 Denis Duflot
Denis Duflot Céline Toubin
Céline Toubin Maurice Monnerville
Maurice Monnerville
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Astron. Space Sci. , 26 March 2021
Sec. Astrochemistry
Volume 8 - 2021 | https://doi.org/10.3389/fspas.2021.645243
This article is part of the Research Topic Exploring the Chemical Universe View all 10 articles
The adsorption of a series of atoms and small molecules and radicals (H, C, N, O, NH, OH, H2O, CH3, and NH3) on hexagonal crystalline and amorphous ice clusters were obtained via classical molecular dynamics and electronic structure methods. The geometry and binding energies were calculated using a QMHigh:QMLow hybrid method on model clusters. Several combination of basis sets, density functionals and semi-empirical methods were compared and tested against previous works. More accurate binding energies were also refined via single point Coupled Cluster calculations. Most species, except carbon atom, physisorb on the surface, leading to rather small binding energies. The carbon atom forms a COH2 molecule and in some cases leads to the formation of a COH-H3O+ complex. Amorphous ices are characterized by slightly stronger binding energies than the crystalline phase. A major result of this work is to also access the dispersion of the binding energies since a variety of adsorption sites is explored. The interaction energies thus obtained may serve to feed or refine astrochemical models. The present methodology could be easily extended to other types of surfaces and larger adsorbates.
Astrochemistry has been an area of intense research in particular with the aim to elucidate the origin of the chemical diversity observed in space. Depending on the evolution stage of the interstellar dust and the region, energetic processes may dominate the chemistry. However, in low UV-photon field regions, as in dense molecular clouds, energetic processes are suppressed and surface reactions of cold molecules become important. It has been demonstrated both experimentally and theoretically that dust grains catalyze the formation of some important molecules, such as H2, H2O, and CO2, organic molecules (H2CO, CH3OH … ) (Ioppolo et al., 2011; Pirim and Krim, 2011; Peters et al., 2013; Rimola et al., 2014; Minissale et al., 2015), or complex/prebiotic molecules (Koch et al., 2008; Rimola and Ugliengo, 2009). Thermally activated surface reactions rarely occur over the activation energy barriers on ice mantles because of their low temperatures (∼ 10 K). In contrast, quantum-tunneling reactions are facilitated in this temperature range. The importance of tunneling effects has been discussed in various studies (Goumans and Kästner, 2010; Hama and Watanabe, 2013; Hickson et al., 2016). Whatever the conditions are, for a reaction to occur on the grain, the molecular species needs to reside or diffuse at the grain surface. Most neutral species can physisorb onto grains through van der Waals interactions at the low temperature conditions of dense molecular clouds. Chemisorption may also occur for radicals or reactive molecules on specific or defective sites. In contrast to chemisorption, physisorption is reversible, the desorption rate being derived from the binding energy and the frequency characterizing the motion of the adsorbed species in the potential energy well. Diffusion is also to be considered when reactions or recombinations occur through Langmuir-Hinshelwood mechanisms. Most astrochemical models (Garrod et al., 2006; Taquet et al., 2013; Ruaud et al., 2016; Cuppen et al., 2017; Wakelam et al., 2017) mimicking the chemical evolution in star forming region have now integrated these gas-grain processes. Among the molecular data used as inputs to these models, the adsorption energy is a key parameter, the diffusion energy being estimated as a fraction (between 0.3 and 1.0) of the binding energy (Ruaud et al., 2016). There is thus a crucial need to improve the binding energies (BE’s) values in the models to better reproduce the astrophysical abundances.
Binding energies depend indeed on the nature of the volatile species, the surface and its coverage. BE’s can be determined experimentally for stable molecules using programmed desorption (TPD) methods (Burke and Brown, 2010; Hama and Watanabe, 2013). Several studies have reviewed known binding energies on various surfaces and more especially water ices (Fraser et al., 2001; Collings et al., 2004; Noble et al., 2012; Hama and Watanabe, 2013; Penteado et al., 2017; Wakelam et al., 2017). However, TPD is not appropriate for reactive atoms or radicals, since they may spontaneously recombine to form stable molecules at the surface before thermal desorption. Consequently, binding energies of atoms or radicals are less documented.
