 Carl J. E. Suster
Carl J. E. Suster David Pham3
David Pham3 Vitali Sintchenko
Vitali Sintchenko- 1Centre for Infectious Diseases and Microbiology – Public Health, Westmead Hospital, Westmead, NSW, Australia
- 2Sydney Infectious Diseases Institute, The University of Sydney, Sydney, NSW, Australia
- 3Centre for Infectious Diseases and Microbiology Laboratory Services, Institute of Clinical Pathology and Medical Research, NSW Health Pathology, Westmead, NSW, Australia
The analysis of microbial genomes has long been recognised as a complex and data-rich domain where artificial intelligence (AI) can assist. As AI technologies have matured and expanded, pathogen genomics has also contended with exponentially larger datasets and an expanding role in clinical and public health practice. In this mini-review, we discuss examples of emerging applications of AI to address challenges in pathogen genomics for precision medicine and public health. These include models for genotyping whole genome sequences, identifying novel pathogens in metagenomic next generation sequencing, modelling genomic information using approaches from computational linguistics, phylodynamic estimation, and using large language models to make bioinformatics more accessible to non-experts. We also examine factors affecting the adoption of AI into routine laboratory and public health practice and the need for a renewed vision for the potential of AI to assist pathogen genomics practice.
1 Introduction
Artificial intelligence (AI) has long been recognised as a powerful tool for analysing genome sequences. The AI boom of the 1980s propelled by the emerging popularity of expert systems saw the development of knowledge-based platforms to assist with experimental planning in genetics (Stefik, 1981; Friedland et al., 1982). The nascent application of AI to molecular biology was viewed with much optimism as a means to make sense of the complex data that were amassing; ambitious visions conceived of a computer intelligence that would not merely process data but would execute the scientific processes of hypothesising, experimental design, and knowledge synthesis (Hunter, 1992; Rawlings et al., 1994). Over the subsequent decade, AI approaches including algorithmic classifiers and artificial neural networks were applied to tasks from distinguishing translational initiation sites in bacteria (Stormo et al., 1982) to predicting protein structure and function (Hunter, 1993). Excitement in AI driving knowledge discovery was tempered by the realisation that contemporary technology and understanding were not sufficiently mature to realise the vision.
In the intervening years, both AI technology and the breadth and scope of pathogen genomics have advanced exponentially. AI has been employed across diverse aspects of the response to the COVID-19 pandemic (Syrowatka et al., 2021; Arora et al., 2021; Chen et al., 2022; Ahmed et al., 2022; Sarmiento Varón et al., 2023; Malhotra and Sodhi, 2023), and in clinical and molecular medicine applications more broadly (Gomes and Ashley, 2023; Haug and Drazen, 2023). The central focus has shifted from integrated knowledge systems to more specialised tools for tasks including predicting antimicrobial resistance (Anahtar et al., 2021) and identifying patterns in larger disease surveillance datasets (Brownstein et al., 2023).
In this mini-review, we focus on innovative applications of AI to pathogen genomics that model diagnostic problems in a novel manner or adopt existing approaches in an unconventional way. We discuss several illustrative examples of how AI can already—or might soon—assist clinical and public health investigations, as summarised in Figure 1 and Table 1. We outline themes shaped by current challenges in the analysis of medically relevant microbial genomic data and the possibilities promised by emerging technologies.
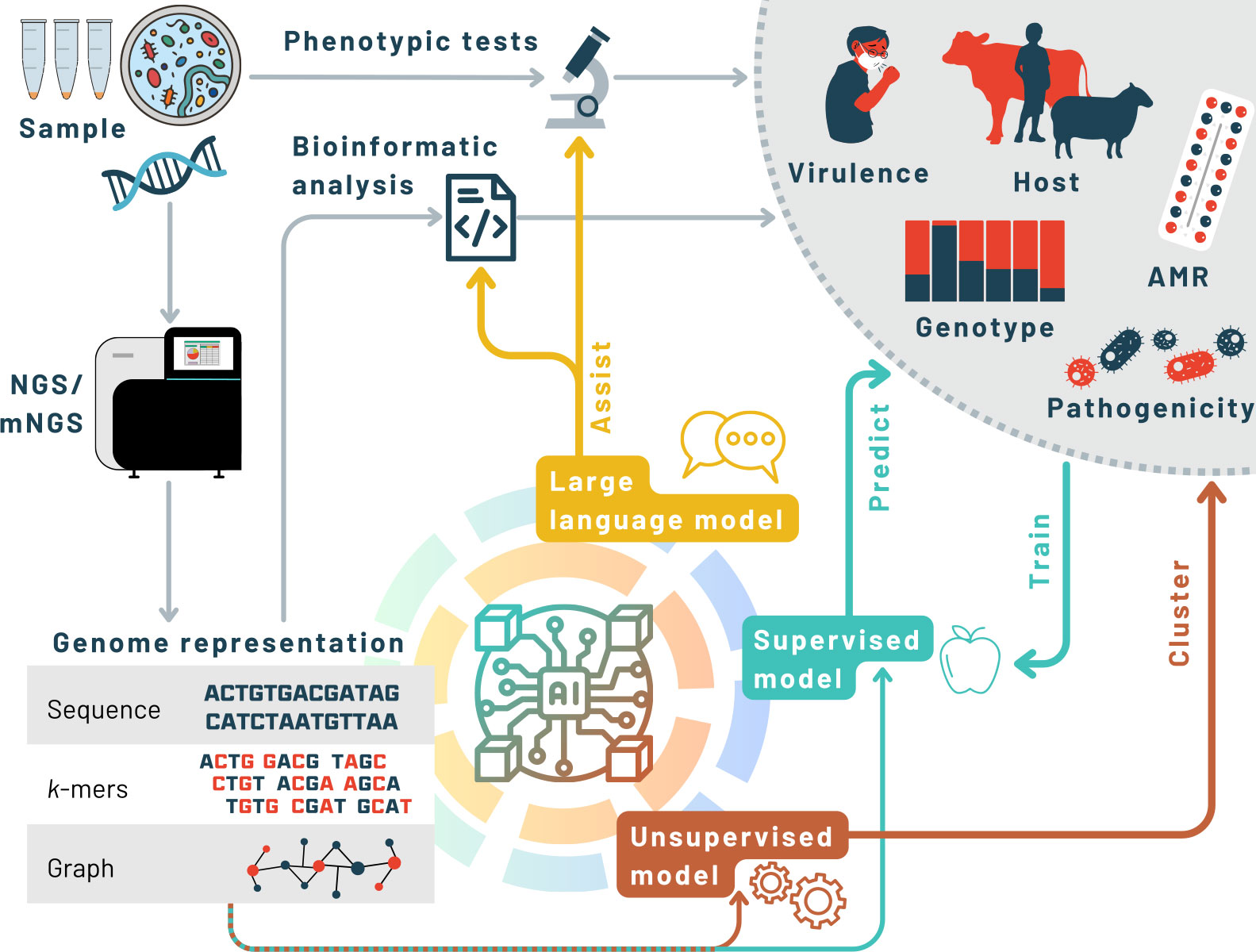
Figure 1 Illustration of the roles of major categories of AI in the workflow from sample to results (grey circle). Conventional workflows (grey arrows) use phenotypic testing or genomic analysis to derive information of public health or clinical relevance. Large language models can assist with generating code, interpreting phenotypic findings, or retrieving information. Supervised ML models are trained with examples to make predictions from genomic data. Unsupervised ML models identify patterns in genomic data without requiring examples. The choice of genome representation is important for AI methods. AMR, antimicrobial resistance; NGS, next generation sequencing; mNGS, metagenomic NGS.
