 Bailey H. Duhon
Bailey H. Duhon Eric C. Bielefeld
Eric C. Bielefeld Yin Ren1,4*
Yin Ren1,4* Jerusha Naidoo
Jerusha Naidoo
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Audiol. Otol., 17 July 2024
Sec. Auditory Science
Volume 2 - 2024 | https://doi.org/10.3389/fauot.2024.1423853
This article is part of the Research TopicAdvancing Audiology Practice and Knowledge at the World Congress of Audiology 2024: A Comprehensive CollectionView all 12 articles
Greater understanding of the molecular intricacies of acquired and hereditary hearing loss has spurred considerable advances in inner ear gene therapy. While approaches like cochlear amplification and cochlear implantation offer varying degrees of efficacy in restoring hearing function, there is an absence of FDA-approved pharmacotherapies targeting the underlying causes of hearing loss. Recent preclinical investigations have demonstrated promising outcomes in murine and non-human primate models, demonstrating efficient transduction and hearing recovery for both acquired and hereditary forms of hearing loss. This review provides a comprehensive analysis of the latest developments in gene therapy for hearing loss. Specifically, we focus on conditions characterized by sensory epithelium and spiral ganglion neuron dysfunction, encompassing both hereditary and acquired etiologies. We discuss recent preclinical advancements in cell-type-specific transduction strategies and highlight key findings from clinical trials exploring gene therapy interventions for hearing loss. Additionally, we address current limitations and offer insights into future directions for advancing gene therapy as a viable treatment option for individuals with hearing loss.
Hearing loss is the most common sensory impairments in humans, affecting more than 1.5 billion people worldwide. Among those, more than 400 million children and adults suffer from severe to profound hearing loss (Collaborators, 2021). Children who are born deaf or have difficulty hearing who do not receive early intervention are at a higher risk for poor literacy outcomes and a lower quality of life (Hrastinski and Wilbur, 2016; Panda et al., 2019; Ronner et al., 2020). Hearing loss in adults has also been associated with higher rates of loneliness, anxiety, and dementia (Huang et al., 2023).
Congenital, or hereditary, hearing loss (HHL) is a well-studied and significant cause of hearing loss in children (Shave et al., 2022), while age-related hearing loss, noise-induced (NIHL), and chemotherapy-induced ototoxicity (CIHL) are significant causes of acquired adult sensorineural hearing loss (SNHL). The primary cause of functional impairment in most cases is the degeneration of sensory epithelium (outer and inner and inner hair cells [OHC/IHCs]), spiral ganglion neurons (SGNs), and the peripheral auditory nerve synapses between them. Despite the high prevalence of these hearing disorders, there are currently no U.S. Food and Drug Administration (FDA)-approved pharmacotherapies available to prevent or reverse SNHL. Clinically, hearing amplification and cochlear implants (CI) are the only treatment options available, however they do not target the underlying molecular driver of the disease.
Gene therapy is a promising therapeutic option for hearing loss that has recently been shown to restore hearing in pediatric patients with monogenic HHL due to OTOF pathogenic variants (Lv et al., 2024; Simons et al., 2024). The unique anatomy of the inner ear lends itself to viral-based gene therapy, is a relatively isolated fluid compartment which lends itself to limited immunogenicity and has a small number of non-dividing target cells. The success of monogenic gene therapy is based on a multitude of preclinical studies in murine and non-human primate (NHP) models, but the focus has primarily been on IHC transduction in monogenic hearing loss. There remains a lack of information regarding treatment for SGN-degenerating conditions. Moreover, only negligible SGN transduction has been accomplished in adult mammals. This deficiency in effective targeting of SGNs limits the potential success of treatment options.
In this review, we aim to explore the use of viral vectors, delivery routes, and clinical implications of gene therapy in the mammalian inner ear. Our objective is to provide a comprehensive evaluation of the available evidence regarding cochlear HC and SGN transduction and the potential benefits and challenges associated with gene therapy for hereditary and acquired hearing loss.
Monogenic, non-syndromic hereditary hearing loss, resulting from singular genetic pathogenic variants, is a prominent pathology within the spectrum of inherited auditory impairments/HHL. Currently, pathogenic variants in approximately 120 genes have been associated with non-syndromic hearing loss in humans (Sharma et al., 2023; Walls et al., 2024). Unraveling the genetic intricacies inherent to the normal development and function of the inner ear and vestibulocochlear nerve has been accelerating at a fast pace, opening new potential therapeutic avenues for gene therapies.
Non-syndromic hearing loss exhibits varying inheritance patterns—autosomal dominant, autosomal recessive, x-linked, and mitochondrial/maternal—with autosomal recessive predominating with more than 70% of known cases (Walls et al., 2024). In addition, de novo pathogenic variants are an important cause of HHL in patients with no familial history (Klimara et al., 2022).
Autosomal dominant, non-syndromic hearing loss (DFNA) typically exhibits heterogeneity, with both males and females being affected from early childhood to adulthood, with the onset of most cases being post-lingual (Aldè et al., 2023). Approximately 80 loci on 50+ genes have been identified, with pathogenic variants in MYO6 (DFNA22), encoding for a crucial motor protein (Oka et al., 2020), and TECTA (DFNA8/12), which encodes for a tectorial membrane component named α-tectorin (Hildebrand et al., 2011), being the most identified in a European cohort (Del Castillo et al., 2022). Additionally, identified genes predominantly affect supporting cell (SC) function (GJB2 [DFNA3A]) (Denoyelle et al., 1998; Wang et al., 2017), HC function with KCNQ4 (DNFA2A) (Arnett et al., 2011) or SGN development and function, such as COCH (DFNA9) (Danial-Farran et al., 2021) and POU4F3 (DFNA15) (Vahava et al., 1998).
Autosomal recessive, non-syndromic hearing loss (DFNB), in contrast to DFNA, is associated with severe to profound hearing loss with most patients having a pre-lingual onset, however some may present with a gradual progression of hearing loss (Sharma et al., 2023). Approximately 85 causative genes have been identified and associated with DFNB, with the GJB2 (DFNB1A) pathogenic variants the most common at an estimated 60% of DFNB cases (Sloan-Heggen et al., 2016; Del Castillo et al., 2022; Walls et al., 2024). GJB2 encodes for connexin 26 (a gap junction protein) in SCs that is crucial for maintaining potassium homeostasis in the HCs. Other causative genes include OTOF (DFNB9) (the most studied gene in gene therapy clinical trials), TMC1 (DFNB7/11), GJB6 (DFNB1B), MYO7A (DFNB2), and SLC26A4 (DFNB4) (Doll et al., 2020; Del Castillo et al., 2022). Specifically, OTOF encodes for otoferlin, a synaptic protein involved in glutamate release from the HCs, thus allowing for electrical conduction of the SGN and propagation of auditory stimuli.
X-linked and mitochondrial inherited pathogenic variations make up a small proportion of non-syndromic hearing loss and have been elegantly discussed elsewhere (Vona et al., 2015; Sloan-Heggen et al., 2016; Walls et al., 2024).
Taken together, non-syndromic hearing loss is a prime candidate for gene therapy applications. By targeting and correcting the specific pathogenic variation responsible for inner ear dysfunction, there is potential to restore normal hearing function. These approaches hold potential to provide treatments tailored to individual patients, representing a significant advancement in the field of audiology and otology. Clinical trials for these applications will be discussed further below.
Syndromic hearing loss is characterized as SNHL that occurs as part of a larger syndrome, often characterized by additional sensory and developmental dysfunction involving the neurological, cardiovascular, renal, and ocular systems. As with non-syndromic causes, most syndromic hearing loss causes are hereditary and can be linked to genetic pathogenic variants that are passed in an autosomal dominant, autosomal recessive, x-linked, or mitochondrial inheritance pattern. Moreover, specific genes, like SLC26A4, have been identified in both syndromic and non-syndromic hearing loss (Honda and Griffith, 2022). While there are a numerous conditions associated with syndromic hearing loss, a few have been well characterized in the literature including Usher, Pendred, and Jervell and Lange-Nielsen. Additional syndromes like neurofibromatosis type two-related schwannomatosis are also associated with syndromic hearing loss but are not primarily characterized by intrinsic inner ear dysfunction (Ren et al., 2021; Mohamed et al., 2023).
Usher syndrome is characterized by bilateral SNHL and retinitis pigmentosa with and without vestibular symptoms (Bonnet et al., 2016; Toms et al., 2020; Delmaghani and El-Amraoui, 2022). In Usher syndrome there is extensive genetic heterogeneity, with three clinically important phenotypes (Type 1, 2, and 3), with further subclassifications of the types based on the causative pathogenic variant, i.e. Usher syndrome Type 1B and MYO7A (Bonnet et al., 2016). The causative genes are broadly associated with ciliary function and are highly expressed in the inner ear and eye, which may explain the clinical phenotypes. Type 1 Usher syndrome (USH1) presents as congenital SNHL, vestibular dysfunction, and impaired visual acuity in adolescence (Blanco-Kelly et al., 2015). A genotype-phenotype relationship has been identified for USH1 with the predominate cause being a pathogenic variant in MYO7A (Le Quesne Stabej et al., 2012; Bonnet et al., 2016). MYO7A encodes for a crucial myosin motor protein that establishes the tip-link tension between the HC stereocilia, allowing for mechano-electrical transduction (Bonnet et al., 2016; Li et al., 2020). USH1C, another causative gene for USH1, encodes for the protein harmonin. It has been the focus of recent preclinical studies for gene therapy using antisense oligonucleotides (Pan et al., 2017; Wang et al., 2020). Harmonin is involved in the tip and lateral links of HC stereocilia and acts as an anchoring scaffold protein. Without tip and lateral links, the HC stereocilia cannot function properly, inhibiting mechano-electrical transduction and causing SNHL. Type 2 Usher syndrome (USH2) is clinically similar to USH1, although notably lacks vestibular findings in most patients. Similar genotype-phenotype relationships were identified and suggest WHRN and USH2A as causative genes (Le Quesne Stabej et al., 2012; Blanco-Kelly et al., 2015; He et al., 2020). WHRN and USH2A, which encode for whirlin and usherin, respectively, are components of the ankle-link protein complex which is crucial for the normal development of HCs (Wang et al., 2023a). Additionally, USH2 is the most common subtype of Usher syndrome, responsible for almost half of all confirmed cases (Bonnet et al., 2016). Type 3 (USH3) is unique in that the onset of hearing loss is post-lingual, with ophthalmologic findings presenting later in life (Delmaghani and El-Amraoui, 2022). Vestibular findings may also be found in USH3 but are clinically heterogenous (Delmaghani and El-Amraoui, 2022). This type of Usher syndrome is mainly associated with CLRN1, encoding for a protein that modulates the transduction efficiency of the excitatory ribbon synapses between HCs and SGNs.
Pendred syndrome is characterized by bilateral, congenital SNHL with an enlarged thyroid gland (goiter) often found in adolescence (Wémeau and Kopp, 2017). Histologically, inner ear malformations include an enlarged vestibular aqueduct (Honda and Griffith, 2022; Saeed et al., 2023). Genetic analysis has demonstrated that pathogenic variations in SLC26A4, FOXI1, and KCNJ10 may be causative (Wu et al., 2022). SLC26A4 encodes for pendrin, a non-specific anion and base (importantly HCO) exchanger protein in the epithelial cells of the inner ear. Pendrin dysfunction may lead to electrolyte and osmotic disturbances, causing enlargement of the vestibular aqueduct and loss of endocochlear potential, thereby causing SNHL.
Patients with Jervell and Lange-Nielsen syndrome (JLNS) develop congenital bilateral SNHL and cardiac arrhythmias due to ion channelopathy resulting in prolonged QT syndrome (Schwartz et al., 2006). Patients with JLNS have a high mortality rate due to risk of ventricular arrythmias, specifically Torsades-de-Pointes (Goldenberg et al., 2006). Pathogenic variations in KCNQ1 and KCNE1, potassium channel-encoding genes, have been identified as causative for JLNS (Vojdani et al., 2019; Walls et al., 2024). These channels are required for potassium movement across the stria vascularis, forming the endocochlear potential.
Currently, preclinical and clinical studies have focused on the non-auditory dysfunction associated with these syndromes, for example, the majority of USH-related studies have focused on ocular gene therapy (Nuzbrokh et al., 2021). Additional studies targeting the inner ear would benefit these patients with SNHL.
NIHL has emerged as a prevalent form of auditory impairment caused by prolonged or acute exposure to acoustic stimuli. Moreover, NIHL is the primary preventable cause of hearing loss (Le et al., 2017). NIHL may be caused by both the classical high intensity exposure (i.e., gunshot, blast exposure, etc.), primarily associated with hair cell degeneration, and more moderate long-term noise exposure (i.e., occupational associated), which might be associated with cochlear synaptopathy and damage to the ribbon synapses. Exposure to high intensity exposure noise, whether singular or continuous, poses a risk for temporary or permanent hearing damage. Temporary NIHL manifests as a reversible temporary threshold shift (TTS), in the absence of neuronal cell death, whereas permanent NIHL leads to permanent threshold shift (PTS) often accompanied by sensory and neuronal cell death within the auditory system (Kujawa and Liberman, 2009; Early et al., 2022). PTS is often due to damage to the HCs, classically for high intensity exposure, and subsequent SGN degeneration (McGill and Schuknecht, 1976; Kujawa and Liberman, 2009). Additionally, exposure to high-intensity noises is thought to cause the HCs to release excess glutamate (Puel et al., 1998), which can lead to excitotoxicity of SGNs, while further impairment to hearing is due to destruction of the ribbon synapses between IHCs and SGNs (Kujawa and Liberman, 2009; Lin et al., 2011; Wagner and Shin, 2019; Hu et al., 2020). Finally, exposure to high-intensity noises can cause damage to the myelin sheath that surrounds the SGNs, also contributing to hearing loss (Brown and Hamann, 2014), particularly the processing of complex sounds in competing background noise. Therefore, in addition to HCs, therapeutic approaches aimed at treating NIHL should focus also on peripheral ribbon synapses and SGNs.
Hearing loss as a side effect of cancer treatment can severely impact the patient's quality of life. Cisplatin is a commonly used chemotherapeutic agent and causes significant ototoxicity in up to 80% of adults and 50% of pediatric patients (Moke et al., 2021). The exact mechanism of cisplatin ototoxicity is still under active investigation, however damage to the stria vascularis and disruption of cochlear energy metabolism, which can then lead to loss of the sensory epithelium, has been implicated in cisplatin- and carboplatin-induced pathophysiology (Liu et al., 2021; Wang et al., 2023b). Additionally, cisplatin has been shown to increase the secretion of cytotoxic inflammatory molecules (So et al., 2007) and creation of reactive oxygen species (Yu et al., 2019), thereby causing DNA damage, all which lead to activation of apoptotic pathways in the cells of the stria vascularis, HCs, and SGNs (Breglio et al., 2017; He et al., 2022). Interestingly, studies have demonstrated that HC and SGN loss may occur at different time points (van Ruijven et al., 2004, 2005). The set of studies published by van Ruijven et al. demonstrated that cisplatin interferes with SGN function and causes aberrant loss of SGNs, resulting in the elevation of compound action potential and auditory brainstem response (ABR) thresholds, as well as detachment of the myelin sheath of the type-I SGNs (a sign of degeneration) within 1 week of treatment initiation (van Ruijven et al., 2004, 2005). Moreover, the early time point (< 1 week) of SGN degeneration and malfunction following cisplatin treatment indicates the importance of the timing of therapeutic intervention (van Ruijven et al., 2004, 2005; Yu et al., 2019).
Human gene therapy for hearing loss involves introduction of genetic material into target cells to address diseases associated with dysfunction of HCs, SGNs or SCs caused either by pathogenic genetic variations or external factors. Gene delivery may be accomplished using viral and non-viral vectors. Among these, viral vectors, particularly adeno-associated viruses (AAVs), have emerged as the most extensively studied and efficacious vectors. AAVs show significant promise because of their higher rates of transduction efficiency than non-viral delivery, lack of pathogenicity, persistence of gene expression, availability of various serotypes (which specifies cellular tropism), and low risk of insertion mutagenesis due to lower host DNA integration (Pupo et al., 2022).
There are three main types of viral vector-based gene therapy: gene editing, gene silencing, and gene replacement (Wang et al., 2019; Petit et al., 2023). Gene replacement involves adding a functional gene into cells with defective or missing copies, aiming to restore normal cellular function (Petit et al., 2023; Lv et al., 2024). Gene editing enables precise modification of genetic material, often utilizing the CRISPR-Cas9 or base editing systems (Noh et al., 2022; Petit et al., 2023). Meanwhile, gene silencing inhibits the translation of a gene into proteins, achievable through mechanisms such as antisense oligonucleotides (Wang et al., 2020; Petit et al., 2023). Of these approaches, gene replacement of a pathogenic variant is the most widely used for treating HHL.
Gene replacement is the primary therapeutic approach studied in murine models of genetic hearing loss. The genes VGLUT3 (Akil et al., 2012; Zhao et al., 2022a; Mathiesen et al., 2023), GJB2 (Yu et al., 2014; Iizuka et al., 2015; Guo et al., 2021), GJB6 (Miwa et al., 2013; Crispino et al., 2017; Zhang et al., 2022), MSRB3 (Kim et al., 2016), TMC1 (Askew et al., 2015; Gao et al., 2018; Nist-Lund et al., 2019; Wu et al., 2021a; Marcovich et al., 2022), KCNQ1 (Chang et al., 2015), USH-associated genes (USH1C (Pan et al., 2017), USH1G (Emptoz et al., 2017), WHRN (Chien et al., 2016; Isgrig et al., 2017)), OTOF (Akil et al., 2019b; Al-Moyed et al., 2019; Rankovic et al., 2020; Tang et al., 2023; Zhang et al., 2023; Qi et al., 2024a,b; Wang et al., 2024), STRC (Shubina-Oleinik et al., 2021), LHFPL5 (György et al., 2017), PJVK (Delmaghani et al., 2015; Lu et al., 2022), KCNE1 (Wu et al., 2021b), and SYNE4 (Taiber et al., 2021) have all been studied for their efficacy and safety as preclinical, viral vector-mediated, gene replacement therapeutic targets.
Viral-mediated gene suppression and editing are other approaches that have been studied as a treatment for deafness related to TMC1 (György et al., 2019b; Yoshimura et al., 2019), KCNQ4 (Noh et al., 2022), and MYO6 (Xue et al., 2022). Noh et al. utilized CRISPR/Cas9 to edit KCNQ4, while Gyorgy et al. utilized CRISPR/Cas9 to disrupt and suppress the mutant allele associated with TMC1. Liposome-mediated delivery (Tao et al., 2023) and systemic delivery of antisense oligonucleotides (Lentz et al., 2013; Wang et al., 2020) have also been studied for both syndromic and non-syndromic HHL.