For species with undefined BE’s, attempts have been made by speculating an additive law: the BE of a given molecule being the sum of the BE’s of the atoms or groups of atoms. This approximation of the BE as implemented in Garrod et al.’s model (Cuppen et al., 2017) gives a rough estimation that can indeed be improved in different manners. Wakelam et al. (Wakelam et al., 2017) have proposed a proportional law that relates the interaction energy between the given species and one water molecule (energy of the dimer) and its binding energy on amorphous solid water. However, the integration of the binding energies computed with this method in the gas-grain code Nautilus (Ruaud et al., 2016) did not improve the prediction of the gas phase abundances. A more precise determination of the BEs and inclusion of nonthermal processes have been invoked as possible improvements. Because of their accuracy, electronic structure calculations appear to be the method of choice to improve the BE determination. However, for computational reasons, this approach often reduces the explicit description of the surface to a small number of atoms or molecules (Koch et al., 2007; Duvernay et al., 2014; Rimola et al., 2014). Thanks to the development of numerical resources, periodic ab initio simulations have been also performed recently to account for larger crystalline ice surfaces though still in a static way neglecting temperature effects and the configurations sampling being limited to the identification of the global minimum (Ferrero et al., 2020a). It should be noted that the size of the crystal and the choice of the functional are critical to surely assess an accurate value of the binding energy (Ferrero et al., 2020b). By contrast, semi-empirical based classical molecular dynamics (MD) simulations account at low computational cost for long range effects through the modeling of quasi-infinite surfaces and offer a full exploration of the possible binding sites providing BE distributions and eventually diffusion barriers (Michoulier et al., 2018). However, classical MD rely strongly on the accuracy, or even availability, of force-field parameters. Such parameters are in particular not available for radicals. Hybrid QM/MM (Quantum Mechanics/Molecular Mechanics) approaches, such as the ONIOM method (Chung et al., 2015), recently used by Sameera et al. (Sameera et al., 2017; Sameera and Maseras, 2018) on crystalline water ices, have the advantage of describing accurately the adsorbate-surface interaction including long range effects, dividing the system into two subunits: the adsorbate and few molecules of the surface treated at a high QM level and the rest of the surface being treated with a MM or lower level QM’ approach. The latter QM/QM’ scheme is the methodology chosen in the present study on ice surfaces generated from classical molecular dynamics simulations, including amorphous ice samples, to quantify the effect of structural disorder.
The binding energy depends on the volatile but also on the grain surface composition. Numerous studies have been conducted to determine the composition of the grains and more especially the grain icy mantle (Whittet et al., 1996; van Dishoeck, 1998; Ehrenfreund and Charnley, 2000; Ehrenfreund et al., 2003; Pontoppidan et al., 2008). They revealed that interstellar ices are mainly composed of water, but may also contain other species like CO, CO2, NH3, and methanol, in less significant quantities. The precise structure of interstellar ice remains an open question, though it is generally admitted that it might be amorphous (Dulieu et al., 2013; Hama and Watanabe, 2013). Amorphous Solid Water (ASW) is known to have physical properties that are distinct from other crystalline states, e.g., a lower thermal conductivity, as well as a larger surface area and higher porosity (Dohnálek et al., 2003). In the spectrum of ASW, a small feature is present on the high wavenumber wing of the OH stretching modes band, due to OH dangling bonds, that are more abundant in the microporous material than in crystalline compact ice (Noble et al., 2014). These unsaturated sites are preferential adsorption sites for incoming molecules (Buch and Devlin, 1991; Michoulier et al., 2020). Proton disorder in amorphous ice was shown to trigger a diversity of adsorption sites and consequently of binding energies for small adsorbates compared to crystalline ice (Kimmel et al., 2001; Kimmel et al., 2001). Even though laboratory ASW or ice grown in MD simulations do not reproduce interstellar icy grains for various factors (growth conditions, porosity, size and time scales … ) (Hama and Watanabe, 2013), characterizing the BEs of various molecules on these model surfaces provide nonetheless valuable insights on the trapping of the gas-phase species, on their availability for surface reactions and on the influence of the surface heterogeneity on the BE values.
The purpose of the present study is to establish a theoretical framework combining a force field based MD sampling and density functional corrected with high level ab initio calculations on some configurations issued from this sampling to accurately describe binding energies (physisorption or chemisorption) of a set of atoms, radicals or molecules on surfaces of interstellar interest. This consists in probing a rich variability of adsorption sites on crystalline or amorphous ices, each of which characterized by a specific binding energy distribution.
A first section presents the details of the methodology and of the performed calculations. Results are then presented. The crystalline ice investigation serves firstly as a benchmark of the model and QM method. Once set, the most accurate combination of methods is then applied on amorphous ices. The obtained values are discussed and compared to published data when available.
