 Mohamed Halawa
Mohamed Halawa Maxwell Akantibila
Maxwell Akantibila Briana E. Reid
Briana E. Reid Valerie J. Carabetta
Valerie J. Carabetta
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Bacteriol., 12 December 2023
Sec. Molecular Bacteriology and Microbiome
Volume 2 - 2023 | https://doi.org/10.3389/fbrio.2023.1304444
This article is part of the Research TopicEditors' Showcase: Molecular Bacteriology and MicrobiomeView all 7 articles
Humans are continually challenged to find new strategies to fight bacterial diseases due to the global health threat of antibiotic resistance. Conventional antibiotics, once hailed as miracle treatments, are progressively losing their effectiveness as drug-resistant bacteria continue to emerge, including multidrug-resistant (MDR) strains. An attractive approach to solving the antibiotic resistance problem is the investigation of other methods, such as therapeutic proteins. Here, we begin with a discussion of antimicrobial resistance and common strategies that bacteria employ to become resistant. Next, we discuss the potential use of antimicrobial peptides and monoclonal antibodies, two types of therapeutic proteins, as possible antimicrobial therapeutics. We discuss their structure, mechanism of action, and highlight examples of possible candidate molecules for clinical use. Finally, we discuss the remaining challenges and explore potential solutions for the implementation of therapeutic proteins into practical clinical use.
Before the turn of the 20th century, the human population was faced with the daunting challenge of dealing with dangerous infectious diseases that often escalated into widespread epidemics, resulting in the loss of countless lives. The efforts made to combat, treat, and control the transmission of these contagious diseases during this time were hindered by a lack of understanding and knowledge. Despite the significant progress made in the field of microscopic investigations in the 17th century, there was still a long way to go in terms of advancements in antimicrobial research. It was not until Alexander Fleming’s groundbreaking discovery in 1928 of the antibiotic properties present in molds that substantial strides were made in this area (Fleming, 1929; Tan and Tatsumura, 2015). The introduction of penicillin in the 1940s marked the beginning of a new era in medicine, often referred to as the golden age of antibiotics. However, with this progress came unforeseen challenges, including the emergence of antimicrobial resistance (AMR) and the development of allergies to specific antibiotics. By the start of the 21st century, the issue of AMR had reached its peak, rendering even the most advanced antibiotics ineffective in clinical settings, and causing an estimated 5 million deaths annually (Daulaire et al., 2015). A sobering analysis of the AMR crisis suggested that this number could potentially double to 10 million deaths per year by 2050 (O'Neill, 2016; Tang et al., 2023). Given the relentless evolution of antibiotic resistance by bacteria, there is a growing interest in exploring alternative treatment options, such as therapeutic proteins like human monoclonal antibodies (mAbs) and antimicrobial peptides (AMPs), which have the potential to revolutionize the field. These therapeutic proteins offer several advantages, including their ability to specifically target pathogenic bacteria, while preserving the normal microbiota. They also have the capacity to activate the immune system, allowing for a broader and more effective response, while also mitigating the toxic effects associated with antibiotic use (Mustafa et al., 2018). This comprehensive review aims to evaluate the potential of therapeutic proteins in the management of antibiotic-resistant bacterial infections, presenting them as a viable solution to this complex problem (Allen, 2017). The subsequent sections will delve into the major mechanisms underlying antibiotic resistance and explore the potential solutions that can be achieved through the use of AMPs and mAbs.
Antibiotic resistance is typically categorized into two main types: intrinsic and acquired. Intrinsic resistance pertains to bacteria that naturally exhibit insensitivity to specific antibiotics (Blair et al., 2015). This is commonly attributed to chromosomal resistance genes, which tend to remain unchanged (Brown-Jaque et al., 2015). For example, some bacterial strains express low affinity penicillin-binding proteins (PBPs) or have a reduced porin content in their outer membranes, such as Pseudomonas aeruginosa. The diminished affinity of PBPs reduces the efficacy of beta-lactam antibiotics, while decreased porin content limits drug access, rendering these strains naturally more resilient to conventional antibiotic treatments (Poole, 2002). On the other hand, acquired resistance results from genetic mutations or horizontal gene transfer, such as transformation, transduction, or conjugation (Abebe et al., 2016). Furthermore, these bacteria can develop resistance to additional drugs through horizontal gene transfer and genetic variation of drug-resistance genes (Xuan et al., 2023). Next, we will discuss the four major mechanisms of antibiotic resistance among bacteria.
The β-lactam family of antibiotics consists of the monobactam aztreonam, penicillins, cephalosporins, and carbapenems. All β-lactam antibiotics encompass a central β-lactam ring, which binds to penicillin-binding proteins (PBPs) and blocks cell wall synthesis, ultimately leading to cell lysis and death. A main mechanism to become resistant to β-lactams is the production of β-lactamases, which are enzymes that cleave and open the lactam ring, rendering the drug ineffective. These enzymes are frequently found on plasmids or transposable elements, so they are easily transferred among bacterial strains and species (Mora-Ochomogo and Lohans, 2021). β-lactamases are divided into four different classes, called Ambler classes, based on their molecular structure and catalytic mechanism (Ali et al., 2018). Ambler classes A, C, and D β-lactamases are serine hydrolases (Wu et al., 2016), while Class B are metallo-enzymes, which need zinc ions for their activity (Sacha et al., 2008). Classes A and D are inhibited by clavulanic acid, while class C enzymes are not (Munita and Arias, 2016). A concerning class of β-lactamases that can hydrolyze cephalosporins, all penicillins, and aztreonam are the extended-spectrum β-lactamases (ESBLs), which can belong to class A, C, or D. Bacteria that harbor one of these genes become resistant to most β-lactam drugs, except for the carbapenems. However, carbapenemases have been identified, such as the Klebsiella pneumoniae carbapenemase (KPC), that render bacteria resistant to all β-lactam drugs (Figure 1).
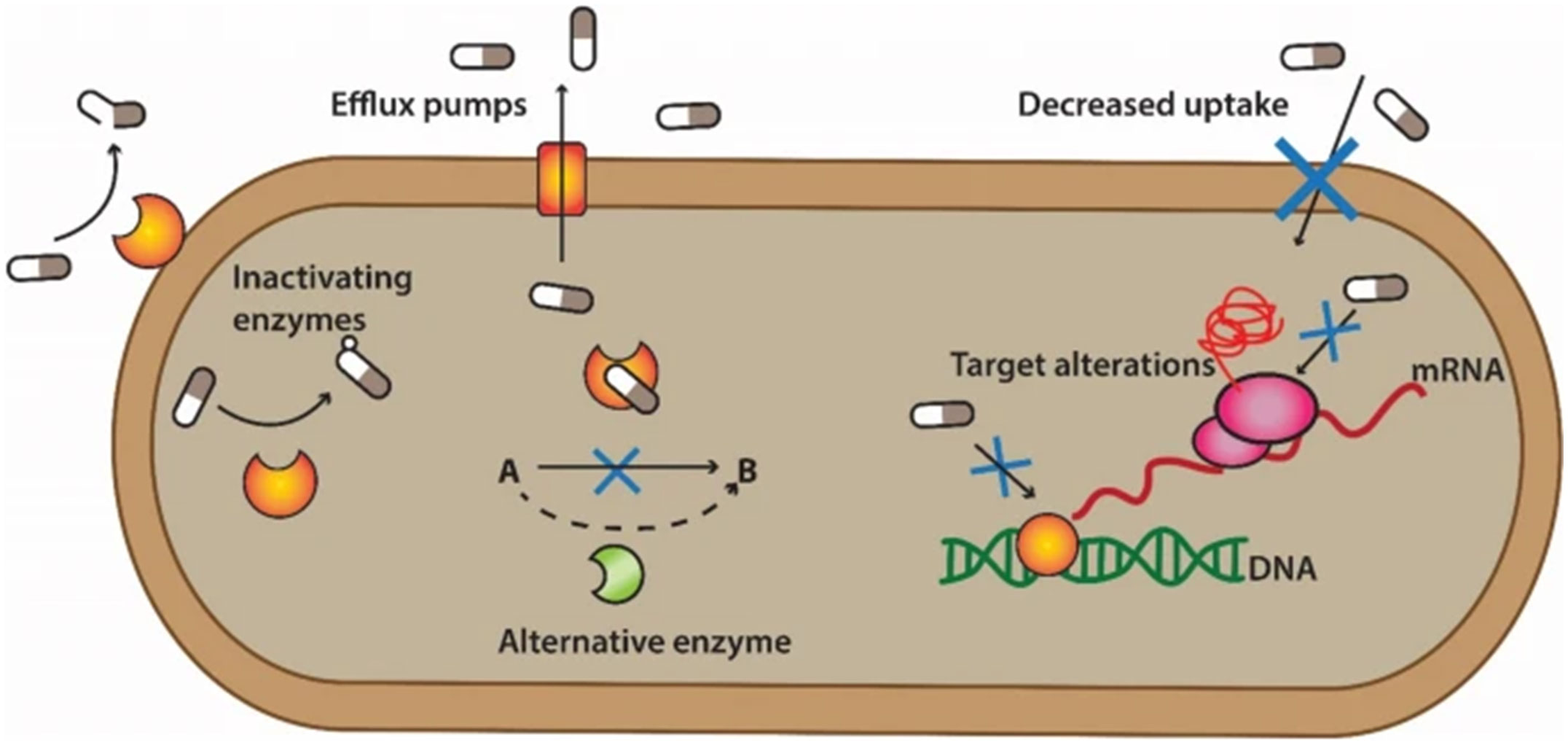
Figure 1 Mechanisms of antibiotic resistance in bacteria. The processes involved in antibiotic resistance entail several common mechanisms. One mechanism is the acquisition of enzymes that render the drugs inactive, like β-lactamases. Another mechanism is overproduction of efflux pumps, which expel specific or multiple drugs from the cell. The substitution of inhibited metabolic pathways with alternative ones is another mechanism. One of the most common, is modification of antibiotic target sites, which results in reduced drug binding affinity, without inactivating the enzyme. Finally, reduced outer membrane permeability prevents the drug from entering the cell and reaching therapeutic levels. Reproduced from (Mutuku et al., 2022), under the Creative Commons CC BY 4.0 license.
The production of the plasmid-mediated aminoglycoside modifying enzymes (AMEs) is the most significant mechanism of resistance to aminoglycosides (Neu, 1989). The antibacterial action of these drugs is lost when they are modified, as they are unable to bind to their ribosomal targets. Acetyltransferases (AAC), phosphotransferases (APH), and adenylases (ANT) are three different types of AMEs. There are even bacterial strains that harbor genes for two or more different types of AMEs (Frase et al., 2012). For instance, P. aeruginosa is capable of producing at least ten active enzymes to develop strong resistance to aminoglycosides (Xuan et al., 2023).
Antibiotics bind to specific amino acids on their protein targets, so mutation of the drug-binding site results in drug resistance. Such mutations typically prevent drug binding but maintain the functionality of the enzyme. For example, the PBPs are the target of β-lactam antibiotics. The two catalytic activities of PBPs that are required for peptidoglycan synthesis are found in a transpeptidase and glycosyltransferase domain (Sauvage and Terrak, 2016). The target for β-lactams is the transpeptidase domain. As a result, when PBPs undergo mutations, the binding affinity the drugs is decreased, but the PBPs function normally (Fishovitz et al., 2014). The production of PBP2a by methicillin-resistant Staphylococcus aureus (MRSA) inhibits β-lactam binding, resulting in transpeptidase activity in the presence of drug and therefore resistance (Gunasekharan et al., 2021). The fluoroquinolones are antibiotics that bind to DNA gyrase and topoisomerase IV and inhibit DNA replication (Drlica et al., 2008). The primary cause of resistance to fluoroquinolones is the occurrence of point mutations in the genes that encode chromosomal topoisomerase (Yamagishi et al., 1996). This resistance is brought about by a reduction in the binding affinity between the drug and the DNA-enzyme complex (Hooper and Jacoby, 2016). The emergence of resistance mutations is primarily due to spontaneous single-gene changes, which typically result in a low level of resistance (Martinez and Baquero, 2000). However, to achieve a high level of resistance, it usually requires the occurrence of two mutations in the topoisomerase gene. Consequently, the prevalence of bacteria that are resistant to fluoroquinolones is relatively low (Tchesnokova et al., 2019).