These studies demonstrated excellent transduction efficiency, low toxicity, and variable recovery of auditory function in animal models. Gene replacement therapies targeting OTOF and SLC17A8-related deafness have been highly successful in a number of preclinical models, with hearing restored to wild-type (WT) ABR thresholds (Akil et al., 2012; Qi et al., 2024b). A few studies, however, did not demonstrate recovery of the ABR Wave 1, suggesting poor survival of SGNs despite general ABR threshold improvement (Akil et al., 2019b; Zhao et al., 2022a). Moreover, some studies involving AAV-mediated TMC1 and OTOF gene replacement demonstrated near complete hearing recovery to WT thresholds in neonatal mice but reported mixed success in adult mouse models (Nist-Lund et al., 2019; Zhang et al., 2023). It is important to note that these efficacy studies were primarily conducted on neonatal mice, which generally, showed better response than mature models. This raises concerns for potential applicability to adult-onset hearing losses. The murine inner ear does not fully develop until around the 2nd week of postnatal life, whereas the human cochlea develops from gestational week 4 to approximately gestational week 30 (Kamiya et al., 2001; Johnson Chacko et al., 2019). Therefore, studies on mature mice may better represent the translational capability for treating postnatal human patients. The neonatal inner ear appears far more amenable to efficient AAV-mediated gene transfer than the adult inner ear (Transduction ranged from 100% of IHCs and ~75%% of SGNs in neonatal mice to 100% of IHCs but < 20% of SGNs in adult mice) (Duarte et al., 2018; Richardson et al., 2021). Those studies that utilized mature murine models for functional gene therapy studies are summarized in Table 1.
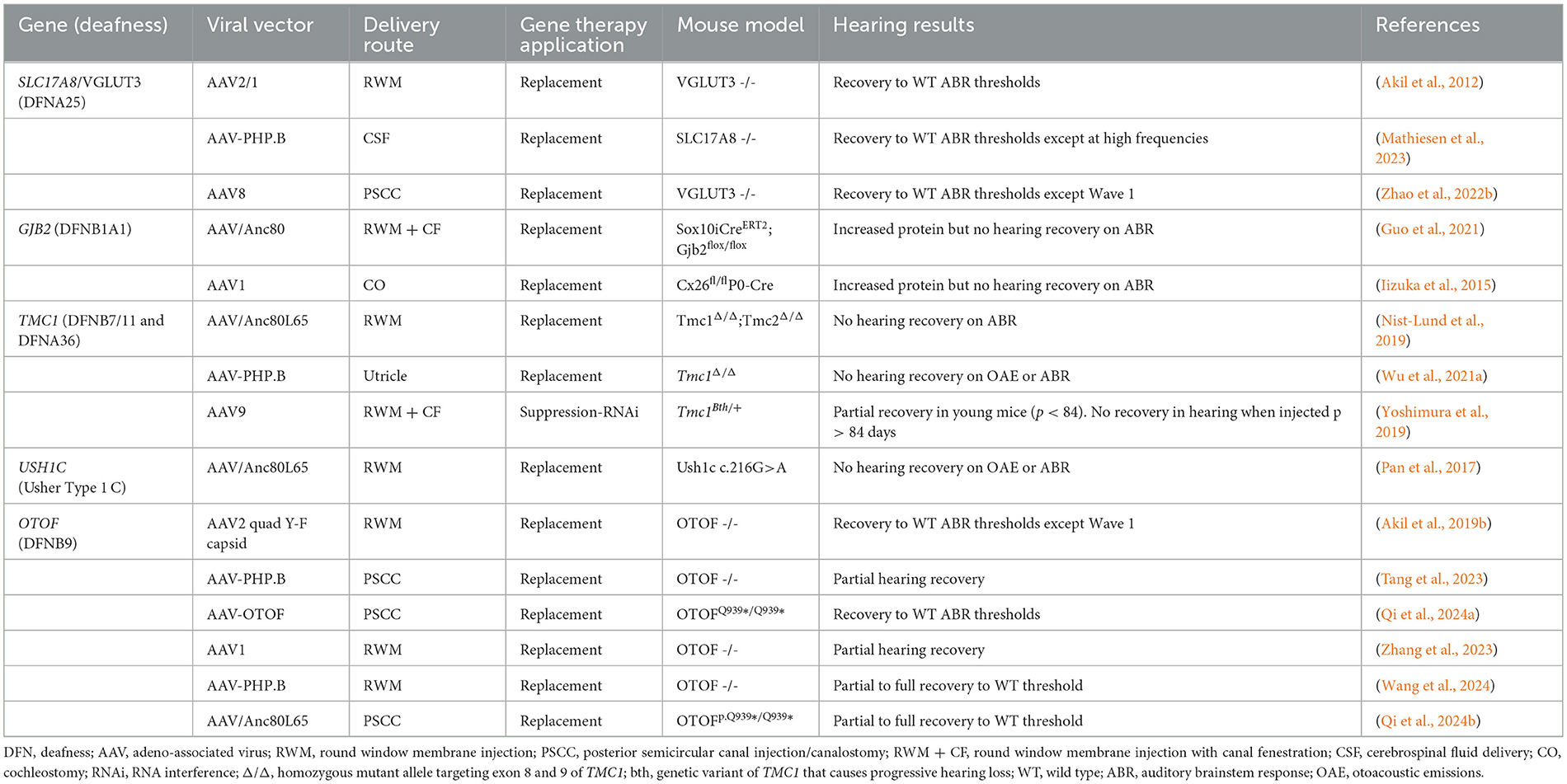
Table 1. Preclinical gene therapy studies for hereditary hearing loss in mature (≥postnatal day 12) murine models.
In acquired hearing loss, the focus is primarily on preventing the degeneration of sensory HCs and SGNs, or regenerating damaged inner ear cells. There are multiple potential targets, and neurotrophins (NTs), proteins that support the growth, survival, and differentiation of hair cells and neurons, are one such avenue to restore or prevent hearing loss.
NTs activate signaling pathways critical for maintaining neuronal function and plasticity (Zigmond et al., 2012) and facilitate proper development of the auditory system (Harasztosi et al., 2020). Among these, brain-derived neurotrophic factor (BDNF), and NT3 are the most well characterized due to their role in cochlea development, with a substantive number of studies also demonstrating the therapeutic potential of glial cell-derived neurotrophic factor (GDNF). A distinction must be made between the developing and mature mammalian cochlea as there are notable differences in both NT availability and receptor expression between the two ages (Johnson Chacko et al., 2017). Specifically, the high affinity receptors for BDNF and NT3, TrkB and TrkC, are strongly expressed in the cochlea from birth to adulthood, but the BDNF ligand itself notably decreases with age (Johnson Chacko et al., 2017). In contrast, GDNF is not present at birth, but appears in the first postnatal week and into adulthood along with its receptor, GFR1a (Stöver et al., 2000). In addition to temporal differences, whether there may be spatial differences in receptor availability along the tonotopic axis of the cochlea and on certain cell types, is yet to be studied. These factors, combined with the interchangeability/cross-reactivity of some NT ligands and receptors, necessitate further studies for the selection of ideal NTs.
Despite these complexities, local exogenous delivery of AAV vectors expressing NTs may provide protective and/or regenerative effects on both SGNs and HCs in the cochlea. Local delivery of the NT proteins themselves has been extensively studied through the use of osmotic minipumps for delivery into the scala tympani (Khalin et al., 2015). However, this delivery method is not likely to be an effective long-term strategy due to transient protection of SGNs due to the relatively short half-lives of the proteins. The protective effects of NTs on SGNs appears to only last for the duration of treatment. These findings have been widely confirmed by studies in other neuronal systems (Montero and Hefti, 1988; Gillespie et al., 2003). Additionally, indwelling cannulas pose significant risk for infection and is a burdensome to the patients. Because it is a one-time treatment, viral vector mediated NT delivery is therefore being intensively investigated (Ramekers et al., 2012). Moreover, Mukherjee et al. demonstrated that local delivery of AAV2 (quad Y-F capsid)-mediated BDNF can recover noise-induced BDNF gene downregulation, ABR wave I amplitude reduction, and synapse loss in a guinea pig model (Mukherjee et al., 2022). Furthermore, AAV-BDNF may be protective in murine models of GJB2-related deafness (DFNB1A) (Takada et al., 2014). Additionally, Hashimoto et al. has showed that AAV-mediated overexpression of NT-3, another NT, in the cochlea following canalostomy injection can provide protection against noise-induced synaptopathy, possibly through the enhancement of synaptic function and the preservation of ribbon synapses between IHCs and SGNs (Hashimoto et al., 2019). However, only AAV-NT3 delivered prior to noise exposure was effective at improving synapse survival. This was corroborated by Bowers et al. who found that NT-3 transduction utilizing a herpes simplex type 1 viral vector attenuated the loss of SGNs following cisplatin therapy (Bowers et al., 2002). Furthermore, Leake et al. provided evidence that AAV-mediated NT gene therapy, specifically using either AAV2-hBDNF can effectively enhance the survival of cochlear SGNs in neonatally deafened cats (Leake et al., 2019). These studies collectively indicate the potential of AAV-mediated NT delivery transduction in the inner ear. However, NT3 delivered onto the RWM without use of a viral vector has shown success in mice, demonstrated improved ABR Wave 1 thresholds and moderate regeneration of the cochlear HC-SGN interface (Suzuki et al., 2016).
Additionally, Fukui et al. demonstrated SGN peripheral fiber growth and epithelial expansion after adenovirus mediated BDNF gene therapy in mature (P28) POU4F3-/- mice, suggesting potential virus-mediated regenerative capacity (Fukui et al., 2012). These findings were corroborated in a deafened murine model that demonstrated SGN fiber regrowth into the basilar membrane following viral-mediated BDNF delivery (Shibata et al., 2010).
Studies have also demonstrated the therapeutic potential of AAV-mediated GDNF expression for mitigating and/or reversing hearing loss in various animal models with NIHL and ototoxicity-mediated hearing loss/CIHL (Shoji et al., 2000; Chen et al., 2003; Shibata et al., 2007; Liu et al., 2008). Not surprisingly, utilization of very high titers of AAV-GDNF of 1.8 × 1011 to 3.6 × 1011 vg (1–2 μL injected) has been reported to cause toxicity in neonatal animals, denoting the importance of sensible dosing strategies, particularly when considering scaling up to larger mammals (Akil et al., 2019a). The study by Leake et al. showed an interesting difference in the efficacy of AAV2-BDNF and AAV5-GDNF. While both vectors elicited a neurotrophic effect on SGN survival in neonatally deafened cats, AAV5-GDNF caused undesirable ectopic fiber sprouting whereas AAV2-BDNF was not associated with ectopic sprouting. Given the lack of an AAV5-BDNF group, it is impossible to draw a clear conclusion about the toxicity of GDNF. Both vectors utilized strong, constituently active promoters (the AAV5 vector utilized the hybrid cytomegalovirus/chicken beta-actin (CBA) promoter and AAV2 utilized the CAG promoter), however the titer differences are significant. AAV2-BDNF was injected at a titer of 3 × 1012 vg/mL (10 μL injected), while AAV5-GDNF was injected at a titer of 1.8 × 1014 vg/mL (10 μL injected). The higher dose of the AAV5-GDNF may explain the adverse events reported in this study (Leake et al., 2019). Moreover, an important factor that is often overlooked is that AAV5 has the capacity to transduce antigen presenting cells in the brain which can lead to a full immune response when a non-self-protein is expressed, whereas AAV2 only transduces neurons, which are not antigen presenting (Samaranch et al., 2017). The Akil et al. (2019a) and Leake at al. (2019) studies involved expression of a human GDNF protein in cats and mice, so it is therefore possible that serotype selection and expression of a non-self-protein could have contributed to these findings. Of note, an immune response following transduction of antigen presenting cells has also been reported with AAV9 (Ciesielska et al., 2013; Samaranch et al., 2014). The human cochlea contains various types of peripheral glial cells that envelop the cochlear nerve, the cell bodies in the spiral ganglia, and the peripheral processes in the osseous spiral lamina. Careful selection of a viral serotype with selective tropism for HCs and SGNs that does not require a high titer for efficient transduction with minimal tropism for antigen presenting cells will be important. Taken together, these results indicate that viral vector mediated NT delivery holds promise for the treatment of acquired hearing loss, but caution is warranted regarding serotype and dose selection.
NTs are not the only potential targets, with viral-mediated delivery of apoptotic inhibitors into the cochlea of rats treated with cisplatin leading to an attenuation of HC degeneration and overall improved ABR thresholds, if administered prophylactically (Cooper et al., 2006). While this study did not perform immunohistochemistry for SGNs specifically, the ABR threshold improvements suggests at least moderate SGN survival (Cooper et al., 2006). Subsequently, the injection of this viral vector through the round window membrane (RWM) in a follow-up study by Chan et al. showed a decrease in efficacy in high-frequency, basal OHCs (Chan et al., 2007). However, administering the treatment vector 2 months prophylactically may pose challenges in clinical scenarios. Bu et al. demonstrated the success of AAV treatment at the same time as cisplatin therapy using a viral vector that expresses c-Myb, a transcription factor involved in cell survival (Bu et al., 2022). The authors found that co-treatment of cisplatin with AAV gene therapy decreased OHC loss in the basal turn of the cochlea, which is known to be commonly affected by cisplatin therapy (Bu et al., 2022). These findings underscore the potential of AAV-mediated gene therapy for preserving auditory function in patients at high risk for noise exposure or undergoing chemotherapy treatments with high ototoxicity.
The delivery of gene therapies into the inner ear requires careful consideration of various factors, including the delivery route, transduction efficiency, potential for immune responses and cellular tropism. Both infection, the ability of the virus to enter and survive in the cell, and transduction, the ability of the viral vector to introduce genetic material into the cell, are important components of transduction efficiency for gene therapy. The inner ear is comprised of two main fluids, perilymph and endolymph (Glueckert et al., 2018). However, these fluid compartments are not as isolated and immunologically privileged as previously thought (Keithley, 2022). First, the inner ear is highly vascularized with arterial flow from the labyrinthine artery but is sequestered by the blood-labyrinth barrier (Mei et al., 2020). Perilymph is connected to the cerebrospinal fluid (CSF)-containing space via the cochlear aqueduct, while endolymph circulates to/from the endolymphatic sac, where it is theorized to interact with the peripheral lymphatic system (Glueckert et al., 2018; Salt and Hirose, 2018). Additionally, the neural component of the inner ear provides a direct route to and from the cochlear nucleus, olivary complex and auditory cortex (Salt and Hirose, 2018).
Viral vectors have been administered via either intracochlear (RWM, cochleostomy) or intralabyrinthine (posterior semi-circular canal (PSCC), endolymphatic sac, utricle, and oval window/stapedectomy) pathways (Figure 1). Despite the success of these delivery routes in neonates, accessing the labyrinth directly is complex, and can lead to the damage and further degeneration of HCs and SGNs. Therapeutic delivery outside of the bony labyrinth, i.e., trans-tympanic and systemic injections, have had minimal success in adult models of hearing loss so far (Shibata et al., 2017). While AAV transduction of HCs has been extensively reported in neonates, most studies report low SGN transduction efficiency. The available data on SGN transduction are summarized in Table 2. The delivery routes, methods used, and other notable factors of delivering therapeutic agents to the inner ear are discussed below through the lens of AAV studies in the cochlea of healthy mice.
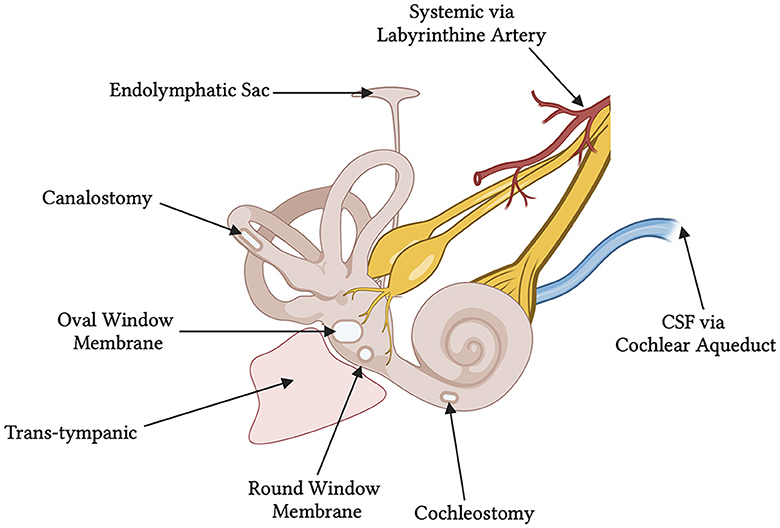
Figure 1. Schematic of the various delivery routes for gene therapy to the human inner ear. At the time of this review, the round window membrane has been utilized in two published clinical trials. The ossicles have been removed from the diagram showing an empty middle ear space and free oval window membrane. CSF, cerebrospinal fluid.
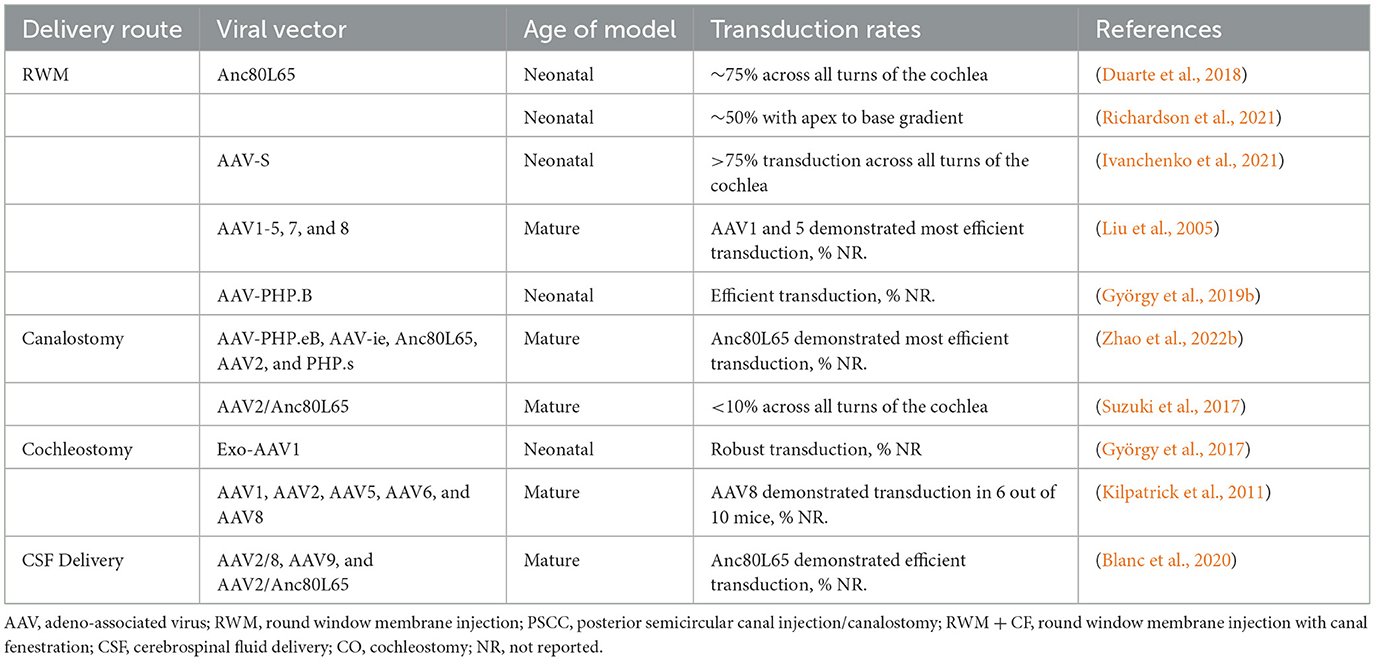
Table 2. SGN transduction based on delivery route and viral vector in murine models with healthy cochlea.