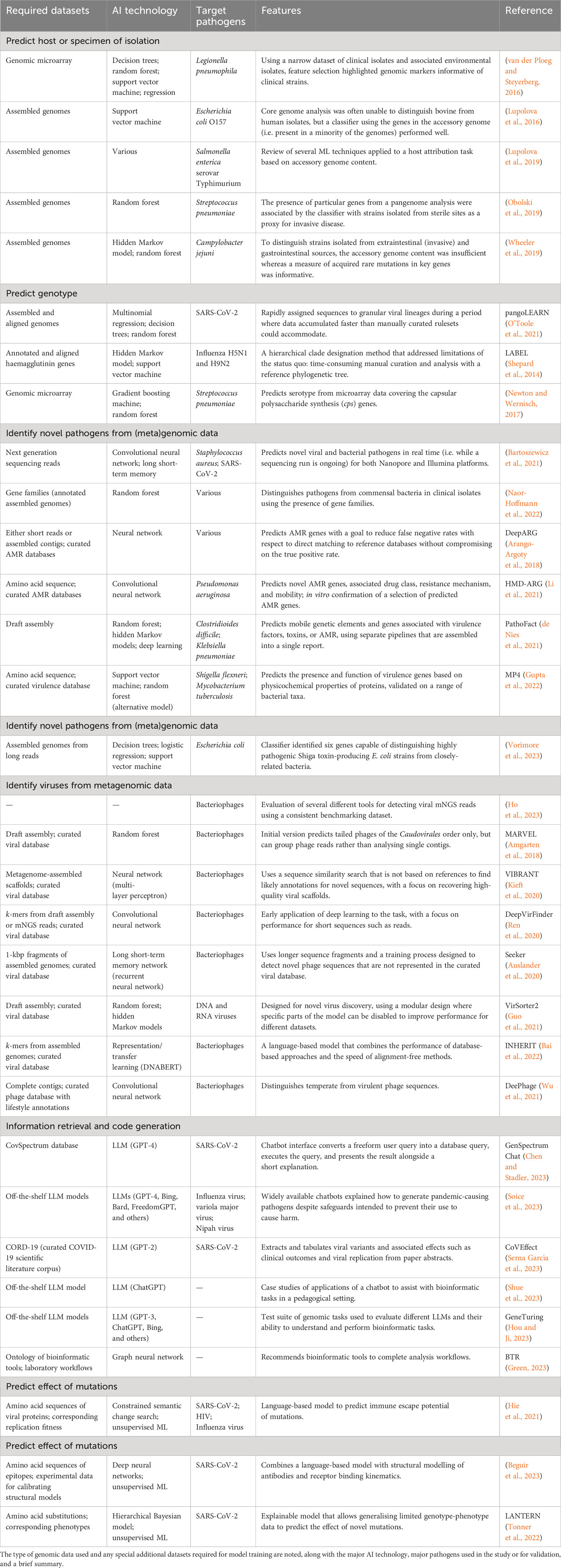
Table 1 Example applications of AI to a selection of problems in pathogen genomics.
2 AI primer
AI broadly encompasses intelligent behaviours exhibited by machines. The field is organised around a long-term goal of producing intelligent and autonomous artificial agents capable of equalling or exceeding human cognitive abilities. Current applications draw on specific facets of intelligence such as reasoning for classification and decision-making, knowledge retrieval and representation, perception, and communication in natural language (Jiang et al., 2022). Machine learning (ML) refers to methods where a task is performed by an algorithm or agent that improves its performance by some measure as it acquires more experience or data (Mitchell, 1997). In general, ML methods are data-driven instead of relying on behaviours fully specified a priori. They are broadly grouped by learning paradigm (Bonaccorso, 2018).
Supervised learning requires labelled examples: a ground truth established by independent means. A candidate model is proposed by relating input features (e.g. attributes of specimens and patients, gene presence, representations of nucleotide or amino acid sequences) to the target variable, which is discrete for classification problems or continuous for regression problems. Learning proceeds iteratively by adjusting the model’s parameters to decrease the error in its predictions. If the model has too closely learned the contours of its training data then it will reproduce spurious correlations and biases present in those examples that do not represent general patterns, and it is said to have overfit its training data. Robust models have sufficient and representative training data and a meaningful cross-validation procedure. Popular supervised models include random forests and support vector machines (Bonaccorso, 2018).
By contrast, unsupervised learning aims to discover structure directly from unlabelled data. This often involves using a similarity measure to cluster the data, and by extension identifying anomalous departures from typical patterns. Common unsupervised techniques include principal component analysis and k-means clustering (Bonaccorso, 2018). Hidden Markov models are a type of latent variable model widely deployed in bioinformatic tools for tasks such as homology detection and sequence alignment, and often trained in an unsupervised manner (Eddy, 2004).
Deep learning refers to the class of models based on artificial neural networks with a multi-layer network topology (Sarker, 2021; Esteva et al., 2019). Training to determine the weights connecting nodes can be supervised, unsupervised, or a combination. In feed-forward networks (e.g. multilayer perceptrons) signals flow through the network in one direction, whereas recurrent networks (e.g. long short-term memory) contain loops allowing long-range patterns to be learned efficiently (Hochreiter and Schmidhuber, 1997). Convolutional layers incorporate two-dimensional relationships in input data, with applications in computer vision (Gu et al., 2018). Many deep learning models (e.g. variational autoencoders) are designed for representation learning: i.e. to take raw data and automatically discover relevant features that would otherwise have been engineered by the analyst (Bengio et al., 2013).
Natural language processing (NLP) is the multidisciplinary area of research concerned with computer analysis of the full gamut of linguistics from the physical manifestations of language to the syntax governing their combination and systems for conveying meaning and style. Recent attention has focused on the advances in large language models (LLMs). These are artificial neural networks with billions of parameters trained using extensive corpora to produce plausible text continuations. Most current LLMs use a transformer architecture based on the ML concept of attention as an alternative to recurrence for capturing long-range patterns (Vaswani et al., 2017; De Santana Correia and Colombini, 2022; Choi and Lee, 2023). Current models can respond to prompts in a way that convincingly suggests the involvement of cognitive skills like comprehension, language production, and reasoning, despite lacking explicit models of grammar or knowledge (Wolfram, 2023; Bubeck et al., 2023).
All applications of AI to genomic data must make a choice of how to represent the genome in a usable form. Models processing raw sequencing reads must cope with uninformative variation including reverse complements, primer adapters, and characteristic errors associated with the sequencing platform. On the other hand, more processed forms such as draft genome assemblies risk discarding potentially relevant information. Many models use k-mers—short overlapping fragments of a small number, k, of nucleotides—as a convenient encoding (Alam and Chowdhury, 2020), while others use larger pieces of genomes. The choice may be influenced by characteristics of the AI techniques employed and the context and quality of data.
