
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Batter. Electrochem., 18 December 2023
Sec. Next Generation Batteries and Technologies
Volume 2 - 2023 | https://doi.org/10.3389/fbael.2023.1293877
This article is part of the Research TopicWomen in Battery Science and TechnologyView all 13 articles
 Laura C. Merrill1*
Laura C. Merrill1* Daniel M. Long2†
Daniel M. Long2† Samantha G. Rosenberg3†Melissa L. Meyerson3Mila Nhu Lam3
Samantha G. Rosenberg3†Melissa L. Meyerson3Mila Nhu Lam3 Katharine L. Harrison1*†
Katharine L. Harrison1*†Li metal anodes are highly sought after for high energy density applications in both primary commercial batteries and next-generation rechargeable batteries. In this research, Li metal electrodes are aged in coin cells for a year with electrolytes relevant to both types of batteries. The aging response is monitored via electrochemical impedance spectroscopy, and Li electrodes are characterized post-mortem. It was found that the carbonate-based electrolytes exhibit the most severe aging effects, despite the use of LiBF4-based carbonate electrolytes in Li/CFx Li primary batteries. Highly concentrated LiFSI electrolytes exhibit the most minimal aging effects, with only a small impedance increase with time. This is likely due to the concentrated nature of the electrolyte causing fewer solvent molecules available to react with the electrode surface. LiI-based electrolytes also show improved aging behavior both on their own and as an additive, with a similar impedance response with time as the concentrated LiFSI electrolytes. Since I− is in its most reduced state, it likely prevents further reaction and may help protect the Li electrode surface with a primarily organic solid electrolyte interphase.
Li metal anodes are highly sought after for high energy density needs due to their high theoretical capacity (3,830 mAh/g) and low reduction potential (−3.04 V vs. SHE). Although recent work has focused largely on integrating Li metal anodes into next-generation rechargeable batteries, Li metal anodes have long been used in commercial Li primary batteries. A challenge relating to Li metal integration is that Li metal is highly susceptible to parasitic reactions that result from the reducing nature of Li. These parasitic reactions form the solid electrolyte interphase (SEI), a mosaic of solvent and salt decomposition products, both during Li electrodeposition/dissolution (charge/discharge) and while the cell is at rest. The successful design of Li metal batteries involves tuning this interphase to prevent continued reaction (or corrosion) of the Li metal.
Ideally, the SEI will passivate Li metal, preventing further reaction between it and the electrolyte. However, most realistic SEIs are “leaky” (allowing transport of electrons through the SEI or electrolyte to penetrate the SEI to the surface of the Li metal), which causes continued reaction between the Li metal and the electrolyte. Extensive parasitic reactions are likely to lead to battery failure due to the consumption of the Li metal anode and/or the electrolyte (Wood et al., 2018). This effect may be accounted for by adding an excess of Li metal or electrolyte to cells, although this compromises the energy density of the battery. Recent Li metal studies emphasize “lean” Li and/or electrolyte systems, with little to no excess materials; however, these studies have largely focused on rechargeability without addressing limitations with the extended aging of the cell (Liu et al., 2019; Niu et al., 2021).
Despite the reactivity of Li as described above, Li primary batteries are rated for long shelf lives while maintaining high energy density. For example, Li/FeS2 and Li/MnO2 commercial primary batteries are designed to have shelf lives of 10–20 years (Lennen et al., 2006; Reddy, 2011). In these cases, the SEI likely stabilizes, passivating the Li metal and preventing further parasitic reactions between the Li metal and the electrolyte. As these chemistries are well established and commercialized, little new research has been published. It is expected that additives are included in the electrolyte formulations for commercial batteries that aid in promoting lasting aging behavior, although exact formulations and additives are not reported in the open literature.
Understanding rechargeable Li metal anode calendar-aging/self-discharge mechanisms has recently gained interest, with a focus largely on the aging of electrodeposited or cycled Li metal. Electrodeposited Li is particularly susceptible to aging as it is highly porous, is composed of many small grains, and suffers from the tendency to form dendritic and other high surface area mossy structures. While bulk Li metal has been investigated within the scope of integrating protection layers or artificial SEIs, these studies are typically restricted to relatively short timeframes of hours to weeks (Yang et al., 2008; Kozen et al., 2017; Lin et al., 2019; Gao et al., 2020; Kolesnikov et al., 2020; Boyle et al., 2021; Merrill et al., 2021; Merrill et al., 2022). Predictive models and computational studies have been used to supplement these studies and can be used to determine limitations over longer term aging (Wood et al., 2018; Dessantis et al., 2023). Despite this, shelf-life is a growing concern with the development of rechargeable Li metal batteries, as commercial batteries may rest from the time when the battery is manufactured to when the final electronic device reaches a consumer. An understanding of the interactions between the electrolyte and Li metal over extended periods of time is needed to design batteries with long shelf lives (Aurbach et al., 1994; Aurbach et al., 1995; Aurbach and Schechter, 2001; Yang et al., 2008; Morales-Ugarte et al., 2019).
We here investigate the aging behavior of Li metal with electrolyte chemistries used with both Li-primary and next-generation rechargeable Li batteries over the course of a year. Li metal symmetric cells were aged with the various electrolyte chemistries while electrochemical impedance spectroscopy (EIS) was used to monitor the interfacial impedance which can be correlated with SEI growth. After the cells were aged, the electrodes were characterized post-mortem through X-ray photoelectron spectroscopy (XPS) and cryo-scanning electron microscopy (SEM)/focused ion-beam milling (FIB). Cells were also cycled after aging and compared to pristine cells to evaluate overpotential trends. We find that highly concentrated electrolytes and LiI additives cause the interfacial impedance to plateau with time, suggesting that the Li electrode eventually becomes passivated in these solutions. Carbonate-based electrolytes are found to form the least stable interphase at the Li electrode surface, evidenced by both a greater magnitude of interfacial impedance and continued increase in impedance with time. We find that aging generally only affects the first nucleation overpotential when the Li symmetric cells are cycled after aging; however, it is likely that this will impact the rate capability of an aged cell or the capacity of a cell with minimal excess Li and electrolyte inventory.
All electrolytes were prepared inside an argon-filled glovebox (less than 0.2 ppm H2O and 0.2 ppm O2). Solvents, or relevant solvent mixtures, were dried over activated alumina for at least 48 h prior to use. Solvents studied include 1,3-dioxolane (DOL, Sigma Aldrich), 1,2-dimethoxyethane (DME, Sigma Aldrich), ethylene carbonate (EC, Sigma Aldrich), ethyl methyl carbonate (EMC, Sigma Aldrich), propylene carbonate (PC, Sigma Aldrich), and vinylene carbonate (VC, Sigma Aldrich). Lithium bis(fluorosulfonyl)imide (LiFSI, Oakwood Chemical) and lithium bis(trifluoromethanesulfonyl)imide (LiTFSI, Sigma Aldrich) were dried at 100 °C under vacuum in a heated antechamber for at least 48 h prior to use. Lithium tetrafluoroborate (LiBF4, Sigma Aldrich) and lithium hexafluorophosphate (LiPF6, Sigma Aldrich) were dried at 60 °C for 48 h in a heated antechamber prior to use. The electrolytes, minus the 4 M LiFSI in DME electrolyte, were prepared by weighing the desired amount of salt to reach the molarity and dissolving it in the corresponding volume of solvent. The 2 wt% VC added to the LiPF6-based electrolyte was determined based on the total solution mass. The 10 mol% LiI was based on the moles of LiTFSI in the electrolyte. The 4 M LiFSI in DME electrolyte was made as described in the literature by weighing the amount of LiFSI and DME to reach a 1:1.4 mol ratio of LiFSI to DME (Qian et al., 2015). The LiI and LiBF4 electrolytes, identified for use with Li primary batteries, were made by dissolving the mass required to reach the desired molarity (0.75 M for LiI and 1 M for LiBF4) in the corresponding volume of the solvent mixtures described in Table 1. Solvent mixtures were made by measuring each solvent to achieve the desired volumetric or gravimetric ratios (described in Table 1).
