 Christian Lund Jakobsen1†Morten Johansen2†Tore Ericsson3Lennart Häggström3Christian Kolle Christensen1Ida Nielsen3
Christian Lund Jakobsen1†Morten Johansen2†Tore Ericsson3Lennart Häggström3Christian Kolle Christensen1Ida Nielsen3 William Robert Brant3
William Robert Brant3 Dorthe Bomholdt Ravnsbæk2*
Dorthe Bomholdt Ravnsbæk2*- 1Department of Physics, Chemistry, and Pharmacy, University of Southern Denmark, Odense, Denmark
- 2Department of Chemistry, Center for Integrated Materials Research, Aarhus University, Aarhus, Denmark
- 3Department of Chemistry, Structural Chemistry, Ångström Laboratories, University of Uppsala, Uppsala, Sweden
Layered Na3Fe3(PO4)4 can function as a positive electrode for both Li- and Na-ion batteries and may hold advantages from both classical layered and phosphate-based electrode materials. Using a combination of ex-situ and operando synchrotron radiation powder X-ray diffraction, void space analysis, and Mössbauer spectroscopy, we herein investigate the structural evolution of the Na3Fe3(PO4)4 framework during Li- and Na-ion intercalation. We show that during discharge, Li- and Na-intercalation into Na3Fe3(PO4)4 occurs via a solid solution reaction wherein Na-ions appear to be preferentially intercalated into the intralayer sites. The intercalation causes an expansion of the unit cell volume, however at open circuit conditions after ion-intercalation (i.e., after battery discharge), Na3+xFe3(PO4)4 and LixNa3Fe3(PO4)4 undergo a structural relaxation, wherein the unit volume contracts below that of the pristine material. Rietveld refinement suggests that the ions intercalated into the intra-layer sites diffuse to the sites in the inter-layer space during the relaxation. This behavior brings new perspectives to understanding structural relaxation and deviations between structural evolution observed under dynamic and static conditions.
1 Introduction
Due to the low price and high global abundance of iron, polyanionic iron-based materials are one of the most attractive electrode material types for future battery applications (Papp et al., 2008; Vaalma et al., 2018). Especially iron phosphates have been given much attention since the discovery of LiFePO4 (Padhi et al., 1997a). Still, several other phosphate frameworks with the active redox couple Fe3+/Fe2+ have also been studied over the years, such as Li3Fe2(PO4)4, LiFeP2O7, Fe3(P2O7), etc. However, these materials provide a lower operating potential than LiFePO4 (Padhi et al., 1997b; Padhi et al., 1997c; Wurm et al., 2002). The appeal of phosphate-based materials lies in the structural robustness (high thermal and chemical stability), e.g., by decreasing the risk of O2-release and thermal runaway (Huang et al., 2016). On the downside, phosphates generally suffer from poor ionic conductivity, which tends to make them unsuitable for high-power applications (Padhi et al., 1997a; Mueller et al., 2011; Jakobsen et al., 2021). Layered transition metal oxides, on the other hand, are much more attractive for high-power batteries as the two-dimensional ion migration in the inter-layer space provides faster kinetics (Kang et al., 2006; Song et al., 2018).
Development of iron-based materials for Li-ion battery cathodes have been quite extensive, and many different compositions and structures have emerged over the years (Padhi et al., 1997b; Padhi et al., 1997c; Wurm et al., 2002). However, the global concern of Li-deposit depletion has moved research toward materials for Na-ion batteries. The sodium iron phosphate, Na3Fe3(PO4)4, exhibits promising properties as a host for both Li- and Na-ions (Trad et al., 2010a; Trad et al., 2010b). The material crystallizes in a monoclinic structure (space group C2/c) and is isostructural to Na3Cr3(PO4)4 (Dyvoire et al., 1980). The material is built from layers of corner-sharing [FeO6]-octahedra (based on two different crystallographic Fe-sites) linked by edge-sharing with phosphate (PO43−) tetrahedra and Na residing in sites between these layers. At least on paper, this creates an ideal framework for ion-intercalation with robustness from the polyanionic framework, potential for fast ion migration in the interlayer spaces, and structural flexibility via contraction/expansion of the interlayer spacing as for the well-known and extensively studied layered transition metal oxides (Weber et al., 2017; Jeong et al., 2019; Quilty et al., 2020; Jakobsen et al., 2022).
Reversible Li- and Na-ion storage in Na3Fe3(PO4)4 has been reported with intercalation of ∼1.8 Na and ∼1.9 Li per Na3Fe3(PO4)4 (Trad et al., 2010a; Trad et al., 2010b). As expected, the intercalation is accompanied by the reduction of Fe3+ to Fe2+. Note that the intercalated ions add to the three Na found in the as-synthesized Na3Fe3(PO4)4. Thus, the material after ion intercalation will henceforth be referred to as LixNa3Fe3(PO4)4 or Na3+xFe3(PO4)4 for Li- and Na-intercalation, respectively. Surprisingly, a significantly larger potential hysteresis has been observed for the intercalation of Li-ions than for Na-ions. This is unexpected considering the larger size of Na+ compared to Li+. The potential hysteresis is corroborated by galvanostatic intermittent titrations revealing that Li-ion intercalation is kinetically limited. The mechanism of ion-intercalation in Na3Fe3(PO4)4 has been visited by powder X-ray diffraction (PXRD), which suggested that the material disorders with intercalation of >1 Na per formula unit. In contrast, Li intercalation leaves the structure relatively unchanged (Trad et al., 2010a; Trad et al., 2010b). However, the details of the structural evolution have yet to be investigated, as this may hold the key to understanding differences between the Li- and Na-ion intercalation mechanisms leading to different kinetics.
To explore and compare the structural changes during the insertion of Li- or Na-ions in Na3Fe3(PO4)4, we have synthesized the material and investigated the ion-intercalation process with ex-situ and operando powder X-ray diffraction (PXRD) experiments. Via void-space analysis in combination with the migration energy barrier profile provided by Shinde et al. (2019), we deduce the interstitial sites in the Na3+xFe3(PO4)4 or LixNa3Fe3(PO4)4 structures at which the Li-/Na-ions are positioned after intercalation. Identifying the intercalation sites has allowed us to reveal post-intercalation structural relaxation involving ion diffusion between vacant sites.
2 Experimental
2.1 Preparation of layered Na3Fe3(PO4)4
In this work, we have synthesized layered Na3Fe3(PO4)4 via a hydrothermal reaction followed by sintering in an oxygen atmosphere. However, several other synthesis routes are known for the material (Trad et al., 2010a; Trad et al., 2010b; David et al., 2016; Liu et al., 2018; Shinde et al., 2019). For the hydrothermal reaction, stoichiometric amounts of Na3PO4 (0.83 g, purity 96%, Sigma-Aldrich/Merck) and FePO4·4 H2O (3.34 g, Sigma-Aldrich/Merck) were mixed in 25 mL demineralized water and stirred for 10 min. The 25 mL solution was sealed in a Teflon™ cup with a total volume of 70 mL. The Telfon™ cup was enclosed in an autoclave made of stainless-steel parts. The autoclave was placed in a cold oven and heated to 200°C for 24 h. The autoclave was naturally cooled in the oven. The product was washed, filtered, and dried. The dried material was pressed into a pellet and sintered at 750°C for 48 h under a constant flow of oxygen. We note that the outcome of the synthesis is very sensitive to the synthesis conditions as illustrated in Supplementary Table S2 and Supplementary Figure S1.