As a defense mechanism, bacteria can counteract antibacterial effects by altering membrane permeability so that antibiotics cannot enter cells to reach their cellular targets (Karnwal et al., 2023). Gram-negative bacteria are the principal hosts of this mechanism. Gram-negative bacteria are protected by an outer membrane barrier composed of phospholipids, lipopolysaccharide (LPS), and membrane proteins, including the porins, which are beta-barrel proteins that serve as pores for nutrients to pass in and out of the cell. The porins are the main route of entry for several classes of antibiotics, including β-lactams. Mutation of porins or reduction of the porin content can contribute to inherent and acquired resistance (Miller, 2016). For example, OmpF and OmpC are two major porins which can form both specific and non-specific channels that can transport nutrients and hydrophilic antibiotics. When a gene mutation results in the loss or reduction of one of these porins, bacteria can develop resistance to β-lactams or other antibiotics because porin-dependent drugs cannot enter the cell. Another example is the loss of the P. aeruginosa-specific porin OprD2, which results in resistance to carbapenems (Delcour, 2009).
Efflux pumps are systems in bacteria that export molecules, including waste products, out of the cell. Some of these efflux systems can also transport antibiotics out of the cytoplasm or periplasm, lowering their effective concentration and conferring resistance. Typically, efflux-mediated drug resistance emerges when these systems are overproduced, either by gene duplication or promoter mutations (Croop et al., 1987). There are six primary categories of efflux pump systems: transporters of ATP-binding cassettes (ABC) are the most common, followed by the major facilitator superfamily (MFS), small multidrug resistance (SMR), proteobacterial antimicrobial compound efflux (PACE) (Hassan et al., 2018) multidrug and toxic compound extrusion (MATE), and resistance nodulation division (RND) superfamily (Sharma et al., 2023). Most efflux pump systems utilize the proton motive force as an energy source, with the exception of the ABC superfamily, which uses ATP (Konings et al., 1997). Bacterial efflux pumps are major contributors to antibiotic resistance, as they can remove a wide range of drugs (Li et al., 2015). For example, efflux pumps have been identified that can remove β-lactams, fluoroquinolones, tetracyclines, macrolides, aminoglycosides, and even non-antibiotic compounds like antiseptics, disinfectants, and metals (Huang et al., 2022). Additionally, some efflux pumps have the capacity to efflux several different drug classes at once, contributing to the emergence of MDR strains (Webber and Piddock, 2003). Designing inhibitors that specifically target these efflux pumps is of great interest and should be a priority, as these drugs could lead to a revitalization of our antiquated antibiotic arsenal by increasing the efficacy of multiple drug classes against specific pathogens. Another strategy to rejuvenate our drug arsenal is to utilize alternative molecules, such as therapeutic proteins, to combat highly drug-resistant infections.
Therapeutic proteins are classified into two broad, distinct categories, namely small and large proteins. The smallest proteins are AMPs, which range from 10 to 50 amino acids in length and are amidated at the C-terminus, a modification that impacts their biological activity (Zasloff, 2002; Pasupuleti et al., 2012). In contrast, large therapeutic proteins, which are utilized in the field of medicine to treat a variety of diseases, are highly complex biological molecules. These therapeutic proteins often have substantial molecular weights, contributing to their intricate structure and function. One specific type of large therapeutic protein is mAbs, which have been widely employed in the field of biomedicine. In the following discussion, we will delve into the distinct properties of both AMPs and mAbs, analyzing their specific characteristics and exploring their potential applications in medicine and disease treatment.
AMPs are small, low-molecular-weight peptides that are essential for innate immunity and can be used to fight infections (Goyal and Mattoo, 2014). These peptides exhibit activity against a broad spectrum of pathogens, such as bacteria, fungi, parasites, and viruses (Goyal and Mattoo, 2014). The Data Repository of Antimicrobial Peptides (DRAMP, available at http://dramp.cpu-bioinfor.org) is a resource for known AMPs. Currently, there are 3791 AMPs reported from six kingdoms, including 2519 animal, 824 plant, 431 bacterial, 7 protozoan, 6 fungal, and 4 archaeal (Patyra and Kwiatek, 2023). In addition to having antibacterial properties, AMPs can also have additional properties, including immune system control, angiogenesis, wound healing, and anticancer activity (Hoskin and Ramamoorthy, 2008). The development of drug resistance as a result of an antibiotic’s single target, and prolonged and extensive use is a significant concern for infection management (Aydin et al., 2015). Comparatively, the use of AMPs presents several distinct advantages that make them a promising therapeutic option. One such advantage lies in their ability to exert their antimicrobial effects on both intracellular and membrane targets within the cell. This broad range of targets allows AMPs to effectively be used against a wide variety of pathogens. By acting on multiple targets, AMPs demonstrate a high level of efficacy, making them a formidable weapon in the fight against infectious diseases (Zhang et al., 2021).
AMPs are classified based on multiple properties, such as sources, manufacturing method, biological potential, amino acid sequence, mechanism of action, and their three-dimensional structure (Wang, 2015). Next, we will discuss the most useful classifications AMPs based upon their structure, activity, and source.
The classification of AMPs based on structural and charge composition are categorized into four groups based on the presence or absence of two secondary structural components: α-helices and β-sheets. These structural components play a pivotal role in determining the overall properties and function of AMPs. The four families of AMPs are linear α-helical peptides, β-sheet containing peptides, peptides that possess both α-helices and β-sheets, and peptides lacking α-helices or β-sheets (Reddy et al., 2004).
The first group pertains to a relatively diverse and extensively researched group of AMPs, namely the α-helical conformational peptides (Figure 2A). From natural sources, hundreds of distinct sequences have been identified (Ebenhan et al., 2014). These peptides typically comprise 12–40 amino acids and are abundant in helix-stabilizing residues, such as alanine, leucine, and lysine (Takahashi et al., 2010). A noteworthy representative of this group is the Magainin-2 family, which is a family of peptides with a broad-spectrum activity that were isolated from the skin of Xenopus laevis (Zasloff et al., 1988). This group can be classified as either anionic or cationic (Zhang et al., 2022). The anionic AMPs have a net charge range of -1 to -8 (Zhang et al., 2021). They interact with microorganisms by forming salt bridges using metal ions and the negatively charged elements of the microbial membrane, which is comparable to the charge-neutralization properties of larger proenzymes (Harris et al., 2009). For instance, the first anionic AMP to be identified, ovine pulmonary surfactant associated anion peptide (SAAP), displayed antibacterial action towards the ovine pathogen Mannheimia haemolytica in a mixture with zinc ions (Zhang et al., 2021). SAAP’s bactericidal action was largely decreased when NaCl and EDTA were added, but recovered when ZnCl2 was added back (Getahun et al., 2022). Additionally, an intrapeptide hydrogen bond between the N-terminal portion of the peptide and the amidated C-terminus of the helical anionic AMP is crucial for stabilizing the tilted helix structure (Mura et al., 2016). The cationic AMPs have a net charge of +2 to +9 and the C-terminus is typically amidated (Huang et al., 2010). While these peptides are disordered in aqueous solutions, they completely or partially change into an α-helical shape in the presence of trifluoroethanol, sodium dodecyl sulfate micelles, phospholipid vesicles, and liposomes. (Pazderková et al., 2019). Additionally, these AMPs often include more than 50% hydrophobic amino acids, allowing them to engage with membranes and form an amphiphilic structure (Zhang et al., 2021).
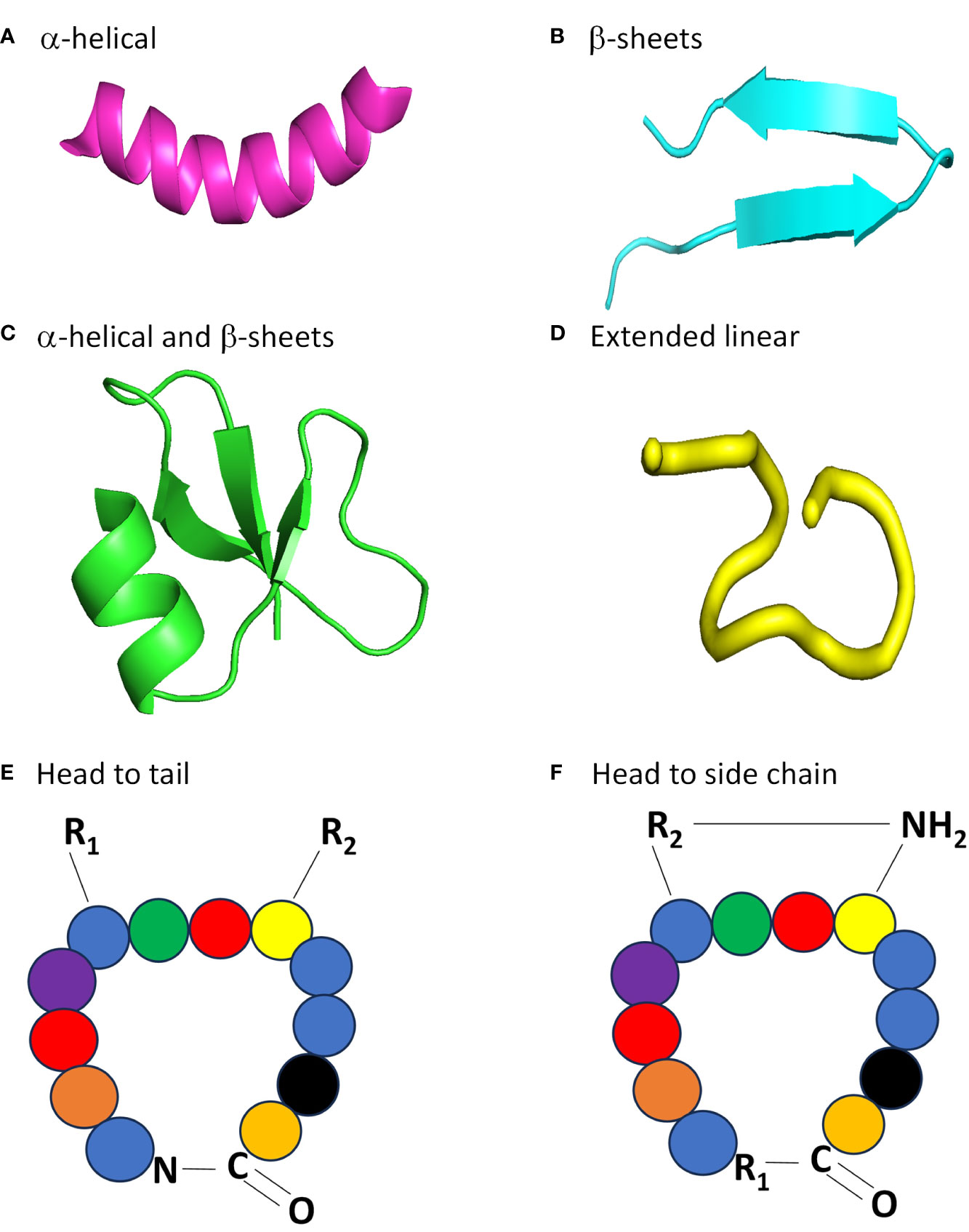
Figure 2 Four major structural classes of AMPs. (A) Clavavin has an α-helical structure, pdb code: 6C41. (B) Protegrin PG-5 is a peptide that has a β-sheet structure, pdb code: 2NC7. (C) Human -defensin 1 has an α-helical and β-sheet containing structure, pdb code:1IJV. (D) The linear extension structure of temporin B, pdb code:6GIL. All models were created using Pymol, with structures obtained from the Protein Data Bank. Example of head to tail (E) and head to side chain (F) cyclic topologies of AMPs. For a head to tail arrangement, an amide bond is formed between the N- and C-terminus of the protein. For head to side chain arrangements, the N-terminus forms an amide bond with an internal carboxyl group (COOH), like from an aspartate or glutamate residue.
