
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Bacteriol., 29 April 2024
Sec. Bacterial Genetics and AI-enhanced Microbial Engineering
Volume 3 - 2024 | https://doi.org/10.3389/fbrio.2024.1339809
 Pacharapong Khrongsee1,2†
Pacharapong Khrongsee1,2† Iris Irby1†
Iris Irby1† Pitchaporn Akaphan1
Pitchaporn Akaphan1 Mariam A. Alami-Rose1
Mariam A. Alami-Rose1 Jedsada Kaewrakmuk3
Jedsada Kaewrakmuk3 Apichai Tuanyok1,4*
Apichai Tuanyok1,4*Introduction: Bacteriophages are known as predators of bacteria and key biological factors influencing genetic recombination through phage transduction in bacteria. Phage transduction is known as one of the most common genetic recombination events found in Burkholderia pseudomallei, a diverse bacterial species and the causative agent of a deadly tropical disease melioidosis. The main objective of this study was to catalog prophages or prophage islands that are common in B. pseudomallei genomes.
Methods: Various bioinformatic tools were used to identify prophages in 106 complete B. pseudomallei genomes, and complete and incomplete genomes in other species within the B. pseudomallei Complex (BPC). Temperate phages were spontaneously induced from selected B. pseudomallei and B. thailandensis strains, and further characterized by transmission electron microscopy and whole genome sequencing.
Results: Nine phage integration hotspots were identified in B. pseudomallei pan-genomes, eight of which were associated with tRNA gene-mediated site-specific recombination (tRNA-SSR) events. These genetic events occurred at various tRNA-genes including tRNA- Phenylalanine (anticodon GAA), - Methionine (CAU), - Proline (UGG), - Arginine (UCU), - Cysteine (GCA), - Arginine (CCG), - Serine (GGA), and – Selenocysteine (UCA) genes. Some of these events were also found in other related species within the B. pseudomallei Complex (BPC). We have demonstrated that lysogenic phages from select BPC strains could use B. pseudomallei strain Bp82 or 576mn as a host. These phages were classified into one of the two major groups, myoviruses or siphoviruses, based on their morphology and genomic composition.
Discussion: We have demonstrated that most B. pseudomallei strains are lysogenic, many containing at least one functional prophage in their genomes. Further investigation of the interactions between B. pseudomallei, bacteriophages, and other environmental and biological factors would provide a bigger picture of genomic diversity, potentially influence on survival of B. pseudomallei in the environment and its pathogenic specialization in hosts.
Burkholderia pseudomallei (B. pseudomallei), a Gram-negative soil bacterium, is the causative agent of melioidosis, a severe tropical disease with a high mortality rate. Melioidosis is endemic to most Asian countries, northern Australia, the Caribbean, and South America. B. pseudomallei is an opportunistic pathogen that can cause disease in both animals and humans from inoculation through skin abrasion, ingestion of contaminated food or water, or inhalation of contaminated dust or aerosol. One factor that influences the frequency and severity of B. pseudomallei outbreaks is its ability to survive in strictly environmental conditions for extended periods of time, including distilled water for at least 16 years (Pumpuang et al., 2011). B. pseudomallei has an open genome that promotes horizontal gene transfers (HGTs) or allow high frequency of recombination of mobile genetic elements (MGEs) to form genomic islands (GIs) which causes genetic diversity among the population (Kim et al., 2005; Tuanyok et al., 2008). This process in B. pseudomallei is not random, as many of these events are associated with specific recombination sites e.g., tRNA genes (Tuanyok et al., 2008).
In general, site-specific recombination is the insertion of foreign DNA into the bacterial genome, which occurs in conserved regions of DNA during chromosomal replication. Typically, this recombination results in the creation of a GI containing inserted genes from external sources, such as plasmids from other bacteria (Hallet and Sherratt, 1997). This system is not only used for insertion of foreign DNA, but is frequently used by temperate bacteriophages, the viruses which infect bacterial cells and enter a lysogenic life cycle leading to the insertion of phage DNA into the bacterial genome. The insertion sites used for site-specific recombination in the bacterial genome are highly similar to attP sequence, the bacteriophages’ own insertion sequences, leading to the insertion of temperate bacteriophages, or prophages, into the genome at the same homologous sites (Gindreau et al., 1997). These prophages have a substantial effect on the phenotypes of the bacteria and increase the diversity of the species. Phage DNA can increase the virulence of a bacterial strain through phage-encoded virulence factors and the expression of antibiotic resistance genes as seen highly efficiently in E. coli O157:H7 (Marinus and Poteete, 2013). Prophages can also influence the evolution of microbial metabolic pathways. Our previous study on the genomic comparison of five B. pseudomallei strains, which causes various clinical presentations in melioidosis patients: K96243, 1710b, 1106a, MSHR668, and MSHR305, identified 71 different GIs. This study also reported the association between about 40-60% of GIs insertions and 13 tRNA locations. The term, “tRNA gene-mediated site-specific recombination” or “tRNA-SSR,” which explained that the locations of these GIs are not random but specific to tRNA gene loci in B. pseudomallei genomes, was proposed (Tuanyok et al., 2008). This process resulted in locating GIs at the 3’ end of tRNA genes and creating short, direct repeats of tRNA genes flanking GIs. In a follow-up study using a pan-genomic approach when 37 genomes were used, we found that gene order was highly conserved among strains, despite the high combination rate as previously observed (Spring-Pearson et al., 2015).
In order to understand the distribution of temperate phages and their impact on the survival and pathogenic specialization of B. pseudomallei, we identified the presence of tRNA-SSR in 106 complete B. pseudomallei genomes and genomes of other members within the Burkholderia pseudomallei complex (BPC), including B. mallei (Nierman et al., 2004), B. thailandensis (Brett et al., 1998), B. oklahomensis (Glass et al., 2006b), B. humptydooensis (Tuanyok et al., 2017), B. singularis (Vandamme et al., 2017), B. savannae and B. mayonis (Hall et al., 2022). The tRNA-SSRs were then identified as putative prophages due to lengths over 27,000 base pairs and containing phage biosynthesis genes. Some of these prophages were characterized for their functionality.
To identify the tRNA-SSRs, we first created a genome database by downloading all complete genomes of B. pseudomallei (N=106) and B. thailandensis (N=19), which were available from GenBank as of January 1st, 2023. Other complete and incomplete BPC species’ genomes including B. mallei (N=108), B. oklahomensis (N=3), B. humptydooensis (N=4), B. savannae (N=1), B. mayonis (N=1), and B. singularis (N=2) were also acquired. All tRNA-SSR events were identified in each genome through local NCBI BLASTn (v 2.2.18) (Camacho et al., 2009) with the tRNA gene sequences from each species’ reference strain (e.g., B. pseudomallei K96243, GenBank accession no. BX571965.1 &BX571966.1; B. mallei, CP000010.1 &CP000011.1; B. thailandensis E264, CP000086.1 &CP000085.1; B. oklahomensis C6786, CP009555.1 &CP009556.1; B. humptydooensis MSMB43, CP013380.1 &CP013382; B. savannae MSMB266, CP013417.1 &CP013418.1; B. mayonis BDU6, CP013386.1 &CP013387.1; and B. singularis LMG 28154, GCA_900176645.1) as the query. With a tabular output, the region between the tRNA genes and their 3’ end direct repeat sequences were sorted and identified based on a length of at least 27,000 base pairs. These coordinates of the direct repeats were compiled into a table used to create BED files, then the BED files were used for extracting a multiFASTA file of each prophage genomes in every tRNA-SSR with the strain names, tRNA repeat, and start and stop points using BEDTools suite – getfasta version 2.30.0 (Quinlan and Hall, 2010). This allows for the sequences to be further analyzed. Supplementary Figure 1 demonstrates the identification of tRNA-SSR and the computational workflow for prophage prediction.
Every prophage underwent further analysis with PHASTER, a swift and specialized tool for phage sequence detection (Zhou et al., 2011; Arndt et al., 2016) to compare with previous BLASTn outcomes. Both BLASTn and PHASTER programs were utilized for predicting prophage regions. PHASTER offers the advantage of providing a brief overview of phage genes within the predicted prophage regions. However, it may sometimes encounter difficulties in precisely identifying the start and stop nucleotides of the prophage regions. To address this limitation, BLASTn was employed as it excels in this aspect. In this process, each prophage was matched to the one it bore the closest resemblance to, based on the inserted sequence. The criterion for this matching required that over 80% of the proteins demonstrate substantial similarity in both the sequence and the query coverage to the target sequence. Subsequently, the affiliation of the identified prophage was traced within the GenBank database, and its confirmation was established through an in-depth analysis of the integrase gene. Lastly, the tRNA-SSR associated prophages identified were further annotated using Genome Annotation services via the Bacterial and Viral Bioinformatics Resource Center (BV-BRC; http://bv-brc.org). This included analyzing prophage features such as genes potentially involved in antimicrobial resistance phenotypes, metabolic or biochemical pathways, or special functions (specialty genes), using the BV-BRC’s tools like the BLAT (BLAST-like alignment tool) (Olson et al., 2023).