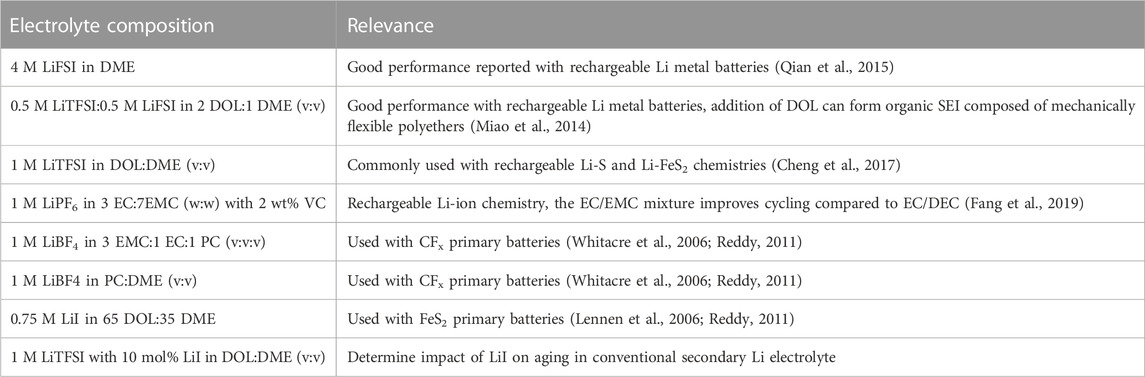
TABLE 1. Electrolyte composition and relevance to primary or secondary Li batteries.
Li/Li symmetric coin cells (SUS316L, 2032, Hohsen) were assembled in an argon-filled glovebox (less than 0.2 ppm H2O and 0.2 ppm O2). The assembled cells contained a wave spring, a 0.2 mm spacer, and two 750 µm Li (Alfa Aesar) electrodes separated by two Celgard 2325 sheets with 80 µL of electrolyte. The Li electrodes used were 16 mm diameter punches and used as received from Alfa Aesar. Cells were made in duplicates to be monitored with age.
Electrochemical impedance spectroscopy (EIS) measurements were taken using a Biologic VMP3 Potentiostat. Frequency was scanned from 1 MHz to 0.1 Hz with an applied AC voltage of 5 mV around 0 V vs. open circuit potential (OCP). Ten data points were taken per decade of frequency and two EIS measurements were taken per measurement. The second EIS spectra were used for analysis. EIS was periodically taken around the same time over the course of a year. The first and second scans of EIS spectra are shown for select cells on Day 0 in Supplementary Figure S1. All cells appear to increase in impedance on the second scan, suggesting that the first scan does not break down the SEI layer and may partly contribute to some minor SEI formation. Cells were aged in duplicates. EIS data were fitted using ZView to determine the interfacial impedance as a function of time.
After aging, one of the duplicate cells was cycled on an Arbin battery cycler at 0.5 mA/cm2 for 2 mAh/cm2 with a cutoff voltage of ±1 V (with negative voltage limits extended to −1.5 V for the LiPF6-containing electrolyte and LiBF4 in PC:DME). Currents and capacities were decreased to 0.05 mA/cm2 for 0.5 mAh/cm2 for some cells as specified below. The other duplicate cell was disassembled inside an argon-filled glovebox using a Hohsen de-crimping die. The electrodes were then extracted, washed in dry solvent for 1 s, and then characterized.
Samples were cut from uncycled Li electrodes after aging and mounted onto a stub in an argon-filled glovebox. Sample transfer was completed using a Leica VCT500 vacuum cryo transfer system. Samples were first transferred to a Leica ACE600 system where they were coated with 10 nm of Pt. The samples were then transferred into a Thermo Fisher Scientific Scios 2 Focused Ion Beam/Scanning Electron Microscope (FIB/SEM) equipped with a Leica cryogenic sample stage cooled to −140°C. Top-down and cross-section SEM images were taken at 5 kV and 50 pA. Cross-sectioning was completed by milling with Ga+ ions at 16 kV at 7.5 nA and 3 nA, followed by polishing at 0.5 nA. An Octane Elite EDAX Energy dispersive X-ray spectroscopy (EDS) detector was used to collect elemental mapping information at 5 kV.
A Kratos Axis Supra XPS system with a monochromatic Al Kα X-ray source operating at a base pressure better than 2x10−9 Torr was used to take XPS measurements. Samples were transferred via an inert transfer arm to prevent ambient air exposure to the samples. Spectra shown in this work are of the pristine surface without sputtering and are taken on uncycled aged Li electrodes. The data was analyzed with CASA XPS by aligning the carbon spectra to 284.8 eV and using relative sensitivity factors (RSF) for the Kratos system, where F 1s is set to 1. Atomic percentages of elements present were calculated from the survey spectra using CASA XPS. The elements present in the electrolyte were selected, although an adequate signal was not always present within the survey spectra compared to the high-resolution spectra. High-resolution spectra were fitted using CASA XPS as well as a Gaussian/Lorentzian (30) line shape with a Shirley-type background. The full-width half maximum was capped at 1.8 due to the monochromatic X-ray source resulting in narrower peaks.
Firstly, the EIS responses of the symmetric Li cells were monitored as functions of time. All cells showed an increase in impedance response with time, albeit to different extents. The increase in the charge transfer resistance/interfacial impedance—represented by an increase in where the semi-circle crosses the x-axis on the right (low frequency) side—suggests a “buildup” of SEI on the Li anode. These are not perfect semi-circles, so it is assumed that the double layer at the interface can be modeled as an imperfect capacitor or a constant phase element (CPE) (Figure 1). Here, we focus only on trends from the Nyquist plots and the interfacial impedance from fits.
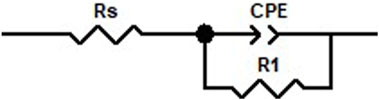
FIGURE 1. Modified Randles circuit used as equivalent circuit to fit the Nyquist plots. Rs represents the bulk solution resistance. Double layer capacitance is modeled using the CPE, and the interfacial impedance is captured by the charge transfer resistance, R1.
We began by analyzing Li/Li symmetric cells with carbonate-containing electrolytes (Figure 2). These electrolytes are typically used with Li-ion chemistries (for the LiPF6 electrolyte) or with primary Li batteries (for the LiBF4 electrolytes, typically used with CFx chemistries) (Xu, 2004; Reddy, 2011). We consider these separately from the ether-only electrolytes as the carbonates are typically reported to have decreased stability against Li metal (Liu et al., 2023). The LiBF4-based electrolytes in carbonates and carbonate-ether mixtures both show significant increases in interfacial impedance and semi-circle sizes with age. This suggests that the Li continues to react with the electrolyte, resulting in a significant aging effect. Carbonate solvents are not typically stable against the Li metal and may accelerate this effect (Aurbach et al., 1994; Aurbach et al., 1995). The “Day 0” EIS spectra for both LiBF4 electrolytes, taken immediately after cell assembly, have interfacial impedances less than 100 Ω—two orders of magnitude less than the cell after a year of aging. Since the LiBF4 electrolytes are suspected to be used with commercial Li/CFx primary batteries, it is expected that additives are incorporated into the battery to help improve aging behavior. Replicate data for the LiBF4 electrolytes are shown in Supplementary Figures S2, S3. While there is some variation from cell to cell, the trends of increasing impedance with time and the order-of-magnitude changes between pristine and aged cells are repeatable.

FIGURE 2. Nyquist plots of Li/Li cells aged in their respective electrolytes over the course of 1 year. Cells shown contain carbonate-type solvents, (A) 1 M LiPF6 in 3 EC:7 EMC with 2 wt% VC, (B) 1 M LiBF4 in 3 EMC:1 EC:1 PC, and (C) 1 M LiBF4 in PC:DME. (D) Interfacial impedance from EIS fits vs. time (in days) for 1 M LiPF6 in 3 EC:7 EMC with 2 wt% VC (pink squares), 1 M LiBF4 in 3 EMC:1 EC:1 PC (blue diamonds), and 1 M LiBF4 in PC:DME (yellow circles). Duplicate measurements are shown in Supplementary Material, Supplementary Figures S2–S4.
The LiPF6-based electrolyte shows an even larger interfacial impedance both in the pristine (Day 0) cell and after aging. Replicate data for the LiPF6 electrolyte is shown in Supplementary Figure S4. LiPF6-based electrolytes are typically used with graphite or silicon anodes (Li-ion chemistries) rather than Li metal itself. The pristine interfacial impedance is around 2000 Ω and increases to about 14,000 Ω, representing a smaller relative increase than the Li electrodes aged in the LiBF4 electrolyte. Since the interfacial impedance was significantly high as early as “Day 0” after no aging, the reaction between the LiPF6 electrolyte and the Li may be very fast. Therefore, it is expected that there is a greater amount of total reaction occurring between the Li surface and the LiPF6 electrolyte, with many of the reactions occurring immediately upon assembly of the cell. The smaller relative increase in interfacial impedance with time suggests that the LiPF6-based electrolyte has a smaller amount of SEI “build-up” over time than the LiBF4-based electrolytes. This may suggest that the initial SEI components that form with the LiPF6-based electrolyte are more passivating and less leaky than the SEI constituents that form with the LiBF4-based electrolyte. This difference may prevent the use of LiPF6-based electrolytes in primary batteries.