The cationic β-family, which constitutes the second group of AMPs, features a minimum of one pair of β-strands [Figure 2B, (Wang, 2015)]. The structural stability of this group of AMPs can be attributed to the presence of cysteine residues in almost all of them, which form one or more disulfide bonds (Nguyen et al., 2011). Consequently, these peptides exhibit enhanced stability in solution and do not elicit significant structural alterations in the membrane environment (Tam et al., 2015). Notable examples of AMPs with a β-sheet structure include bovine lactoferrin, human defensins, and protegrin (Reddy et al., 2004). The third group of AMPs contain both α-helical and β-sheet components are present not only in humans and other mammals, but also in invertebrates and plants (Wang, 2014). The arrangement of three to five disulfide bonds is an important feature of these AMPs, including the cis defensins (small cysteine-rich cationic proteins) superfamily (Figure 2C). These defensins have bactericidal activity due to their positive charge and hydrophobic amino acids, which interact with phosphatidyl chains to destroy bacterial membranes (Parisi et al., 2019).
Some AMPs do not adopt any specific 3D structure, neither in solution nor in contact with membranes. These AMPs are commonly referred to as extended linear structures (Figure 2D). Typically, these peptides are devoid of α-helices and β-sheets and are usually rich in amino acids such as glycine, proline, tryptophan, and histidine (Koehbach and Craik, 2019). The categorization method is deemed as an ad-hoc tactic as there are less than 400 recognized 3D configurations among more than 2600 recognized AMPs belonging to this family (Xuan et al., 2023). A new set of AMPs, the fifth group, has recently been suggested for AMPs with complex and cyclic topologies. These peptides differ from those in the previous four categories as they do not have a linear structure but have a “head to tail” or “head to side chain” cyclic topology (Figures 2E, F). The preservation of their structure is achieved by disulfide bonds or thioether bridges (Deshayes et al., 2022).
The second classification of AMPs is based on their cellular target. AMPs can be divided into two categories: those that have intracellular targets and those that target and damage the cell membrane (Lei et al., 2019). Many models have been proposed to explain the extracellular AMPs mechanism of action (Figure 3). For the barrel-stave model (Figure 3A), the AMPs cluster together, break down the lipid bilayer as multimers, and create channels that allow the cytoplasm to leak out (Huan et al., 2020). Thus, the AMPs cause cell death by collapsed membranes and subsequent lysis (Giuliani et al., 2008). The AMP alamethicin acts via this mechanism (Bessin et al., 2004). For the carpet model (Figure 3B), the AMPs are positioned parallel to the cell membrane. Their hydrophilic end is directed towards the solvent and their hydrophobic end is directed towards the phospholipid bilayer. The membrane surface is covered by AMPs like a carpet and they act as a detergent to break down the cell membrane (Sato and Feix, 2006). The human cathelicidin LL-37 is an example that works using this mechanism (Xhindoli et al., 2016). The toroidal pore model (Figure 3C), proposes that AMPs are vertically implanted to generate a ring gap in the outer membrane (Ahmed and Hammami, 2019). Arenicin, lacticin Q, and magainin 2 are examples of AMPs that function by this mechanism (Patra et al., 2022). Furthermore, fluid domains produced by cationic peptides like BP2, TC19, and TC84 further damage the membrane barrier. Some peptides are effective on targets that are both intracellular and extracellular, and they can change their mode of action based on the concentration of the peptide, membrane structure, and the growth stage of the pathogen (Yount and Yeaman, 2005). These dual function AMPs that interact with intracellular enzymes typically first disrupt microbial membranes, which allows them to gain entry into the cell. These non-membrane mechanisms inhibit various processes, such as protein and nucleic acid biosynthesis, protease activity, and cell division. AMPs inhibit these processes by interfering with related enzymes (Scocchi et al., 2016). For example, the enterobacterial AMP microcin was the first identified that blocks DNA gyrase, comparable to the fluoroquinolones, and blocks the formation of the gyrase-DNA complex, preventing DNA replication (Le et al., 2017).
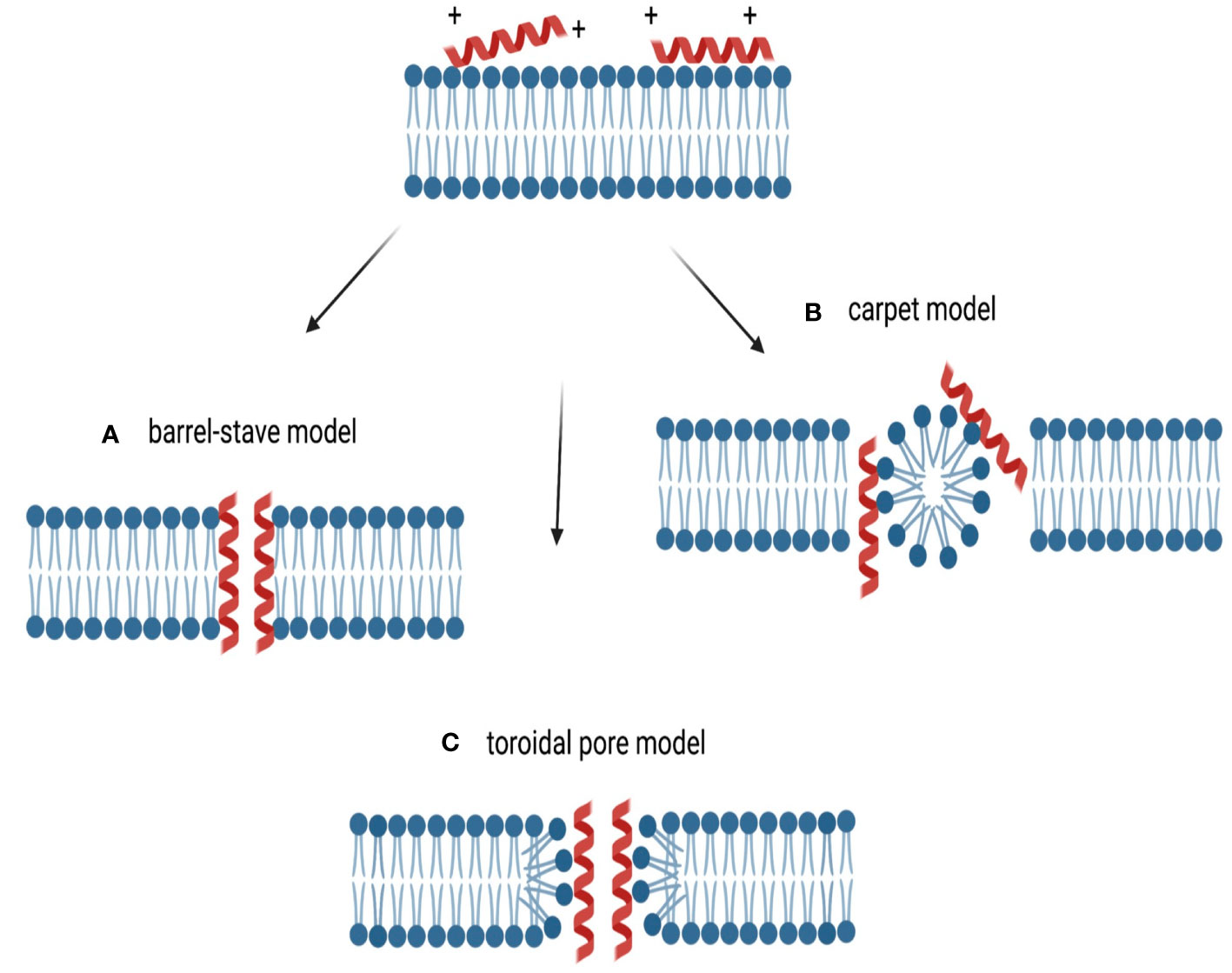
Figure 3 Types of membrane targeting actions of cationic AMPs. The cationic nature of the peptides allows for interaction with the negatively charged membrane. (A) The barrel-stave model, where AMPs aggregate and create a hole, with hydrophilic portions on the inside and hydrophobic residues contacting the membrane lipids. (B) In the carpet-like model, AMPs accumulate at the plasma membrane, acting like detergents that alter and destroy the membrane. (C) For the toroidal pore model, AMPs perpendicularly enter the membrane, altering the lipid structure and creating a ring pore. Reproduced under open access Creative Commons CC BY 4.0 license from (Zupin et al., 2022).
The origins of AMPs can be classified into several categories, including mammalian, amphibian, microbial, and insect (Huan et al., 2020). Additionally, the AMPs discovered in marine environments have garnered considerable interest in recent years due to their rich biodiversity, unique adaptations to extreme conditions, and the pressing need for novel drugs in the face of increasing resistance (Bertrand and Munoz-Garay, 2019). AMPs from mammalian sources typically come from humans, sheep, cattle, and other vertebrates (Huttner and Bevins, 1999). Human host defense peptides (HDPs) are effective against microbial infections, but they take diverse forms depending on the developmental growth stage (Yeung et al., 2011). Many areas of the body, including the skin, eyes, ears, mouth, lung, intestine, and urethra, contain HDPs (Silva et al., 2018). HDPs found in human breast milk are important for the establishment of the newborns innate immune system and lowers the risk of infection (Marszalek and Lodish, 2005; Cacho and Lawrence, 2017). Furthermore, it has been reported that HDPs, such as the cathelicidins and defensins, exhibit a substantial influence on immunological regulation, apoptosis, and wound healing, in addition to their antibacterial characteristics (Brown and Hancock, 2006).
AMPs produced by amphibians safeguard them against pathogens, including those that have contributed to the worldwide decline in their populations (Groner et al., 2014). The primary source of amphibian AMPs is the frog, with Magainin being the most promising among them, as they have potent, broad-spectrum activity, making them potential candidates for therapeutic development (Rinaldi, 2002). AMPs are widely distributed in the skin secretions of frogs from the genera Xenopus, Silurana, Hymenochirus, and Pseudhymenochirus (Owolabi et al., 2017). The AMPs produced by insects are primarily synthesized within the adipose tissues and hemocytes of insects, and contribute to their remarkable adaptability for survival (Eleftherianos et al., 2021). The various families of insect AMPs are widely distributed in species such as the guppy silkworm, bees, and fruit flies (Huan et al., 2020). Notably, cecropin A exhibits potent anti-inflammatory and anticancer activities (Brady et al., 2019).
Plant stems, seeds, and leaves have also been used to extract and isolate AMPs. These compounds are divided into several categories, such as thionins, defensins, and snakins (Ferdes, 2018). Recently identified AMPs from marine origins have contributed to the rising value of marine resources (Wu et al., 2021). Several of the reported marine AMPs have shown promising results in vivo, even though most of these AMPs have only been experimentally validated in vitro. For instance, As-CATH4 has an immune stimulating effect in vivo and can improve the anti-infective properties of medications when used in combination. These anti-infective properties include enhancing phagocytosis, activating innate immune responses, and promoting wound healing. A prospective substitute for antibiotics is myticusin-beta, an immune-related AMP from Mytilus coruscus (Semreen et al., 2018), due to its broad-spectrum activity, minimal resistance development potential, and natural origin (Oh et al., 2020). AMPs can also be produced by bacteria, fungi, and other microorganisms. Examples include nisin and gramicidin from Lactococcus lactis, Bacillus subtilis, and Bacillus brevis (Yazici et al., 2018).
