 and 13 in Figure 1C). We were able to obtain results within 24 hours of obtaining the Varroa mites in the laboratory.
The sequencing data from the Cas9-targeted library had substantial enrichment in three out of the eight targeted mitochondrial fragments—specifically, regions A, B, and C—across the three colonies under investigation (Figure 6A). Significant enrichment was observed only in fragments where the gRNAs were oriented such that the PAM sequences were directly adjacent to the enriched fragment on both ends. The coverage depth of three enriched regions ranged from 400 to 3000X across the three colonies. The remaining five fragments displayed limited enrichment in all cases. Consensus sequences for enriched regions A, B, and C were generated for each colony (Supplementary 2).
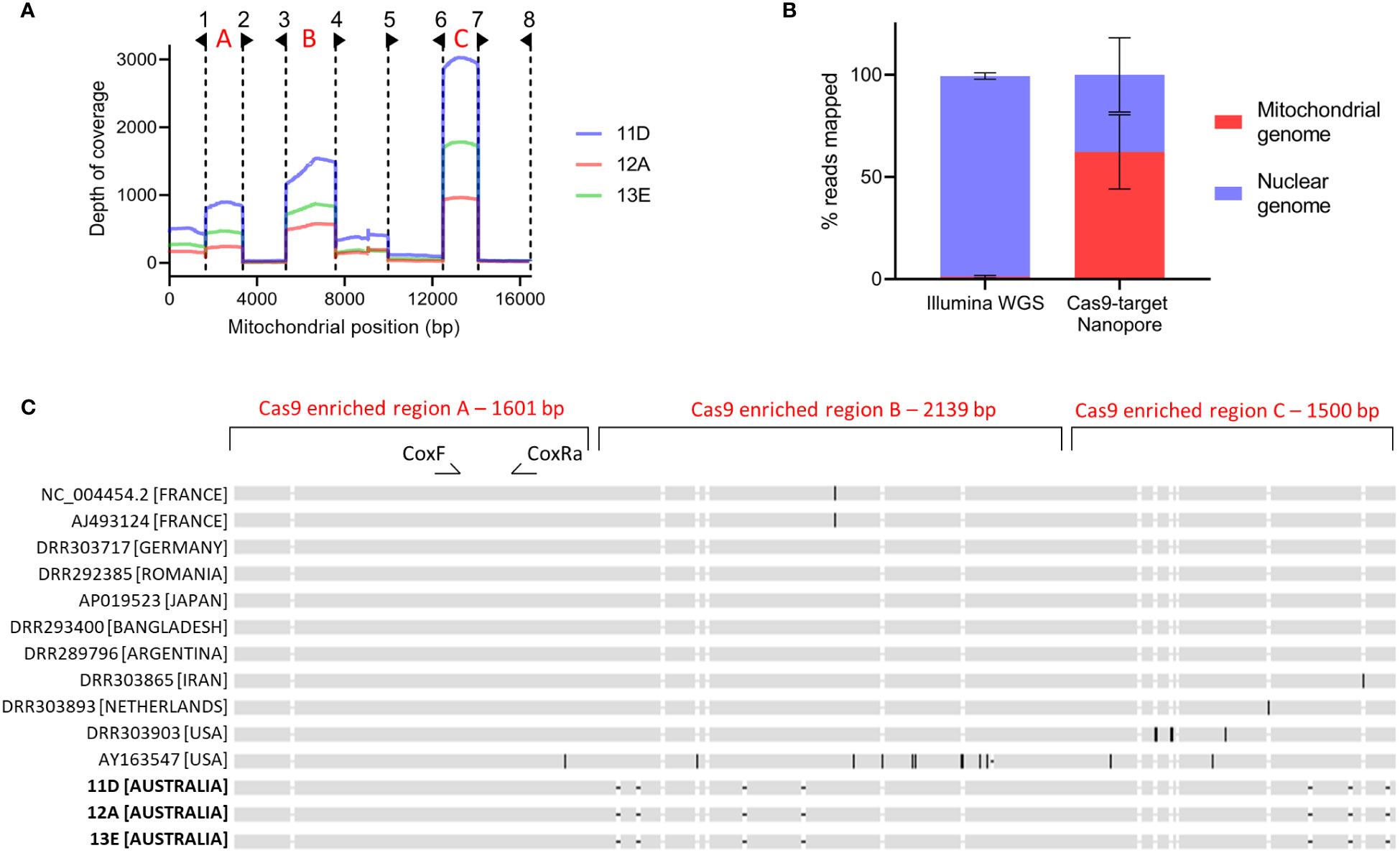
Figure 6 Multiplexed Cas9-targeted Nanopore sequencing of Varroa destructor mitochondrial genome (A) Depth of coverage across Varroa destructor mitochondrial genome for colonies 11D, 12A and 13E. Dotted lines and flags indicate the eight gRNA locations and directions. Red letters indicate three highly enriched regions used for concatenation, alignment and phylogenetic tree formation. (B) Mean percentage of reads mapped to Varroa destructor nuclear and mitochondrial genomes when using Illumina whole genome sequencing (WGS; n=3) or Cas9-target Nanopore sequencing (n=3). Error bars are standard deviation. (C) MAFFT sequence alignment of the 5240 bp concatenated region from three Australian colonies and 11 additional Varroa destructor sequences. All data sets were of the Korean haplogroup based on the conventional 458 bp COXI region used for amplicon sequencing. See Supplementary 2 for nucleotide level alignment.