3 Virulence and genotype prediction
The characterisation of medically-relevant pathogens—as distinct from non-virulent colonising organisms—is one of the most urgent tasks in clinical microbiology. This task includes the identification of pathogens present in a specimen, their type based on antigenic markers or genomic features (Ramadan, 2022), and their virulence and antimicrobial resistance (AMR) phenotypes. These findings inform clinical management, public health responses to outbreaks, and the objectives of surveillance programs including risk planning and antimicrobial stewardship. Analysis of microbial whole genome sequences (WGS) has been increasingly complementing or in some instances replacing traditional microbiological assays in this setting (Gilbert, 2002). The greater depth of biologically meaningful information contained in WGS enables considerable nuance in investigations and commensurate challenges for interpretation.
With a suitably large collection of related microbial genomes, it becomes possible to search for genomic determinants of specific phenotypes. Supervised ML classifiers are well-suited to this task as they can uncover weak signals that correlate with membership in particular classes. A number of studies have examined surveillance datasets consisting of genomes isolated from both clinical and environmental samples, using Legionella pneumophila genome microarray data (van der Ploeg and Steyerberg, 2016) and WGS for Escherichia coli (Lupolova et al., 2016) and Salmonella enterica (Lupolova et al., 2019). These studies trained ML classifiers to predict which sequences were associated with clinical sources. This information by itself is of limited value since it is known to the laboratory in most circumstances. Instead, it is the ability to interrogate the workings of the classifiers that turns out to be useful. Identifying genomic features that the classifiers relied upon can direct subsequent experimental confirmation of virulence determinants. Examples where the classifiers incorrectly predict environmental isolates to be clinical are suggestive of enhanced zoonotic potential, and could prompt further investigation.
Public health and clinical laboratories may not have ready access to environmental samples if they are processed by independent pathology providers. Similar studies have trained ML models to distinguish isolates collected from sterile and non-sterile sites for Streptococcus pneumoniae (Obolski et al., 2019) and Campylobacter jejuni (Wheeler et al., 2019). Collection from sterile sites was used as a proxy for invasive disease. Examination of the models revealed genes associated with invasive disease that could then be compared to experimentally verified virulence determinants. More generally, genome-wide association studies (GWAS) using ML for identifying virulence markers may incorporate in vivo or in vitro empirical measures of virulence, clinical data such as disease severity and therapeutic responses, and statistical correction for population structure confounding associations (Allen et al., 2021). By combining genomic data with detailed phenotypic and clinical information, ML can discover nuanced relationships between microbial virulence factors and host risk factors to inform personalised treatment (Recker et al., 2017).
SARS-CoV-2 lineage designations have been a crucial tool in the COVID-19 pandemic response, including for disease surveillance. Being a novel virus, lineage definitions were developed de novo based on complete viral genomes: the challenge was in updating the scheme to keep pace with viral evolution and the deluge of reported sequences. The dynamic Pangolin scheme emerged as the de facto international standard, accompanied by the pangoLEARN tool to predict lineages using an ML classifier (O’Toole et al., 2021). Over time the classifier was changed from multinomial logistic regression to decision trees, and then to a random forest with version 4 of Pangolin (O’Toole et al). The classifier operated on a representation of the complete aligned genome with uninformative sites removed. In principle, lineage designation could have used a manually curated set of rules based on the presence of expected mutations, but in practice this was not feasible given the weekly update cadence adopted to maintain relevancy. A revised ML model could be trained in 30 minutes and could call lineages for 1000 genomes in 25 seconds, meaning that it offered a practical solution. Subsequent analysis reported that pangoLEARN was less accurate and less stable across versions compared to an alternative approach using phylogenetic placement (Schneider A de et al., 2023).
4 Pathogen discovery from metagenomic data
Metagenomic next generation sequencing (mNGS)—the minimally biased recovery and analysis of all nucleic acid from a clinical specimen—has emerging applications in clinical (Chiu and Miller, 2019) and public health (Ko et al., 2022) microbiology. Their inclusive approach to sequencing allows metagenomic methods to be highly sensitive and to avoid specifying details of the target organism a priori as would be required in amplicon sequencing. As a consequence, analyses must contend with a large number of mNGS reads from the host organism, laboratory environment, and other non-target sources (Salter et al., 2014). Reads that cannot be assigned to known reference sequences constitute the “microbial dark matter”: fastidious organisms that are not readily cultured in standard media, or organisms that are otherwise absent from or underrepresented in genomic databases.
One option for progress is to painstakingly shed light on the dark matter as is done by a number of cataloguing projects, novel and more inclusive culturing approaches, and culture-independent sequencing experiments (Lok, 2015; Jiao et al., 2021). In parallel, AI-based approaches have sought to transfer hard-earned experimental data about known microorganisms to under-characterised or novel organisms by identifying structural similarities in genomes (Hoarfrost et al., 2022). Deep learning models are well-suited to this problem of learning subtle patterns from vast accumulations of mNGS reads. They have been deployed to identify novel pathogens (Bartoszewicz et al., 2021; Naor-Hoffmann et al., 2022) as well as genes associated with AMR (Arango-Argoty et al., 2018) or virulence (de Nies et al., 2021; Gupta et al., 2022) in bacteria. Li et al. (2021) experimentally confirmed in vitro resistance for some examples of Pseudomonas aeruginosa AMR genes predicted by their model in cases where direct sequence similarity methods would have failed due to low nucleotide identity with existing databases.
A class of ML-based methods is devoted to identifying viruses or viral fragments from metagenomic data, including methods with a specific focus on bacteriophages (Ho et al., 2023; Amgarten et al., 2018; Kieft et al., 2020; Auslander et al., 2020; Guo et al., 2021; Bai et al., 2022). The principle underpinning these methods is that fragments of a viral genome share more features with other viruses than they do with non-viral organisms present in the library. Subtle differences in nucleotide or k-mer frequency, or particular motifs might provide enough clues for a model to identify a viral read even when it is quite dissimilar to its nearest relative in reference databases. Other tools sub-categorise viral reads including to distinguish temperate phages from virulent phages (Wu et al., 2021).
AI techniques have also been applied to biome source tracking: the task of identifying source microbial communities that contributed to a specimen (Zha et al., 2022). Rather than searching for fragments of genomes to explain a pathology, this task is concerned with grouping all of the data into likely communities of organisms. Shenhav et al. (2019) developed an unsupervised learning approach based on expectation maximisation, which they used to predict the contribution of maternal microflora to infant microbiome, to identify evidence of food and soil contaminants in longitudinal samples from a household, and to distinguish gut microbiota of critically ill patients from those of healthy adults. Deep learning approaches have been successfully applied to classify human microbiomes by the associated disease group with high accuracy and reduced computational requirements for prediction compared to existing approaches (Chong et al., 2022).
5 The genome as biological language
One approach to genome modelling derives from early in the history of genomic sequencing and uses the framework of computational linguistics to make rigorous the metaphor of genomes as a biological language (Brendel and Busse, 1984; Searls, 2002; Searls, 2013). This tradition views RNA secondary structure, transcription, translation, regulation, expression, protein folding, and various other biological processes as adhering to particular formal grammars and therefore amenable to analysis with tools and insights developed in the context of natural and computer languages. Protein language models such as ESMFold (Lin et al., 2023) are an alternative to protein structure prediction methods that use multiple sequence alignments, such as the successful AlphaFold2 model (Bertoline et al., 2023).
