
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Audiol. Otol., 18 June 2024
Sec. Auditory Science
Volume 2 - 2024 | https://doi.org/10.3389/fauot.2024.1400991
This article is part of the Research TopicThe Noncoding Genome in Auditory Function and Hearing LossView all 3 articles
 Yu Huang1,2†
Yu Huang1,2† Jia Geng1,2†Yang Long3Wenyu Xiong1,2
Jia Geng1,2†Yang Long3Wenyu Xiong1,2 Lu Kang1,2Meilin Chen1,2Ting Tang1,2Mingjun Zhong1,2
Lu Kang1,2Meilin Chen1,2Ting Tang1,2Mingjun Zhong1,2 Fengxiao Bu1,2
Fengxiao Bu1,2 Yu Lu1,2
Yu Lu1,2 Jing Cheng1,2*Huijun Yuan1,2*
Jing Cheng1,2*Huijun Yuan1,2*Introduction: Waardenburg syndrome (WS) is a genetic disorder characterized by hearing loss, hypopigmentation, and distinct facial features. Despite > 60% molecular diagnosis rate for WS patients, pathogenic variants within coding regions are predominant, with few non-coding copy number variations (CNVs) reported.
Methods: In this study, we performed whole genome sequencing (WGS) on 59 undiagnosed WS patients and analyzed the CNVs within the promoter and enhancer regions of the SOX10 gene.
Results: We identified five novel pathogenic deletions ranging from 448 bp to 70 kb upstream of SOX10. Two deletions were in the enhancer region, while three were in the promoter and 5'UTR region. These CNVs manifested as WS type II in eight patients from five unrelated families, demonstrating phenotypic heterogeneity. Furthermore, analysis of CNV1 within the enhancer region suggested a potential mechanism involving Alu-mediated non-allelic homologous recombination (NAHR).
Conclusion: Our findings extend the mutation spectrum of the SOX10 gene and elucidate the pathogenic role of CNVs in cis-regulatory elements, particularly variations in enhancer and promoter regions, thereby enhancing clinical gene detection and interpretation of non-coding regions.
Waardenburg syndrome (WS) is a rare auditory-pigmentary disorder characterized by various combinations of sensorineural hearing loss and pigmentation anomalies. It is estimated to affect approximately 1 in 42,000 live births, representing 1–3% of all congenital deafness patients (Read and Newton, 1997). Four subtypes have been clinically defined based on the presence or absence of additional symptoms, with related causal genes reported: WS1 (OMIM # 193500) or WS3 (OMIM # 148820), resulting from pathogenic variants in PAX3 (Foy et al., 1990; Tassabehji et al., 1992), marked by dystopia canthorum; WS2 (OMIM # 193510, 608890, 611584), caused by mutations in SOX10 (Bondurand et al., 2007), KITLG (Ogawa et al., 2017), MITF (Hughes et al., 1994; Tassabehji et al., 1994), or SNAI2 (Sánchez-Martín et al., 2002); and WS4 (OMIM # 277580, 613265, 613266), arising from mutations in SOX10 (Pingault et al., 1998), EDN3 (Edery et al., 1996), or EDNRB (Attié et al., 1995; Van Camp et al., 1995), often accompanied by Hirschsprung disease (HSCR).
Current diagnoses of these genes explain approximately 61%−71% of WS cases (Ideura et al., 2019; Batissoco et al., 2022; Wang et al., 2022). Within WS patients who undergo molecular diagnoses, the detection of pathogenic variants in the SOX10 gene constitutes a significant proportion, approximately 1/3. While most studies have focused on mutations in the coding region of the genome, non-coding variations may also play an important role. Bondurand et al. (2012) and Lecerf et al. (2014) observed that deletions in the SOX10 enhancers could contribute to the development of WS4 (Bondurand et al., 2012) or HSCR (Lecerf et al., 2014). In vitro experiments demonstrated a significant reduction in SOX10 transactivation capacity with the enhancer mutant construct compared to the wild-type (Lecerf et al., 2014).
Predicting the effects of non-coding variants poses challenges. Given that non-coding regions comprise about 98% of the genome, pinpointing functionally significant genomic areas becomes particularly daunting (Spielmann and Mundlos, 2016). Non-coding variants influence gene expression levels and cellular processes by modulating regulatory elements like promoters, enhancers, and non-coding RNAs. Previous investigations have examined and compared SOX10 enhancers across various species, including humans, mouse, and zebrafish, revealing 12 enhancers located upstream of SOX10 and 2 downstream of the gene (Werner et al., 2007; Antonellis et al., 2008; Betancur et al., 2010). These identified SOX10-related regulatory elements facilitate comprehension of variant pathogenicity within these regions.
Approximately 50% of the non-coding region of the human genome consists of repetitive elements such as LINE-1 and Alu sequences, which exhibit high homology and may disrupt genomic stability, leading to CNV formation through mechanisms like non-allelic homologous recombination (NAHR) or non-homologous end joining (NHEJ). The deletion involved these repeat elements and NAHR of TMC1 has been reported in non-syndromic hearing loss (NSHL) patients (Shearer et al., 2014).
Evaluation of gene dosage sensitivity is crucial for classifying associated CNVs. Heterozygous deletions of SOX10 detected in WS2 and WS4 patients, along with mutations resulting in mRNA reduction via nonsense-mediated decay (NMD) pathway reported in WS4 patients, confirm the haploinsufficiency (HI) of SOX10 in relation to WS (Pingault et al., 1998; Southard-Smith et al., 1999; Inoue et al., 2004).
In this study, we utilized WGS data from 59 WS patients to investigate CNVs affecting upstream regulatory elements of the SOX10 gene. We identified five non-coding CNVs in enhancer and promoter regions, further demonstrating the pathogenicity of non-coding CNVs in WS and proving to be indispensable for WGS-based patient reporting.
Initiated in 2013, the CDGC (n = 20,666) project is dedicated to elucidating the genetic underpinnings of hearing loss and associated syndromes. The project enlisted individuals with severe HL (pure tone audiometry, PTA > 40 dB) from specialized educational institutions, centers for deaf children rehabilitation, and hospitals across mainland China. Prior to participation, all patients or guardians provided written informed consent, which was sanctioned by the ethical committees of West China Hospital and Southwest Hospital. Clinical information and peripheral blood samples were collected from probands and their family members.
