 Xiangyu Wang
Xiangyu Wang Yi Yang1,2
Yi Yang1,2 Yongxin Lv
Yongxin Lv Weishu Zhao
Weishu Zhao- 1State Key Laboratory of Microbial Metabolism, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai, China
- 2International Center for Deep Life Investigation (IC-DLI), Shanghai Jiao Tong University, Shanghai, China
- 3Southern Marine Science and Engineering Guangdong Laboratory (Zhuhai), Zhuhai, China
D-amino acids (D-AAs) have been produced both in organisms and in environments via biotic or abiotic processes. However, the existence of these organic materials and associated microbial degradation activity has not been previously investigated in subduction zones where tectonic activities result in the release of hydrothermal organic matter. Here, we isolated the bacterium Halomonas sp. LMO_D1 from a sample obtained from the Mariana trench, and we determined that this isolate utilized 13 different D-AAs (D-Ala, D-Glu, D-Asp, D-Ser, D-Leu, D-Val, D-Tyr, D-Gln, D-Asn, D-Pro, D-Arg, D-Phe, and D-Ile) in the laboratory and could grow on D-AAs under high hydrostatic pressure (HHP). Moreover, the metabolism of L-AAs was more severely impaired under HHP conditions compared with that of their enantiomers. The essential function gene (Chr_2344) required for D-AA catabolism in strain LMO_D1 was identified and confirmed according to the fosmid library method used on the D-AAs plate. The encoded enzyme of this gene (DAADH_2344) was identified as D-amino acid dehydrogenase (DAADH), and this gene product supports the catabolism of a broad range of D-AAs. The ubiquitous distribution of DAADHs within the Mariana Trench sediments suggests that microorganisms that utilize D-AAs are common within these sediments. Our findings provide novel insights into the microbial potential for utilizing abiotic enantiomers of amino acids within the subduction zone of the Mariana trench under HHP, and our results provide an instructive significance for understanding these abiotic enantiomers and allow for insights regarding how organisms within extraterrestrial HHP environments can potentially cope with toxic D-AAs.
Introduction
Among all three domains of life, proteins are composed of 19 L-form amino acids (along with the achiral glycine), and these are known as the 20 proteinogenic amino acids (Burton and Berger, 2018). The use of one type of enantiomers instead of enantiomers mixtures is considered to be the foundation for the stability of biomolecules (Hein and Blackmond, 2012). Although single handedness has been selected for use in proteins, both of the mirror-image forms of amino acids can be generated via abiotic or biotic processes. The simulated scenarios in the context of laboratory abiotic synthesis of these chiral molecules, including spark discharge experiments (Miller, 1953), the Strecker reaction (Peltzer et al., 1984), and other hydrothermal reactions (Kebukawa et al., 2017) have demonstrated that racemic mixtures are always yielded. Racemization is another non-negligible process that can generate enantiomer mixtures. The estimated racemization half-lives of amino acids are 0.1–1 million years at temperatures comparable with that of the surface of the Earth (Bada and Miller, 1987). Natural abiotic synthesis products are primarily identified in meteorites, where the vast majority of chiral molecules are racemic (Pizzarello, 2006). To date, 14 of the 20 proteinogenic amino acids (Ala, Asp, Glu, Gly, Ile, Leu, Pro, Ser, Thr, Val, Tyr, Phe, Met, and Trp) have been detected in meteorites or identified in abiotic synthesis (Menez et al., 2018; Glavin et al., 2020). The amounts of amino acids can reach the order of ppm (parts per million) and can vary according to the types of amino acids or meteorites (Pizzarello et al., 2012; Glavin et al., 2020). In addition to their generation via biotic processes, certain D-AAs have also been discovered in some functional structures present in prokaryotes, eukaryotes, and viruses. For example, D-AAs (usually D-Ala, D-Glu, D-Ser, and D-Asp) are components of the cell walls of bacteria (Vollmer et al., 2008). D-Ser acts as a neurotransmitter in the brain (Snyder and Kim, 2000). These D-AAs are generated from L-AAs by enzymatic racemization that occurs via racemases (Zhang and Sun, 2014) and are released into the environment by direct secretion or through cell lysis (Zhang et al., 2015).
As enantiomers of proteinogenic amino acids, D-AAs present within various environments are toxic to organisms due to their ability to interfere with translation by binding to tRNA (Soutourina et al., 2004) and to interfere with the formation of peptidoglycans (Caparros et al., 1992). Therefore, the catabolism of D-AAs is a significant component of the detoxification process, and this process can be summarized into two aspects that include the direct oxidization of D-AAs to α-keto acids and the transformation of D-AAs into L-enantiomers in a manner that is coupled with L-AA catabolism (Zhang et al., 2021). Related enzymes involved in D-AA catabolism can be divided into broad-spectrum oxidase/dehydrogenases, substrate-specific oxidases, and racemases. DAADHs are the most commonly used broad-spectrum oxidases for D-AA direct oxidation to the corresponding α-keto acids and ammonia in prokaryotes, and have been identified in various species such as Pseudomonas aeruginosa (Marshall and Sokatch, 1968), Escherichia coli (Olsiewski et al., 1980), and Helicobacter pylori (Tanigawa et al., 2010). D-AA oxidases catalyze the oxidative deamination of uncharged aliphatic, aromatic, and polar D-AAs to produce α-keto acids, ammonia, and hydrogen peroxide that have been observed in most eukaryotic organisms and in some bacteria (Pollegioni et al., 2008). The substrate-specific oxidases include D-aspartate oxidase, D-Ser dehydratase, D-Cys desulfhydrase, and D-proline dehydrogenase. Conversely, amino acid racemases that function to mediate the interconversion of amino acid enantiomers are ubiquitous in bacteria and are consequently considered to play an important role in preventing the toxicity of D-AAs (Zhang and Sun, 2014). In archaea, a PLP-dependent racemase enables Pyrococcus horikoshii OT-3 to offset the essential L-amino acid requirement by adding D-type enantiomers (Kawakami et al., 2015). These enzymes are related to either D-AA oxidation or to D-AA transformation to the L-enantiomers and can also enable the utilization of D-AAs as carbon or nitrogen sources. The bioavailability of D-AAs has been verified in a number of microbes that have been isolated from a number of sources including soils, the rhizosphere (Radkov et al., 2016), seawater, and sediments (Fu et al., 2016; Kubota et al., 2016; Yu et al., 2019). As the formation of D-AAs by biotic racemization is a ubiquitous process, the utilization of D-AAs may not be limited to the above ecosystems. In this study, we primarily aim to extend the knowledge of the catabolic processes of D-AAs to the deepest area on Earth (the hadal trench).
The hadal trench possesses a water depth of greater than 6,000 m and contains complex processes that occur at different scales, such as geology, physical dynamics, geochemistry, and biology, and this is currently the least studied area within the ocean (Jamieson, 2011). The subduction zones in the hadal trench was formed as a result of oceanic plate converging with continental plate in combination with multiple extreme characteristics, such as extremely deep water depth, high pressure, cold, and geological activity. Organic matter arrived in the hadal trench through deposition from upper water or due to migration from steep slopes that benefit from the unique funnel-like shape of the trench and frequent seismic activity (Jamieson, 2011; Luo et al., 2017). During the descent of the oceanic plate, materials within the subduction slab and the seawater are transported to the overlying mantle (Plumper et al., 2017) or reach the oceanic mantle through cracks generated by slab flexing (Zhou et al., 2015). The slab hydration and serpentinization processes liberated mantle carbon and, by reducing power, generated alkaline fluids that erupted through slab faults and fueled the ecosystem (Ohara et al., 2012). Evidence of abiotically formatted amino acids beneath the Atlantis Massif during the alteration of serpentinites indicate the potential for fluid–rock interactions that may allow for abiotic formation of amino acids in the oceanic lithosphere (Menez et al., 2018). No available data have indicated the existence of D-AAs within the Mariana trench; however, it is still reasonable to assume that both abiotic and biotic racemization from L-AAs and also the abiotic formation of amino acids can generate D-AAs within the hadal environment.
Here, we report a bacterial strain (Halomonas sp. LMO_D1) that possesses the ability to utilize D-AAs, and this bacterial strain was enriched and isolated from subduction zones within the Mariana trench. Cultivation experiments demonstrated that the LMO_D1 strain could grow on D-AAs even under HHP conditions. One of the essential enzymes for D-AA utilization (DAADH) has been confirmed to function well both in vivo and in vitro. Our results revealed the capability of one microbe to utilize D-AAs under HHP conditions within the subduction zone, and these results extended our knowledge of the microbes within the hadal trench and provided a valuable microbiological material for abiotic enantiomer utilization.