DNABERT builds on the BERT language representation model by treating DNA sequences as sentences composed of individual k-mer “words” (Ji et al., 2021). DNABERT first learns the basic syntax of this language through exposure to DNA sequences with sections masked, attempting to predict the missing sections. The training is then transferred and specialised for specific applications. This approach was used in the INHERIT model to identify bacteriophage sequences: Bai et al. (2022) selected 6-mers (i.e. k-mers 6 nucleotides in length) as their unit of analysis and pre-trained their model separately on examples of bacterial genomes and phages. The pre-trained models were then fine-tuned to perform the phage classification task. The final model out-performed several other ML tools across multiple measures of accuracy when compared by the authors.
Protein language models have been applied to predict the effect of novel mutations on viral epitopes. Hie et al. (2021) developed one such method to predict the immune escape potential of mutations in several viral antigens. Models for specific viral proteins were trained using corpora of amino acid sequences and experimental data quantifying the replication fitness of different mutations. Using a framework developed for NLP, amino acids were treated as words and replication fitness was modelled as “grammaticality”. Although the models were provided with no specific data about immune escape—their training was unsupervised in this respect—they successfully predicted amino acid residues with elevated potential for immune escape including the receptor binding domain of the SARS-CoV-2 spike protein. This was done by enumerating possible mutations and searching for examples that were grammatical (i.e. without a substantial fitness cost) but conferred a “semantic” (i.e. functional) change. Clustering based on the embedding learned by the models showed high correlation with host species.
6 Phylogenetic and phylodynamic inference
In public health settings, pathogen genomics plays in increasingly pivotal role alongside traditional epidemiology in detecting and investigating infectious disease outbreaks (Sintchenko and Holmes, 2015). The prevailing paradigm applies statistical models to infer likely phylogenies from sequence data. Phylodynamic approaches use molecular clock and nucleotide substitution models to produce time-scaled phylogenies, and to these fit epidemiological models (e.g. compartmental or birth-death models). This yields estimates of population dynamics parameters such as basic reproduction numbers and generation times, or inferred transmission trees (Grenfell et al., 2004; Attwood et al., 2022; Stockdale et al., 2022). In addition to genome sequences and sample collection dates, phylodynamic approaches can incorporate epidemiological data to infer host-related events such as disease introductions into discrete geographic regions, or to constrain transmission hypotheses with contact data (Volz et al., 2013; Ingle et al., 2021). Phylodynamic inference is computationally expensive and requires the explicit formulation of a likelihood appropriate for the available data and model assumptions (Voznica et al., 2022).
Recent work has shown that likelihood-free deep learning models can converge on similar epidemiological parameter estimates to equivalent phylodynamic models in a fraction of the computational time (Voznica et al., 2022; Kupperman et al., 2022; Thompson et al., 2023). The incentive is that AI can overcome fundamental scalability issues in likelihood-based models both in terms of computational tractability with large datasets, and the requirement for increasingly complex likelihoods as models are made more biologically realistic. The incentive is that AI can overcome fundamental scalability issues in likelihood-based models both in terms of computational tractability with large datasets, and the requirement for increasingly complex likelihoods as models are made more biologically realistic. On the other hand, phylodynamic models are attractive because they are inferential frameworks where such assumptions are explicit and inspectable, and the uncertainty of parameter estimates can be rigorously computed. As an emerging area of research, much remains to be understood about the feasibility and limitations of AI approaches for phylodynamic estimation.
7 Automating genomic analysis
Recent advancements in LLMs have led to an explosion of interest in their potential to perform cognitively demanding tasks that have traditionally been out of reach for AI. The technology powering the models is undergoing rapid development to the extent that it is challenging for researchers outside the field to assess its genuine capabilities. Many models are part of tools published by for-profit entities such as ChatGPT (OpenAI) (OpenAI, 2023), Bard (Google), and LLaMa (Meta), which have an interest in developing markets for their technology.
In microbial genomics research, a natural question to consider is whether LLMs are capable of directly performing bioinformatic analysis of sequencing data. General-purpose language models have been shown to be weak at certain tasks that are simple for humans, including counting the occurrence of a letter or word in an example text (Wang H. et al., 2023), and models are limited in the amount of input they can process. This suggests that current models are poorly suited to sequence analysis tasks such as computing nucleotide frequencies. AI platforms are evolving rapidly and addressing some practical limitations, however significant obstacles remain (Wang L. et al., 2023). There will often be no means to verify quantitative results proposed by LLMs without independently conducting the computation.
While LLMs might not be suitable for computation, many are designed to be able to produce computer source code. Preliminary studies have explored the limitations and possibilities of using LLMs for bioinformatic coding tasks (Piccolo et al., 2023; Shue et al., 2023; Hou and Ji, 2023). Current generation models perform well at generating functional code, but often contain subtle errors due to a lack of understanding about the context in which the code will be used. Without specific prompting, they will often propose code that imperfectly implements routines such as parsing the FASTA file format, overlooking the existence of well-tested implementations in specialised software libraries such as Biopython (Cock et al., 2009). Despite the need for caution and verification, LLMs can be useful for increasing productivity and expanding the accessibility of routine bioinformatic tasks to students or non-experts.
LLMs excel at responding to queries in natural language and extracting information from text. This capacity has been used to provide an experimental query interface to the CovSpectrum database, allowing users to ask for up-to-date details of circulating SARS-CoV-2 variants in natural language (Chen and Stadler, 2023). A pedagogical exercise has illustrated that general-purpose LLMs can be coerced into providing relevant information to non-experts requesting instructions for the synthesis of biothreat agents despite safeguards intended to prevent this (Soice et al., 2023). LLMs have also been used to automatically extract information about characterised SARS-CoV-2 mutations from published scientific literature (Serna García et al., 2023). Their ability to generate plausible text has implications for academic publication and translational research, with associated bioethical considerations that remain to be explored in detail (Page et al., 2023; Coiera et al., 2023).
8 Discussion
AI offers exciting possibilities for diverse practices within the field of microbial genomics. There are plentiful demonstrations in research settings of its capabilities for analysis of genomic and associated data that exceed what can be achieved within a realistic amount of time and effort by human experts. Despite this, AI for pathogen genomics is not yet ubiquitous in public health and diagnostic laboratory operations.
As a prerequisite for adoption of AI, a case must be developed in economic terms for its impact on day-to-day operations in settings where pathogen WGS or mNGS is established practice. Indeed in clinical microbiology laboratories more broadly, AI is already gaining traction as a consequence of increasing laboratory workflow automation and robotics (Naugler and Church, 2019; Bailey et al., 2019; Lakbar et al., 2023). Such a case would address expected utility: the perceived benefits of AI must justify the burden of implementation and validation before introducing a meaningful dependency on the technology. Training in clinical and laboratory science does not typically emphasise the skills required to robustly use even simpler classes of AI, whereas AI experts may face difficulty accessing the data and contexts relevant to health practitioners in specific settings. Workforce capacity is an important barrier to uptake.