Within the CDGC cohort, 290 patients from 228 families were diagnosed with WS. Diagnosis followed the clinical criteria outlined by the Waardenburg Consortium (Milunsky, 1993). Major criteria include congenital sensorineural hearing loss, white forelock, iris pigmentation abnormalities, dystopia canthorum (W index > 1.95), and affected first-degree relatives. Minor criteria comprised skin hypopigmentation, synophrys, broad nasal root, underdeveloped alae nasi, and premature graying of hair (age < 30 years). A diagnosis of WS1 required two major criteria or one major and two minor criteria in a proband; WS2 excluded dystopia canthorum; WS3 included musculoskeletal upper limb abnormalities in addition to WS1 criteria; WS4 included aganglionic megacolon in addition to WS2 criteria.
Genomic DNA was isolated from whole blood samples utilizing the QIAamp DNA Blood Kit (Qiagen, Limburg, the Netherlands). Exons and 50 flanking base pairs of 157 targeted genes in the TGS-HL Panel including SOX10, PAX3, MITF, EDNRB, EDN3, SNAI2 and KITLG, were captured and enriched using Agilent SureSelect Target Enrichment Kit (Agilent Technologies Inc, Santa Clara, CA, USA). High throughput sequencing was performed as 2 × 150 bp paired-end reads, following the manufacture's protocol, on Illumina HiSeq 2000 sequencers (Illumina Inc, San Diego, CA, USA). The CNVplex® assay (Genesky Diagnostics Inc., Suzhou, Jiangsu, China) was used to validate the potential copy number variation (CNV) across hearing loss genes (Zhang et al., 2015). All variants underwent systematic evaluation and classification into five categories: (1) pathogenic, (2) likely pathogenic (3), uncertain significance, (4) likely benign, or (5) benign, in accordance with the ACMG/AMP Variant Interpretation Guidelines for Genetic Hearing Loss (Oza et al., 2018).
For this study, we enrolled 59 WS patients from 51 families lacking genetic diagnoses within the coding regions of SOX10, PAX3, MITF, EDNRB, EDN3, SNAI2, and KITLG, and workflow performed in Figure 1. Additionally, 8,674 hearing loss patients from the CDGC cohort, who lacked genetic diagnoses among the 157 HL-related genes, were recruited.
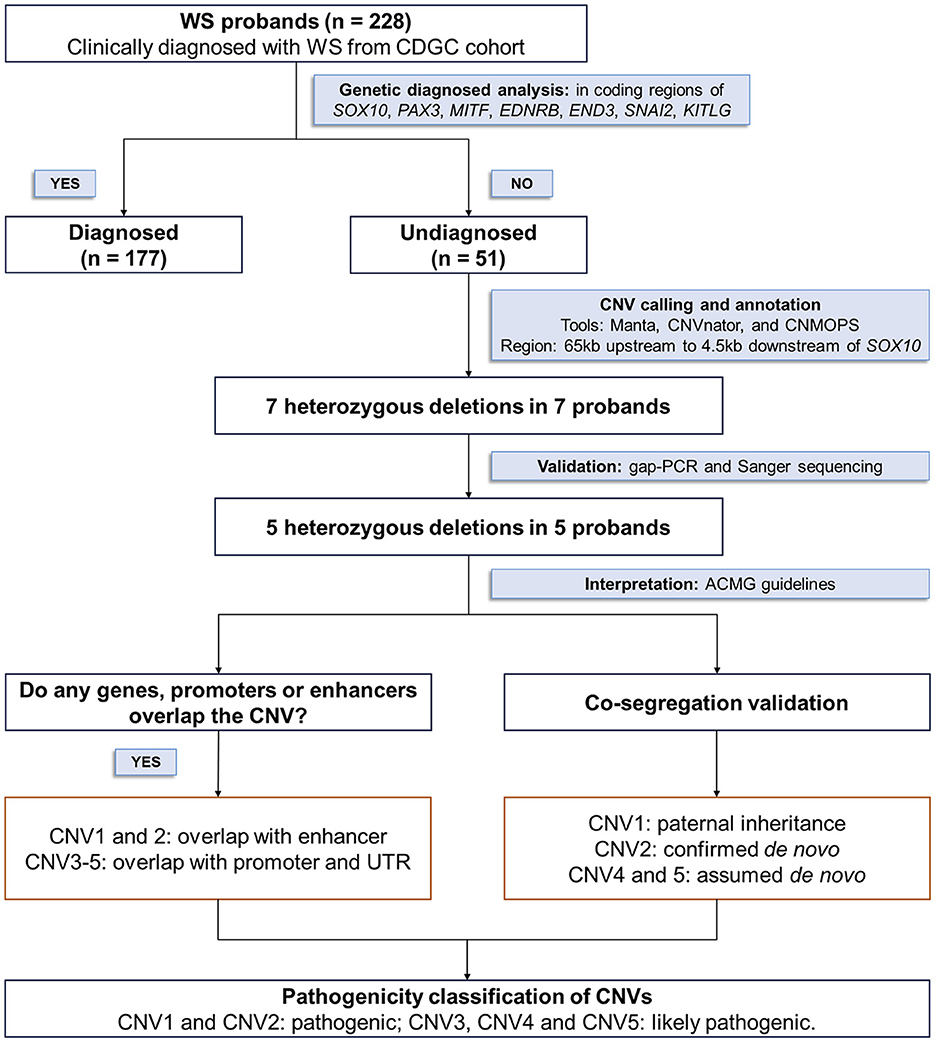
Figure 1. Strategy for investigating the pathogenicity of SOX10 non-coding CNVs.
Following this, DNA fragments from probands were size-selected within the range of 300-420 bp using the LE220-plus Focused-ultrasonicator and Agilent 4200 Bioanalyzer. A library was prepared to generate DNA nano balls (DNBs) according to the manufacturer's instructions from the DNBSEQ-T7RS High-throughput Sequencing Set V2.0 using PE150 (MGI, Shenzhen, Guangdong, China). WGS was performed on the MGI DNBSEQ-T7 sequencer at the Genomic Center, Research Core Facility of West China Hospital. To generate clean reads, the sequencing output Fastq file underwent Trimmomatic (v0.36) for the removal of adapters and low-quality bases. The cleaned reads were then mapped to the Human genome (Hg38/GRCh38) using the Burrows Wheeler Aligner (BWA, v0.5.10).