To evaluate the effectiveness of Cas9-mediated enrichment, we compared the reads from the three Cas9-targeted Nanopore V. destructor libraries with those from three conventional Illumina whole genome V. destructor libraries. The comparison involved mapping these reads to the complete V. destructor genome, inclusive of the mitochondrial genome and calculating the percentage of total reads mapped to either the nuclear or mitochondrial genome. As shown in Figure 6, an average of 62.3% of the total reads from the Cas9-targeted Nanopore library, compared to only 1.2% of total reads from the conventional Illumina whole genome library, mapped to the V. destructor mitochondrial genome.
The enriched regions designated A, B, and C in Figure 6A, made up 31.8% of the mitochondrial genome. We concatenated sequences from these regions to generate a unified 5,240 bp sequence for each colony, all of which were identical across the three colonies. The concatenated sequences from colonies 11, 12, and 13 were aligned with the equivalent regions from eleven publicly available V. destructor datasets belonging to the Korean haplogroup (Figure 6C). While no sequence variation was detected within the conventional 458 bp COXI amplicon region—defined by the CoxF and CoxRa primers—our analysis uncovered divergences in sequences beyond this locus. A phylogenetic tree, illustrated in Figure 7, was constructed based on this alignment. This dataset enabled a more refined phylogenetic resolution within the Korean haplogroup of V. destructor.

Figure 7 A phylogenetic tree based on the Varroa destructor alignment shown in Figure 6C. DRR283612, belonging to the Japan/Thailand haplogroup is included for comparison. The tree was generated using a simple Jukes-Cantor UPGMA model to prevent overfitting of the data. The 5240 bp enriched region was able to distinguish between some members of the Korean haplogroup. Locations within square brackets indicate the origin of the sample.
Discussion
In this case study, we evaluated two targeted Nanopore DNA sequencing methods for rapid species identification and haplogroup determination of V. destructor amidst an incipient outbreak in Australia. The first detection of the pest occurred in June 2022 within government-managed sentinel colonies located at the Port of Newcastle. Samples from parasitized colonies were promptly sent to NSW DPI’s Elizabeth Macarthur Agricultural Institute for analysis. Results were obtained within 24-hours using either Nanopore sequencing of a 458 bp amplicon of the mitochondrial COXI gene or Cas9-targeted Nanopore sequencing of 5420 bp of the mitochondrial genome. Both techniques had specific merits and limitations.
Employing PCR-based amplicon sequencing of the COXI gene yielded rapid results, thanks in part to a well-established PCR protocol by Anderson and Fuchs (1998) that requires minimal DNA input requirements. The data enabled species identification (Figure 4) and supported the construction of a phylogenetic tree (Figure 5), which placed all ten evaluated samples within the widely distributed Korean haplogroup. The results substantiated the incursion response of the NSW DPI, which included the destruction of parasitized colonies and restrictions on colony movement. Although critical information was obtained from PCR-amplicon sequencing, the technique has intrinsic limitations in terms of adaptability to new targets, multiplexing capabilities, and data output (Holm et al., 2019; Meek and Larson, 2019). Data output, in particular, constrains this method’s utility for epidemiological investigation.