2.2 Void-space analysis and structural drawings
Previous investigations of migration energy barrier profiles inspired the void-space analysis (Shinde et al., 2019). The void-space analysis was performed using the software CrystalExplorer (Spackman et al., 2021), employing an isovalue of 0.004 e∙Å−3. The Na-ions in the structure of the as-synthesized (i.e., charged) material were not removed before the void-space analysis. The structural drawings are made using the software Diamond (v. 4.6.6) and VESTA (Bergerhoff et al., 1996; Pennington, 1999; Momma and Izumi, 2011).
2.3 Electrochemical characterization
The pristine material was mixed with conductive carbon (Graphite C-Nergy SFG6L, 99.5%, Imerys Graphite, and Carbon. Acetylene Black VXC72, Cabot Corporation in 1:1 wt%) and polyvinylidene fluoride Kynar® binder (HSC900, MTI Corporation) all in 6:3:1 mass ratio. The powders were dispersed in ∼1 mL acetone and mixed well on a vortex-mixer for 1 min. The suspension was spread on a glass plate and collected after evaporation of the acetone. In a Specac® mini-pellet press, Ø7 mm free-standing pellets were pressed uniaxial with a mass of 8–12 mg each under 1.8 metric tonnes (Christensen et al., 2018; Henriksen et al., 2020). The prepared pellets were mounted in a Swagelok®-type cell made of Teflon™ on stainless-steel cylinders. For the galvanostatic charge-discharge samples, a Na-disc served as anode. An ion-permeable microporous Whatman GF/B glass fiber separator was wetted in a 1 M solution of sodium perchlorate in propylene carbonate, serving as the electrolyte. The cells were cycled between 1.5–3.0 V with a C/10 or C/25 current rate. The discharged Li-cell was assembled as the Na-cells, except for the use of a lithium-disc as anode and lithium hexafluorophosphate (LiPF6) in dimethyl carbonate and ethylene carbonate, (1 M LiPF6, 50/50 vol%, Solvionic 99.9%). The cycling rates assume that one mobile ion can be intercalated per formula unit for all measurements. The samples for Mössbauer spectroscopy and ex-situ PXRD after relaxation were prepared by discharging the cells to the lower potential limit as described above, whereafter the cells were disassembled in the glovebox, the electrode removed and washed with electrolyte solvent and left in the glovebox for ∼30 days before the measurements were conducted.
2.4 Ex-situ PXRD
The pristine material was mounted in a Kapton® polyimide capillary at PETRA III on beamline P02.1 at DESY in Hamburg (Schökel et al., 2021). The sample was exposed to the beam (λ = 0.20734 Å) for 30 s. The data was recorded on a 2D Varex XRD 4343CT (150 μm2 × 150 μm2 pixel size, 2880 × 2880 pixel area) detector with a CsI scintillator. Furthermore, two samples in the discharged state, with either Li- or Na-ions intercalated, named LixNa3Fe3(PO4)4 and Na3+xFe3(PO4)4, respectively, were extracted from the battery cell and sealed in Kapton® polyimide tape. The two pellets were mounted on the DanMAX beamline at MAX IV in Lund, Sweden, and exposed to the beam (λ = 0.3541Å) for 10 s. The data was recorded on a DECTRIS PILATUS3 × 2M CdTe 2D area detector (172 μm2 × 172 μm2 pixel size, 1,475 × 1,679 pixel area). All the data were integrated using the Data Analysis WorkbeNch software (Filik et al., 2017) developed by Basham et al. (2015). For the Rietveld refinements an academic version of Topas 6 (Coelho, 2018) and the FullProf suite (Rodriguez-Carvajal, 2001) were used. The background was described by scaling a measurement of empty Kapton® tape collected for each beamtime. Furthermore, a Chebyshev polynomial of 6th order was applied to the refinement to describe any additional background effects in TOPAS. The background was described using linear extrapolation between carefully selected background points when using FullProf. The observed diffraction peaks were described with a Thompson-Cox-Hasting pseudo-Voigt profile function. The instrumental profile parameters were obtained by refining a measurement of CeO2 and fixing these during the refinement of the powder patterns. The structural information for the layered sodium iron (III) phosphate (Na3Fe3(PO4)4) was obtained from Lajmi et al. (2002). Furthermore, small amounts of iron (III) oxide (hematite, α-Fe2O3) with trigonal symmetry (space group R-3c), described by Blake et al. (1966) was used to describe the data fully. For the Rietveld refinement, the lattice parameters, scale factor, sodium occupancies, zero-offset, and overall thermal vibrational factor were refined.
2.5 Operando PXRD
The material was investigated during operation both as a Na- and Li-ion battery cathode material at PETRA III beamline P02.1, DESY in Hamburg. The pellets were prepared as described in the section Electrochemical Characterization. The cells were assembled in an Ar-filled glovebox in AMPIX battery test cells (Borkiewicz et al., 2012). To physically separate the Ø11 mm Na-/Li-disc anodes from the cathode-pellet, a Whatman GF/B microporous glass fiber, permeable to ions, was utilized. For the Li-cell, a 1 M solution of LiPF6 in dimethyl carbonate and ethylene carbonate, 50/50 v/v (99.9%, Solvionic), served as the electrolyte. Whereas, for the Na-ion battery, an electrolyte composed of a 1 M sodium bistrifluoromethane sulfonyl-imide (NaTFSI) solution, using propylene carbonate (PC) as solvent. The two cells were mounted on a homemade sample holder for eight AMPIX cells and mounted at the beamline. The two cells were cycled between 4.5–2.0 V and 3.0–1.5 V for the Li- and Na-cell, respectively. The cells were connected to a BioLogic™ battery cycler and a constant current with a C/10 rate applied, assuming one Li/Na per formula unit to be inserted into the structure. The cells were exposed to the beam (λ = 0.20734 Å) for 30 s per scan and a scan was collected every ∼6.67 min. The data was recorded on a 2D area detector Varex XRD 4343CT (150 μm2 × 150 μm2 pixel size, 2880 × 2880 pixel area) with a CsI scintillator. The integration was done as described for the ex-situ data but with masking of single crystal diffraction peaks and spots originating from the metallic anodes. The data was sequential Rietveld refined, using the model described by Lajmi et al. (2002). The background was described by modelling two artificial broad peaks at position 2θ = 3.37° and 5.72° (Q = 1.78 Å−1 and 3.02 Å−1, respectively) together with a 9th-order Chebyshev polynomial. Furthermore, a Pawley fit for the graphite phase (mainly attributed to the glassy carbon windows) and the Na metal for the Na cell were used. The lattice parameters, scale factors, an overall thermal vibration factor, particle size, and a 2nd order spherical harmonics were refined for both samples.