In common with other applications of AI in the medical domain, high-stake clinical decisions have ethical and medico-legal consequences that demand defensibility. Research done to develop explainable AI for transparency of decision-making (Wadden, 2022; Durán and Jongsma, 2021; Minh et al., 2022) and to understand algorithmic discrimination (Obermeyer et al., 2019; Heinrichs, 2022) is underdeveloped in many relevant applications. It remains important to be cognisant of how AI tools are designed so that their outputs are interpreted in the context of their limitations and strengths (Scott et al., 2021; Couckuyt et al., 2022; Sokhansanj and Rosen, 2022). Regulatory frameworks will need to contend with these issues before widespread adoption of AI in clinical diagnostics is appropriate.
Even where stakes are potentially lower, the reliability of AI must be demonstrated. ML models are developed using specific datasets for training and validation and it is challenging to evaluate the extent to which they will reliably generalise. Cross-validation using available data has little bearing on performance with qualitatively different data including in different health systems, cohorts, and populations (Futoma et al., 2020). Tools may also perform poorly with different sequencing platforms, as has been noted with viral detection models in metagenomic experiments aimed at taxonomic characterisation of microbiomes when comparing short- and long-read data (Zaragoza-Solas et al., 2022). In general, local validation of AI tools is necessary. As microbial populations, public datasets, and disease epidemiology evolve, models require regular maintenance to ensure their performance.
Current genomic data are heavily biased towards geographic regions with sequencing capacity (Brito et al., 2022). The development of AI tools requires large datasets; equitable access to health technologies including AI requires that low- and middle-income countries be in a position to directly benefit from sequence data that they contribute to global efforts. This constitutes a key principle identified by the World Health Organization in its guidance for sharing pathogen genomic data (World Health Organization, 2022).
In this mini-review, we have presented examples of emerging applications of AI technology to tasks concerning pathogen genomics in clinical and public health settings. Approaches along these lines will play an increasing role in diagnostic and public health laboratories as well as in microbial genomics research, with expanding access to rich genomic, epidemiological, and laboratory data. Some of the obstacles that curbed early enthusiasm for the potential of artificial intelligence are being overcome by maturing technology, however we now ask that AI is not merely technically capable, but that it operates in accordance with our core principles of ethics, equity, and reliability.
Author contributions
CS: Investigation, Methodology, Visualization, Writing – original draft, Writing – review & editing. DP: Writing – review & editing. JK: Writing – review & editing. VS: Conceptualization, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the Prevention Research Support Program funded by the New South Wales Ministry of Health.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ahmed A., Boopathy P., S S. R. (2022). Artificial intelligence for the novel corona virus (COVID-19) pandemic: opportunities, challenges, and future directions. Int. J. E-Health Med. Commun. IJEHMC 13, 1–21. doi: 10.4018/IJEHMC.20220701.oa5
Alam M., Chowdhury U. F. (2020). Short k-mer abundance profiles yield robust machine learning features and accurate classifiers for RNA viruses. PloS One 15, e0239381. doi: 10.1371/journal.pone.0239381
Allen J. P., Snitkin E., Pincus N. B., Hauser A. R. (2021). Forest and trees: exploring bacterial virulence with genome-wide association studies and machine learning. Trends Microbiol. 29, 621–633. doi: 10.1016/j.tim.2020.12.002
Amgarten D., Braga L. P. P., da Silva A. M., Setubal J. C. (2018). MARVEL, a tool for prediction of bacteriophage sequences in metagenomic bins. Front. Genet. 9. doi: 10.3389/fgene.2018.00304
Anahtar M. N., Yang J. H., Kanjilal S. (2021). Applications of machine learning to the problem of antimicrobial resistance: an emerging model for translational research. J. Clin. Microbiol. 59, e01260-20. doi: 10.1128/JCM.01260-20
Arango-Argoty G., Garner E., Pruden A., Heath L. S., Vikesland P., Zhang L. (2018). DeepARG: a deep learning approach for predicting antibiotic resistance genes from metagenomic data. Microbiome 6, 23. doi: 10.1186/s40168-018-0401-z
Arora G., Joshi J., Mandal R. S., Shrivastava N., Virmani R., Sethi T. (2021). Artificial intelligence in surveillance, diagnosis, drug discovery and vaccine development against COVID-19. Pathogens 10, 1048. doi: 10.3390/pathogens10081048
Attwood S. W., Hill S. C., Aanensen D. M., Connor T. R., Pybus O. G. (2022). Phylogenetic and phylodynamic approaches to understanding and combating the early SARS-CoV-2 pandemic. Nat. Rev. Genet. 23, 547–562 doi: 10.1038/s41576-022-00483-8
Auslander N., Gussow A. B., Benler S., Wolf Y. I., Koonin E. V. (2020). Seeker: alignment-free identification of bacteriophage genomes by deep learning. Nucleic Acids Res. 48, e121–e121. doi: 10.1093/nar/gkaa856
Bai Z., Zhang Y., Miyano S., Yamaguchi R., Fujimoto K., Uematsu S., et al. (2022). Identification of bacteriophage genome sequences with representation learning. Bioinformatics 38, 4264–4270. doi: 10.1093/bioinformatics/btac509
Bailey A. L., Ledeboer N., Burnham C.-A. D. (2019). Clinical microbiology is growing up: the total laboratory automation revolution. Clin. Chem. 65, 634–643. doi: 10.1373/clinchem.2017.274522
Bartoszewicz J. M., Genske U., Renard B. Y. (2021). Deep learning-based real-time detection of novel pathogens during sequencing. Brief Bioinform. 22, bbab269. doi: 10.1093/bib/bbab269
Beguir K., Skwark M. J., Fu Y., Pierrot T., Carranza N. L., Laterre A., et al. (2023). Early computational detection of potential high-risk SARS-CoV-2 variants. Comput. Biol. Med. 155, 106618. doi: 10.1016/j.compbiomed.2023.106618
Bengio Y., Courville A., Vincent P. (2013). Representation learning: A review and new perspectives. IEEE Trans. Pattern Anal. Mach. Intell. 35, 1798–1828. doi: 10.1109/TPAMI.2013.50
Bertoline L. M. F., Lima A. N., Krieger J. E., Teixeira S. K. (2023). Before and after AlphaFold2: An overview of protein structure prediction. Front. Bioinforma 3. doi: 10.3389/fbinf.2023.1120370
Bonaccorso G. (2018). Machine Learning Algorithms: Popular algorithms for data science and machine learning. 2nd Edition (Birmingham, UK: Packt Publishing Ltd), 514.