The Cas9-targeted Nanopore sequencing method can provide richer data within an equivalent time frame, affording opportunities for more nuanced epidemiological investigation. Requiring a higher DNA input (~400 ng), this approach required DNA from four mites per experiment, as opposed to the single-mite basis in PCR-amplicon sequencing, thus providing colony-specific rather than mite-specific data. Despite the absence of detectable genetic variation among the three Australian samples (Figure 6C), the 5420 bp region of the V. destructor mitochondrial genome demonstrated greater discriminative power within the Korean haplogroup (Figure 7). The 5420 bp enriched region was formed from successful targeting of three out of eight mitochondrial regions, suggesting that future protocol adjustments could reduce the gRNA to a set of six (gRNAs 1-4, 6, 7) as gRNAs 5 and 8 were not useful for enrichment purposes. Cas9-targeting efficacy was further highlighted by the depth of coverage across the three enriched regions, ranging from 400-3000X in samples from colonies 11, 12 and 13 (Figure 6A). When compared with conventional Illumina whole-genome sequencing, the level of enrichment was striking. As shown in Figure 6B, an average of 62.3% of the total reads from the Cas9-targeted Nanopore library mapped to the V. destructor mitochondrial genome. In contrast, only 1.2% of the total reads obtained from Illumina whole-genome sequencing were mapped to the V. destructor mitochondrial genome. These findings compellingly substantiate the utility of Cas9-targeting for library enrichment, reinforcing its potential as a valuable tool for future genomic studies and biosecurity investigations. Additionally, mutations in mitochondrial DNA related to synthetic acaricide resistance have been found, which can be of great concern for biosecurity control schemes (Strachecka et al., 2015).
One key observation from our analysis was that gRNA orientation played a crucial role in fragment enrichment. Specifically, all three fragments that were enriched featured gRNAs oriented in a manner that placed the PAM sequences directly adjacent to both ends of the enriched fragments. Such orientation bias has been reported in prior Cas9-targeted experiments and is often attributed to the Cas9 enzyme remaining bound to the non-PAM side of the cleaved DNA (Gilpatrick et al., 2020; López-Girona et al., 2020; Stangl et al., 2020). We hypothesised that treatment with Proteinase K, followed by heat treatment at 80°C, would dislodge any bound Cas9, thereby allowing for adapter ligation at both newly formed termini for each cleavage site. Both Proteinase K and 80°C heat treatment have previously been used effectively remove bound Cas9 from DNA (Anders & Jinek, 2014; Mehravar et al., 2019; David et al., 2022). Our findings, however, corroborated by existing scientific literature, lead us to conclude that post-cleavage 5′→3′ degradation activity by Cas9 on the non-PAM side inhibits adapter ligation, rather than the enzyme remaining bound. The phenomena of Cas9 preferentially generating staggered-ended DNA products on the non-PAM side, was highlighted by Stephenson et al. (2018), and has implications for Cas9-targeted library preparation, limiting the effectiveness of sequencing adapter ligation. Future protocol iterations could include end-repair of these ‘chewed-back’ DNA sequences prior to adapter ligation as a possible solution, aiming to achieve bi-directional sequencing at cleavage sites.
Within the 5420 bp sequence alignment, schematically shown in Figure 6C (refer to Supplementary 2 for nucleotide-level alignment), it is evident that three sequence gaps, observed solely in the three Australian samples and absent in the other eleven, are located in homopolymer regions composed of eight or more thymine nucleotides. These gaps are likely artefacts attributable to the inherent challenges Nanopore sequencing faces when sequencing homopolymer regions (Wang et al., 2021). To help reduce this frequently observed issue, Oxford Nanopore Technologies has updated its flow cell chemistry to V14 and pore engineering to R10.4.1 since the completion of this study (Ni et al., 2023). Anticipated ongoing advancements in both flow cell design and base-calling software aim to further mitigate this challenge.
Our analysis of thirteen V. destructor samples, sourced from five geographically distinct locations, revealed an absence of genetic diversity in all mitochondrial genome regions sequenced. This can be attributed to three primary factors: the high levels of inbreeding inherent to these mites that makes them less susceptible to genetic variation, the genetic bottleneck associated with an incursion event, and the early detection of the mites in Australia, thereby limiting the time frame for any evolutionary genetic changes to manifest (Rosenkranz et al., 2010; Dynes et al., 2017). Applying Cas9-targeted mitochondrial sequencing to species with an exogamy reproductive behaviour would likely yield more informative tracing data compared to the endogamy arrhenotokous parthenogenetic behaviour exhibited by V. destructor. NSW DPI, in collaboration with other partner organisations, is now utilising whole-genome sequencing to better understand the origin of the Australian V. destructor incursion and the pest’s movement patterns within Australia.