Today, there are several AMPs that have been approved for clinical use as antibiotic alternatives, including nisin, polymixins, and daptomycin [for a recent review, see (Dijksteel et al., 2021)]. In addition, there are several examples of AMPs that have been used in recent clinical trials. One such example is the synthetic, cysteine-rich AMP Iseganan (IB-367), which has been investigated for its ability to prevent oral mucositis and surgical site infections. This promising AMP was well absorbed and tolerated in phase I clinical trials (Rizzetto et al., 2022). Its application has been studied in phase III clinical trials in a variety of prophylactic applications, including following cancer treatment, where a significant reduction in oral aerobic bacterial and yeast load was observed (Mosca et al., 2000; Rizzetto et al., 2023). Importantly, there was no change in the minimum inhibitory concentration after one month, meaning resistance to IB-367 did not emerge (Giles et al., 2004). However, there is still a need to evaluate the efficacy of this AMP against MDR strains. Omiganan (MBI-226), another synthetic AMP, has also been investigated as a topical agent for the prevention and treatment of skin infections and catheter-associated infections. Its effectiveness and safety in these applications were evaluated in clinical trials, but in phase III trials it failed to demonstrate superiority over standard treatments (Dijksteel et al., 2021). LTX-109, a novel synthetic peptide, was developed to treat skin and soft tissue infections, or decolonize nasal passages of S. aureus (Isaksson et al., 2011; Saravolatz et al., 2017). LTX-109 was well tolerated and safe in phase I/IIa studies (Mercer and O'Neil, 2020). With an excellent safety profile, this AMP is promising for the treatment bacterial infections and its efficacy against MDR bacterial infections should be determined. The use of AMPs as an inhaled treatment could be revolutionary in the treatment of severe respiratory infections, including chronically infected cystic fibrosis patients. Preclinical studies have demonstrated the antimicrobial activity of some AMPs, but more studies are required. It is essential to demonstrate safety and efficacy of inhaled formulations to provide justification for clinical trials (Li et al., 2023). Finally, the use of AMPs for novel medical device coatings has also been extensively explored (Copling et al., 2023). These coatings would be designed to lower the risk of device-related infections, without contributing to the rising resistance problem (Drexelius and Neundorf, 2021). Overall, AMPs represent a promising alternative therapy to reduce antibiotic use and combat the resistance problem. The volume of potential future applications is numerous, which justifies continued AMP research and product development.
Monoclonal antibodies (mAbs) are one of the most promising groups of biological therapeutics and are a potential treatment option for a variety of diseases (Ecker et al., 2015). As many bacterial strains become highly-drug resistant, some infections become untreatable. mAbs are a new therapeutic option that might substitute for antibiotics (Saylor et al., 2009). The limitations of the early technologies, such as cross-reactivity and low stability, underlined the need for new methods to enhance the functional characteristics of already known mAbs and to find exceptionally potent new ones as therapies (Weiner, 2015).
Between the years 1990 and 2000, there was an expansion in molecular biology methodologies and structural-based theories, opening previously unheard-of opportunities to design antibody molecules, which paved the way for the engineering of the fragment antigen binding (Fab) and fragment crystallizable (Fc) regions. The affinity for the target antigen could now be increased through Fab engineering (Chiu et al., 2019). To accomplish this, two primary strategies are used: mutational screening of the Fab regions, and engineering of the Fab region to increase affinity to specific antigens, which is based on the structure of mAbs (Roque et al., 2004). The random mutation approach is based on the generation of libraries using an error- prone DNA polymerase for polymerase chain reaction (PCR) that introduces random mutations throughout the variable Fab region. This approach was successfully used to construct a panel of toxin-neutralizing antibodies against Bacillus anthracis (Maynard et al., 2002). In contrast, the structure-based method for Fab engineering and affinity enhancement relies on the analyses of complex antibody-antigen structures and the modification of specific contact sites in the variable region (Ewert et al., 2004). The structure-based technique is often used in the development of mAbs against bacterial pathogens, such as the Streptococci. This strategy requires machine and deep learning approaches for structural prediction and computational design of antibody nanocages, which are nanoscale structures coated with antibodies that allow for selective targeting and delivery to specific cells or tissues (Goulet et al., 2022).
Another molecular biology technique used to increase antibody efficacy and the spectrum of protection is the formation of an antibody-antibiotic conjugate [AAC, (Mariathasan and Tan, 2017)]. AACs were introduced in 2015 as a promising treatment option for bacterial infections. In this method, the main features of an antibody and antibiotic are integrated into a single molecule (Mariathasan and Tan, 2017). Recently, an AAC was shown to have promise for the management of S. aureus, which was more effective than vancomycin for the treatment of bacteremia. mAbs capable of binding to surface antigens of S. aureus were purified from infected patients. These mAbs were highly specific and had affinity for cell membrane preparations. Antibiotics with bactericidal or bacteriostatic action against S. aureus, such as vancomycin or daptomycin, could be used as the antibiotic load. The AAC works by tagging bacteria for opsonization and once inside the phagocyte, proteases release the drug and enhance killing (Lehar et al., 2015). This technology could be widely applicable to many bacterial pathogens and is completely customizable. An additional benefit is that only pathogens are targeted, which should leave the normal microbiota intact. Due to antibody specificity, this should also reduce the spread of resistance genes. Further exploration and expansion of this technology is warranted.
mAbs function by diverse mechanisms for the treatment of bacterial infections. One significant mechanism involves the Fc region of the antibody, which undergoes a conformational change upon binding to its target surface antigen. This change enhances the interaction between the antigen and the antibody, which activates the complement system, ultimately leading to the formation of the membrane attack complex and the subsequent bacterial lysis [Figure 4A, (Woof and Burton, 2004)]. Another mechanism involves the interaction of the Fc region with corresponding receptors on phagocytic cells. When an antibody bound to an exposed bacterial antigen engages with these receptors, it promotes opsonophagocytosis of the bacterial cell (Figure 4B). Finally, antibodies that target bacterial virulence factors can disrupt their function by preventing their binding to cellular targets or interfering with their multimerization (McConnell, 2019). While such toxin-targeted antibodies may not have strong antibacterial effects on the infecting organism, they provide the host with the opportunity to mount a robust immune response, while limiting tissue damage caused by toxins. Interestingly, the mechanism of action for all three approved antibacterial mAbs involves neutralization [Figure 4C, (Nagy et al., 2017)]. For example, the drugs raxibacumab and obiltoxaximab work by binding to the protective antigen component of the B. anthracis toxin, preventing its interaction with its target, the capillary morphogenesis protein 2 (Manish et al., 2020). This also inhibits the other toxin components, like edema factor and lethal factor, from entering cells and exerting their detrimental effects (Vacca et al., 2022). A similar mode of action applies to bezlotoxumab, which is used to prevent recurrent Clostridioides difficile infections, where antibody binding hinders the toxin’s interaction with its cellular receptor on the intestinal mucosa (Wilcox et al., 2017).
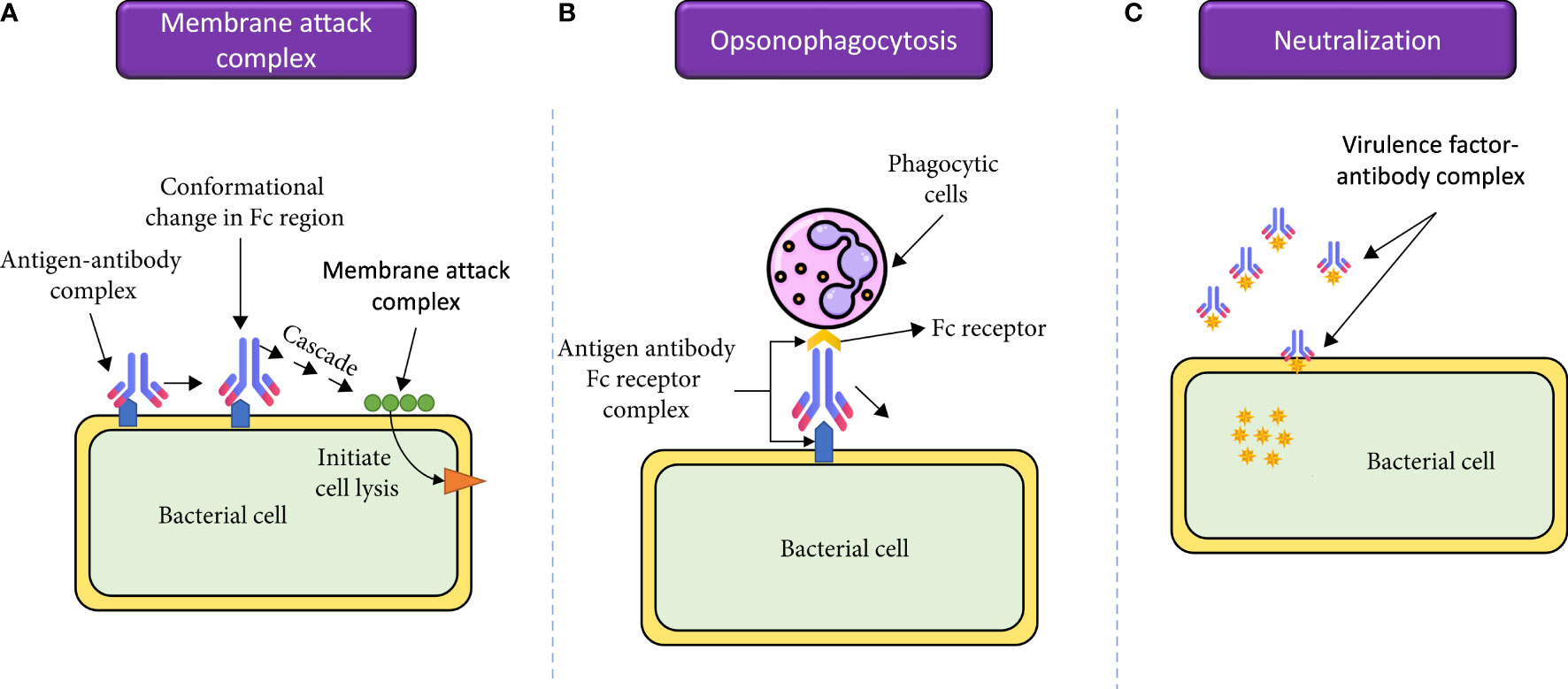
Figure 4 The molecular mechanisms by which mAbs can be used for treatment of bacterial infections. (A) mAbs are involved in activation of the complement cascade and therefore, complement-mediated killing of pathogens. (B) mAbs bind to their target antigens and recruit phagocytic cells, leading to opsonophagocytosis of the pathogens. (C) mAbs can bind and neutralize bacterial virulence factors, which will reduce inflammatory immune responses and limit tissue damage. Reproduced with modifications under open access Creative Commons CC BY 4.0 License from (Ahmed et al., 2023).
mAbs exhibit properties that render them promising candidates for addressing infections caused by MDR bacteria, as well as other antibiotic-resistant infections. First, antibacterial mAbs often target toxins or surface-exposed antigens that are not typical targets of currently available antibiotics (Nie et al., 2020). Consequently, the existing resistance mechanisms are unlikely to diminish their efficacy. Second, non-targeted bacterial species, including those constituting the normal microbiota, are less susceptible to the effects of mAbs compared to antibiotics, particularly broad-spectrum antibiotics that significantly impact beneficial bacteria residing in humans and animals (Jones-Nelson et al., 2020).
A wide variety of mAb-based medicines have emerged as effective therapeutic alternatives. Several of these antibodies have advanced to early-stage clinical investigations; however, many of them are still in preclinical development. Raxibacumab, a mAb was approved by the U.S Food and Drug Administration (FDA) for the treatment and prevention of inhalational anthrax caused by B. anthracis in 2012. The mechanism of action of raxibacumab depends on its ability to bind to the protective antigen component of the anthrax toxin, which blocks toxin function (Tsai and Morris, 2015). Biltoxaximab, another antibody used to manage and prevent inhalational anthrax works by a similar mechanism (Yamamoto et al., 2016). The targeted mAb fragment avidocin was developed to specifically target P. aeruginosa. Its effect involves binding to specific components of LPS in the outer membrane, disrupting the cell wall (Phanchana et al., 2021). MEDI3902, a novel bispecific mAb, targets two polysaccharide antigens Psl and PcrV of P. aeruginosa, which significantly lowers the possibility of resistance development as multiple mutations would be required (McConnell, 2019). Another mAb with FDA approval, bezlotoxumab, is recommended for the treatment of recurrent C. difficile infection. Bezlotoxumab targets the inflammatory enterotoxin B, which reduces the chance of further tissue damage (Navalkele and Chopra, 2018). Notably, all Bezlotoxumab-like mAbs work by neutralizing bacterial toxins, reducing tissue damage, and eventually improving patient outcomes. These types of mAbs have also been effective in treating MRSA infections (Martens and Demain, 2017).
Severe bacterial pathogens have emerged as a global concern, with existing methods to combat them often carrying adverse side effects and contributing to the proliferation of antibiotic resistance, thus posing risks to human health. In response, therapeutic proteins have been suggested as an innovative means of infection control due to their safety and direct targeting of specific pathogens. However, uncertainties persist concerning their efficacy and practical implementation. Moreover, challenges related to the stability, solubility, large-scale production, and purification of therapeutic proteins for widespread medical use need to be addressed. Next, we will discuss the largest hurdles for the practical implementation of therapeutic proteins into clinical practice.