Conventional PCR was used to amplify six integrase genes known to be associated with prophages in 321 B. pseudomallei strains from 142 clinical and 179 environmental sources, collected from a local endemic area of southern Thailand (Kaewrakmuk et al., 2023), and 9 B. thailandensis strains. Each PCR assay contained a 10 µL reaction comprising 1X Chai Sahara Hot Start Master Mix (Chai Bio Inc., Santa Clara, CA, USA), 2 µM of each oligonucleotide primer, and 1 µL of 10 ng of genomic bacterial DNA. Thermocycling conditions, conducted in 8-well PCR strips thermal cycle, were as follow: hot start at 95 C for 5 minutes; 35 cycles of denaturation at 95°C for 45 seconds, annealing at 52°C for 45 seconds, and extension at 72°C for 1 minute; and final step to allow extension at 72 C for 5 minutes. Then, the PCR products were visualized by SYBR Safe stain in 2% agarose gel-electrophoresis running with 120 Volts 500 Amperes for 35 minutes. Details of PCR primers, expected amplicon sizes and their targeting genes are summarized in Supplementary Table 1.
Temperate phages were isolated from three selected BPC strains including B. pseudomallei Bp82, and B. thailandensis E264 and TXDOH. Bacterial host strains B. pseudomallei Bp82 (Propst et al., 2010) and 576mn (Norris et al., 2017), the biosafe non-select agent strains and the representatives of serotype A and B, were used. Briefly, an overnight culture of each tested bacterial strain and each of the two host strains in LB broth was sub-cultured into a 3 mL fresh LB broth and grown at 37°C with shaking at 250 rpm for 5 hours. Cultures of both biosafe strains were supplemented with 0.8 µg/mL of adenine (Propst et al., 2010). A volume of 1 mL of overnight culture of each tested strain was centrifuged at 16,000 x g for 1 minute. The resulting supernatant was then filtered through a 0.45-micron (dia.) syringe filter into a sterile tube. Bacterial host soft agar was prepared by mixing 100 µL each of the fresh culture of Bp82 or 576mn with 4.5 mL of molten LB adenine soft agar (0.35% w/v) in 5 ml sterile tube. The mixture was then poured onto an LB agar plate supplemented with 0.80 µg/mL of adenine. Once the agar solidified, 100 µL of the filtered supernatant was added on top of the bacterial soft agar plate. The plate was briefly rotated to allow the phage solution to spread across the agar’s surface and was left to dry for 10 minutes. After drying, the plate was inverted and incubated at 37°C in an incubator overnight. The plaque formations were subsequently observed.
We used PCR to guide the identification of temperate phages present in B. pseudomallei Bp82. Conventional PCR assays were used to amplify three different phage integrase genes uniquely presented in different prophages. Each PCR had a 10 µL reaction containing 1X Chai Sahara Hot Start Master Mix (Chai Bio Inc., Santa Clara, CA, USA), 2 µM of each oligonucleotide primer, and 1 µL of phage solution diluted from each plaque. The primer sequences for each phage integrase assay are as follows: i) tRNA-SSR-Pro (UGG) assay targeting gene BP1026B_I2161; forward primer, 5’-GTGGCCGAAGTATGGAGATTAC-3’, and reverse primer. 5’-CGATCCACGTCTGACCATTT-3’; ii) tRNA-SSR-Arg (CCG) assay targeting gene BP1026B_I13636; forward primer, 5’-TGCTGTCGAATGGGAATGG-3’, and reverse primer, 5’-AACCGCGGTAAACGAACA-3’; and iii) tRNA-SSR-Phe (GAA) assay targeting gene BPSL0129; forward primer, 5’-AAGGTTGCGAGCGATTACTG-3’, and reverse primer, 5’-GCCATGCGTGGTTGTAGAT-3’. Thermocycling conditions included hot start at 95°C for 5 minutes, and 35 cycles of denaturation at 95°C for 45 seconds, annealing at 52°C for 45 seconds, and extension at 72°C for 1 minute, and the final extension at 72°C for 5 minutes. PCR products were visualized by gel electrophoresis operated at 120 Volts and 500 Amperes for 35 minutes. Expected PCR product sizes for these three assays were 438, 362, and 676 bp, respectively.
A volume of 100 mL of filtered plate lysate phage suspension in SM buffer was subjected to filtration using a 0.22-micron (dia.) bottle-top vacuum filter to eliminate any potential residual bacterial cells. Subsequently, the resulting mixture underwent centrifugation at 25,000 × g for a duration of 60 minutes. Following this centrifugation step, the supernatant was discarded, revealing a noticeable yellow spot, which constituted the phage pellet. To resuspend the pellet, 1 mL of LB broth was added. To facilitate the extraction of phage DNA, 500 µL of concentrated lysate was combined with 6.25 of μL 1 M MgCl2, 0.4 μL of DNase I (1000 U/mL), and 5 μL of RNase A (20 mg/mL) was introduced into the mixture. This mixture was then incubated at 37°C for 60 minutes. Subsequent steps involved the destabilization of phage capsids and inactivation of DNase I by the addition of 25 μL of 10% SDS, 20 μL of 0.5 M EDTA (pH 8.0), and 1.25 µL of proteinase K (20 mg/mL). The incubation was continued at 55°C for another 60 minutes. DNA extraction was executed by introducing an equal volume of phenol-chloroform-isoamyl alcohol (PCI) in a ratio of 25:24:1, followed by vigorous mixing and centrifugation at 13,000 × g for 5 minutes. The resulting aqueous phase was carefully transferred to a new tube. The DNA extraction process was repeated twice using PCI and once using chloroform. To precipitate the DNA, an equal volume of absolute ethanol containing 50 µL of 3 M sodium acetate at pH 5.2 was added and incubated at -20°C overnight. The resulting DNA pellet was obtained by centrifugation at 13,000 × g for 10 minutes. Afterward, the pellet was washed twice with 1 mL of 70% ethanol and air-dried within the confines of a biosafety cabinet. Finally, the pellet was dissolved in 50 µL of DNase-free sterile water.
DNA samples were subjected to sequencing using Illumina sequencing techniques. Illumina sequencing libraries were created using the tagmentation-based and PCR-based Illumina DNA Prep kit, along with custom IDT 10 bp unique dual indices (UDI), targeting an insert size of 320 bp. No additional DNA fragmentation or size selection steps were included in the process. The Illumina sequencing was conducted using an Illumina NovaSeq 6000 sequencer in multiplexed shared-flow-cell runs, generating 2x151 paired-end reads. Following sequencing, demultiplexing, quality control, and adapter trimming were carried out using bcl-convert version v4.1.5, a software tool developed by Illumina Inc. in San Diego, California, USA. Genomic assemblies were performed utilizing Phage genome assembler v2021.03 workflow within the CPT Phage Galaxy platform, a web-based framework for biological computation hosted at the Center for Phage Technology (CPT), which was established by the Texas A&M University (Ramsey et al., 2020). Genome annotations were performed using online tools via BV-BRC as described above (Olson et al., 2023).
A 100 µL volume of phage sample containing a minimum of 108 plaque forming units/mL concentration was diluted with 900 µL of SM buffer. Subsequently, 10 µL of this phage solution was carefully applied to a glow-discharged copper grid stabilized with carbon and allowed to incubate for 5 minutes. Following this incubation period, the grid was briefly exposed to a 1% (w/v) uranyl acetate solution for 1 minute to enable staining. The prepared grids were then subjected to visualization by FEI -Tecnai G2 Spirit Twin Transmission Electron Microscope (Thermo Fisher Scientific Inc., Hillsboro, OR, USA) housed at the University of Florida’s Interdisciplinary Center for Biotechnology Research. Phage sizes were determined based on the average measurements obtained from five independent phages, providing valuable insights into their structural characteristics.
The frequency of tRNA-SSRs within the pangenome of B. pseudomallei was examined, revealing a total of 185 occurrences across 106 complete genomes. These tRNA-SSR events were linked to 15 distinct tRNA gene sequences. Among them, 53 integrated DNA segments exhibited sizes exceeding 27,000 base pairs and were predicted to be potential prophages using the PHASTER tool (Zhou et al., 2011; Arndt et al., 2016). Notably, these prophage predictions were associated with specific tRNA-SSRs located at seven different tRNA genes such as tRNA-Phenylalanine (anticodon GAA), -Methionine (CAU), -Proline (UGG), -Arginine (UCU), -Cysteine (GCA), and -Arginine (CCG) on chromosome 1, and as well as tRNA-Selenocystiene (UCA) on chromosome 2. Frequencies of these tRNA-SSR events along with the identified prophages in B. pseudomallei pangenome are shown in Figure 1. Furthermore, in order to validate the prophage identity of the insertions at these tRNA genes, an analysis was conducted on the direct repeat sequences which are identical to 3’end sequence of tRNA genes, downstream of the integration sites. These short sequences were believed to be generated during phage transduction and consistently displayed identical length and sequence characteristics for each tRNA-SSR instance associated with each respective tRNA gene. This observation underscores the presence of similar prophages from the same phage group at each tRNA gene location. Supplementary Table 2 (2.1) summarizes the presence of these tRNA-SSR events and the associated direct repeats for each B. pseudomallei genome.