CNVs in WGS data were identified using Manta, CNVnator, and CNMOPS algorithms. Annotation of CNVs was subsequently carried out using ANNOVAR, with reference to public databases including RefSeq, gnomAD (CNVs v4.0), the 1000 Genomes Project, and the Database of Genomic Variants (DGV). Filtering criteria comprised the following: (1) allele frequencies < 1% in gnomAD and the 1000 Genomes Project; (2) < 50% reciprocal overlap with known DGV events of the same type; (3) < 25% overlap with repetitive and low-complexity regions (RLCRs); and 4) CNV read depth ranging between 10X and 300X. Visual confirmation of CNVs was carried out using the IGV_2.16.2 tool to visualize BAM format files derived from WGS data. Interpretation of CNVs adhered to the guidelines established by the American College of Medical Genetics and Genomics (ACMG) in 2020 (Riggs et al., 2020).
CNV breakpoints and co-segregation in pedigrees were additionally confirmed through gap-PCR and Sanger sequencing. Gap-PCR was carried out utilizing the TaKaRa Taq™ Hot Start Version (Takara, Japan) on the SimpliAmp™ Thermal Cycler (Applied Biosystems, Foster City, CA, USA). Primer sequences for both gap-PCR and sequencing are detailed in Supplementary Table 1. Sequencing data were precisely interpreted by alignment with reference sequences sourced from the UCSC Genome Browser (https://genome.ucsc.edu/) using SnapGene v4.1.9 software.
A total of 59 WS patients from 51 Chinese families, previously undiagnosed by known WS genes, were recruited for this study. These patients were selected from a sub cohort of 290 WS patients in the CDGC who met the clinical diagnostic criteria. WGS was employed to detect CNVs within regulator regions spanning from 65kb upstream to 4.5kb downstream of the SOX10 gene, which includes the promoter, 5'UTR, and enhancers (U1/MCS7, U2/MCS5, U3/MCS4, U4, U5/MCS2, D6, D7, MCS1, MCS3, MCS6, MCS8, and MCS9, as illustrated in Figure 2A). The enhancer elements MCS1-9 and U1-5, D6, and D7 were all derived from previous studies, where these enhancers were identified in the genomes of mouse and zebrafish, respectively. We aligned these elements with the human genome using sequence conservation.
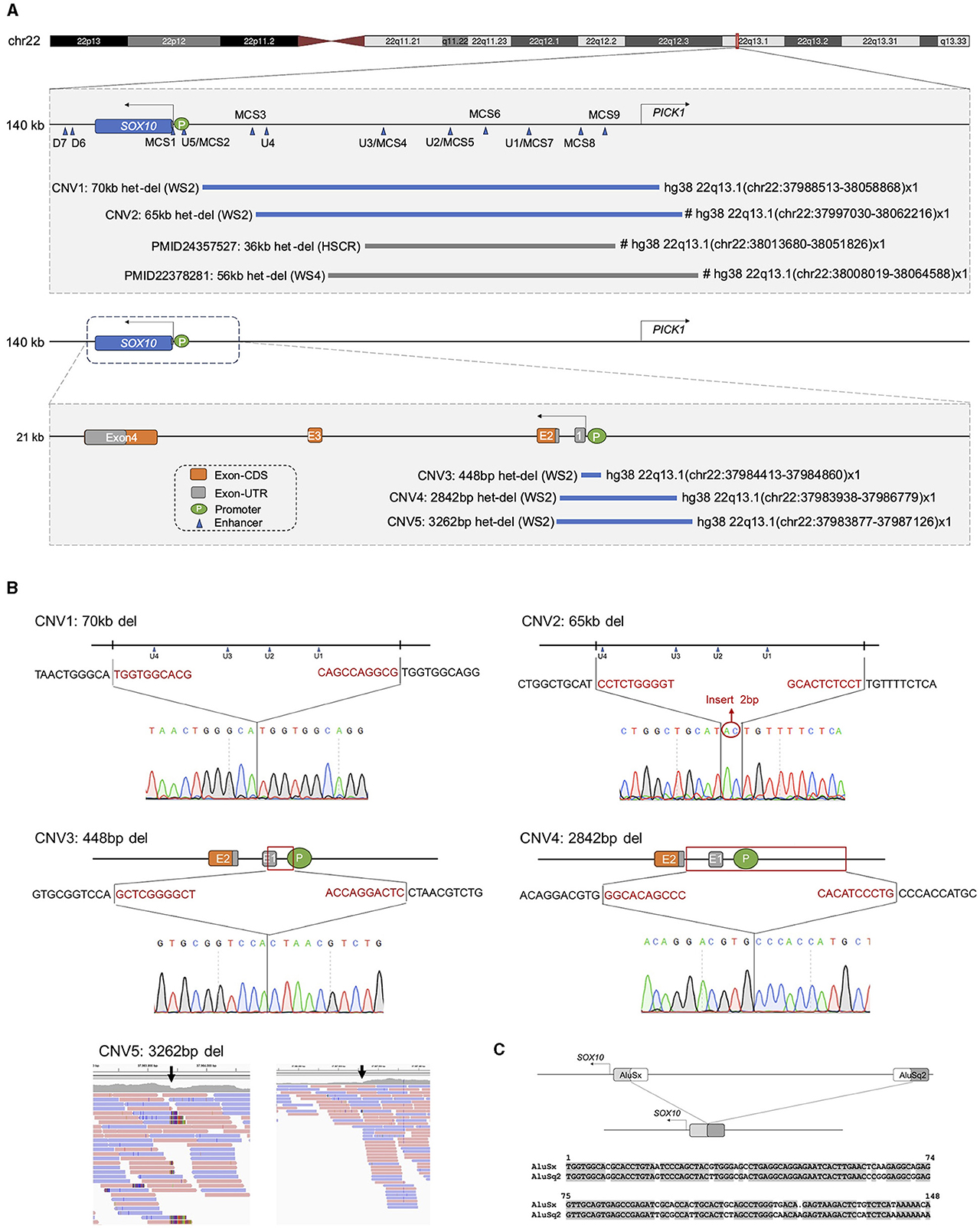
Figure 2. Identification and validation of deletions affecting SOX10 regulators in 5 families. (A) Schematic diagram illustrating the region spanning from the centromere to the telomere, which encompasses the SOX10 and PICK1 genes. Blue triangles mark the enhancers, where enhancers consist of two types reported in previous literature (MCS indicates multiple-species conserved sequences of regulatory regions; U/D represents upstream and downstream enhancers). Five heterozygous deletions varied from 448bp to 70 kb. De novo deletion is indicated with “#.” “het-del” means “heterozygous deletion”; “WS2” means “Waardenburg syndrome Type2”; “HSCR” means “Hirschsprung's disease”; “WS4” means “Waardenburg syndrome Type4.” (B) Breakpoint junction sequence of five probands with deletion of SOX10. CNV1-4: sanger sequencing; CNV5: BAM file screenshot from WGS. An insertion of 2bp was also identified in CNV2. (C) Top: Schematic representation of AluSx and AluSq2 from the centromere to telomere, and deletion detected in CNV1. Fragments of AluSx and AluSq2 maintained during the deletion event are indicated in gray. Bottom: Alignment of AluSx and AluSq2 sequences. Gray highlights indicate conserved nucleotides.