As a start, it is important to agree on the definition of the BE of a species on a substrate. Throughout the paper, BE will be defined as the positive quantity according to the following equation:
Where
Geometry optimization were carried out in Cartesian coordinates on the QMHigh part, the QMLow zone remaining frozen. Default convergence thresholds on gradients and displacements were used. It should be noticed that no BSSE effect was considered in the present work. For crystalline (Ih) ices, the geometries were obtained from the supplementary material of Sameera et al. (Sameera et al., 2017). They are derived from Karseemeijer et al. (Karssemeijer et al., 2012) and were obtained at the ONIOM(DFT:AMOEBA) level. Thus, two ice models with 4 sites differing by the surface dangling bonds ordering were used. In model A, 162 water molecules are present, with 24 in the QMHigh region. Model B comprises 165 water molecules, the QMHigh zone containing 20 molecules (see Figure 2 in Sameera et al. for a graphical representation). For OH, HCO, and CH3, the Sameera et al. geometries were used as starting guesses. For the 6 other adsorbates, the initial geometry was generated manually. The harmonic frequencies were obtained at the ωB97X-D/6-31+G**:PM6 level only. The resulting total zero-point vibrational energy correction (ZPE) was then scaled using a factor of 0.9523 taken from the NIST database (https://cccbdb.nist.gov/vsfx.asp). This ZPE correction was then added to the pure electronic BE’s obtained will all the different levels of theory employed:
For amorphous cases, a different strategy was adopted. An amorphous ice slab was generated by classical molecular dynamics simulations following the procedure detailed in Michoulier et al. (Michoulier et al., 2018), using the TIP4P/2005 water model (Abascal and Vega, 2005; Vega and Abascal, 2011). Classical molecular dynamics trajectories have been used to model the adsorption of a single water molecule, described as well with the TIP4P/2005 model, on the generated ice slab at T = 77 K. The goal of these independent trajectories is to sample a variety of adsorption sites that will serve as guess sites for the other adsorbates. The MD simulation details can be found in (Michoulier et al., 2018). The amorphous ice slab contains here 1344 molecules in a simulation box of dimensions: 25.89 × 29.95 × 90.28 Å. The initial position of the adsorbed water molecule is randomly chosen for each trajectory. After a 1 ns trajectory with a 1 fs timestep at T = 77 K, the final configuration of each trajectory is collected, the conservation of total energy and the convergence of the usual parameters being carefully checked for each trajectory.
The obtained adsorption geometries were cut into 3 different layers. The first one defining the QMHigh part, consists in the absorbed molecule surrounded by a sphere containing about 20 other water molecules. Then, about 120–150 molecules in a slightly larger sphere were selected for the QMLow part and finally the remaining ∼1200 ones were removed. The radii of the spheres were adjusted to obtain approximately the same number of molecules as in the crystalline case. The typical radius of these spheres is about 7–8 Å for the High-level part and about 12–15 Å for the Low level one. The reduction of the number of water molecules from the slab to a reduced cluster is illustrated in Figure 1. The same ONIOM procedure used for the crystalline phase was applied to these clusters. For reasons explained below, these calculations were performed at the ωB97X-D/6-31+G**:PM6 level only. The other adsorption cases were treated by replacing the water molecule by the corresponding adsorbate and reoptimizing the geometries. This method proved to give reasonable guess structures for the ONIOM geometry optimization, despite the different nature of the interaction between the various species and the surface. As in the crystalline case, the QMHigh part without the adsorbate (bare ice surface) was optimized separately as well.
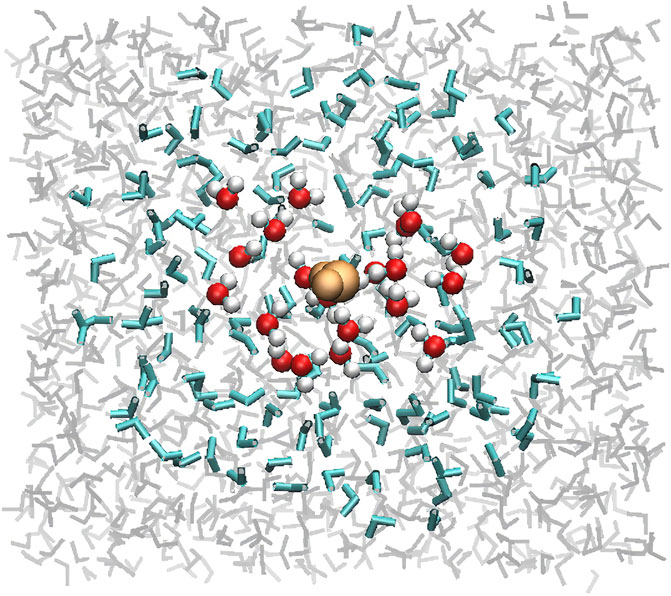
FIGURE 1. Top view of a snapshot of amorphous ice issued from a MD trajectory at 77 K. The layers for the ONIOM calculation are represented accordingly: adsorbed H2O (beige), QMHigh (red/white), QMLow (blue), and the remaining water molecules (grey).
The procedure described in the previous paragraphs gives geometry and binding energies at the DFT level. In order to obtain more accurate binding energies, the ωB97X-D/6-31+G**:PM6 QMHigh partial geometries were extracted and used as inputs for a CBS/DLPNO-CCSD(T) (Guo et al., 2018) single point calculation, using the ORCA code (Neese, 2018; Neese et al., 2020). Resolution of the identity (RIJK) was used (Weigend et al., 2009) with the auxiliary basis set from Weigend (Weigend, 2008). For DLPNO-CCSD(T), the normal criteria were used as discussed by Liakos et al. (Liakos et al., 2015). The auxiliary basis set for correlation is taken from Hellweg et al. (Hellweg et al., 2007). For Complete Basis Set (CBS) extrapolation, def2-TZVPP and def2-QVZPP basis sets were employed (Weigend and Ahlrichs, 2005). The Hartree-Fock and the correlation energies were extrapolated separately as shown in (Neese and Valeev, 2011) with α = 7.880 for Hartree-Fock and β = 2.970 for correlation. The three (def2-TZVPP, def2-QVZPP and CBS) resulting DLPNO-CCSD(T) energies then replaced the DFT part of the total ONIOM extrapolated energy. The most sophisticated BE can thus be considered as CBS/DLPNO-CCSD(T):PM6//ωB97X-D/6-31+G**:PM6, corrected with the ωB97X-D/6-31+G**:PM6 ZPE. CBS results should also be free of BSSE (Basis Set Superposition Error), but it should also be noted that BSSE effects were calculated to be small by Sameera et al. (Sameera et al., 2017).