The most studied delivery route to the cochlea is through the RWM, which provides access to the perilymph fluid and cells of the cochlea. Due to the relatively low calcification of the temporal bone in neonatal mice vs. adults, the otic bulla may be pierced utilizing various techniques. In neonates, the bulla can be accessed with only a needle (Richardson et al., 2021), although drilling with a scalpel or a diamond drill is required for adult mice (Yoshimura et al., 2018). After gaining access to the otic bulla, the RWM can be easily penetrated using a variety of injection techniques (discussed further below). This method of accessing the RWM is similar to that used in other species like guinea pigs and even large mammals like NHPs (Wang et al., 2012; György et al., 2019a; Ivanchenko et al., 2020; Andres-Mateos et al., 2022). Micromanipulators with a micropump syringe attached to the micropipette allow for the highest level of control during the injection, although pressurizing the system with a Hamilton syringe has been done in other preclinical model studies (Richardson et al., 2021). For NHPs, a cortical mastoidectomy must be performed to expose the facial recess to allow for RWM access (György et al., 2019a,b; Ivanchenko et al., 2020; Andres-Mateos et al., 2022). György et al. (2019a) and Ivanchenko et al. (2020) have successfully used a 29-gauge needle to make the incision through the RWM. To inject the solution, a Hamilton syringe may be attached to the needle with a silicone tube. RWM delivery is associated with operative risks, most notably damage to the facial nerve, that must be considered when translating these findings into human studies. Approaches into the middle ear space where the RWM can be accessed with an intratympanic injection do not require a mastoidectomy.
While RWM delivery provides direct access to the perilymph in the scala tympani, care must be taken to avoid damaging the surrounding structures. Because the inner ear is not entirely a closed-fluid system, transient increases in pressure following injection of the viral vectors can lead to further HC and SGN degeneration. The closed bony cochlea is sensitive to pressure fluctuations and fluid shifts, which may be significant when performing a RWM injection. To combat this, multiple investigators have opted to create openings in the PSCC, to expose perilymph and allow for the dispersion of excess pressure (Suzuki et al., 2017; Yoshimura et al., 2018; Richardson et al., 2021). Regardless of technique used for fenestration of the canal, all investigators observed the efflux of perilymph fluid before injecting the viral vector through the RWM. For NHPs, fenestration of the oval window is favored over the PSCC, as it is thought to allow for fluid displacement and create a general flow of the administered vector toward the helicotrema (Andres-Mateos et al., 2022). A transcanal approach to RWM with stapes fenestration is has been demonstrated to be safe in the OTOF clinical trials (ChiCTR- 2200063181). Accessing the perilymph fluid through RWM injections, with or without further bony fenestrations, has the potential to deliver the viral vectors into the central nervous system via the cochlear aqueduct which connects the perilymph and is connected to the CSF. However, this may likely only an issue in rodents where the aqueduct remains patent, compared to in humans where the patency is theorized to decrease with age. Some investigators have minimized this risk by using HC-specific promoters (Ranum et al., 2023; Wang et al., 2024).
AAV delivery with the RWM injection has been well explored, providing high transduction rates for HCs although SGN rates vary significantly based on vector serotype. Here, we briefly discuss the use of commonly studied AAVs, including Anc80L65, AAV9-PHP.B, and AAV2, in the transduction of HCs and SGNs.
Anc80L65, an ancestral capsid discovered through computational approaches, is one of the most promising serotypes in inner ear gene therapy and has been predominately studied using RWM injections. This vector was first described by Landegger et al. (2017) demonstrating robust transduction of the IHCs and OHCs when injected at P1in mice. In this study, Anc80L65 injected at a dose of 1.7 × 109 vg in 1 μL outperformed AAV1, 2, 6, and 8, at lower doses than AAV1 and AAV2 (3.5 × 1010 vg in 1 μL and 6.0 × 109 vg in 1 μL). Interestingly, the authors noted Anc80L65 transduction in the contralateral uninjected cochlea in a subset of animals and cerebellar transduction, possibly reaching these areas via the cochlear aqueduct (Landegger et al., 2017). In an additional study utilizing neonatal mice, the Anc80L65 vector successfully transduces over 90% of IHCs across all cochlear turns with a relatively low titer (1.40 × 109 vg in 1 μL) (Yoshimura et al., 2018). In non-neonatal mice (P49), the transduction rate of IHCs was near 100%. No information is available regarding Anc80L65 mediated transduction of the cochlea when administered to mice >3 months old, when they are considered mature adults equivalent to humans aged 20 years old. Similarly, in guinea pigs, OHC transduction by Anc80L65 decreased from 90% to near < 70% from base to apex at a titer of 8.5 × 109 vg in 5 μL) (Wang et al., 2022). Further studies are needed to determine whether increased titers can facilitate transduction of OHCs throughout the cochlea in older mice and guinea pigs. The apex-to-base gradient of HC transduction with Anc80L65 has also been observed in adult NHPs aged 3–4 years. Andres-Mateo et al. noted a near 100% transduction rate of IHCs at the apex with a sharp decline to around 20% transduction rate at the base. In addition, transducing NHP OHCs was much less successful (< 5 positive OHCs observed in the 3,000 Hz section) and also exhibited the apex-to-base gradient of transduction (Andres-Mateos et al., 2022). The cause of this discrepancy is unknown; however, it is hypothesized that the decrease in basal OHC transduction is due to difficulty of the vector in crossing Reissner's membrane (from the scala vestibuli), requiring the vector to travel to the scala tympani where the vector can cross the basilar membrane. Another explanation is that viral tropism changes as a result of cell surface receptor differences between the base and apex, affecting the AAV's entry into the cell. Additionally, due to the tonotopic organization of the cochlea, the intrinsic cellular differences (metabolic activity, gene expression profiles, morphology, etc) between the apex and base as well as cellular specificity (tropism) of the vector itself could lead to the transduction gradient observed.
The AAV9-PHP.B vector has a modified capsule to increase transduction and reduce immunogenicity compared to the original AAV9 (Deverman et al., 2016). It has been demonstrated to effectively infect neonatal mouse HCs, with studies reporting between 60%−80% effectiveness for IHCs and between 40–60% for OHCs (György et al., 2019a; Ivanchenko et al., 2020). Infection of AAV9-PHP.B following RWM injection in a mature cochlea has been studied in OTOF-/- mice, and this approach demonstrated an infection rate of 80 to near 100% of IHCs (Wang et al., 2024). Two studies in juvenile NHPs have demonstrated transduction of HCs between 90–100% at relatively high doses of above 3–7 × 1011 vg (10–20μL injected), but there is a steep decline in transduction efficiency of ~50% at lower titers at 1 × 1011 vg (10μL injected) (György et al., 2019a; Ivanchenko et al., 2020).
AAV serotype 2 (AAV2) is the most extensively characterized vectors clinically for central nervous system disorders due to its high efficiency transduction of neurons (Christine et al., 2009; Pearson et al., 2021). In addition, AAV2 is one of the most common serotypes used in pseudotyping to create recombinant AAVs (rAAV), including the Anc80L65 discussed previously. Yoshimura et al. found that in neonatal mice, the transduction efficiency of HCs with AAV2 is titer dependent. The authors observed near 95% IHC transduction efficiency across all turns of the cochlea with a high titer of 3.9 × 1010 vg (1μL injected), however this was reduced to < 20% at titer of 1.4 × 109 vg (1μL injected) (Yoshimura et al., 2018). Using similar rAAVs at a titer of 1.0 × 1012 vg/mL (5–10μL injected onto the gelfoam), Wang et al. reported that the rAAV2 introduced through digestion of the RWM transduces almost 100% of IHCs in a guinea pig at the apex but was not successful at transducing IHCs at the base. In addition, < 20% of the OHCs of the guinea pig were transduced at the apex and close to 0% at the base (Wang et al., 2012). In the direct comparison of AAV9 and AAV2/Anc80L65 done by Yoshimura and colleagues, the Anc80L65 vector was clearly the more effective at transducing HCs in the neonatal mice at the same titer, although this study notedly lack a reporting of SGN transduction (Yoshimura et al., 2018). Additionally, Landegger et al. demonstrated that Anc80L65 was superior to AAV2 even when injected at a significantly lower titer (Landegger et al., 2017).
A variety of naturally occurring AAVs and novel AAV capsids have been used in inner ear gene therapy, including: AAV5, AAV8, and AAV-inner ear (AAV-i.e.,). The novel capsid AAV-i.e., described by Tan et al. (2019) successfully transduces almost all IHCs and a majority of OHCs across the neonatal mouse cochlea at a titer of 1 × 1010 vg (1.5 μL injected), but not at 3.6 × 109 vg (1.5 μL injected). This novel rAAV utilizes varying peptide sequences, including a peptide from the AAV.PHP.eB vector. Notably, this study observed high rates of transduction of cochlear support cells and vestibular cells, however the authors found that Anc80L65 was more efficient at transducing both HCs and SGNs in the neonatal model (Tan et al., 2019). Despite the success of the AAV-i.e., in neonatal mice, there were no complementary studies evaluating transduction efficiency completed in adult mammals. In a study conducted by Liu et al., AAV1-5, 7, and 8, were analyzed for their success rate in transducing adult mice HCs and SGNs (Liu et al., 2005). Out of the seven vectors used, AAV3 was the best at transducing IHCs. However, SGN rates have not been addressed in most publications, despite them being one of the vitally impacted cell types.
SGN transduction rates after RW delivery vary considerably across studies and are often not reported in other studies. Duarte et al. reported a high level of SGN transduction (74%) with Anc80L65 across the cochlea in neonatal mice (Duarte et al., 2018). Interestingly, the dose used by Duarte et al. (2018) (2.2 × 108 vg in 1μL) was significantly lower than that of Richardson et al. (2021) (9.23 × 108 vg in 1μL) who reported worse SGN transduction rates of approximately 50% with an apex-to-base decreasing gradient. The utilization of the RWM injection compared to a canalostomy injection might explain the difference in transduction rates seen in the study by Suzuki et al. (2017). It is likely that the RWM technique offers greater transduction of SGNs as compared to canalostomy. However, the studies by Richardson et al. and Duarte et al. may suggest a negative correlation with titer and SGN transduction with RWM injections.
SGN transduction is < 20% in adult mice with Anc80L65 (Richardson et al., 2021). In NHPs and guinea pigs, SGN transduction was even lower, with no transduction being detected at vector doses of 2.5 × 1011 vg (30 μL injected) and 27.69 × 108 vg (3 μL injected), respectively (Richardson et al., 2021; Andres-Mateos et al., 2022). Moderate titer levels of 2.2 × 1011-8.33 × 1012 vg/mL were utilized in these studies. It is unclear whether a higher titer could lead to higher SGN transduction in adult mammals, although in our experience higher titer Anc80L65 preparations have not been available via commercial suppliers.
Other vectors which have performed well in HC transduction, include AAV-PHP.B, which exhibits low SGN transduction rates even in neonatal mice. Moreover, the rates vary greatly in juvenile NHPs even with similar titers of 3–3.5 × 1011 vg (Ivanchenko et al. only injected 1.2μL v. Gyorgy et al. who injected 10μL) (György et al., 2019a; Ivanchenko et al., 2020). A study by Liu et al. comparing AAV1-5, AAV7, and AAV8 via RWM delivery, demonstrated that the AAV5 vector more efficiently transduced SGNs than the other serotypes (Liu et al., 2005). This finding is consistent with robust neuronal transduction observed with AAV5 with direct brain administration (Samaranch et al., 2017). Moreover, rAAV2 infused via RWM digestion in a guinea pig demonstrated >90% transduction efficiency for SGNs at the 1st turn of the cochlea and retained >80% throughout the whole cochlea (Wang et al., 2012). However, the age of the animals are unknown. Thus far it appears that Anc80L65 most potently transduces SGNs via RWM injection, although questions remain regarding the applicability to adult mammals including the effect of increasing titer.
Canalostomy can be used to create an opening in one of the semicircular canals, most often the PSCC due to the ease of access in rodents (Suzuki et al., 2017; Zhao et al., 2022b). The rationale behind this approach is that by accessing the semicircular canals, and not the cochlea itself, there may be less iatrogenic hearing loss caused by the injection procedure. A postauricular incision and blunt dissection allow for exposure of the bone overlying the PSCC in neonatal mice. Fenestration of the PSCC and direct injection can then be accomplished. To puncture the bone overlying the PSCC, 25–29-gauge hypodermic needles and a Bonn micro probe have been described (Suzuki et al., 2017; Tao et al., 2018; Isgrig et al., 2019; Zhu et al., 2021). For vector delivery, both polyimide and polyethylene tubes connected to glass micropipettes and Nanoliter Microinjection Systems have been used. Suzuki et al. connected the polyimide and polyethylene tubes in series for injection (Suzuki et al., 2017). To minimize perilymph leakage, many preclinical studies have utilized cyanoacrylate glue to seal the injection site. However, due to the small size of the PSCC and limited viewing window, there is a possibility for the injection needle to pierce the membranous labyrinth in addition to the bony labyrinth. This would then expose the vector to the endolymph and perilymph, instead of just the perilymph. Although canalostomy has been successfully used in a number of preclinical studies, one study reported that the procedure significantly increased the ABR threshold, which was also affected by perilymph leakage and speed of injection (Zhu et al., 2021). The PSCC is not as easily accessible in NHPs and humans due to its anatomic location and requires a mastoidectomy. The lateral/horizontal semicircular canal (LSCC) would be more easily accessible following a mastoidectomy in NHPs and humans, however a canalostomy of the LSCC has not been well studied in preclinical murine models due to anatomic constraints.
Similar to other delivery strategies, canalostomy delivery of AAV2/Anc80L65 transduces 100% of IHCs in mature mice (aged 6 to 10 weeks) (Suzuki et al., 2017; Tao et al., 2018). Anc80L65-injected cochleae also demonstrated a 70% OHC infection rate in these mice (Suzuki et al., 2017). Moreover, Isgrig et al. found that AAV2.7m8, a novel synthetic vector, transduced over 80% of IHCs and 70% of OHCs in neonatal mice, without causing an increase in ABR thresholds compared to non-injected control mice (Isgrig et al., 2019). Both Anc80L65 and AAV2.7m8 outperformed the other studied AAV serotypes including AAV1, AAV2, AAV8BP2, AAV8, AAV9, AAV.PHP, and AAV.ie (Suzuki et al., 2017; Tao et al., 2018; Isgrig et al., 2019; Zhu et al., 2021; Zhao et al., 2022b). However, the efficiency of AAV2.7m8 following a canalostomy vector infusion compared to the other vectors could be confounded due to the differing age of the models studied.
Suzuki et al. (2017) demonstrated < 10% SGN transduction with using AAV2/Anc80L65. Additionally, Zhao et al. (2022b) demonstrated that AAV-PHP.eB, AAV-i.e., AAV/Anc80L65, and AAV-PHP.S, but not AAV2, were able to infect SGNs at a moderate rate, despite no formal measurements. Other studies examined SCs but did not address SGNs.
The oval window is another entry point to deliver gene therapy vectors into the inner ear. However, it is mainly accessible in humans and NHPs. In mice, the oval window is covered by the stapedial artery, limiting the oval window's use in preclinical models of gene therapy for hearing loss. Although, Wang et al. (2022) was successful in transducing the inner ear of guinea pigs (>75% of IHCs) following oval window injection. Currently, the oval window has been limited to sites of fenestration, not injection, in NHP, preclinical studies (Ivanchenko et al., 2021; Andres-Mateos et al., 2022). The clinical trial CGF166, discussed below, is the only clinical trial that utilizes this route.
The cochleostomy delivery route provides direct access to the endolymph fluid in the scala media. Injection of viral vectors directly into the endolymph may assist with the transduction of HCs and SCs that are bathed in it. To perform cochleostomy in adult mice, the otic bulla must be surgically exposed before drilling into the bony portion of the lateral wall (Chien et al., 2015; Shu et al., 2016). Two approaches have been described, one being a similar surgical approach to the RWM injection site with a postauricular incision (Shu et al., 2016), and the second being a more caudal incision extending from the mandible to the clavicular area (Chien et al., 2015). As described earlier, the otic bulla may be perforated using a surgical drill at the space between the basal turn of the cochlea and the RWM. Once the periosteum at the base of the cochlea is removed, the scala media may be accessed without damaging the membranous portion of the lateral wall with the drill. In neonatal mice, the otic bulla can be pierced directly without need for drilling (Shu et al., 2016). Despite the ease of access to the endolymphatic fluid in the scala media of both models, the site of the injection invariably can lead to damage to the surrounding structures, including the stria vascularis. Destruction of the delicate cochlear structures during the injection and surgical approach, paired with leakage of the endolymph following breach of the scala media, can lead to the potential loss of endolymph, reduction of the endocochlear potential, and cellular injury (Kilpatrick et al., 2011; Chien et al., 2015; Shu et al., 2016). It is important to note that the scala media is a smaller fluid compartment compared to the perilymphatic space, limiting the volume of vector that can be administered.
AAV administration via cochleostomy has resulted in promising infection rates of both HCs and SCs, despite reports of moderate postoperative hearing loss due to iatrogenic trauma (Chien et al., 2015). In a direct comparison of RWM to cochleostomy delivery with AAV8 in mice aged 1–2 months, Chien et al. (2015) demonstrated modest infection rates of HCs and SCs between the two injection techniques, with IHCs vastly predominating (~30% of IHCs compared to ~12% of OHCs and SCs in cochleostomy). The infection rates were modest ~ 30% of IHC transduction at the base with a sharp reduction toward the apical turns of the cochlea at early time points following vector infusion, by 4 weeks the majority of HCs (only 32 IHCs per 400 mm-section examined were remaining) in the basal turns of the cochlea had died in the cochleostomy cohort. This loss of IHCs and OHCs was reflected hearing function of the mice, with the cochleostomy cohort demonstrating over a 20 dB increase in high-frequency ABR thresholds compared to the RWM cohort. These findings are corroborated in a study by Shu et al. (2016) that studied the infection rates of numerous AAVs via cochleostomy delivery in both neonatal and adult mice. In this study, the adult mice demonstrated extensive loss of OHCs at the basal turns of the cochlea, regardless of vector serotype. However, this did not occur in neonatal mice, who demonstrated minimal changes in ABR thresholds, suggesting the mature cochlea is more susceptible to iatrogenic injury during cochleostomy delivery. While the findings of Kilpatrick et al. may initially appear contradictory, as they indicate no significant changes in post-injection ABR of mature mice at low frequencies, a notable increase in ABR threshold (>20 dB) was observed in the high frequencies (Kilpatrick et al., 2011). This increase suggests potential damage to the basal turn HCs, aligning with the conclusions drawn by Shu et al. and Chien et al.
Both AAV8 and Anc80L65 serotypes have transduced the inner ear with high efficiency via cochleostomy delivery (Kilpatrick et al., 2011; Chien et al., 2015; Shu et al., 2016; Gu et al., 2019). Anc80L65 infected near 100% of IHCs and 90% of OHCs across all turns of the cochlea in neonate mice, however, infection of SC was < 30% at the same titer (Gu et al., 2019). AAV8 and Anc80L65 vectors have been delivered by cochleostomy in a number of notable preclinical studies in murine models of NSHL investigating deafness associated with GJB2 (Iizuka et al., 2015), MYO6 (Xue et al., 2022), KCNQ1 (Chang et al., 2015), and LHFPL5 (György et al., 2017). These studies demonstrated significant hearing recovery in neonatal mice, with the KCNQ1-/- mice demonstrating complete recovery comparable to WT mice ABR thresholds.