Materials and methods
Sample collection
The sediment sample MT086 from the Mariana rench was used in this study for enrichment and strain isolation. MT086 was collected in the Challenger Deep (11°11.6988′N, 141°48.7008′E) at a depth of 8,636 m during cruise TS01 using a submersible manipulator in August of 2016. The collected samples were stored in sterile pressurized vessels at 45 MPa and 4°C for further processing.
Enrichment, isolation, and cultivation using defined medium
The medium used in this study was based on an artificial seawater (ASW) and composed of the following components per liter: 26.0 g of NaCl, 5.0 g of MgCl2·6H2O, 1.06 g of CaCl2, 4.0 g of NaSO4, 0.3 g of NH4Cl, 0.1 g of KH2PO4, 0.5 of g KCl, and 2.52 g of NaHCO3. Additionally, ASW was supplemented with 1 ml of a trace element mixture, a vitamin mixture, a vitamin B12 solution, and a thiamine solution as described previously (Widdel et al., 2006). The pH of the medium was adjusted to 7.0 using 1 M HCl solution. A mixture of 19 D-AAs (0.01% w/v for each) was supplemented as the sole carbon source for enrichment and isolation of microbes with corresponding metabolic potential. The cultures were incubated at 8°C, and this was designed based on the optimal growth temperature of the majority of obligate piezophiles (2°C–10°C). To test the bioavailability of different amino acids, the cells were harvested, washed three times with PBS buffer, and then inoculated into ASW medium that was supplemented with 6 mM amino acids. When amino acids served as the sole nitrogen source, NH4Cl was removed, and 6 mM glucose was added.
Enrichment and cultivation under various pressures were both performed as previously described (Wang et al., 2021). Sediments (0.15% w/v) or pure-cultured cells (initial OD600 = 0.01) were inoculated into the defined medium. The cultures were transferred into sterile plastic syringes and sealed with a rubber cover. The syringes were placed into a stainless pressure vessel (Feiyu Science and Technology Exploitation Co. Ltd., Nantong, China) and compressed as described in a previous study (Wang et al., 2021). Each culture was repeated in triplicate.
DNA extraction and PCR/qPCR amplification of 16S rRNA genes
Total DNA from sediments and enrichment samples was extracted and purified using the FastDNA™ Spin Kit for Soil (MP Biomedicals, USA) following the manual protocol. Genomic DNA from strain LMO_D1 was extracted using the phenol–chloroform method. The full-length bacterial 16S rRNA genes were amplified using primers 27F (5-AGAGTTTGATCCTGGCTCAG-3) and 1492R (5-GGTTACCTTGTTACGACTT-3). The amplified products were ligated into the pMD18-T vector (TaKaRa, Tokyo, Japan) for sequencing. To determine the cell density of the sediment samples and enrichment culture, 16S rRNA genes from bacteria and archaea were quantified by qPCR using primers 341F/519R (5-CCTACGGGWGGCWGCA-3/5-TTACCGCGGCKGCTG-3) and 519F/908R (5-CAGCMGCCGCGGTAA-3/5-CCCGCCAATTCCTTTAAGTT-3), respectively. Plasmids possessing archaea and bacterial 16S rRNA genes were used for standard curve construction. The quantification standard consisted of a dilution series of 1 × 103 and 1 × 109 copies/μl. To analyze the microbial composition, the V4 and V4–V5 regions of the 16S rRNA genes were amplified with primers 515F (5-GTGCCAGCMGCCGCGGTAA-3) and 806R (5-GGACTACHVGGGTWTCTAAT-3) for bacteria and 516F (5-TGYCAGCCGCCGCGGTAAHACCVGC-3) and 855R (5-TCCCCCGCCAATTCCTTTAA-3) for archaea.
Genome and 16S rRNA gene amplicon sequencing and data analysis
Genomic DNA was sequenced using an Illumina HiSeq (400 bp pair-end library; Illumina, USA) and PacBio (20-kb insert library; Pacific Biosciences, USA) platform at Shanghai Personal Biotechnology Co., Ltd. (Shanghai, China). The raw sequence reads were trimmed with SOAPec v2.0 based on the k-mer length at 17 (Luo et al., 2012). The trimmed sequences from the Illumina HiSeq platform were assembled using A5-miseq version 20160825 (Tritt et al., 2012) and SPAdes genome assembler v3.11.1 (Bankevich et al., 2012). Sequences from the PacBio platform were assembled using HGAP4 (Chin et al., 2016) and CANU V1.6 (Koren et al., 2017). Misassemblies were detected using MUMmer v3 (Delcher et al., 1999) and corrected using Pilon version 1.22 (Walker et al., 2014). Open reading frames (ORFs) were predicted using GeneMarkS version 4.32 (Besemer et al., 2001). The annotation and alignment of ORFs were both performed using Diamond v0.9.10.111 (Buchfink et al., 2015) against the NCBI NR database using an E-value cutoff of less than 10−6.
The 16S rRNA gene amplicons with different barcodes were mixed together at the same amounts and sequenced on an Illumina MiSeq platform using 2 × 250-bp cycles. For raw sequence data, low-quality reads (average quality value <20) and lengths <150 bp and also any ambiguities were excluded from the analysis. Further analysis was performed using the QIIME2 version 2020.11 software pipeline (Bolyen et al., 2019). Taxonomic assignment was conducted using the SILVA database release 138 (Quast et al., 2013).
Fosmid library construction, screening, and identification
The genomic library of the LMO_D1 strain was constructed using the CopyControl™ HTP Fosmid Library Production Kit (Cat. No. CCFOS059) according to the instructions of the manufacturer. High-molecular-weight genomic DNA was sheared randomly, ligated into the fosmid vector pCC2FOS™, and transfected into E. coli strain EPI300™-T1R. Then clones were inoculated onto M9 minimal agar that was supplemented with a D-AAs mixture (0.01% w/v for each), 12.5 μg ml−1 of chloramphenicol, and 0.1% CopyControl™ induction solution to induce clones with high copy numbers. Fosmid DNA from positive clones was extracted using the E. Z.N.A.™ plasmid mini kit (Omega Bio-Tek, USA). The flanking sequences of the cloned insert DNA were sequenced using pCC2FOS forward and reverse sequencing primers (5′-GTACAACGACACCTAGAC-3′ and 5′-CAGGAAACAGCCTAGGAA-3′) and blasted with the genome of strain LMO_D1 to identify the position of the insert DNA within the genome.
Protein expression, purification, and enzyme assay
The DAADH gene (Chr_2344) was cloned into the pET-28a vector between the NdeI and HindIII sites and expressed heterologously in E. coli BL21 (DE3). The recombinant E. coli strain was induced with isopropyl-β-D-thiogalactoside (0.1 mM) at 16°C for 20 h. Cells were harvested by centrifugation at 3,000 × g for 3 min and suspended in Buffer A containing 20 mM Tris-HCl (pH 8.0), 0.3 M NaCl, and 10% glycerol. Cells were lysed by sonication, and cell debris was removed by centrifugation at 8,000 × g for 40 min. The soluble and membrane fractions were separated by ultra-speed centrifugation at 160,000 × g for 60 min. The membrane fraction was suspended in buffer A that was supplemented with 0.03% Triton X-100. Sonication and ultra-speed centrifugation were repeated to obtain solubilized membrane proteins. The target protein was purified using Ni2+-NTA resin and eluted with Buffer B containing 20 mM Tris-HCl (pH 8.0), 0.3 M NaCl, 0.3 M imidazole, and 10% glycerol.
Enzyme activity was detected using a modified method as described previously (Tanigawa et al., 2010). The reaction mixture was composed of 50 mM Tris-HCl (pH 8.0), 20 mM D-AA, 10 μM flavin adenine dinucleotide (FAD), 1.8 μM phenazine methosulfate (PMS), 0.5 mM 2,6-dichlorophenolindophenol (DCIP), and an appropriate amount of enzyme. The reaction was initiated by the addition of substrate, subsequently performed at 37°C, and ultimately terminated by the addition of 0.25% SDS. Enzyme activity was quantified by measuring the reduction of DCIP at 600 nm. As PMS could be reduced nonenzymatically by Cys, the substrate specificity for Cys was detected using the 3-methyl-2-benzothiazolinone hydrazine (MBTH) method as described previously (Yu et al., 2019).
