 Yi Yuan
Yi Yuan Denise Yan
Denise Yan Jeffrey Skidmore1
Jeffrey Skidmore1 Prem Chapagain
Prem Chapagain Xuezhong Liu
Xuezhong Liu Shuman He
Shuman He
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Audiol. Otol. , 07 July 2023
Sec. Clinical Research in Auditory Implants and Hearing Aids
Volume 1 - 2023 | https://doi.org/10.3389/fauot.2023.1213323
Introduction: This preliminary study identified a missense variant in ACTG1 (NM_001614.5) in a family with autosomal dominant non-syndromic hearing loss (ADNSHL). The responsiveness of the electrically-stimulated cochlear nerve (CN) in two implanted participants with this missense change was also evaluated and reported.
Methods: Genetic testing was done using a custom capture panel (MiamiOtoGenes) and whole exome sequencing. The responsiveness of the electrically-stimulated CN was evaluated in two members of this family (G1 and G4) using the electrically evoked compound action potential (eCAP). eCAP results from these two participants were compared with those measured three implanted patient populations: children with cochlear nerve deficiency (CND), children with idiopathic hearing loss and normal-sized cochlear nerves (NSCN), and post-lingually deafened adults.
Results: Sequencing of ACTG1 identified a missense c.737A>T (p. Gln246Leu) variant in ACTG1 (NM_001614.5) which is most likely the genetic cause of ADNSHL in this family. eCAP results measured in these two participants showed substantial variations.
Discussion: The results indicated the missense c.737A>T (p. Gln246Leu) variant in ACTG1 (NM_001614.5) co-segregated with hearing loss in this family. The responsiveness of the electrically-stimulated CN can vary among patients with the same genetic variants, which suggests the importance of evaluating the functional status of the CN for individual CI patients.
Autosomal dominant non-syndromic hearing loss (ADNSHL) accounts for approximately 20% of non-syndromic hereditary hearing loss cases, and is often characterized by a post-lingual onset and progressive hearing loss (Shearer et al., 1993). It has a broad phenotypic spectrum which includes considerable variation in age of onset, type of hearing loss configurations (i.e., low-frequency, mid-frequency, high-frequency, and all-frequency), and the rate of progression (see review by Pennings et al., 2003). Today, around 51 genes and 68 chromosomal loci have been associated with ADNSHL (Hereditary Hearing Loss Homepage; http://hereditaryhearingloss.org, 2021). These deafness genes encode a myriad of proteins that have a specialized role in various cell types, structures, and processes in the cochlea (Hilgert et al., 2009).
Much of the function of the inner ear relies on the hair bundle, a cluster of actin-filled stereocilia for transduction of auditory and vestibular stimuli into neural impulses. Mutations that alter the amino acid structure of actin are generally not tolerated, because actin has a large repertoire of interacting ligands and actin-binding partners to perform various functions, including movement, cellular structures, redistribution of surface receptors for regulation of cellular responses and other processes (e.g., Kovar et al., 2006; Ferron et al., 2007). Specifically, depending on the organization of the filaments, actin can be deployed for support of a variety of cellular processes. Microfilament functions include cell motility or control of cytokinesis (Pollard and Cooper, 2009); cells also use actin to adopt and maintain specific cellular structures (e.g., Michelot and Drubin, 2011). In the apical region of the hair cell, parallel bundles of actin fibers with identical polarity mediate protrusion of stereocilia (Tilney et al., 1983); in the cuticular plate lying beneath the apical surface, a dense meshwork of actin filaments anchors the stereocilia (DeRosier and Tilney, 1989) and the adherents junction encircling the hair cell next to the apical region also contains antiparallel actin filaments (Hirokawa and Tilney, 1982). Actin structures that support the plasma membrane of cells and provide a track for the passage of synaptic vesicles are also found in the basolateral region of the hair cell.
There are six actin genes in the human genome. Three alpha [αskeletal-actin 1 (ACTA1), αsmooth-actin 2 (ACTA2), αcardiac-actin 1 (ACTC1)], one beta [βcyto-actin (ACTB)], two gamma [γcyto-actin 1 (ACTG1), γsmooth-actin 2 (ACTG2 The γ-actin ACTG1 isoform is predominantly expressed in the auditory hair cell, and β-actin (ACTB) is an isoform at the front of the cell where actin filaments polymerize. The β- and γ-actin isoforms are present in non-muscle cells and differ at only four amino acids positions at the N-terminus. These actin isoforms are highly conserved at the protein level, with >90% homology between the single yeast actin (ACT1) gene and the human γ-actin. Vertebrate γ-actin amino acids are identical between humans, mice, cattle, and chickens (Sheterline et al., 1998).
ACTG1 mutations were identified as responsible for either non-syndromic deafness or Baraitser-Winter syndrome, a disorder characterized by distinct craniofacial features, ocular colobomata and neuronal migration defect and variable hearing loss (e.g., Rivière et al., 2012). It is believed that mutations in γ-actin cause hearing loss mainly by impairing the function and/or viability of hair cells (Van Wijk et al., 2003; Rendtorff et al., 2006). DFNA20 (MIM 604717) and DFNA26 (MIM 604717) are loci linked to dominant, non-syndromic, progressive hearing loss that map to chromosome 17q25.3 (Morell et al., 2000; Yang and Smith, 2000). Individuals with the DFNA20 and DFNA26 (DFNA20/26) exhibit sensorineural hearing loss that, very similar to age related hearing loss, affects initially only the high frequencies and steadily progresses to include all frequencies (Yang and Smith, 2000; Elfenbein et al., 2001). Distortion product otoacoustic emission (DPOAE) assessments are consistent with a cochlear site of lesion (Elfenbein et al., 2001). However, Teig (1968) reported pathological tone decay at 2 and 4 kHz when hearing loss is more than 50 dB in patients with ACTG1 missense mutations. These results suggested potential neural lesions along the auditory pathway in patients with ACTG1 missense mutations.
Theoretically, neural encoding and processing of electrical stimulation in implanted patients who carry the ACTG1 missense mutation should not be compromised since this genetic lesion mainly affects the hair cells which are “bypassed” by the cochlear implant (CI). As a result, patients with ACTG1 missense mutation are expected to have good CI clinical outcomes. However, this theoretical possibility has not been evaluated in human CI users, which represents a knowledge gap in the field of cochlear implantation. The responsiveness of the electrically-stimulated CN has been suggested to be important for hearing outcomes in CI users (e.g., Long et al., 2014; He et al., 2018; Skidmore et al., 2021). Therefore, it was selected as the functional, phenotypic readout in this preliminary study.
In this study, we ascertained a five-generation family with progressive ADNSHL. We used our targeted custom MiamiOtoGenes panel containing 230 hearing loss-associated genes to analyze DNA samples from two affected family members (G2 and G4) and sample from one patient (G3) was subjected to whole exome sequencing (WES). Using this strategy, we identified a missense c.737A>T (p. Gln246Leu) variant in ACTG1 (NM_001614.5) that co-segregated with hearing loss and most likely to be the genetic cause of ADNSHL in this family. This genetic finding provided an opportunity to explore the phenotypic effect of this missense change on the responsiveness of the CN to electrical stimulation. The responsiveness of the electrically stimulated CN was also evaluated using electrophysiological measures of the electrically evoked compound action potential (eCAP) in this preliminary study. The eCAP is a near-field measure of synchronized neural responses generated by a group of CN fibers. eCAP measures that can be used to assess the responsiveness of the CN to electrical stimulation include slope of the eCAP amplitude growth function (AGF), the absolute refractory period (t0), and the speed of recovery from neural adaption (τ3). The eCAP AGF represents a series of eCAP amplitudes recorded at corresponding stimulation levels (Skidmore et al., 2021). The rate at which the eCAP amplitude increases with increasing stimulation level (i.e., slope) has been shown to be associated with the responsiveness of CN fibers, with greater slopes indicating better CN responsiveness (He et al., 2018, 2020). t0 is an estimate of the absolute recovery period of the CN fibers based on the eCAP refractory recovery function (RRF). It represents the minimum time interval that is needed to elicit the eCAP to a second pulse (i.e., probe pulse) after a suprathreshold masking pulse (He et al., 2018; Skidmore et al., 2021). Smaller t0 values indicate better neural responsiveness in both animal models (Shepherd et al., 2004) and human CI users (He et al., 2018). τ3 has been shown to be associated with spiral ganglion neuron (SGN) survival in animal models (Ramekers et al., 2015). In human CI users, patients with longer τ3s tend to have poorer speech perception performance (He et al., 2022).