By combining the outcomes of three analysis software tools (Manta, CNVnator, and CNMOPS), we detected 7 heterozygous deletions in 10 WS patients from 7 families. Validation experiments were conducted to verify the reliability of WGS BAM files, gap-PCR, and co-segregation Sanger sequencing in the families. Subsequent analysis revealed that two CNVs were false positives in the WGS BAM files. Therefore, only 5 deletions (Table 1 and Figure 2A) underwent further scrutiny for pathogenicity, following the ACMG & ClinGen technical standards.
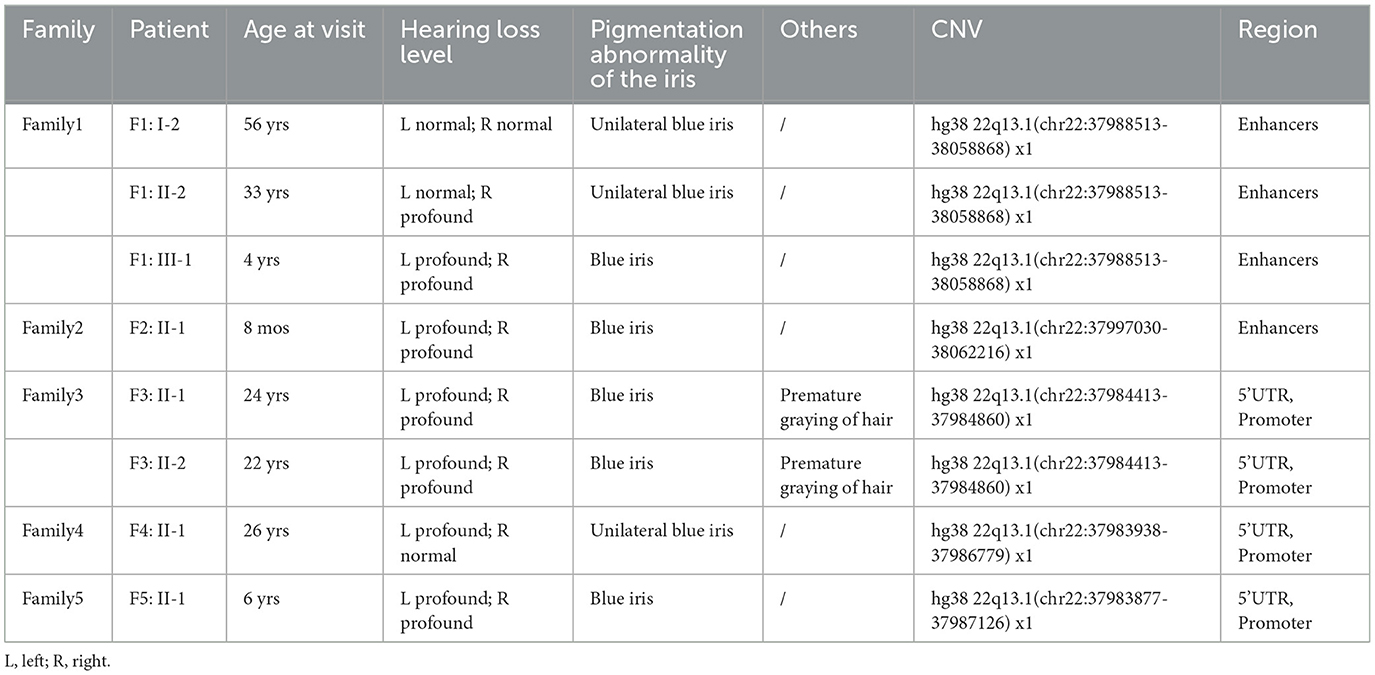
Table 1. Clinical and molecular genetic characteristics of eight WS patients with SOX10 non-coding pathogenic CNVs.
Among the identified CNVs, CNV1; chr22:37988513-38058868 in Family 1 and CNV2; chr22:37997030-38062216 in Family 2, were identified, spanning regions containing four homologous enhancers (U1, U2/MCS5, U3, and U4). These CNVs showed complete overlap with reported pathogenic CNVs within the SOX10 enhancer subregions. Moreover, CNV3; chr22:37984413-37984860 in Family 3, CNV4; chr22:37983938-37986779 in Family 4, and CNV5; chr22:37983877-37987126 in Family 5 were identified. These variants were located within the 5'UTR, spanning exon 1 and part of exon 2, as well as the promoter region of the SOX10 gene. The breakpoints associated with these copy number losses were confirmed via gap-PCR, except for CNV5, which could only be demonstrated by BAM file owing to DNA sample depletion (Figure 2B). Importantly, these CNV events were absent in both the gnomAD (CNVs v4.0) and DGV databases.
All families were followed up to confirm the presence of relatives with WS-related phenotypes and to collect blood samples from parents, siblings, or other related individuals. In Family 1, three consecutive generations exhibited autosomal dominant inheritance of WS, with CNV1 inherited from the proband's grandmother. Notably, none of the parents in Families 2-5 displayed WS-related phenotypes. Additionally, CNV2 in Family 2 was confirmed as de novo through gap-PCR, while parental samples from Families 3-5 were unavailable. Based on the ACMG guidelines modifier, we classified 5 CNVs as pathogenic/likely pathogenic, with detailed evidence and final pathogenicity classification in Table 2.