Results
Enriching and screening of a microorganism possessing D-amino acid metabolic potential
We conducted two sets of enrichment experiments that were supplemented with D-AAs as the sole carbon source. Each set of enrichment experiments was performed using two independent groups that differed in regard to cultivation pressure, and these groups were denoted as GA (at 0.1 MPa) and GH (at 70 MPa). As low temperature and HHP resulted in a reduced growth rate, the enrichment was continued for 69 days. After enrichment, the biomass yields of the GA and GH groups were estimated by quantification of 16S rRNA that was increased to 4.87 × 108 and 2.61 × 105 cells/ml, respectively, from an initial biomass of 1.0 × 103 cells/ml. The structures of the microbial communities for these two enrichments were analyzed and compared with that of the original sediment via 16S rRNA amplicon analysis (Materials and methods section). The enrichment samples were dominated by Halomonas, and the abundances of this species were increased to 95.5% and 73.9% in the GA and GH groups from 1.9% in the sediment sample MT086. The abundance of Macellibacteroides was also increased to 0.9% and 11.7% in the GA and GH groups, and this value was less than 0.001% in MT086 (Figure 1).
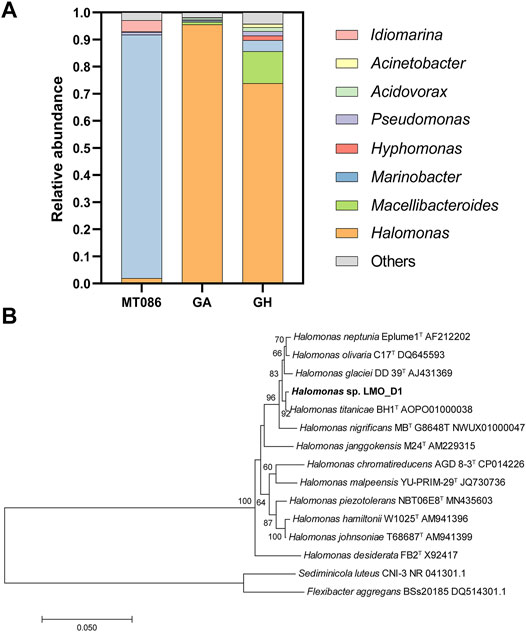
FIGURE 1. Enrichments and isolations of microorganisms with the D-amino acid (D-AA) utilization capability. (A) Microbial community composition of MT086 sediment sample, enrichment sample with D-AAs as sole carbon source under 0.1 MPa (GA) and 70 MPa (GH) at genus level. (B) Neighbor-joining phylogenetic tree based on 16S rRNA gene sequences showing the phylogenetic position of strain LMO_D1 and other closely related species. Percentage bootstrap values above 60% (1,000 replicates) are shown at branch nodes. Sediminicola luteus CNI-3 and Flexibacter aggregans BSs20185 were used as an outgroup. Bar, 0.05 substitutions per nucleotide position.
Strain isolation was performed by spreading sediment or enrichment samples onto the plates. The growth was monitored, and visible clones were present after a 2-week incubation at 8°C. When the screening plates were supplemented with all 19 D-AAs as the sole carbon source, strain LMO_D1 derived from the sediment sample MT086 was identified as Halomonas (Figure 1B). Strains were also isolated from D-AA-enriched samples of the GA and GH groups, and these were all identified as the same species as strain LMO_D1.
D-amino acid utilization by strain LMO_D1 under atmospheric pressure
When supplied as nutrients, D-AAs can be utilized as carbon or nitrogen sources, and these were separately tested in this study. The capacity of strain LMO_D1 to utilize each individual D-AA as a sole carbon and/or sole nitrogen source was investigated by cultivation under atmospheric pressure (0.1 MPa) (Figure 2A). For comparison, 20 proteinogenic amino acids (L-type) were also used as the sole carbon/nitrogen sources to measure the growth of the LMO_D1 strain. Each individual amino acid at a concentration of 6 mM was tested as the sole nitrogen source (6 mM D-glucose supplemented as a carbon source). Modified ASW medium (NH4Cl removed) without the addition of any nitrogen source was used as a negative control. Individually, all 19 D-AAs were able to independently support the growth of strain LMO_D1, as indicated by an obviously increasing biomass compared with that of the negative control. Similarly, 18L-AAs and Gly could also be utilized as a nitrogen source by strain LMO_D1, with the exception of L-Cys. L-Cys cannot support the growth of strain LMO_D1, while D-Cys does support the growth of this strain. For the other 18 amino acids where both the L and D-types can be used as sole nitrogen sources, an overall improved growth was observed in response to supplementation with L-AAs as the sole nitrogen source compared with that observed in response to supplementation with D-AAs, and this was particularly true for Phe, His, Lys, Glu, Asp, Gln, and Asn (Figure 2).
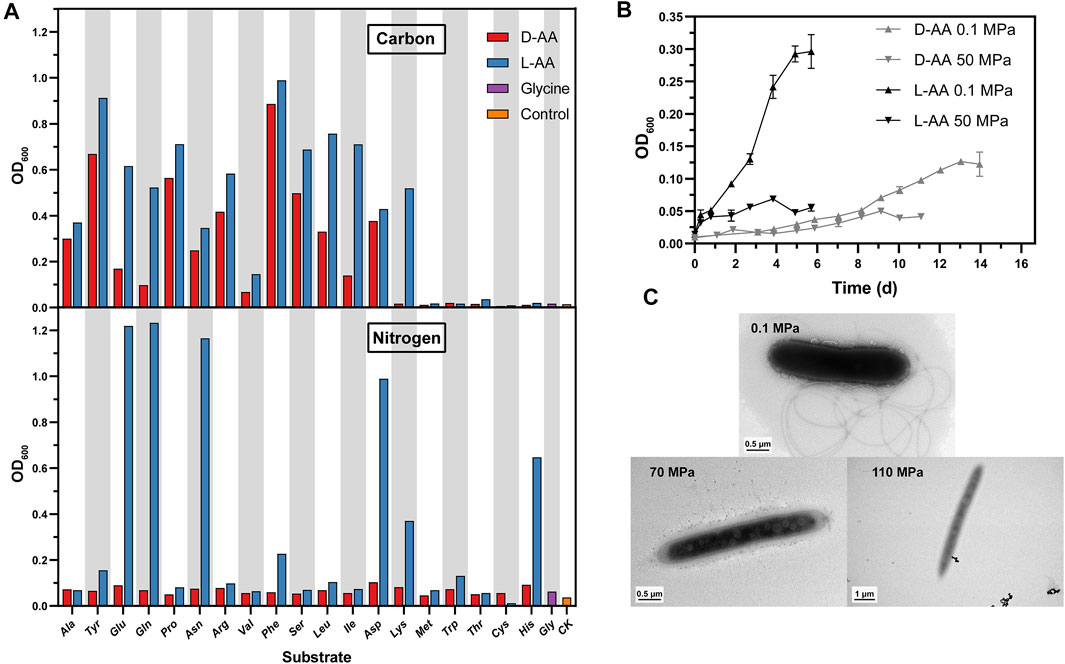
FIGURE 2. Physiology and morphology of strain LMO_D1. (A) Amino acids as sole carbon or nitrogen source. CK, negative control without adding any carbon or nitrogen source. (B) Growth curve of strain LMO_D1 supplemented with the mixture of 19 D-AAs or 19 L-AAs as sole carbon source under 0.1 MPa or 50 MPa. Error bars represent the standard deviations among triplicates. (C) Transmission electron microscopic image of strain LMO_D1 cultivated under various pressure conditions (i.e., 0.1, 70, and 110 MPa).
When each individual amino acid at a concentration of 6 mM was tested as a sole carbon source (5.6 mM NH4Cl was used as the nitrogen source), ASW medium without the addition of any carbon source was used as a negative control. Strain LMO_D1 exhibited broad utilization capability for these D-AAs (13 individuals in total), including four common D-AAs (D-Ala, D-Glu, D-Asp, and D-Ser) that are components of peptidoglycans in most bacteria and have been widely reported to be present in marine systems (McCarthy et al., 1998) and nine uncommon D-AAs (D-Leu, D-Val, D-Tyr, D-Gln, D-Asn, D-Pro, D-Arg, D-Phe, D-Ile) that are rarely detected in environments (McCarthy et al., 1998; Kaiser and Benner, 2008) (Figure 2A). A similar trend was also observed for L-AA utilization in strain LMO_D1. Individual L-types of 13 AAs (Ala, Glu, Asp, Ser, Leu, Val, Tyr, Gln, Asn, Pro, Arg, Phe, and Ile) can support the growth in a manner similar to that of their D-types, thus indicating the existence of consistent catabolic pathways for amino acid enantiomers. Another L-AA (L-Lys) can support the growth of strain LMO_D1, while D-Lys cannot, thus, indicating the presence of an L-Lys utilization pathway in LMO_D1 and a lack of transfer enzymes between L-Lys and D-Lys in this strain.