We next examined the Nyquist plots of the electrolytes under investigation that are based on ether-type solvents. These include a dual salt-based electrolyte (0.5 M LiTFSI:0.5 M LiFSI in 2 DOL:1 DME, “bisalt”) and 4 M LiFSI electrolyte (Figures 3B, C), which have been demonstrated to exhibit good Li metal cycling performance (Miao et al., 2014; Qian et al., 2015; Merrill et al., 2021). The 1 M LiTFSI in DOL:DME electrolyte, which is commonly used with secondary Li-S and Li-FeS2 batteries, is shown in Figure 3A (Li et al., 2015). Figure 3D shows EIS spectra of 0.75 M LiI in 65 DOL:35 DME, which is used with commercial primary Li-FeS2 batteries (Lennen et al., 2006; Reddy, 2011). Replicate EIS data for the ether-based electrolytes are shown in Supplementary Figures S5–S8. The bisalt and LiTFSI electrolytes exhibit very similar aging behavior: both have a comparable increase in interfacial impedance with time, although the interfacial impedance of the cell with the LiTFSI electrolyte appears to begin plateauing around 10 months.
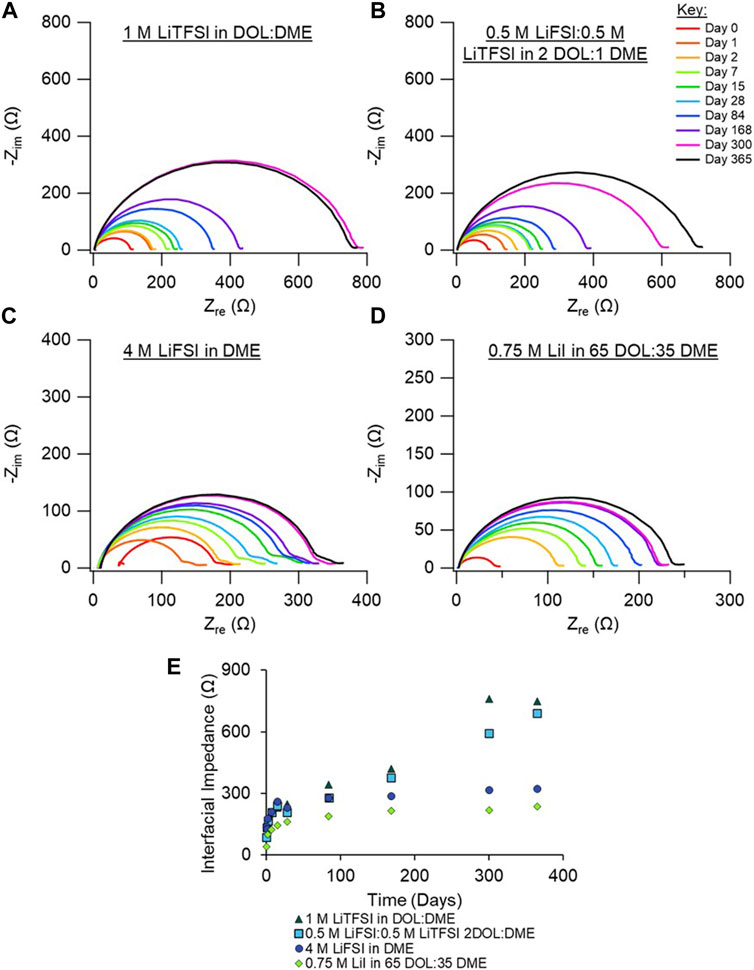
FIGURE 3. Nyquist plots of Li/Li symmetric cells aged in their respective electrolytes over the course of 1 year. Cells shown contain ether-based electrolytes, (A) 1 M LiTFSI in DOL:DME, (B) 0.5 M LiFSI:0.5 M LiTFSI in 2 DOL:1 DME, (C) 4 M LiFSI in DME, and (D) 0.75 M LiI in 65 DOL: 35 DME. (E) Interfacial impedance from EIS fits vs. time (in days) for 1 M LiTFSI in DOL:DME (dark green triangles), 0.5 M LiFSI:0.5 M LiTFSI in 2 DOL:1 DME (turquoise squares), 4 M LiFSI in DME (blue circles), and 0.75 M LiI in 65 DOL: 35 DME (green diamonds). Duplicate measurements are shown in Supplementary Material, Supplementary Figures S5–S8.
The 4 M LiFSI and the LiI electrolytes exhibit similar impedance responses, with a lower increase in interfacial impedance compared to the other electrolytes studied here. It is expected that the highly concentrated nature of the 4 M LiFSI prevents excess reaction between the electrolyte and the Li surface, considering that nearly all DME molecules in the solution are coordinated with an Li ion. Therefore, there are fewer species readily available to react with the Li metal. We note that the leftward shift of the full EIS spectra for the cell with the 4 M LiFSI electrolyte between Days 0 and 1 is likely an artifact of poor wetting of the separator, given that the semi-circle’s size is constant. After 1 day, the system appears to be fully wetted, as indicated by a decrease in the solution resistance. For the cell with the LiI electrolyte, the low interfacial impedance is likely due to the anion I− already being in its most reduced state, preventing further reaction with the Li metal. Although both DOL and DME could react with the Li surface, the extent of reaction is small given the small impedance rise with time. From the XPS analysis, discussed below, little I- species are present in the SEI, suggesting that the interfacial impedance increase is largely due to solvent breakdown on Li. Altogether, the ethereal solvents have greater reductive stabilities compared to the carbonate electrolytes; therefore, they should have better aging behavior than the carbonate electrolytes, as observed here.
Given the improved impedance response observed of the cell with the LiI electrolyte, we also investigated the addition of LiI to the LiTFSI electrolyte. The resulting EIS behavior, shown in Figure 4 and with replicate data shown in Supplementary Figure S9, is very similar to LiI alone, with low overall impedance and a minimal relative increase in impedance (compared with the LiTFSI electrolyte shown in Figure 3A). This suggests that the addition of LiI may help create a more preferential or protective interphase and/or prevent the decomposition of LiTFSI at the Li surface. LiI has been reported as an additive with LiTFSI-based electrolytes for Li-S flow batteries to improve performance, although LiI becomes depleted during cycling (Meyerson et al., 2022). We note that I− oxidizes at about 3.6 V versus Li/Li+, so it would be more appropriate as an additive to promote a passivating and stable SEI than as a salt in a rechargeable full cell battery with cathodes that operate near or above the I− oxidation potential. LiI has been demonstrated as an electrolyte additive in prior literature for Li-ion and Li/S batteries to improve interfaces (Komaba et al., 2003; Wu et al., 2015; Meyerson et al., 2022).
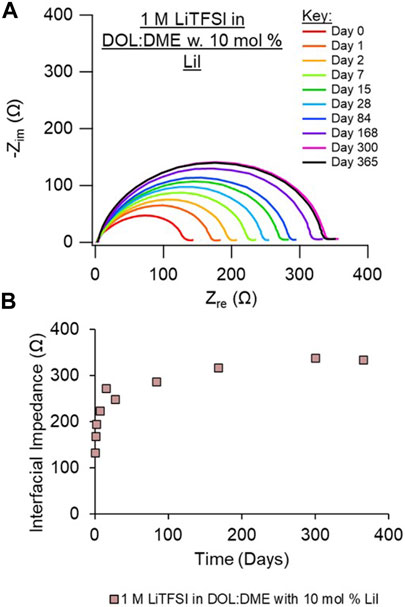
FIGURE 4. (A) Nyquist plots of an Li/Li symmetric cell aged over the course of 1 year. (B) Interfacial impedance from EIS fits vs. time (in days). Electrolyte investigated is 1 M LiTFSI in DOL/DME with 10 mol% LiI. Duplicate measurements are shown in Supplementary Material, Supplementary Figure S9.
Ultimately, the increase in interfacial impedance with time only partially affects Li cycling, as shown in Figure 5, which compares the cycling of the year-aged Li/Li cells (black) to pristine cells that rested only 24 h (red). The cells were generally cycled at a rate of 0.5 mA/cm2 to 2 mAh/cm2, with the first 10 cycles shown below in Figure 5. This moderate-to-low current was chosen to enable the aged cells to support cycling, and high rates were deemed unlikely since many cells showed significant impedance rise during aging and significant overpotentials upon the initial nucleation at the start of cycling. Limited sample numbers prevented the evaluation of rate capabilities after aging. The cell aged in the LiPF6-containing electrolyte could not support cycling at 0.5 mA/cm2 as the voltage immediately polarized to the −1.5 V voltage limit. Instead, the aged and pristine cells with the LiPF6-containing electrolyte were cycled at 0.05 mA/cm2 for 0.5 mAh/cm2. Considering that the LiPF6 electrolyte had the highest observed interfacial impedance of all the cells, it is not surprising that the higher current could not be delivered within the potential limits set. Although the pristine cell containing the LiPF6 electrolyte is shown cycling at a low rate in Figure 5, a pristine cell can support Li cycling at 0.5 mA/cm2 (Supplementary Figure S10). This suggests that, over time, the impedance rise in cells aged with LiPF6 electrolyte could degrade the rate capability of an Li battery.