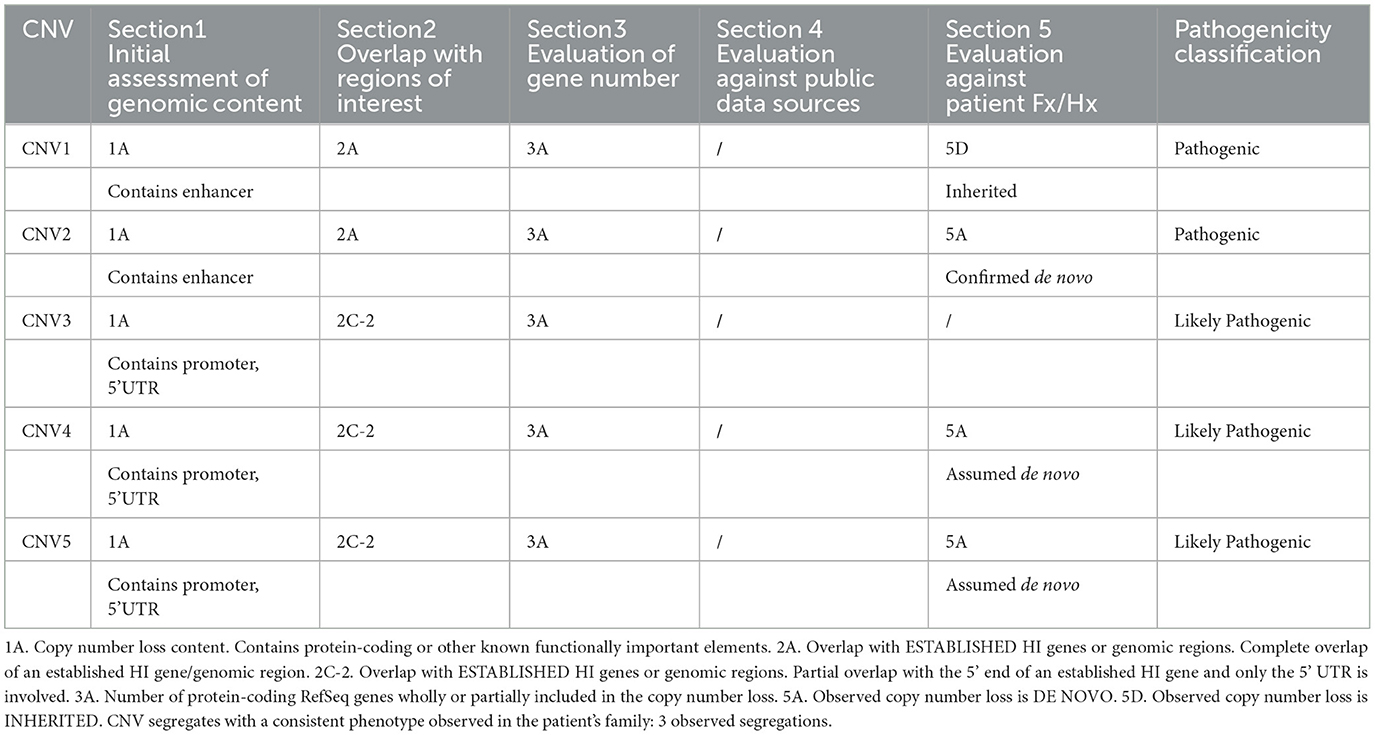
Table 2. Available evidence and final classification of the detected CNVs.
Analysis of sequences surrounding each CNV breakpoints using Repeat Masker (http://www.repeatmasker.org/) revealed significant similarity only for CNV1 breakpoints, corresponding to Alu repeats. These Alu repeats belong to distinct Alu families: AluSx and AluSq2. Further analysis revealed an 85.8% similarity between AluSx and AluSq2 (see Figure 2C), suggesting that the CNV was initially generated by Alu-mediated NAHR.
Considering the phenotypic heterogeneity of WS2 and its potential to manifest as NSHL, we expanded our analysis to include CNVs in the non-coding regions of SOX10 among 8,674 hearing loss patients with WGS. Among these patients, only one heterozygous deletion was detected overlapping with SOX10 enhancers. Specifically, CNV6 encompassing genomic region 22q13.1 (chr22: 37992678-38003872) was identified in two unrelated NSHL patients. However, this CNV did not segregate with the disease in either family. In Family S1, exhibiting an autosomal dominant inheritance pattern, the proband did not inherit the CNV from the affected mother, while in Family S2, the proband inherited the variant from an unaffected mother. These data suggest CNV6 is a likely benign variant (see Supplementary Figure 1).
There was phenotypic heterogeneity associated with CNVs identified in this study (Figures 3A–C). In Family 1, the proband (F1: III-1) displayed bilateral profound hearing loss and bilateral blue irises, while the father (F1: II-2) presented with sensorineural hearing loss in the right ear and a blue iris in the right eye. The grandmother (F1: I-1) only displayed a blue iris in the right eye with normal hearing levels. Upon examination via computed tomography (CT) scan, the proband revealed an enlarged vestibular pool, with a narrow and insufficiently developed lumen of the lateral semicircular canal (Figure 3D). Patients in Families 2 and 5 both manifested bilateral profound hearing loss and bilateral blue irises. In Family 3, siblings displayed premature graying during their teenage years, along with bilateral profound hearing loss and blue iris pigmentation. The proband of Family 4 exhibited unilateral sensorineural deafness and unilateral blue iris. Notably, none of these patients exhibited congenital megacolon.