The efforts in New South Wales to eradicate V. destructor have been monumental; however, despite significant investment with a budget of $132 million, eradication efforts pivoted towards a management strategy in September 2023 (Department of Agriculture Fisheries and Forestry, 2023a, Department of Agriculture Fisheries and Forestry, 2023b). Varroa destructor has now cemented its status as a global threat to the European honey bee. Moving forward, Australian initiatives will focus on containment strategies to prevent further geographical spread. The rapid genetic diagnostic methods developed in this study could provide an integral component for monitoring and identification within a comprehensive integrated pest management system for V. destructor. These methods could be adopted not only across Australia but also internationally.
In summary, this case study brings to the fore two Nanopore sequencing techniques aimed at rapid species identification and haplogroup determination within a biosecurity context. Our results demonstrate that Nanopore amplicon sequencing is a robust tool for rapid species confirmation of invertebrate pests. This study marks the first application of Cas9-targeted Nanopore sequencing in biosecurity, and given its promising outcomes, it is unlikely to be its last use. The technique can yield rapid results, offers longer read lengths, and is more easily adaptable, as the inclusion of additional gRNAs for new genomic targets is generally simpler and quicker than designing, validating, and optimising a new set of primers for inclusion in an existing multiplexed PCR reaction. Our findings contribute valuable insights to the future development of Cas9-targeted Nanopore sequencing, especially in a biosecurity setting where timely and precise diagnosis is imperative.
Data availability statement
Raw sequencing data generated in this project can be found under the NCBI BioProject ID: PRJNA1062103. See Supplementary Material for a complete list of data sources and accession numbers of the publicly available data used in this study.
Author contributions
GM: Conceptualization, Formal analysis, Investigation, Methodology, Visualization, Writing – original draft, Writing – review & editing. KR: Formal analysis, Investigation, Methodology, Writing – review & editing. KW: Investigation, Writing – review & editing. JW: Data curation, Formal analysis, Investigation, Methodology, Writing – review & editing. LD: Methodology, Investigation, Writing – review & editing. LB: Investigation, Writing – review & editing. AW: Investigation, Writing – review & editing. BO’R: Funding acquisition, Writing – review & editing. DB: Conceptualization, Funding acquisition, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was partly funded by Plant Health Australia and the NSW Department of Primary Industries led National Varroa Mite (Varroa destructor) Response Plan.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/frbee.2024.1334543/full#supplementary-material
References
Abeynayake S. W., Fiorito S., Dinsdale A., Whattam M., Crowe B., Sparks K., et al. (2021). A rapid and cost-effective identification of invertebrate pests at the borders using minION sequencing of DNA barcodes. Genes 12, 1138. doi: 10.3390/genes12081138
Anders C., Jinek M. (2014). In vitro enzymology of Cas9. Methods Enzymol 546, 1–20. doi: 10.1016/b978-0-12-801185-0.00001-5
Anderson D. L., Fuchs S. (1998). Two genetically distinct populations of Varroa jacobsoni with contrasting reproductive abilities on Apis mellifera. J. Apicultural Res. 37, 69–78. doi: 10.1080/00218839.1998.11100957
Bahreini R., Nasr M., Docherty C., de Herdt O., Muirhead S., Feindel D. (2020). Evaluation of potential miticide toxicity to Varroa destructor and honey bees, Apis mellifera, under laboratory conditions. Sci. Rep. 10, 21529. doi: 10.1038/s41598-020-78561-2
Baker A. S. (1999). Mites and Ticks of Domestic Animals: An Identification Guide and Information Source (London: The Natural History Museum, The Stationery Office).
Bankevich A., Nurk S., Antipov D., Gurevich A. A., Dvorkin M., Kulikov A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
David S. R., Maheshwaram S. K., Shet D., Lakshminarayana M. B., Soni G. V. (2022). Temperature dependent in vitro binding and release of target DNA by Cas9 enzyme. Sci. Rep. 12, 15243. doi: 10.1038/s41598-022-19485-x
Department of Agriculture Fisheries and Forestry (2023a) National Management Group Communiqué: Varroa destructor – 27 June 2023. Available online at: https://www.agriculture.gov.au/about/news/stay-informed/communiques/varroa-destructor-27-june-2023.
Department of Agriculture Fisheries and Forestry (2023b) Varroa destructor – 19 September 2023. Available online at: https://www.agriculture.gov.au/about/news/stay-informed/communiques/varroa-destructor-19-sept-2023#daff-page-main.