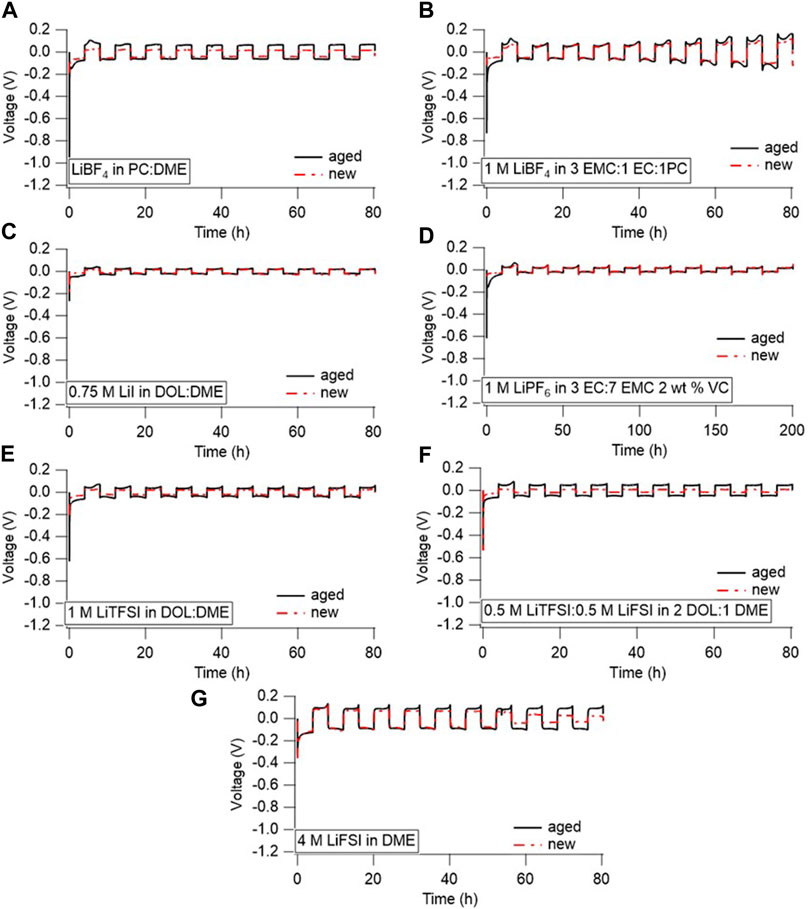
FIGURE 5. Li/Li symmetric cells cycled in (A) LiBF4 in PC:DME, (B) 1 M LiBF4 in 3 EMC:1 EC:1 PC, (C) 0.75 M LiI in 65 DOL:35 DME, (D) 1 M LiPF6 in 3 EC:7 EMC with 2 wt% VC, (E) 1 M LiTFSI in DOL:DME, (F) 0.5 M LiTFSI-0.5 M LiFSI in 2 DOL:1 DME, and (G) 4 M LiFSI in DME. All cells were cycled at 0.5 mA/cm2 for 2 mAh/cm2, except for (D), which was cycled at 0.05 mA/cm2 for 0.5 mAh/cm2.
The rest of the cells cycled at 0.5 mA/cm2 exhibited changes in overpotential between new and aged cells that reflect the impedance measurements. The most significant changes were observed with the LiTFSI and LiBF4 containing electrolytes, which align well with the EIS results. The overpotential of the LiBF4 in PC/DME was the lowest, just negative of −1 V, but the pristine cell’s overpotential was around −0.2 V. However, the initial resistance is likely simply related to breaking through that initial SEI layer, as the overpotential for the following cycles remains relatively stable without a large rise in resistance. In fact, the cycling behavior (after the initial nucleation overpotential) between the new and aged cells is very similar, if not the same, for many of the chemistries. Only the bisalt electrolyte had a significant decrease in overpotential during cycling between the aged and pristine cells, possibly due to changes in electrode surface area with cycling, as observed by Merrill et al. (2023) with cycled Li with similar LiFSI/LiTFSI bisalt electrolytes. The lower overpotential with the pristine cell compared to the aged cell suggests that the SEI that formed with the aged cell may be more stable and therefore contributed to more internal resistance across the cell during cycling. We have previously shown that the SEI formed during aging differs from the SEI formed during continuous cycling (Merrill et al., 2021). Therefore, these data suggest that the accumulated SEI formed during the year-long aging most severely impacts performance in the cycle immediately following the rest, and that low-rate performance is restored after that first cycle. It is possible that high-rate performance may still be impacted by the rest, which may not be apparent by these relatively low current density tests. We note that the cells in this work were aged with excess Li metal and electrolyte. The impedance rise during rest likely accompanies loss of electrolyte and/or Li inventory. In cells with lean electrolyte or without excess Li metal, the loss of electrolyte and Li during rest may have manifested in reduced capacity during cycling (Liu et al., 2019). Extended cycling of the aged cells beyond the first 10 cycles is shown in Supplementary Figure S11. Electrolytes that typically exhibit effective Li cycling (such as the 4 M LiFSI in DME or the bisalt electrolyte) (Miao et al., 2014; Qian et al., 2015; Merrill et al., 2021) exhibit stable cycles beyond the first 10 cycles, whereas the electrolytes typically used with Li primary chemistries do not. Whether the deviation from stable cycling is due to the aging or the performance of the actual electrolyte is unclear and would need further investigation to ascertain this. The cell containing the LiTFSI-based electrolyte appeared to short, presumably due to cell handling, after the first 10 cycles. This appeared to resolve with cycling and the cell was able to cycle normally, albeit with slight polarization, over time.
XPS was completed on a select number of electrodes to determine which species were present in the SEI, and to draw correlations between SEI composition and aging behavior. The electrodes chosen were aged in LiBF4 in PC:DME, 4 M LiFSI in DME, LiTFSI in DOL:DME, and LiI in DOL:DME. Each of these electrolytes spans a range of aging behaviors from relatively minimal aging (4 M LiFSI, LiI electrolytes) to more significant aging (LiBF4 and LiTFSI electrolytes). The atomic percentages of expected elements based on electrolyte composition were calculated from survey spectra (Figure 6). Corresponding high-resolution spectra of elements of interest are shown in Figures 7, 8. The electrode with the highest percentage of Li was from the cell aged in 4 M LiFSI in DME, likely due to the large amount of LiF observed in the high-resolution spectra (Figure 8). The electrodes with the least amount of Li-containing species (indicated by pink in Figure 6) present on the surfaces were aged in LiI-only electrolyte and the LiTFSI with the LiI additive electrolyte. Therefore, an increase in Li species on the surface of the electrode does not necessarily correlate with improved aging behavior as determined by EIS.
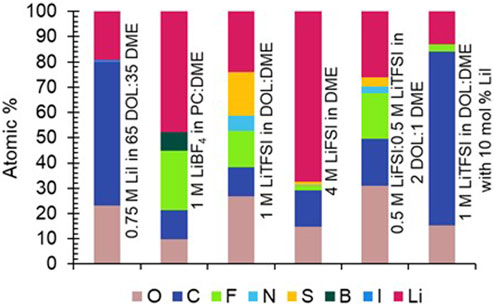
FIGURE 6. Atomic percentages taken from XPS survey spectra of uncycled Li electrodes aged for 1 year in cells with 0.75 M LiI in 65 DOL:35 DME, 1 M LiBF4 in PC:DME, 1 M LiTFSI in DOL:DME, 4 M LiFSI in DME, 0.5 M LiFSI:0.5 M LiTFSI in 2 DOL:1 DME, and 1 M LiTFSI in DOL:DME with 10 mol% LiI.
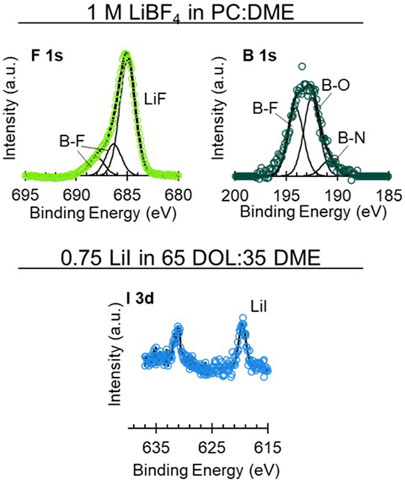
FIGURE 7. High-resolution XPS spectra on uncycled Li electrodes aged in cells with 1 M LiBF4 in PC:DME and 0.75 M LiI in 65 DOL:35 DME. Upper spectra show: F 1s (left, green) and B 1s (right, dark green). Lower spectra show I 3d (light blue).