Brendel V., Busse H. G. (1984). Genome structure described by formal languages. Nucleic Acids Res. 12, 2561–2568. doi: 10.1093/nar/12.5.2561
Brito A. F., Semenova E., Dudas G., Hassler G. W., Kalinich C. C., Kraemer M. U. G., et al. (2022). Global disparities in SARS-CoV-2 genomic surveillance. Nat. Commun. 13, 7003. doi: 10.1038/s41467-022-33713-y
Brownstein J. S., Rader B., Astley C. M., Tian H. (2023). Advances in artificial intelligence for infectious-disease surveillance. N Engl. J. Med. 388, 1597–1607. doi: 10.1056/NEJMra2119215
Bubeck S., Chandrasekaran V., Eldan R., Gehrke J., Horvitz E., Kamar E., et al. (2023). Sparks of artificial general intelligence: early experiments with GPT-4. arXiv [Preprint]. doi: 10.48550/ARXIV.2303.12712
Chen C., Stadler T. (2023). GenSpectrum chat: data exploration in public health using large language models. arXiv [Preprint]. doi: 10.48550/arXiv.2305.13821
Chen J., Li K., Zhang Z., Li K., Yu P. S. (2022). A survey on applications of artificial intelligence in fighting against COVID-19. ACM Comput. Surv 54, 1–32. doi: 10.1145/3465398
Chiu C. Y., Miller S. A. (2019). Clinical metagenomics. Nat. Rev. Genet. 20, 341–355. doi: 10.1038/s41576-019-0113-7
Choi S. R., Lee M. (2023). Transformer architecture and attention mechanisms in genome data analysis: A comprehensive review. Biology 12, 1033. doi: 10.3390/biology12071033
Chong H., Zha Y., Yu Q., Cheng M., Xiong G., Wang N., et al. (2022). EXPERT: transfer learning-enabled context-aware microbial community classification. Brief Bioinform. 23, bbac396. doi: 10.1093/bib/bbac396
Cock P. J. A., Antao T., Chang J. T., Chapman B. A., Cox C. J., Dalke A., et al. (2009). Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25, 1422–1423. doi: 10.1093/bioinformatics/btp163
Coiera E. W., Verspoor K., Hansen D. P. (2023). We need to chat about artificial intelligence. Med. J. Aust. 219, 98–100. doi: 10.5694/mja2.51992
Couckuyt A., Seurinck R., Emmaneel A., Quintelier K., Novak D., Van Gassen S., et al. (2022). Challenges in translational machine learning. Hum. Genet. 141, 1451–1466. doi: 10.1007/s00439-022-02439-8
de Bernardi Schneider A., Su M., Hinrichs A. S., Wang J., Amin H., Bell J., et al. (2023). SARS-CoV-2 lineage assignments using phylogenetic placement/UShER are superior to pangoLEARN machine learning method. Virus Evolution 10, 1. doi: 10.1093/ve/vead085
de Nies L., Lopes S., Busi S. B., Galata V., Heintz-Buschart A., Laczny C. C., et al. (2021). PathoFact: a pipeline for the prediction of virulence factors and antimicrobial resistance genes in metagenomic data. Microbiome 9, 49. doi: 10.1186/s40168-020-00993-9
De Santana Correia A., Colombini E. L. (2022). Attention, please! A survey of neural attention models in deep learning. Artif. Intell. Rev. 55, 6037–6124. doi: 10.1007/s10462-022-10148-x
Durán J. M., Jongsma K. R. (2021). Who is afraid of black box algorithms? On the epistemological and ethical basis of trust in medical AI. J. Med. Ethics 47, 329–335. doi: 10.1136/medethics-2020-106820
Eddy S. R. (2004). What is a hidden Markov model? Nat. Biotechnol. 22, 1315–1316. doi: 10.1038/nbt1004-1315
Esteva A., Robicquet A., Ramsundar B., Kuleshov V., DePristo M., Chou K., et al. (2019). A guide to deep learning in healthcare. Nat. Med. 25, 24–29. doi: 10.1038/s41591-018-0316-z
Friedland P., Kedes L., Brutlag D., Iwasaki Y., Bach R. (1982). GENESIS, a knowledge-based genetic engineering simulation system for represntation of genetic data and experiment planning. Nucleic Acids Res. 10, 323–340. doi: 10.1093/nar/10.1.323
Futoma J., Simons M., Panch T., Doshi-Velez F., Celi L. A. (2020). The myth of generalisability in clinical research and machine learning in health care. Lancet Digit Health 2, e489–e492. doi: 10.1016/S2589-7500(20)30186-2
Gilbert G. L. (2002). Molecular diagnostics in infectious diseases and public health microbiology: cottage industry to postgenomics. Trends Mol. Med. 8, 280–287. doi: 10.1016/S1471-4914(02)02349-3
Gomes B., Ashley E. A. (2023). Artificial intelligence in molecular medicine. N Engl. J. Med. 388, 2456–2465. doi: 10.1056/NEJMra2204787
Green R. (2023). Applying Deep Learning Techniques to Assist Bioinformatics Researchers in Analysis Pipeline Composition (Cincinnati (OH): University of Cincinnati). Available at: http://rave.ohiolink.edu/etdc/view?acc_num=ucin1684776249065885.
Grenfell B. T., Pybus O. G., Gog J. R., Wood J. L. N., Daly J. M., Mumford J. A., et al. (2004). Unifying the epidemiological and evolutionary dynamics of pathogens. Science 303, 327–332. doi: 10.1126/science.1090727
Gu J., Wang Z., Kuen J., Ma L., Shahroudy A., Shuai B., et al. (2018). Recent advances in convolutional neural networks. Pattern Recognit 77, 354–377. doi: 10.1016/j.patcog.2017.10.013
Guo J., Bolduc B., Zayed A. A., Varsani A., Dominguez-Huerta G., Delmont T. O., et al. (2021). VirSorter2: a multi-classifier, expert-guided approach to detect diverse DNA and RNA viruses. Microbiome 9, 37. doi: 10.1186/s40168-020-00990-y
Gupta A., Malwe A. S., Srivastava G. N., Thoudam P., Hibare K., Sharma V. K. (2022). MP4: a machine learning based classification tool for prediction and functional annotation of pathogenic proteins from metagenomic and genomic datasets. BMC Bioinf. 23, 507. doi: 10.1186/s12859-022-05061-7
Haug C. J., Drazen J. M. (2023). Artificial intelligence and machine learning in clinical medicine, 2023. N Engl. J. Med. 388, 1201–1208. doi: 10.1056/NEJMra2302038
Heinrichs B. (2022). Discrimination in the age of artificial intelligence. AI Soc. 37, 143–154. doi: 10.1007/s00146-021-01192-2
Hie B., Zhong E. D., Berger B., Bryson B. (2021). Learning the language of viral evolution and escape. Science 371, 284–288. doi: 10.1126/science.abd7331
Ho S. F. S., Wheeler N. E., Millard A. D., Van Schaik W. (2023). Gauge your phage: benchmarking of bacteriophage identification tools in metagenomic sequencing data. Microbiome 11, 84. doi: 10.1186/s40168-023-01533-x
Hoarfrost A., Aptekmann A., Farfañuk G., Bromberg Y. (2022). Deep learning of a bacterial and archaeal universal language of life enables transfer learning and illuminates microbial dark matter. Nat. Commun. 13, 1–12. doi: 10.1038/s41467-022-30070-8
Hochreiter S., Schmidhuber J. (1997). Long short-term memory. Neural Comput. 9, 1735–1780. doi: 10.1162/neco.1997.9.8.1735
Hou W., Ji Z. (2023). GeneTuring tests GPT models in genomics. bioRxiv [Preprint]. doi: 10.1101/2023.03.11.532238
Hunter L. (1992). “Artificial intelligence and molecular biology,” in Proceedings of the AAAI Conference on Artificial Intelligence. 866–868 (AAAI Press).
Hunter L. (Ed.) (1993). Artificial intelligence and molecular biology (Cambridge, Massachusetts, USA: AAAI Press/The MIT Press).