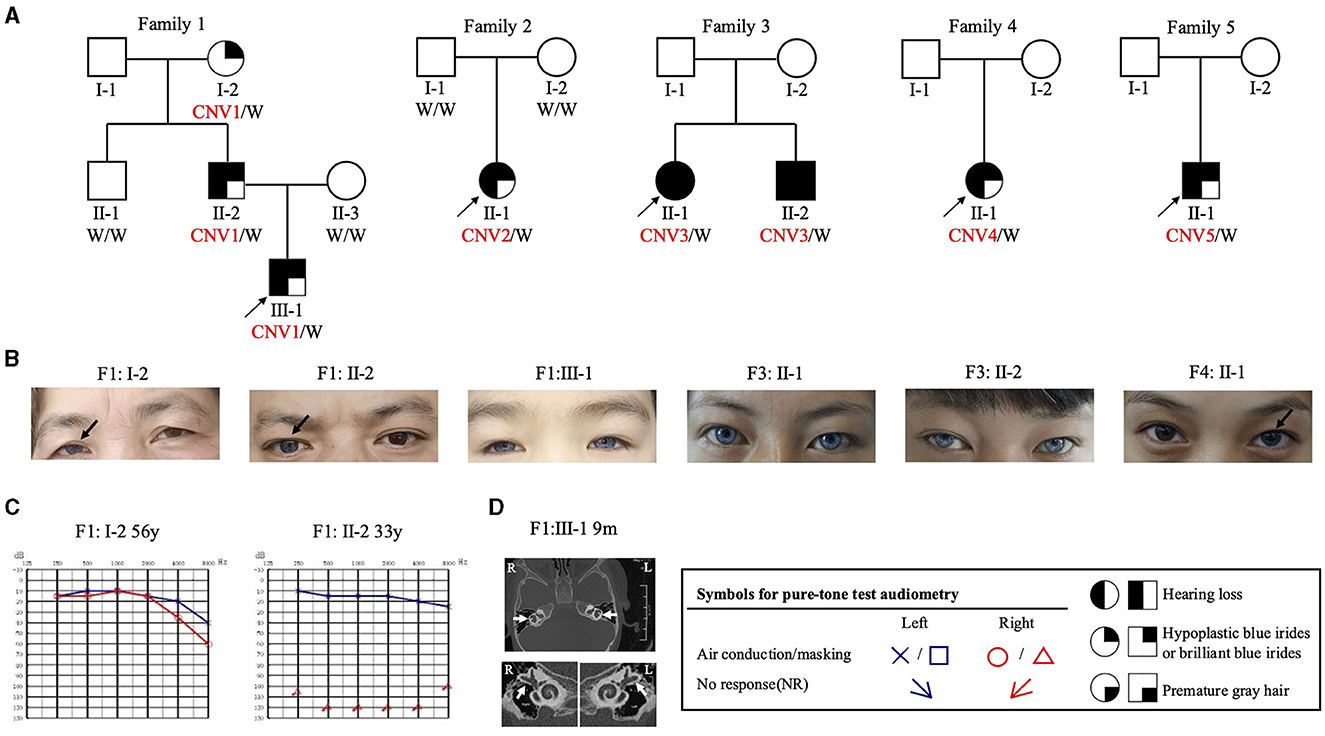
Figure 3. Pedigree and clinical features of WS families identified in the CDGC cohort. (A) Pedigrees of the 5 unrelated families. Black arrows, probands; “W” in black means “wild type” and CNV1-5 are in red. (B) Photograph of the irises of patients with WS. Black arrows, unilateral blue iris; the remaining three had bilateral blue irises. (C) Pure-tone audiometry results of subjects F1: I-2 and F1: II-2. “y” means “year.” (D) CT scan of subject F1: III-1. White arrow, abnormal development of the bilateral semicircular canals. “R” means “right side,” “L” means “left side”.
In this study, we conducted WGS on 59 patients with WS and examined CNVs within the regulatory region of the SOX10 gene. Previously, the CDGC cohort collected 290 clinically diagnosed WS patients from 228 families. Among them, 77.6% (177/228) were diagnosed with pathogenic variants in the coding regions of SOX10, PAX3, MITF, or EDNRB genes through TGS (these data will be presented in separate article). Here, our investigation identified five pathogenic deletions within the non-coding region of the SOX10 gene, including the pathogenic CNVs in the promoter and 5'UTR reported for the first time. This led to a 2.2% (5/228) increase in molecular diagnostic rates, underscoring the importance of rare non-coding variations in gene identification. Therefore, these findings indicate that non-coding CNVs play an important role in WS and should be considered in WGS-based diagnostics.
Evaluating the pathogenicity of variants in non-coding regions presents a significant challenge in clinical genetic testing. Guidelines provided by ACMG/AMP and ClinGen offer valuable insights for this purpose. According to ACMG guidelines (Riggs et al., 2020), copy number loss, as outlined in Section 2, pertains to “Overlap with Established/Predicted HI or Established Benign Genes/Genomic Regions.” Bondurand et al. (2012) and Lecerf et al. (2014) reported clinical patients involving SOX10 enhancer deletions (Werner et al., 2007; Ellingford et al., 2022). CNV; chr22:38013680-38051826 resulted in HSCR, while CNV; chr22:38008019-38064588 led to WS4. Both deletions were identified de novo through comparative genomic hybridization (CGH)-array analysis. Based on these findings, the minimal overlap region between all pathogenic CNVs upstream of SOX10 gene should be considered an established HI genomic region. CNV1 and CNV2 in this study contain extensions of established SOX10 enhancers that completely overlap with the previously identified HI region; therefore, we applied criterion 2A to CNV1 and CNV2. CNVs 3-5 occur within the SOX10 promoter and 5' UTR region. Given the critical role of both the promoter and UTR in transcriptional regulation, and the emphasis on promoter and UTR variants in the clinical interpretation of non-coding region variants, as highlighted by Ellingford et al. (2022), we used criterion 2C-2, which specifies “Partial overlap with the 5' end of an established HI gene and only the 5' UTR is involved.” CNV6, which occurs within the upstream enhancer region of SOX10 and meets criterion 1A, was detected in two unrelated NSHL patients but did not segregate in two separate pedigrees (in accordance with 5E), leading to its classification as likely benign. This suggests that heterozygous deletions involving only enhancers MCS3 (zebrafish) and U4 (mouse) may be tolerated.
In our study, we identified CNV1, which resulted from Alu-mediated NAHR. Alu elements are transposable elements that constitute approximately 10% of the human genome (Kazazian and Moran, 2017). Due to their abundance and sequence homogeneity, Alu elements frequently participate in genomic rearrangements. These rearrangements can have significant implications for human health as ~0.1% of human genetic disorders are linked to Alu-mediated genomic rearrangements (Deininger and Batzer, 1999).