In this preliminary and descriptive study, we reported the identification of the missense c.737A>T (p. Gln246Leu) variant in ACTG1 (NM_001614.5), and the responsiveness of the electrically stimulated CN evaluated in two participants with this missense change. In addition, their electrophysiological results were compared with three implanted patient populations: children with cochlear nerve deficiency (CND), children with idiopathic sensorineural hearing loss and normal-sized cochlear nerves (NSCNs) and postlingually deafened adult CI users with various etiologies of hearing loss, including trauma, noise exposure, Meniere's disease, autoimmune disease, vestibular schwannoma, sudden hearing loss.
We ascertained a five-generation American family with ADSNHL. Saliva samples were collected, and DNA was extracted from six available family members, of whom five were affected. All clinical data of affected members in this family were obtained from questionnaires and included age at onset, degree, evolution, and symmetry of the hearing loss, presence of tinnitus, and other relevant clinical manifestations. None of the patients had any history of chronic noise exposure or ototoxic drugs. The progressiveness of HL was determined based on patients' medical records and their answers on the questionnaires. The hearing levels of the participants were evaluated by pure-tone audiometry (details shown in Figure 1). The pure-tone average (PTA) was calculated by averaging the audiometric thresholds across four specific frequencies (0.5, 1, 2, and 4 kHz). The severity of hearing loss (HL) was categorized as follows: mild (PTA: 21–40 dB HL), moderate (41–70 dB HL), severe (71–95 dB HL), and profound (>95 dB HL). Detailed demographic information of this family is listed in Table 1.
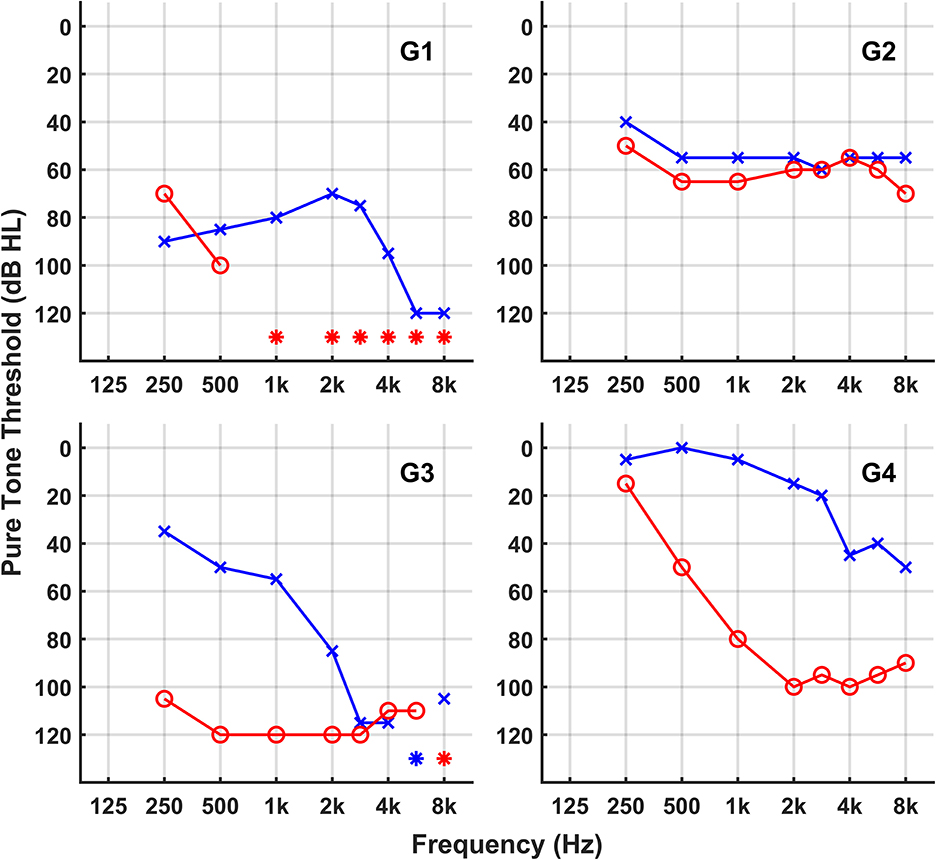
Figure 1. Audiograms of the family members. *Symbol stars indicate no response.
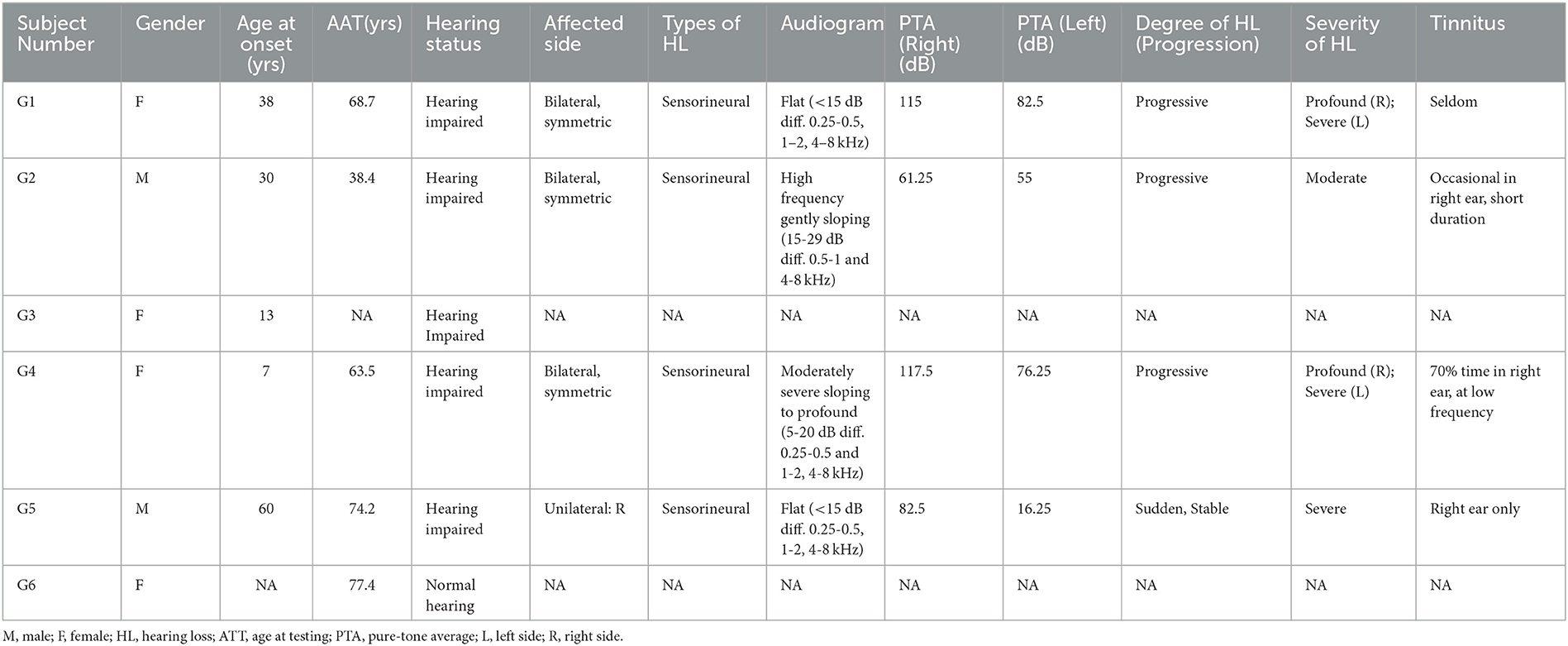
Table 1. Demographic information of genetic testing participants.
Among the five affected participants, only G1 and G4 are CI users. As a result, electrophysiological evaluations were only conducted in these two participants. Both participants were implanted with a Cochlear™ Nucleus® (Cochlear Ltd., Macquarie, NSW, Australia) CI [24RE(CA)] with a full electrode array insertion in the test ear. The Cochlear™ Nucleus® CI has 22 CI electrodes, with electrode 1 typically located at the base and electrode 22 located toward the apex of the cochlea. G1 was implanted in both ears and G4 was unilaterally implanted in the right ear. All three implanted ears were assessed in this study. Electrode locations tested in each participant for each eCAP measure and speech perception scores obtained from their most recent clinical reports are listed in Table 2.