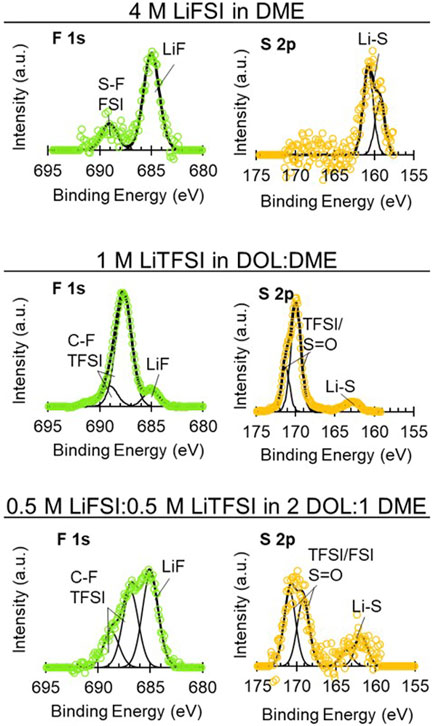
FIGURE 8. High-resolution XPS spectra on uncycled Li electrodes aged in cells with 4 M LiFSI in DME, 1 M LiTFSI in DOL:DME, and 0.5 M LiFSI:0.5 M LiTFSI in 2 DOL:1 DME. Spectra show: F 1s (left, green) and S 2p (right, yellow).
From the high-resolution spectra, the Li surface exposed to the LiBF4 electrolyte exhibits species like LiF (F 1s 685 eV), Li2CO3 (C 1s 289–291 eV, O 1s 531–533 eV), LiBF4 (F 1s 688 eV, B 1s 196.5 eV), Li2O (O 1s 528 eV), as well as various organic containing species present in the C 1s and O 1s spectra (Moulder et al., 1992; Verma et al., 2010). F 1s and B 1s spectra are shown in Figure 7, and C 1s, O 1s, and Li 1s spectra are shown in Supplementary Figure S12. Each of these species is expected based on prior surface studies of Li in LiBF4 and corroborated by the XPS (Kanamura et al., 1995; Jurng et al., 2018). Other B species that could be present include BN and B2O3 (Moulder et al., 1992).
The Li exposed to the LiI electrolyte had small amounts of I species and a significant presence of various C-O species (Figure 7 and Supplementary Figure S12). The C-O species are likely polyethers as a result of DOL decomposition (Verma et al., 2010; Fiedler et al., 2017). The I peak around 619 eV is likely due to LiI and the secondary peak around 631 eV is due to the orbital splitting. The peak in the Li 1s spectra around 49 eV is not due to an Li species but rather to the I 4d peak from the LiI. The small amount of I likely results from I- being in its fully reduced form in the LiI salt, leading to a largely organic SEI evident by primarily solvent decomposition products on the Li surface. High-resolution spectra of the LiTFSI in DOL:DME with 10 mol% LiI are shown in Supplementary Figure S13. This is further demonstrated by Figure 6, in which the survey spectra of the LiTFSI in DOL:DME with the LiI additive exhibit minimal S, N, and F species compared to the LiTFSI in DOL:DME alone without any LiI additive. This spectrum appears like the LiI-only spectra shown in Figure 7, with S 2p and F 1s signal just above the noise. This suggests that the LiI helps inhibit, though not completely prevent, the decomposition of the LiTFSI, resulting in the improved aging behavior observed in the EIS results.
XPS of the Li electrodes aged in the LiFSI, LiTFSI, and bisalt electrolytes are shown in Figure 8 and Supplementary Figure S12. For the Li aged in LiFSI, the SEI that formed is largely composed of inorganic species, as typically reported in the literature, which is likely due to the cleavage of the S-F bond in the FSI anion (Camacho-Forero and Balbuena, 2017; Merrill et al., 2021). It is notable that, in the F 1s and S 2p spectra, there are significant quantities of LiF (685 eV) and LixSy (∼162 eV) species but little-to-no S-F (687–689 eV) or -SO2 (∼170 eV) species. Organic species that are observed in the C 1s and O 1s spectra—ether and alkoxide species—are due to solvent decomposition (Fiedler et al., 2017). The literature suggests that the inorganic SEI species (e.g., LiF) are more electronically passivating than the organic species present in the Li exposed to LiTFSI, leading to a less “leaky” SEI and, thereby, an emphasis on electrolytes that form LiF-rich SEIs for improved Li cycling (Fang et al., 2019; Ren et al., 2019; Hobold et al., 2021). From the perspective of aging, this would translate to the lower interfacial impedance observed with the LiFSI electrolyte compared to the LiTFSI electrolyte and smaller relative changes in interfacial impedance. It is possible that, as with Li cycling, the more inorganic/electronically passivating SEI species (e.g., LiF, Li2O) contribute to the improved aging behavior. However, the ability of an SEI to passivate Li and minimize impedance rise during aging is likely more complicated than simply organic versus inorganic character. While a primarily inorganic SEI formed from aging Li in the 4 M LiFSI DME electrolyte leads to a low impedance rise during aging; the primarily organic SEI formed from aging in the LiI 65 DOL:35 DME electrolyte leads to a similarly low impedance rise. In addition to inorganic versus organic considerations, we suspect that the highly concentrated nature of the 4 M LiFSI in DME electrolyte also contributes to improved aging due to the fewer solvent molecules that are able to interact with the electrode surface. However, we do not have a direct comparison to ascertain the correlation of salt concentration with the impedance aging behavior.
The 1 M LiTFSI in DOL:DME and 0.5 M LiFSI:0.5 M LiTFSI in 2 DOL:DME exhibited similar aging responses per the EIS, as described above. The interfacial impedance increase with time for both electrolytes was greater than the 4 M LiFSI in DME and 0.75 M LiI electrolytes but less than the carbonate-containing electrolytes. The high-resolution XPS for both electrolytes is shown in Figure 8. The Li exposed to the LiTFSI electrolyte in Figure 8 has several commonly reported decomposition species. This includes -CF species present in both the F 1s (687–688 eV) and C 1s (292 eV) spectra. A small amount of the inorganic LiF species (685 eV) is present as well, although to a lesser extent than the organic fluorine species (-CF, -SF). The organic F species is likely a result of cleavage of the S-N or S-C bonds in the TFSI anion, whereas LiF would result from cleavage of the C-F bond. The O 1s spectra contain the mostly organic oxygen-containing species 532–534 eV, which are often alkoxide and ether species. Additionally, a small amount of Li2CO3 could also be present. The S 2p spectra contain a small amount of polysulfide type species (∼162 eV) but have more general sulfur decomposition species (∼170 eV). It has previously been suggested that, with fewer electrons, the S-N and C-S are most likely to break in the decomposition of the LiTFSI salt, so the high binding energy peaks thus likely result from fragments containing S (=O)2 moieties such as CF3SO2 and SO2 (Camacho-Forero and Balbuena, 2017).
Bisalt electrolytes containing mixtures of LiFSI and LiTFSI are reported to form SEIs with mixtures of decomposition products from both salts on electrochemically deposited Li—notably increased LiF compared to LiTFSI alone and increased organic species and other salt decomposition products relative to LiFSI alone (Miao et al., 2014; Merrill et al., 2021), consistent with the results in Figure 8. We note that the total Li-ion concentration in the bisalt electrolyte is the same as the LiTFSI electrolyte (although the solvent ratios are different)—1 M. This trends with the EIS response of the Li symmetric cell with the bisalt electrolyte, which exhibits similar behavior to the cell containing the LiTFSI electrolyte. The organic SEI formed with the bisalt and LiTFSI electrolytes is different from that formed by the LiI electrolyte due to the differences between the anions (FSI− and TFSI− vs. I−). The SEI formed with the bisalt and LiTFSI is composed of both solvent and salt decomposition products; the anion is able to continue reacting at the Li surface whereas the SEI formed with the LiI electrolyte is composed primarily of solvent decomposition products, and the anion (I−) cannot be reduced further. However, the addition of LiI to the LiTFSI electrolyte appears to prevent the reaction of the TFSI anion with the Li surface (Supplementary Figure S13).