In the following, binding energies are expressed in eV. They can be converted in Kelvins using: 1.000 eV = 11604.59 K = 96.4853 kJ/mol.
The study of the crystalline structures was performed to benchmark the various DFT/ONIOM models employed in the present work. Furthermore, the crystal results may be compared with the amorphous ones as well. We have adopted the nomenclature of Sameera et al. (Sameera et al., 2017) who distinguish 8 different adsorption sites (A1, A2, A3, A4, B1, B2, B3, and B4) depending on the different dangling bonds arrangement at the surface. A detailed comparison with Sameera et al. for the eight sites is given in the supplementary material. In the perspective of studying amorphous ices, we will only report here four characteristic BE’s for each species: the minimum, the maximum, the average values and the standard deviation σ, respectively. In the following, we present in the figures the geometries corresponding to the most stable configuration (largest BE) obtained at ONIOM(ωB97X-D/6-31+G**:PM6) level only. It should be noticed that the most stable geometry depends on the employed functionals, basis sets and semi-empirical methods, as emphasized in the supplementary material. A general trend is that the most stable situation generally corresponds to a A-type site, i.e., above the center of a hexagon. There are however some exceptions. For clarity, only the QMHigh part is represented in the figures.
We start by examining the results obtained with the 6 combinations of basis sets, functionals and semi-empirical methods.
As displayed in Table 1, the binding energy of the hydrogen atom appears to be very small, in a consistent pattern for all levels of calculations. The slightly negative values correspond to an artefact due to the convergence threshold of the geometry optimization process. It is possible that using tighter convergence criteria on the geometry optimization process would solve this issue. However, the Potential Energy Surface (PES) is indeed very flat, with the H atom roaming almost freely above the surface (see e.g., Figure 2). Hence the convergence of the SCF process can be very slow, even using the expensive quadratic convergence algorithm (Schlegel and McDouall, 1991) and the computational cost is quite high.
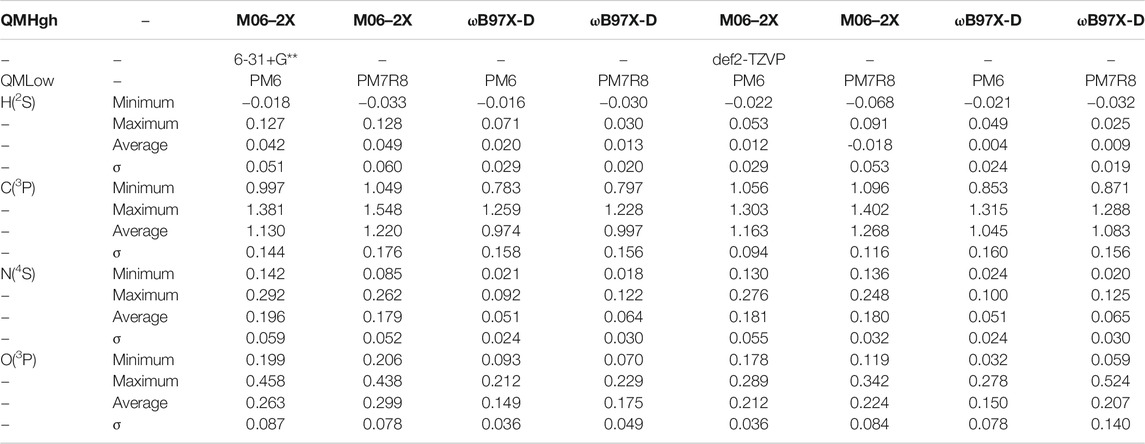
TABLE 1. Calculated BE’s at ONIOM level for atomic species.
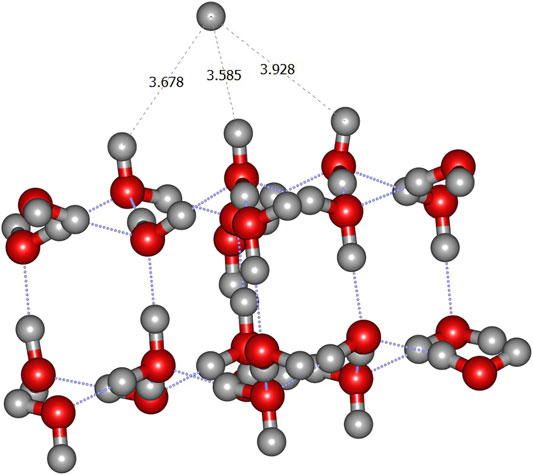
FIGURE 2. ONIOM(ωB97X-D/6–31+G**:PM6) most stable geometry (A1 site) for hydrogen atom on crystalline ice.