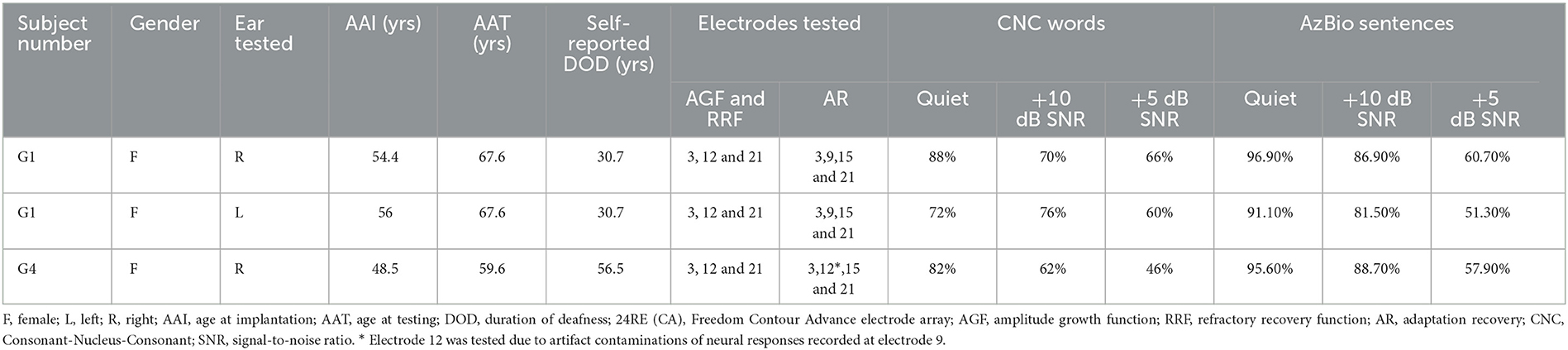
Table 2. Electrophysiological testing information of study participants.
The study was approved by the ethics committee of The Ohio State University (2017H0131 and 2018H0587). Written informed consent was obtained from all participants prior to saliva sample collection and electrophysiological evaluation.
Genomic DNA was extracted from saliva samples using the commercial kit Oragene™ (DNA Genotek OG-500; Ottawa, Ontario, Canada). Agarose gel and Qubit (Life Technologies, CA, USA) were used for DNA quantity and quality assessment. DNA samples from 2 affected individuals (G2 and G4) were analyzed using a SureSelect custom next generation sequencing (NGS) gene panel (MiamiOtoGenes) of 230 known and candidate genes associated with sensorineural hearing loss (Yan et al., 2016). This panel was designed to include all exons, 5' UTRs and 3' UTRs of the genes and covered a target size of approximately 1.598 Mbp encompassing 4,422 regions. The targeted sequencing was processed at the Hussman Institute for Human Genomics (HIHG) Sequencing core, University of Miami. The SureSelectTarget Enrichment (Agilent, Santa Clara, CA, USA) of coding exons and flanking intronic sequences in-solution hybridization capture system was used according to the manufacturer's standard protocol. Adapter sequences for the Illumina HiSeq 2000 were ligated, and the enriched DNA samples were prepared using the standard methods for the HiSeq 2000 instrument (Illumina, San Diego, CA, USA). The average insert size of 180 bp and paired-end sequencing reads were used. To rule out any potential unreported deafness causative gene, the sample from G3 was subjected to whole exome sequencing simultaneously. The enrichment of coding exons and flanking intronic regions was performed using a solution hybrid selection method with the SureSelectXT human all exon 60 Mb v6 kit (Agilent Technologies) following the manufacturer's standard protocol.
Bioinformatics processing and data analysis were performed as previously reported (Yan et al., 2016). In brief, The Illumina CASAVA v1.8 pipeline was used to assemble 99 bp sequence reads. Burrows-Wheeler Aligner (BWA) was applied for alignment of sequence reads to the human reference genome (hg19) (Li and Durbin, 2009), and variants were called using FreeBayes (Garrison and Marth, 2012). KGGSeq (http://pmglab.top/kggseq/) was then used for variant filtering based on quality/score read depth and minor allele frequency (MAF thresholds of 0.0005 for ADNSHL variants) as reported in dbSNP141, the National Heart, Lung, and Blood Institute Exome Sequencing Project Exome Variant Server, Seattle, WA Project (Exome Variant Server, 2023), the Genome Aggregation Database (gnomAD; https://gnomad.broadinstitute.org) and the 1000 Genome Project Database. Variants were further analyzed based on their presence and pathogenicity information in Human Gene Mutation Database (HGMD; http://www.hgmd.cf.ac.uk), the Deafness Variation Database (DVD) (deafnessvariationdatabase.org), and ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/). Copy number variations (CNV) calling was performed using an R-based tool (Nord et al., 2011). This method normalizes read-depth data by sample batch and compares median read-depth ratios using a sliding-window approach. Variants meeting these criteria were further annotated according to specific standard terminology of American College of Medical Genetics and Genomics (ACMG) guidelines (Richards et al., 2015) that recommends classification of DNA variants into five categories including pathogenic, likely-pathogenic, variant of uncertain significance (VUS), likely-benign, and benign. To analyze the possible functional pathogenic effects of the missense variants, two types of prediction programs, SIFT, and Polyphen score were used. The Human Gene Mutation Database (http://www.hgmd.cf.ac.uk), the Deafness Variation Database (DVD) (deafnessvariationdatabase.org), and Clinvar (http://www.ncbi.nlm.nih.gov/clinvar/) were used as references to determine the novelty and probable pathogenicity of the allelic variations detected in our sequencing approach.
The candidate variation was confirmed via Sanger sequencing. Primer3, v. 0.4.0 (http://primer3.ut.ee) was used for primer design. Polymerase chain reaction (PCR) reactions were performed with 40–60 ng of genomic DNA and Taq DNA polymerase (Sigma) using standard protocols and following primers: Forward: GACATCAAGGAGAAGCTGTGC; Reverse: GCTGTGTCACCGAGGATGTA. PCR products were purified with Qiagen Qiaquick purification kit and bidirectionally sequenced using the ABI PRISM Big Dye Terminator Cycle Sequencing V3.1 Ready Reaction Kit and passed on the ABIPRISM 3730 DNA Analyzer (Applied Biosystems). DNA sequence analysis was performed with DNASTAR Lasergene software.
All eCAP measures were conducted using the Advanced Neural Response Telemetry function via the Custom Sound EP (v. 5.2) software interface (Cochlear Ltd, Sydney, NSW, Australia). For measuring the eCAP AGF and RRF, the stimulus was a symmetric, cathodic-leading, biphasic pulse with a pulse phase duration of 25 s/phase. The presentation rate of the stimulus was 15 Hz. For measuring τ3, the probe was the single pulse as that described for measuring the eCAP AGF and RRF. The masker stimulus was a train of biphasic pulses with the same characteristics as those of the single-pulse stimulus. The duration of this masker pulse train was 100 ms with a carrier rate of 900 pulses per second (pps) per channel. The presentation rate of the masker pulse train and the probe pulse was 2 Hz. Other recording parameters included an amplifier gain of 50 dB, a sampling delay of 122 s, an effective sampling rate of 20 kHz, and 50 sweeps per averaged eCAP response. The stimulus was presented to individual CI electrodes in a monopolar-coupled configuration via a N6 sound processor that was connected to a programming pod.
For eCAP AGF and RRF measures, electrode locations tested in children with NSCNs and postlingually deafened adult CI users were the same as those tested in G1 and G4. In children with CND, three electrodes across electrode array with measurable eCAPs were tested. In this study, these electrodes were considered as the “basal”, the “middle,” and the “apical” electrode based on their relative locations among electrodes with measurable eCAPs. Details for selecting these electrode locations in children with CND were reported in He et al. (2020). For τ3 estimation, electrode locations tested in G1 and G4 were similar with those tested in other postlingually deafened adult CI users with various etiologies for hearing loss.
Detailed approaches used to measure the eCAP AGF and estimate slope have been reported in Skidmore et al. (2021). Briefly, the eCAP AGF was obtained using the forward-masking-paradigm. The probe pulse was initially presented at 10 clinical unit levels (CLs) lower than the maximum comfortable level (C level) and decreased by 1 CL for at least five consecutive measurements. Subsequently, the stimulation level of the probe pulse systematically decreased in steps of 5 CLs until no eCAP response could be visually identified. The stimulation level was then increased in steps of 1 CL until at least five consecutive eCAPs were measured using this small step size. In this test, the masker pulse was always presented at 10 CLs higher than the probe pulse. The eCAP AGF was obtained by plotting the eCAP amplitude as a function of stimulation level. The slope of the eCAP AGF was estimated using the “Window method” (Skidmore et al., 2022) at each electrode location listed in Table 2.