Kilpatrick et al. studied the infection rates of AAV1, AAV2, AAV5, AAV6, and AAV8 on SGNs in both healthy, WT mice and ototoxic drug-exposed, deafened mice (Kilpatrick et al., 2011). AAV8 infected SGNS in 6 out of 10 (60%) healthy WT mice aged 2 to 12 months, with an approximately 35% infection rate reported across all AAV serotypes studied. Furthermore, the infection rate of SGNs using all AAVs increased in the deafened cohort, with an average of 52% of mice demonstrating SGN infection. However, this study did not quantify SGN transduction itself, but counted the number of mice that demonstrated green fluorescent protein (GFP) expression in the SGNs following transduction with the AAV-GFP vectors of varying serotypes. György et al. (2017) studied exosome mediated AAV delivery via a cochleostomy procedure and demonstrated significant transduction of SGNs as well. Similarly, these authors did not quantify the SGN transduction, although representative images suggest robust (>75%) transduction efficiency.
While the cochleostomy procedure in mice has only demonstrated moderate SGN infection rates, it has been additionally studied in adult guinea pig models following viral vector delivery to both the scala tympani (perilymph) and scala media (endolymph). Studies administering AAV2 (Budenz et al., 2015; Pfingst et al., 2017) and rAAV8 (Chen et al., 2018) expressing NT-3 demonstrated variable SGN transduction efficiency from near 0% to >75% in at least one turn of the cochlea. Moreover, Chen et al. demonstrated significant ribbon synapse survival rates following noise-induced toxicity with rAAV8-NT-3 (Chen et al., 2018).
Administration of the vector through the endolymphatic sac and the utricle are two additional routes that provide access to the endolymph. However, these studies have not been as extensively studied as canalostomy delivery. The utricle, an otolith organ, is theorized to allow access to the endolymph while avoiding the iatrogenic cochlear damage associated with canalostomy. This route has only been studied in young (postnatal age ≤ 16 days) mice where the utricle could be pierced directly with the injection needle (Lee et al., 2020) so questions remain about the translatability to large mammals.
The endolymphatic sac can be accessed following drilling to expose both the dura and the sigmoid sinus. The vector can subsequently be administered directly into either the endolymphatic duct or sac (Yamasoba et al., 1999).
Lee et al. (2020) demonstrated near 100% infection of IHCs and OHCs in all turns of the cochlea following utricle injection of AAV9-PHP.B in young mice, which was more efficient than their RWM injection controls at an average of 80%−90%. At P16, AAV9-PHP.B outperformed Anc80L65 in terms of IHC transduction efficiency, especially at the basal turns of the cochlea (Lee et al., 2020). Consistent with these findings, AAV9-PHP.B utricle injections by Wu et al. (2021a) in TMC1-/- mice demonstrated similar HC transduction rates.
Direct adenoviral vector injection into the endolymphatic duct/sac has proven feasible in a study by Yamasoba et al. (1999). Cochlear transduction was only noted in the mice that exhibited intraoperative swelling of the endolymphatic sac during the injection, suggesting a successful injection technique. While the HC transduction was not quantified, the authors noted infection of HCs that bordered the endolymph, in addition to infection of the vestibular end organs (Yamasoba et al., 1999). Both the utricle and endolymphatic sac injection are technically more challenging than other delivery methods, with a higher risk of iatrogenic hearing loss associated with the delivery process.
Given the challenges in effective HC and SGN transduction particularly across the tonotopic axis of the adult mammalian cochlea with cochlear delivery, and the connection between CSF and inner ear, CSF delivery has been investigated recently to deliver genes to the inner ear.
Given that it is difficult to limit CNS transduction following injection into the CSF, hair cell-specific promotors, such as MYO7 have been investigated to limit transduction to hair cells. Blanc et al. reported that injection of viral vectors (AAV2/8, AAV9, and AAV2/Anc80L65) into the cisterna magna (CM) of mature mice results moderate transduction of IHCs bilaterally, however the efficiency differs across the tonotopic axis (Blanc et al., 2020). AAV2/Anc80L65 was the most efficient serotype, transfecting nearly 90% of the IHCs at the base but close to 0% at the apex. Moreover, a significant portion of the SGNs were transduced, however the percentage transduction was not reported. Auditory and vestibular studies revealed that the CSF delivery did not affect hearing or vestibular function compared to the cochleostomy approach (Blanc et al., 2020). In another study by Mathiesen et al. (2023), AAV-PHP.B-CBA-VGLUT3-WPRE injected in the cisterna magna injected at a relatively high dose of 2.27 × 1011 vg (10 μL of a 2.27 × 1013 vg/mL solution) restored hearing in SLC17A8-/- mice, which lack vesicular glutamate transporters and are therefore unable to release the neurotransmitter, in all frequencies except 40 kHz. The authors report minimal off-target VGLUT3 transgene expression in the brain (specifically the cortex, hippocampal CA2 region and cerebellum) and no expression in the liver. These findings are contradictory to many rodent and NHP studies which have consistently demonstrated widespread brain and spinal cord transduction following CSF delivery of AAV-PHP.B (Liguore et al., 2019; Arotcarena et al., 2021; Chatterjee et al., 2022). Moreover, this study was conducted utilizing mice aged 4 to 15 weeks old (P28-P105) at the time of injection, suggesting CSF delivery as a potential route for transduction of mature cochlea (Mathiesen et al., 2023).
Notably absent from this study was an assessment of presence of viral vector genomes in the liver, assessment of spinal cord transduction and screening of dorsal root ganglia (DRG) for an immune response. A recent meta-analysis of AAV delivered by CSF for CNS indications revealed DRG pathology in 83% of NHP regardless of the AAV serotype or promoter used (a total of 213 NHP which received intracisternal AAV) (Hordeaux et al., 2020). Those findings are consistent with those by Samaranch et al. who observed widespread brain, spinal cord and DRG transduction with intra-CM delivered AAV9 and AAV7 in NHPs (Samaranch et al., 2013). Consistent with Mathiesen et al., Samaranch et al. did not report any significant peripheral organ transduction, however transduction of both astrocytes and neurons was reported.
Efficient cochlea transduction following CSF delivery has also recently been reported in NHPs by Ranum et al. (2023) who explored cochlear transduction following intracerebroventricular injection of AAV9, and novel capsid variants of AAV1, 2, and 9. The AAV1 and AAV2 capsid variants, AAV1.RPG.mNG and AAV2.HDG.mTFP, transduced nearly all IHCs. Alteration of the AAV9 capsid appeared to affect transduction efficiency along the cochlea tonotopic axis in rhesus macaques. Cochlea transduction with AAV9.eGFP was strongest at the apex, while animals injected with the AAV9 capsid variant AAV9.KGG.eGFP demonstrated strongest gene expression at the base. This finding suggests that the apex to base transduction gradient observed in many studies could be due to varied expression of receptors required for viral entry into the cell across the tonotopic axis of the cochlea. Interestingly AAV9 performed better at IHC transduction in the rhesus macaque compared to the African green monkey, further implicating cellular physiological differences in transduction efficiency (Ranum et al., 2023). No SGN transduction was reported in this study, however it is possible that use of serotypes with high neuronal tropism (e.g., AAV5 or AAV-PHP.B) could improve SGN transduction with this delivery route.
Taken together, these studies suggest CSF delivery through CM and intracerebroventricular injections is an efficient delivery method for gene therapy to the inner ear. However, caution is warranted regarding the potential for off-target delivery to other organs, particularly when considering upscale to larger mammals and humans. Additionally, significantly more vector is required for CM injections compared to intracochlear injections due to potential to dilute the vector within the CSF. A relatively high dose of 2.27 × 1011 vg and 4.7 × 1011 vg (both in 10μL) was used in the functional and GFP studies (Mathiesen et al., 2023). Development of novel AAV serotypes specifically designed to target inner ear cells and de-target peripheral organs is an essential next step to see this route of administration enter clinical applications.
In addition to CSF delivery, injection of viral vectors directly to the brainstem may represent another potential route. AAV vectors exhibit natural tropism and route of axonal transport (anterograde, retrograde, and bi-directional) to specific cell types, which can be further modified through genetic engineering to allow for transport of genetic material along axonal pathways (Gao et al., 2002; Cearley and Wolfe, 2007; Salegio et al., 2013). The use of AAV vectors for axonal transport is a robust method that can achieve transgene expression in brain regions beyond the direct site of injection in adult NHPs (Samaranch et al., 2017; Naidoo et al., 2018). Anterograde transport involves entry of the AAV vector into cell bodies at the infusion site. Intact virions are transported along axons that originate within the site of vector delivery, are released at the axon terminal, and then transduce neighboring cells that synapse onto the dendrites (Salegio et al., 2013; Green et al., 2016). In contrast, retrograde transport requires uptake of the AAV vector by axonal projections, followed by transport to the distally located soma where it transduces the host cell nucleus, allowing for identification of neurons that project to a specific brain region (Salegio et al., 2013; Green et al., 2016). Harnessing axonal transport of AAVs to transduce various regions of the brain has also been shown as effective and safe in human trials for various diseases (Pearson et al., 2021; Rocco et al., 2022). Brainstem delivery of AAV into the cochlear nucleus could therefore use both anterograde and retrograde transport to infect the cochlea using the efferent and afferent cochlear nerve fibers, respectively (Figure 2). The brainstem delivery route has yet to be studied for its efficacy for transducing the cells of the inner ear. However, given the success with axonal transport in adult mammals within the brain, it could be expected that brainstem delivery might be a prime method for improving SGN transduction. The brainstem delivery route is a more invasive delivery route, however intraparenchymal AAV delivery has a clinical track record of safety, including in the upper brainstem in regions in close proximity to the Superior Olivary Complex and Cochlear Nucleus (Pearson et al., 2021). Gadoteridol co-infusion with AAV, and intraoperative MRI allows for real-time infusion of vector, and precise cannula placement avoiding major blood vessels and nuclei responsible for vital life functions (Lonser et al., 2020). Moreover, surgical biopsies of tumors from the proposed regions of the brainstem are routinely performed in patients with low morbidity (Lonser et al., 2020). The benefits in quality of life therefore present a potential case for development of this approach for some forms of hearing loss (Lonser et al., 2020; Pearson et al., 2021).
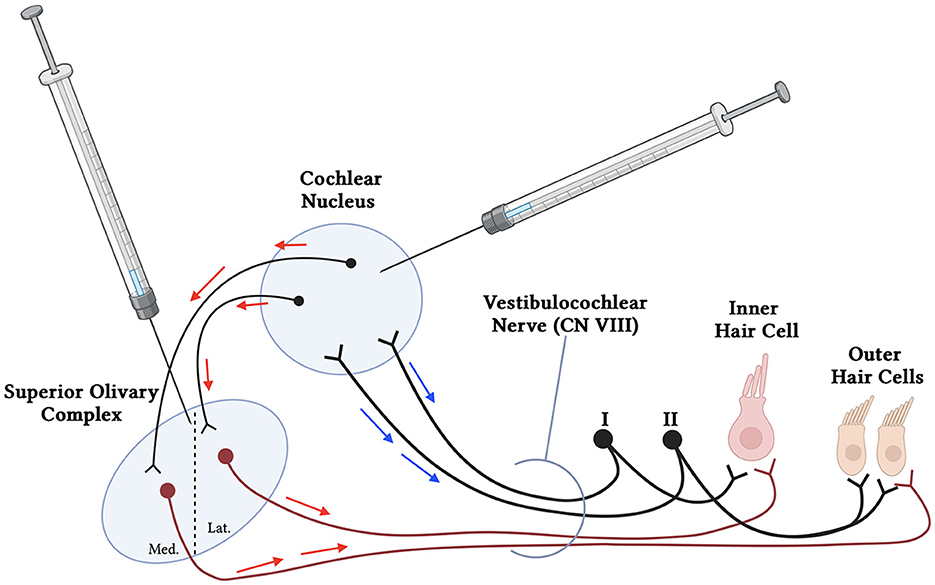
Figure 2. Simplified schematic of proposed brainstem delivery of viral vector to the cochlea using anterograde and retrograde transport. Viral vectors may be administered using MRI-guided intraparenchymal injections to the cochlear nucleus in the brainstem. The AAVs may utilize both anterograde (red arrows) and retrograde (blue arrows) transport through the afferent (Type I and II spiral ganglion neurons) (black), interneurons to the Superior Olivary Complex, and efferent neuronal fibers (maroon) to reach both the inner and outer hair cells of the cochlea. Additional neurons communicating with and from the cochlear nucleus and superior olivary complex have been removed for simplicity.
Promoter selection is an important consideration when designing a gene therapy as gene expression in non-target cells can cause an immune response or significantly alter cellular homeostasis and result in undesirable clinical effects. While the majority of preclinical and clinical studies have opted to use strong, constitutively active promoters such as cytomegalovirus (CMV), the chicken beta-actin (CBA), and the combination CMV-enhancer/CBA promoter (CAG) promoters (Liu et al., 2007; Gu et al., 2019; Rambeau et al., 2024), hair cell specific promoters have shown great promise to limit gene expression to HCs for diseases which only affect HCs (Lv et al., 2024). CMV, CBA, CAG, elongation factor 1α (EF-1α), and hair-cell specific promoters such as Myo15 and Myo7a have all shown durable gene expression in the inner ear. A comparison of the CAG promoter and Myo7A promoters by Liu et al. (2007), showed that the CAG promoter elicited the highest expression in IHCs, while the Myo7A promoter demonstrated the most specific expression of HCs. The constitutive CMV and the CAG promoters have both demonstrated robust gene expression in HCs and SCs in preclinical studies (Gu et al., 2019; Rambeau et al., 2024). Moreover, Myo15 was demonstrated to direct gene expression in IHCs and OHCs effectively, showing promise in the development of a treatment of HHL in OTOF-/- models (Wang et al., 2024) and is also being utilized in a clinical trial initiated by Eye and ENT Hospital of Fudan University which has demonstrated remarkable safety and efficacy in patients with OTOF pathogenic variants thus far (Lv et al., 2024). Given that AAVs induce persistent gene expression in target cells, a balance must be struck between the strength and cell specify of the promoter, AAV titer and serotype such that transgene expression levels are as close as possible to physiological levels for the disease being treated.
Significant progress has been made over recent years in the clinical development of gene therapies for congenital hearing genetic disorders, however little progress has been made in the translation of therapies for adult-onset hearing loss or treatment for non-genetic causes of hearing loss.
A Phase 1/2 clinical trial sponsored by Novartis Pharmaceuticals for severe-to-profound unilateral or bilateral hearing loss without a known genetic cause was completed in 2021 (Clinicaltrials.gov Identifier: NCT02132130). This study included 22 patients who received CGF166, a recombinant adenovirus (rAd5) to deliver ATOH1 to the inner ear via the oval window injection. Based on preclinical studies, it was expected that forced ATOH1 expression in HL patients may transdifferentiate remaining SC to functional HC, leading to rescue of HL. At baseline, the 22 patients had a recorded average pure tone audiometric threshold of 69.3 dB (±15.4). At the culmination of the study, the 19 remaining patients (2 lost to follow-up) had an average PTA of 78.7 dB (±18.8), demonstrating hearing status that did not show significant improvement following treatment. There were no serious adverse events noted. The reasons for failure of this trial are unclear but could include inefficient gene delivery across the cochlear axis, choice of vector, titer, patient inclusion criteria and lack of reinnervation of HCs with SGNs. The gene target ATOH1 is a key regulator for HC regeneration, however, is not associated with SGN regeneration. Therefore, patients with profound hearing loss with secondary degeneration of the ribbon synapses and SGNs may not benefit.
There have been exciting breakthroughs for gene therapy for genetic congenital HHL in pediatric patients. Gene therapy clinical trials for OTOF-related (DFNB9) deafness are currently enrolling by Akouos/Eli Lilly and Company, Decibel Therapeutics/Regeneron, Sensorion, Otovia Therapeutics, as well as universities and research centers. A summary of clinical trial data is shown in Table 3.
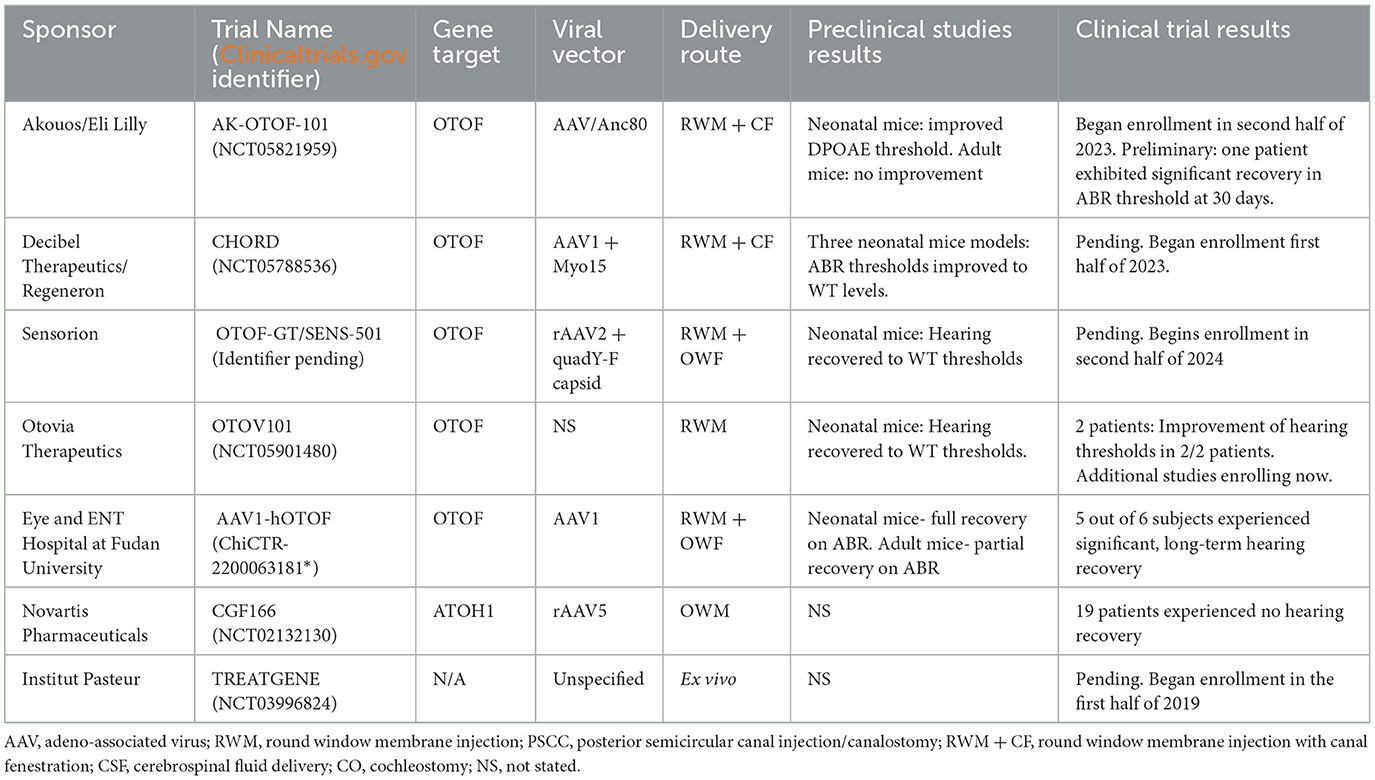
Table 3. Summary of available human clinical trial data regarding viral vector gene therapy for hearing loss.