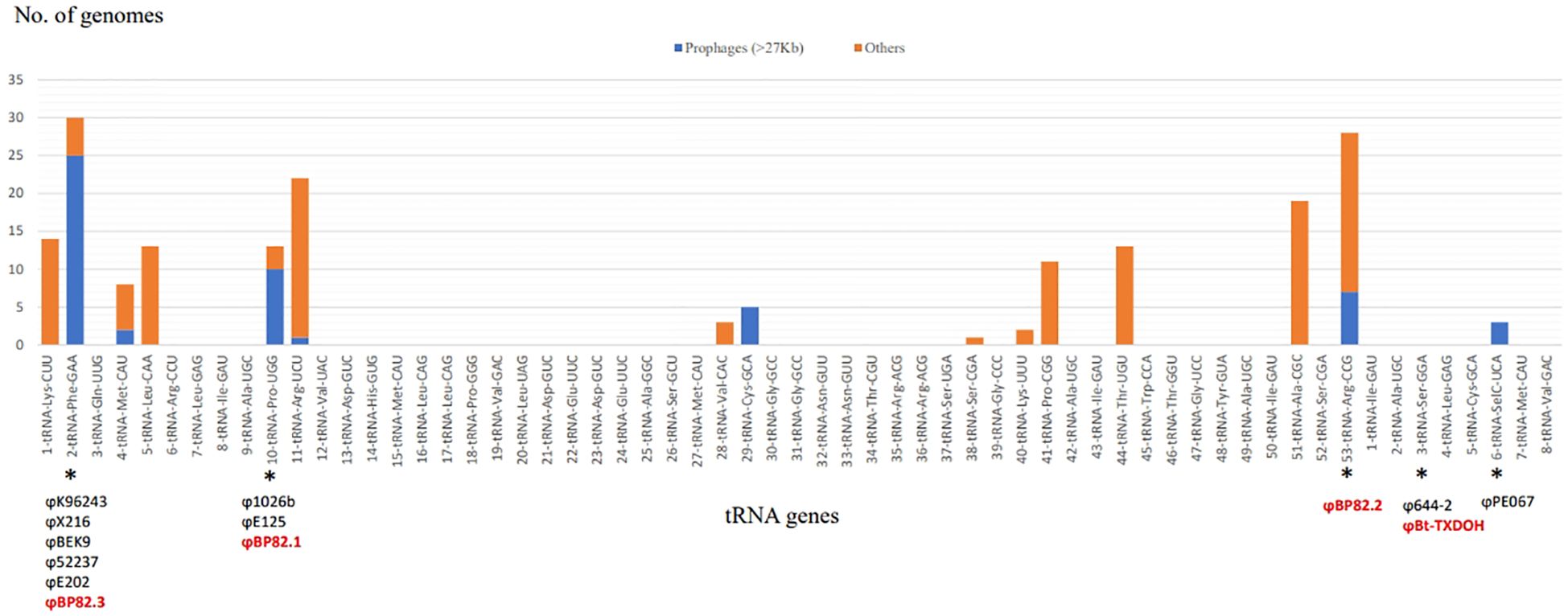
Figure 1 Distribution of tRNA-SSR sites in B. pseudomallei Genomes. The bar chart illustrates the count of genomes with either anticipated prophages (blue bars) or alternate exogenous DNA sequences (orange bars) associated with specific tRNA genes. Asterisks mark the prophage sites that have undergone characterization, highlighting the names of the temperate phages isolated for this research (red text) as well as those identified in prior research (black text).
Our investigation extended to seven additional species within the BPC including B. mallei, B. thailandensis, B. oklahomensis, B. humptydooensis, B. sevannae, B. mayonis, and B. singularis. Within the genomes of B. thailandensis, we identified a total of 69 instances of tRNA-SSRs across 19 complete genomes. These occurrences were distributed among 19 distinct tRNA gene loci, with two of them—tRNA-Pro (UGG) in strain USAMRU Malaysia#20 and tRNA-Ser (GGA) in strain TXDOH—being associated with prophage elements. Similar prophage associations were observed in B. oklahomensis genomes, specifically at tRNA-Arg (UCU) in strain C6786, tRNA-Phe (GAA) in strain E0147, and tRNA-Arg (CCG) in strain FDAARGOS_900. Notably, in both B. humtydooensis strains MSMB121 and MSMB122, we found a putative prophage linked to tRNA-SSR at tRNA-Leu (UAG), a novel prophage location unique to B. humptydooensis among BPC species. For B. savannae, B. mayonis, and B. singularis, which had a limited number of available genomes, we identified two putative prophages associated with tRNA-SSR at tRNA-Arg (UCU). One was located in B. savannae MSMB266 genome, while the other was found in B. mayonis BDU6. Although tRNA-SSR events occurred in B. singularis LMG 28154 and TSV85, none of them exhibited a prophage association. Our analysis also encompassed 108 complete genomes of B. mallei, revealing a single type of tRNA-SSR event linked to tRNA-Pro (CGG), which is equivalent to BPSLt41 locus in B. pseudomallei K96243 and also known as BMA_tRNA_Pro-5 in B. mallei ATCC 23344, in most B. mallei strains (data not shown). This integration site, spanning approximately 4.6 kb, featured insertion sequence elements, specifically IS407A, as previously described (Tuanyok et al., 2008). These tRNA-SSR occurrences and their prophage associations in BPC genomes were summarized in Supplementary Tables 2 (2.1 to 2.7). Details of the 3’end tRNA-gene direct repeat sequences associated with tRNA-SSR events at each tRNA gene are presented in Supplementary Table 3.
In this investigation, we focused on the identification of tRNA-SSR and phage attachment (attP) sequences within publicly accessible phage genomes associated with BPC species. We undertook this endeavor using 20 available BPC phage genomes, the majority of which are lysogenic phages that have been previously documented in various studies, as indicated in Table 1. Our research builds upon insights from previous genomic island research (Tuanyok et al., 2008) and our current findings have revealed that phage integrations are not random; rather, they exhibit a high degree of site-specificity. Among the 20 phage genomes we analyzed, 17 of them were found to have known attP sequences. Notably, 13 of these attP sequences were found to be identical to the 3’ end sequences of five different tRNA genes. To illustrate: 1) the genomes of φE125, φ1026b, and φBP82.1 featured a 49 bp attP sequence identical to the 3’ end sequence of tRNA-Pro (UGG); 2) φ52237, φE202, φX216, φBEK9, and φBp82.3 genomes contained a 45 bp attP sequence identical to the 3’ end sequence of tRNA-Phe (GAA); 3) φBp644.2 and φBt-TXDOH genomes had a 30 bp attP sequence identical to the 3’ end sequence of tRNA-Ser (GGA); 4) φE094 and φBP82.2 contained a 45 bp attP sequence identical to the 3’ end sequence of tRNA-Arg (CCG), and 5) φPE067 possessed a 16 bp attP sequence, 5’-CGGCTGCCTTCCGCCA-3’, identical to the 3’ end sequence of tRNA-SelC (UCA) as described in (Hammerl et al., 2020). These tRNA genes are corresponding to BPSLt10, BPSLt02, BPSSt03, BPSLt53, and BPSSt06 locus tags annotated in B. pseudomallei K96243 genome (Holden et al., 2004). It’s worth noting that φPE067 was previously identified to have a 19 bp attP sequence, 5’-CGGCTGCCTTCCGCCACAA-3’, identical to a chromosomal region responsible for selenocysteine biosynthesis on chromosome 2 of B. thailandensis E067 (Hammerl et al., 2020). Among the remaining four phage genomes, two, namely φE131 and φE058, were identified in the same study to have an 18 bp attP sequence identical to a chromosomal region of B. thailandensis E058. The other two genomes, φE12-2 and φPK23, contained an 18 bp attP sequence identical to a genomic island repeat sequence associated with GI15 of B. pseudomallei K96243. Further details regarding these publicly available BPC phage genomes and their corresponding attP sequences are presented in Table 1. Building on previous findings of seven distinct tRNA gene loci associated with prophages and the identification of the 30 bp attP sequence of φBp644-2, which matches the 3’end sequence of tRNA-Ser (GGA), this study uncovered a total of eight primary sites of phage integration in B. pseudomallei. These genetic events have been observed at the tRNA- Phe (GAA), - Met (CAU), - Proline (UGG), - Arg (UCU), - Cys (GCA), - Arg (CCG), - Ser (GGA), and – SelC (UCA) genes. Genomic locations of these tRNA-SSR events are illustrated in Figure 2.