The carbon atom case is also specific since all calculations, with all 6 levels of theory, give a rather large BE, above 1 eV (Table 1). The length of the C–O bond, is about 1.6 Å (see Figure 3A). This is in agreement with recent theoretical work on crystalline (Ferrero et al., 2020b) and amorphous (Shimonishi et al., 2018) ices who conclude on a chemisorption rather than physisorption. Indeed, in the triplet state of gas phase OCH2, the molecule is slightly pyamidalized and the C–O bond is elongated to a larger value of ∼1.88 Å (Hickson et al., 2016). Ferrero et al. also find a transfer of a hydrogen atom to a nearby water molecule to form a COH radical. Furthermore, a Mulliken population analysis shows that this radical has a negative charge. Our own calculations show a slightly different picture: a Wiberg bond index analysis (Wiberg, 1968) confirms a weak covalent bond between C and O (0.73 for C for A2 site). Furthermore, a NBO population analysis (Weinhold and Carpenter, 1988), shows that the COH2 remains essentially neutral. The only exception is the A4 site, for which, at all levels of calculation, the adsorption of the carbon atom leads to the loss of a H atom from the attacked H2O (Figure 3B). The NBO analysis this time confirms that COH fragment carries a total charge of −0.733 while the H3O part has a +0.650 positive charge (Figure 3C). Thus, this geometry corresponds to the formation of a H3O+–COH− complex, very much like in a solvated situation and in agreement with Ferrero et al. (Ferrero et al., 2020b).
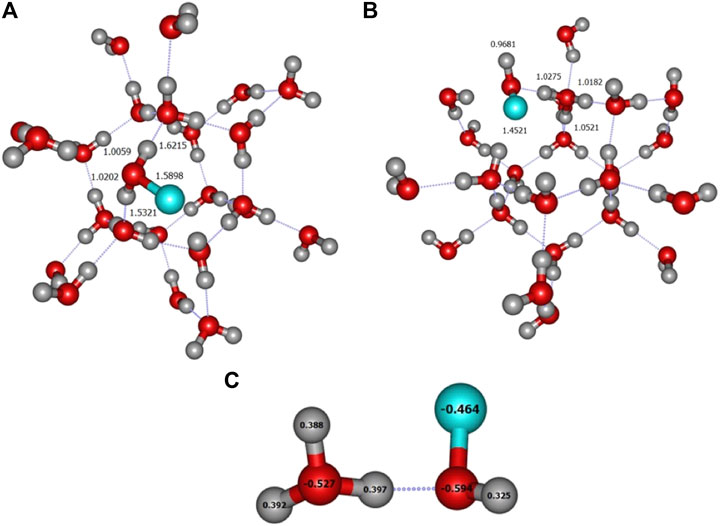
FIGURE 3. (A) ONIOM(ωB97X-D/6-31+G**:PM6) most stable geometry (A2 site) for carbon atom. (B) Formation of the H3O+COH− complex (A4 site) (C) NBO charges of the H3O+COH− complex. These geometries were obtained on crystalline ice.
On the other hand, the last two studied atoms, namely N and O in their respective ground state, have weak interaction energies, characteristic of physisorption (Table 1). The most stable geometries are displayed in Figures 4A,B, respectively, Table 1 also shows a remarkably large difference between the M06–2X and ωB97X-D, independent of the basis sets and semi-empirical methods used. The M06–2X are about twice larger, despite the fact that the geometries are very similar for all sites.
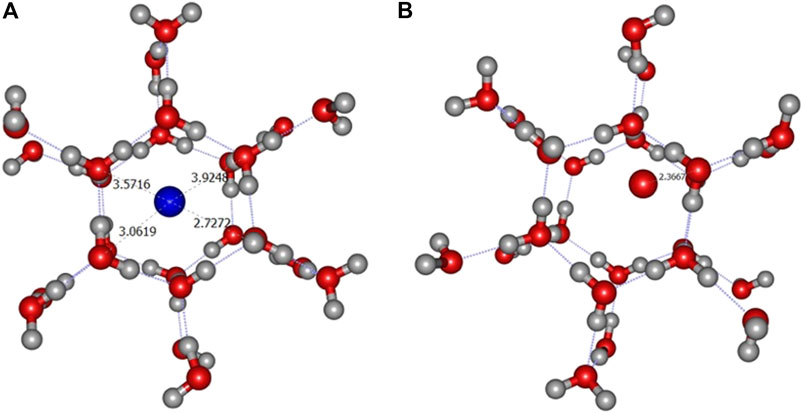
FIGURE 4. ONIOM(ωB97X-D/6-31+G**:PM6) most stable geometry for N (A) and O (B) atoms (A2 site) on crystalline ice.