Supplementary Figures S2–S9 show photographs of opened cells. We note that the Li electrodes aged in the bisalt electrolyte contain large divots in the center of each electrode. This suggests significant Li corrosion in these electrolytes. This is not evident in cycling behavior due to the excess Li and electrolyte in these cells that enable them to cycle at the programmed capacity despite losses, but this would likely lead to capacity fade in cells with lean electrolyte or without excess Li (Liu et al., 2019). This same behavior is not seen with the Li electrodes from the LiTFSI or with the LiFSI electrolytes. However, most electrodes show a subtle amount of surface roughening towards the center of the Li electrodes, regardless of electrolyte chemistry. We hypothesize that this is due to slight increases in pressure toward the center of the cell compared to the edges due to the slightly convex nature of punched Li metal electrodes. The Li electrode aged in the LiI electrolyte does not appear to greatly deviate from the macroscopic behavior observed with the other electrodes; however, discoloration of the electrolyte is observed, indicated by a yellow color on the separator. We note that the pristine electrolyte is colorless; therefore, it is expected that the LiI decomposed to form iodine, creating the yellow solution. This may be accelerated by the presence of the Li electrode. It is notable that the Li aged in 1 M LiBF4 in 3 EMC:EC:PC has brown discoloration on the surface, whereas the electrode aged in 1 M LiBF4 in PC:DME is also slightly brown (though less severe). The presence of DME in the latter electrolyte may prevent reaction due to the increased reductive stability of DME. Nevertheless, the corrosion of the Li electrode observed here corroborates the EIS findings discussed earlier.
CryoSEM top-down and cross-sectional images were taken of the Li from the cells after the year of aging. An SEM image and corresponding EDS of the pristine Li foil are shown in Supplementary Figure S14. The pristine surface shows that the starting Li foil has some cracks and texture, likely due to the material’s processing. The pristine surface has a few sites where some oxygen is present, possibly due to the presence of Li2O, and a small amount of carbon present across the surface of the Li. Carbon and oxygen impurities are typical of commercial Li films and have been reported to be at the 100 ppm level in commercial Li (Ho et al., 2022). Corresponding EDS is shown in Supplementary Figures S15–S22. The top-down images (Figure 9) were mostly taken to examine the Li surface and morphology after the extended exposure. The cross-sectional images in Figure 10 were used to compare SEI thicknesses. The top-down images show a variety of textures between the electrolytes, and they do not appear to correlate between electrolytes of similar chemistries. The Li aged in each of the LiBF4 electrolytes shows some cracking on the Li surface, which may be caused by brittle SEI components. The cracking may also be due to the disassembly and post-mortem sample preparation processes. The Li surface exposed to the LiI electrolyte showed some striations due to some oxygen-containing species per the EDS spectra with small amounts of iodine-containing species on the surface. The LiPF6 electrolyte appeared to cause sporadic texture on the Li surface, with the lighter areas likely a result of salt and/or salt decomposition products, based on the EDS mapping.

FIGURE 9. Top-down SEM images of uncycled Li electrodes after aging in an Li/Li symmetric cell in the respective electrolyte for 1 year. (A) LiBF4 in PC:DME, (B) LiBF4 in 3 EMC:EC:PC, (C) 0.75 M LiI in 65 DOL:35 DME, (D) 1 M LiPF6 in 3 EC:7 EMC with 2 wt% VC, (E) 1 M LiTFSI in DOL:DME, (F) 0.5 M LiTFSI-0.5 M LiFSI in 2 DOL:DME, (G) 4 M LiFSI in DME, and (H) 1 M LiTFSI with 10 mol% LiI in DOL:DME.

FIGURE 10. Cross-sectional SEM images of Li electrode after aging in an Li/Li symmetric cell in the respective electrolyte for 1 year. (A) LiBF4 in PC:DME, (B) LiBF4 in 3 EMC:EC:PC, (C) 0.75 M LiI in 65 DOL:35 DME, (D) 1 M LiPF6 in 3 EC:7 EMC with 2 wt% VC, (E) 1 M LiTFSI in DOL:DME, (F) 0.5 M LiTFSI-0.5 M LiFSI in 2 DOL:DME, (G) 4 M LiFSI in DME, and (H) 1 M LiTFSI with 10 mol% LiI in DOL:DME.
The Li surface exposed to the LiTFSI electrolyte had some cracks, but it was qualitatively different from the LiBF4 electrolyte, appearing to be more layered. The patterning observed on the Li exposed to the LiTFSI electrolyte was similar to the Li exposed to the LiTFSI electrolyte with the LiI additive, despite the chemical differences present in the XPS spectra. It is notable that the Li exposed to the bisalt electrolyte showed very different behavior from both the LiFSI and LiTFSI electrolytes, despite the same constituents from each being present. The Li surface here appeared to have tree-like structures, which do not appear to correlate with a particular element or decomposition product per the EDS (Supplementary Figure S17) and may be the result of salt crystals after the solvent dried, despite the washing procedure. The Li aged in the LiFSI electrolyte appears to have the least amount of texture on the surface. The SEM image of the pristine Li (Supplementary Figure S14) shows more texture than the Li aged with the LiFSI, suggesting that the SEI that formed in the presence of the LiFSI electrolyte may have been able to homogenize the surface. The visibility of these features may be due to fewer decomposition products accumulating on the surface, based on the surface chemistry determined by the XPS and the small impedance rise, as described previously.
The comparison of the SEI thicknesses in Figure 10 was slightly limited due to the small sample size and to the SEI itself being fairly thin and heterogeneous. The Li metal underneath the SEI generally appears darker in the secondary electron SEM images than the SEI formed during aging. There does not appear to be a large difference in thickness between the SEIs of each of the samples, despite the changes in EIS behavior. Where differences are qualitatively observed, there is no correlation with the EIS results; in fact, it is the opposite in some cases. For example, the Li that was exposed to LiI cross section (Figure 10C) is qualitatively thicker than the Li that was exposed to LiBF4 (Figures 10A, B). The layer on the LiI looks very porous, suggesting that the SEI could be more prone to the continued transfer of electrolyte species to the Li surface. One possibility, when comparing the LiBF4 and the LiI, is that, because we see the breakdown of the salt species with the LiBF4 and the breakdown of the solvent species with LiI, the more inorganic interface is actually simply more electronically passivating than the LiI, enabling continuous electron leakage throughout aging but porosity that enables little impact on Li+ transport. However, the LiI electrolyte exhibited a very small impedance rise, which implies facile Li+ transfer kinetics after aging and would normally be assumed to be associated with minimal growth of the SEI due to a favorable and passivating SEI. We note that LiI in 65 DOL:35 DME is commercialized in primary Li/FeS2 batteries that exhibit 20-year shelf lives. The long shelf life is not surprising given the low impedance rise observed in our study with the LiI in 65 DOL:35 DME electrolyte, but it is surprising given the thick and porous SEI observed in cryo-SEM images. One area that we cannot address is the degree of SEI damage (breaking/cracking, solubilizing in the electrolyte) either in situ or even during processing for the cryoSEM, and these ex-situ images may not be fully representative of the SEIs before cell disassembly, washing, and imaging.
It is likely that, because each of the SEI components has different degrees of electronic passivation, porosity in the SEI film, and electrolyte uptake, the relationship between impedance response and SEI thickness will be very different between these different compounds. Regardless, it is likely that the increase in the EIS observed over time is related to the buildup of the SEI layer, and this increase in EIS is relative to each individual cell such that both the initial impedance and the rise over time should be considered when evaluating aging. We also note that the LiTFSI electrolyte with LiI added appears to have reacted with the Li beneath the SEI layer such that corrosion occurred not only at the surface but within the bulk of the Li. This is evident from the void space in the bulk Li metal. This case is counterintuitive to the EIS results, suggesting that this Li electrode experienced minimal aging. It is possible, despite the minimal increase in interfacial impedance, that the Li was still reacting and that the reaction products were not sufficiently electronically passivating to have a significant impact on the EIS. It is also possible that the reactivity below the surface increased the interfacial area such that the overall impedance was lower due to increased available surface sites for reaction. These effects would be more obvious in cells with minimal electrolyte or Li metal rather than the excesses used here.
We evaluated the aging behavior of Li metal in several electrolytes commonly used in the literature in rechargeable and Li primary batteries. We found that the aging of Li in LiBF4 and LiPF6 in carbonates was the most severe, followed by the LiTFSI and bisalt electrolytes in DOL:DME, and then the 4 M LiFSI in DME and LiI in DOL:DME electrolytes. The carbonate-based electrolytes are expected to have the most severe aging behavior as the carbonate solvents are less stable against Li than the ether solvents. Despite this, LiBF4-based electrolytes are used in commercial primary Li batteries and therefore probably have additives to improve aging behavior. The 4 M LiFSI in DME electrolyte is expected to have improved behavior due to its highly concentrated nature (fewer free solvent molecules available to react with Li) and a largely inorganic, passivating SEI. Although the LiI electrolyte is not concentrated like the 4 M LiFSI in DME electrolyte, the I anion’s condition in its most reduced state likely contributed to the improved aging behavior. We note that the LiI in the DOL DME electrolyte forms a mostly organic SEI, and the 4 M LiFSI in the DME electrolyte forms a mostly inorganic SEI. Despite these very different SEI compositions, these two electrolytes exhibit the lowest impedance during aging. However, SEM suggests that the LiI in the DOL:DME electrolyte may form a thicker and more porous SEI than 4 M LiFSI in DME, suggesting that the highly concentrated solution or inorganic SEI associated with 4 M LiFSI in DME may be more passivating towards reaction with Li metal during long-term aging.