Dietemann V., Nazzi F., Martin S. J., Anderson D. L., Locke B., Delaplane K. S., et al. (2013). Standard methods for varroa research. J. Apicultural Res. 52, 1–54. doi: 10.3896/IBRA.1.52.1.09
Dynes T. L., De Roode J. C., Lyons J. I., Berry J. A., Delaplane K. S., Brosi B. J. (2017). Fine scale population genetic structure of Varroa destructor, an ectoparasitic mite of the honey bee (Apis mellifera). Apidologie 48, 93–101. doi: 10.1007/s13592-016-0453-7
Giliba R. A., Mpinga I. H., Ndimuligo S. A., Mpanda M. M. (2020). Changing climate patterns risk the spread of Varroa destructor infestation of African honey bees in Tanzania. Ecol. Processes 9, 48. doi: 10.1186/s13717-020-00247-4
Gilpatrick T., Lee I., Graham J. E., Raimondeau E., Bowen R., Heron A., et al. (2020). Targeted nanopore sequencing with Cas9-guided adapter ligation. Nat. Biotechnol. 38, 433–438. doi: 10.1038/s41587-020-0407-5
Hall C., Zascavage R. R., Sedlazeck F., Planz J. (2020). Potential applications of nanopore sequencing for forensic analysis. Forensic Sci. Rev. 32, 23–54.
Holm J. B., Humphrys M. S., Robinson C. K., Settles M. L., Ott S., Fu L., et al. (2019). Ultrahigh-Throughput multiplexing and sequencing of <500-base-pair amplicon regions on the illumina hiSeq 2500 platform. mSystems 4(1). doi: 10.1128/msystems.00029-19
Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J. A., Charpentier E. (2012). A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. science 337, 816–821. doi: 10.1126/science.1225829
Katoh K., Standley D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4), 772–80. doi: 10.1093/molbev/mst010
Krantz G. W., Walter D. E. (2009) A manual of acarology. Available online at: http://books.google.com/books?id=x00gAQAAMAAJ.
Li H. (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 34(18), 3094–3100. doi: 10.1093/bioinformatics/bty191
Li W., Wang C., Huang Z. Y., Chen Y., Han R. (2019). Reproduction of distinct varroa destructor genotypes on honey bee worker brood. Insects 10, 372. doi: 10.3390/insects10110372
Lin Z., Qin Y., Page P., Wang S., Li L., Wen Z., et al. (2018). Reproduction of parasitic mites Varroa destructor in original and new honeybee hosts. Ecol. Evol. 8, 2135–2145. doi: 10.1002/ece3.3802
López-Girona E., Davy M. W., Albert N. W., Hilario E., Smart M. E., Kirk C., et al. (2020). CRISPR-Cas9 enrichment and long read sequencing for fine mapping in plants. Plant Methods 16, 121. doi: 10.1186/s13007-020-00661-x
McDonald T. L., Zhou W., Castro C. P., Mumm C., Switzenberg J. A., Mills R. E., et al. (2021). Cas9 targeted enrichment of mobile elements using nanopore sequencing. Nat. Commun. 12, 3586. doi: 10.1038/s41467-021-23918-y
McFarlane G. R., Whitelaw C. B. A., Lillico S. G. (2023). Gene drive: Past, present and future roads to vertebrate biocontrol. Appl. Biosci. 2, 52–70. doi: 10.3390/applbiosci2010006
Md V., Misra S., Li H., Aluru S. (2019). Efficient architecture-aware acceleration of BWA-MEM for multicore systems. ArXiv. doi: 10.48550/arXiv.1907.12931
Meek M. H., Larson W. A. (2019). The future is now: Amplicon sequencing and sequence capture usher in the conservation genomics era. Mol. Ecol. Resour. 19, 795–803. doi: 10.1111/1755-0998.12998
Mehravar M., Shirazi A., Mehrazar M. M., Nazari M. (2019). In vitro pre-validation of gene editing by CRISPR/cas9 ribonucleoprotein. Avicenna J. Med. Biotechnol. 11, 259–263.
Ni Y., Liu X., Simeneh Z. M., Yang M., Li R. (2023). Benchmarking of Nanopore R10.4 and R9.4.1 flow cells in single-cell whole-genome amplification and whole-genome shotgun sequencing. Comput. Struct. Biotechnol. J. 21, 2352–2364. doi: 10.1016/j.csbj.2023.03.038
Oudemans A. C. (1904). On a new genus and species of Parasitic Acari. Notes Leyden Museum 24, 216–222.