The Akouos/Eli Lilly and Company AK-OTOF-101 trial (Clinicaltrials.gov Identifier: NCT05821959) commenced enrollment of pediatric subjects aged 2 to 17 years in a Phase 1/2 trial in September 2023, subsequent to receiving Investigational New Drug (IND) approval from the FDA in 2022. This trial employs AAVAnc80-hOTOF, a dual AAVAnc80 vector approach incorporating 6 kB of human otoferlin cDNA controlled by an un-named, ubiquitous promoter, with the aim of producing functional otoferlin in the inner ear. A dual-vector system is utilized as the size of the OTOF gene surpasses the packaging capacity of most AAVs. Utilizing two AAV cassettes, each containing different segments of the OTOF coding sequence, facilitates the in vivo assembly of a full-length otoferlin protein. Because of the size of the otoferlin gene, a dual vector approach is utilized in all the clinical trials for OTOF-related deafness. The downside of this approach is that it necessitates two vectors to enter each cell in order to produce functional protein and also exposes the patients to a greater viral load than if a single AAV was used. Vector delivery is performed via RWM injection with concomitant fenestration of the stapes footplate using a patented needle device. Intracochlear administration of AK-OTOF to 3-week-old OTOF-/- mice yielded both short-term (15 days) and long-term (>6 months) hearing restoration, as evidenced by ABR threshold reductions (Hickox et al., 2021; Gao et al., 2023). Subsequent analysis revealed robust (20%−80% of IHCs) human otoferlin expression in the IHCs of injected mice. However, the efficacy of ABR restoration in OTOF-/- mice was contingent upon pre-injection distorted product otoacoustic emissions (DPOAE) levels and, therefore, the age of the model. Less than 20% of OTOF-/- mice aged 7.5 months or older exhibited recovery of OAEs, irrespective of preoperative hearing level. Additionally, among mice aged 7.5 months with no baseline OAE, representative of severe hearing loss, none achieved hearing levels comparable to control WT mice. Safety studies conducted on OTOF-/- mice and NHPs revealed no significant adverse findings associated with intracochlear injection of AK-OTOF (Hickox et al., 2022). The NHP studies revealed minimal vector persisting in the liver, spleen, and lymph nodes at 6 months post injection. However, organ weights and brain histopathology, representative of systemic pathology, were normal at this time frame (Hickox et al., 2022; Gao et al., 2023). Regarding human subjects, preliminary reports demonstrate that one patient (11 years-old) with profound hearing loss demonstrated significant hearing recovery at 30 days post injection, reaching thresholds of 35 and 60 dB on the ABR (Simons et al., 2024). Moreover, no serious adverse events or toxicity were recorded.
Decibel Therapeutics'/Regeneron's CHORD trial (Clinicaltrials.gov Identifier: NCT05788536), is a phase 1/2 trial which began in May 2023, is actively enrolling pediatric subjects (< 18 years) with biallelic OTOF pathogenic variants. This study cohort mirrors that of AK-OTOF-101, with a notable inclusion of infants aged 6 months. Employing the DB-OTO dual AAV1 vector system featuring a hair cell-specific promoter, Myo15, and the human OTOF v5 transgenes, the CHORD trial aims to address OTOF-related deafness, DFNB9. Preclinical investigations utilized murine models, including Q828x, R1934Q, and Deaf5, and utilized treatment with a dual AAV vector system, administered via PSCC injection (Chung et al., 2023; Valayannopoulos, 2023). The Q28x murine model (OTOFQ28x/Q28x) hosts an orthologous, functionally null allele that represents the pGln829* pathogenic variant found in Latin American and Spanish populations (Migliosi et al., 2002). The R1394Q and Deaf5 murine models are missense mutations, significantly decreasing gene function, in the p.Arg1934Gln and p.Ile318Asn loci, respectively (Longo-Guess et al., 2007; Kim et al., 2018). Four weeks post-injection, all Otoferlin-deficient models treated with AAV1-Myo15-myc-mOTOF demonstrated ABR levels comparable to WT mice, while vehicle-treated OTOF-/- controls exhibited no detectable ABR. Immunohistochemical analysis further revealed over 50% transduction of IHCs in all mouse cochleae. Notably, in NHPs, RWM injection of AAV1-Myo15 (DB-OTO) with LSCC fenestration, demonstrated no serious adverse events or immunological reactions when dosed based on total perilymph volume (Koehler et al., 2023; Valayannopoulos, 2023). Furthermore, NHP studies utilizing DB-OTO showcased transduction of over 75% of IHCs, with minimal vector dispersion observed in the brain and spinal cord. Results from this trial have not yet been released.
Sensorion's OTOF-GT/SENS-501 program is poised to commence enrollment of pediatric patients aged 6 to 36 months for a phase 1/2 clinical trial in the second half of 2024. OTOF-GT employs a dual vector AAV cassette system encapsulated within a recombinant AAV2 quadY-F capsid, addressing the challenge posed by the OTOF gene's size, which surpasses the packaging capacity of most single-cassette AAV therapies. Surgical intervention will involve the administration of OTOF-GT via the RWM concurrently with stapedectomy/oval window fenestration, as outlined in their NHP studies (Rambeau et al., 2024). Preclinical investigations of OTOF-GT utilized a dual AAV2 vector, recombinant AAV2 quadY-F capsid, and chimeric CAG promoter for OTOF coding sequence delivery. Notably, modifications to the AAV2 capsid, informed by previous studies on the retina, were implemented to enhance gene transfer efficiency (Petrs-Silva et al., 2011). OTOF-/- mice injected at P10 exhibited hearing recovery similar to WT mice up to 54 weeks post injection, the last measured time point. Subsequent analysis of cochleae from mice injected at P17 and P30 revealed significantly higher numbers of ribbon synapses in transduced IHCs compared to non-transduced counterparts (Akil et al., 2019b). However, the number of ribbon synapses in OTOF-/- mice remained markedly reduced compared to age-matched WT mice, regardless of transduction status (Akil et al., 2019b). WT NHP preclinical studies (N=6/dose) demonstrated IHC transduction, yet no significant changes in ABR or DPOAE were observed. Suggesting no viral-mediated ototoxicity or iatrogenic damage. Additionally, viral vector biodistribution in NHPs was predominantly confined to the injected ear structure (Rambeau et al., 2024).
A study sponsored by Otovia Therapeutics based in Jinan, Shandong, China began enrolling subjects in mid-2023, and the investigators recently published a report of two cases (Qi et al., 2024a). This study utilized a dual vector AAV system, termed OTOV101 (also termed AAV-OTOF), which used a hair cell-specific promotor. A 5-year-old participant received a unilateral injection of the AAV-OTOF, while an 8-year-old participant received bilateral injections (Qi et al., 2024a). The viral vector was administered through the RWM following a mastoidectomy (Figure 3). At around 1 month post injection, the 5-year-old participant's ABR threshold was near 30 dB HL, comparable to the hearing of a similar patient with no pathogenic OTOF variants. Further, at 3 months post injection, the patient was able to respond to voiced questions without the use of any assisted amplification devices in the injected ear. The 8-year-old patient with bilateral injections demonstrated similar improvements in ABR thresholds, reaching near 30 dB and 50 dB in the right and left ears respectively, at 3 months post injection. This 8-year-old participant is the oldest documented patient to have experienced improvement in hearing following intracochlear gene therapy, to our knowledge. No systemic toxicities or serious adverse events were noted in these two patients. The Otovia therapeutics-sponsored study (Clinicaltrials.gov Identifier: NCT05901480) is set to continue participant enrollment until the end of 2024 with an estimated enrollment of five patients. However, the Otovia Therapeutics study will not pursue the mastoidectomy approach as was used in the first two patients, but instead administer the vector through the tympanic membrane/external auditory canal approach.
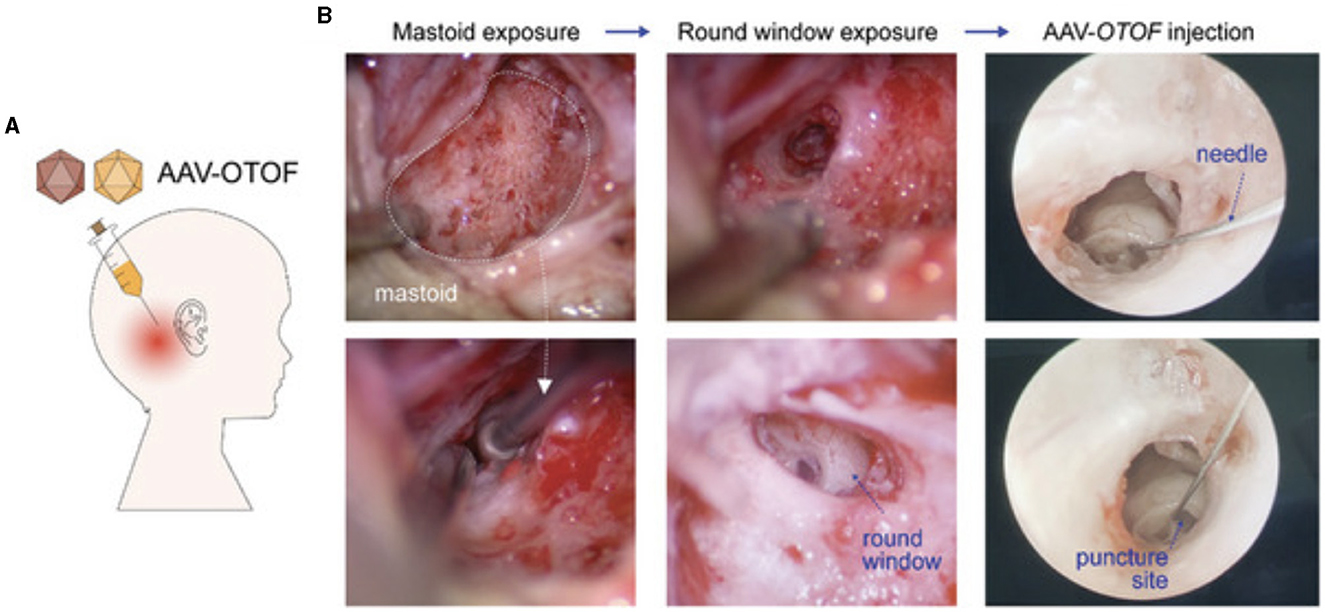
Figure 3. (A) Schematic of experimental design for injecting viral vectors in human patients. (B) Representative intraoperative images during the mastoidectomy procedure and subsequent AAV injection through the round window membrane (Figure reproduced from Qi et al. (2024a,b) under the open access license agreement; https://creativecommons.org/licenses/by/4.0/).
The first in-human gene therapy clinical trial for a hearing disorder was recently completed. Results from a single-arm, single-center study based at the Eye and ENT Hospital of Fudan University, which began enrollment in December 2022, were recently published (Lv et al., 2024). This study utilized AAV1-hOTOF, a dual vector AAV system with a Myo15 promoter. Preclinical studies on AAV1-hOTOF demonstrated a dose-responsive effect on hearing preservation following RWM injection in neonatal OTOF-/- mice (Zhang et al., 2023). The high dose OTOF-/- group (6 × 1010 vg/cochlea) exhibited ABR thresholds similar to WT mice injected with vehicle controls, and otoferlin protein was detected in all turns of the cochleae of these mice. When AAV1-hOTOF was injected in adult mice, those that received doses >3 × 1010 demonstrated long-term (>6 months) partial recovery in hearing levels. Moreover, the safety of AAV1-hOTOF was examined when RWM injection in WT mice led to no difference in ABR function, behavior, or pathology (Zhang et al., 2023). In NHPs, no toxicity following a high dose of AAV1-hOTOF was noted. However, viral genomes were detected at minimal levels in the brain, spinal cord, and liver (Zhang et al., 2023). These preclinical studies supported the clinical trial data that were published in The Lancet in early 2024 (Lv et al., 2024). Six subjects (Average age: 4.1 years, Range 1.0 to 6.2 years) diagnosed with DFNB9 and complete hearing loss (no ABR at baseline, 3/6 had pure-tone audiometry recordings (all greater than 100 dB), and 4/6 had unilateral CI) were prospectively enrolled in the study. The participants were administered 9 × 1011 vg/cochlea (n = 1) or 1.5 × 1012 vg/cochlea (n = 5) of AAV1-hOTOF unilaterally via a transcanal RWM injection with stapes fenestration. Concurrent systemic corticosteroids and antibiotics were administered. There were no serious adverse events or dose-limiting toxicities noted throughout the 26-week-long study. Moreover, vestibular function (n = 4) and otoscopic examination (n = 6) were normal at the long-term follow up (>26 weeks). Five out of six participants exhibited significant long-term (>26 weeks) hearing recovery (at least 10-dB threshold reduction on the ABR) that was first detected between 4–6 weeks post injection. Additionally, speech perception in 3 of the 4 CI users was also significantly improved. Overall, this prospective study of six patients demonstrated significant hearing recovery in 5/6 participants with no serious adverse events at the long-term follow-up. This clinical trial is groundbreaking with extremely promising results. Moreover, this trial suggests that success in animal models can be translated to similar degrees of success in human models, paving the way for rapid clinical translation of other gene targets.
To our knowledge, there are no active gene therapy clinical trials involving for deafness related to other genetic pathogenic variants, such as GJB2, or gene delivery of neurotrophic factors for the treatment of NIHL and CIHL. However, Decibel Therapeutics is conducting an IND-enabling study, AAV.103, that will evaluate the safety of AAV vectors for GJB2-associated deafness in preparation for human trials.
Given the fundamental differences in the timing of inner ear development and onset of hearing between rodent models and humans, there may be limited success in the translation of gene therapy treatments from mice to humans. The age of the preclinical model presents a significant limitation in inner ear transduction and the translation of gene therapy for adult humans. Neonatal mice exhibit high transduction efficiency in both HCs and SGNs, due to, in part, the regenerative capacity of the cells during early development. In mice, the HCs are mostly matured by P7, a process that starts as early as embryonic day 14 (Kolla et al., 2020). While the IHCs and OHCs may be distinct at P7, the ribbon synapses do not reach adult configuration until at least P14 when the cochlea matures (Michanski et al., 2019). In humans, IHCs begin to develop in gestational week 10 (GW10), with the fetus able to respond to low frequency tones at GW19 (Hepper and Shahidullah, 1994; Johnson Chacko et al., 2019). Fetal response to all frequencies, representing IHC maturation and mature connections between the HCs, ribbon synapses, and SGNs, was observed at GW35, demonstrating complete cochlear maturation embryonically (Hepper and Shahidullah, 1994). The later GW35 timepoint may better represent maturation of neural components, as studies have demonstrated IHCs and OHCs across all turns of the cochlea at GW24, a much earlier timepoint (Kelley, 2007; Johnson Chacko et al., 2019). In addition to cochlear maturation, the critical time period for auditory development varies between humans and mice. The murine critical period ends at approximately P28 while the human critical period extends through the first few years of life (Kral, 2013). Due to this maturation timeline, gene therapy for mature models must overcome substantial hurdles, particularly concerning SGNs, where transduction efficiency can plummet to as low as 10% in mature mice (Table 2). Additionally, variation in transduction efficiency is pronounced in both IHCs and OHCs, with notable apex-to-base gradients complicating uniform therapeutic delivery. These challenges are attributed to age-related changes in cellular physiology, expression of receptors required for AAV entry, including the transient regenerative capacity of cochlear cells, which do not become fully quiescent and developed until around the 2nd week of postnatal development. This age limitation underscores the need for innovative approaches to enhance transduction efficiency and bridge the gap between neonatal and mature models in gene therapy research.
An important consideration for future clinical trials is identifying the ideal patient population to study and subsequently determining the availability of suitable animal models. This currently presents a major challenge for gene therapy advancements. While rare HHL diseases like Usher syndrome and OTOF-related deafness have provided valuable proof-of-principle studies, they affect a relatively small number of patients. Moreover, the therapeutic window for intervention is narrow, as demonstrated in studies showing minimal success in mature preclinical and clinical models. Additionally, the ideal patient population requires a specific auditory profile, having absent ABR but intact DPOAE, indicating the presence of IHCs available for transduction, and preferably without CIs. However, this patient population is almost non-existent in the United States, as congenitally deaf individuals typically receive CIs early in life (most by 1 year of age), and those who do not, often opt for communication using American Sign Language (ASL). These patients within the culturally Deaf community are less likely to desire gene therapy approaches to mediate hearing loss. While genetic mouse models are valuable for testing viral transduction efficiency and hearing restoration, NHP models are better suited for assessing clinical feasibility. Unfortunately, there are almost no models of hearing loss in NHPs, leading to reliance on studies using GFP to only index infection and transduction rates instead.
To maximize the impact of gene therapy interventions, it is crucial to expand the target population to include adult patients with non-genetic forms of hearing loss, such as NIHL and age-related hearing loss (presbycusis), which account for the majority of cases of SNHL. Consequently, approaches that are gene-agnostic, such as NTs like GDNF and BDNF, and potentially even transcription factor ATOH1, are likely to be the future focus of research and development in the field of gene therapy for acquired hearing loss.
Auditory neuropathy may consist of damage or dysfunction of any portion of the pathway from the cochlea to the brain, with HCs, ribbon synapses, and SGNs representing the major anatomical components. As discussed previously, pathogenic variants in genes such as SLC17A8 and OTOF have implications as targets for SNHL related to auditory neuropathy, with OTOF being extensively studied. However, current studies have demonstrated inefficient infection rates of SGNs (Table 2) and have instead focused on HC transduction. This limitation severely affects the translational capacity of these studies for use in SNHL associated with degeneration of the SGNs with or without cochlear damage. NT delivery using viral vectors have established a neuroprotective role following NIHL and CIHL but have yet to demonstrate significant regenerative capacity.
Recent advancements in both preclinical studies and clinical trials have highlighted the potential of gene therapy in treating sensorineural hearing loss. Notably, HHL, particularly associated with pathogenic variants in the OTOF gene, has emerged as a focal point of clinical discoveries. The first clinical trial for HHL has demonstrated robust hearing recovery in pediatric subjects, marking a significant milestone in the field.
However, gene therapy for degenerative conditions such as NIHL and CIHL is still in its early stages. Addressing these gaps necessitates a focus on mature models, with a specific emphasis on enhancing SGN transduction. Further preclinical investigations are essential to bridge these disparities. Despite these challenges, the rapid progress in utilizing gene therapy for both acquired and HHL holds great promise for improving patient outcomes.
BD: Conceptualization, Data curation, Writing – original draft, Writing – review & editing. EB: Conceptualization, Writing – review & editing. YR: Conceptualization, Data curation, Writing – review & editing. JN: Conceptualization, Data curation, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by seed grants from the Ohio State University Neurological Research Institute and President's Research Excellence (PRE) Accelerator Programs (EB, YR, and JN).