Therapeutic proteins are subject to enzymatic and pH-dependent degradation in the gastrointestinal tract (GIT), which affects the pharmacodynamic properties like intestinal absorption, bioavailability, distribution, half-life, renal clearance, and elimination, all of which are important issues to address (Soltani et al., 2021). This is a problem for AMPs like bacteriocins. Fortunately, it was found that chemically produced bacteriocins incorporating d-amino acids are less sensitive to proteolytic cleavage in the GIT. In addition to d-amino acids, parenteral dosing may provide some protection against proteolytic breakdown of bacteriocins, particularly in cases of systemic infection (Kaur and Kaur, 2015). When directly compared to antibiotics, bacteriocins are less effective. Due to their vulnerability to proteases in vivo, bacteriocins have a shorter half-life than antibiotic equivalents (Bagley, 2014). Furthermore, bacteriocins are less labile at high temperatures and in high pH conditions than antibiotics. Bacteriocin stability is related to their varied structure and the quantity of post-translational modifications required for activity, like cyclization, disulfide bridges, and nonconventional amino acid incorporation (Grosu-Tudor et al., 2014). Biocompatibility studies have revealed that class II bacteriocins, nisin, and other lantipeptides are non-cytotoxic to several eukaryotic cell lines at doses 100-fold higher than their bactericidal concentrations (Meade et al., 2020). More research is needed to determine how to improve the stability and potency of bacteriocins for medical purposes.
For mAbs, their pharmacokinetics can change based on the specific mAb, the target bacterial strain, the infection site, and patient-specific characteristics, such as renal function, all of which are important considerations. It is possible that a single mAb will not be effective against all strains or serotypes of a given disease. Targeting a single molecule may not be adequate because different species may have different variants of the antigen present on their surfaces (De Vos et al., 2014). The use of recombinant polyclonal antibodies or the co-administration of mAb combinations may be a solution. For instance, against S. aureus, it was demonstrated that combining mAbs targeting a specific virulence factor produced better outcomes than monotherapy, leading to increased efficacy and greater strain coverage. This strategy contained a cocktail of mAbs, each directed against clumping factor A (ClfA), a key virulence factor in S. aureus bloodstream infections (Tkaczyk et al., 2012). The need for numerous methods of production and the requirement to clinically evaluate each individual mAb are challenges that antibody combinations will face during the clinical development process. Thus, from a production standpoint, it still may be more practical to create a single, diverse molecule (Struble et al., 2023).
Finally, the effector mechanism of the mAb might be considerably influenced by the type of the bound epitope. It is interesting that several mAbs that target the same antigen can activate different effector pathways. The spatial arrangement of the epitope and the target cell membrane is one explanation for this action. Fc-mediated mechanisms may be affected by where the bound mAb is in relation to the cell surface. For example, the brief half-life of the active components of the complement cascade is a key element in the setting of complement-dependent cytotoxicity. The possibility of the activated complement components successfully attaching to the bacterial cell surface is decreased if the cascade is initiated far from the target cell membrane. Therefore, the effectiveness in stimulating the entire cycle, including formation of the membrane attack complex, decreases with increasing distance from the cell surface. These findings highlight the complex nature of mAb interactions with bacterial pathogens and the significance of considering all variables in order to maximize their therapeutic effectiveness (Acharya et al., 2014).
Recombinant techniques are often used in the generation of therapeutic proteins, including solid or liquid phase synthesis, intravenous injection, and in vivo biotechnology. However, due to the various physiochemical properties of each therapeutic protein and the requirement to modify the conditions for every protein, such as expression conditions and purification method, the process can become rather expensive and difficult for large-scale manufacturing (Tripathi, 2016). A large portion of these costs are attributed to instrumentation and chemicals utilized in every therapeutic protein production process. More specifically, costs for mAb production can be attributed as one-third to cell culture, one-third to purification, and one-third to support (Li et al., 2010). It is estimated that process development and clinical manufacturing costs will represent 40–60% of developmental costs, potentially exceeding the cost of clinical trials. To reduce manufacturing costs, the search for alternative production strategies has been on the rise, including transgenic expression systems, or utilizing Escherichia coli or yeast to produce antibody fragments (Roque et al., 2004). The transgenic expression system includes genetically modified plants, such as tobacco and corn (Yao et al., 2015). E. coli and yeast have been utilized as host organisms to produce antibody fragments due to their rapid proliferation, efficient genetic tools, and cost-effectiveness. Rapid production of large quantities of recombinant antibody fragments can be achieved by using microorganism expression strategies (Mahdavi et al., 2022). These systems will need to be optimized and further developed to meet the demands of production for clinical use.
Patients may encounter challenges in accepting therapeutic proteins due to factors like administration methods (e.g., injections), potential immunogenicity, and the perception of genetic modification, which can make them seem unfamiliar or complex compared to traditional drug therapies. Due to stability and absorption concerns, many AMPs and mAbs cannot be taken orally. They may only be convenient and available to patients if administered intravenously or topically (Cleland et al., 2001). In most outpatient clinics, intravenous medication is not ideal; however, this may not be a problem for hospitalized patients, who are the most likely the target group for therapeutic protein use (Brobst and Borger, 2021). Patients must be informed of the benefits of starting or transitioning to therapeutic proteins and have any worries about doing so allayed (Xu et al., 2013). In fact, the FDA supports that patients need to be fully educated in order to make choices based on current knowledge and available data (Hinestrosa et al., 2007).
Therapeutic proteins may cause nocebo effects in patients, which are only noticeable to the patient and may have an impact on quality of life and treatment adherence (Rezk and Pieper, 2018). Nocebo effects are the worsening or incitation of symptoms caused by a negative attitude towards specific treatment (Hohenschurz-Schmidt et al., 2022). Nocebo reactions to therapeutic proteins may be brought on by a variety of interrelated, patient-related factors and psychological mechanisms influenced by both the therapeutic environment and the information given to patients (Kravvariti et al., 2018). Personal experiences, media coverage, and search engine results may also affect how people perceive therapeutic proteins (Faasse and Petrie, 2013). Nocebo effects should be considered during patient communication and diminished by using positive framing, contextualized informed consent, and united messaging (Zech et al., 2022). Even though patients are using the internet more frequently for health information, a survey found that perceptions of a doctor’s quality had a greater impact on treatment compliance than perceptions of the quality of internet health information, underscoring the significance of effective communication with healthcare providers. Additionally, as acknowledged in the oncology setting, educational materials created by medical societies or governmental bodies in collaboration with patient groups may be crucial in boosting patient education regarding therapeutic proteins (Lagassé et al., 2017).
Only a few AMPs have successfully completed clinical trials and the FDA has just recently approved three mAbs (Butler and Paterson, 2020). The difficulty in translating results from in vivo studies to clinical trials is one of the main causes of antibacterial mAbs and peptides clinical trial failures (Vacca et al., 2022). Data obtained using animal models does not always correspond to therapeutic trials conducted in people, because human genetic and immunological backgrounds differ from those of animal models. It is essential to select animal species that are pharmacologically relevant in order to comprehend and anticipate potential harmful toxic effects in humans (Prior et al., 2020). Non-human primates are frequently the only pharmacologically relevant species, rather than mice or rabbits, because of the great specificity that distinguishes human mAbs. For example, there are a number of problems in selecting animal models for sexually transmitted diseases whose primary site of infection is the urogenital tract (Chapman et al., 2009). Animals have a different estrous cycle and gestational time, and the anatomical site of infection may not be like that of humans, including the lack of human-specific receptors for bacterial adhesion and invasion. However, human infection-like mouse models have been developed and used for some mAb testing. In order to test mAb efficacy, an animal model of genital tract infections for the strictly human-adapted Neisseria gonorrhoeae was created in female mice treated with estrogen (Ivanov et al., 2021). A mAb that recognized a gonococcal lipo-oligosaccharide was tested and the authors concluded that animal models can be useful for analyzing the Fc-mediated effects of human IgG1 mAbs (Gulati et al., 2015). As a result, despite their anatomical differences, mouse models can be utilized to assess and provide some indications about whether mAbs function in vivo, which will be useful further pre-clinical research.
The immunogenicity that mAbs and AMPs generate is important for the practical application of these substances (Pérez de la Lastra et al., 2022). The term “immunogenicity” describes a foreign substance’s capacity to trigger an immunological response in the body, such as a therapeutic mAb or an AMP (Silberstein et al., 2015). Patients who receive mAbs and AMPs are at risk for an immune response against these “foreign antigens” and may develop antibodies against them. Preclinical and clinical research must evaluate the potential development of antidrug antibodies (ADAs) and infusion reactions. ADAs can block the mAb’s or AMP’s therapeutic action, lowering their efficacy. ADAs can potentially cause allergic responses or other immune-related side effects as well (Pratt, 2018). Infusion reactions may exhibit a spectrum of manifestations, ranging from mild indications, such as fever and chills, to more severe anaphylactic responses. These responses may occur when the patient’s immune system recognizes the mAb or AMP itself as an external entity or responds to impurities or aggregates in the formulation (Cheifetz and Mayer, 2005).
Therapeutic proteins remain a promising option for overcoming antibiotic resistance and fighting bacterial infections, despite the previously mentioned challenges. There are multiple possible approaches to further improve the feasibility of therapeutic proteins for clinical use. Targeting protein-protein interactions (PPIs), which are involved in a number of physiological processes and have emerged as a viable target for the treatment of diseases, is one such approach (Carro, 2018). In contrast to typical targets like G protein-coupled receptors (GPCRs), enzymes, and ion channels, which have distinct binding areas, PPI interfaces have a flat, expansive, and hydrophilic binding surface. This makes them challenging targets. It is generally accepted that not all amino acid residues at the interface contribute equally to binding and a few specific locations provide almost all the binding energy. Thus, it is possible to rationally design novel therapeutic proteins capable of preventing these crucial interactions by focusing on these “druggable” areas (Wang et al., 2011).
Another strategy for addressing the issue of antibiotic resistance lies in the exploration of alternative approaches, such as the utilization of machine and deep learning methods (Maruthamuthu et al., 2020). These techniques can be harnessed for the prediction of interactions between mAbs and surface-exposed targets. By doing so, it becomes feasible to identify antigens that can elicit a protective antibody response, thereby serving as promising therapeutic candidates. At the end of 2020, a program called AlphaFold2 was created for protein structural prediction using machine and deep learning (Jumper et al., 2021). In recent years, technologies such as AlphaFold-multimer (March 2021), Absolut! (July 2022), and AbAdapt (September 2022) were developed with the purpose of enhancing the prediction of antibody-antigen interactions to make machine and deep learning approaches appropriate for achieving this goal. The de novo design of antibody-like domains has recently been performed using computer models for high-resolution structural validation. To avoid the limitations of mAb engineering, Chidyausiku and colleagues devised a strategy that involved creating scaffolds that resembled antibodies but were artificially created to graft loops containing the complementarity determining regions of functional antibodies onto them. A critical turning point in the field of mAb discovery, design, and development was the advance in understanding the three-dimensional structure of proteins (Chidyausiku et al., 2022). Another method to identify potential targets for the development of therapeutic proteins is to use mass-spectrometry based proteomics. One technique that may be valuable is isobaric tags for relative and absolute quantification (iTRAQ). iTRAQ utilizes isobaric tags to label primary amines of proteins and has the capacity to be multiplexed for direct comparisons of protein levels between samples or treatment conditions. This technology could be used to simultaneously identify proteins present in outer membrane fractions and their relative expression levels, which could be useful in the evaluation of the efficacy and spectrum of mAbs (Wu et al., 2020).