Parker J., Helmstetter A. J., Devey D., Wilkinson T., Papadopulos A. S. T. (2017). Field-based species identification of closely-related plants using real-time nanopore sequencing. Sci. Rep. 7, 8345. doi: 10.1038/s41598-017-08461-5
Quan J., Langelier C., Kuchta A., Batson J., Teyssier N., Lyden A., et al. (2019). FLASH: A next-generation CRISPR diagnostic for multiplexed detection of antimicrobial resistance sequences. Nucleic Acids Res. 47, e83. doi: 10.1093/nar/gkz418
Roberts J. M. K., Anderson D. L., Durr P. A. (2017). Absence of deformed wing virus and Varroa destructor in Australia provides unique perspectives on honeybee viral landscapes and colony losses. Sci. Rep. 7, 6925. doi: 10.1038/s41598-017-07290-w
Roberts J. M. K., Anderson D. L., Tay W. T. (2015). Multiple host shifts by the emerging honeybee parasite, Varroa jacobsoni. Mol. Ecol. 24, 2379–2391. doi: 10.1111/mec.13185
Rosenkranz P., Aumeier P., Ziegelmann B. (2010). Biology and control of Varroa destructor. J. Invertebrate Pathol. 103, S96–S119. doi: 10.1016/j.jip.2009.07.016
Rubben K., Tilleman L., Deserranno K., Tytgat O., Deforce D., Van Nieuwerburgh F. (2022). Cas9 targeted nanopore sequencing with enhanced variant calling improves CYP2D6-CYP2D7 hybrid allele genotyping. PloS Genet. 18, e1010176. doi: 10.1371/journal.pgen.1010176
Sneath P. H., Sokal R. R. (1973). “Numerical taxonomy,” in The principles and practice of numerical classification. (San Fancisco: W.H.Freeman & Co Ltd)
Stamatakis A. (2014). RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Stangl C., de Blank S., Renkens I., Westera L., Verbeek T., Valle-Inclan J. E., et al. (2020). Partner independent fusion gene detection by multiplexed CRISPR-Cas9 enrichment and long read nanopore sequencing. Nat. Commun. 11, 2861. doi: 10.1038/s41467-020-16641-7
Stephenson A. A., Raper A. T., Suo Z. (2018). Bidirectional degradation of DNA cleavage products catalyzed by CRISPR/Cas9. J Am Chem Soc 140(10), 3743–3750. doi: 10.1021/jacs.7b13050
Strachecka A., Borsuk G., Olszewski K., Paleolog J. (2015). A new detection method for a newly revealed mechanism of pyrethroid resistance development in Varroa destructor. Parasitol. Res. 114, 3999–4004. doi: 10.1007/s00436-015-4627-4
Traynor K. S., Mondet F., de Miranda J. R., Techer M., Kowallik V., Oddie M. A., et al. (2020). Varroa destructor: A complex parasite, crippling honey bees worldwide. Trends Parasitol. 36, 592–606. doi: 10.1016/j.pt.2020.04.004
Wang Y., Zhao Y., Bollas A., Wang Y., Au K. F. (2021). Nanopore sequencing technology, bioinformatics and applications. Nat. Biotechnol. 39, 1348–1365. doi: 10.1038/s41587-021-01108-x
Keywords: Varroa, nanopore, CRISPR, Cas9, amplicon, sequencing, pest, invasive
Citation: McFarlane GR, Robinson KL, Whitaker K, Webster J, Drysdale L, Brancalion L, Webster A, O’Rourke B and Bogema DR (2024) Amplicon and Cas9-targeted nanopore sequencing of Varroa destructor at the onset of an outbreak in Australia. Front. Bee Sci. 2:1334543. doi: 10.3389/frbee.2024.1334543
Received: 07 November 2023; Accepted: 30 January 2024;
Published: 16 February 2024.
Edited by:
M. Marta Guarna, Agriculture and Agri-Food Canada (AAFC), CanadaReviewed by:
Solenn Patalano, Alexander Fleming Biomedical Sciences Research Center, GreeceNuria Morfin, British Columbia Technology Transfer Program, Canada
Copyright © 2024 McFarlane, Robinson, Whitaker, Webster, Drysdale, Brancalion, Webster, O’Rourke and Bogema. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gus R. McFarlane, Z3VzLm1jZmFybGFuZUBkcGkubnN3Lmdvdi5hdQ==