Detailed approaches used to measure the eCAP RRF and estimate t0 have been reported in our previous studies (He et al., 2018; Skidmore et al., 2021). Briefly, the eCAP RRF was obtained using a modified template subtraction method. A series of eCAPs were recorded as the masker-probe-interval (MPI) was systematically increased from 0.40 to 10.0 ms. The eCAP RRF was obtained by plotting normalized eCAP amplitudes (re: the eCAP amplitude measured at the MPI of 10 ms) as a function of MPI. An estimate of t0 was found using statistical modeling with an exponential decay function. For each test ear, t0s measured at three electrode locations that are listed in Table 2.
The recovery speed from neural adaptation of the CN fibers (i.e., τ3) was measured using the same method as that reported in He et al. (2022). Briefly, eCAPs evoked by the probe pulse presented at different time intervals after the cessation of the last pulse of the masker pulse train were measured. Time intervals tested in this study included 1.054, 2, 4, 8, 16, 32, 64, 128 and 256 ms. To obtain the adaptation recovery function, normalized eCAP amplitudes (re: the eCAP amplitude measured at the MPI of 256 ms) were plotted as a function of tested time interval. τ3 was estimated based on the adaptation recovery function using a mathematical model with up to three exponential components. Details of this mathematical model have been reported in He et al. (2022).
The family spanning five generations showed a typical autosomal dominant inheritance pattern of non-syndromic hearing loss (Figure 2). There was no vestibular dysfunction or other clinical abnormalities indicating syndromic hearing loss. Audiological assessments of the patients revealed bilateral, sensorineural hearing loss that began in the early or mid- 30s affecting all genders that progressed to profound hearing loss by age 50–60 involving all frequencies.
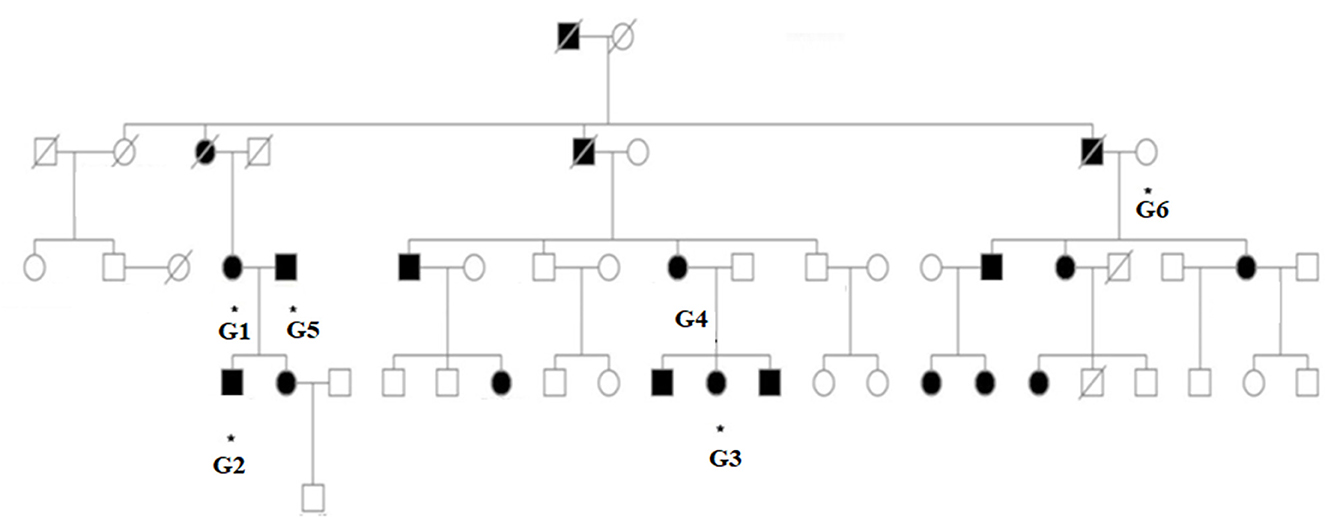
Figure 2. Pedigree of the family with ADSNHL. The pedigree of the five-generation family with the p. Gln246Leu variant in the ACTG1 gene. Filled symbols represent the affected individuals.
In this study, we ascertained a five-generation family with progressive ADNSHL. We used our targeted custom MiamiOtoGenes panel containing 230 hearing loss-associated genes to analyze DNA samples of 2 (G2 and G4) affected family members and sample of 1 patient (G3) was subjected to whole exome sequencing (WES). We then used KGGSeq (http://pmglab.top/kggseq/) for variant filtering based on quality/score read depth according to the autosomal dominant inheritance model, functional significance (i.e., non-sense, missense, or splice site), single-nucleotide variants shared by the 3 affected individuals and minor allele frequency (MAF) thresholds of 0.0005 for ADNSHL variants as reported in public databases. Only 1 heterozygous variant shared by the 3 patients analyzed by NGS was identified using this simple filtering strategy. This sequence change is located in the ACTG1 (hg19) gene on chromosome 17q25.3 at 7,9478,279, c.737A>T (NM_001614.5) leading to p. Gln246Leu (P63261). The identified variant (rs1568061110) has not been published as a pathogenic variant, nor has it been reported as a benign variant to our knowledge. The variant is not observed in large population cohorts (gnomAD). The results of Sanger sequencing analysis (Figures 3A, B), revealed that all of the available affected family members (G1–G4) were heterozygous for this variant, while it was not observed in G6, a family member with normal hearing and also absent in G5, an affected individual related to the family by marriage. The residue glutamine at the position 246 is highly conserved across the species (Figure 3C) and the evolutionary conservation of the involved amino acid is further corroborated by genomic evolutionary rate profiling conservation score (GERP) and PhastCons analysis. Moreover, the observed Gln to Leu amino acid change at position 246 is predicted to be damaging according to PolyPhen, MutationTaster, MutationAssessor, LRT, FATHMM, GERP+, VEST3, PROVEAN, MetaSVM_pred, MetaLR_pred and phastCons. In silico deleterious prediction analysis indicated that the identified variant was potentially deleterious. The characteristics are summarized in Table 3.
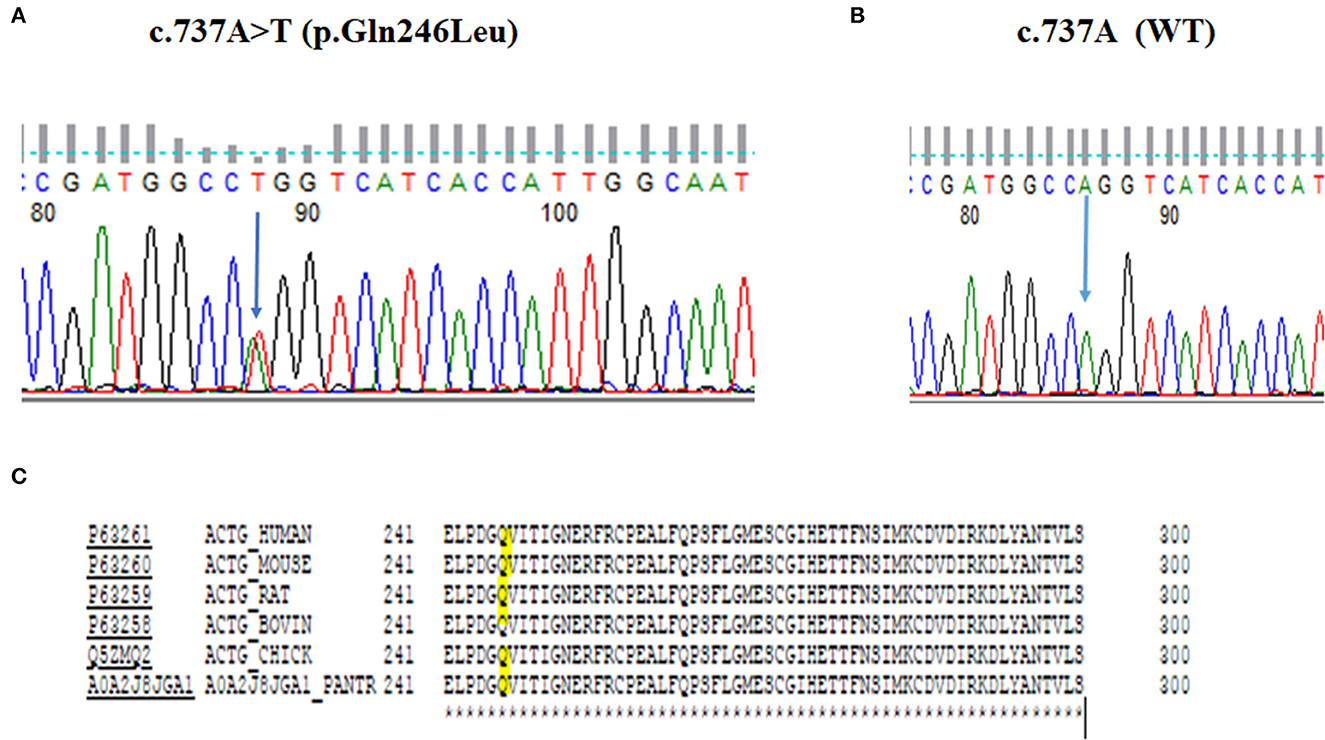
Figure 3. DNA sequencing chromatograms. DNA sequencing chromatograms show the c.737A>T (p.Gln246Leu) variant in ACTG1 in a heterozygous state (A), while normal hearing member has wild-type allele (B). The residue Q (highlighted) is perfectly preserved across the species (C).