Overall, better growth was observed when the medium was supplemented with L-type AA as the sole carbon or nitrogen source compared with growth in response to supplementation with the D-type. Strain LMO_D1 can utilize a greater number of D-AAs as a nitrogen source than they can as a carbon source, thus, indicating that D-AAs are more easily used as nitrogen sources. This is consistent with the results of a previous study (Wang et al., 2020).
Utilization of D-amino acids by strain LMO_D1 under high hydrostatic pressure
Considering that strain LMO_D1 was isolated from the hadal zone with a water depth of 8,636 m (equal to ∼86 MPa) where the HHP caused by in situ water depth typically affects the metabolism of microorganisms, we also conducted D-AA utilization experiments under HHP to test if D-AA utilization can occur in the in situ environment (Figure 2B). The enrichment results indicated that the utilization of D-AAs occurred under 70 MPa conditions. However, the pre-experiments revealed that it is difficult to monitor and quantify the growth in a relatively short time under 70 and 110 MPa conditions, despite the integral cells being observed under these two pressures (Figure 2C). Thus, we chose a moderate pressure of 50 MPa that represents an upper limit that inhibits the growth of most piezosensitive microorganisms (Bartlett, 2002) to perform physiological experiments under HHP.
Strain LMO_D1 was then cultivated by supplementing individual L- or D-AAs as sole carbon sources under 50 MPa, and these were compared with cultivations performed under 0.1 MPa (Figure 2B). In general, the growth yields under HHP were lower than those under 0.1 MPa, and this was consistent with the enrichment results. When supplemented with D-AAs, growth under two different pressures exhibited a similar trend during the early stage. Subsequently, the maximum biomass yields were observed after approximately 9 and 13 days of incubation under 50 and 0.1 MPa, respectively, with a 2.52-fold higher biomass yield under 0.1 MPa. However, when cultivated with L-AAs, strain LMO_D1 exhibited a significantly higher growth rate and biomass yield under 0.1 MPa compared with these values in response to cultivation under 50 MPa. The maximum biomass under 0.1 MPa was 4.29-fold higher than that under 50 MPa. When comparing the cultivation results in response to supplementation with L-AAs and D-AAs, better growth was observed under both 0.1 and 50 MPa when L-AAs were supplemented. The growth variances under two different pressures were more significant when L-AAs were supplemented, and this indicated that the influence of HHP was different for the utilization of different amino acid enantiomers. In our experimental conditions, L-AA metabolism was more severely impaired under HHP compared with this value using their enantiomers.
Genomic evidence of D-amino acid catabolism
To investigate the catabolism of D-AAs, the complete genome of Halomonas sp. LMO_D1 was acquired using Illumina Hiseq and Pacbio platforms and subsequently analyzed (Materials and methods section ). The complete genome of strain LMO_D1 contained a circular chromosome possessing a sequence length of 5.41 Mb and GC content of 54.77%. The genome possessed the highest identity with Halomonas sp. KHS3 (average nucleotide identity = 99.12% with 91.57% sequence alignment) that was isolated from the seawater of Mar del Plata. This bacterial strain harbors a strong ability to degrade different types of aromatic compounds (Corti Monzon et al., 2018), and no reports have been published on the capacity of this strain to utilize D-AAs.
According to the genomic information, strain LMO_D1 contained a complete glycolysis/glycogenesis pathway and TCA cycle that enabled the catabolism of the carbon skeleton of D-AAs. Four types of D-AA catabolism genes were identified in the strain LMO_D1 genome, including eight amino acid racemases, four DAADHs, one D-Ser dehydratase, and one D-Cys desulfhydrase (Table 1). Racemase mediates the interconversion of enantiomers. The annotated amino acid racemases present in strain LMO_D1 included alanine racemase, glutamate racemase, Asp/Glu racemase, and proline racemase. DAADHs are FAD-dependent oxidases that catalyze the deamination of D-AAs to the corresponding α-keto acids and ammonia. Four genes (Chr_2344, Chr_3502, Chr_3902, and Chr_4236) were annotated as DAADH within the genome of strain LMO_D1 based on the KEGG and COG databases (Galperin et al., 2021; Kanehisa et al., 2021). However, the proteins encoded by these genes shared very low identity (22.07–28.77%), thus, suggesting potentially different functions. D-Ser dehydratase transforms D-Ser into pyruvate, and ammonia D-Cys desulfhydrase transforms D-Cys into hydrogen sulfide, ammonia, and pyruvate. The abundant genes involved in D-AA catabolism may be attributed to the broad utilization of D-AAs in strain LMO_D1. Although the genome annotation revealed a small number of candidates for D-AA utilization in strain LMO_D1, it remains unclear as to which gene(s) are essential.
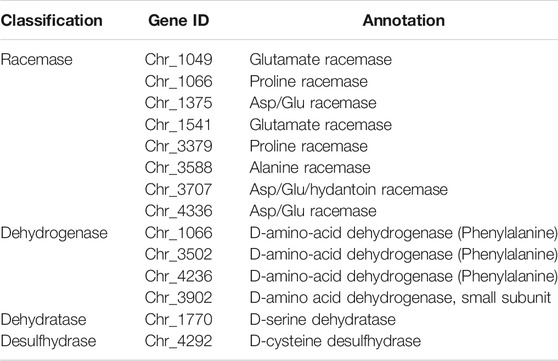
TABLE 1. Annotated genes involved in D-AAs metabolism.
Functional gene screening for D-amino acid catabolism via fosmid library
To further identify key enzymes functioning in D-AA metabolism, a genomic DNA fosmid library of strain LMO_D1 in the heterologous host E. coli EPI300™–T1R was screened based on functions, where target clones grew on a mixture of 19 D-AAs as the sole carbon source with NH4Cl as the nitrogen source. The host cells were confirmed to be unable to subsist on D-AAs. The titer of the constructed library was estimated to be 50,000 colony-forming units (cfu) per milliliter. The number of clones required to ensure a 99% probability of a given DNA sequence for strain LMO_D1 (genome = 5.4 Mb) contained within a fosmid library composed of 30-kb inserts was calculated as N = ln (1–0.99)/ln [1—(30 × 103 bases/5.4 × 106 bases)] = 827 clones. A total of 3,168 clones (containing ∼95.0 Mb sequence) from the library were inoculated on the D-AA screening plate, and 13 clones grew on the plate after 72-h incubation (Supplementary Figure S1).
The sequencing results revealed that there is a 9,203-bp sequence (the location in the genome is 2,544,984–2,554,187) containing seven coding sequences (Supplementary Figure S2) that are mutual for the 13 positive clones. Among them, five coding sequences were annotated with a definite function, and only one of them (Chr_2344) was annotated as a D-AA dehydrogenase (DAADH _2344) that may be related to D-AA utilization. The fosmid DNA of all 13 positive clones covered an overall 64.4-kb sequence (the location in the genome is 2,515,951–2,579,915) containing the coding sequences from Chr_2310 to Chr_2371. With the exception of the encoding gene for DAADH_2344 (Chr_2344), none of the other genes were annotated as related genes for D-AA metabolism. Specifically, the clones containing DAADH_2344 were able to grow on the D-AAs plate; other genes involved in D-AA catabolism were not included in the insert DNA of the positive clones. Our findings revealed a key role for DAADH _2344 in D-AA utilization in strain LMO_D1.
DAADH_2344 exhibited low identity (<28.34%) with all reviewed DAADHs according to the Swiss-Prot database (UniProt, 2021) (Supplementary Figure S3). Among the DAADHs whose functions have been identified, DAADH_2344 possessed the highest identity (26.00%) with DAADH from Pseudomonas aeruginosa PAO1 (NCBI accession NP_253,991.1). These low identities compared with the known DAADH indicate that the function of DAADH_2344 should be further confirmed.
Purification and identification of key enzyme D-amino acid dehydrogenase_2344 for D-amino acid metabolism
To further identify the function of DAADH_2344, we expressed its encoding gene (Chr_2344) in recombinant E. coli. Although DAADH has been reported as a membrane-bond protein in previous studies (Olsiewski et al., 1980; Tanigawa et al., 2010), we observed that DAADH_2344 was primarily distributed in the soluble fraction, thus, indicating that DAADH_2344 is a novel type DAADH compared with the previously reported enzyme (Supplementary Figure S4). The apparent molecular mass of the purified enzyme possessing two copies of the His6-tag was approximately 47 kDa according to SDS-PAGE, and this was consistent with the calculated value of 47.27 kDa for DAADH_2344 (Supplementary Figure S4).