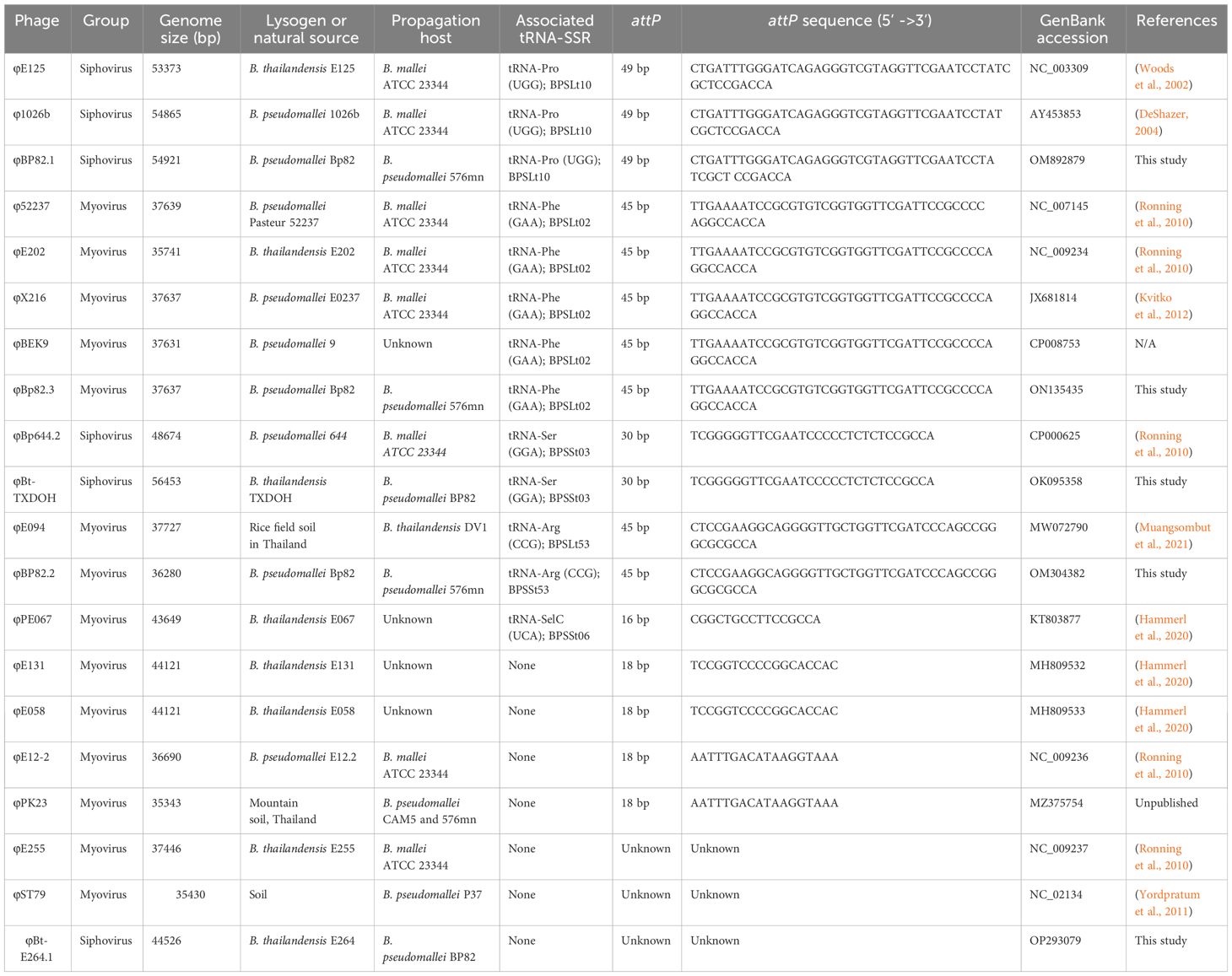
Table 1 Known or predicted attP sequences identified in Burkholderia phage genomes.
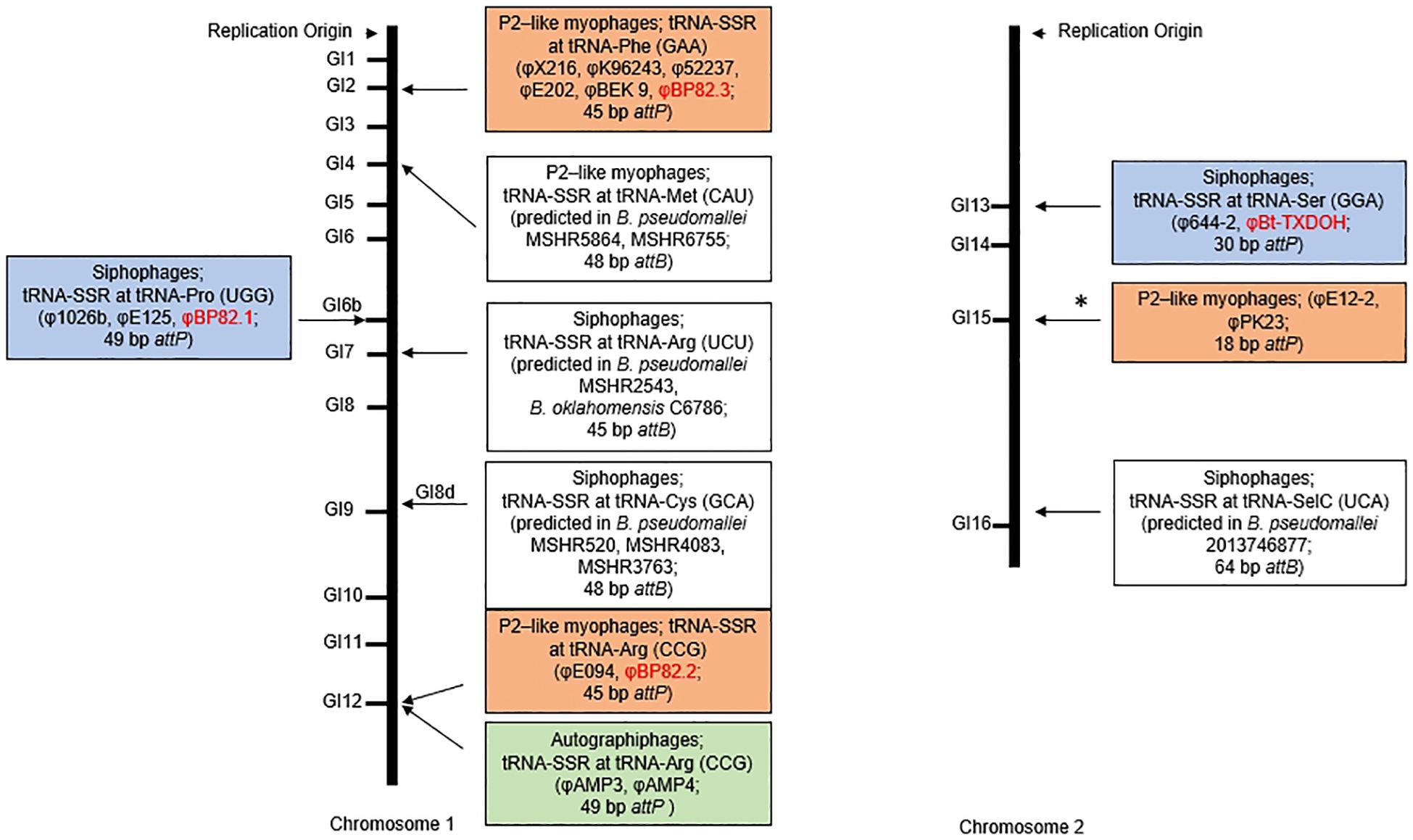
Figure 2 Illustrating the chromosomal locations of the prophages identified in B. pseudomallei genomes. This graphic depicts the chromosomal positions of prophages in B. pseudomallei genomes, corresponding to genomic island (GI) sites on B. pseudomallei K96243’s two chromosomes. There are eight sites with either predicted or confirmed prophages linked to tRNA-SSR sites at specified tRNA genes. An additional site correlates with GI15 (denoted by an asterisk). The annotations in each box detail the phage group (myovirus in orange, siphovirus in blue, autographivirus in green), the type of tRNA-SSR, and names of recognized phages or predicted prophages in BPC strains. Names of the temperate phages investigated in this study are in red text.