The authors would like to thank Krystof Bankiewicz, MD, PhD. (Chief Scientific Officer, Gene Therapy Institute at The Ohio State University) and Russell Lonser, MD (Director, Gene Therapy Institute at The Ohio State University) for their continued support. Figures were created with Biorender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Akil, O., Blits, B., and Lustig, L. R. Leake, P. A. (2019a). Virally mediated overexpression of glial-derived neurotrophic factor elicits age- and dose-dependent neuronal toxicity and hearing loss. Hum. Gene. Ther. 30, 88–105. doi: 10.1089/hum.2018.028
Akil, O., Dyka, F., Calvet, C., Emptoz, A., Lahlou, G., Nouaille, S., et al. (2019b). Dual AAV-mediated gene therapy restores hearing in a DFNB9 mouse model. Proc. Natl. Acad. Sci. USA. 116, 4496–4501. doi: 10.1073/pnas.1817537116
Akil, O., Seal, R. P., Burke, K., Wang, C., Alemi, A., During, M., et al. (2012). Restoration of hearing in the VGLUT3 knockout mouse using virally mediated gene therapy. Neuron 75, 283–293. doi: 10.1016/j.neuron.2012.05.019
Aldè, M., Cantarella, G., Zanetti, D., Pignataro, L., La Mantia, I., Maiolino, L., et al. (2023). Autosomal dominant non-syndromic hearing loss (DFNA): a comprehensive narrative review. Biomedicines 11:1616. doi: 10.3390/biomedicines11061616
Al-Moyed, H., Cepeda, A. P., Jung, S., Moser, T., Kügler, S., and Reisinger, E. (2019). A dual-AAV approach restores fast exocytosis and partially rescues auditory function in deaf otoferlin knock-out mice. EMBO Mol. Med. 11:e9396. doi: 10.15252/emmm.201809396
Andres-Mateos, E., Landegger, L. D., Unzu, C., Phillips, J., Lin, B. M., Dewyer, N. A., et al. (2022). Choice of vector and surgical approach enables efficient cochlear gene transfer in nonhuman primate. Nat. Commun. 13:1359. doi: 10.1038/s41467-022-28969-3
Arnett, J., Emery, S. B., Kim, T. B., Boerst, A. K., Lee, K., Leal, S. M., et al. (2011). Autosomal dominant progressive sensorineural hearing loss due to a novel mutation in the KCNQ4 gene. Arch. Otolaryngol. Head Neck Surg. 137, 54–59. doi: 10.1001/archoto.2010.234
Arotcarena, M. L., Dovero, S., Biendon, N., Dutheil, N., Planche, V., Bezard, E., et al. (2021). Pilot Study assessing the impact of intrathecal administration of variants AAV-PHP.B and AAV-PHP.eB on brain transduction in adult rhesus macaques. Front. Bioeng Biotechnol. 9:762209. doi: 10.3389/fbioe.2021.762209
Askew, C., Rochat, C., Pan, B., Asai, Y., Ahmed, H., Child, E., et al. (2015). Tmc gene therapy restores auditory function in deaf mice. Sci. Transl. Med. 7:295ra108. doi: 10.1126/scitranslmed.aab1996
Blanc, F., Mondain, M., Bemelmans, A. P., Affortit, C., Puel, J. L., and Wang, J. (2020). rAAV-mediated cochlear gene therapy: prospects and challenges for clinical application. J. Clin. Med. 9:589. doi: 10.3390/jcm9020589
Blanco-Kelly, F., Jaijo, T., Aller, E., Avila-Fernandez, A., López-Molina, M. I., Giménez, A., et al. (2015). Clinical aspects of Usher syndrome and the USH2A gene in a cohort of 433 patients. JAMA Ophthalmol. 133, 157–164. doi: 10.1001/jamaophthalmol.2014.4498
Bonnet, C., Riahi, Z., Chantot-Bastaraud, S., Smagghe, L., Letexier, M., Marcaillou, C., et al. (2016). An innovative strategy for the molecular diagnosis of Usher syndrome identifies causal biallelic mutations in 93% of European patients. Eur. J. Hum. Genet. 24, 1730–1738. doi: 10.1038/ejhg.2016.99
Bowers, W. J., Chen, X., Guo, H., Frisina, D. R., Federoff, H. J., and Frisina, R. D. (2002). Neurotrophin-3 transduction attenuates cisplatin spiral ganglion neuron ototoxicity in the cochlea. Mol. Ther. 6, 12–18. doi: 10.1006/mthe.2002.0627
Breglio, A. M., Rusheen, A. E., Shide, E. D., Fernandez, K. A., Spielbauer, K. K., McLachlin, K. M., et al. (2017). Cisplatin is retained in the cochlea indefinitely following chemotherapy. Nat. Commun. 8:1654. doi: 10.1038/s41467-017-01837-1
Brown, A. M., and Hamann, M. (2014). Computational modeling of the effects of auditory nerve dysmyelination. Front. Neuroanat. 8:73. doi: 10.3389/fnana.2014.00073
Bu, C., Xu, L., Han, Y., Wang, M., Wang, X., Liu, W., et al. (2022). c-Myb protects cochlear hair cells from cisplatin-induced damage via the PI3K/Akt signaling pathway. Cell Death Discov. 8:78. doi: 10.1038/s41420-022-00879-9
Budenz, C. L., Wong, H. T., Swiderski, D. L., Shibata, S. B., Pfingst, B. E., and Raphael, Y. (2015). Differential effects of AAV.BDNF and AAV.Ntf3 in the deafened adult guinea pig ear. Sci. Rep. 5:8619. doi: 10.1038/srep08619
Cearley, C. N., and Wolfe, J. H. (2007). A single injection of an adeno-associated virus vector into nuclei with divergent connections results in widespread vector distribution in the brain and global correction of a neurogenetic disease. J. Neurosci. 27, 9928–9940. doi: 10.1523/JNEUROSCI.2185-07.2007
Chan, D. K., Lieberman, D. M., Musatov, S., Goldfein, J. A., Selesnick, S. H., and Kaplitt, M. G. (2007). Protection against cisplatin-induced ototoxicity by adeno-associated virus-mediated delivery of the X-linked inhibitor of apoptosis protein is not dependent on caspase inhibition. Otol. Neurotol. 28, 417–425. doi: 10.1097/01.mao.0000247826.28893.7a
Chang, Q., Wang, J., Li, Q., Kim, Y., Zhou, B., Wang, Y., et al. (2015). Virally mediated Kcnq1 gene replacement therapy in the immature scala media restores hearing in a mouse model of human Jervell and Lange-Nielsen deafness syndrome. EMBO Mol. Med. 7, 1077–1086. doi: 10.15252/emmm.201404929
Chatterjee, D., Marmion, D. J., McBride, J. L., Manfredsson, F. P., Butler, D., Messer, A., et al. (2022). Enhanced CNS transduction from AAV.PHP.eB infusion into the cisterna magna of older adult rats compared to AAV9. Gene Ther. 29, 390–397. doi: 10.1038/s41434-021-00244-y
Chen, H., Xing, Y., Xia, L., Chen, Z., Yin, S., and Wang, J. (2018). AAV-mediated NT-3 overexpression protects cochleae against noise-induced synaptopathy. Gene Ther. 25, 251–259. doi: 10.1038/s41434-018-0012-0
Chen, X. W., Li, H., Cao, K. L., Wei, C. G., and Jin, X. (2003). The transduction of neurotrophin-3/GDNF attenuates the SGNC damage in deaf mice. Zhonghua Yi Xue Za Zhi 83, 1517–1520.
Chien, W. W., Isgrig, K., Roy, S., Belyantseva, I. A., Drummond, M. C., May, L. A., et al. (2016). Gene therapy restores hair cell stereocilia morphology in inner ears of deaf whirler mice. Mol. Ther. 24, 17–25. doi: 10.1038/mt.2015.150
Chien, W. W., McDougald, D. S., Roy, S., Fitzgerald, T. S., and Cunningham, L. L. (2015). Cochlear gene transfer mediated by adeno-associated virus: comparison of two surgical approaches. Laryngoscope 125, 2557–2564. doi: 10.1002/lary.25317
Christine, C. W., Starr, P. A., Larson, P. S., Eberling, J. L., Jagust, W. J., Hawkins, R. A., et al. (2009). Safety and tolerability of putaminal AADC gene therapy for Parkinson disease. Neurology 73, 1662–1669. doi: 10.1212/WNL.0b013e3181c29356
Chung, Y., Fucchi, Q., Koehler, S., Pan, N., Kim, Y., Young Yi, E., et al. (2023). Restoration of Auditory Function with OTOF Gene Transfer Therapy in Nonsense (Q828X) and Missense (R1934Q and Deaf5) Models of Otoferlin Deficiency. Los Angeles, CA: American Society of Gene and Cell Therapy Annual Meeting.
Ciesielska, A., Hadaczek, P., Mittermeyer, G., Zhou, S., Wright, J. F., Bankiewicz, K. S., et al. (2013). Cerebral infusion of AAV9 vector-encoding non-self proteins can elicit cell-mediated immune responses. Mol. Ther. 21, 158–166. doi: 10.1038/mt.2012.167
Collaborators, G. H. L. (2021). Hearing loss prevalence and years lived with disability, 1990-2019: findings from the Global Burden of Disease Study 2019. Lancet 397, 996–1009. doi: 10.1016/S0140-6736(21)00516-X
Cooper, L. B., Chan, D. K., Roediger, F. C., Shaffer, B. R., Fraser, J. F., Musatov, S., et al. (2006). AAV-mediated delivery of the caspase inhibitor XIAP protects against cisplatin ototoxicity. Otol. Neurotol. 27, 484–490. doi: 10.1097/00129492-200606000-00009
Crispino, G., Galindo Ramirez, F., Campioni, M., Zorzi, V., Praetorius, M., Di Pasquale, G., et al. (2017). In vivo genetic manipulation of inner ear connexin expression by bovine adeno-associated viral vectors. Sci. Rep. 7:6567. doi: 10.1038/s41598-017-06759-y
Danial-Farran, N., Chervinsky, E., Nadar-Ponniah, P. T., Cohen Barak, E., Taiber, S., Khayat, M., et al. (2021). Homozygote loss-of-function variants in the human COCH gene underlie hearing loss. Eur. J. Hum. Genet. 29, 338–342. doi: 10.1038/s41431-020-00724-6
Del Castillo, I., Morín, M., Domínguez-Ruiz, M., and Moreno-Pelayo, M. A. (2022). Genetic etiology of non-syndromic hearing loss in Europe. Hum. Genet. 141, 683–696. doi: 10.1007/s00439-021-02425-6
Delmaghani, S., Defourny, J., Aghaie, A., Beurg, M., Dulon, D., Thelen, N., et al. (2015). Hypervulnerability to sound exposure through impaired adaptive proliferation of peroxisomes. Cell 163, 894–906. doi: 10.1016/j.cell.2015.10.023
Delmaghani, S., and El-Amraoui, A. (2022). The genetic and phenotypic landscapes of Usher syndrome: from disease mechanisms to a new classification. Hum. Genet. 141, 709–735. doi: 10.1007/s00439-022-02448-7
Denoyelle, F., Lina-Granade, G., Plauchu, H., Bruzzone, R., Chaïb, H., Lévi-Acobas, F., et al. (1998). Connexin 26 gene linked to a dominant deafness. Nature 393, 319–320. doi: 10.1038/30639
Deverman, B. E., Pravdo, P. L., Simpson, B. P., Kumar, S. R., Chan, K. Y., Banerjee, A., et al. (2016). Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat. Biotechnol. 34, 204–209. doi: 10.1038/nbt.3440
Doll, J., Vona, B., Schnapp, L., Rüschendorf, F., Khan, I., Khan, S., et al. (2020). Genetic spectrum of syndromic and non-syndromic hearing loss in pakistani families. Genes 11:1329. doi: 10.3390/genes11111329
Duarte, M. J., Kanumuri, V. V., Landegger, L. D., Tarabichi, O., Sinha, S., Meng, X., et al. (2018). Ancestral adeno-associated virus vector delivery of opsins to spiral ganglion neurons: implications for optogenetic cochlear implants. Mol. Ther. 26, 1931–1939. doi: 10.1016/j.ymthe.2018.05.023
Early, S., Du, E., Boussaty, E., and Friedman, R. (2022). Genetics of noise-induced hearing loss in the mouse model. Hear. Res. 425:108505. doi: 10.1016/j.heares.2022.108505
Emptoz, A., Michel, V., Lelli, A., Akil, O., Boutet de Monvel, J., Lahlou, G., et al. (2017). Local gene therapy durably restores vestibular function in a mouse model of Usher syndrome type 1G. Proc. Natl. Acad. Sci. USA. 114, 9695–9700. doi: 10.1073/pnas.1708894114
Fukui, H., Wong, H. T., Beyer, L. A., Case, B. G., Swiderski, D. L., Di Polo, A., et al. (2012). BDNF gene therapy induces auditory nerve survival and fiber sprouting in deaf Pou4f3 mutant mice. Sci. Rep. 2:838. doi: 10.1038/srep00838
Gao, G. P., Alvira, M. R., Wang, L., Calcedo, R., Johnston, J., and Wilson, J. M. (2002). Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc. Natl. Acad. Sci. USA. 99, 11854–11859. doi: 10.1073/pnas.182412299
Gao, X., Tao, Y., Lamas, V., Huang, M., Yeh, W. H., Pan, B., et al. (2018). Treatment of autosomal dominant hearing loss by in vivo delivery of genome editing agents. Nature 553, 217–221. doi: 10.1038/nature25164
Gao, Y., Francis, S. P., Ng, R., Asai, Y., Darcy, Y., Lenz, D., et al. (2023). “Preclinical Development of a Genetic Medicine for Otoferlin Gene-mediated Hearing Loss: AK-OTOF,” in Association for Research in Otolaryngology 46th Annual Mid-Winter Meeting, Orlando, FL.
Gillespie, L. N., Clark, G. M., Bartlett, P. F., and Marzella, P. L. (2003). BDNF-induced survival of auditory neurons in vivo: Cessation of treatment leads to accelerated loss of survival effects. J. Neurosci. Res. 71, 785–790. doi: 10.1002/jnr.10542
Glueckert, R., Johnson Chacko, L., Rask-Andersen, H., Liu, W., Handschuh, S., and Schrott-Fischer, A. (2018). Anatomical basis of drug delivery to the inner ear. Hear. Res. 368, 10–27. doi: 10.1016/j.heares.2018.06.017
Goldenberg, I., Moss, A. J., Zareba, W., McNitt, S., Robinson, J. L., Qi, M., et al. (2006). Clinical course and risk stratification of patients affected with the Jervell and Lange-Nielsen syndrome. J. Cardiovasc. Electrophysiol. 17, 1161–1168. doi: 10.1111/j.1540-8167.2006.00587.x
Green, F., Samaranch, L., Zhang, H. S., Manning-Bog, A., Meyer, K., Forsayeth, J., et al. (2016). Axonal transport of AAV9 in nonhuman primate brain. Gene Ther. 23, 520–526. doi: 10.1038/gt.2016.24
Gu, X., Chai, R., Guo, L., Dong, B., Li, W., Shu, Y., et al. (2019). Transduction of adeno-associated virus vectors targeting hair cells and supporting cells in the neonatal mouse cochlea. Front. Cell. Neurosci. 13:8. doi: 10.3389/fncel.2019.00008
Guo, J., Ma, X., Skidmore, J. M., Cimerman, J., Prieskorn, D. M., Beyer, L. A., et al. (2021). gene therapy and conditional deletion reveal developmental stage-dependent effects on inner ear structure and function. Mol. Ther. Methods Clin. Dev. 23, 319–333. doi: 10.1016/j.omtm.2021.09.009
György, B., Meijer, E. J., Ivanchenko, M. V., Tenneson, K., Emond, F., Hanlon, K. S., et al. (2019a). Gene transfer with AAV9-PHP.B rescues hearing in a mouse model of usher syndrome 3A and transduces hair cells in a non-human primate. Mol. Ther. Methods Clin. Dev. 13, 1–13. doi: 10.1016/j.omtm.2018.11.003
György, B., Nist-Lund, C., Pan, B., Asai, Y., Karavitaki, K. D., Kleinstiver, B. P., et al. (2019b). Allele-specific gene editing prevents deafness in a model of dominant progressive hearing loss. Nat. Med. 25, 1123–1130. doi: 10.1038/s41591-019-0500-9
György, B., Sage, C., Indzhykulian, A. A., Scheffer, D. I., Brisson, A. R., Tan, S., et al. (2017). Rescue of hearing by gene delivery to inner-ear hair cells using exosome-associated AAV. Mol. Ther. 25, 379–391. doi: 10.1016/j.ymthe.2016.12.010
Harasztosi, C., Wolter, S., Gutsche, K., Durán-Alonso, M. B., López-Hernández, I., Pascual, A., et al. (2020). Differential deletion of GDNF in the auditory system leads to altered sound responsiveness. J. Neurosci. Res. 98, 1764–1779. doi: 10.1002/jnr.24544
Hashimoto, K., Hickman, T. T., Suzuki, J., Ji, L., Kohrman, D. C., Corfas, G., et al. (2019). Protection from noise-induced cochlear synaptopathy by virally mediated overexpression of NT3. Sci. Rep. 9:15362. doi: 10.1038/s41598-019-51724-6
He, C., Liu, X., Zhong, Z., and Chen, J. (2020). Mutation screening of the USH2A gene reveals two novel pathogenic variants in Chinese patients causing simplex usher syndrome 2. BMC Ophthalmol. 20:70. doi: 10.1186/s12886-020-01342-y
He, Y., Zheng, Z., Liu, C., Li, W., Zhao, L., Nie, G., et al. (2022). Inhibiting DNA methylation alleviates cisplatin-induced hearing loss by decreasing oxidative stress-induced mitochondria-dependent apoptosis. Acta Pharm. Sin. B, 12, 1305–1321. doi: 10.1016/j.apsb.2021.11.002
Hepper, P. G., and Shahidullah, B. S. (1994). Development of fetal hearing. Arch. Dis. Child. Fetal Neonatal Ed. 71, F81–F87. doi: 10.1136/fn.71.2.F81
Hickox, A., Gao, Y., Francis, S. P., Asai, Y., Ng, R., Chiang, H., et al. (2021). Nonclinical In Vivo Expression, Durability of Effect, Biodistribution/Shedding, and Safety Evaluations Support Planned Clinical Development of AK-OTOF (AAVAnc80-hOTOF Vector) for OTOF-mediated Hearing Loss. Washington, DC: American Society of Gene and Cell Therapy.
Hickox, A. E., Gao, Y., Yu, F. f., Darcy, Y., Reape, K., Francis, S. P., et al. (2022). Durable Recovery of Auditory Function Following Intracochlear Delivery of AK-OTOF (AAVAnc80-hOTOF) in a Translationally Relevant Mouse Model of Otoferline Gene (OTOF)-mediated Hearing Loss. Baltimore, MD: American Society for Gene and Cell Therapy.