The purified DAADH_2344 exhibited the specific activity of this enzyme in the presence of cofactor FAD and possessed a broad substrate scope according to the results of substrate specificity measurements using 19 D-AAs. In total, DAADH_2344 exhibited enzymatic activity against 16 D-AAs. By measuring the reduction of DCIP, the highest specific activity was observed for D-Met and D-Phe with specific activities of 2.20 and 2.18 μmol min−1 mg−1, respectively, and this was followed by D-His, D-Val, and D-Leu with ∼50% specific activity compared with that against D-Met. D-Trp, D-Thr, and D-Ile with ∼30% specific activity compared with that against D-Met. Relatively low activities were observed for D-Ala, D-Asn, D-Ser, D-Arg, D-Gln, D-Asp, and D-Lys (<15% compared with D-Met). In contrast, no activity was observed for D-Pro, D-Glu, and D-Cys (Table 2). Additionally, DAADH_2344 exhibited strict stereospecificity, and none of the L-AAs could be used as a substrate.
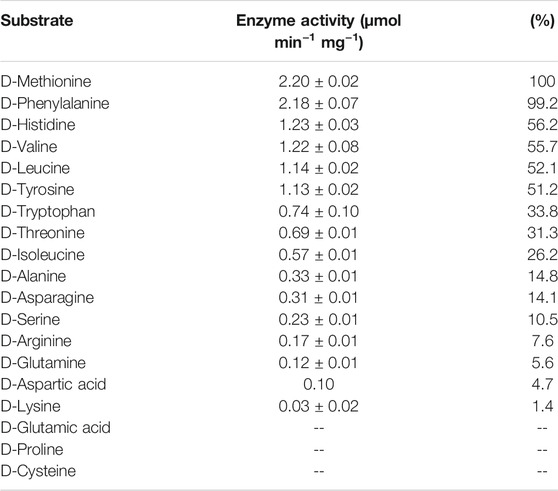
TABLE 2. Substrate specificity of DAADH_2344.
Interestingly, despite the ability of the enzyme DAADH_2344 to function well against D-Met, D-Trp, D-Thr, and D-His in vitro, the strain LMO_D1 can only utilize D-Met, D-Trp, D-Thr, and D-His physiologically (the same as their L-type) as a nitrogen source and not as a carbon source. The failure to utilize these amino acids was assumed to be restricted by the catabolism of corresponding α-keto acids (4-methylthio-2-oxobutanoic acid, indolepyruvate, 2-oxobutanoate, urocanate), and the pathways for these compounds to enter the TCA cycle were incomplete in the genome of strain LMO_D1. Thus, neither the D nor the L-type of these amino acids was bioavailable as a carbon source. In contrast, strain LMO_D1 could utilize D-Pro, D-Glu, and D-Cys as carbon or nitrogen sources for which DAADH exhibited no activity. Hence, the catabolism of D-Pro, D-Glu, and D-Cys may be conducted by other D-AA catabolic enzymes, such as racemase, desulfhydrase, or other DAADHs encoded by the LMO_D1 strain (Table 1). The other 11 D-AAs (D-Ala, D-Asp, D-Ser, D-Leu, D-Val, D-Tyr, D-Gln, D-Asn, D-Arg, D-Phe, and D-Ile) that can support the growth of strain LMO_D1 could be deaminated by DAADH_2344. Based on the genomic evidence, the deaminization product of 3 D-AAs [pyruvate (D-Ala and D-Ser) and oxaloacetate (D-Asp)] could enter the TCA cycle directly. Among the α-keto acids of other D-AAs, phenylpyruvate (D-Phe), 4-methyl-2-oxopentanoate (D-Leu), and (S)-3-Methyl-2-oxopentanoic acid (D-Ile) could be transformed into acetyl-CoA, 4-hydroxyphenylpyruvate (D-Tyr) could be transformed to fumarate, and 3-methyl-2-oxobutanoic acid (D-Val) could be transformed to succinate. The 5-guanidino-2-oxopentanoate (D-Arg) could be transformed to L-Arg via L-arginine:pyruvate aminotransferase, and this could later be catabolized to succinate. The 2-oxoglutarate (D-Gln) and 2-oxosuccinamate could be transformed to 2-oxoglutarate and oxaloacetate, respectively, via the function of omega-amidase.
D-amino acid dehydrogenase distribution in Mariana trench sediment samples
DAADH_2344 was observed to strongly facilitate D-AA catabolism in strain LMO_D1 and enabled E. coli to grow with D-AAs as a carbon source. In regard to the essential function, the abundance of DAADH can represent the abundance of the microorganisms possessing a relatively strong adaptation to the toxic D-AAs environment. To explore the potential D-AA utilization activity in the hadal trench, we further investigated the abundance of DAADHs in sediment samples collected at a water depth of 5,481–10,893 m and those were distributed in different layers of the push cores (Figure 3) based on the metagenomic data of the sediment samples in the Mariana trench (unpublished data from our lab and public database). To compare the relative abundance of DAADHs in the different samples, the read counts for DAADHs (KO number K00285) were normalized to transcripts per million (TPM) counts. The relative abundance of homologous sequences of DAADH_2344 (identity >30%, e-value <10−5) was also investigated. Overall, DAADHs were widespread within the sediment samples in the Mariana trench, and their abundance ranged from 27.60 to 206.20 TPM. Similarly, homologs (identity >30%) of DAADH_2344 were also common in the majority of the investigated samples, with the exception of one sample (Mariana_7939) that exhibited an abundance ranging from 0 to 40.47 TPM. An increase in the abundance of DAADH in conjunction with sediment depth was evident, particularly in samples D3 and D93 (Figure 3A). A similar trend was observed for the distribution of proteins that are closely related to DAADH_2344. However, there was no clear tendency for sediment samples at deeper water depths to harbor higher abundances of DAADHs. (Figure 3B).
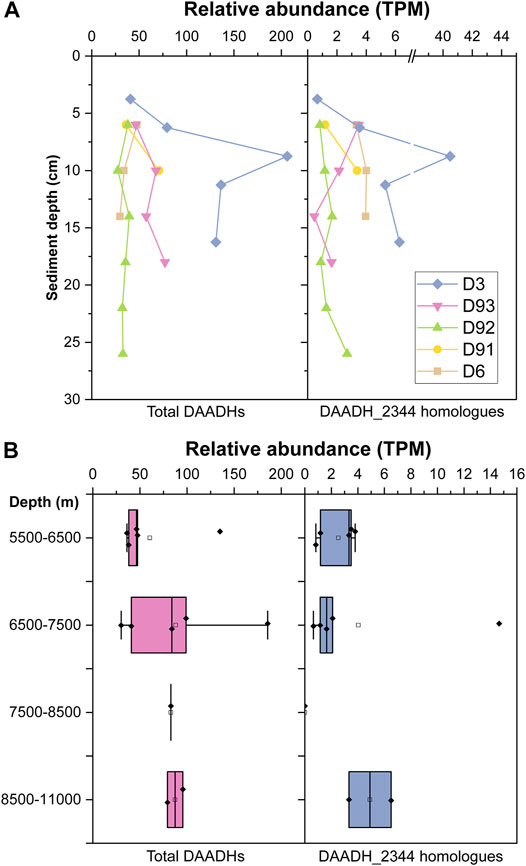
FIGURE 3. Relative abundance [transcripts per million (TPM)] of total D-amino acid dehydrogenase (DAADHs) and DAADH_2344 homologs (identity >30%) in Mariana trench sediment samples from different sediment layers (A) and water depth (B). Boxplots represent the statistical analysis among metagenome data of sediment samples from a certain range of water layers. Solid squares represent TPM value of an individual sample; Hollow squares represent the means; bars inside the boxes represent the medians; the hinges out of the boxes represent the lower and upper quartiles; the whiskers extend to the most extreme values (no more than 1.5 times the interquartile range from the box).