Ingle D. J., Howden B. P., Duchene S. (2021). Development of phylodynamic methods for bacterial pathogens. Trends Microbiol. 29, 788–797. doi: 10.1016/j.tim.2021.02.008
Ji Y., Zhou Z., Liu H., Davuluri R. V. (2021). DNABERT: pre-trained Bidirectional Encoder Representations from Transformers model for DNA-language in genome. Bioinformatics 37, 2112–2120. doi: 10.1093/bioinformatics/btab083
Jiang Y., Li X., Luo H., Yin S., Kaynak O. (2022). Quo vadis artificial intelligence? Discovery Artif. Intell. 2, 4. doi: 10.1007/s44163-022-00022-8
Jiao J.-Y., Liu L., Hua Z.-S., Fang B.-Z., Zhou E.-M., Salam N., et al. (2021). Microbial dark matter coming to light: challenges and opportunities. Natl. Sci. Rev. 8, nwaa280. doi: 10.1093/nsr/nwaa280
Kieft K., Zhou Z., Anantharaman K. (2020). VIBRANT: automated recovery, annotation and curation of microbial viruses, and evaluation of viral community function from genomic sequences. Microbiome 8, 90. doi: 10.1186/s40168-020-00867-0
Ko K. K. K., Chng K. R., Nagarajan N. (2022). Metagenomics-enabled microbial surveillance. Nat. Microbiol. 7, 486–496. doi: 10.1038/s41564-022-01089-w
Kupperman M. D., Leitner T., Ke R. (2022). A deep learning approach to real-time HIV outbreak detection using genetic data. PloS Comput. Biol. 18, e1010598. doi: 10.1371/journal.pcbi.1010598
Lakbar I., Singer M., Leone M. (2023). 2030: will we still need our microbiologist? Intensive Care Med. 49, 1232–1234 doi: 10.1007/s00134-023-07186-6
Li Y., Xu Z., Han W., Cao H., Umarov R., Yan A., et al. (2021). HMD-ARG: hierarchical multi-task deep learning for annotating antibiotic resistance genes. Microbiome 9, 1–12. doi: 10.1186/s40168-021-01002-3
Lin Z., Akin H., Rao R., Hie B., Zhu Z., Lu W., et al. (2023). Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123–1130. doi: 10.1126/science.ade2574
Lupolova N., Dallman T. J., Matthews L., Bono J. L., Gally D. L. (2016). Support vector machine applied to predict the zoonotic potential of E. coli O157 cattle isolates. Proc. Natl. Acad. Sci. 113, 11312–11317. doi: 10.1073/pnas.1606567113
Lupolova N., Lycett S. J., Gally D. L. (2019). A guide to machine learning for bacterial host attribution using genome sequence data. Microb. Genomics 5. doi: 10.1099/mgen.0.000317
Malhotra D., Sodhi G. K. (2023). “A Survey on the role of ML and AI in fighting Covid-19,” in 2023 International Conference on Advancement in Computation & Computer Technologies (InCACCT). (New York City: Institute of Electrical and Electronics Engineers (IEEE)) 27–32. doi: 10.1109/InCACCT57535.2023.10141732
Minh D., Wang H. X., Li Y. F., Nguyen T. N. (2022). Explainable artificial intelligence: a comprehensive review. Artif. Intell. Rev. 55, 3503–3568. doi: 10.1007/s10462-021-10088-y
Naor-Hoffmann S., Svetlitsky D., Sal-Man N., Orenstein Y., Ziv-Ukelson M. (2022). Predicting the pathogenicity of bacterial genomes using widely spread protein families. BMC Bioinf. 23, 253. doi: 10.1186/s12859-022-04777-w
Naugler C., Church D. L. (2019). Automation and artificial intelligence in the clinical laboratory. Crit. Rev. Clin. Lab. Sci. 56, 98–110. doi: 10.1080/10408363.2018.1561640
Newton R., Wernisch L. (2017). A comparison of machine learning and Bayesian modelling for molecular serotyping. BMC Genomics 18, 606. doi: 10.1186/s12864-017-3998-6
Obermeyer Z., Powers B., Vogeli C., Mullainathan S. (2019). Dissecting racial bias in an algorithm used to manage the health of populations. Science 366, 447–453. doi: 10.1126/science.aax2342
Obolski U., Gori A., Lourenço J., Thompson C., Thompson R., French N., et al. (2019). Identifying genes associated with invasive disease in S. pneumoniae by applying a machine learning approach to whole genome sequence typing data. Sci. Rep. 9, 1–9. doi: 10.1038/s41598-019-40346-7
O’Toole Á, Scher E., Rambaut A. pangoLEARN description. Available online at: https://cov-lineages.org/resources/pangolin/pangolearn.html (Accessed October 4, 2023).
O’Toole Á, Scher E., Underwood A., Jackson B., Hill V., McCrone J. T., et al. (2021). Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. Virus Evol. 7, veab064. doi: 10.1093/ve/veab064
Page A. J., Tumelty N. M., Sheppard S. K. (2023). Navigating the AI frontier: ethical considerations and best practices in microbial genomics research. Microb. Genomics 9. doi: 10.1099/mgen.0.001049
Piccolo S. R., Denny P., Luxton-Reilly A., Payne S., Ridge P. G. (2023). Evaluating a large language model’s ability to solve programming exercises from an introductory bioinformatics course. PLoS Comput Biol 19 (9), e1011511. doi: 10.1371/journal.pcbi.1011511
Ramadan A. A. (2022). Bacterial typing methods from past to present: A comprehensive overview. Gene Rep. 29, 101675. doi: 10.1016/j.genrep.2022.101675
Rawlings C. J., Fox J. P., Thompson E. A., Robson B. (1994). Artificial intelligence in molecular biology: A review and assessment. Philos. Trans. Biol. Sci. 344, 353–363. doi: 10.1098/rstb.1994.0074
Recker M., Laabei M., Toleman M. S., Reuter S., Saunderson R. B., Blane B., et al. (2017). Clonal differences in Staphylococcus aureus bacteraemia-associated mortality. Nat. Microbiol. 2, 1381–1388. doi: 10.1038/s41564-017-0001-x
Ren J., Song K., Deng C., Ahlgren N. A., Fuhrman J. A., Li Y., et al. (2020). Identifying viruses from metagenomic data using deep learning. Quant Biol. 8, 64–77. doi: 10.1007/s40484-019-0187-4
Salter S. J., Cox M. J., Turek E. M., Calus S. T., Cookson W. O., Moffatt M. F., et al. (2014). Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 12, 87. doi: 10.1186/s12915-014-0087-z
Sarker I. H. (2021). Deep learning: A comprehensive overview on techniques, taxonomy, applications and research directions. SN Comput. Sci. 2, 420. doi: 10.1007/s42979-021-00815-1
Sarmiento Varón L., González-Puelma J., Medina-Ortiz D., Aldridge J., Alvarez-Saravia D., Uribe-Paredes R., et al. (2023). The role of machine learning in health policies during the COVID-19 pandemic and in long COVID management. Front. Public Health 11. doi: 10.3389/fpubh.2023.1140353
Scott I., Carter S., Coiera E. (2021). Clinician checklist for assessing suitability of machine learning applications in healthcare. BMJ Health Care Inform 28, e100251. doi: 10.1136/bmjhci-2020-100251