Ultimately, the impact of the aging on cycling appears only to impact the first nucleation overpotential in Li/Li symmetric cells with excess Li and electrolyte. Once this regime is passed, cycling behavior is generally nearly identical to the pristine cells, assuming that only the Li/electrolyte interface is affected. However, the consequence of this is that, in a full Li battery, the cell may not actually cycle unless the rate is dropped below some threshold where the initial resistance across the cell is not limiting. It is also likely that cycling at higher rates will show increased overpotentials in aged cells relative to unaged cells, but low current density cycling may have minimal impact after the initial overpotential. Beyond the impacts to overpotential and rate capability, a full cell will likely experience more complex aging behaviors due to the non-zero electrochemical potential across the cell. Possible aging mechanisms to consider when evaluating the long-term aging of a full cell could include current collector corrosion (particularly with LiFSI- and LiTFSI-based electrolytes), electrolyte degradation and/or cell instability due to increased voltages, and cathode materials leaching into the electrolyte. The symmetric cells studied in this work also contained excess Li and electrolyte such that reservoirs were available during cycling experiments after aging. Therefore, decreased capacity after aging would not be expected, despite the Li corrosion and electrolyte depletion likely associated with impedance rise and SEI growth during aging. In contrast, cells with lean electrolyte and no excess Li would likely exhibit capacity loss during the cycling test that followed the year of aging due to the irreversible consumption of Li inventory and electrolyte (Wood et al., 2018; Liu et al., 2019). In cells with excess Li and electrolyte, we were able to monitor aging trends through EIS over a year without sacrificing a cell during the aging study due to irreversible consumption of the Li/electrolyte. Although this is not practical for next-generation cell designs, Li-primary cells have been reported to have excess electrolyte which may contribute to their long shelf lives (Reddy, 2011; Ziesche et al., 2020). Extended aging in cells with lean electrolyte/Li will be the topic of future work.
Interestingly, the thickness of the SEI did not appear to correlate with the EIS spectra, and it is expected that this is related to the actual speciation and electrical resistance of the SEI components. For example, the LiBF4 electrolytes had SEIs that were rich in inorganic species (Li2CO3, LiBF4, LiF) and had very large increases in resistance with time, but they did not exhibit a very thick SEI layer from the cross-sectional images. The LiI in DOL DME electrolyte, on the other hand, appeared to have a relatively thicker SEI but exhibited a lower increase in impedance with time. This may indicate that Li+ transport through the SEI is facile in the organic SEI formed from the LiI in the DOL DME electrolyte, even if the SEI thickness grows with aging; this may be consistent with the porosity observed in SEM images. The 4 M LiFSI in DME electrolyte ultimately had very favorable results, including nearly identical cycling behavior between the aged and pristine cells, minimal increase in impedance, and the highest Li present in the XPS spectra. This suggests that the highly concentrated electrolyte coupled with a largely inorganic SEI will have decreased aging effects.
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding authors.
LM: data curation, formal analysis, investigation, methodology, writing–original draft. DL: data curation, writing–review and editing. SR: data curation, writing–review and editing. MM: data curation, writing–review and editing. ML: data curation, writing–review and editing. KH: conceptualization, funding acquisition, supervision, writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Laboratory Directed Research and Development (LDRD) program at Sandia National Laboratories.
The authors thank Bryan Wygant for helpful discussions and Benjamin Warren for laboratory assistance. Sandia National Laboratories is a multi-mission laboratory managed and operated by National Technology and Engineering Solutions of Sandia, LLC (NTESS), a wholly owned subsidiary of Honeywell International Inc., for the U.S. Department of Energy’s National Nuclear Security Administration (DOE/NNSA) under contract DE-NA0003525. This written work is authored by an employee of NTESS. The employee, not NTESS, owns the right, title, and interest in and to the written work and is responsible for its contents. Any subjective views or opinions that might be expressed in the written work do not necessarily represent the views of the U.S. Government. The publisher acknowledges that the U.S. Government retains a non-exclusive, paid-up, irrevocable, world-wide license to publish or reproduce the published form of this written work or allow others to do so for U.S. Government purposes. The DOE will provide public access to results of federally sponsored research in accordance with the DOE Public Access Plan. This work was performed, in part, at the Center for Integrated Nanotechnologies, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science. This work was authored in part by the National Renewable Energy Laboratory, operated by Alliance for Sustainable Energy, LLC, for the U.S. Department of Energy (DOE) under Contract No. DE-AC36-08GO28308.
Author DML is employed by UES Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, editors, and reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbael.2023.1293877/full#supplementary-material
Aurbach, D., Ein-Ely, Y., and Zaban, A. (1994). The surface chemistry of lithium electrodes in alkyl carbonate solutions. J. Electrochem. Soc. 141, L1–L3. doi:10.1149/1.2054718
Aurbach, D., and Schechter, A. (2001). Changes in the resistance of electrolyte solutions during contact with lithium electrodes at open circuit potential that reflect the Li surface chemistry. Electrochimica Acta 46, 2395–2400. doi:10.1016/s0013-4686(01)00428-5
Aurbach, D., Zaban, A., Schechter, A., Ein-Eli, Y., Zinigrad, E., and Markovsky, B. (1995). The study of electrolyte solutions based on ethylene and diethyl carbonates for rechargeable Li batteries: I . Li metal anodes. J. Electrochem. Soc. 142, 2873–2882. doi:10.1149/1.2048658
Boyle, D. T., Huang, W., Wang, H., Li, Y., Chen, H., Yu, Z., et al. (2021). Corrosion of lithium metal anodes during calendar ageing and its microscopic origins. Nat. Energy 6, 487–494. doi:10.1038/s41560-021-00787-9
Camacho-Forero, L., and Balbuena, P. B. (2017). Elucidating electrolyte decomposition under electron-rich environments at the lithium-metal anode. Phys. Chem. Chem. Phys. 19, 30861–30873. doi:10.1039/c7cp06485c
Cheng, S., Wang, J., Lin, H., Li, W., Qiu, Y., Zheng, Z., et al. (2017). Improved cycling stability of the capping agent-free nanocrystalline FeS2 cathode via an upper cut-off voltage control. J. Mater Sci. 52, 2442–2451. doi:10.1007/s10853-016-0538-8
Dessantis, D., Di Prima, P., Versaci, D., Amici, J., Francia, C., Bodoardo, S., et al. (2023). Aging of a lithium-metal/LFP cell: predictive model and experimental validation. Batteries 9, 146. doi:10.3390/batteries9030146
Fang, C., Li, J., Zhang, M., Zhang, Y., Yang, F., Lee, J. Z., et al. (2019). Quantifying inactive lithium in lithium metal batteries. Nature 572, 511–515. doi:10.1038/s41586-019-1481-z
Fiedler, C., Luerssen, B., Rohnke, M., Sann, J., and Janek, J. (2017). XPS and SIMS analysis of solid electrolyte interphases on lithium formed by ether-based electrolytes. J. Electrochem. Soc. 164, A3742–A3749. doi:10.1149/2.0851714jes
Gao, Y., Rojas, T., Wang, K., Liu, S., Wang, D., Chen, T., et al. (2020). Low-temperature and high-rate-charging lithium metal batteries enabled by an electrochemically active monolayer-regulated interface. Nat. Energy 5, 534–542. doi:10.1038/s41560-020-0640-7
Ho, A. S., Westover, A. S., Browning, K., Maslyn, J. A., Parkinson, D. Y., Sahore, R., et al. (2022). Comparing the purity of rolled versus evaporated lithium metal films using X-ray microtomography. ACS Energy Lett. 7, 1120–1124. doi:10.1021/acsenergylett.2c00255
Hobold, G. M., Lopez, J., Guo, R., Minafra, N., Banerjee, A., Shirley Meng, Y., et al. (2021). Moving beyond 99.9% Coulombic efficiency for lithium anodes in liquid electrolytes. Nat. Energy 6, 951–960. doi:10.1038/s41560-021-00910-w
Jurng, S., Brown, Z. L., Kim, J., and Lucht, B. L. (2018). Effect of electrolyte on the nanostructure of the solid electrolyte interphase (SEI) and performance of lithium metal anodes. Energy and Environ. Sci. 11, 2600–2608. doi:10.1039/c8ee00364e
Kanamura, K., Tamura, H., Shiraishi, S., and Takehara, Z.-I. (1995). XPS analysis of lithium surfaces following immersion in various solvents containing LiBF4. J. Electrochem. Soc. 142, 340–347. doi:10.1149/1.2044000
Kolesnikov, A., Kolek, M., Dohmann, J. F., Horsthemke, F., Börner, M., Bieker, P., et al. (2020). Galvanic corrosion of lithium-powder-based electrodes. Adv. Energy Mater. 10, 2000017. doi:10.1002/aenm.202000017
Komaba, S., Kaplan, B., Ohtsuka, T., Kataoka, Y., Kumagai, N., and Groult, H. (2003). Inorganic electrolyte additives to suppress the degradation of graphite anodes by dissolved Mn (II) for lithium-ion batteries. J. Power Sources 119-121, 378–382. doi:10.1016/s0378-7753(03)00224-6
Kozen, A. C., Lin, C.-F., Zhao, O., Lee, S.-B., Rubloff, G. W., and Noked, M. (2017). Stabilization of lithium metal anodes by hybrid artificial solid electrolyte interphase. Chem. Mater. 29, 6298–6307. doi:10.1021/acs.chemmater.7b01496
Lennen, R. M., Poese, B. A., Swank, J. A., and Goldberg, A. B. (2006). “Active Li/FeS2 battery performance and aging study for army munitions applications,” in Proceedings of the 42nd Power Sources Conference, Philadelphia, PA, June 2006.