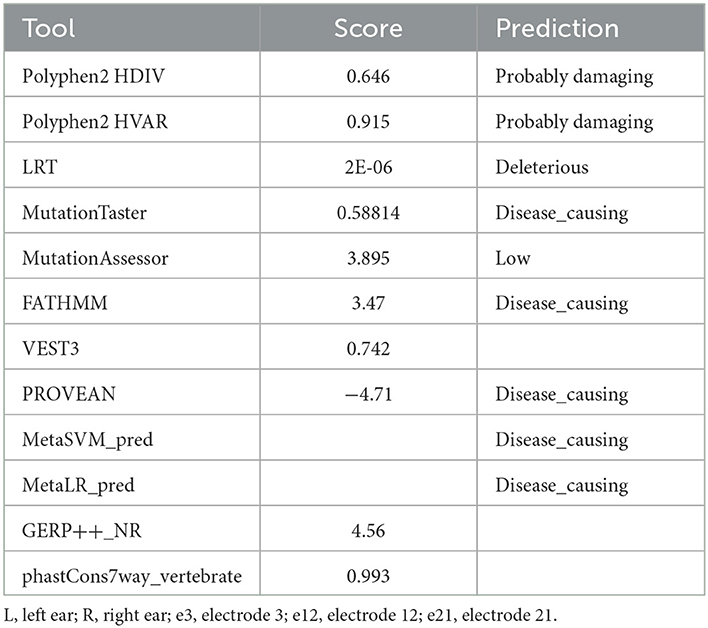
Table 3. Scores and predicted effects of the observed Gln to Leu amino acid change at position 246 determined using different tools.
Human cytoplasmic actomyosin complex based on the cryo-EM structure (pdb id: 5jlh), shows five γ-Actin chains wrapped by Tropomyosin α-3 helices, and two myosin chains outside of the Tropomyosin α-3 helices. p. Gln246 residue shown in VDW representation (Figure 4A) and important residues, including the mutation p. Gln246Leu, at the interface between two γ-Actin chains are shown as sticks (Figure 4B). The mutation was modeled with Chimera (Pettersen et al., 2004) and the structure was rendered with VMD (Humphrey et al., 1996).
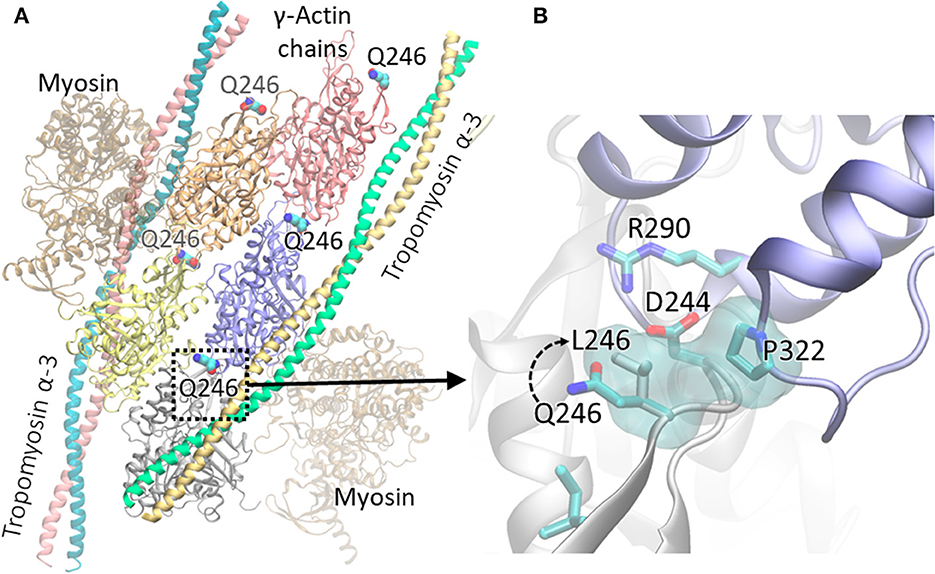
Figure 4. Human cytoplasmic actomyosin complex based on the cryo-EM structure (pdb id: 5jlh), showing γ-Actin chains wrapped by Tropomyosin α-3 helices. The p. Gln246 residue shown in VDW representation (A); neighboring residues including the p. Gln246Leu, at the interface between two γ-Actin chains are shown as sticks (B). The variant was modeled with Chimera (Pettersen et al., 2004) and the structure was rendered with VMD (Humphrey et al., 1996).
In the cytoplasmic actomyosin complex, residue p. Gln246 lies at the interface between γ-Actin chains. As shown in Figure 4, Anp244 in one chain makes an ionic interaction with Arg290 of the other chain. The sidechain of p. Gln246 is in the close proximity of Asp244 in the Wild type. The mutation p. Gln246Leu creates a non-specific hydrophobic contact between Leu246 and Pro322, and possibly affects proper orientation of Asp244. Therefore, this mutation causes significant changes at the interfacial interactions between two γ-Actin chains.
For each eCAP measure, results measured at different electrode locations in G1 and G4 are listed in Tables 4, 5. In addition, their results were compared with 24 implanted children with CND, 32 implanted children with idiopathic sensorineural hearing loss and NSCNs, and 43 postlingually deafened adult CI users with various etiologies of hearing loss. Detailed demographic information, including individual patients' etiologies of hearing loss, and study results of these three comparative implanted patient populations have been reported in our previous studies (He et al., 2018, 2022; Skidmore et al., 2021), and were not re-stated here.
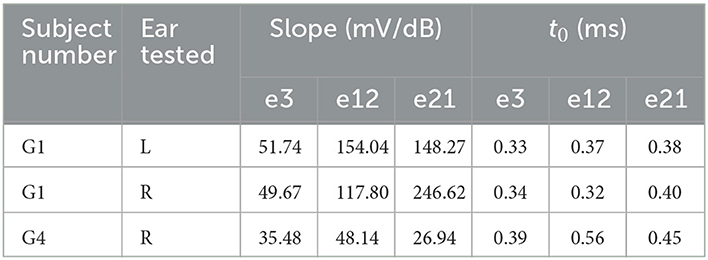
Table 4. Slopes of the electrically evoked compound action potential amplitude growth function and absolute refractory periods (i.e., t0s) measured in G1 and G4.
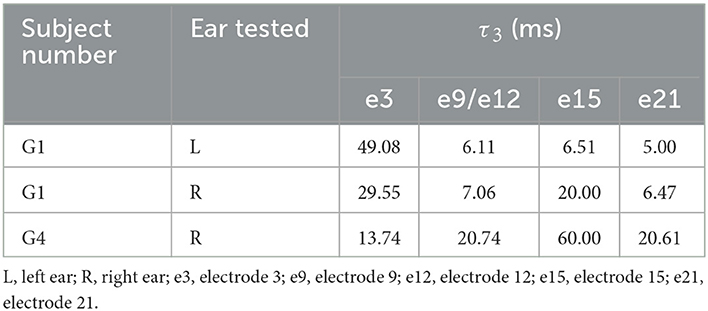
Table 5. τ3s measured in G1 and G4 at four electrode locations.