All 63 prophages identified as being associated with tRNA-SSR events across multiple strains within the BPC species were further annotated for gene features. Their sizes ranged from 27 kb, with 37 protein-coding sequences (CDSs) in a prophage associated with tRNA-Phe (GAA) gene of B. pseudomallei BPs111, to 61 kb, with 81 CDSs in a prophage associated with tRNA-Pro (UGG) gene in B. pseudomallei 1710b. Details of these prophages, including sizes and genomic identifier - locus tags and their sequences are available in Supplementary Table 5.1 – 5.6, Supplementary Text 1, respectively. Based on the annotation findings (Supplementary Table 6.1), seven specialty genes encoding phage holins were identified. Five of them were found in the prophages associated with tRNA-Cys (GCA) gene in B. pseudomallei strain MSHR520, MSHR305, MSHR3763, MSHR4083, and BDP, while other two were found in the prophages associated with tRNA-Arg (UCU) gene in B. pseudomallei 3000015486 and B. oklahomensis C6786, as detailed in Supplementary Table 6.2. Furthermore, biochemical pathway analysis has shown that some prophage genes are involved in purine and pyrimidine metabolism, such as those encoding for DNA-directed DNA polymerases, enzyme commission (EC) 2.7.7.6 and 2.7.7.7). These genes were found in the prophages associated with tRNA-Arg (UCU) gene and tRNA-Leu (UAG) gene. Interestingly, a prophage gene (CDS: 32008.1054.peg.132) in a prophage of B. humptydooensis MSMB122 was annotated to encode a metabolic enzyme, 7-cyano-7-deazaguanine synthase (EC 6.3.4.20), responsible for metabolism of cofactors and vitamins. Details of these genes and their biochemical pathways are available in Supplementary Table 6.3. Notably, none of the prophage genes identified in this study are coded for virulence or antimicrobial resistance determinants.
Based on the analysis of prophage profiles, it was found that B. pseudomallei 1026b, a prototypic strain, harbored three potential prophages associated with tRNA-SSR sites at tRNA-Pro (UGG), tRNA-Arg (CCG), and tRNA-Phe (GAA), shown in a schematic diagram in Supplementary Figure 1B. In a previous study (DeShazer, 2004), a well-characterized temperate phage, φ1026b, was isolated from this particular B. pseudomallei strain, forming plaques on a B. mallei ATCC23344’s lawn. It was established that this temperate phage was associated with a prophage caused by the tRNA-SSR at tRNA-Pro (UGG). Given that this strain contained two additional prophages, the question arose regarding their functionality, as only a temperate phage had been isolated in the previous study. To investigate this further, we used strain Bp82, a virulence-attenuated mutant of B. pseudomallei 1026b, to propagate the temperate phages from Bp82 using the host strain 576mn, which is also a virulence-attenuated strain suitable for a BSL-2 laboratory setting. Subsequently, we employed PCR assays targeting specific integrase genes associated with each tRNA-SSR to guide the isolation of distinct phages. As a result of these efforts, three different phage types were successfully isolated, named φBP82.1, φBP82.2, and φBP82.3. Transmission electron microscopy (TEM) was utilized to visualize the morphology of these phages. Notably, φBP82.1 exhibited characteristic similar to a lambda-like tailed phage belonging to the siphovirus group, while both φBP82.2 and φBP82.3 were identified as a P2-like phage with a contractile tail, classified within the myovirus group. TEM images of these three phages are presented in Figure 3.
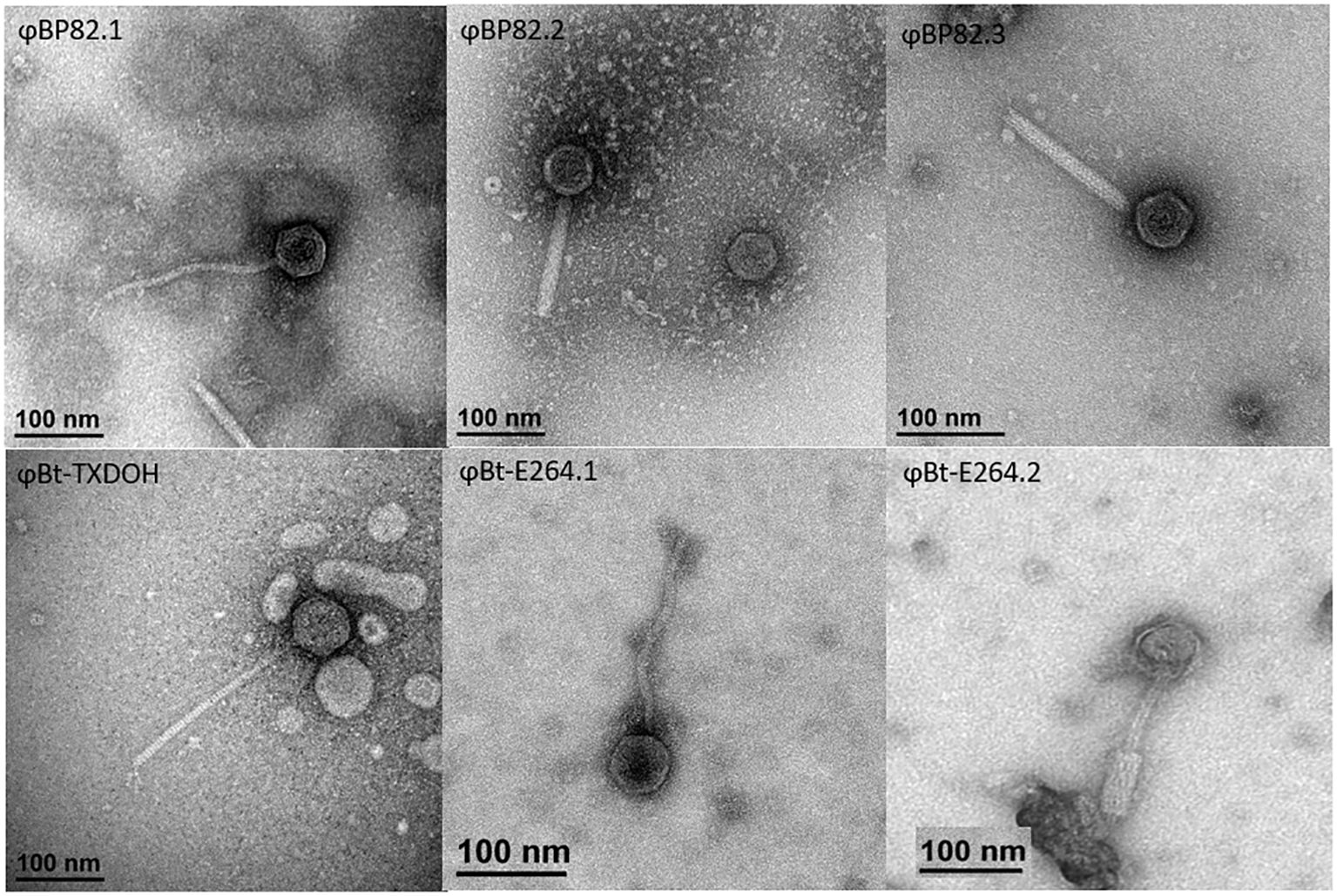
Figure 3 Transmission electron microscopy depictions of six characterized temperate phages. The upper section displays phages φBP82.1, φBP82.2, and φBP82.3, which were naturally induced from B pseudomallei Bp82 and purified from plaques on B pseudomallei 576mn lawn cultures. The lower section shows phages φBt-TXDOH, φBt-E264.1, and φBt-E264.2, originating from B thailandensis strains TXDOH and E264. Phages φBt-TXDOH and φBt-E264.1 were extracted from B pseudomallei Bp82 lawn cultures, whereas φBt-E264.2 was obtained from B pseudomallei 576mn lawns. Phages φBP82.1, φBt-TXDOH, and φBt-E264.1 belong to the siphovirus group. In contrast, phages with contractile tail, φBP82.2, φBP82.3, and φBt-E264.2 are P2-like viruses classified under the myovirus group.
Furthermore, we successfully isolated temperate phages from two different B. thailandensis strains, TXDOH (also known as 2003015869 (Glass et al., 2006a)), and E264 (Brett et al., 1998). In the case of TXDOH, it contained a putative prophage with an approximate size of 56.4 kb, identified to be associated with a tRNA-SSR at tRNA-Ser (GGA) located on its chromosome 2. This tRNA gene corresponds to the BTH_II2014 locus on chromosome 2 of B. thailandensis E264 and the BPSSt03 locus on chromosome 2 of B. pseudomallei K96243. From TXDOH, we isolated a temperate phage named φBt-TXDOH, which formed plaques on a B. pseudomallei Bp82 culture, while another host strain, 576mn, did not yield any plaques. Further analysis confirmed the morphology of φBt-TXDOH as a lambda-like tailed phage, classifying it within the siphovirus group.