Therapeutic proteins have the potential to become an important weapon in the battle against drug-resistant bacterial infections if they are properly developed and are highly effective. To achieve this goal, significant research must be carried out to overcome the significant challenges, such as stability and efficacy (Mellstedt, 2013). These proteins are distinguished from standard antibiotics through their unique mechanisms of action, sources, and specificity. Unlike traditional antibiotics with broad-spectrum coverage, therapeutic proteins operate with greater specificity. For instance, mAbs can selectively bind to specific bacterial components, possibly even at the level of a specific strain, neutralizing their effects. The advanced biotechnological methods employed in therapeutic protein production enable precise design and control over their properties, minimizing the risk of impacting beneficial bacteria and reducing the likelihood of resistance development. Appropriate animal models will need to be standardized and extensive clinical trials are required to determine the safety and efficacy of therapeutic proteins. Guidelines for research development, quality, production, and distribution will need to be created and enforced by policy makers and governing bodies. Furthermore, as this technology is relatively new, educational materials will be required to give prescribers and patients confidence in the strictness involved in the development process, production standards, and acceptance of therapeutic proteins as equivalent effective and safe medicines. As more therapeutic proteins become available, being aware of these concepts may help physicians avoid potential errors linked with the quality level of the therapeutic proteins and the consistency of the manufacturers making them (O'Callaghan et al., 2019). As our antibiotic arsenal continues to shrink due to widespread resistance, therapeutic proteins may represent our best option for the future.
MH: Conceptualization, Visualization, Writing – original draft, Writing – review & editing. MA: Writing – original draft, Writing – review & editing. BR: Writing – original draft, Writing – review & editing. VC: Funding acquisition, Resources, Supervision, Visualization, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by grant GM138303 awarded to VC. Research reported in this publication was supported by the National Institute Of General Medical Sciences of the National Institutes of Health under Award Number T34GM136492. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) VC declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abebe E., Tegegne B., Tibebu S. (2016). A review on molecular mechanisms of bacterial resistance to antibiotics. Eur. J. Appl. Sci. 8, 301–310.
Acharya P., Tolbert W. D., Gohain N., Wu X., Yu L., Liu T., et al. (2014). Structural definition of an antibody-dependent cellular cytotoxicity response implicated in reduced risk for HIV-1 infection. J. Virol. 88, 12895–12906.
Ahmed S., Ahmed M. Z., Rafique S., Almasoudi S. E., Shah M., Jalil N. A. C., et al. (2023). Recent approaches for downplaying antibiotic resistance: Molecular mechanisms. BioMed. Res. Int. 2023, 5250040. doi: 10.1155/2023/5250040
Ahmed T. A. E., Hammami R. (2019). Recent insights into structure–function relationships of antimicrobial peptides. J. Food Biochem. 43, e12546.
Ali T., Ali I., Khan N. A., Han B., Gao J. (2018). The growing genetic and functional diversity of extended spectrum beta-lactamases. BioMed. Res. Int. 2018, 2018.
Allen H. (2017). “Alternatives to antibiotics: Why and how,” in NAM perspectives (Washington, DC: National Academy of Medicine). Discussion Paper.
Aydin S., Ince B., Ince O. (2015). Development of antibiotic resistance genes in microbial communities during long-term operation of anaerobic reactors in the treatment of pharmaceutical wastewater. Water Res. 83, 337–344.
Bagley C. P. (2014). Potential role of synthetic antimicrobial peptides in animal health to combat growing concerns of antibiotic resistance-a review. Wyno. Acad. J. Agric. Sci. 2, 19–28.
Bertrand B., Munoz-Garay C. (2019). Marine antimicrobial peptides: a promising source of new generation antibiotics and other bio-active molecules. Int. J. Pept. Res. Ther. 25, 1441–1450.
Bessin Y., Saint N., Marri L., Marchini D., Molle. G. (2004). Antibacterial activity and pore-forming properties of ceratotoxins: a mechanism of action based on the barrel stave model. Biochim. Biophys. Acta (BBA)-Biomembranes. 1667, 148–156.
Blair J. M. A., Webber M. A., Baylay A. J., Ogbolu D. O., Piddock L. J. V. (2015). Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 13, 42–51.
Brady D., Grapputo A., Romoli O., Sandrelli F. (2019). Insect cecropins, antimicrobial peptides with potential therapeutic applications. Int. J. Mol. Sci. 20, 1–22. doi: https://www.mdpi.com/1422-0067/20/23/5862
Brobst B., Borger J. (2021). Benefits and risks of administering monoclonal antibody therapy for coronavirus (COVID-19). In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing.
Brown K. L., Hancock R. E. W. (2006). Cationic host defense (antimicrobial) peptides. Curr. Opin. Immunol. 18, 24–30.
Brown-Jaque M., Calero-Cáceres W., Muniesa M. (2015). Transfer of antibiotic-resistance genes via phage-related mobile elements. Plasmid 79, 1–7.
Butler M. S., Paterson D. L. (2020). Antibiotics in the clinical pipeline in October 2019. J. Antibiot. 73, 329–364.
Carro L. (2018). Protein–protein interactions in bacteria: a promising and challenging avenue towards the discovery of new antibiotics. Beilstein. J. Organic. Chem. 14, 2881–2896.
Chapman K., Pullen N., Coney L., Dempster M., Andrews L., Bajramovic J., et al. (2009). “Preclinical development of monoclonal antibodies: considerations for the use of non-human primates,”. MAbs (Taylor & Francis) 1 (5), 505–516.
Cheifetz A., Mayer L. (2005). Monoclonal antibodies, immunogenicity, and associated infusion reactions. Mount. Sinai. J. Med. New York. 72, 250–256.
Chidyausiku T. M., Mendes S. R., Klima J. C., Nadal M., Eckhard U., Roel-Touris J., et al. (2022). De novo design of immunoglobulin-like domains. Nat. Commun. 13, 5661.
Chiu M. L., Goulet D. R., Teplyakov A., Gilliland G. L. (2019). Antibody structure and function: the basis for engineering therapeutics. Antibodies 8, 55.
Cleland J. L., Daugherty A., Mrsny R. (2001). Emerging protein delivery methods. Curr. Opin. Biotechnol. 12, 212–219.
Copling A., Akantibila M., Kumaresan R., Fleischer G., Cortes D., Tripathi R. S., et al. (2023). Recent advances in antimicrobial peptide hydrogels. Int. J. Mol. Sci. 24 (8), 7563.
Croop J. M., Guild B. C., Gros P., Housman D. E. (1987). Genetics of multidrug resistance: relationship of a cloned gene to the complete multidrug resistant phenotype. Cancer Res. 47, 5982–5988.
Daulaire N., Bang A., Tomson G., Kalyango J. N., Cars O. (2015). Universal access to effective antibiotics is essential for tackling antibiotic resistance. J. Law. Med. Ethics. 43, 17–21.
Delcour A. H. (2009). Outer membrane permeability and antibiotic resistance. Biochim. Biophys. Acta (BBA)-Proteins. Proteomics 1794, 808–816.
Deshayes C., Arafath Md N., Apaire-Marchais V., Roger E. (2022). Drug delivery systems for the oral administration of antimicrobial peptides: Promising tools to treat infectious diseases. Front. Med. Technol. 3, 778645.
De Vos S., Forero-Torres A., Ansell S. M., Kahl B., Cheson B. D., Bartlett N. L., et al. (2014). A phase II study of dacetuzumab (SGN-40) in patients with relapsed diffuse large B-cell lymphoma (DLBCL) and correlative analyses of patient-specific factors. J. Hematol. Oncol. 7, 1–9.
Dijksteel G. S., Ulrich M. M. W., Middelkoop E., Boekema B. (2021). Review: Lessons learned from clinical trials using antimicrobial peptides (AMPs). Front. Microbiol. 12, 616979.
Drexelius M. G., Neundorf I. (2021). Application of antimicrobial peptides on biomedical implants: Three ways to pursue peptide coatings. Int. J. Mol. Sci. 22 (24), 13212.
Drlica K., Malik M., Kerns R. J., Zhao X. (2008). Quinolone-mediated bacterial death. Antimicrob. Agents chemother. 52, 385–392.
Ebenhan T., Gheysens O., Kruger H. G., Zeevaart J. R., Sathekge M. M. (2014). Antimicrobial peptides: their role as infection-selective tracers for molecular imaging. BioMed. Res. Int 2014, 867381.
Ecker D. M., Jones S. D., Levine H. L. (2015). “The therapeutic monoclonal antibody market,”. MAbs (Taylor & Francis) 7 (1), 9–14. doi: 10.4161/19420862.2015.989042
Eleftherianos I., Zhang W., Heryanto C., Mohamed A., Contreras G., Tettamanti G., et al. (2021). Diversity of insect antimicrobial peptides and proteins-A functional perspective: A review. Int. J. Biol. Macromol. 191, 277–287.
Ewert S., Honegger A., Plückthun A. (2004). Stability improvement of antibodies for extracellular and intracellular applications: CDR grafting to stable frameworks and structure-based framework engineering. Methods 34, 184–199.
Faasse K., Petrie K. J. (2013). The nocebo effect: patient expectations and medication side effects. Postgraduate. Med. J. 89, 540–546.
Ferdes M. (2018). Fighting Antimicrobial Resistance. Budimir N. Chapter: Antimicrobial Compounds from plants (Zagreb, Croatia: IAPC-OBP), 243–271.
Fishovitz J., Hermoso J. A., Chang M., Mobashery S. (2014). Penicillin-binding protein 2a of methicillin-resistant Staphylococcus aureus. IUBMB Life 66, 572–577.
Fleming A. (1929). On the antibacterial action of cultures of a penicillium, with special reference to their use in the isolation of B. influenzae. Br. J. Exp. Pathol. 10, 226.
Frase H., Toth M., Vakulenko S. B. (2012). Revisiting the nucleotide and aminoglycoside substrate specificity of the bifunctional aminoglycoside acetyltransferase (6′)-Ie/aminoglycoside phosphotransferase (2 ″)-Ia enzyme. J. Biol. Chem. 287, 43262–43269.
Getahun Y. A., Ali D. A., Taye B. W., Alemayehu Y. A. (2022). Multidrug-resistant microbial therapy using antimicrobial peptides and the CRISPR/Cas9 system. Vet. Med.: Res. Rep. 13, 173–190.
Giles F. J., Rodriguez R., Weisdorf D., Wingard J. R., Martin P. J., Fleming T. R., et al. (2004). A phase III, randomized, double-blind, placebo-controlled, study of iseganan for the reduction of stomatitis in patients receiving stomatotoxic chemotherapy. Leuk. Res. 28, 559–565.
Giuliani A., Pirri G., Bozzi A., Giulio A.Di, Aschi M., Rinaldi. A. C. (2008). Antimicrobial peptides: natural templates for synthetic membrane-active compounds. Cell. Mol. Life Sci. 65, 2450–2460.
Goulet A., Joos R., Lavelle K., Sinderen D. V., Mahony J., Cambillau. C. (2022). A structural discovery journey of streptococcal phages adhesion devices by AlphaFold2. Front. Mol. Biosci. 9, 960325.
Goyal R. K., Mattoo A. K. (2014). Multitasking antimicrobial peptides in plant development and host defense against biotic/abiotic stress. Plant Sci. 228, 135–149.
Groner M. L., Rollins-Smith L. A., Reinert L. K., Hempel J., Bier M. E., Relyea R. A. (2014). Interactive effects of competition and predator cues on immune responses of leopard frogs at metamorphosis. J. Exp. Biol. 217, 351–358.
Grosu-Tudor S.-S., Stancu M.-M., Pelinescu D., Zamfir M. (2014). Characterization of some bacteriocins produced by lactic acid bacteria isolated from fermented foods. World J. Microbiol. Biotechnol. 30, 2459–2469.
Gulati S., Mu X., Zheng B., Reed G. W., Ram S., Rice P. A. (2015). Antibody to reduction modifiable protein increases the bacterial burden and the duration of gonococcal infection in a mouse model. J. Infect. Dis. 212, 311–315.
Gunasekharan M., Choi T.-I., Rukayadi Y., Latif M. A. M., Karunakaran T., Faudzi S. M. M., et al. (2021). Preliminary insight of pyrrolylated-chalcones as new anti-methicillin-resistant Staphylococcus aureus (Anti-MRSA) agents. Molecules 26, 5314.
Harris F., Dennison S. R., Phoenix D. A. (2009). Anionic antimicrobial peptides from eukaryotic organisms. Curr. Protein Pept. Sci. 10, 585–606.
Hassan K. A., Liu Q., Elbourne L. D. H., Ahmad I., Sharples D., Naidu V., Chan C. K., et al (2018). Pacing across the membrane: the novel PACE family of efflux pumps is widespread in Gram-negative pathogens. Res Microbiol 169 (7-8), 450–454.