Data listed in Table 4 clearly show that slopes of the eCAP AGF measured in G1 and G4 varied substantially and demonstrated different trends. Specifically, slopes measured in both ears of G1 increased as the stimulating electrode moved from the base (i.e., electrode 3) to more apical locations of the cochlea (i.e., electrode 12 and electrode 21). This trend was not observed in results measured in G4. In addition, slopes measured in both ears of G1 were substantially larger than those measured in G4 at all three electrode locations.
The top row of Figure 5 depicts results measured in these two participants and the means and standard deviations of slopes measured in three comparative CI patient populations. It is apparent that slopes measured at the middle-array and the apical electrodes in both ears of G1 were substantially larger than those measured in all three CI patient populations. In contrast, slopes measured in G4 were smaller than the averaged slopes measured in implanted children with NSCNs (86.23–83.19 mV/dB) and other adult CI users (42.18–60.56 mV/dB) and appeared to be similar with those measured in implanted children with CND (43.23–50.00 mV/dB).
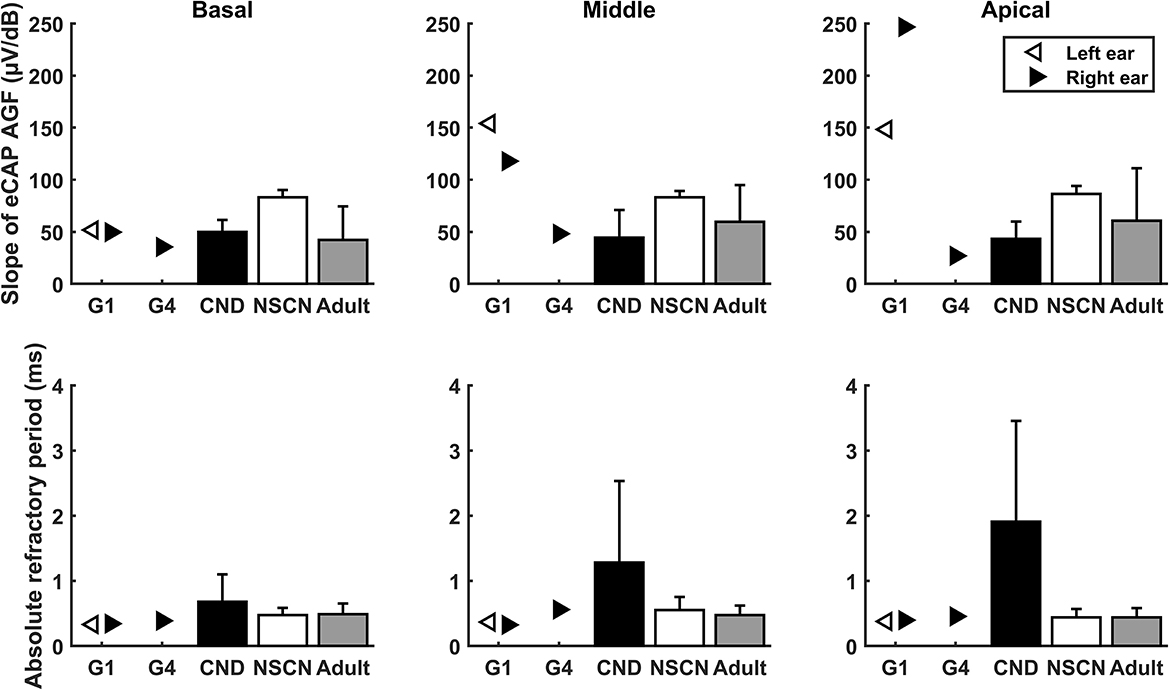
Figure 5. Results of slope of the eCAP AGF (top row) and absolute refractory period of the eCAP RRF (bottom row) measured in G1 and G4, along with the means and standard deviations of the results from the three comparative CI patient populations (CND: N = 24; NSCN: N = 32; Adult: N = 43).
Compared with slopes of the eCAP AGF, t0s measured these two participants (Table 4) show much less variations between participants. In addition, unlike slopes of the eCAP AGF, t0s measured in these two participants do not show any apparent trend that was associated with electrode location.
The bottom row of Figure 5 shows t0s measured in these two participants and the means and the standard deviations of results measured in three CI patient populations. These data show that children with CND clearly had much longer t0s (p < 0.001) than other CI users. t0s measured in all electrode locations in both ears of G1 were shorter than the means of t0s measure in implanted children with CND (0.68–1.91 ms), implanted children with NSCNs (0.44–0.55 ms), and other adult CI users (0.44–0.49 ms). In contrast, t0s measured in G4 were similar with those measured in implanted children with NSCNs and other adult CI users but smaller than those measured in implanted children with CND.
τ3 s measured at different electrode locations in G1 and G4 are listed in Table 5. Similar to slopes of the eCAP AGF, substantial variations in τ3 were observed for results measured at different electrodes within the same participants as well as between these two participants. In both ears of G1, τ3 measured at the basal electrode location was substantially longer than those measured at all other electrode locations. This data trend was not observed for results measured in G4.
Figure 6 shows the comparison between results measured in these two participants and those estimated for other adult CI user with different etiologies. τ3s measured at all except for the basal electrode location were much shorter than the mean of τ3s (48.72–52.74 ms) measured in other postlingually deafened adult CI users. τ3 s measured in G4 are within the same range as those measured in other adult CI users.
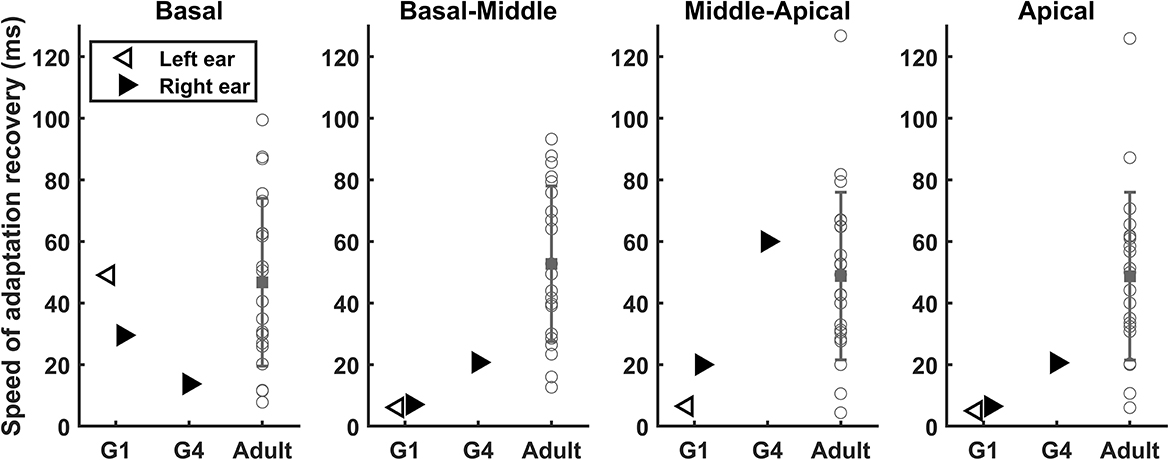
Figure 6. Results of speeds of adaptation recovery measured inG1 and G4, along with the means and standard deviations of the data from the comparative adult cochlear implant user group (Adult: N = 22).
The present study extended the clinical features of hearing loss patients with ACTG1 variants by identification and characterization of the missense p. Gln246Leu variant in ACTG1. Furthermore, this preliminary study incorporated electrophysiological measures of the eCAP to explore the direct link between this genetic variant and the responsiveness of the CN to electrical stimulation in human listeners. Our limited results showed substantial variations in different eCAP measures in two participants with this missense change. Overall, these limited results highlight the importance of evaluating the ability of the peripheral auditory system to respond to electrical stimulation in individual CI users with different genetic profiles.
Up to now, more than 30 mutations in the ACTG1 gene have been reported (Figure 7). Several mutations in the ACTG1 gene were found to cause ADNSHL linked to the DFNA20/26 locus (Van Wijk et al., 2003; Rendtorff et al., 2006). For example, Van Wijk et al. (2003) identified the c.833C>T (p.Thr278Ile) mutation in the gene encoding cytoplasmic ⋎-1-actin (ACTG1) in a Dutch family with ADNSHL (DFNA20/26 locus). Rendtorff et al. (2006) also found a missense mutation (c.1109T>C; p.Val370Ala) in ACTG1 in a Norwegian DFNA20/26 family.