In contrast, B. thailandensis E264, a reference laboratory strain used in many studies, did not show any prophages associated with tRNA genes based on its tRNA-SSR profile. Surprisingly, we identified two distinct temperate phage types from this strain. One of them, designated φBt-E264.1, exhibited characteristics resembling a lambda-like tailed phage and belonged to the siphovirus group. The other, named φBt-E264.2, was a P2-like phage with a contractile tail, categorized within the myovirus group. Both φBt-E264.1 and φBt-E264.2 were isolated, with φBt-E264.1 originating from the lawn of Bp82 and φBt-E264.2 from 576mn. Detailed TEM images of these three phages from B. thailandensis can be found in Figure 3.
In this research, we conducted the sequencing of five distinct temperate phages, φBP82.1, φBP82.2, φBP82.3, φBt-TXDOH, and φBt-E264.1, utilizing Illumina technology. We have attempted to sequence φBt-E264.2, but were unsuccessful due to its low yield in its host strain (B. pseudomallei 576mn). The specific details of these sequenced genomes and their corresponding GenBank accession numbers can be found in Table 1. Subsequently, we performed a comparative analysis of these genome sequences with known temperate phages within the context of B. pseudomallei, employing Easyfig (Sullivan et al., 2011), shown in Figure 4. Our investigation unveiled pronounced dissimilarities between the genomes of the two phage families, characterized by variations in their sizes. Notably, siphophages φBP82.1, φBt-TXDOH, and φBt-E264 exhibited genome sizes ranging from 44.5 kb to 56.5 kb, while myophages φBP82.2 and φBP82.3 possessed genome sizes of approximately 36.3 kb and 37.6 kb, respectively.
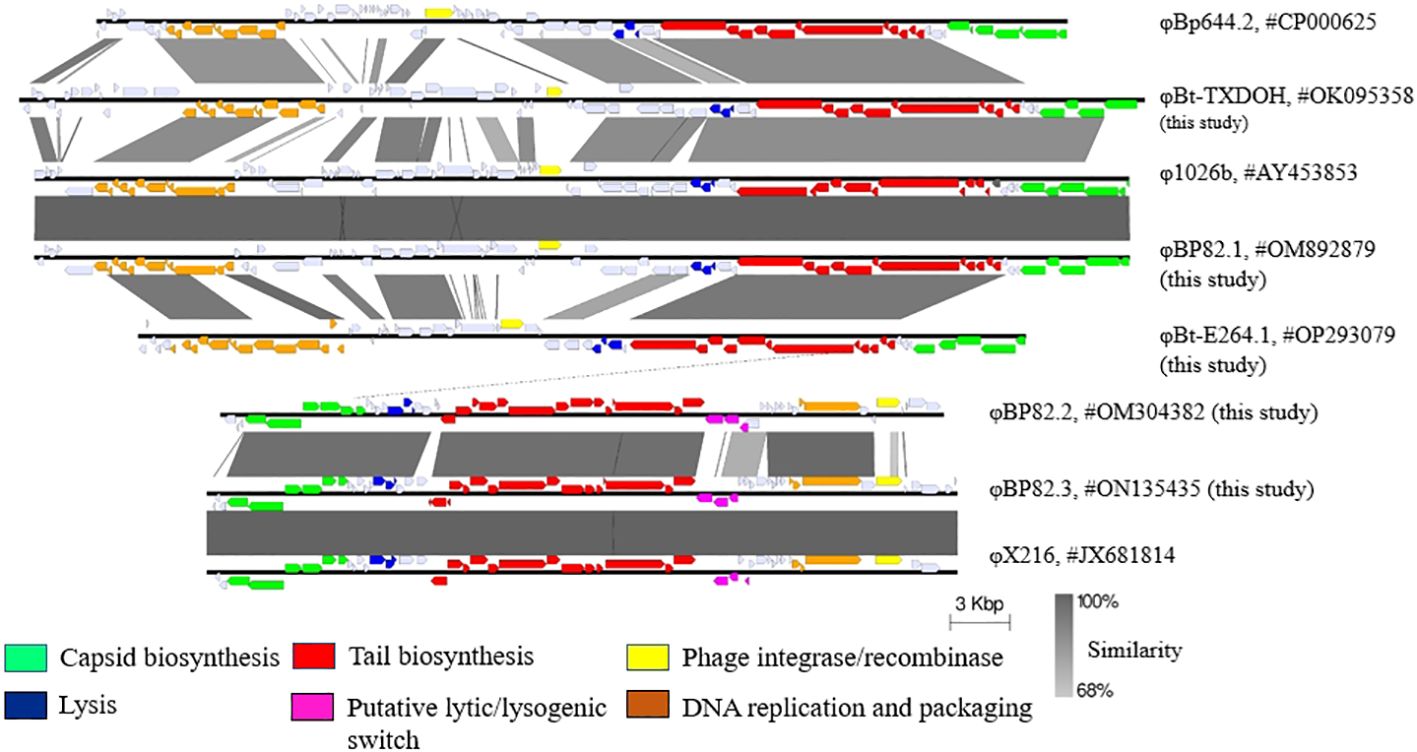
Figure 4 Genomic comparison of newly sequenced temperate bacteriophages. This analysis encompasses the genomes of five recently sequenced phages: φBt-TXDOH, φBP82.1, φBt-E264.1, φBP82.2, and φBP82.3, alongside known phages from the siphovirus and myovirus groups. The gene sequences associated with the formation of phage capsids and tails, as well as those involved in DNA replication and packaging, exhibit similarities within each family group. In contrast, genes implicated in the lytic/lysogenic cycle transition and phage integrase functions show variability, particularly when linked with distinct integration sites. Unexpectedly, φBP82.3 genome is almost identical to the genome of φX216 as previously described (Kvitko et al., 2012), even though both phages were induced from different B pseudomallei strains. We noted that the capsid biosynthesis genes in φBp644.2 and φBt-E264.1 are different to those in other siphoviruses being compared.
Investigating this, we employed PCR assays targeting six distinct phage integrase genes, each uniquely associated with a prophage genomic location. This study encompassed 321 B. pseudomallei strains and 9 B. thailandensis strains originating from southern Thailand. These prophages were associated with tRNA-genes such as tRNA-Phe (GAA), -Pro (UGG), -Arg (CCG), -Arg (UCU) and -Ser (GGA), and the prophage associated with the temperate phage φBt-E264.1 in B. thailandensis E264, as described above. Our findings revealed that 165 (51.4%) of the B. pseudomallei strains tested yielded positive results in at least one of the PCR assays, with 47 of them exhibiting positivity in two different assays. Specifically, 108, 45, and 31 strains tested positive in the PCR assays targeting prophages associated with tRNA-SSR at tRNA-Phe (GAA), -Pro (UGG), and -Arg (CCG), respectively. Interestingly, 19 B. pseudomallei strains tested positive for the integrase gene BTH_II1011, associated with the prophage linked to φBt-E264.1 in B. thailandensis E264. Additionally, 31 strains tested positive in two PCR assays targeting prophages at tRNA-Phe (GAA) and tRNA-Pro (UGG), while none of the B. pseudomallei strains displayed positive results in PCR assays targeting prophages associated with tRNA-SSR at tRNA-Arg (UCU) or Ser (GGA). Furthermore, among the nine B. thailandensis strains included in our study, four of them tested positive in the PCR assay targeting a functional prophage linked to tRNA-Ser (GGA) in B. thailandensis TXDOH and φBt-TXDOH. The remaining B. thailandensis strains displayed positivity in various other PCR assays, except for the one targeting the prophage at tRNA-Pro (UGG). PCR results are summarized in Supplementary Table 4.