Discussion
In this study, we isolated Halomonas sp. LMO_D1 that possesses a broad range of D-AA utilization capability from the sample collected from subduction zones in the Mariana trench. In previous studies, the catabolism of D-AAs was primarily examined in soils, surface marine zones, or sediments within a 1,500-m water depth (Kubota et al., 2016). Our study extends the current knowledge base to the deep trench, where both racemization and abiotic synthesis of amino acids are expected to occur in this rarely explored environment. The occurrence of D-AA catabolism is prompted by the adaptation of organisms to toxic D-AA environment. D-AA utilizers are typically discovered in environments where a high abundance of D-AAs is detected (Kawasaki and Benner, 2006; Vranova et al., 2011). Therefore, the capability of D-AA catabolism by Halomonas sp. LMO_D1 indicated a potentially high abundance of D-AAs in the hadal trench. Additionally, the hadal trench is an oligotrophic environment in which the catabolism of D-AAs is a strategy for better use of limited nutrients.
The isolate in this study, Halomonas sp. LMO_D1 is the reported microorganism that can utilize the broadest range of D-AAs. When comparing the bioavailability of D-AAs and L-AAs, the usable D-AAs for strain LMO_D1 are less abundant than are L-AAs as carbon sources, and this indicates that the carbon skeleton degradation of D-AAs shared a similar pathway to that of their L-enantiomer. However, when D-AAs are utilized as a nitrogen source, one more D-AA (D-Cys) can support cell growth, while their L-enantiomers cannot. In the genome of strain LMO_D1, the existence of D-Cys desulfhydrase that converts D-Cys into hydrogen sulfide, ammonia, and pyruvate can explain the utilization of D-Cys. However, it is difficult to determine why L-Cys cannot be utilized in the context of a general Cys desulfhydrase. This is likely correlated with the transcription level of genes or the tolerance upper limit of the substrates. A similar phenomenon in which D-AAs supported higher biomass yield than L-enantiomers has also been reported previously in experiments examining Raoultella ornithinolytica A25; however, the mechanism remains unclear (Naganuma et al., 2018). Further investigation may be required to fill the knowledge gap in regard to the metabolism of amino acid enantiomers.
In the genome of strain LMO_D1, the enzymes involved in D-AA catabolism covered all three types of enzymes, including racemases, substrate-specific oxidases, and broad-spectrum oxidase/dehydrogenases. Amino acid racemases participate in both the synthetic and catabolic processes of D-AAs (Zhang et al., 2021). Biosynthesis racemases are universal in bacteria that contain Ala, Glu, Asp, Ser, Lys, Pro, and broad-spectrum racemase (Hernandez and Cava, 2016), while catabolic racemases are only reported to function on Ala and are inducible by L-AAs (Walsh, 1989). For substrate-specific oxidase in the LMO_D1 genome, D-Ser dehydratase and D-Cys desulfhydrase only act on D-Ser and D-Cys, respectively. Therefore, the broad D-AA catabolism in LMO_D1 relies on the broad-spectrum oxidase/dehydrogenase DAADH. We confirmed the essential function of DAADH_2344 according to the ability of E. coli clones containing DAADH_2344 to grow on the D-AAs plate. The enhanced ability to subsist on D-AAs supported by DAADH_2344 and not by the other enzymes can be explained by the broad substrate specificity of the enzyme and a possible D-AA-induced transcriptional mechanism. The relative activity of DAADH_2344 against the primary components of peptidoglycans (D-Ala, D-Glu, D-Ser, and D-Asp) was much lower (<15%) than that of the optimal substrate D-Met. A similar low activity against D-Glu and D-Asp was also observed in DAADH_PA (highest identity to DAADH_2344 among all identified DAADHs) from Pseudomonas aeruginosa PAO1 (He et al., 2011). However, DAADH_PA exhibited higher relative activity against D-Ala and D-Ser. These results revealed that with the exception of the D-AAs from peptidoglycans, DAADH_2344 exhibits a greater preference for D-AAs that are less common in peptidoglycans and instead are likely generated through abiotic processes or other biotic processes such as secretions by microorganisms for physiological regulation (Lam et al., 2009).
Based on the various production mechanisms of D-AAs within the environment, we believe that D-AAs exert an impact on the hadal trench ecosystem. However, we only obtained Halomonas in the enrichment and isolation experiments. The substrate concentration supplied in this study (100 mg/L of each D-AA) was much higher than the concentration of free D-AAs in the environment (Lomstein et al., 2006), and this may have resulted in a selective growth advantage for the dominant group and the inhibition of the growth of microbes possessing lower tolerances to D-AAs. As two catabolic strategies for coping with D-AAs, racemases are universal in bacteria and represent a common adaptation to the toxic D-AA environment. Moreover, oxidase/dehydrogenases allow microorganisms to possess an enhanced ability to utilize D-AAs. We attempted to fully understand these microorganisms based on metagenomic data. DAADHs are widespread in all samples examined in this study, and this indicates that D-AA utilizers are common in the environment. D-AA-utilizing species are likely far more diverse than the limited species that we isolated in our current study. In addition to the limitations of our isolation methods, some amino acid auxotrophs may be able to utilize D-AAs as a carbon/nitrogen source but not as their sole source. Additionally, we can search for more D-AA utilizers based on metagenome data. The discovery of an increasing abundance of DAADH in conjunction with sediment depth is consistent with the increasing proportion of D-AAs with sediment depth that was reported in a previous study (Pedersen et al., 2001), likely due to the observation that D-AAs were retained for a longer period compared with L-AAs and also likely due to the racemization process. As the generation of D-AAs is mediated by both biotic and abiotic processes, more sample sites are required to clarify the connection between the abundance of DAADH and the water depth of samples and the contribution of hydrothermal activities in regard to D-AA input in the subduction zone.
In this study, the cultivation experiments under HHP demonstrated that the strain was able to perform D-AA utilization in the original environment. The enrichment experiment was performed at 70 MPa, and the increased biomass suggested that D-AA utilization can occur at least at a water depth of 7,000 m. Under these conditions, the growth of most mesophilic microorganisms, such as E. coli, from the surface is completely inhibited (Bartlett, 2002). The strain LMO_D1 continued to grow at such a high hydrostatic pressure, and this indicated that it performed as a piezotolerant bacterium that should not originate from surface water. We observed that HHP exerted different degrees of impact on the metabolism of amino acid enantiomers. Under HHP, the metabolism of L-AAs was impaired more severely than that of D-AAs. A likely reason for this is that DAADH mediated the catabolic reaction using quinone rather than oxygen as an electron acceptor, thus, avoiding the production of H2O2 that would diminish cellular oxidative stress and benefit growth under HHP (Xiao et al., 2021). Whether the phenomenon of HHP in reducing the difference between L-AAs and D-AAs in regard to supporting growth is common or exclusive in strain LMO_D1 required further investigation in other deep-sea inhabitants. The study of how pizeotolerant or piezophilic life copes with toxic D-AAs here on Earth will inspire speculation regarding how extraterrestrial life may be sustained in the icy moons of Jupiter or Saturn and the subsurface environment in Mars, where HHP would also exert a large impact on the potential life. The expanded knowledge regarding the different physiological characteristics required in response to L-AAs and D-AAs will aid in refining life detection experiments (Navarro-Gonzalez et al., 2003; Zhang et al., 2021).
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA744311 and https://www.ncbi.nlm.nih.gov/, SRX5177670 and https://www.ncbi.nlm.nih.gov/, SRR15098654 and https://www.ncbi.nlm.nih.gov/, SRR15098655.
Author Contributions
XW, XX, and WZ designed this study. XW and YY performed the experiments. XW and YL contributed to data analysis. XW and WZ wrote the article. XX revised the article.