Lin, D., Liu, Y., Li, Y., Li, Y., Pei, A., Xie, J., et al. (2019). Fast galvanic lithium corrosion involving a Kirkendall-type mechanism. Nat. Chem. 11, 382–389. doi:10.1038/s41557-018-0203-8
Liu, J., Bao, Z., Cui, Y., Dufek, E. J., Goodenough, J. B., Khalifah, P., et al. (2019). Pathways for practical high-energy long-cycling lithium metal batteries. Nat. Energy 4, 180–186. doi:10.1038/s41560-019-0338-x
Liu, J., Ihuaenyi, S., Kuphal, R., Salinas, J., Xie, L., Yang, L., et al. (2023). A comparison of carbonate-based and ether-based electrolyte systems for lithium metal batteries. J. Electrochem. Soc. 170, 010535. doi:10.1149/1945-7111/acb3fe
Li, W., Yao, H., Yan, K., Zheng, G., Liang, Z., Chiang, Y. M., et al. (2015). The synergetic effect of lithium polysulfide and lithium nitrate to prevent lithium dendrite growth. Nat. Commun. 6, 7436. doi:10.1038/ncomms8436
Merrill, L. C., Gannon, R. N., Jungjohann, K. L., Randolph, S. J., Goriparti, S., Zavadil, K. R., et al. (2023). Evaluation of lithium metal anode volumetric expansion through laser plasma focused ion beam cross-sectional imaging. J. Electrochem. Soc. 170, 080527. doi:10.1149/1945-7111/acf162
Merrill, L. C., Long, D. M., Small, K. A., Jungjohann, K. L., Leung, K., Bassett, K. L., et al. (2022). Role of coatings as artificial solid electrolyte interphases on lithium metal self-discharge. J. Phys. Chem. C 126, 17490–17501. doi:10.1021/acs.jpcc.2c05385
Merrill, L. C., Rosenberg, S. G., Jungjohann, K. L., and Harrison, K. L. (2021). Uncovering the relationship between aging and cycling on lithium metal battery self-discharge. ACS Appl. Energy Mater. 4, 7589–7598. doi:10.1021/acsaem.1c00874
Meyerson, M. L., Rosenberg, S. G., and Small, L. J. (2022). A mediated Li–S flow battery for grid-scale energy storage. ACS Appl. Energy Mater. 5, 4202–4211. doi:10.1021/acsaem.1c03673
Miao, R., Yang, J., Feng, X., Jia, H., Wang, J., and Nuli, Y. (2014). Novel dual-salts electrolyte solution for dendrite-free lithium-metal based rechargeable batteries with high cycle reversibility. J. Power Sources 271, 291–297. doi:10.1016/j.jpowsour.2014.08.011
Morales-Ugarte, J. E., Benayad, A., Santini, C. C., and Bouchet, R. (2019). Electrochemical impedance spectroscopy and X-ray photoelectron spectroscopy study of lithium metal surface aging in imidazolium-based ionic liquid electrolytes performed at open-circuit voltage. ACS Appl. Mater Interfaces 11, 21955–21964. doi:10.1021/acsami.9b00753
Moulder, J. F., Stickle, W. F., Sobol, P. E., and Kbomben, K. D. (1992). Handbook of X-ray photoelectron spectroscopy: a reference book of standard spectra for identification and interpretation of XPS data. Waltham: Perkin-Elmer Corporation.
Niu, C., Liu, D., Lochala, J. A., Anderson, C. S., Cao, X., Gross, M. E., et al. (2021). Balancing interfacial reactions to achieve long cycle life in high-energy lithium metal batteries. Nat. Energy 6, 723–732. doi:10.1038/s41560-021-00852-3
Qian, J., Henderson, W. A., Xu, W., Bhattacharya, P., Engelhard, M., Borodin, O., et al. (2015). High rate and stable cycling of lithium metal anode. Nat. Commun. 6, 6362. doi:10.1038/ncomms7362
Ren, X., Zou, L., Cao, X., Engelhard, M. H., Liu, W., Burton, S. D., et al. (2019). Enabling high-voltage lithium-metal batteries under practical conditions. Joule 3, 1662–1676. doi:10.1016/j.joule.2019.05.006
Verma, P., Maire, P., and Novak, P. (2010). A review of the features and analyses of the solid electrolyte interphase in Li-ion batteries. Electrochimica Acta 55, 6332–6341. doi:10.1016/j.electacta.2010.05.072
Whitacre, J., Yazami, R., Hamwi, A., Smart, M. C., Bennett, W., Surya Prakash, G., et al. (2006). Low operational temperature Li–CFx batteries using cathodes containing sub-fluorinated graphitic materials. J. Power Sources 160, 577–584. doi:10.1016/j.jpowsour.2006.01.045
Wood, S. M., Fang, C., Dufek, E. J., Nagpure, S. C., Sazhin, S. V., Liaw, B., et al. (2018). Predicting calendar aging in lithium metal secondary batteries: the impacts of solid electrolyte interphase composition and stability. Adv. Energy Mater. 8, 1801427. doi:10.1002/aenm.201801427
Wu, F., Lee, J. T., Nitta, N., Kim, H., Borodin, O., and Yushin, G. (2015). Lithium iodide as a promising electrolyte additive for lithium–sulfur batteries: mechanisms of performance enhancement. Adv. Mater. 27, 101–108. doi:10.1002/adma.201404194
Xu, K. (2004). Nonaqueous liquid electrolytes for lithium-based rechargeable batteries. Chem. Rev. 104, 4303–4418. doi:10.1021/cr030203g
Yang, L., Smith, C., Patrissi, C., Schumacher, C. R., and Lucht, B. L. (2008). Surface reactions and performance of non-aqueous electrolytes with lithium metal anodes. J. Power Sources 185, 1359–1366. doi:10.1016/j.jpowsour.2008.09.037
Keywords: lithium, calendar aging, battery, electrolyte, solid electrolyte interphase
Citation: Merrill LC, Long DM, Rosenberg SG, Meyerson ML, Lam MN and Harrison KL (2023) Real time lithium metal calendar aging in common battery electrolytes. Front. Batteries Electrochem. 2:1293877. doi: 10.3389/fbael.2023.1293877
Received: 13 September 2023; Accepted: 27 November 2023;
Published: 18 December 2023.
Edited by:
Linqin Mu, Arizona State University, United StatesReviewed by:
Dong Hou, University of Louisiana at Lafayette, United StatesCopyright © 2023 Merrill, Long, Rosenberg, Meyerson, Lam and Harrison. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laura C. Merrill, bGNtZXJyaUBzYW5kaWEuZ292; Katharine L. Harrison, a2F0aWUuaGFycmlzb25AbnJlbC5nb3Y=
†Present addresses: Daniel M. Long, Air Force Research Laboratory, Materials and Manufacturing Directorate, AFRL/RX, Dayton, OH, United States; UES Inc., Dayton, OH, United States
Samantha G. Rosenberg, Lockheed Martin Space, Palo Alto, CA, United States
Katharine L. Harrison, National Renewable Energy Laboratory, Golden, CO, United States
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.