It is well-recognized that B. pseudomallei is one of the most successful bacterial pathogens in causing severe infections in humans and many animal species, and indeed it has remarkable ability to survive in diverse environments. The acquisition of genetic materials through horizontal gene transfer and the resulting genomic plasticity significantly contribute to adaptability and pathogenicity in bacteria. This process enable not only B. pseudomallei but also other pathogenic bacteria to thrive in various ecological niches and contributes to their success as a pathogens. In the current study, we identified tRNA-SRR as a major mechanism that shaped up individual B. pseudomallei genomes and this mechanism was also found in its closely relative species within the BPC. We believe that phage transduction and the lysogenic state could have contributed to the fitness of B. pseudomallei. Here are some major points to discuss:
We have established a connection between tRNA-SSR events and a minimum of 15 tRNA gene loci within the pangenome of B. pseudomallei. Among these, seven loci are particularly noteworthy: tRNA-Phe (GAA), tRNA-Met (CAU), tRNA-Pro (UGG), tRNA-Arg (UCU), tRNA-Cys (GCA), tRNA-Arg (CCG), and tRNA-SelC (UCA). This association is drawn from an in-depth analysis of 106 complete genomes of B. pseudomallei, revealing the presence of potential prophages within these tRNA loci. Furthermore, our investigation uncovered an additional tRNA-SSR event involving tRNA-Ser (GGA), which is linked to a functional prophage in B. thailandensis TXDOH. This discovery was made through the examination of the temperate phage φBt-TXDOH. Conclusively, these eight tRNA genes are identified as the hotspots for phage integration (Figure 2). Historically, our understanding of the tRNA gene’s involvement in phage integration began in 2002 when Woods and colleagues (Woods et al., 2002) demonstrated that the temperate siphophage φE125, spontaneously produced by B. thailandensis E125, integrated its genome into B. mallei ATCC 23344. This integration occurred at a short 49 bp homologous 3’ end sequence of the tRNA-Pro (UGG) gene. A follow-up study by DeShazer (2004) revealed a similar case with the temperate siphophage φ1026b from B. pseudomallei 1026b, which shared a genome sequence with φE125 and utilized the same attP sequence, a 49 bp homologous sequence at the 3’ end of the tRNA-Pro (UGG) gene. A few years later, Ronning and colleagues conducted an analysis of 37 prophages and prophage-like elements from six different Burkholderia species (Ronning et al., 2010). Among them, five temperate phages (φ52237, φE12-2, φ644-2, φE202, and φE255) were spontaneously isolated from lysogenic B. pseudomallei and B. thailandensis host strains. Three of these phages were found to have attachment or integration sites (attP) associated with 3’ end sequences of tRNA genes. Specifically, φ52237 and φE202 featured a 45 bp attP identical to the 3’ end sequence of tRNA-Phe (GAA), while φ644-2 possessed a 30 bp attP sequence identical to that of tRNA-Ser (GGA).
In addition, a recent study by Hammerl and colleagues characterized three temperate phages (φE067, φE131, φE058) from lysogenic B. thailandensis strains E067, E131, and E058 (Hammerl et al., 2020). Upon analysis, φE067 was found to have a 16 bp attP sequence identical to tRNA-SelC (UCA) in B. pseudomallei K96243. The cumulative findings from our research and others have suggested that these eight tRNA locations serve as hotspots for phage integrations in B. pseudomallei, and some of these hotspots in other BPC species also harbor prophages. Charactering these prophages in these species warrants further investigation.
This study has established that three phages, φBP82.1, φBP82.2, and φBP82.3, were naturally released from corresponding prophages within B. pseudomallei Bp82, a strain derived from B. pseudomallei 1026b lacking the purM gene (Propst et al., 2010). We employed PCR techniques, focusing on unique integrase genes in each tRNA-SSR from these prophages, to confirm their presences in many small plaques formed on a B. pseudomallei 576mn bacterial lawn. Further examination revealed that φBP82.1 is a match for φ1026b, a previously identified siphophage in B. pseudomallei 1026b, and the two other phages were developed from the two suspected prophages associated with tRNA-SSR events. Moreover, the study extended to explore the prevalence of such prophages among B. pseudomallei strains from clinical and environmental sources in a region of southern Thailand, with over half of the strains testing positive for at least one prophage. These findings indicate a widespread lysogenic state within the B. pseudomallei population. The study also noted the commonality of prophages linked to specific tRNA-SSR events, although some associated with certain tRNA genes were absent in the Thai strains, raising the question of geographic specificity of prophage presence. Despite their rarity, the prophage associated with tRNA-Arg (UCU) was identified in BPC strains from diverse geographical locations, including B. pseudomallei MSHR2543 and B. savannae MSMB266 from Australia and B. oklahomensis C6786 from the USA. These prophages are classified within the siphovirus group based on their genomic composition. However, due to the unavailability of these strains in public repositories, it was not possible to confirm their functionality in relation to the tRNA-SSR. Interestingly, a rare prophage associated with tRNA-Ser (GGA) in B. pseudomallei 644 (Ronning et al., 2010) was present in several B. thailandensis strains, and we successfully induced φBt-TXDOH from a functional prophage correlating with this tRNA-Ser site. This finding suggests potential avenues for further research into the distribution of this prophage among B. thailandensis populations and the biological significance of its temperate phage, since they infected B. pseudomallei BP82 in our experiments.
During the peer review of this article, a question was raised about whether the presence of prophages contribute to virulence, antimicrobial resistance, and or fitness of B. pseudomallei, particularly since the genomes analyzed originated from diverse sources, including human and animal cases, as well as the environment. To date, no virulence or antimicrobial resistance genes have been identified in the 63 prophages from the genomes of B. pseudomallei and other BPC species. Notably, a higher frequency (43/91, 47.3%) of prophages was observed in strains from human and animal cases compared to environmental strains (2/10, 20%), according to Supplementary Table 5.1. It is important to note that most genomes used in this study were from the clinical B. pseudomallei strains and two of them, 1026b and BGR, contained up to three prophages. Furthermore, the presence of specialty genes, such as those encoding holins (phage-encoded membrane proteins involved in bacterial cell lysis in the final stage of the bacteriophage reproductive cycle (Abeysekera et al., 2022)), in some prophages are of particular interest. These genes are notably present in two rare prophages: one associated with the tRNA-Cys (GCA) gene found only in Australian B. pseudomallei strains, and the other was associated with tRNA-Arg (UCU) in B. oklahomensis C6786 (as detailed in Supplementary Table 6.2). Additionally, we have identified several prophage genes involved in biochemical pathways, particularly in purine and pyrimidine metabolism in B. humptydooensis MSMB121, MSMB122, and B. savannae MSMB266, as well as in folate biosynthesis in a prophage of B. oklahomensis C6786 (refer to Supplementary Table 6.3). These genes could enhance the fitness of the bacterial host strains. We believe that the possession of prophage genes could confer benefit to their bacterial hosts at certain points, which warrants further investigation. These studies would include mutagenesis of prophage genes, phage transduction, and virulence determination in animal models.
In summary, the data presented here displays that there is a clear association between specific tRNA genes and the presence of prophages, and that this presence is not random. Additionally, due to the high number of tRNA-SSR events in BPC genomes associated with various MGEs including prophages, we conclude that these MGEs contribute to an increase in diversity of the BPC species similar to other bacterial species that have an open-genome and not related to BPC genomic plasticity.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, OM892879 https://www.ncbi.nlm.nih.gov/genbank/, ON135435, https://www.ncbi.nlm.nih.gov/genbank/, OM304382 https://www.ncbi.nlm.nih.gov/genbank/, OK095358, https://www.ncbi.nlm.nih.gov/genbank/, OP293079.
PK: Data curation, Investigation, Methodology, Writing – review & editing, Formal analysis. II: Data curation, Investigation, Methodology, Writing – original draft, Formal analysis. PA: Investigation, Methodology, Writing – review & editing. MA-R: Investigation, Methodology, Writing – review & editing. JK: Resources, Writing – review & editing. AT: Funding acquisition, Supervision, Writing – original draft, Writing – review & editing, Conceptualization, Formal analysis, Project administration.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was funded in full by Tuanyok Burkholderia Laboratory’s salary saving funds. AT was supported in part by the University of Florida - Preeminent Professorship Program and the Defense Threat Reduction Agency grant no. HDTRA12110029.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbrio.2024.1339809/full#supplementary-material
Supplementary Text 1 | DNA sequences of tRNA-SSR associated prophages in B. pseudomallei and other BPC species.