Table 2 summarizes the results for the 4 studied diatomic and triatomic molecules (NH, OH, HCO and H2O). The corresponding lowest lying equilibrium geometries are displayed in Figures 5, 6. The maximum BE of NH is about 50% larger than for OH, which is typical of hydrogen bonding (David et al., 2017). For OH, the maximum BE, around 0.7 eV depending on the level of calculation, is typical of the formation of two hydrogen bonds, in agreement with previous theoretical works (Du et al., 2006; Guedes et al., 2003), as shown in Figure 5B.
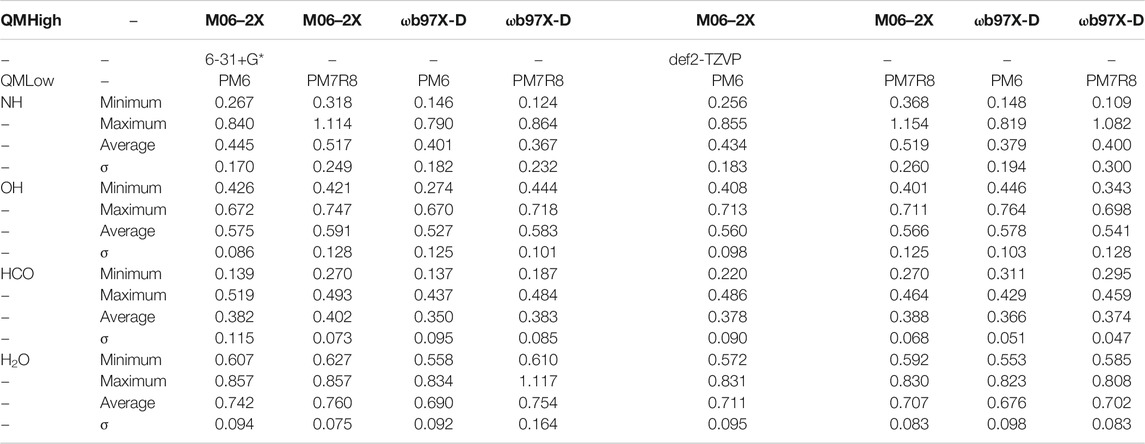
TABLE 2. Calculated BE’s at ONIOM level for diatomic and triatomic systems.
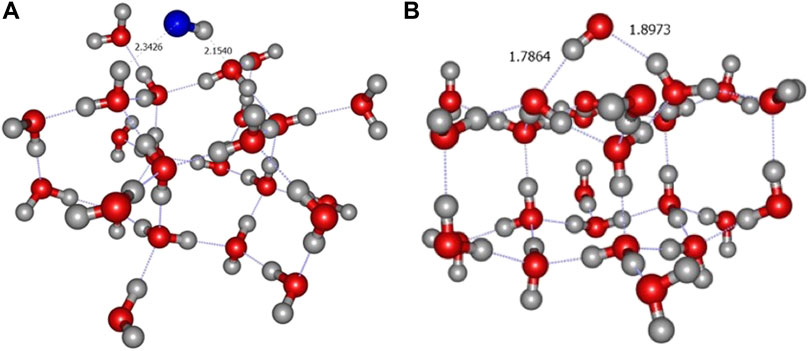
FIGURE 5. ONIOM(ωB97X-D/6-31+G**:PM6) most stable geometry for NH (A) and OH (B) molecules (A2 site) on crystalline ice.
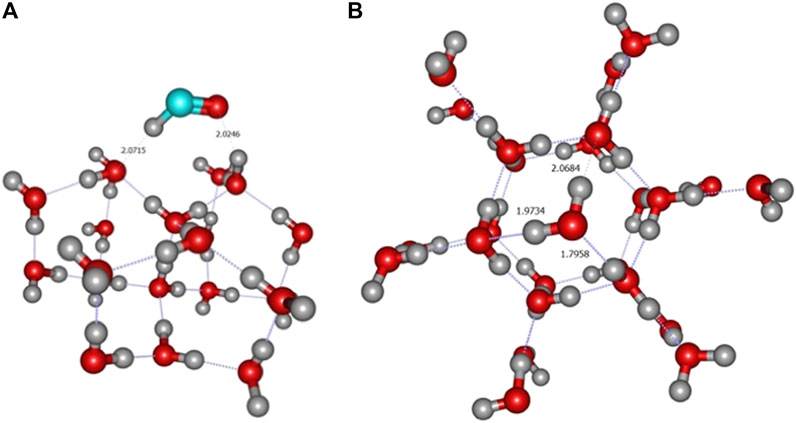
FIGURE 6. ONIOM(ωB97X-D/6-31+G**:PM6) most stable geometry for HCO (B3 site) (A) and H2O (A2 site) (B) molecules on crystalline ice.
The situation is very similar in the HCO case, which creates two hydrogen bonds as well (Figure 6A) although the BE is slightly lower (about 0.5 eV). For H2O, the rather high value (about 0.6 eV according to Table 2), is consistent with the building of three hydrogen bonds in the most favorable conformation shown in Figure 6B.