Hildebrand, M. S., Morín, M., Meyer, N. C., Mayo, F., Modamio-Hoybjor, S., Mencía, A., et al. (2011). DFNA8/12 caused by TECTA mutations is the most identified subtype of nonsyndromic autosomal dominant hearing loss. Hum. Mutat. 32, 825–834. doi: 10.1002/humu.21512
Honda, K., and Griffith, A. J. (2022). Genetic architecture and phenotypic landscape of SLC26A4-related hearing loss. Hum. Genet. 141, 455–464. doi: 10.1007/s00439-021-02311-1
Hordeaux, J., Buza, E. L., Dyer, C., Goode, T., Mitchell, T. W., Richman, L., et al. (2020). Adeno-associated virus-induced dorsal root ganglion pathology. Hum. Gene Ther. 31, 808–818. doi: 10.1089/hum.2020.167
Hrastinski, I., and Wilbur, R. B. (2016). Academic achievement of deaf and hard-of-hearing students in an ASL/English bilingual program. J. Deaf Stud. Deaf Educ. 21, 156–170. doi: 10.1093/deafed/env072
Hu, N., Rutherford, M. A., and Green, S. H. (2020). Protection of cochlear synapses from noise-induced excitotoxic trauma by blockade of Ca. Proc. Natl. Acad. Sci. USA. 117, 3828–3838. doi: 10.1073/pnas.1914247117
Huang, A. R., Jiang, K., Lin, F. R., Deal, J. A., and Reed, N. S. (2023). Hearing loss and dementia prevalence in older adults in the US. JAMA, 329, 171–173. doi: 10.1001/jama.2022.20954
Iizuka, T., Kamiya, K., Gotoh, S., Sugitani, Y., Suzuki, M., Noda, T., et al. (2015). Perinatal Gjb2 gene transfer rescues hearing in a mouse model of hereditary deafness. Hum. Mol. Genet. 24, 3651–3661. doi: 10.1093/hmg/ddv109
Isgrig, K., McDougald, D. S., Zhu, J., Wang, H. J., Bennett, J., and Chien, W. W. (2019). AAV2.7m8 is a powerful viral vector for inner ear gene therapy. Nat. Commun. 10, 427. doi: 10.1038/s41467-018-08243-1
Isgrig, K., Shteamer, J. W., Belyantseva, I. A., Drummond, M. C., Fitzgerald, T. S., Vijayakumar, S., et al. (2017). Gene therapy restores balance and auditory functions in a mouse model of usher syndrome. Mol. Ther. 25, 780–791. doi: 10.1016/j.ymthe.2017.01.007
Ivanchenko, M. V., Hanlon, K. S., Devine, M. K., Tenneson, K., Emond, F., Lafond, J. F., et al. (2020). Preclinical testing of AAV9-PHP.B for transgene expression in the non-human primate cochlea. Hear. Res. 394:107930. doi: 10.1016/j.heares.2020.107930
Ivanchenko, M. V., Hanlon, K. S., Hathaway, D. M., Klein, A. J., Peters, C. W., Li, Y., et al. (2021). AAV-S: a versatile capsid variant for transduction of mouse and primate inner ear. Mol. Ther. Methods Clin. Dev. 21, 382–398. doi: 10.1016/j.omtm.2021.03.019
Johnson Chacko, L., Blumer, M. J. F., Pechriggl, E., Rask-Andersen, H., Dietl, W., Haim, A., et al. (2017). Role of BDNF and neurotrophic receptors in human inner ear development. Cell Tissue Res. 370, 347–363. doi: 10.1007/s00441-017-2686-9
Johnson Chacko, L., Wertjanz, D., Sergi, C., Dudas, J., Fischer, N., Eberharter, T., et al. (2019). Growth and cellular patterning during fetal human inner ear development studied by a correlative imaging approach. BMC Dev. Biol. 19:11. doi: 10.1186/s12861-019-0191-y
Kamiya, K., Takahashi, K., Kitamura, K., Momoi, T., and Yoshikawa, Y. (2001). Mitosis and apoptosis in postnatal auditory system of the C3H/He strain. Brain Res. 901, 296–302. doi: 10.1016/S0006-8993(01)02300-9
Keithley, E. M. (2022). Inner ear immunity. Hear. Res. 419:108518. doi: 10.1016/j.heares.2022.108518
Kelley, M. W. (2007). Cellular commitment and differentiation in the organ of Corti. Int. J. Dev. Biol. 51, 571–583. doi: 10.1387/ijdb.072388mk
Khalin, I., Alyautdin, R., Kocherga, G., and Bakar, M. A. (2015). Targeted delivery of brain-derived neurotrophic factor for the treatment of blindness and deafness. Int. J. Nanomed. 10, 3245–3267. doi: 10.2147/IJN.S77480
Kilpatrick, L. A., Li, Q., Yang, J., Goddard, J. C., Fekete, D. M., and Lang, H. (2011). Adeno-associated virus-mediated gene delivery into the scala media of the normal and deafened adult mouse ear. Gene Ther. 18, 569–578. doi: 10.1038/gt.2010.175
Kim, B. J., Jang, J. H., Han, J. H., Park, H. R., Oh, D. Y., Lee, S., et al. (2018). Mutational and phenotypic spectrum of OTOF-related auditory neuropathy in Koreans: eliciting reciprocal interaction between bench and clinics. J. Transl. Med. 16:330. doi: 10.1186/s12967-018-1708-z
Kim, M. A., Cho, H. J., Bae, S. H., Lee, B., Oh, S. K., Kwon, T. J., et al. (2016). Methionine sulfoxide reductase B3-targeted in utero gene therapy rescues hearing function in a mouse model of congenital sensorineural hearing loss. Antioxid. Redox Signal. 24, 590–602. doi: 10.1089/ars.2015.6442
Klimara, M. J., Nishimura, C., Wang, D., Kolbe, D. L., Schaefer, A. M., Walls, W. D., et al. (2022). De novo variants are a common cause of genetic hearing loss. Genet. Med. 24, 2555–2567. doi: 10.1016/j.gim.2022.08.028
Koehler, S., Chung, Y., Pan, N., Becker, L., Cancelarich, S., Thompson, C., et al. (2023). Nonclinical Pharmacology, Biodistribution, and Safety Studies Supporting the Clinical Development of DB-OTO (AAV1-Myo15-hOTOFv5) for Hearing Loss due to Genetic Otoferlin Protein Deficiency. Orlando, FL: Association for Research in Otolaryngology Annual Mid-Winter Meeting.
Kolla, L., Kelly, M. C., Mann, Z. F., Anaya-Rocha, A., Ellis, K., Lemons, A., et al. (2020). Characterization of the development of the mouse cochlear epithelium at the single cell level. Nat. Commun. 11:2389. doi: 10.1038/s41467-020-16113-y
Kral, A. (2013). Auditory critical periods: a review from system's perspective. Neuroscience 247, 117–133. doi: 10.1016/j.neuroscience.2013.05.021
Kujawa, S. G., and Liberman, M. C. (2009). Adding insult to injury: cochlear nerve degeneration after “temporary” noise-induced hearing loss. J. Neurosci. 29, 14077–14085. doi: 10.1523/JNEUROSCI.2845-09.2009
Landegger, L. D., Pan, B., Askew, C., Wassmer, S. J., Gluck, S. D., Galvin, A., et al. (2017). A synthetic AAV vector enables safe and efficient gene transfer to the mammalian inner ear. Nat. Biotechnol. 35, 280–284. doi: 10.1038/nbt.3781
Le Quesne Stabej, P., Saihan, Z., Rangesh, N., Steele-Stallard, H. B., Ambrose, J., Coffey, A., et al. (2012). Comprehensive sequence analysis of nine Usher syndrome genes in the UK National collaborative usher study. J. Med. Genet. 49, 27–36. doi: 10.1136/jmedgenet-2011-100468
Le, T. N., Straatman, L. V., Lea, J., and Westerberg, B. (2017). Current insights in noise-induced hearing loss: a literature review of the underlying mechanism, pathophysiology, asymmetry, and management options. J. Otolaryngol. Head Neck Surg. 46:41. doi: 10.1186/s40463-017-0219-x
Leake, P. A., Rebscher, S. J., Dore., C, and Akil, O. (2019). AAV-mediated neurotrophin gene therapy promotes improved survival of cochlear spiral ganglion neurons in neonatally deafened cats: comparison of AAV2-hBDNF and AAV5-hGDNF. J. Assoc. Res. Otolaryngol. 20, 341–361. doi: 10.1007/s10162-019-00723-5
Lee, J., Nist-Lund, C., Solanes, P., Goldberg, H., Wu, J., Pan, B., et al. (2020). Efficient viral transduction in mouse inner ear hair cells with utricle injection and AAV9-PHP.B. Hear. Res. 394:107882. doi: 10.1016/j.heares.2020.107882
Lentz, J. J., Jodelka, F. M., Hinrich, A. J., McCaffrey, K. E., Farris, H. E., Spalitta, M. J., et al. (2013). Rescue of hearing and vestibular function by antisense oligonucleotides in a mouse model of human deafness. Nat. Med. 19, 345–350. doi: 10.1038/nm.3106
Li, S., Mecca, A., Kim, J., Caprara, G. A., Wagner, E. L., Du, T. T., et al. (2020). Myosin-VIIa is expressed in multiple isoforms and essential for tensioning the hair cell mechanotransduction complex. Nat. Commun. 11:2066. doi: 10.1038/s41467-020-15936-z
Liguore, W. A., Domire, J. S., Button, D., Wang, Y., Dufour, B. D., Srinivasan, S., et al. (2019). AAV-PHP.B administration results in a differential pattern of cns biodistribution in non-human primates compared with mice. Mol. Ther. 27, 2018–2037. doi: 10.1016/j.ymthe.2019.07.017
Lin, H. W., Furman, A. C., Kujawa, S. G., and Liberman, M. C. (2011). Primary neural degeneration in the Guinea pig cochlea after reversible noise-induced threshold shift. J. Assoc. Res. Otolaryngol. 12, 605–616. doi: 10.1007/s10162-011-0277-0
Liu, W., Xu, L., Wang, X., Zhang, D., Sun, G., Wang, M., et al. (2021). PRDX1 activates autophagy via the PTEN-AKT signaling pathway to protect against cisplatin-induced spiral ganglion neuron damage. Autophagy 17, 4159–4181. doi: 10.1080/15548627.2021.1905466
Liu, Y., Okada, T., Nomoto, T., Ke, X., Kume, A., Ozawa, K., et al. (2007). Promoter effects of adeno-associated viral vector for transgene expression in the cochlea in vivo. Exp. Mol. Med. 39, 170–175. doi: 10.1038/emm.2007.19
Liu, Y., Okada, T., Sheykholeslami, K., Shimazaki, K., Nomoto, T., Muramatsu, S., et al. (2005). Specific and efficient transduction of Cochlear inner hair cells with recombinant adeno-associated virus type 3 vector. Mol. Ther. 12, 725–733. doi: 10.1016/j.ymthe.2005.03.021
Liu, Y., Okada, T., Shimazaki, K., Sheykholeslami, K., Nomoto, T., Muramatsu, S. I., et al. (2008). Protection against aminoglycoside-induced ototoxicity by regulated AAV vector-mediated GDNF gene transfer into the cochlea. Mol. Ther. 16, 474–480. doi: 10.1038/sj.mt.6300379
Longo-Guess, C., Gagnon, L. H., Bergstrom, D. E., and Johnson, K. R. (2007). A missense mutation in the conserved C2B domain of otoferlin causes deafness in a new mouse model of DFNB9. Hear. Res. 234, 21–28. doi: 10.1016/j.heares.2007.09.005
Lonser, R. R., Akhter, A. S., Zabek, M., Elder, J. B., and Bankiewicz, K. S. (2020). Direct convective delivery of adeno-associated virus gene therapy for treatment of neurological disorders. J. Neurosurg. 134, 1751–1763. doi: 10.3171/2020.4.JNS20701
Lu, Y. C., Tsai, Y. H., Chan, Y. H., Hu, C. J., Huang, C. Y., Xiao, R., et al. (2022). Gene therapy with a synthetic adeno-associated viral vector improves audiovestibular phenotypes in Pjvk-mutant mice. JCI Insight 7:152941. doi: 10.1172/jci.insight.152941
Lv, J., Wang, H., Cheng, X., Chen, Y., Wang, D., Zhang, L., et al. (2024). AAV1-hOTOF gene therapy for autosomal recessive deafness 9: a single-arm trial. Lancet. 403, 2317–2325. doi: 10.1016/S0140-6736(23)02874-X
Marcovich, I., Baer, N. K., Shubina-Oleinik, O., Eclov, R., Beard, C. W., and Holt, J. R. (2022). Optimized AAV vectors for TMC1 gene therapy in a humanized mouse model of DFNB7/11. Biomolecules 12:914. doi: 10.3390/biom12070914
Mathiesen, B. K., Miyakoshi, L. M., Cederroth, C. R., Tserga, E., Versteegh, C., Bork, P. A. R., et al. (2023). Delivery of gene therapy through a cerebrospinal fluid conduit to rescue hearing in adult mice. Sci. Transl. Med. 15:eabq3916. doi: 10.1126/scitranslmed.abq3916
McGill, T. J., and Schuknecht, H. F. (1976). Human cochlear changes in noise induced hearing loss. Laryngoscope 86, 1293–1302. doi: 10.1288/00005537-197609000-00001
Mei, X., Glueckert, R., Schrott-Fischer, A., Li, H., Ladak, H. M., Agrawal, S. K., et al. (2020). Vascular supply of the human spiral ganglion: novel three-dimensional analysis using synchrotron phase-contrast imaging and histology. Sci. Rep. 10:5877. doi: 10.1038/s41598-020-62653-0
Michanski, S., Smaluch, K., Steyer, A. M., Chakrabarti, R., Setz, C., Oestreicher, D., et al. (2019). Mapping developmental maturation of inner hair cell ribbon synapses in the apical mouse cochlea. Proc. Natl. Acad. Sci. USA. 116, 6415–6424. doi: 10.1073/pnas.1812029116
Migliosi, V., Modamio-Høybjør, S., Moreno-Pelayo, M. A., Rodríguez-Ballesteros, M., Villamar, M., Tellería, D., et al. (2002). Q829X, a novel mutation in the gene encoding otoferlin (OTOF), is frequently found in Spanish patients with prelingual non-syndromic hearing loss. J. Med. Genet. 39, 502–506. doi: 10.1136/jmg.39.7.502
Miwa, T., Minoda, R., Ise, M., Yamada, T., and Yumoto, E. (2013). Mouse otocyst transuterine gene transfer restores hearing in mice with connexin 30 deletion-associated hearing loss. Mol. Ther. 21, 1142–1150. doi: 10.1038/mt.2013.62
Mohamed, T., Melfi, V., Colciago, A., and Magnaghi, V. (2023). Hearing loss and vestibular schwannoma: new insights into Schwann cells implication. Cell Death Dis. 14:629. doi: 10.1038/s41419-023-06141-z
Moke, D. J., Luo, C., Millstein, J., Knight, K. R., Rassekh, S. R., Brooks, B., et al. (2021). Prevalence and risk factors for cisplatin-induced hearing loss in children, adolescents, and young adults: a multi-institutional North American cohort study. Lancet Child Adolesc. Health 5, 274–283. doi: 10.1016/S2352-4642(21)00020-1
Montero, C. N., and Hefti, F. (1988). Rescue of lesioned septal cholinergic neurons by nerve growth factor: specificity and requirement for chronic treatment. J. Neurosci. 8, 2986–2999. doi: 10.1523/JNEUROSCI.08-08-02986.1988
Mukherjee, S., Kuroiwa, M., Oakden, W., Paul, B. T., Noman, A., Chen, J., et al. (2022). Local magnetic delivery of adeno-associated virus AAV2(quad Y-F)-mediated BDNF gene therapy restores hearing after noise injury. Mol. Ther. 30, 519–533. doi: 10.1016/j.ymthe.2021.07.013
Naidoo, J., Stanek, L. M., Ohno, K., Trewman, S., Samaranch, L., Hadaczek, P., et al. (2018). Extensive transduction and enhanced spread of a modified AAV2 capsid in the non-human primate CNS. Mol. Ther. 26, 2418–2430. doi: 10.1016/j.ymthe.2018.07.008
Nist-Lund, C. A., Pan, B., Patterson, A., Asai, Y., Chen, T., Zhou, W., et al. (2019). Improved TMC1 gene therapy restores hearing and balance in mice with genetic inner ear disorders. Nat. Commun. 10:236. doi: 10.1038/s41467-018-08264-w
Noh, B., Rim, J. H., Gopalappa, R., Lin, H., Kim, K. M., Kang, M. J., et al. (2022). outer hair cell gene editing ameliorates progressive hearing loss in dominant-negative. Theranostics 12, 2465–2482. doi: 10.7150/thno.67781
Nuzbrokh, Y., Ragi, S. D., and Tsang, S. H. (2021). Gene therapy for inherited retinal diseases. Ann. Transl. Med. 9:1278. doi: 10.21037/atm-20-4726
Oka, S. I., Day, T. F., Nishio, S. Y., Moteki, H., Miyagawa, M., Morita, S., et al. (2020). Clinical characteristics and in vitro analysis of MYO6 variants causing late-onset progressive hearing loss. Genes 11:273. doi: 10.3390/genes11030273
Pan, B., Askew, C., Galvin, A., Heman-Ackah, S., Asai, Y., Indzhykulian, A. A., et al. (2017). Gene therapy restores auditory and vestibular function in a mouse model of Usher syndrome type 1c. Nat. Biotechnol. 35, 264–272. doi: 10.1038/nbt.3801
Panda, S., Sikka, K., Singh, V., Agarwal, S., Kumar, R., Thakar, A., et al. (2019). Comprehensive analysis of factors leading to poor performance in prelingual cochlear implant recipients. Otol. Neurotol. 40, 754–760. doi: 10.1097/MAO.0000000000002237
Pearson, T. S., Gupta, N., San Sebastian, W., Imamura-Ching, J., Viehoever, A., Grijalvo-Perez, A., et al. (2021). Gene therapy for aromatic L-amino acid decarboxylase deficiency by MR-guided direct delivery of AAV2-AADC to midbrain dopaminergic neurons. Nat. Commun. 12:4251. doi: 10.1038/s41467-021-24524-8
Petit, C., Bonnet, C., and Safieddine, S. (2023). Deafness: from genetic architecture to gene therapy. Nat. Rev. Genet. 24, 665–686. doi: 10.1038/s41576-023-00597-7
Petrs-Silva, H., Dinculescu, A., Li, Q., Deng, W. T., Pang, J. J., Min, S. H., et al. (2011). Novel properties of tyrosine-mutant AAV2 vectors in the mouse retina. Mol. Ther. 19, 293–301. doi: 10.1038/mt.2010.234
Pfingst, B. E., Colesa, D. J., Swiderski, D. L., Hughes, A. P., Strahl, S. B., Sinan, M., et al. (2017). Neurotrophin gene therapy in deafened ears with cochlear implants: long-term effects on nerve survival and functional Measures. J. Assoc. Res. Otolaryngol. 18, 731–750. doi: 10.1007/s10162-017-0633-9
Puel, J. L., Ruel, J., Gervais d'Aldin, C., and Pujol, R. (1998). Excitotoxicity and repair of cochlear synapses after noise-trauma induced hearing loss. Neuroreport 9, 2109–2114. doi: 10.1097/00001756-199806220-00037
Pupo, A., Fernández, A., Low, S. H., François, A., Suárez-Amarán, L., and Samulski, R. J. (2022). AAV vectors: the Rubik's cube of human gene therapy. Mol. Ther. 30, 3515–3541. doi: 10.1016/j.ymthe.2022.09.015
Qi, J., Tan, F., Zhang, L., Lu, L., Zhang, S., Zhai, Y., et al. (2024a). AAV-mediated gene therapy restores hearing in patients with DFNB9 deafness. Adv. Sci. 11:e2306788. doi: 10.1002/advs.202306788
Qi, J., Zhang, L., Tan, F., Zhang, Y., Zhou, Y., Zhang, Z., et al. (2024b). Preclinical efficacy and safety evaluation of AAV-OTOF in DFNB9 mouse model and nonhuman primate. Adv. Sci. 11:e2306201. doi: 10.1002/advs.202306201
Rambeau, P., Olivier, G., Ba, C. T. V., Barrot, L., Boudra, R., Reis, J. D. D., et al. (2024). Preclinical Development of SENS-501 as a Treatment for Autosomal Recessive Non-Syndromic Deafness 9 (DFNB9) Using An Adeno Associated Vector-Based Gene Therapy. Anaheim, CA: Association for Research in Otolaryngology Annual Mid-Winter Meeting.