Searls D. B. (2013). A primer in macromolecular linguistics. Biopolymers 99, 203–217. doi: 10.1002/bip.22101
Serna García G., Al Khalaf R., Invernici F., Ceri S., Bernasconi A. (2023). CoVEffect: interactive system for mining the effects of SARS-CoV-2 mutations and variants based on deep learning. GigaScience 12, giad036. doi: 10.1093/gigascience/giad036
Shenhav L., Thompson M., Joseph T. A., Briscoe L., Furman O., Bogumil D., et al. (2019). FEAST: fast expectation-maximization for microbial source tracking. Nat. Methods 16, 627–632. doi: 10.1038/s41592-019-0431-x
Shepard S. S., Davis C. T., Bahl J., Rivailler P., York I. A., Donis R. O. (2014). LABEL: fast and accurate lineage assignment with assessment of H5N1 and H9N2 influenza A hemagglutinins. PloS One 9, e86921. doi: 10.1371/journal.pone.0086921
Shue E., Liu L., Li B., Feng Z., Li X., Hu G. (2023). Empowering beginners in bioinformatics with ChatGPT. Quant Biol. 11, 105–108. doi: 10.15302/J-QB-023-0327
Sintchenko V., Holmes E. C. (2015). The role of pathogen genomics in assessing disease transmission. BMJ 350, h1314–h1314. doi: 10.1136/bmj.h1314
Soice E. H., Rocha R., Cordova K., Specter M., Esvelt K. M. (2023). Can large language models democratize access to dual-use biotechnology? arXiv [Preprint]. doi: 10.48550/arXiv.2306.03809
Sokhansanj B. A., Rosen G. L. (2022). Mapping data to deep understanding: making the most of the deluge of SARS-coV-2 genome sequences. mSystems 7, e00035–e00022. doi: 10.1128/msystems.00035-22
Stefik M. (1981). Planning with constraints (MOLGEN: part 1). Artif. Intell. 16, 111–139. doi: 10.1016/0004-3702(81)90007-2
Stockdale J. E., Liu P., Colijn C. (2022). The potential of genomics for infectious disease forecasting. Nat. Microbiol. 7, 1736–1743. doi: 10.1038/s41564-022-01233-6
Stormo G. D., Schneider T. D., Gold L., Ehrenfeucht A. (1982). Use of the ‘Perceptron’ algorithm to distinguish translational initiation sites in E. coli. Nucleic Acids Res. 10, 2997–3011. doi: 10.1093/nar/10.9.2997
Syrowatka A., Kuznetsova M., Alsubai A., Beckman A. L., Bain P. A., Craig K. J. T., et al. (2021). Leveraging artificial intelligence for pandemic preparedness and response: a scoping review to identify key use cases. NPJ Digit Med. 4, 96. doi: 10.1038/s41746-021-00459-8
Thompson A., Liebeskind B., Scully E. J., Landis M. (2024). Deep learning and likelihood approaches for viral phylogeography converge on the same answers whether the inference model is right or wrong. Systematic Biology, syad074. doi: 10.1101/2023.02.08.527714. preprint.
Tonner P. D., Pressman A., Ross D. (2022). Interpretable modeling of genotype–phenotype landscapes with state-of-the-art predictive power. Proc. Natl. Acad. Sci. 119, e2114021119. doi: 10.1073/pnas.2114021119
van der Ploeg T., Steyerberg E. W. (2016). Feature selection and validated predictive performance in the domain of Legionella pneumophila: a comparative study. BMC Res. Notes 9, 147. doi: 10.1186/s13104-016-1945-2
Vaswani A., Shazeer N., Parmar N., Uszkoreit J., Jones L., Gomez A. N., et al. (2017)Attention is All you Need Advances in Neural Information Processing Systems (Curran Associates, Inc). Available online at: https://proceedings.neurips.cc/paper_files/paper/2017/hash/3f5ee243547dee91fbd053c1c4a845aa-Abstract.html (Accessed December 20, 2023).
Volz E. M., Koelle K., Bedford T. (2013). Viral phylodynamics. PloS Comput. Biol. 9, e1002947. doi: 10.1371/journal.pcbi.1002947
Vorimore F., Jaudou S., Tran M.-L., Richard H., Fach P., Delannoy S. (2023). Combination of whole genome sequencing and supervised machine learning provides unambiguous identification of eae-positive Shiga toxin-producing Escherichia coli. Front. Microbiol. 14. doi: 10.3389/fmicb.2023.1118158
Voznica J., Zhukova A., Boskova V., Saulnier E., Lemoine F., Moslonka-Lefebvre M., et al. (2022). Deep learning from phylogenies to uncover the epidemiological dynamics of outbreaks. Nat. Commun. 13, 3896. doi: 10.1038/s41467-022-31511-0
Wadden J. J. (2022). Defining the undefinable: the black box problem in healthcare artificial intelligence. J. Med. Ethics 48, 764–768. doi: 10.1136/medethics-2021-107529
Wang L., Ge X., Liu L., Hu G. (2023). Code interpreter for bioinformatics: are we there yet? Ann. BioMed. Eng. doi: 10.1007/s10439-023-03324-9
Wang H., Luo X., Wang W., Yan X. (2023). Bot or human? Detecting chatGPT imposters with A single question. arXiv [Preprint]. doi: 10.48550/arXiv.2305.06424
Wheeler N. E., Blackmore T., Reynolds A. D., Midwinter A. C., Marshall J., French N. P., et al. (2019). Genomic correlates of extraintestinal infection are linked with changes in cell morphology in Campylobacter jejuni. Microb. Genomics 5. doi: 10.1099/mgen.0.000251
Wolfram S. (2023). What Is ChatGPT Doing … and Why Does It Work? (Champaign, Illinois: Wolfram Media, Inc).
World Health Organization (2022). WHO guiding principles for pathogen genome data sharing (Geneva: World Health Organization). Available at: https://iris.who.int/handle/10665/364222.
Wu S., Fang Z., Tan J., Li M., Wang C., Guo Q., et al. (2021). DeePhage: distinguishing virulent and temperate phage-derived sequences in metavirome data with a deep learning approach. GigaScience 10, giab056. doi: 10.1093/gigascience/giab056
Zaragoza-Solas A., Haro-Moreno J. M., Rodriguez-Valera F., López-Pérez M. (2022). Long-read metagenomics improves the recovery of viral diversity from complex natural marine samples. mSystems 7, e00192–e00122. doi: 10.1128/msystems.00192-22
Keywords: pathogen genomics, laboratory diagnosis, public health, decision support, machine learning, artificial intelligence
Citation: Suster CJE, Pham D, Kok J and Sintchenko V (2024) Emerging applications of artificial intelligence in pathogen genomics. Front. Bacteriol. 3:1326958. doi: 10.3389/fbrio.2024.1326958
Received: 24 October 2023; Accepted: 23 February 2024;
Published: 06 March 2024.
Edited by:
Daniel Yero, Autonomous University of Barcelona, SpainReviewed by:
Ruian Ke, Los Alamos National Laboratory (DOE), United StatesSamantha J. Lycett, University of Edinburgh, United Kingdom
Copyright © 2024 Suster, Pham, Kok and Sintchenko. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carl J. E. Suster, Y2FybC5zdXN0ZXJAaGVhbHRoLm5zdy5nb3YuYXU=; Vitali Sintchenko, dml0YWxpLnNpbnRjaGVua29Ac3lkbmV5LmVkdS5hdQ==