The BE of CH3 is consistently predicted to be around 0.3 eV (Table 3) and this radical is only bound by a weak interaction as can be seen in Figure 7A. In contrast, for ammonia, the calculated binding energies appear to be rather high, between 0.8 and 1.2 eV depending on the level of theory (Table 3). This is correlated to the existence of two hydrogen bonds between NH3 and two water molecules as displayed in Figure 7B. This is consistent with the intermolecular energy obtained for the isolated NH3-H2O dimer (0.473 eV) (Wakelam et al., 2017).
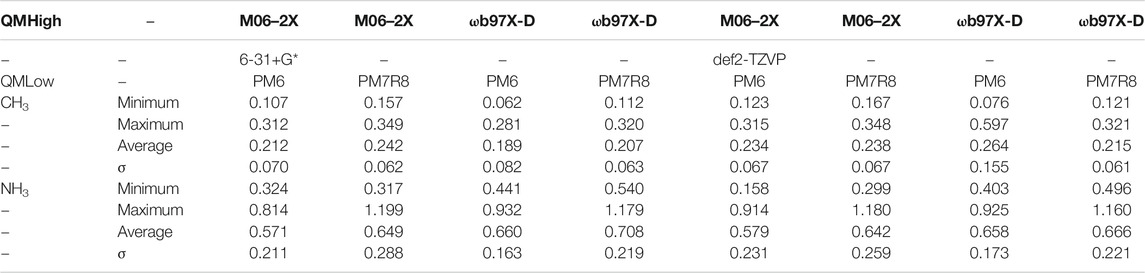
TABLE 3. Calculated BE’s at ONIOM level for tetratomic systems.
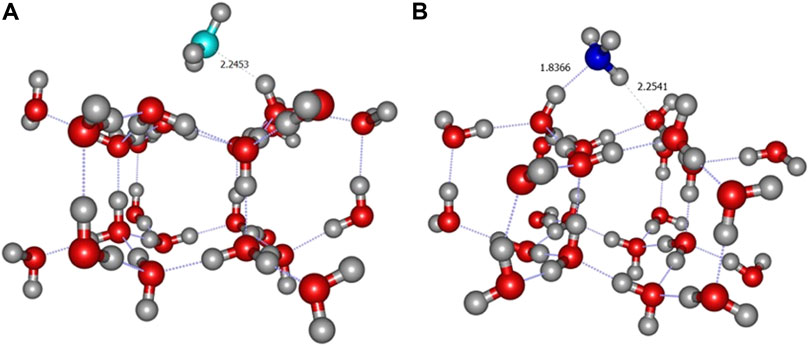
FIGURE 7. ONIOM(ωB97X-D/6-31+G**:PM6) most stable geometry for CH3 (A2 site) (A) and NH3 (A3 site) (B) molecules on crystalline ice.
From the inspection of the average energies given in Tables 1–3, and the fact that all methods happened to give very similar geometries, we have selected the ωB97X-D/6-31+G**:PM6 combination to obtain the ZPE correction and apply the single point CBS correction. Indeed, the M06–2X gives too large values for N and O atoms compared to ωB97X-D. This latter functional, being range-separated describes more accurately long-range interactions. Basis sets effects seem to be small and the 6-31+G** one is much less demanding. Finally, PM6 was selected over PM7R8 since this latter method, although available in the Gaussian 16 code and detailed in the PhD work of Throssell (Throssell, 2017), does not seem to have been published yet.
Table 4 shows the summary of the results obtained for crystalline ices at our best level of calculations, i.e., CBS/DLPNO-CCSD(T):PM6//ΟΝΙΟΜ(ωB97X-D/6-31+G**:PM6) including scaled ZPE correction calculated at the ΟΝΙΟΜ(ωB97X-D/6-31+G**:PM6) level. A graphical representation is shown in Figure 8. The impact of ZPE on BE’s is shown in Figure 8A Except for the case of CH3, a linear fit gives a correlation coefficient larger than 0.96. As expected, ZPE effects are larger for polyatomic molecules. For C, N, and O, the BE tends to increase by 3–7 %. For molecules, the ZPE lowers the BE’s, the drop varying from 10 to 20% with the notable exception of CH3 where it reaches 33%. Recently, Ferrero et al. (Ferrero et al., 2020a) have obtained the relation BE0 = 0.854 BEe using a fragment approach to calculate ZPE effects on crystalline ice. Their set of species does not include any atomic system but contains larger molecules, leading to much larger ZPE’s.