2.6 Mössbauer spectroscopy
Mössbauer measurements were carried out at room temperature on a spectrometer with a constant acceleration type of vibrator and a 57CoRh source. Three samples are investigated: the pristine Na3Fe3(PO4)4, an electrode discharged vs. Li and Na. Based on columbic counting, the compositions of the discharged samples are and Li2.2Na3Fe3(PO4)4 and Na4.7Fe3(PO4)4. The electrodes were discharged using current rates of C/50 or C/25, removed from the batteries, and enclosed in sealed Kapton pockets. The so-formed absorbers had a sample concentration of ∼6 mg/cm2. Calibration spectra were recorded at 295 K using natural Fe metal foil as a reference absorber. The spectra were folded and fitted using the least square Mössbauer fitting program Recoil to obtain the values of the center shift (CS), the magnitude of the electric quadrupole splitting IQSI, the full width at half maximum W of the Lorentzian absorption lines and the spectral area A.
3 Results and discussion
3.1 Structural characterization of Na3Fe3(PO4)4
As expected, Rietveld refinement (Figure 1A) of the PXRD data of the pristine (as-synthesized) material confirms that Na3Fe3(PO4)4 crystallizes in a monoclinic unit cell (space group C2/c) with FeO6-PO4 layers in the bc-plane (Figure 2A). The structure has two crystallographic distinct Na-sites. Na1 (Wyckoff site 4e) sits in the inter-layer space in distorted prismatic coordination with Na-O distances of 2.47–2.50 Å, while Na2 (Wyckoff site 8f) sits on the edge of the FeO6-PO4 layers in distorted 6-fold coordination to oxygen with Na-O distances ranging from 2.31–2.86 Å.
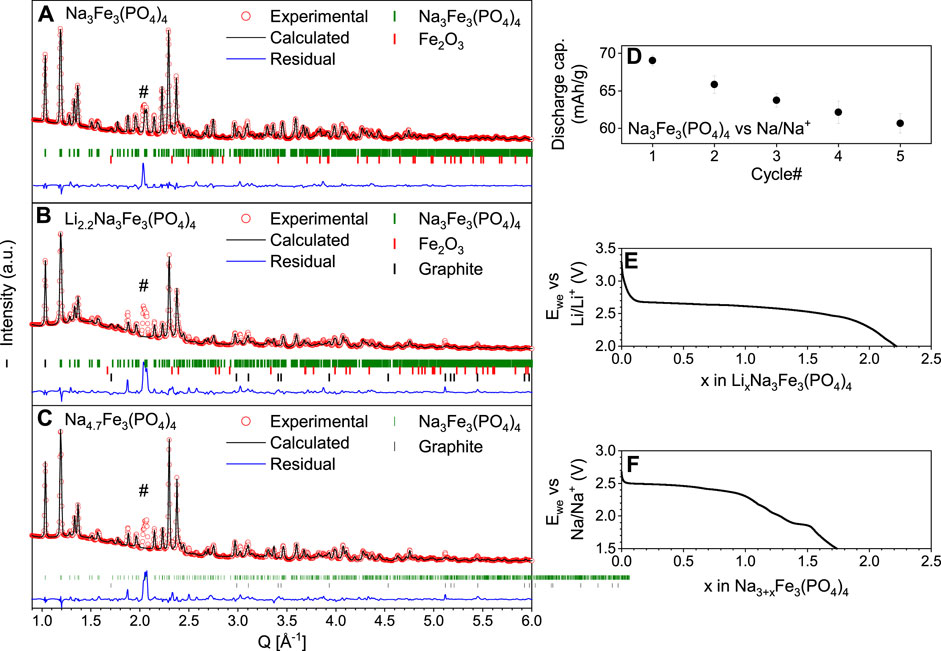
Figure 1. Rietveld refinement of (A) pristine Na3Fe3(PO4)4, (B) the electrode discharged vs. Li, i.e., Li2.2Na3Fe3(PO4)4 and (C) the electrode discharged vs. Na, i.e., Na4.7Na3Fe3(PO4)4. The collected PXRD data are depicted in red circles, the calculated patterns in black line, and the residual in blue. Furthermore, the Bragg peak position of Na3Fe3(PO4)4, hematite (αFe2O3), and graphite are shown as vertical green, red, and black ticks, respectively. # is assigned to a minor unidentified phase being at a constant position ∼2.07 Å−1. (D) Average discharge capacity (mAh/g) vs. cycle number for three identical half-cells with Na3Fe3(PO4)4 vs. Na-metal. (E) Discharge potential profile of LixNa3Fe3(PO4)4 vs. Li. (F) discharge profile of Na3+xFe3(PO4)4.
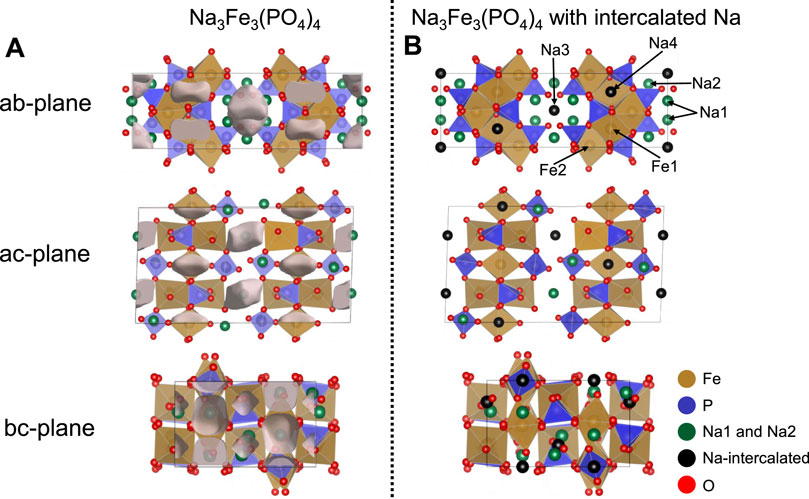
Figure 2. (A) Structural illustrations of Na3Fe3(PO4)4 with identified void spaces depicted as semi-transparent grey clouds. (B) Structural illustrations of Na3Fe3(PO4)4 showing the Na intercalation sites identified by void space analysis in (A). For both (A,B) sodium atoms in the pristine Na3Fe3(PO4)4 are displayed in green spheres, oxygen in red spheres, iron in brown octahedra, and phosphates in blue tetrahedra. The intercalation sites in (B) are depicted as black spheres.