Figure 7. Schematic representation of the full length ACTG1 gene showing the six exons. The mutations associated with DFNA20/26 are indicated above the gene and those linked to Baraitser –Winter syndrome are represented below the exons.
Missense mutations are the most common and so far only one frameshift substitution (p.Val209Alafs*73) has been reported (Sommen et al., 2016) Most of the ACTG1 mutations are “private”, with the exception of three DFNA20/26 associated mutations (Lys118Met, Glu241Lys, and Leu299Val) that were identified in at least two families. The Lys118Met mutation in ACTG1 has been reported in four families and is thus the most common mutation. In addition, a mutation involving the same residue (Lys118Asn) has also been identified in a Spanish family. So far, at least five of the mutations identified in DFNA20/26 families worldwide have been found to carry mutation involving the residue Lys118, suggesting that the amino acid residue Lys118 of ACTG1 may represent a mutational hot spot (Wang et al., 2018) that can be used as the key screening site of the ACTG1 gene detection and the target of gene therapy in the future.
Studies of γcyto-actin null mice model revealed that cyto-actin is necessary to maintain cytoskeletal integrity and function (Belyantseva et al., 2009). In parallel, the dominant effects on hearing phenotypes caused by haploinsufficiency that occurs through several mechanisms are well described in patients with autosomal dominant hearing loss. The mechanism of haploinsufficiency in the present family may result in an instability of the protein as suggested by the structural modeling study. The loss of single functional copy of ACTG1 would have led to the insufficient maintenance of normal function similarly to the γcyto-actin null mouse model.
The present work explored the functional, phenotypic effect of this ACTG1 missense variant on the responsiveness of the CN to electrical stimulation. Decline in CN responsiveness in implanted patients with ACTG1 missense variant was not expected because the primary targeting site of this nucleotide change is believed to be the sensory partition (organ of Corti and synapse) of the cochlea. Nevertheless, eCAP results measured in the two participant who carry the same missense variant showed substantial variations and an inconsistency in alignment with expected results. Specifically, slopes of the eCAP AGF measured in both ears of G1 and in other adult CI users gradually increased as the stimulating electrode moved toward more apical locations within the cochlea (Figure 5). This increase can be due to the gradually reduced diameter of the cochlear turn toward the apical end, the distance between the electrode and the local CN fibers is smaller for more apical electrode locations than for electrodes near basal regions. In addition, it has been shown that neural structures at the apex tend to have better neural survival or be healthier than those located at more basal region of the cochlea in typical listeners with sensorineural hearing loss (Zimmermann et al., 1995). Both factors can lead larger slopes measured at more apical electrode locations (e.g., Zimmermann et al., 1995). More importantly, slopes measured at the middle-array and the apical electrode location were substantially larger than any other CI patient populations reported in this study (Figure 5). Results of τ3 measured in both ear of G1 (Figure 6) were consistent with the slope data in terms of the expected CN responsiveness. Finally, t0s measured in all electrode locations in both ears of G1 were shorter than results measured in all three comparative CI patient populations. Overall, these results suggest robust responsiveness of the electrically stimulated CN in G1, which aligns with the expected results.
In contrast, the electrode-location associated data trend in slope or τt3 was not observed in the results measured in the right ear of G4 (Figure 5). As a matter of fact, slopes measured in G4 appeared to be similar with those measured in implanted children with CND, a patient population with damages in the CN. t0s and τ3s measured in G4 were similar with those measured in implanted children with NSCNs and/or other adult CI users. Overall, these results failed to support the idea that this missense variant does not affect responsiveness of the CN to electrical stimulation in human CI users.
Due to the extremely limited demographic and audiologic data, factors leading to these inconsistent results remain unknown. We can only speculate that the difference in duration of deafness might have, at least partially, contributed to the inconsistency in eCAP results measured in G1 and G4. Specifically, the self-reported duration of deafness of G4 was 56.5 years, which is much longer than that reported by G1 (i.e., 30.7 years). In animal models, longer duration of deafness have been shown to lead to greater functional (e.g., altered spontaneous activity, increased absolute refractory period) and anatomical deterioration (demyelination, loss of peripheral axons and SGNs) of the CN (e.g., Shepherd et al., 2004). These deteriorations will reduce the CN responsiveness to electrical stimulation as measured using the eCAP. In a separate project, the annual noise exposure was assessed and quantified using Noise Exposure Questionnaire (Johnson et al., 2017). The scores for G1 and G4 were 77 and 72, respectively. Therefore, the difference in eCAP results between G1 and G4 is unlikely due to the difference in the amount of annual noise exposure.
Even though the inconsistency in eCAP results measured in G1 and G4 is disappointing and somewhat confusing at this point, these results do suggest the importance of 1) evaluating auditory function for individual patients even with the same genetic profiles, and 2) incorporating patient-specific factors when interpreting and utilizing genetic results for patient care.
The missense c.737A>T (p. Gln246Leu) variant in ACTG1 (NM_001614.5) co-segregated with hearing loss and the most likely to be the genetic cause of ADNSHL in a five-generation American family. Different functional, phenotypic results can be observed in patients with the same genetic variant, which suggests the importance of assessing the functional status of the CN for individual CI users. Further studies with more participants are warranted to test the feasibility of accurately predicting the responsiveness of the electrically stimulated CN based on genetic profiles and patient-specific factors in human CI users.
The electrophysiological data presented in the study are deposited in the Dryad repository, accession number doi: 10.5061/dryad.z8w9ghxj7. All relevant genetic data is contained within the article.
The studies involving human participants were reviewed and approved by the Ethics Committee of The Ohio State University (2017H0131 and 2018H0587). The patients/participants provided their written informed consent to participate in this study.
YY and DY: data curation, formal analysis, writing—original draft preparation, reviewing, and editing. JS: formal analysis, writing—original draft preparation, reviewing, and editing. PC: data curation and formal analysis. XL and SH: conceptualization, funding acquisition, investigation, writing—original draft preparation, reviewing, and editing. All authors contributed to the article and approved the submitted version.
This work was supported by the R01 grants (R01 DC005575 and R01 DC012115) from the National Institutes of Health/National Institute on Deafness and Other Communication Disorders and the Department of Defense grant (RH220053) awarded to XL, as well as the R01 (R01DC017846) from the National Institute on Deafness and Other Communication Disorders and the R01 grant (R01DC016038) from the National Institute on Deafness and Other Communication Disorders and the National Institute of General Medical Sciences awarded to SH.
XL declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Belyantseva, I. A., Perrin, B. J., Sonnemann, K. J., Zhu, M., Stepanyan, R., McGee, J., et al. (2009). γ-Actin is required for cytoskeletal maintenance but not development. Proc. Nat. Acad. Sci. 106, 9703–9708. doi: 10.1073/pnas.0900221106
DeRosier, D. J., and Tilney, L. G. (1989). The structure of the cuticular plate, an in vivo actin gel. J. Cell Biol. 109, 2853–2867. doi: 10.1083/jcb.109.6.2853
Elfenbein, J. L., Fisher, R. A., Wei, S., Morell, R. J., Stewart, C., Friedman, T. B., et al. (2001). Audiologic aspects of the search for DFNA20: A gene causing late-onset, progressive, sensorineural hearing loss. Ear Hearing 22, 279–288. doi: 10.1097/00003446-200108000-00003
Exome Variant Server (2023). NHLBI GO Exome Sequencing Project (ESP), Seattle, WA. Available online at: http://evs.gs.washington.edu/EVS/ (accessed May 24, 2023).