Ramekers, D., Versnel, H., Grolman, W., and Klis, S. F. (2012). Neurotrophins and their role in the cochlea. Hear. Res. 288, 19–33. doi: 10.1016/j.heares.2012.03.002
Rankovic, V., Vogl, C., Dörje, N. M., Bahader, I., Duque-Afonso, C. J., Thirumalai, A., et al. (2020). Overloaded adeno-associated virus as a novel gene therapeutic tool for otoferlin-related deafness. Front. Mol. Neurosci. 13:600051. doi: 10.3389/fnmol.2020.600051
Ranum, P. T., Tecedor, L., Keiser, M. S., Chen, Y. H., Leib, D. E., Liu, X., et al. (2023). Cochlear transduction via cerebrospinal fluid delivery of AAV in non-human primates. Mol. Ther. 31, 609–612. doi: 10.1016/j.ymthe.2022.12.018
Ren, Y., Chari, D. A., Vasilijic, S., Welling, D. B., and Stankovic, K. M. (2021). New developments in neurofibromatosis type 2 and vestibular schwannoma. Neurooncol. Adv. 3:vdaa153. doi: 10.1093/noajnl/vdaa153
Richardson, R. T., Thompson, A. C., Wise, A. K., Ajay, E. A., Gunewardene, N., O'Leary, S. J., et al. (2021). Viral-mediated transduction of auditory neurons with opsins for optical and hybrid activation. Sci. Rep. 11:11229. doi: 10.1038/s41598-021-90764-9
Rocco, M. T., Akhter, A. S., Ehrlich, D. J., Scott, G. C., Lungu, C., Munjal, V., et al. (2022). Long-term safety of MRI-guided administration of AAV2-GDNF and gadoteridol in the putamen of individuals with Parkinson's disease. Mol. Ther. 30, 3632–3638. doi: 10.1016/j.ymthe.2022.08.003
Ronner, E. A., Benchetrit, L., Levesque, P., Basonbul, R. A., and Cohen, M. S. (2020). Quality of life in children with sensorineural hearing loss. Otolaryngol. Head Neck Surg. 162, 129–136. doi: 10.1177/0194599819886122
Saeed, H. S., Fergie, M., Mey, K., West, N., Caye-Thomasen, P., Nash, R., et al. (2023). Enlarged vestibular aqueduct and associated inner ear malformations: hearing loss prognostic factors and data modeling from an international cohort. J. Int. Adv. Otol. 19, 454–460. doi: 10.5152/iao.2023.231044
Salegio, E. A., Samaranch, L., Kells, A. P., Mittermeyer, G., San Sebastian, W., Zhou, S., et al. (2013). Axonal transport of adeno-associated viral vectors is serotype-dependent. Gene Ther. 20, 348–352. doi: 10.1038/gt.2012.27
Salt, A. N., and Hirose, K. (2018). Communication pathways to and from the inner ear and their contributions to drug delivery. Hear. Res. 362, 25–37. doi: 10.1016/j.heares.2017.12.010
Samaranch, L., Blits, B., San Sebastian, W., Hadaczek, P., Bringas, J., Sudhakar, V., et al. (2017). MR-guided parenchymal delivery of adeno-associated viral vector serotype 5 in non-human primate brain. Gene Ther. 24, 253–261. doi: 10.1038/gt.2017.14
Samaranch, L., Salegio, E. A., San Sebastian, W., Kells, A. P., Bringas, J. R., Forsayeth, J., et al. (2013). Strong cortical and spinal cord transduction after AAV7 and AAV9 delivery into the cerebrospinal fluid of nonhuman primates. Hum. Gene Ther. 24, 526–532. doi: 10.1089/hum.2013.005
Samaranch, L., Sebastian, W. S., Kells, A. P., Salegio, E. A., Heller, G., Bringas, J. R., et al. (2014). AAV9-mediated expression of a non-self protein in nonhuman primate central nervous system triggers widespread neuroinflammation driven by antigen-presenting cell transduction. Mol. Ther. 22, 329–337. doi: 10.1038/mt.2013.266
Schwartz, P. J., Spazzolini, C., Crotti, L., Bathen, J., Amlie, J. P., Timothy, K., et al. (2006). The Jervell and Lange-Nielsen syndrome: natural history, molecular basis, and clinical outcome. Circulation 113, 783–790. doi: 10.1161/CIRCULATIONAHA.105.592899
Sharma, N., Kumari, D., Panigrahi, I., and Khetarpal, P. (2023). A systematic review of the monogenic causes of Non-Syndromic Hearing Loss (NSHL) and discussion of Current Diagnosis and Treatment options. Clin. Genet. 103, 16–34. doi: 10.1111/cge.14228
Shave, S., Botti, C., and Kwong, K. (2022). Congenital sensorineural hearing loss. Pediatr. Clin. North Am. 69, 221–234. doi: 10.1016/j.pcl.2021.12.006
Shibata, S. B., Cortez, S. R., Beyer, L. A., Wiler, J. A., Di Polo, A., Pfingst, B. E., et al. (2010). Transgenic BDNF induces nerve fiber regrowth into the auditory epithelium in deaf cochleae. Exp. Neurol. 223, 464–472. doi: 10.1016/j.expneurol.2010.01.011
Shibata, S. B., Osumi, Y., Yagi, M., Kanda, S., Kawamoto, K., Kuriyama, H., et al. (2007). Administration of amitriptyline attenuates noise-induced hearing loss via glial cell line-derived neurotrophic factor (GDNF) induction. Brain Res. 1144, 74–81. doi: 10.1016/j.brainres.2007.01.090
Shibata, S. B., Yoshimura, H., Ranum, P. T., Goodwin, A. T., and Smith, R. J. H. (2017). Intravenous rAAV2/9 injection for murine cochlear gene delivery. Sci. Rep. 7:9609. doi: 10.1038/s41598-017-09805-x
Shoji, F., Yamasoba, T., Magal, E., Dolan, D. F., Altschuler, R. A., and Miller, J. M. (2000). Glial cell line-derived neurotrophic factor has a dose dependent influence on noise-induced hearing loss in the guinea pig cochlea. Hear. Res. 142, 41–55. doi: 10.1016/S0378-5955(00)00007-1
Shu, Y., Tao, Y., Wang, Z., Tang, Y., Li, H., Dai, P., et al. (2016). Identification of adeno-associated viral vectors that target neonatal and adult mammalian inner ear cell subtypes. Hum. Gene Ther. 27, 687–699. doi: 10.1089/hum.2016.053
Shubina-Oleinik, O., Nist-Lund, C., French, C., Rockowitz, S., Shearer, A. E., and Holt, J. R. (2021). Dual-vector gene therapy restores cochlear amplification and auditory sensitivity in a mouse model of DFNB16 hearing loss. Sci. Adv. 7:eabi7629. doi: 10.1126/sciadv.abi7629
Simons, E. J., Germiller, J. A., Haag, O., Wu, C.-C., Hickox, A. E., Valero, M. D., et al. (2024). Clinical development of AK-OTOF gene therapy for OTOF -mediated hearing loss. Anaheim, CA: Association for Research in Otolaryngology Mid-Winter Meeting.
Sloan-Heggen, C. M., Bierer, A. O., Shearer, A. E., Kolbe, D. L., Nishimura, C. J., Frees, K. L., et al. (2016). Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum. Genet. 135, 441–450. doi: 10.1007/s00439-016-1648-8
So, H., Kim, H., Lee, J. H., Park, C., Kim, Y., Kim, E., et al. (2007). Cisplatin cytotoxicity of auditory cells requires secretions of proinflammatory cytokines via activation of ERK and NF-kappaB. J. Assoc. Res. Otolaryngol. 8, 338–355. doi: 10.1007/s10162-007-0084-9
Stöver, T., Gong, T. L., Cho, Y., Altschuler, R. A., and Lomax, M. I. (2000). Expression of the GDNF family members and their receptors in the mature rat cochlea. Brain Res. Mol. Brain Res. 76, 25–35. doi: 10.1016/S0169-328X(99)00328-9
Suzuki, J., Corfas, G., and Liberman, M. C. (2016). Round-window delivery of neurotrophin 3 regenerates cochlear synapses after acoustic overexposure. Sci. Rep. 6:24907. doi: 10.1038/srep24907
Suzuki, J., Hashimoto, K., Xiao, R., Vandenberghe, L. H., and Liberman, M. C. (2017). Cochlear gene therapy with ancestral AAV in adult mice: complete transduction of inner hair cells without cochlear dysfunction. Sci. Rep. 7:45524. doi: 10.1038/srep45524
Taiber, S., Cohen, R., Yizhar-Barnea, O., Sprinzak, D., Holt, J. R., and Avraham, K. B. (2021). Neonatal AAV gene therapy rescues hearing in a mouse model of SYNE4 deafness. EMBO Mol. Med. 13:e13259. doi: 10.15252/emmm.202013259
Takada, Y., Beyer, L. A., Swiderski, D. L., O'Neal, A. L., Prieskorn, D. M., Shivatzki, S., et al. (2014). Connexin 26 null mice exhibit spiral ganglion degeneration that can be blocked by BDNF gene therapy. Hear. Res. 309, 124–135. doi: 10.1016/j.heares.2013.11.009
Tan, F., Chu, C., Qi, J., Li, W., You, D., Li, K., et al. (2019). AAV-ie enables safe and efficient gene transfer to inner ear cells. Nat. Commun. 10:3733. doi: 10.1038/s41467-019-11687-8
Tang, H., Wang, H., Wang, S., Hu, S. W., Lv, J., Xun, M., et al. (2023). Hearing of Otof-deficient mice restored by trans-splicing of N- and C-terminal otoferlin. Hum. Genet. 142, 289–304. doi: 10.1007/s00439-022-02504-2
Tao, Y., Huang, M., Shu, Y., Ruprecht, A., Wang, H., Tang, Y., et al. (2018). Delivery of adeno-associated virus vectors in adult mammalian inner-ear cell subtypes without auditory dysfunction. Hum. Gene Ther. 29, 492–506. doi: 10.1089/hum.2017.120
Tao, Y., Lamas, V., Du, W., Zhu, W., Li, Y., Whittaker, M. N., et al. (2023). Treatment of monogenic and digenic dominant genetic hearing loss by CRISPR-Cas9 ribonucleoprotein delivery in vivo. Nat. Commun. 14:4928. doi: 10.1038/s41467-023-40476-7
Toms, M., Pagarkar, W., and Moosajee, M. (2020). Usher syndrome: clinical features, molecular genetics and advancing therapeutics. Ther. Adv. Ophthalmol. 12:2515841420952194. doi: 10.1177/2515841420952194
Vahava, O., Morell, R., Lynch, E. D., Weiss, S., Kagan, M. E., Ahituv, N., et al. (1998). Mutation in transcription factor POU4F3 associated with inherited progressive hearing loss in humans. Science 279, 1950–1954. doi: 10.1126/science.279.5358.1950
Valayannopoulos, V. (2023). Nonclinical Pharmacology, Biodistribution, and Safety Studies Supporting the Clinical Development of DB- OTO (AAV1-Myo15-hOTOFv5) for Hearing Loss Due to Genetic Otoferlin Protein Deficiency. Los Angeles, CA: American Society of Gene and Cell Therapy Annual Meeting.
van Ruijven, M. W., de Groot, J. C., Klis, S. F., and Smoorenburg, G. F. (2005). The cochlear targets of cisplatin: an electrophysiological and morphological time-sequence study. Hear. Res. 205, 241–248. doi: 10.1016/j.heares.2005.03.023
van Ruijven, M. W., de Groot, J. C., and Smoorenburg, G. F. (2004). Time sequence of degeneration pattern in the guinea pig cochlea during cisplatin administration. A quantitative histological study. Hear. Res. 197, 44–54. doi: 10.1016/j.heares.2004.07.014
Vojdani, S., Amirsalari, S., Milanizadeh, S., Molaei, F., Ajalloueyane, M., Khosravi, A., et al. (2019). Mutation screening of KCNQ1 and KCNE1 genes in Iranian patients with jervell and lange-nielsen syndrome. Fetal Pediatr. Pathol. 38, 273–281. doi: 10.1080/15513815.2019.1585500
Vona, B., Nanda, I., Hofrichter, M. A., Shehata-Dieler, W., and Haaf, T. (2015). Non-syndromic hearing loss gene identification: a brief history and glimpse into the future. Mol. Cell. Probes, 29, 260–270. doi: 10.1016/j.mcp.2015.03.008
Wagner, E. L., and Shin, J. B. (2019). Mechanisms of hair cell damage and repair. Trends Neurosci. 42, 414–424. doi: 10.1016/j.tins.2019.03.006
Walls, W., Azaiez, H., and Smith, R. (2024). Hereditary Hearing Loss Homepage. Available online at: https://hereditaryhearingloss.org (accessed April 24, 2024).
Wang, D., Tai, P. W. L., and Gao, G. (2019). Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 18, 358–378. doi: 10.1038/s41573-019-0012-9
Wang, H., Du, H., Ren, R., Du, T., Lin, L., Feng, Z., et al. (2023a). Temporal and spatial assembly of inner ear hair cell ankle link condensate through phase separation. Nat. Commun. 14:1657. doi: 10.1038/s41467-023-37267-5
Wang, H., Murphy, R., Taaffe, D., Yin, S., Xia, L., Hauswirth, W. W., et al. (2012). Efficient cochlear gene transfection in guinea-pigs with adeno-associated viral vectors by partial digestion of round window membrane. Gene Ther. 19, 255–263. doi: 10.1038/gt.2011.91
Wang, H., Wu, K., Yu, L., Xie, L., Xiong, W., Wang, D., et al. (2017). A novel dominant GJB2 (DFNA3) mutation in a Chinese family. Sci. Rep. 7:34425. doi: 10.1038/srep34425
Wang, H., Xun, M., Tang, H., Zhao, J., Hu, S., Zhang, L., et al. (2024). Hair cell-specific Myo15 promoter-mediated gene therapy rescues hearing in DFNB9 mouse model. Mol. Ther. Nucleic Acid 35:2135. doi: 10.1016/j.omtn.2024.102135
Wang, J., Zhao, L., Gu, X., Xue, Y., Wang, S., Xiao, R., et al. (2022). Efficient delivery of adeno-associated virus into inner ear. Hum. Gene Ther. 33, 719–728. doi: 10.1089/hum.2021.236
Wang, L., Kempton, J. B., Jiang, H., Jodelka, F. M., Brigande, A. M., Dumont, R. A., et al. (2020). Fetal antisense oligonucleotide therapy for congenital deafness and vestibular dysfunction. Nucleic Acids Res. 48, 5065–5080. doi: 10.1093/nar/gkaa194
Wang, X., Zhou, Y., Wang, D., Wang, Y., Zhou, Z., Ma, X., et al. (2023b). Cisplatin-induced ototoxicity: From signaling network to therapeutic targets. Biomed. Pharmacother. 157:114045. doi: 10.1016/j.biopha.2022.114045
Wémeau, J. L., and Kopp, P. (2017). Pendred syndrome. Best Pract. Res. Clin. Endocrinol. Metab. 31, 213–224. doi: 10.1016/j.beem.2017.04.011
Wu, J., Cao, Z., Su, Y., Wang, Y., Cai, R., Chen, J., et al. (2022). Molecular diagnose of a large hearing loss population from China by targeted genome sequencing. J. Hum. Genet. 67, 643–649. doi: 10.1038/s10038-022-01066-5
Wu, J., Solanes, P., Nist-Lund, C., Spataro, S., Shubina-Oleinik, O., Marcovich, I., et al. (2021a). Single and dual vector gene therapy with AAV9-PHP.B rescues hearing in TMC1 mutant mice. Mol. Ther. 29, 973–988. doi: 10.1016/j.ymthe.2020.11.016
Wu, X., Zhang, L., Li, Y., Zhang, W., Wang, J., Cai, C., et al. (2021b). Gene therapy via canalostomy approach preserves auditory and vestibular functions in a mouse model of Jervell and Lange-Nielsen syndrome type 2. Nat. Commun. 12:697. doi: 10.1038/s41467-020-20808-7
Xue, Y., Hu, X., Wang, D., Li, D., Li, Y., Wang, F., et al. (2022). Gene editing in a Myo6 semi-dominant mouse model rescues auditory function. Mol. Ther. 30, 105–118. doi: 10.1016/j.ymthe.2021.06.015
Yamasoba, T., Yagi, M., Roessler, B. J., Miller, J. M., and Raphael, Y. (1999). Inner ear transgene expression after adenoviral vector inoculation in the endolymphatic sac. Hum. Gene Ther. 10, 769–774. doi: 10.1089/10430349950018526
Yoshimura, H., Shibata, S. B., Ranum, P. T., Moteki, H., and Smith, R. J. H. (2019). Targeted Allele Suppression Prevents Progressive Hearing Loss in the Mature Murine Model of Human TMC1 Deafness. Mol. Ther. 27, 681–690. doi: 10.1016/j.ymthe.2018.12.014
Yoshimura, H., Shibata, S. B., Ranum, P. T., and Smith, R. J. H. (2018). Enhanced viral-mediated cochlear gene delivery in adult mice by combining canal fenestration with round window membrane inoculation. Sci. Rep. 8:2980. doi: 10.1038/s41598-018-21233-z
Yu, Q., Wang, Y., Chang, Q., Wang, J., Gong, S., Li, H., et al. (2014). Virally expressed connexin26 restores gap junction function in the cochlea of conditional Gjb2 knockout mice. Gene Ther. 21, 71–80. doi: 10.1038/gt.2013.59
Yu, X., Man, R., Li, Y., Yang, Q., Li, H., Yang, H., et al. (2019). Paeoniflorin protects spiral ganglion neurons from cisplatin-induced ototoxicity: possible relation to PINK1/BAD pathway. J. Cell. Mol. Med. 23, 5098–5107. doi: 10.1111/jcmm.14379
Zhang, L., Wang, H., Xun, M., Tang, H., Wang, J., Lv, J., et al. (2023). Preclinical evaluation of the efficacy and safety of AAV1-hOTOF in mice and nonhuman primates. Mol. Ther. Methods Clin. Dev. 31:101154. doi: 10.1016/j.omtm.2023.101154
Zhang, L., Wang, W., Kim, S. M., Wang, J., Zhou, B., Kong, W., et al. (2022). Virally mediated connexin 26 expression in postnatal scala media significantly and transiently preserves hearing in connexin 30 null mice. Front. Cell Dev. Biol. 10:900416. doi: 10.3389/fcell.2022.969989
Zhao, X., Liu, H., Cai, R., and Wu, H. (2022a). Gene therapy restores auditory functions in an adult vglut3 knockout mouse model. Hum. Gene Ther. 33, 729–739. doi: 10.1089/hum.2022.062
Zhao, Y., Zhang, L., Wang, D., Chen, B., and Shu, Y. (2022b). Approaches and vectors for efficient cochlear gene transfer in adult mouse models. Biomolecules 13:38. doi: 10.3390/biom13010038
Zhu, J., Choi, J. W., Ishibashi, Y., Isgrig, K., Grati, M., Bennett, J., et al. (2021). Refining surgical techniques for efficient posterior semicircular canal gene delivery in the adult mammalian inner ear with minimal hearing loss. Sci. Rep. 11:18856. doi: 10.1038/s41598-021-98412-y
Keywords: gene therapy, sensorineural hearing loss, AAV vectors, clinical trials, spiral ganglion neurons, hair cells
Citation: Duhon BH, Bielefeld EC, Ren Y and Naidoo J (2024) Gene therapy advancements for the treatment of acquired and hereditary hearing loss. Front. Audiol. Otol. 2:1423853. doi: 10.3389/fauot.2024.1423853
Received: 26 April 2024; Accepted: 27 June 2024;
Published: 17 July 2024.
Edited by:
Xuezhong Liu, University of Miami, United StatesReviewed by:
Jun Suzuki, Tohoku University, JapanCopyright © 2024 Duhon, Bielefeld, Ren and Naidoo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jerusha Naidoo, amVydXNoYS5uYWlkb29Ab3N1bWMuZWR1; Yin Ren, eWluLnJlbkBvc3VtYy5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.