Understanding the pathogenicity of non-coding variants is crucial for identifying potential disease-causing mechanisms, improving disease risk prediction, and developing targeted therapies (Mattick et al., 2018). However, interpreting the pathogenicity of non-coding variants poses several challenges. Firstly, their wide-ranging and intricate nature makes precise annotation of regulatory elements difficult. Regulatory elements often display cell-type-specific activity, resulting in varying effects across different tissues and cell types. Analyzing non-coding variants in relevant cell types is essential for accurately assessing their functional impact. Secondly, limited data on non-coding CNV allele frequency pose challenges to achieving statistical power. Accumulating sufficient data for the analysis of rare variants requires collaboration and large-scale genomic projects. Thirdly, experimental validation of the functional impact of non-coding variants is still immature and technically challenging. In vivo experiments on the impact of non-coding region variants on gene function are even more challenging.
This study identifies five SOX10 non-coding CNVs in WS patients, lacking direct SOX10 expression data in human cochlea or skin tissue. Subsequent investigations should utilize cell models or organoid models to explore the impact of SOX10 non-coding CNVs on gene function. For the remaining 51 undiagnosed WS patients, will continue to analyze the pathogenicity of CNVs and other types of variants (such as SNVs and InDels) in non-coding regions of other known WS genes and explore reliable experimental validation methods. Additionally, ongoing work includes identifying new genes causing WS.
The CNVs we identified align with previously documented regions but manifest distinct phenotypes. Specifically, while all five CNVs identified in our study correlate with WS2, previously reported patients exhibited a megacolon phenotype, leading to diagnoses of WS4 or HSCR. The underlying mechanism behind this phenotypic variation remains poorly understood.
In summary, our study unveiled five novel non-coding deletions within the SOX10 gene, spanning the 5'UTR, promoter, and enhancer regions. These findings aid in diagnosing more WS patients and provide valuable guidance for their genetic counseling. Moreover, our discoveries broaden the spectrum of pathogenic variants within the non-coding regions of the SOX10 gene and established a framework for interpreting non-coding regions in other genes.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding authors.
The studies involving humans were approved by Ethics Committee on Biomedical Research, West China Hospital of Sichuan University. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants' legal guardians/next of kin. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
YH: Conceptualization, Formal analysis, Writing—original draft, Writing—review & editing. JG: Writing—original draft, Writing—review & editing. YLo: Resources, Writing—review & editing. WX: Writing—review & editing, Resources. LK: Data curation, Writing—review & editing. MC: Writing—review & editing, Software. TT: Writing—review & editing. MZ: Writing—review & editing. FB: Formal analysis, Writing—review & editing. YLu: Resources, Writing—review & editing. JC: Conceptualization, Supervision, Writing—original draft, Writing—review & editing. HY: Supervision, Writing—review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the 1·3·5 project for the disciplines of excellence-Interdisciplinary innovation project, West China Hospital, Sichuan University (No. ZYJC20002) and National Natural Science Foundation of China (No. 82202046).
We extend our sincere gratitude to all the participating families for their invaluable contribution and support to this study. Additionally, we would like to acknowledge the helpful discussions with members of the laboratory.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor HA declared a past co-authorship with the authors FB, MZ, YLu, JC, and HY.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fauot.2024.1400991/full#supplementary-material
Antonellis, A., Huynh, J. L., Lee-Lin, S. Q., Vinton, R. M., Renaud, G., Loftus, S. K., et al. (2008). Identification of neural crest and glial enhancers at the mouse Sox10 locus through transgenesis in zebrafish. PLoS Genet. 4:e1000174. doi: 10.1371/journal.pgen.1000174
Attié, T., Till, M., Pelet, A., Amiel, J., Edery, P., Boutrand, L., et al. (1995). Mutation of the endothelin-receptor B gene in Waardenburg-Hirschsprung disease. Hum. Mol. Genet. 4, 2407–2409. doi: 10.1093/hmg/4.12.2407
Batissoco, A. C., Pedroso-Campos, V., Pardono, E., Sampaio-Silva, J., Sonoda, C. Y., Vieira-Silva, G. A., et al. (2022). Molecular and genetic characterization of a large Brazilian cohort presenting hearing loss. Hum. Genet. 141, 519–538. doi: 10.1007/s00439-021-02372-2
Betancur, P., Bronner-Fraser, M., and Sauka-Spengler, T. (2010). Genomic code for Sox10 activation reveals a key regulatory enhancer for cranial neural crest. Proc. Natl. Acad. Sci. U. S. A. 107, 3570–3575. doi: 10.1073/pnas.0906596107
Bondurand, N., Dastot-Le Moal, F., Stanchina, L., Collot, N., Baral, V., Marlin, S., et al. (2007). Deletions at the SOX10 gene locus cause Waardenburg syndrome types 2 and 4. Am. J. Hum. Genet. 81, 1169–1185. doi: 10.1086/522090
Bondurand, N., Fouquet, V., Baral, V., Lecerf, L., Loundon, N., Goossens, M., et al. (2012). Alu-mediated deletion of SOX10 regulatory elements in Waardenburg syndrome type 4. Eur. J. Hum. Genetics 20, 990–994. doi: 10.1038/ejhg.2012.29
Deininger, P. L., and Batzer, M. A. (1999). Alu repeats and human disease. Mol. Genet. Metab. 67, 183–193. doi: 10.1006/mgme.1999.2864
Edery, P., Attié, T., Amiel, J., Pelet, A., Eng, C., Hofstra, R. M., et al. (1996). Mutation of the endothelin-3 gene in the Waardenburg-Hirschsprung disease (Shah-Waardenburg syndrome). Nat. Genet. 12, 442–444. doi: 10.1038/ng0496-442
Ellingford, J. M., Ahn, J. W., Bagnall, R. D., Baralle, D., Barton, S., Campbell, C., et al. (2022). Recommendations for clinical interpretation of variants found in non-coding regions of the genome. Genome Med. 14, 1–14. doi: 10.1186/s13073-022-01073-3
Foy, C., Newton, V., Wellesley, D., Harris, R., and Read, A. P. (1990). Assignment of the locus for Waardenburg syndrome type I to human chromosome 2q37 and possible homology to the Splotch mouse. Am. J. Hum. Genet. 46, 1017–1023.