Funding
This study was financially supported by the following funding: the Natural Science Foundation of China (grant numbers 41921006, 41530967, and 41776173). the National Key R and D project of China (grant number 2018YFC0309800).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are grateful to all the team members on the R/V Tansuo01 for their invaluable efforts in the sampling cruise.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fspas.2021.741053/full#supplementary-material
References
Bada, J. L., and Miller, S. L. (1987). Racemization and the Origin of Optically Active Organic Compounds in Living Organisms. Biosystems 20 (1), 21–26. doi:10.1016/0303-2647(87)90016-5
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a New Genome Assembly Algorithm and its Applications to Single-Cell Sequencing. J. Comput. Biol. 19 (5), 455–477. doi:10.1089/cmb.2012.0021
Bartlett, D. H. (2002). Pressure Effects on In Vivo Microbial Processes. Biochim. Biophys. Acta 1595 (1), 367–381. doi:10.1016/S0167-4838(01)00357-0
Besemer, J., Lomsadze, A., and Borodovsky, M. (2001). GeneMarkS: a Self-Training Method for Prediction of Gene Starts in Microbial Genomes. Implications for Finding Sequence Motifs in Regulatory Regions. Nucleic Acids Res. 29 (12), 2607–2618. doi:10.1093/nar/29.12.2607
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., Al-Ghalith, G. A., et al. (2019). Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 37 (8), 852–857. doi:10.1038/s41587-019-0209-9
Buchfink, B., Xie, C., and Huson, D. H. (2015). Fast and Sensitive Protein Alignment Using DIAMOND. Nat. Methods 12 (1), 59–60. doi:10.1038/nmeth.3176
Burton, A., and Berger, E. (2018). Insights into Abiotically-Generated Amino Acid Enantiomeric Excesses Found in Meteorites. Life 8 (2), 14. doi:10.3390/life8020014
Caparrós, M., Pisabarro, A. G., and de Pedro, M. A. (1992). Effect of D-Amino Acids on Structure and Synthesis of Peptidoglycan in Escherichia coli. J. Bacteriol. 174 (17), 5549–5559. doi:10.1128/jb.174.17.5549-5559.1992
Chin, C.-S., Peluso, P., Sedlazeck, F. J., Nattestad, M., Concepcion, G. T., Clum, A., et al. (2016). Phased Diploid Genome Assembly with Single-Molecule Real-Time Sequencing. Nat. Methods 13 (12), 1050–1054. doi:10.1038/nmeth.4035
Corti Monzón, G., Nisenbaum, M., Herrera Seitz, M. K., and Murialdo, S. E. (2018). New Findings on Aromatic Compounds' Degradation and Their Metabolic Pathways, the Biosurfactant Production and Motility of the Halophilic Bacterium Halomonas Sp. KHS3. Curr. Microbiol. 75 (8), 1108–1118. doi:10.1007/s00284-018-1497-x
Delcher, A. L., Kasif, S., Fleischmann, R. D., Peterson, J., White, O., and Salzberg, S. L. (1999). Alignment of Whole Genomes. Nucleic Acids Res. 27 (11), 2369–2376. doi:10.1093/nar/27.11.2369
Fu, Y., Wang, R., Zhang, Z., and Jiao, N. (2016). Complete Genome Sequence of the D -Amino Acid Catabolism Bacterium Phaeobacter Sp. Strain JL2886, Isolated from Deep Seawater of the South China Sea. Genome Announc 4 (5). doi:10.1128/genomeA.00913-16
Galperin, M. Y., Wolf, Y. I., Makarova, K. S., Vera Alvarez, R., Landsman, D., and Koonin, E. V. (2021). COG Database Update: Focus on Microbial Diversity, Model Organisms, and Widespread Pathogens. Nucleic Acids Res. 49 (D1), D274–D281. doi:10.1093/nar/gkaa1018
Glavin, D. P., Burton, A. S., Elsila, J. E., Aponte, J. C., and Dworkin, J. P. (2020). The Search for Chiral Asymmetry as a Potential Biosignature in Our Solar System. Chem. Rev. 120 (11), 4660–4689. doi:10.1021/acs.chemrev.9b00474
He, W., Li, C., and Lu, C.-D. (2011). Regulation and Characterization of the dadRAX Locus for D -Amino Acid Catabolism in Pseudomonas aeruginosa PAO1. J. Bacteriol. 193 (9), 2107–2115. doi:10.1128/JB.00036-11
Hein, J. E., and Blackmond, D. G. (2012). On the Origin of Single Chirality of Amino Acids and Sugars in Biogenesis. Acc. Chem. Res. 45 (12), 2045–2054. doi:10.1021/ar200316n
Hernández, S. B., and Cava, F. (2016). Environmental Roles of Microbial Amino Acid Racemases. Environ. Microbiol. 18 (6), 1673–1685. doi:10.1111/1462-2920.13072
Jamieson, A. J. (2011). “Ecology of Deep Oceans: Hadal Trenches,” in eLS. (Chichester: John Wiley and Sons, Ltd). doi:10.1002/9780470015902.a0023606
Kaiser, K., and Benner, R. (2008). Major Bacterial Contribution to the Ocean Reservoir of Detrital Organic Carbon and Nitrogen. Limnol. Oceanogr. 53 (1), 99–112. doi:10.4319/lo.2008.53.1.0099
Kanehisa, M., Furumichi, M., Sato, Y., Ishiguro-Watanabe, M., and Tanabe, M. (2021). KEGG: Integrating Viruses and Cellular Organisms. Nucleic Acids Res. 49 (D1), D545–D551. doi:10.1093/nar/gkaa970
Kawakami, R., Ohmori, T., Sakuraba, H., and Ohshima, T. (2015). Identification of a Novel Amino Acid Racemase from a Hyperthermophilic Archaeon Pyrococcus Horikoshii OT-3 Induced by D-Amino Acids. Amino Acids 47 (8), 1579–1587. doi:10.1007/s00726-015-2001-6
Kawasaki, N., and Benner, R. (2006). Bacterial Release of Dissolved Organic Matter during Cell Growth and Decline: Molecular Origin and Composition. Limnol. Oceanogr. 51 (5), 2170–2180. doi:10.4319/lo.2006.51.5.2170
Kebukawa, Y., Chan, Q. H. S., Tachibana, S., Kobayashi, K., and Zolensky, M. E. (2017). One-pot Synthesis of Amino Acid Precursors with Insoluble Organic Matter in Planetesimals with Aqueous Activity. Sci. Adv. 3 (3), e1602093. doi:10.1126/sciadv.1602093
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M. (2017). Canu: Scalable and Accurate Long-Read Assembly via Adaptivek-Mer Weighting and Repeat Separation. Genome Res. 27 (5), 722–736. doi:10.1101/gr.215087.116
Kubota, T., Kobayashi, T., Nunoura, T., Maruyama, F., and Deguchi, S. (2016). Enantioselective Utilization of D-Amino Acids by Deep-Sea Microorganisms. Front. Microbiol. 7, 511. doi:10.3389/fmicb.2016.00511
Lam, H., Oh, D.-C., Cava, F., Takacs, C. N., Clardy, J., de Pedro, M. A., et al. (2009). D-amino Acids Govern Stationary Phase Cell wall Remodeling in Bacteria. Science 325 (5947), 1552–1555. doi:10.1126/science.1178123
Lomstein, B. A., Jørgensen, B. B., Schubert, C. J., and Niggemann, J. (2006). Amino Acid Biogeo- and Stereochemistry in Coastal Chilean Sediments. Geochimica Et Cosmochimica Acta 70 (12), 2970–2989. doi:10.1016/j.gca.2006.03.015
Luo, M., Gieskes, J., Chen, L., Shi, X., and Chen, D. (2017). Provenances, Distribution, and Accumulation of Organic Matter in the Southern Mariana Trench Rim and Slope: Implication for Carbon Cycle and Burial in Hadal Trenches. Mar. Geology. 386, 98–106. doi:10.1016/j.margeo.2017.02.012
Luo, R., Liu, B., Xie, Y., Li, Z., Huang, W., Yuan, J., et al. (2012). SOAPdenovo2: an Empirically Improved Memory-Efficient Short-Read De Novo Assembler. GigaSci 1 (1), 18. doi:10.1186/2047-217X-1-18
Marshall, V. P., and Sokatch, J. R. (1968). Oxidation of D -Amino Acids by a Particulate Enzyme from Pseudomonas aeruginosa. J. Bacteriol. 95 (4), 1419–1424. doi:10.1128/jb.95.4.1419-1424.1968
McCarthy, M. D., Hedges, J. I., and Benner, R. (1998). Major Bacterial Contribution to marine Dissolved Organic Nitrogen. Science 281 (5374), 231–234. doi:10.1126/science.281.5374.231