Ferron, F., Rebowski, G., Lee, S. H., and Dominguez, R. (2007). Structural basis for the recruitment of profilin–actin complexes during filament elongation by Ena/Vasp. The EMBO J. 26, 4597–4606. doi: 10.1038/sj.emboj.7601874
Garrison, E., and Marth, G. (2012). Haplotype-based variant detection from short-read sequencing. arXiv [Preprint]. arXiv: 1207.3907. Available online at: https://arxiv.org/pdf/1207.3907.pdf
He, S., Shahsavarani, B. S., McFayden, T. C., Wang, H., Gill, K. E., Xu, L., et al. (2018). Responsiveness of the electrically stimulated cochlear nerve in children with cochlear nerve deficiency. Ear Hearing 39, 238–250. doi: 10.1097/AUD.0000000000000467
He, S., Skidmore, J., Carter, B. L., Lemeshow, S., and Sun, S. (2022). Postlingually deafened adult cochlear implant users with prolonged recovery from neural adaptation at the level of the auditory nerve tend to have poorer speech perception performance. Ear Hearing 11, 10–1097. doi: 10.1097/AUD.0000000000001244
He, S., Xu, L., Skidmore, J., Chao, X., Jeng, F. C., Wang, R., et al. (2020). The effect of Interphase Gap on neural response of the electrically stimulated cochlear nerve in children with cochlear nerve deficiency and children with normal-sized cochlear nerves. Ear Hearing 41, 918–934. doi: 10.1097/AUD.0000000000000815
Hilgert, N., Smith, R., and Camp, G. (2009). Function and expression pattern of non-syndromic deafness genes. Current Mol. Med. 9, 546–564. doi: 10.2174/156652409788488775
Hirokawa, N., and Tilney, L. G. (1982). Interactions between actin filaments and between actin filaments and membranes in quick-frozen and deeply etched hair cells of the chick ear. J. Cell Biol. 95, 249–261. doi: 10.1083/jcb.95.1.249
Humphrey, W., Dalke, A., and Schulten, K. (1996). VMD: visual molecular dynamics. J. Mol. Graphics 14, 33–38. doi: 10.1016/0263-7855(96)00018-5
Johnson, T. A., Cooper, S., Stamper, G. C., and Chertoff, M. (2017). Noise exposure questionnaire: a tool for quantifying annual noise exposure. J. Am. Acad. Audiol. 28, 14–35. doi: 10.3766/jaaa.15070
Kovar, D. R., Harris, E. S., Mahaffy, R., Higgs, H. N., and Pollard, T. D. (2006). Control of the Assembly of ATP- and ADP-actin by Formins and Profilin. Cell 124, 423–435. doi: 10.1016/j.cell.2005.11.038
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Long, C. J., Holden, T. A., McClelland, G. H., Parkinson, W. S., Shelton, C., Kelsall, D. C., et al. (2014). Examining the electro-neural interface of cochlear implant users using psychophysics, CT scans, and speech understanding. J. Assoc. Otolaryngol. 15, 293–304. doi: 10.1007/s10162-013-0437-5
Michelot, A., and Drubin, D. G. (2011). Building distinct actin filament networks in a common cytoplasm. Current Biol. 21, R560–R569. doi: 10.1016/j.cub.2011.06.019
Morell, R. J., Friderici, K. H., Wei, S., Elfenbein, J. L., Friedman, T. B., Fisher, R. A., et al. (2000). A new locus for late-onset, progressive, hereditary hearing loss DFNA20 maps to 17q25. Genomics 63, 1–6. doi: 10.1006/geno.1999.6058
Nord, A. S., Lee, M., King, M.-.C, and Walsh, T. (2011). Accurate and exact CNV identification from targeted high-throughput sequence data. BMC Genomics 12(1). doi: 10.1186/1471-2164-12-184
Pennings, R., Huygen, P., Van Camp, G., and Cremers, C. (2003). A review of progressive phenotypes in non-syndromic autosomal dominant hearing impairment. Audiol. Med. 1, 47–55. doi: 10.1080/16513860310003085
Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., et al. (2004). UCSF chimera – a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. doi: 10.1002/jcc.20084
Pollard, T. D., and Cooper, J. A. (2009). Actin, a central player in cell shape and Movement. Science 326, 1208–1212. doi: 10.1126/science.1175862
Ramekers, D., Versnel, H., Strahl, S. B., Klis, S. F., and Grolman, W. (2015). Temporary neurotrophin treatment prevents deafness-induced auditory nerve degeneration and preserves function. J. Neurosci. 35, 12331–12345. doi: 10.1523/JNEUROSCI.0096-15.2015
Rendtorff, N. D., Zhu, M., Fagerheim, T., Antal, T. L., Jones, M. P., Teslovich, T. M., et al. (2006). A novel missense mutation in ACTG1 causes dominant deafness in a Norwegian DFNA20/26 family, but ACTG1 mutations are not frequent among families with hereditary hearing impairment. Eur. J. Hum. Genetics 14, 1097–1105. doi: 10.1038/sj.ejhg.5201670
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genetics Med. 17, 405–424. doi: 10.1038/gim.2015.30
Rivière, J. B., van Bon, B. W., Hoischen, A., Kholmanskikh, S. S., O'Roak, B. J., and Gilissen, C. (2012). De novo mutations in the actin genes ACTB and ACTG1 cause Baraitser-Winter syndrome. Nat. Genet. 44, 440–444. doi: 10.1038/ng.1091
Shearer, A. E., Hildebrand, M. S., and Smith, R. J. (1993). “Hereditary hearing loss and deafness overview,” in GeneReviews®, eds M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. Stephens, et al. (Seattle, WA: University of Washington).
Shepherd, R. K., Roberts, L. A., and Paolini, A. G. (2004). Long-term sensorineural hearing loss induces functional changes in the rat auditory nerve. Eur. J. Neurosci. 20, 3131–3140. doi: 10.1111/j.1460-9568.2004.03809.x
Skidmore, J., Ramekers, D., Colesa, D. J., Schvartz-Leyzac, K. C., Pfingst, B. E., He, S., et al. (2022). A broadly applicable method for characterizing the slope of the electrically evoked compound action potential amplitude growth function. Ear Hearing 43, 150–164. doi: 10.1097/AUD.0000000000001084
Skidmore, J., Xu, L., Chao, X., Riggs, W. J., Pellittieri, A., Vaughan, C., et al. (2021). Prediction of the functional status of the cochlear nerve in individual cochlear implant users using machine learning and electrophysiological measures. Ear Hearing 42, 180–192. doi: 10.1097/AUD.0000000000000916
Sommen, M., Schrauwen, I., Vandeweyer, G., Boeckx, N., Corneveaux, J. J., van den Ende, J., et al. (2016). DNA diagnostics of hereditary hearing loss: a targeted resequencing approach combined with a mutation classification system. Hum. Mut. 37, 812–819. doi: 10.1002/humu.22999
Teig, E. (1968). Hereditary progressive perceptive deafness in a family of 72 patients. Acta Oto-Laryngol. 65, 365–372. doi: 10.3109/00016486809120977
Tilney, L. G., Egelman, E. H., DeRosier, D. J., and Saunder, J. C. (1983). Actin filaments, stereocilia, and hair cells of the bird cochlea. II. packing of actin filaments in the stereocilia and in the cuticular plate and what happens to the organization when the stereocilia are bent. J. Cell Biol. 96, 822–834. doi: 10.1083/jcb.96.3.822
Van Wijk, E., Krieger, E., and Kemperman, M. H. (2003). A mutation in the gamma actin 1 (ACTG1) gene causes autosomal dominant hearing loss (DFNA20/26). J. Med. Genetics 40, 879–884. doi: 10.1136/jmg.40.12.879
Wang, L., Yan, D., Qin, L., Li, T., Liu, H., Li, W., et al. (2018). Amino acid 118 in the deafness causing (DFNA20/26) ACTG1 gene is a mutational hot spot. Gene Rep. 11, 264–269. doi: 10.1016/j.genrep.2018.04.011
Yan, D., Tekin, D., Bademci, G., Foster, J., Cengiz, F. B., and Kannan-Sundhari, A. (2016). Spectrum of DNA variants for nonsyndromic deafness in a large cohort from multiple continents. Hum. Genet. 135, 953–961. doi: 10.1007/s00439-016-1697-z
Yang, T., and Smith, R. (2000). A novel locus DFNA 26 maps to chromosome 17q25 in two unrelated families with progressive autosomal dominant hearing loss. Am. J. Hum. Genet. 67, 300–300.
Keywords: electrical stimulation, missense variant, genetic profile, auditory nerve, electrically evoked auditory compound action potentials
Citation: Yuan Y, Yan D, Skidmore J, Chapagain P, Liu X and He S (2023) Responsiveness of the electrically stimulated cochlear nerve in patients with a missense variant in ACTG1: preliminary results. Front. Audiol. Otol. 1:1213323. doi: 10.3389/fauot.2023.1213323
Received: 27 April 2023; Accepted: 15 June 2023;
Published: 07 July 2023.
Edited by:
Z. Jason Qian, Stanford University, United StatesReviewed by:
Kazuaki Homma, Northwestern University, United StatesCopyright © 2023 Yuan, Yan, Skidmore, Chapagain, Liu and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xuezhong Liu, WC5MaXUxQG1lZC5taWFtaS5lZHU=; Shuman He, U2h1bWFuLkhlQG9zdW1jLmVkdQ==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.