Hinestrosa M.C., Dickersin K., Klein P., Mayer M., Noss K., Slamon D., et al. (2007). Shaping the future of biomarker research in breast cancer to ensure clinical relevance. Nat. Rev. Cancer 7, 309–315.
Hohenschurz-Schmidt D., Thomson O. P., Rossettini G., Miciak M., Newell D., Roberts L., et al. (2022). Avoiding nocebo and other undesirable effects in chiropractic, osteopathy and physiotherapy: An invitation to reflect. Musculoskeletal. Sci. Pract. 62, 102677.
Hooper D. C., Jacoby G. A. (2016). Topoisomerase inhibitors: fluoroquinolone mechanisms of action and resistance. Cold Spring Harbor Perspect. Med. 6 (9), a025320.
Hoskin D. W., Ramamoorthy A. (2008). Studies on anticancer activities of antimicrobial peptides. Biochim. Biophys. Acta (BBA)-Biomembranes. 1778, 357–375.
Huan Y., Kong Q., Mou H., Yi H. (2020). Antimicrobial peptides: classification, design, application and research progress in multiple fields. Front. Microbiol. 11, 2559.
Huang Y., Huang J., Chen Y. (2010). Alpha-helical cationic antimicrobial peptides: relationships of structure and function. Protein Cell 1, 143–152.
Huang L., Wu C., Gao H., Xu C., Dai M., Huang L., et al. (2022). Bacterial multidrug efflux pumps at the frontline of antimicrobial resistance: An overview. Antibiotics 11, 520.
Huttner K. M., Bevins C. L. (1999). Antimicrobial peptides as mediators of epithelial host defense. Pediatr. Res. 45, 785–794.
Isaksson J., Brandsdal B. O., Engqvist M., Flaten G. E., Svendsen J. S., Stensen W. (2011). A synthetic antimicrobial peptidomimetic (LTX 109): stereochemical impact on membrane disruption. J. Med. Chem. 54, 5786–5795.
Ivanov S. S., Castore R., Rodriguez M. D. J., Circu M., Dragoi. A.-M. (2021). Neisseria gonorrhoeae subverts formin-dependent actin polymerization to colonize human macrophages. PloS Pathog. 17, e1010184.
Jones-Nelson O., Tovchigrechko A., Glover M. S., Fernandes F., Rangaswamy U., Liu H., et al. (2020). Antibacterial monoclonal antibodies do not disrupt the intestinal microbiome or its function. Antimicrob. Agents chemother. 64 (5), e02347-19. doi: 10.1128/aac.02347-19
Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589.
Karnwal A., Kumar G., Pant G., Hossain K., Ahmad A., Alshammari. M. B. (2023). Perspectives on usage of functional nanomaterials in antimicrobial therapy for antibiotic-resistant bacterial infections. ACS Omega. 8, 13492–13508.
Koehbach J., Craik D. J. (2019). The vast structural diversity of antimicrobial peptides. Trends Pharmacol. Sci. 40, 517–528.
Konings W. N., Lolkema J. S., Bolhuis H., Van Veen H. W., Poolman B., Driessen A. J. M. (1997). The role of transport processes in survival of lactic acid bacteria, energy transduction and multidrug resistance. Antonie. van. leeuwenhoek. 71, 117–128.
Kravvariti E., Kitas G. D., Mitsikostas D. D., Sfikakis P. P. (2018). Nocebos in rheumatology: emerging concepts and their implications for clinical practice. Nat. Rev. Rheumatol. 14, 727–740.
Lagassé H. A. D., Alexaki A., Simhadri V. L., Katagiri N. H., Jankowski W., Sauna Z. E., et al. (2017). Recent advances in (therapeutic protein) drug development. F1000Research 6, 113.
Le C.-F., Fang C.-M., Sekaran S. D. (2017). Intracellular targeting mechanisms by antimicrobial peptides. Antimicrob. Agents chemother. 61 (4). doi: 10.1128/aac.02340-16
Lehar S. M., Pillow T., Xu M., Staben L., Kajihara K. K., Vandlen R., et al. (2015). Novel antibody–antibiotic conjugate eliminates intracellular S. aureus. Nature 527, 323–328.
Lei J., Sun L., Huang S., Zhu C., Li P., He J., et al. (2019). The antimicrobial peptides and their potential clinical applications. Am. J. Trans. Res. 11, 3919.
Li X.-Z., Plésiat P., Nikaido H. (2015). The challenge of efflux-mediated antibiotic resistance in Gram-negative bacteria. Clin. Microbiol. Rev. 28, 337–418.
Li F., Vijayasankaran N., Shen A., Kiss R., Amanullah A. (2010). “Cell culture processes for monoclonal antibody production,”. MAbs (Taylor & Francis) 2 (5), 466–479.
Li J., Zheng H., Leung S. S. Y. (2023). Pulmonary delivery of emerging antibacterials for bacterial lung infections treatment. Pharm. Res. 40, 1057–1072.
Mahdavi S. Z. B., Oroojalian F., Eyvazi S., Hejazi M., Baradaran B., Pouladi N., et al. (2022). An overview on display systems (phage, bacterial, and yeast display) for production of anticancer antibodies; advantages and disadvantages. Int. J. Biol. Macromol. 208, 421–442.
Manish M., Verma S., Kandari D., Kulshreshtha P., Singh S., Bhatnagar. R. (2020). Anthrax prevention through vaccine and post-exposure therapy. Expert Opin. Biol. Ther. 20, 1405–1425.
Mariathasan S., Tan M.-W. (2017). Antibody–antibiotic conjugates: a novel therapeutic platform against bacterial infections. Trends Mol. Med. 23, 135–149.
Marszalek J. R., Lodish H. F. (2005). Docosahexaenoic acid, fatty acid–interacting proteins, and neuronal function: breastmilk and fish are good for you. Annu. Rev. Cell Dev. Biol. 21, 633–657.
Martens E., Demain A. L. (2017). The antibiotic resistance crisis, with a focus on the United States. J. Antibiot. 70, 520–526.
Martinez J. L., Baquero F. (2000). Mutation frequencies and antibiotic resistance. Antimicrob. Agents chemother. 44, 1771–1777.
Maruthamuthu M. K., Rudge S. R., Ardekani A. M., Ladisch M. R., Verma M. S. (2020). Process analytical technologies and data analytics for the manufacture of monoclonal antibodies. Trends Biotechnol. 38, 1169–1186.
Maynard J. A., Maassen C. B. M., Leppla S. H., Brasky K., Patterson J. L., Iverson B. L., et al. (2002). Protection against anthrax toxin by recombinant antibody fragments correlates with antigen affinity. Nat. Biotechnol. 20, 597–601.
McConnell M. J. (2019). Where are we with monoclonal antibodies for multidrug-resistant infections? Drug Discovery Today 24, 1132–1138.
Meade E., Slattery M. A., Garvey M. (2020). Bacteriocins, potent antimicrobial peptides and the fight against multi drug resistant species: resistance is futile? Antibiotics 9, 32.
Mercer D. K., O'Neil D. A. (2020). Innate inspiration: antifungal peptides and other immunotherapeutics from the host immune response. Front. Immunol. 11, 2177.
Miller S. I. (2016). Antibiotic resistance and regulation of the Gram-negative bacterial outer membrane barrier by host innate immune molecules. MBio 7 (5), e01541-16. doi: 10.1128/mbio.01541-16
Mora-Ochomogo M., Lohans C. T. (2021). β-Lactam antibiotic targets and resistance mechanisms: from covalent inhibitors to substrates. RSC. Med. Chem. 12, 1623–1639.
Mosca D. A., Hurst M. A., So W., Viajar B. S., Fujii C. A., Falla T. J. (2000). IB-367, a protegrin peptide with in vitro and in vivo activities against the microflora associated with oral mucositis. Antimicrob. Agents Chemother. 44, 1803–1808.
Munita J. M., Arias C. A. (2016). Mechanisms of antibiotic resistance. Microbiol. Spectr. 4 (2), 10.1128/microbiolspec.VMBF-0016-2015. doi: 10.1128/microbiolspec.VMBF-0016-2015
Mura M., Wang J., Zhou Y., Pinna M., Zvelindovsky A. V., Dennison S. R., et al. (2016). The effect of amidation on the behaviour of antimicrobial peptides. Eur. Biophys. J. 45, 195–207.
Mustafa S., Balkhy H., Gabere M. N. (2018). Current treatment options and the role of peptides as potential therapeutic components for Middle East Respiratory Syndrome (MERS): a review. J. Infect. Public Health 11, 9–17.
Mutuku C., Gazdag Z., Melegh S. (2022). Occurrence of antibiotics and bacterial resistance genes in wastewater: resistance mechanisms and antimicrobial resistance control approaches. World J Microbiol Biotechnol. 38 (9), 152. doi: 10.1007/s11274-022-03334-0
Nagy E., Nagy G., Power C. A., Badarau A., Szijártó V. (2017). Anti-bacterial monoclonal antibodies. Recombinant. Antibodies. Infect. Dis. 1053, 119–153.
Navalkele B. D., Chopra T. (2018). Bezlotoxumab: an emerging monoclonal antibody therapy for prevention of recurrent Clostridium difficile infection. Biol.: Targets Ther. 12, 11–21.
Neu H. C. (1989). Overview of mechanisms of bacterial resistance. Diagn. Microbiol. Infect. Dis. 12, 109–116.
Nguyen L. T., Haney E. F., Vogel H. J. (2011). The expanding scope of antimicrobial peptide structures and their modes of action. Trends Biotechnol. 29, 464–472.
Nie D., Hu Y., Chen Z., Li M., Hou Z., Luo X., et al. (2020). Outer membrane protein A (OmpA) as a potential therapeutic target for Acinetobacter baumannii infection. J. Biomed. Sci. 27, 1–8.
O'Callaghan J., Barry S. P., Bermingham M., Morris J. M., Griffin B. T. (2019). Regulation of biosimilar medicines and current perspectives on interchangeability and policy. Eur. J. Clin. Pharmacol. 75, 1–11.
O'Neill J. (2016). Tackling drug-resistant infections globally: final report and recommendations (Government of the United Kingdom). Available at: https://apo.org.au/node/6398
Oh R., Lee M. J., Kim Y.-O., Nam B.-H., Kong H. J., Kim J.-W., et al. (2020). Myticusin-beta, antimicrobial peptide from the marine bivalve, Mytilus coruscus. Fish. Shellfish. Immunol. 99, 342–352.
Owolabi B. O., Musale V., Ojo O. O., Charlotte Moffett R., McGahon M. K., Curtis T. M., et al. (2017). Actions of PGLa-AM1 and its [A14K] and [A20K] analogues and their therapeutic potential as anti-diabetic agents. Biochimie 138, 1–12.
Parisi K., Shafee T. M. A., Quimbar P., van der Weerden N. L., Bleackley M. R., Anderson. M. A. (2019). “The evolution, function and mechanisms of action for plant defensins,” in Semin Cell Dev Biol (Elsevier) 88, 107–118. doi: 10.1016/j.semcdb.2018.02.004
Pasupuleti M., Schmidtchen A., Malmsten M. (2012). Antimicrobial peptides: key components of the innate immune system. Crit. Rev. Biotechnol. 32, 143–171.
Patra A., Das J., Agrawal N. R., Kushwaha G. S., Ghosh M., Son Y.-O. (2022). Marine antimicrobial peptides-based strategies for tackling bacterial biofilm and biofouling challenges. Molecules 27, 7546.
Patyra E., Kwiatek K. (2023). Insect meals and insect antimicrobial peptides as an alternative for antibiotics and growth promoters in livestock production. Pathogens 12, 854.
Pazderková M., Maloň P., Zíma V., Hofbauerová K., Kopecký V. Jr, Kočišová E., et al. (2019). Interaction of halictine-related antimicrobial peptides with membrane models. Int. J. Mol. Sci. 20, 631.
Pérez de la Lastra J. M., Anand U., González-Acosta S., López M. R., Dey A., Bontempi E., et al. (2022). Antimicrobial resistance in the COVID-19 landscape: is there an opportunity for anti-infective antibodies and antimicrobial peptides? Front. Immunol. 13, 921483.