As an intercalation cathode, Na3Fe3(PO4)4 is synthesized in the charged state, i.e., additional ions can be intercalated into the structure, resulting in compositions of LixNa3Fe3(PO4)4 or Na3+xFe3(PO4)4 depending on whether Li- or Na-ions are intercalated. Thus, additional sites must exist in the Na3Fe3(PO4)4 structure. Using void-space analysis (Spackman et al., 2021), we have located areas in the structure that are lower in electron density and thus likely to represent the ion-intercalation sites. These lower electron density areas are illustrated as semi-transparent grey clouds in Figure 2A. This corresponds well with the i1 and i2 interstitial Na-sites determined by Shinde et al. (2019) using migration barrier energy profiles derived from bond valence site energy models for Na diffusion. The identified intercalation sites have been added to the structural model for Na3Fe3(PO4)4 as Na3/Li3 on Wyckoff position 4e and Na4/Li4 on Wyckoff position 4c. This is shown in Figure 2B; Supplementary Table S1. The Na4-site is positioned inside the FeO6-PO4 layers in a distorted [NaO8] cube with Na-O distances ranging from 2.26–3.01 Å. The Na3-site sits in the middle of the interlayer space. The coordination is also distorted cubic, but with Na-O distances of 2.64–3.38 Å, it is substantially larger than the Na4-site. An Na-atom on the Na3-site is likely to vibrate significantly. Rietveld refinement of the Na-occupancies on the intercalation positions in the pristine material shows that no Na is placed on these positions in the as-synthesized material. This refinement, however, suggests that the Na1- and Na2-sites are only occupied ∼70% and 83%, respectively, bringing the composition to Na2.86Fe3(PO4)4 and the average Fe-valence state to +3.05.
3.2 Electrochemical characterization
Trad et al. (Trad et al., 2010a; Trad et al., 2010b) previously studied Na3Fe3(PO4)4 both as a Li- and Na-ion battery material, showing that just under two ions can be intercalated into Na3Fe3(PO4)4. Ideally, reduction of all Fe3+ to Fe2+ in the Na3Fe3(PO4)4 should be possible, which would provide a capacity of ∼120 mAh/g (based on the pristine Na3Fe3(PO4)4 material), equivalent to intercalating three alkali ions. However, we find, in line with the results by Trad et al., that even using a low current rate (C/25 per single ion intercalated, i.e., 1.7 mA/g), only intercalates 2.2 Li-ions (88 mAh/g) and 1.7 Na-ions (69 mAh/g) according to Columbic counting (Figures 1E, F).
As the material is cycled against Na, the discharge capacity drops more than 10% after just five cycles (Figure 1D). The fact that the capacity during charge is larger than that during discharge points to electrolyte degradation, which is likely since ClO4− in propylene carbonate is more prone to form CO2, HClO4, and acetone radicals compared to other electrolytes (Leggesse et al., 2013). However, the drop in capacity may also be attributed to change in the crystal structural trapping Na- or Li-ions. During discharge vs. Na, Na3+xFe3(PO4)4 shows a relatively small potential hysteresis, i.e., <50 mV for some regions (Supplementary Figure S2). Low hysteresis for Na-ion intercalation is also reported by Trad and co-workers, who additionally show that the intercalation of Li-ion creates a much more significant hysteresis, suggesting a lower overpotential and less kinetic limitations in the Na-cell (Trad et al., 2010b).
3.3 57Fe Mössbauer spectroscopy
It has previously been speculated that intercalation of Li-ions causes reduction of iron at both iron sites, while intercalation of Na-ions preferentially reduces iron on the Fe2-site. To investigate which iron site participates in the redox reaction process, we performed 57Fe Mössbauer spectroscopy (Figure 3) of the pristine Na3Fe3(PO4)4 and the samples prepared by discharging vs. Li and Na, i.e., Li2.2Na3Fe3(PO4)4 and Na4.7Fe3(PO4)4 (same samples as investigated by PXRD in Figure 1). In all Mössbauer spectra, a small magnetic pattern (spectral area ∼5%) was observed due to the hematite, α-Fe2O3, impurity. The hyperfine parameters obtained from simulating the patterns can be found in Supplementary Table S5.
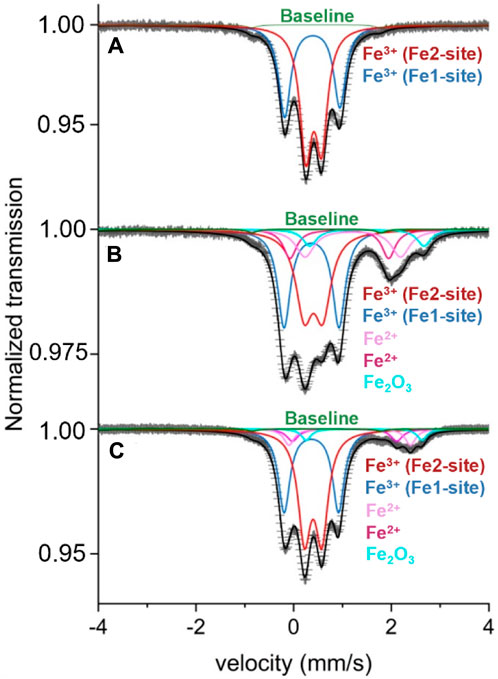
Figure 3. Mössbauer spectra of (A) Na3Fe3(PO4)4, (B) Na4.7Fe3(PO4)4 (discharged using C/25), and (C) Li2.2Na3Fe3(PO4)4 (discharged using C/50). The data is shown as black scatter points, the baseline in green, Fe3+ doublets in red (Fe2-site) and blue (Fe1-site), Fe.
As expected from the composition, the pristine Na3Fe3(PO4)4 yields a spectrum with two distinct Fe3+ doublets with slightly asymmetric doublet lines. The two Fe3+ doublets can be assigned to the crystallographic 4d and 8f sites, i.e., Fe1 and Fe2, respectively (Figure 3A). The spectral area ratio, Aratio = A (4d)/A (8f), is 0.63 (4). An additional spectrum recorded at 82 K gave Aratio = 0.52 (4), which is close to the expected ratio of 0.50 for fully Fe-occupied 4d and 8f sites. The reason for different experimental spectral area ratios originates from different temperature-dependent f-factors for Fe at the 4d and 8f sites. At lower temperatures, the f-factors tend to be similar (Ericsson and Wäppling, 1976).
The discharged samples reveal again two Fe3+ doublets (i.e., for the Fe3+1- and Fe3+2-sites) as well as three Mössbauer patterns attributed to Fe2+ (Figures 3B, C). The compositions obtained from columbic counting, i.e., Na4.7Fe3(PO4)4 and Li2.2Na3Fe3(PO4)4, suggest average Fe oxidation states of 2.43 and 2.26, respectively. However, based on the observed spectral areas, the Mössbauer data suggests average Fe-oxidation states of 2.70 and 2.85. This difference may be caused either by a lower degree of ion intercalation during discharge than suggested by columbic counting, e.g., due to electrochemical-driven side reactions or by post-discharge oxidation.