Abeysekera G. S., Love M. J., Manners S. H., Billington C., Dobson R. C. J. (2022). Bacteriophage-encoded lethal membrane disruptors: Advances in understanding and potential applications. Front. Microbiol. 13, 1044143. doi: 10.3389/fmicb.2022.1044143
Arndt D., Grant J. R., Marcu A., Sajed T., Pon A., Liang Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Brett P. J., DeShazer D., Woods D. E. (1998). Burkholderia Thailandensis sp. nov., a Burkholderia pseudomallei-like species. Int. J. Syst. Bacteriol. 48 Pt 1, 317–320. doi: 10.1099/00207713-48-1-317
Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., et al. (2009). BLAST+: architecture and applications. BMC Bioinf. 10, 421. doi: 10.1186/1471-2105-10-421
DeShazer D. (2004). Genomic diversity of Burkholderia pseudomallei clinical isolates: subtractive hybridization reveals a Burkholderia mallei-specific prophage in B. pseudomallei 1026b. J. Bacteriol. 186, 3938–3950. doi: 10.1128/JB.186.12.3938-3950.2004
Gindreau E., Torlois S., Lonvaud-Funel A. (1997). Identification and sequence analysis of the region encoding the site-specific integration system from Leuconostoc oenos (OEnococcus oeni) temperate bacteriophage phi 10MC. FEMS Microbiol. Lett. 147, 279–285. doi: 10.1016/S0378-1097(96)00540-X
Glass M. B., Gee J. E., Steigerwalt A. G., Cavuoti D., Barton T., Hardy R. D., et al. (2006a). Pneumonia and septicemia caused by Burkholderia Thailandensis in the United States. J. Clin. Microbiol. 44, 4601–4604. doi: 10.1128/JCM.01585-06
Glass M. B., Steigerwalt A. G., Jordan J. G., Wilkins P. P., Gee J. E. (2006b). Burkholderia oklahomensis sp. nov., a Burkholderia pseudomallei-like species formerly known as the Oklahoma strain of Pseudomonas pseudomallei. Int. J. Syst. Evol. Microbiol. 56, 2171–2176. doi: 10.1099/ijs.0.63991-0
Hall C. M., Baker A. L., Sahl J. W., Mayo M., Scholz H. C., Kaestli M., et al. (2022). Expanding the Burkholderia pseudomallei Complex with the Addition of Two Novel Species: Burkholderia mayonis sp. nov. and Burkholderia savannae sp. nov. Appl. Environ. Microbiol. 88, e0158321. doi: 10.1128/AEM.01583-21
Hallet B., Sherratt D. J. (1997). Transposition and site-specific recombination: adapting DNA cut-and-paste mechanisms to a variety of genetic rearrangements. FEMS Microbiol. Rev. 21, 157–178. doi: 10.1111/j.1574-6976.1997.tb00349.x
Hammerl J. A., Volkmar S., Jacob D., Klein I., Jäckel C., Hertwig S. (2020). The burkholderia Thailandensis phages ΦE058 and ΦE067 represent distinct prototypes of a new subgroup of temperate burkholderia myoviruses. Front. Microbiol. 11, 1120. doi: 10.3389/fmicb.2020.01120
Holden M. T., Titball R. W., Peacock S. J., Cerdeno-Tarraga A. M., Atkins T., Crossman L. C., et al. (2004). Genomic plasticity of the causative agent of melioidosis, Burkholderia pseudomallei. Proc. Natl. Acad. Sci. USA 101, 14240–14245. doi: 10.1073/pnas.0403302101
Kaewrakmuk J., Chusri S., Hortiwakul T., Kawila S., Patungkaro W., Jariyapradub B., et al. (2023). Under-reporting cases and deaths from melioidosis: A retrospective finding in Songkhla and Phatthalung province of southern Thailand, 2014-2020. Trop. Med. Infect. Dis. 8, 1–11. doi: 10.3390/tropicalmed8050286
Kim H. S., Schell M. A., Yu Y., Ulrich R. L., Sarria S. H., Nierman W. C., et al. (2005). Bacterial genome adaptation to niches: divergence of the potential virulence genes in three Burkholderia species of different survival strategies. BMC Genomics 6, 1–13. doi: 10.1186/1471-2164-6-174
Kvitko B. H., Cox C. R., DeShazer D., Johnson S. L., Voorhees K. J., Schweizer H. P. (2012). φX216, a P2-like bacteriophage with broad Burkholderia pseudomallei and B. mallei strain infectivity. BMC Microbiol. 12, 289. doi: 10.1186/1471-2180-12-289
Marinus M. G., Poteete A. R. (2013). High efficiency generalized transduction in Escherichia coli O157:H7. F1000Res 2, 7. doi: 10.12688/f1000research
Muangsombut V., Withatanung P., Chantratita N., Chareonsudjai S., Lim J., Galyov E. E., et al. (2021). Rapid clinical screening of burkholderia pseudomallei colonies by a bacteriophage tail fiber-based latex agglutination assay. Appl. Environ. Microbiol. 87, e0301920. doi: 10.1128/AEM.03019-20
Nierman W. C., DeShazer D., Kim H. S., Tettelin H., Nelson K. E., Feldblyum T., et al. (2004). Structural flexibility in the Burkholderia mallei genome. Proc. Natl. Acad. Sci. USA 101, 14246–14251. doi: 10.1073/pnas.0403306101
Norris M. H., Rahman Khan M. S., Schweizer H. P., Tuanyok A. (2017). An avirulent Burkholderia pseudomallei ΔpurM strain with atypical type B LPS: expansion of the toolkit for biosafe studies of melioidosis. BMC Microbiol. 17, 132. doi: 10.1186/s12866-017-1040-4
Olson R. D., Assaf R., Brettin T., Conrad N., Cucinell C., Davis J. J., et al. (2023). Introducing the Bacterial and Viral Bioinformatics Resource Center (BV-BRC): a resource combining PATRIC, IRD and ViPR. Nucleic Acids Res. 51, D678–d689. doi: 10.1093/nar/gkac1003
Propst K. L., Mima T., Choi K. H., Dow S. W., Schweizer H. P. (2010). A Burkholderia pseudomallei delta-purM mutant is a avirulent in immune competent and immune deficient animals: candidate strain for exclusion from select agent lists. Infect. Immun. 78, 3136–3143. doi: 10.1128/IAI.01313-09
Pumpuang A., Chantratita N., Wikraiphat C., Saiprom N., Day N. P. J., Peacock S. J., et al. (2011). Survival of Burkholderia pseudomallei in distilled water for 16 years. Trans. R. Soc. Trop. Med. Hygiene 105, 598–600. doi: 10.1016/j.trstmh.2011.06.004
Quinlan A. R., Hall I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. doi: 10.1093/bioinformatics/btq033
Ramsey J., Rasche H., Maughmer C., Criscione A., Mijalis E., Liu M., et al. (2020). Galaxy and Apollo as a biologist-friendly interface for high-quality cooperative phage genome annotation. PloS Comput. Biol. 16, e1008214. doi: 10.1371/journal.pcbi.1008214
Ronning C. M., Losada L., Brinkac L., Inman J., Ulrich R. L., Schell M., et al. (2010). Genetic and phenotypic diversity in Burkholderia: contributions by prophage and phage-like elements. BMC Microbiol. 10, 202. doi: 10.1186/1471-2180-10-202
Spring-Pearson S. M., Stone J. K., Doyle A., Allender C. J., Okinaka R. T., Mayo M., et al. (2015). Pangenome Analysis of Burkholderia pseudomallei: Genome Evolution Preserves Gene Order despite High Recombination Rates. PloS One 10, e0140274. doi: 10.1371/journal.pone.0140274
Sullivan M. J., Petty N. K., Beatson S. A. (2011). Easyfig: a genome comparison visualizer. Bioinformatics 27, 1009–1010. doi: 10.1093/bioinformatics/btr039
Tuanyok A., Leadem B. R., Auerbach R. K., Beckstrom-Sternberg S. M., Beckstrom-Sternberg J. S., Mayo M., et al. (2008). Genomic islands from five strains of Burkholderia pseudomallei. BMC Genomics 9, 566. doi: 10.1186/1471-2164-9-566
Tuanyok A., Mayo M., Scholz H., Hall C. M., Allender C. J., Kaestli M., et al. (2017). Burkholderia humptydooensis sp. nov., a New Species Related to Burkholderia Thailandensis and the Fifth Member of the Burkholderia pseudomallei Complex. Appl. Environ. Microbiol. 83, e02802-16. doi: 10.1128/AEM.02802-16
Vandamme P., Peeters C., De Smet B., Price E. P., Sarovich D. S., Henry D. A., et al. (2017). Comparative Genomics of Burkholderia singularis sp. nov., a Low G+C Content, Free-Living Bacterium That Defies Taxonomic Dissection of the Genus Burkholderia. Front. Microbiol. 8, 1679. doi: 10.3389/fmicb.2017.01679
Woods D. E., Jeddeloh J. A., Fritz D. L., DeShazer D. (2002). Burkholderia Thailandensis E125 harbors a temperate bacteriophage specific for Burkholderia mallei. J. Bacteriol. 184, 4003–4017. doi: 10.1128/JB.184.14.4003-4017.2002
Yordpratum U., tattawasart U., Wongratanacheewin S., Sermswan R. W. (2011). Novel lytic bacteriopphages from soil that lyse Burkholderia pseudomallei. FEMS Microbiol. Lett. 314, 81–88. doi: 10.1111/fml.2010.314.issue-1
Keywords: Burkholderia pseudomallei, melioidosis, prophage, temperate phage, tRNA-SSR
Citation: Khrongsee P, Irby I, Akaphan P, Alami-Rose MA, Kaewrakmuk J and Tuanyok A (2024) A comprehensive study of prophage islands in Burkholderia pseudomallei complex. Front. Bacteriol. 3:1339809. doi: 10.3389/fbrio.2024.1339809
Received: 16 November 2023; Accepted: 15 April 2024;
Published: 29 April 2024.
Edited by:
Antony T. Vincent, Laval University, CanadaReviewed by:
Sohinee Sarkar, Royal Children’s Hospital, AustraliaCopyright © 2024 Khrongsee, Irby, Akaphan, Alami-Rose, Kaewrakmuk and Tuanyok. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Apichai Tuanyok, dHVhbnlva0B1ZmwuZWR1
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.