Phanchana M., Harnvoravongchai P., Wongkuna S., Phetruen T., Phothichaisri W., Panturat S., et al. (2021). Frontiers in antibiotic alternatives for Clostridioides difficile infection. World J. Gastroenterol. 27, 7210.
Poole K. (2002). Outer membranes and efflux: the path to multidrug resistance in Gram-negative bacteria. Curr. Pharm. Biotechnol. 3, 77–98.
Pratt K. P. (2018). Anti-drug antibodies: emerging approaches to predict, reduce or reverse biotherapeutic immunogenicity. Antibodies 7, 19.
Prior H., Haworth R., Labram B., Roberts R., Wolfreys A., Sewell. F. (2020). Justification for species selection for pharmaceutical toxicity studies. Toxicol. Res. 9, 758–770.
Reddy K. V. R., Yedery R. D., Aranha C. (2004). Antimicrobial peptides: premises and promises. Int. J. Antimicrob. Agents 24, 536–547.
Rezk M. F., Pieper B. (2018). To see or NOsee: the debate on the nocebo effect and optimizing the use of biosimilars. Adv. Ther. 35, 749–753.
Rinaldi A. C. (2002). Antimicrobial peptides from amphibian skin: an expanding scenario: Commentary. Curr. Opin. Chem. Biol. 6, 799–804.
Rizzetto G., Gambini D., Maurizi A., Candelora M., Molinelli E., Cirioni O., et al. (2022). Our experience over 20 years: Antimicrobial peptides against Gram positives, Gram negatives, and fungi. Pharmaceutics 15.
Rizzetto G., Gambini D., Maurizi A., Molinelli E., Simoni E. De, Pallotta F., et al. (2023). The sources of antimicrobial peptides against Gram-positives and Gram-negatives: our research experience. Infez Med. 31 (3), 306–322.
Roque A., Cecília A., Lowe C. R., Taipa M. Â. (2004). Antibodies and genetically engineered related molecules: production and purification. Biotechnol. Prog. 20, 639–654.
Sacha P., Wieczorek P., Hauschild T., Zórawski M., Olszańska D., Tryniszewska. E. (2008). Metallo-beta-lactamases of Pseudomonas aeruginosa–a novel mechanism resistance to beta-lactam antibiotics. Folia Histochem. Cytobiol. 46, 137–142.
Saravolatz L. D., Pawlak J., Martin H., Saravolatz S., Johnson L., Wold H., et al. (2017). Postantibiotic effect and postantibiotic sub-MIC effect of LTX-109 and mupirocin on Staphylococcus aureus blood isolates. Lett. Appl. Microbiol. 65, 410–413.
Sato H., Feix J. B. (2006). Peptide–membrane interactions and mechanisms of membrane destruction by amphipathic α-helical antimicrobial peptides. Biochim. Biophys. Acta (BBA)-Biomembranes. 1758, 1245–1256.
Sauvage E., Terrak M. (2016). Glycosyltransferases and transpeptidases/penicillin-binding proteins: valuable targets for new antibacterials. Antibiotics 5, 12.
Saylor C., Dadachova E., Casadevall A. (2009). Monoclonal antibody-based therapies for microbial diseases. Vaccine 27, G38–G46.
Scocchi M., Mardirossian M., Runti G., Benincasa M. (2016). Non-membrane permeabilizing modes of action of antimicrobial peptides on bacteria. Curr. Topics. med. Chem. 16, 76–88.
Semreen M. H., El-Gamal M. I., Abdin S., Alkhazraji H., Kamal L., Hammad S., et al. (2018). Recent updates of marine antimicrobial peptides. Saudi. Pharm. J. 26, 396–409.
Sharma S., Kaushik V., Kulshrestha M., Tiwari V. (2023). Different efflux pump systems in Acinetobacter baumannii and their role in multidrug resistance. Adv. Microbiol. Infect. Dis. Public Health 17, 155–168.
Silberstein S., Lenz R., Xu C. (2015). Therapeutic monoclonal antibodies: what headache specialists need to know. Headache.: J. Head Face. Pain 55, 1171–1182.
Silva O. N., Porto W. F., Ribeiro S. M., Batista I., Franco O. L. (2018). Host-defense peptides and their potential use as biomarkers in human diseases. Drug Discovery Today 23, 1666–1671.
Soltani S., Hammami R., Cotter P. D., Rebuffat S., Said L. B., Gaudreau H., et al. (2021). Bacteriocins as a new generation of antimicrobials: toxicity aspects and regulations. FEMS Microbiol. Rev. 45, fuaa039.
Struble E. B., Rawson J. M. O., Stantchev T., Scott D., Shapiro M. A. (2023). Uses and challenges of antiviral polyclonal and monoclonal antibody therapies. Pharmaceutics 15, 1538.
Takahashi D., Shukla S. K., Prakash Om, Zhang G. (2010). Structural determinants of host defense peptides for antimicrobial activity and target cell selectivity. Biochimie 92, 1236–1241.
Tam J. P., Wang S., Wong K., Tan W. L. (2015). Antimicrobial peptides from plants. Pharmaceuticals 8, 711–757.
Tan S. Y., Tatsumura Y. (2015). Alexander Fleming, (1881–1955): discoverer of penicillin. Singapore. Med. J. 56, 366.
Tang Ka W. K., Millar B. C., Moore J. E. (2023). Antimicrobial resistance (AMR). Br. J. Biomed. Sci. 80, 11387.
Tchesnokova V., Radey M., Chattopadhyay S., Larson L., Weaver J. L., Kisiela D., et al. (2019). Pandemic fluoroquinolone resistant Escherichia coli clone ST1193 emerged via simultaneous homologous recombinations in 11 gene loci. Proc. Natl. Acad. Sci. 116, 14740–14748.
Tkaczyk C., Hua L., Varkey R., Shi Y., Dettinger L., Woods R., et al. (2012). Identification of anti-alpha toxin monoclonal antibodies that reduce the severity of Staphylococcus aureus dermonecrosis and exhibit a correlation between affinity and potency. Clin. Vaccine Immunol. 19, 377–385.
Tripathi N. K. (2016). Production and purification of recombinant proteins from Escherichia coli. ChemBioEng. Rev. 3, 116–133.
Tsai C.-W., Morris S. (2015). Approval of raxibacumab for the treatment of inhalation anthrax under the US Food and Drug Administration “Animal Rule”. Front. Microbiol. 6, 1320.
Vacca F., Sala C., Rappuoli R. (2022). Monoclonal antibodies for bacterial pathogens: Mechanisms of action and engineering approaches for enhanced effector functions. Biomedicines 10, 2126.
Wang G. (2015). Improved methods for classification, prediction, and design of antimicrobial peptides. Comput. Peptidol. 1268, 43–66.
Wang M., Medeiros B. C., Erba H. P., DeAngelo D. J., Giles F. J., Swords R. T. (2011). Targeting protein neddylation: a novel therapeutic strategy for the treatment of cancer. Expert Opin. Ther. Targets 15, 253–264.
Webber M. A., Piddock L. J. V. (2003). The importance of efflux pumps in bacterial antibiotic resistance. J. Antimicrob. chemother. 51, 9–11.
Weiner G. J. (2015). Building better monoclonal antibody-based therapeutics. Nat. Rev. Cancer 15, 361–370.
Wilcox M. H., Gerding D. N., Poxton I. R., Kelly C., Nathan R., Birch T., et al. (2017). Bezlotoxumab for prevention of recurrent Clostridium difficile infection. New Engl. J. Med. 376, 305–317.
Woof J. M., Burton D. R. (2004). Human antibody–Fc receptor interactions illuminated by crystal structures. Nat. Rev. Immunol. 4, 89–99.
Wu R., Patocka J., Nepovimova E., Oleksak P., Valis M., Wu W., et al. (2021). Marine invertebrate peptides: Antimicrobial peptides. Front. Microbiol. 12, 785085.
Wu H., Wang M., Liu Y., Wang X., Wang Y., Lu J., et al. (2016). Characterization of antimicrobial resistance in Klebsiella species isolated from chicken broilers. Int. J. Food Microbiol. 232, 95–102.
Wu H., Zhang X.-Y., Niu M., Li F.-F., Gao S., Wei W., et al. (2020). Isobaric tags for relative and absolute quantitation in proteomic analysis of potential biomarkers in invasive cancer, ductal carcinoma in situ, and mammary fibroadenoma. Front. Oncol. 10, 574552.
Xhindoli D., Pacor S., Benincasa M., Scocchi M., Gennaro R., Tossi. A. (2016). The human cathelicidin LL-37—A pore-forming antibacterial peptide and host-cell modulator. Biochim. Biophys. Acta (BBA)-Biomembranes. 1858, 546–566.
Xu Z., Davis H. M., Zhou H. (2013). Rational development and utilization of antibody-based therapeutic proteins in pediatrics. Pharmacol. Ther. 137, 225–247.
Xuan J., Feng W., Wang J., Wang R., Zhang B., Bo L., et al. (2023). Antimicrobial peptides for combating drug-resistant bacterial infections. Drug Resistance. Updates. 68, 100954.
Yamagishi J., Kojima T., Oyamada Y., Fujimoto K., Hattori H., Nakamura S., et al. (1996). Alterations in the DNA topoisomerase IV grlA gene responsible for quinolone resistance in Staphylococcus aureus. Antimicrob. Agents chemother. 40, 1157–1163.
Yamamoto B. J., Shadiack A. M., Carpenter S., Sanford D., Henning L. N., Gonzales N., et al. (2016). Obiltoxaximab prevents disseminated Bacillus anthracis infection and improves survival during pre-and postexposure prophylaxis in animal models of inhalational anthrax. Antimicrob. Agents chemother. 60, 5796–5805.
Yao J., Weng Y., Dickey A., Wang K. Y. (2015). Plants as factories for human pharmaceuticals: applications and challenges. Int. J. Mol. Sci. 16, 28549–28565.
Yazici A., Ortucu S., Taskin M., Marinelli L. (2018). Natural-based antibiofilm and antimicrobial peptides from microorganisms. Curr. Topics. med. Chem. 18, 2102–2107.
Yeung A. T. Y., Gellatly S. L., Hancock R. E. W. (2011). Multifunctional cationic host defence peptides and their clinical applications. Cell. Mol. Life Sci. 68, 2161–2176.
Yount N. Y., Yeaman M. R. (2005). Immunocontinuum: perspectives in antimicrobial peptide mechanisms of action and resistance. Protein Pept. Lett. 12, 49–67.
Zasloff M., Martin B., Chen H.-C. (1988). Antimicrobial activity of synthetic magainin peptides and several analogues. Proc. Natl. Acad. Sci. 85, 910–913.
Zech N., Schrödinger M., Hansen E. (2022). Avoidance of nocebo effects by coincident naming of treatment benefits during the medical interview for informed consent—Evidence from dynamometry. Front. Psychol. 13, 923044.
Zhang Q.-Y., Yan Z.-B., Meng Y.-M., Hong X.-Y., Shao G., Ma J.-J., et al. (2021). Antimicrobial peptides: mechanism of action, activity and clinical potential. Military. Med. Res. 8, 1–25.
Zhang H., Zheng J., Cheng W., Mao Y., Yu X. (2022). Antibacterial activity of an anti-lipopolysaccharide factor (MjALF-D) identified from kuruma prawn (Marsupenaeus japonicus). Fish. Shellfish. Immunol. 127, 295–305.
Keywords: antimicrobial peptide, monoclonal antibodies, bacteria, AMR, AMP, antibiotic resistance
Citation: Halawa M, Akantibila M, Reid BE and Carabetta VJ (2023) Therapeutic proteins have the potential to become new weapons in the fight against antibiotic resistance. Front. Bacteriol. 2:1304444. doi: 10.3389/fbrio.2023.1304444
Received: 29 September 2023; Accepted: 28 November 2023;
Published: 12 December 2023.
Edited by:
Yanjiao Zhou, UCONN Health, United StatesReviewed by:
Timothy Meredith, The Pennsylvania State University (PSU), United StatesCopyright © 2023 Halawa, Akantibila, Reid and Carabetta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Valerie J. Carabetta, Y2FyYWJldHRhQHJvd2FuLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.