The Mössbauer data also provides valuable information about the redox activity of the Fe1 and Fe2 sites. For the Na4.7Fe3(PO4)4 sample (Figure 3B), the spectral Fe3+ Aratio changes to 0.95 (4). This indicates that the Fe3+ on the Fe2-site has been significantly reduced as compared to the Fe1-site, i.e., the Fe2-site is more active during the intercalation of Na-ions. For Li2.2Na3Fe3(PO4)4, a similar effect is observed, but the preferred reduction of the Fe2-site is less pronounced compared with Na intercalation. These discoveries are in good agreement with the findings by Trad et al. (Trad et al., 2010a; Trad et al., 2010b). Assigning the Fe2+ patterns to the Fe1 (4d) or Fe2 (8f) sites has not been possible, but according to the above-found preferential Fe3+ reduction of the Fe2 site, a major part of these Fe2+ patterns can be ascribed to the 8f sites. Tentatively, the third doublet with a deviating central shift is attributed to Fe2+ at the 4d site (Supplementary Table S5).
3.4 Structural evolution during ion-intercalation
To elaborate on the structural evolution during ion intercalation in Na3Fe3(PO4)4, we investigated the processes of both Li- and Na-ion intercalation with operando PXRD (Figures 4A–F). Previous PXRD investigations have shown that Na-ion intercalation into the structure causes a cell expansion of ∼50 Å3, and it has been suggested that the material disorders structurally upon intercalation of Na-ions when reaching ∼2.36 V, while insertion of Li-ions barely causes any structural changes (Trad et al., 2010a). It has been speculated that the origin of this difference, besides the difference in the size of the intercalated ions, derives from the Na-ions in the synthesized material not taking part in the mobilization of the Li-ions during operation, whereas they do upon Na-ion intercalation (Trad et al., 2010a; Trad et al., 2010b). This might explain why the potential hysteresis of ion intercalation differs between Li- and Na-ions.
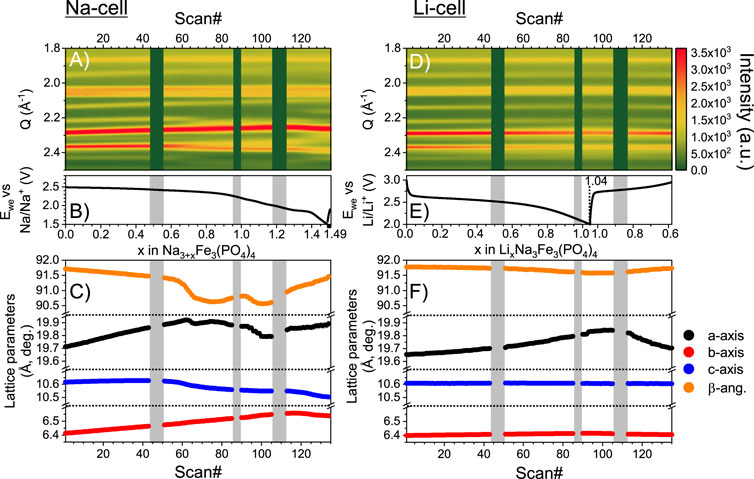
Figure 4. (A) Contour plot of the operando PXRD data from intercalation of Na into Na3Fe3(PO4)q. (B) Potential profile collected simultaneously with the operando PXRD data. (C) Rietveld refined lattice parameters evolution during intercalation of Na. (D–F) Same types of plots as (A-C) but for Li intercalation into the same Na3Fe3(PO4)4 framework. Grey vertical bars shown on both data sets illustrate loss of beam.
In line with previous findings, our investigation shows that the structure of Na3Fe3(PO4)4 evolves like a solid solution during the intercalation of both Li- and Na-ions (Figures 4A, E) (Trad et al., 2010a; Trad et al., 2010b). Hence, for the sequential refinement of the operando PXRD, only one phase has been employed to obtain good descriptions of the data for the entire cycling processes (see agreement factors in Supplementary Figure S3). Nearing the end of discharge, the agreement factors increase ∼1% for the Na-cell, which may be linked to some reflections becoming more diffuse at this stage. However, we find no significant change in the scale factor between the first PXRD data before discharge (Scan# 1) and the PXRD data taken at the end of discharge for the Na-cell (Scan# 133), which suggests that the reflected intensity, and thereby the fraction of crystalline material, is not decreasing, i.e., structural disorder is not induced by intercalation.
The evolution of the lattice parameters is shown in Figures 4C, F. During the intercalation of Li-ions, the a-axis, i.e., the interlayer space, increases continuously. At the same time, no significant changes are observed for the b- and c-axis, i.e., the structure of the layers remain relatively unchanged. For the Na-intercalation, the a- and b-axis expand during intercalation of the first ∼0.7 Na-ions, while the c-axis remains relatively constant. Upon continued Na intercalation, the a- and c-axis begin to decrease slightly, while the b-axis continues to increase. Expansion of the structure is expected due to the reduction of iron and the occupation of the intercalation sites, while contraction may appear as the intercalated ions shield the repulsion within the framework. The overall volume change during discharge is ∼1.6 and ∼2.6 vol% against Li and Na, respectively. Such relatively small volume changes could prove as an advantage in, e.g., solid state batteries.
Unfortunately, the quality of the operando PXRD data does not allow refinement of the Na-occupancies. However, insights can be gained by comparing the intensity ratio of the (110)- and overlapping (002)-(−111)-(111)-peaks at ∼1.0 and 1.2 Å−1 in the data to those in simulated patterns with full occupancies on the Na3-, Na4- and both sites (see Supplementary Figures S5, S6). In the pristine material, i.e., where the Na3- and Na4-sites are empty, these peaks display a 3:4 intensity ratio. During discharge, the intensities approach a 1:1 ratio, which, based on comparison to the simulated patterns, suggests a preference for intercalation on the Na4-site, i.e., preferential intercalation on the intralayer sites. Intercalation on the interlayer Na3-site would cause the intensity ratio to decrease.
3.5 Structural relaxation and ion-diffusion after discharge
Operando PXRD revealed that ion intercalation results in a unit cell expansion of ∼1 and ∼2% for the Li- and Na-ion cells, respectively (Table 1; Supplementary Figure S4), consistent with previous reports (Trad et al., 2010a; Trad et al., 2010b). Besides the samples studied by operando PXRD, two discharged electrodes were also investigated by ex-situ PXRD. These were prepared by discharging vs. Li and Na yielding compositions of Li2.2Na3Fe3(PO4)4 and Na4.7Fe3(PO4)4 according to columbic counting. The electrodes were retrieved from the cells, and ex-situ PXRD was collected after letting the electrodes relax in the glovebox for ∼30 days after the discharge was performed. Rietveld refinement of ex-situ PXRD from the discharged-relaxed electrodes (Figures 1B, C) reveals a ∼0.6 vol% contraction of the unit cell volumes of Li2.2Na3Fe3(PO4)4 and Na4.7Fe3(PO4)4 as compared to the pristine Na3Fe3(PO4)4 (Table 1; Supplementary Table S1, S3, S4). Hence, the unit cell volumes of the discharged materials, i.e., Li2.2Na3Fe3(PO4)4 and Na4.7Fe3(PO4)4, contract ∼1.6 and ∼2.6 vol% after the dynamic discharge (based on refinement of the operando PXRD at the end of discharge). Thus, structural relaxation must occur at open circuit conditions after discharge.