Ménez, B., Pisapia, C., Andreani, M., Jamme, F., Vanbellingen, Q. P., Brunelle, A., et al. (2018). Abiotic Synthesis of Amino Acids in the Recesses of the Oceanic Lithosphere. Nature 564 (7734), 59–63. doi:10.1038/s41586-018-0684-z
Miller, S. L. (1953). A Production of Amino Acids under Possible Primitive Earth Conditions. Science 117 (3046), 528–529. doi:10.1126/science.117.3046.528
Naganuma, T., Iinuma, Y., Nishiwaki, H., Murase, R., Masaki, K., and Nakai, R. (2018). Enhanced Bacterial Growth and Gene Expression of D-Amino Acid Dehydrogenase with D-Glutamate as the Sole Carbon Source. Front. Microbiol. 9, 2097. doi:10.3389/fmicb.2018.02097
Navarro-González, R., Rainey, F. A., Molina, P., Bagaley, D. R., Hollen, B. J., de la Rosa, J., et al. (2003). Mars-like Soils in the Atacama Desert, Chile, and the Dry Limit of Microbial Life. Science 302 (5647), 1018–1021. doi:10.1126/science.1089143
Ohara, Y., Reagan, M. K., Fujikura, K., Watanabe, H., Michibayashi, K., Ishii, T., et al. (2012). A Serpentinite-Hosted Ecosystem in the Southern Mariana Forearc. Proc. Natl. Acad. Sci. 109 (8), 2831–2835. doi:10.1073/pnas.1112005109
Olsiewski, P. J., Kaczorowski, G. J., and Walsh, C. (1980). Purification and Properties of D-Amino Acid Dehydrogenase, an Inducible Membrane-Bound Iron-Sulfur Flavoenzyme from Escherichia coli B. J. Biol. Chem. 255 (10), 4487–4494. doi:10.1016/S0021-9258(19)85517-5
Pedersen, A.-G. U., Thomsen, T. R., Lomstein, B. A., and Jørgensen, N. O. G. (2001). Bacterial Influence on Amino Acid Enantiomerization in a Coastal marine Sediment. Limnol. Oceanogr. 46 (6), 1358–1369. doi:10.4319/lo.2001.46.6.1358
Peltzer, E. T., Bada, J. L., Schlesinger, G., and Miller, S. L. (1984). The Chemical Conditions on the Parent Body of the Murchison Meteorite: Some Conclusions Based on Amino, Hydroxy and Dicarboxylic Acids. Adv. Space Res. 4 (12), 69–74. doi:10.1016/0273-1177(84)90546-5
Pizzarello, S., Schrader, D. L., Monroe, A. A., and Lauretta, D. S. (2012). Large Enantiomeric Excesses in Primitive Meteorites and the Diverse Effects of Water in Cosmochemical Evolution. Proc. Natl. Acad. Sci. USA 109 (30), 11949–11954. doi:10.1073/pnas.1204865109
Pizzarello, S. (2006). The Chemistry of Life's Origin: a Carbonaceous Meteorite Perspective. Acc. Chem. Res. 39 (4), 231–237. doi:10.1021/ar050049f
Plümper, O., King, H. E., Geisler, T., Liu, Y., Pabst, S., Savov, I. P., et al. (2017). Subduction Zone Forearc Serpentinites as Incubators for Deep Microbial Life. Proc. Natl. Acad. Sci. USA 114 (17), 4324–4329. doi:10.1073/pnas.1612147114
Pollegioni, L., Molla, G., Sacchi, S., Rosini, E., Verga, R., and Pilone, M. S. (2008). Properties and Applications of Microbial D-Amino Acid Oxidases: Current State and Perspectives. Appl. Microbiol. Biotechnol. 78 (1), 1–16. doi:10.1007/s00253-007-1282-4
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 41 (Database issue), D590–D596. doi:10.1093/nar/gks1219
Radkov, A. D., McNeill, K., Uda, K., and Moe, L. A. (2016). D-amino Acid Catabolism Is Common Among Soil-Dwelling Bacteria. Microb. Environ. 31 (2), 165–168. doi:10.1264/jsme2.ME15126
Snyder, S. H., and Kim, P. M. (2000). D-amino Acids as Putative Neurotransmitters: Focus on D-Serine. Neurochem. Res. 25 (5), 553–560. doi:10.1023/A:1007586314648
Soutourina, O., Soutourina, J., Blanquet, S., and Plateau, P. (2004). Formation of D-Tyrosyl-tRNATyr Accounts for the Toxicity of D-Tyrosine toward Escherichia coli. J. Biol. Chem. 279 (41), 42560–42565. doi:10.1074/jbc.M402931200
Tanigawa, M., Shinohara, T., Saito, M., Nishimura, K., Hasegawa, Y., Wakabayashi, S., et al. (2010). D-amino Acid Dehydrogenase from Helicobacter pylori NCTC 11637. Amino Acids 38 (1), 247–255. doi:10.1007/s00726-009-0240-0
Tritt, A., Eisen, J. A., Facciotti, M. T., and Darling, A. E. (2012). An Integrated Pipeline for De Novo Assembly of Microbial Genomes. PLoS One 7 (9), e42304. doi:10.1371/journal.pone.0042304
UniProt, C. (2021). UniProt: the Universal Protein Knowledgebase in 2021. Nucleic Acids Res. 49 (D1), D480–D489. doi:10.1093/nar/gkaa1100
Vollmer, W., Blanot, D., and de Pedro, M. A. (2008). Peptidoglycan Structure and Architecture. FEMS Microbiol. Rev. 32 (2), 149–167. doi:10.1111/j.1574-6976.2007.00094.x
Vranova, V., Zahradnickova, H., Janous, D., Skene, K. R., Matharu, A. S., Rejsek, K., et al. (2011). The Significance of D-Amino Acids in Soil, Fate and Utilization by Microbes and Plants: Review and Identification of Knowledge Gaps. Plant Soil 354 (1-2), 21–39. doi:10.1007/s11104-011-1059-5
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS One 9 (11), e112963. doi:10.1371/journal.pone.0112963
Walsh, C. T. (1989). Enzymes in the D-Alanine branch of Bacterial Cell wall Peptidoglycan Assembly. J. Biol. Chem. 264 (5), 2393–2396. doi:10.1016/s0021-9258(19)81624-1
Wang, H., Zhang, Y., Bartlett, D. H., and Xiao, X. (2021). Transcriptomic Analysis Reveals Common Adaptation Mechanisms under Different Stresses for Moderately Piezophilic Bacteria. Microb. Ecol. 81 (3), 617–629. doi:10.1007/s00248-020-01609-3
Wang, R., Zhang, Z., Sun, J., and Jiao, N. (2020). Differences in Bioavailability of Canonical and Non-canonical D-Amino Acids for marine Microbes. Sci. Total Environ. 733, 139216. doi:10.1016/j.scitotenv.2020.139216
Widdel, F., Boetius, A., and Rabus, R. (2006). Anaerobic Biodegradation of Hydrocarbons Including Methane. The prokaryotes 2, 1028–1049. doi:10.1007/0-387-30742-7_33
Xiao, X., Zhang, Y., and Wang, F. (2021). Hydrostatic Pressure Is the Universal Key Driver of Microbial Evolution in the Deep Ocean and beyond. Environ. Microbiol. Rep. 13 (1), 68–72. doi:10.1111/1758-2229.12915
Yu, Y., Yang, J., Zheng, L.-Y., Sheng, Q., Li, C.-Y., Wang, M., et al. (2019). Diversity of D-Amino Acid Utilizing Bacteria from Kongsfjorden, Arctic and the Metabolic Pathways for Seven D-Amino Acids. Front. Microbiol. 10, 2983. doi:10.3389/fmicb.2019.02983
Zhang, G., and Sun, H. J. (2014). Racemization in Reverse: Evidence that D-Amino Acid Toxicity on Earth Is Controlled by Bacteria with Racemases. PLoS One 9 (3), e92101. doi:10.1371/journal.pone.0092101
Zhang, L., Zeng, F., McKay, C. P., Navarro-González, R., and Sun, H. J. (2021). Optimizing Chiral Selectivity in Practical Life-Detection Instruments. Astrobiology 21 (5), 505–510. doi:10.1089/ast.2020.2381
Zhang, Z., Zheng, Q., and Jiao, N. (2015). Microbial D-Amino Acids and marine Carbon Storage. Sci. China Earth Sci. 59 (1), 17–24. doi:10.1007/s11430-015-5155-x
Keywords: D-amino acids, Mariana trench, high hydrostatic pressure, D-amino acids dehydrogenase, abiotic enantiomers
Citation: Wang X, Yang Y, Lv Y, Xiao X and Zhao W (2021) The Capability of Utilizing Abiotic Enantiomers of Amino Acids by Halomonas sp. LMO_D1 Derived From the Mariana Trench. Front. Astron. Space Sci. 8:741053. doi: 10.3389/fspas.2021.741053
Received: 14 July 2021; Accepted: 29 October 2021;
Published: 14 December 2021.
Edited by:
Yiliang Li, The University of Hong Kong, Hong Kong SAR, ChinaReviewed by:
Henry Sun, Desert Research Institute (DRI), United StatesZhiyong Huang, Tianjin Institute of Industrial Biotechnology (CAS), China
Copyright © 2021 Wang, Yang, Lv, Xiao and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weishu Zhao, endzaDg4QHNqdHUuZWR1LmNu