Hughes, A. E., Newton, V. E., Liu, X. Z., and Read, A. P. A. (1994). gene for Waardenburg syndrome type 2 maps close to the human homologue of the microphthalmia gene at chromosome 3p12-p14.1. Nat. Genet. 7, 509–512. doi: 10.1038/ng0894-509
Ideura, M., Nishio, S. Y., Moteki, H., Takumi, Y., Miyagawa, M., Sato, T., et al. (2019). Comprehensive analysis of syndromic hearing loss patients in Japan. Sci. Rep. 9:11976. doi: 10.1038/s41598-019-47141-4
Inoue, K., Khajavi, M., Ohyama, T., Hirabayashi, S., Wilson, J., Reggin, J. D., et al. (2004). Molecular mechanism for distinct neurological phenotypes conveyed by allelic truncating mutations. Nat. Genet. 36, 361–369. doi: 10.1038/ng1322
Kazazian, H. H., and Moran, J. V. (2017). Mobile DNA in health and disease. N. Engl. J. Med. 377, 361–370. doi: 10.1056/NEJMra1510092
Lecerf, L., Kavo, A., Ruiz-Ferrer, M., Baral, V., Watanabe, Y., Chaoui, A., et al. (2014). An impairment of long distance SOX10 regulatory elements underlies isolated hirschsprung disease. Hum. Mutation 35, 303–307. doi: 10.1002/humu.22499
Mattick, J. S., Dinger, M., Schonrock, N., and Cowley, M. (2018). Whole genome sequencing provides better diagnostic yield and future value than whole exome sequencing. Med. J. Aust. 209, 197–199. doi: 10.5694/mja17.01176
Milunsky, J. M. (1993). “Waardenburg syndrome type I,” in GeneReviews(®), eds. M. P. Adam, J. Feldman, G. M. Mirzaa, R. A. Pagon (Seattle, WA: University of Washington).
Ogawa, Y., Kono, M., and Akiyama, M. (2017). Pigmented macules in Waardenburg syndrome type 2 due to KITLG mutation. Pigment. Cell. Melanoma Res. 30, 501–504. doi: 10.1111/pcmr.12597
Oza, A. M., DiStefano, M. T., Hemphill, S. E., Cushman, B. J., Grant, A. R., Siegert, R. K., et al. (2018). Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum. Mutation 39, 1593–1613. doi: 10.1002/humu.23630
Pingault, V., Bondurand, N., Kuhlbrodt, K., Goerich, D. E., Préhu, M. O., Puliti, A., et al. (1998). SOX10 mutations in patients with Waardenburg-Hirschsprung disease. Nat. Genet. 18, 171–173. doi: 10.1038/ng0298-171
Read, A. P., and Newton, V. E. (1997). Waardenburg syndrome. J. Med. Genet. 34, 656–665. doi: 10.1136/jmg.34.8.656
Riggs, E. R., Andersen, E. F., Cherry, A. M., Kantarci, S., Kearney, H., Patel, A., et al. (2020). Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the american college of medical genetics and genomics (ACMG) and the clinical genome resource (ClinGen). Genet. Med. 22, 245–257. doi: 10.1038/s41436-019-0686-8
Sánchez-Martín, M., Rodríguez-García, A., Pérez-Losada, J., Sagrera, A., Read, A. P., Sánchez-García, I. S. L. U. G., et al. (2002). (SNAI2) deletions in patients with Waardenburg disease. Hum. Mol. Genet. 11, 3231–3236. doi: 10.1093/hmg/11.25.3231
Shearer, A. E., Kolbe, D. L., Azaiez, H., Sloan, C. M., Frees, K. L., Weaver, A. E., et al. (2014). Copy number variants are a common cause of non-syndromic hearing loss. Genome Med. 6, 37. doi: 10.1186/gm554
Southard-Smith, E. M., Angrist, M., Ellison, J. S., Agarwala, R., Baxevanis, A. D., Chakravarti, A., et al. (1999). The Sox10 (Dom) mouse: modeling the genetic variation of Waardenburg-Shah (WS4) syndrome. Genome Res. 9, 215–225. doi: 10.1101/gr.9.3.215
Spielmann, M., and Mundlos, S. (2016). Looking beyond the genes: the role of non-coding variants in human disease. Hum. Mol. Genet. 25, R157–r65. doi: 10.1093/hmg/ddw205
Tassabehji, M., Newton, V. E., and Read, A. P. (1994). Waardenburg syndrome type 2 caused by mutations in the human microphthalmia (MITF) gene. Nat. Genet. 8, 251–255. doi: 10.1038/ng1194-251
Tassabehji, M., Read, A. P., Newton, V. E., Harris, R., Balling, R., Gruss, P., et al. (1992). Waardenburg's syndrome patients have mutations in the human homologue of the Pax-3 paired box gene. Nature 355, 635–636. doi: 10.1038/355635a0
Van Camp, G., Van Thienen, M. N., Handig, I., Van Roy, B., Rao, V. S., Milunsky, A., et al. (1995). Chromosome 13q deletion with Waardenburg syndrome: further evidence for a gene involved in neural crest function on 13q. J. Med. Genet. 32, 531–536. doi: 10.1136/jmg.32.7.531
Wang, G., Li, X., Gao, X., Su, Y., Han, M., Gao, B., et al. (2022). Analysis of genotype-phenotype relationships in 90 Chinese probands with Waardenburg syndrome. Hum. Genet. 141, 839–852. doi: 10.1007/s00439-021-02301-3
Werner, T., Hammer, A., Wahlbuhl, M., Bösl, M. R., and Wegner, M. (2007). Multiple conserved regulatory elements with overlapping functions determine Sox10 expression in mouse embryogenesis. Nucleic Acids Res. 35, 6526–6538. doi: 10.1093/nar/gkm727
Keywords: SOX10, Waardenburg syndrome, non-coding, enhancer, promoter, CNV
Citation: Huang Y, Geng J, Long Y, Xiong W, Kang L, Chen M, Tang T, Zhong M, Bu F, Lu Y, Cheng J and Yuan H (2024) Five novel cis-regulatory deletions of SOX10 cause Waardenburg syndrome type II. Front. Audiol. Otol. 2:1400991. doi: 10.3389/fauot.2024.1400991
Received: 14 March 2024; Accepted: 30 May 2024;
Published: 18 June 2024.
Edited by:
Hela Azaiez, The University of Iowa, United StatesReviewed by:
Monika Oldak, Institute of Physiology and Pathology of Hearing (IFPS), PolandCopyright © 2024 Huang, Geng, Long, Xiong, Kang, Chen, Tang, Zhong, Bu, Lu, Cheng and Yuan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huijun Yuan, eXVhbmhqMzAxQHdjaHNjdS5jbg==; Jing Cheng, Y2hlbmdqaW5nQHdjaHNjdS5jbg==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.