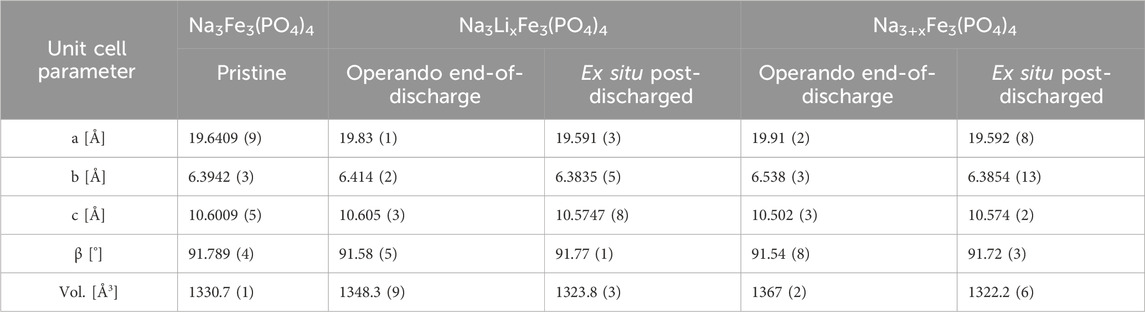
Table 1. Comparison of the unit cell parameters for the pristine Na3Fe3(PO4)4.
Rietveld refinement of the ex-situ PXRD data of the electrodes post-discharge also reveals that the intercalated Na-ions occupy both the interlayer Na3- and intralayer Na4-sites. However, there is a preference for the interlayer Na3-site, i.e., the occupancy of the Na4:Na3 sites shows a 1:2.7 ratio (Supplementary Table S4). This is consistent with the intensity ratio of the (110) and overlapping (002)-(−111)-(111)-peaks being 3:4. Note also that no significant changes in the occupancy of the Na-sites (Na1 and Na2) are observed as compared to the pristine Na3Fe3(PO4)4 (Supplementary Table S4). Hence, during the relaxation after discharge, some Na-ions diffuse from the Na4-site to the Na3-site, which results in a contraction of the unit cell parameters likely due to Na-ions on the Na3-site shielding the repulsion between the FeO6-PO4 layers.
The indication of post-discharge ion diffusion suggests that similar phenomena may occur in other intercalation electrodes and cause structural relaxation at open-circuit conditions. In this study, observation of Na-diffusion is enabled by the detectable difference in the PXRD data between different Na-site occupations. This is often not possible in studies of Li-ion electrode materials due to the low X-ray scattering power of Li. Hence, the phenomena may, for good reasons, be missed. One such case is LixV2(PO4)3, where operando PXRD reveals that the formation of LiV2(PO4)3 by electrochemical Li-intercalation in V2(PO4)3 (i.e., discharge) causes an increase in the unit cell volume of ∼5% (Sørensen et al., 2018). In contrast, at quasi-equilibrium conditions (i.e., after chemical lithiation), LiV2(PO4)3 has a unit cell volume ∼2.5% smaller than that of V2(PO4)3 (Yin et al., 2003). Hence, structural relaxation may be caused by Li-diffusion between vacant sites in the structure due to site preferences. In fact, cases are also known wherein host lattice atoms participates in the migration, e.g., in spinel LixFe2O3, Li-insertion pushes Fe from the 8a to the 16c site, however, due to site preferences, during relaxation Fe returns to the 8a site, indicating that Li moves to the 16c site (Park et al., 2011).
Within the topic of structural relaxation, lithium iron phosphate, LiFePO4, is a well-known case, wherein metastable LixFePO4 (x = 0–1) solid solutions form during fast discharge (i.e., high current rates). Upon relaxation, the LixFePO4 solid solutions change composition (evidenced from changes in the lattice parameters) towards LiFePO4 and FePO4 (Liu et al., 2014). Hence, Li-ions must diffuse in the FePO4 framework to cause changes in the composition and phase segregation. However, it is important to note that although the process involves Li-ion diffusion at open-circuit conditions, the phenomenon occurs at a macroscopic scale and likely across crystallite boundaries in LiFePO4. In contrast, it seemingly only occurs at an atomistic local scale within Na3+xFe3(PO4)4 and LixV2(PO4)3. Also, the driving forces are most likely different, i.e., in the Na3+xFe3(PO4)4 and LixV2(PO4)3 it occurs due to site preferences, while in LiFePO4, the driving force is the compositional metastability of LixFePO4 solid solutions. This difference, however, underlines that understanding structural relaxation during open circuit conditions is not only important for two-phase systems but should also be considered for single-phase (i.e., solid solution) systems, as the electrochemical properties may be significantly affected by intermittent charge or discharge and the duration of open circuit conditions.
4 Conclusion
Layered sodium iron phosphate, Na3Fe3(PO4)4, was prepared via hydrothermal synthesis and studied as a battery electrode against both Li- and Na-ion intercalation. Galvanostatic discharge provided intercalation of ∼2.2 Li and ∼1.7 Na (based on columbic counting) at potential plateaus around ∼2.5 V. The redox process occurs, according to Mössbauer spectroscopy, primarily at one of the two Fe-sites during Na-intercalation, while there appears to be little site preference for the Fe-activity during Li-intercalation. During cycling, operando PXRD shows that the structure of Na3+xFe3(PO4)4 and LixNa3Fe3(PO4)4 behaves as solid solutions and only undergoes minor structural changes. Interestingly, the data indicate that during dynamic discharge, Na-ions are intercalated into the intralayer sites within the FeO6-PO4 layers, while after relaxation, the ions to some extent diffuse to the interlayer sites, which also cause a ∼1.6 and ∼2.6 vol% contraction of the unit cell volume for LixNa3Fe3(PO4)4 and Na3+xFe3(PO4)4, respectively.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author contributions
CJ: Conceptualization, Formal Analysis, Investigation, Methodology, Writing–original draft. MJ: Formal Analysis, Investigation, Methodology, Validation, Writing–review and editing. TE: Formal Analysis, Investigation, Writing–review and editing. LH: Formal Analysis, Investigation, Writing–review and editing. CC: Supervision, Writing–review and editing. IN: Formal Analysis, Validation, Writing–review and editing. WB: Project administration, Validation, Writing–review and editing. DR: Conceptualization, Formal Analysis, Funding acquisition, Investigation, Project administration, Resources, Supervision, Validation, Visualization, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. The work presented in this was funded by the Carlsberg Foundation (Grant No. CF17-0823) and the Novo Nordisk Foundation (Grant DEMBATT, Grant no. NNF20OC0062068). Travelling expenses related to beamtimes were reimbursed by the Danish instrument center DanScatt. We acknowledge MAX IV Laboratory for time on Beamline DanMAX under Proposal 20211012. Research conducted at MAX IV is supported by the Swedish Research council under contract 2018-07152, the Swedish Governmental Agency for Innovation Systems under contract 2018-04969, and Formas under contract 2019-02496. DanMAX is funded by the NUFI grant no. 4059-00009B.
Acknowledgments
The authors thank the Carlsberg Foundation (Grant No. CF17-0823) and the Novo Nordisk Foundation (Grant no. NNF20OC0062068) for the financial support of this research. We thank the Danish Agency for Science, Technology, and Innovation for funding the instrument center DanScatt. Furthermore, the authors acknowledge DESY (Hamburg, Germany), a member of the Helmholtz Association HGF, for the provision of experimental facilities at PETRA III (Dippel et al., 2015) and would like to thank Volodymyr Baran for assistance in using beamline P02.1.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbael.2024.1433241/full#supplementary-material
References
Basham, M., Filik, J., Wharmby, M. T., Chang, P. C. Y., El Kassaby, B., Gerring, M., et al. (2015). Data analysis WorkbeNch (DAWN). J. Synchrotron Radiat. 22 (3), 853–858. doi:10.1107/s1600577515002283
Bergerhoff, G., Berndt, M., and Brandenburg, K. (1996). Evaluation of crystallographic data with the program DIAMOND. J. Res. Natl. Inst. Stand. Technol. 101 (3), 221. doi:10.6028/jres.101.023
Blake, R. L., Hessevick, R. E., Zoltai, T., and Finger, L. W. (1966). Refinement of the hematite structure. Am. Mineralogist 51 (1-2), 123–129.
Borkiewicz, O. J., Shyam, B., Wiaderek, K. M., Kurtz, C., Chupas, P. J., and Chapman, K. W. (2012). The AMPIX electrochemical cell: a versatile apparatus for in situ X-ray scattering and spectroscopic measurements. J. Appl. Cryst. 45 (6), 1261–1269. doi:10.1107/S0021889812042720
Christensen, C. K., Sørensen, D. R., Hvam, J., and Ravnsbæk, D. B. (2018). Structural evolution of disordered LixV2O5 bronzes in V2O5 cathodes for Li-ion batteries. Chem. Mater. 31 (2), 512–520. doi:10.1021/acs.chemmater.8b04558
Coelho, A. A. (2018). TOPAS and TOPAS-Academic: an optimization program integrating computer algebra and crystallographic objects written in C++. J. Appl. Crystallogr. 51 (1), 210–218. doi:10.1107/s1600576718000183
David, R., Pautrat, A., Kabbour, H., and Mentré, O. (2016). Common building motifs in Ba2Fe3(PO4)4·2H2O, BaFe3(PO4)3, and Na3Fe3(PO4)4: labile Fe2+/Fe3+ ordering and charge-dependent magnetism. Inorg. Chem. 55 (9), 4354–4361. doi:10.1021/acs.inorgchem.6b00071
Dippel, A.-C., Liermann, H.-P., Delitz, J. T., Walter, P., Schulte-Schrepping, H., Seeck, O. H., et al. (2015). Beamline P02.1 at PETRA III for high-resolution and high-energy powder diffraction. J. synchrotron Radiat. 22 (3), 675–687. doi:10.1107/S1600577515002222
Dyvoire, F., Pintard-screpel, M., and Bretey, E. (1980). The Phosphates Na3M2(III)(PO4)3 (M=Cr,Fe) and Na3Cr3(PO4)4 - crystallographic data and ionic-conductivity. Comptes Rendus Hebd. Des. Seances De. L Acad. Des. Sci. Ser. C 290 (10), 185–188. doi:10.1002/chin.198030016
Ericsson, T., and Wäppling, R. (1976). Texture effects in 3/2-1/2 mössbauer spectra. J. Phys. Colloques 37 (C6), C6-719–C6-723. doi:10.1051/jphyscol:19766150
Filik, J., Ashton, A. W., Chang, P. C. Y., Chater, P. A., Day, S. J., Drakopoulos, M., et al. (2017). Processing two-dimensional X-ray diffraction and small-angle scattering data in DAWN 2. J. Appl. Crystallogr. 50 (3), 959–966. doi:10.1107/s1600576717004708
Henriksen, C., Mathiesen, J. K., and Ravnsbæk, D. B. (2020). Improving capacity and rate capability of Li-ion cathode materials through ball milling and carbon coating – best practice for research purposes. Solid State Ionics 344, 115152. doi:10.1016/j.ssi.2019.115152
Huang, Y., Lin, Y.-C., Jenkins, D. M., Chernova, N. A., Chung, Y., Radhakrishnan, B., et al. (2016). Thermal stability and reactivity of cathode materials for Li-ion batteries. ACS Appl. Mater. Interfaces 8 (11), 7013–7021. doi:10.1021/acsami.5b12081
Jakobsen, C. L., Nielsen, K. S., Morgen, P., Bøjesen, E. D., and Ravnsbæk, D. B. (2021). Polymorphic purity and structural charge–discharge evolution of β-LiVOPO4 cathodes. J. Phys. Chem. C 125 (44), 24301–24309. doi:10.1021/acs.jpcc.1c05494
Jakobsen, C. L., Brighi, M., Andersen, B. P., Ducrest, G., Černý, R., and Ravnsbæk, D. B. (2022). Expanded solid-solution behavior and charge-discharge asymmetry in NaxCrO2 Na-ion battery electrodes. J. Power Sources 535, 231317. doi:10.1016/j.jpowsour.2022.231317
Jeong, M., Lee, H., Yoon, J., and Yoon, W.-S. (2019). O3-type NaNi1/3Fe1/3Mn1/3O2 layered cathode for Na-ion batteries: Structural evolution and redox mechanism upon Na (de) intercalation. J. Power Sources 439, 227064. doi:10.1016/j.jpowsour.2019.227064
Kang, K., Meng, Y. S., Breger, J., Grey, C. P., and Ceder, G. (2006). Electrodes with high power and high capacity for rechargeable lithium batteries. Science 311 (5763), 977–980. doi:10.1126/science.1122152
Lajmi, B., Hidouri, M., Rzeigui, M., and Ben Amara, M. (2002). Reinvestigation of the binary diagram Na3PO4–FePO4 and crystal structure of a new iron phosphate Na3Fe3(PO4)4. Mater. Res. Bull. 37 (15), 2407–2416. doi:10.1016/S0025-5408(02)00940-6
Leggesse, E. G., Lin, R. T., Teng, T.-F., Chen, C.-L., and Jiang, J.-C. (2013). Oxidative decomposition of propylene carbonate in lithium ion batteries: a DFT study. J. Phys. Chem. A 117 (33), 7959–7969. doi:10.1021/jp403436u
Liu, H., Strobridge, F. C., Borkiewicz, O. J., Wiaderek, K. M., Chapman, K. W., Chupas, P. J., et al. (2014). Capturing metastable structures during high-rate cycling of LiFePO4 nanoparticle electrodes. Science 344 (6191), 1252817. doi:10.1126/science.1252817
Liu, R., Liu, H., Sheng, T., Zheng, S., Zhong, G., Zheng, G., et al. (2018). Novel 3.9 V layered Na3V3(PO4)4 cathode material for sodium ion batteries. ACS Appl. Energy Mater. 1 (8), 3603–3606. doi:10.1021/acsaem.8b00889
Momma, K., and Izumi, F. (2011). VESTA3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 44 (6), 1272–1276. doi:10.1107/S0021889811038970
Mueller, T., Hautier, G., Jain, A., and Ceder, G. (2011). Evaluation of tavorite-structured cathode materials for lithium-ion batteries using high-throughput computing. Chem. Mater. 23 (17), 3854–3862. doi:10.1021/cm200753g
Padhi, A. K., Nanjundaswamy, K. S., and Goodenough, J. B. (1997a). Phospho-olivines as positive-electrode materials for rechargeable lithium batteries. J. Electrochem. Soc. 144 (4), 1188–1194. doi:10.1149/1.1837571
Padhi, A. K., Nanjundaswamy, K. S., Masquelier, C., and Goodenough, J. B. (1997b). Mapping of transition metal redox energies in phosphates with NASICON structure by lithium intercalation. J. Electrochem. Soc. 144 (8), 2581–2586. doi:10.1149/1.1837868
Padhi, A. K., Nanjundaswamy, K. S., Masquelier, C., Okada, S., and Goodenough, J. B. (1997c). Effect of structure on the Fe3+∕Fe2+ redox couple in iron phosphates. J. Electrochem. Soc. 144 (5), 1609–1613. doi:10.1149/1.1837649
Papp, J. F., Bray, E. L., Edelstein, D. L., Fenton, M. D., Guberman, D. E., Hedrick, J. B., et al. (2008). Factors that influence the price of Al, Cd, Co, Cu, Fe, Ni, Pb, rare earth elements, and Zn. Open-File Rep. doi:10.3133/ofr20081356
Park, S., Oda, M., and Yao, T. (2011). Relaxation structure analysis of Li inserted γ-Fe2O3. Solid State Ionics 203 (1), 29–32. doi:10.1016/j.ssi.2011.09.014
Pennington, W. T. (1999). DIAMOND– Visual crystal structure information system. J. Appl. Crystallogr. 32 (5), 1028–1029. doi:10.1107/S0021889899011486
Quilty, C. D., Bock, D. C., Yan, S., Takeuchi, K. J., Takeuchi, E. S., and Marschilok, A. C. (2020). Probing sources of capacity fade in LiNi0.6Mn0.2Co0.2O2 (NMC622): an operando XRD study of Li/NMC622 batteries during extended cycling. J. Phys. Chem. C 124 (15), 8119–8128. doi:10.1021/acs.jpcc.0c00262
Rodriguez-Carvajal, J. (2001). Recent developments of the program FULLPROF, commission on powder diffraction. IUCr Newsl. 26.
Schökel, A., Etter, M., Berghäuser, A., Horst, A., Lindackers, D., Whittle, T. A., et al. (2021). Multi-analyser detector (MAD) for high-resolution and high-energy powder X-ray diffraction. J. Synchrotron Radiat. 28 (1), 146–157. doi:10.1107/S1600577520013223
Shinde, G. S., Gond, R., Avdeev, M., Ling, C. D., Rao, R. P., Adams, S., et al. (2019). Revisiting the layered Na3Fe3(PO4)4 phosphate sodium insertion compound: structure, magnetic and electrochemical study. Mater. Res. Express 7 (1), 014001. doi:10.1088/2053-1591/ab54f4
Song, L., Tang, F., Xiao, Z., Cao, Z., Zhu, H., and Li, A. (2018). Enhanced electrochemical properties of polyaniline-coated LiNi0.8Co0.1Mn0.1O2 cathode material for lithium-ion batteries. J. Electron. Mater. 47 (10), 5896–5904. doi:10.1007/s11664-018-6453-9
Sørensen, D. R., Mathiesen, J. K., and Ravnsbæk, D. B. (2018). Dynamic charge-discharge phase transitions in Li3V2(PO4)3 cathodes. J. Power Sources 396, 437–443. doi:10.1016/j.jpowsour.2018.06.023
Spackman, P. R., Turner, M. J., McKinnon, J. J., Wolff, S. K., Grimwood, D. J., Jayatilaka, D., et al. (2021). CrystalExplorer: a program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 54 (3), 1006–1011. doi:10.1107/s1600576721002910
Trad, K., Carlier, D., Croguennec, L., Wattiaux, A., Lajmi, B., Ben Amara, M., et al. (2010a). A layered iron(III) phosphate phase, Na3Fe3(PO4)4: synthesis, structure, and electrochemical properties as positive electrode in sodium batteries. J. Phys. Chem. C 114 (21), 10034–10044. doi:10.1021/jp100751b
Trad, K., Carlier, D., Wattiaux, A., Ben Amara, M., and Delmas, C. (2010b). Study of a layered iron(III) phosphate phase Na3Fe3(PO4)4 used as positive electrode in lithium batteries. J. Electrochem. Soc. 157 (8), A947–A952. doi:10.1149/1.3442365
Vaalma, C., Buchholz, D., Weil, M., and Passerini, S. (2018). A cost and resource analysis of sodium-ion batteries. Nat. Rev. Mater. 3 (4), 18013. doi:10.1038/natrevmats.2018.13
Weber, R., Fell, C. R., Dahn, J. R., and Hy, S. (2017). Operando X-ray diffraction study of polycrystalline and single-crystal LixNi0.5Mn0.3Co0.2O2. J. Electrochem. Soc. 164 (13), A2992–A2999. doi:10.1149/2.0441713jes
Wurm, C., Morcrette, M., Rousse, G., Dupont, L., and Masquelier, C. (2002). Lithium insertion/extraction into/from LiMX2O7 compositions (M = Fe, V; X = P, as) prepared via a solution method. Chem. Mater. 14 (6), 2701–2710. doi:10.1021/cm020168e
Keywords: Li- and Na-ion batteries, cathode materials, structural relaxation, operando PXRD, polyanionic electrode, ion-intercalation
Citation: Jakobsen CL, Johansen M, Ericsson T, Häggström L, Christensen CK, Nielsen I, Brant WR and Ravnsbæk DB (2024) Ion-intercalation mechanism and structural relaxation in layered iron phosphate Na3Fe3(PO4)4 cathodes. Front. Batteries Electrochem. 3:1433241. doi: 10.3389/fbael.2024.1433241
Received: 16 May 2024; Accepted: 22 July 2024;
Published: 07 August 2024.
Edited by:
Christopher Johnson, Argonne National Laboratory (DOE), United StatesReviewed by:
John Vaughey, Argonne National Laboratory (DOE), United StatesJacob Olchowka, UMR5026 Institut de Chimie de la Matière Condensée de Bordeaux (ICMCB), France
Copyright © 2024 Jakobsen, Johansen, Ericsson, Häggström, Christensen, Nielsen, Brant and Ravnsbæk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dorthe Bomholdt Ravnsbæk, ZG9ydGhlQGNoZW0uYXUuZGs=
†These authors have contributed equally to this work