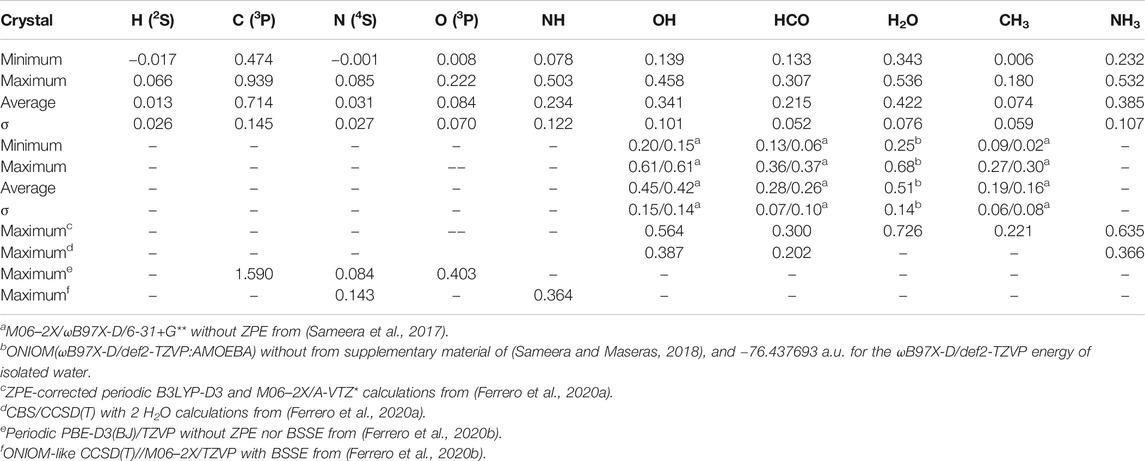
TABLE 4. Calculated BE’s at CBS/DLPNO-CCSD(T):PM6//ONIOM(ωB97X-D/6–31+G**:PM6) including ZPE for crystalline structures.
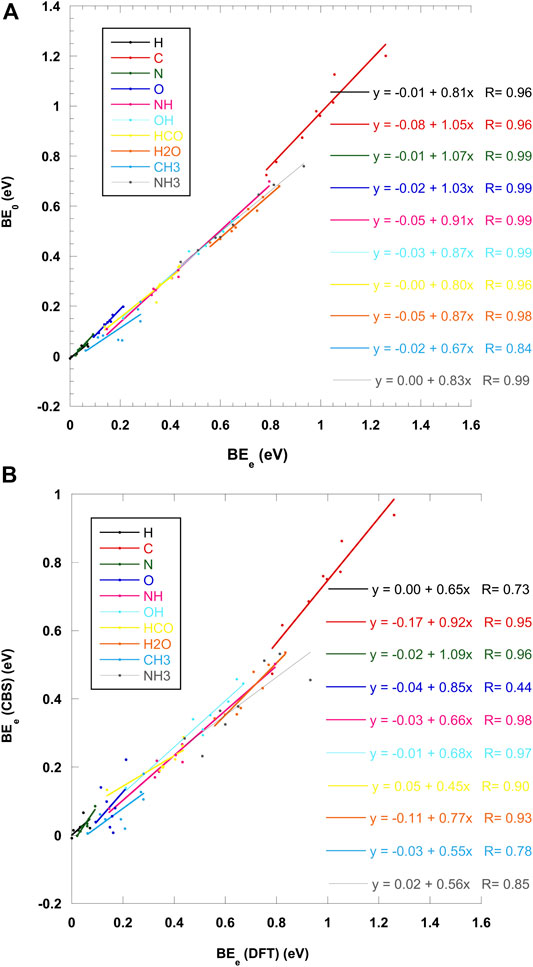
FIGURE 8. Effect of scaled ZPE on ONIOM(ωB97X-D/6-31+G**:PM6) BE’s for crystalline ices (A) Effect of CBS single point on ZPE-corrected BE’s for crystalline ices (B).
In addition, single point CBS/DLPNO-CCSD(T) calculations have also some effect on the BE’s, as illustrated in Figure 8B. The correction is more erratic, with a bad (< 0.9) correlation coefficient for H, O, CH3, and NH3. Except for the nitrogen atom where CBS tends to augment the BE by 9%, the other values are lowered, sometimes considerably (up to 55% for HCO for example). More details on ZPE and CCSD(T) corrections for each of the eight sites can be found in the supplementary material file.
Table 4 also compares when available (i.e., OH, HCO, H2O, and CH3) the present results to the single point DFT calculations on Mechanical Embedding (ME) ONIOM(DFT/AMOEBA) geometries of Sameera et al. (Sameera et al., 2017; Sameera and Maseras, 2018). It should be noticed that since these calculations do not include any ZPE, the BE’s are larger than the present results. In addition, the maximum values given in Table 4 correspond to the most stable configuration on a perfect crystalline ice. Our results can also be compared to the recent values obtained by Ferrero et al. (Ferrero et al., 2020a; Ferrero et al., 2020b) using periodic DFT methods and given at the bottom of Table 5. Except for N atom, our values for C and O are much lower than the periodic PBE-D3(BJ)/TZVP of Ferrero et al. (Ferrero et al., 2020b). The recent study of Martinez-Bachs et al. (Ferrero et al., 2020b) performed in an ONIOM-like CCSD(T)//M06–2X/TZVP scheme provides a rather high value for N (0.143 eV) compared to our maximum (0.083 eV). On the other hand, their value for NH (0.364 eV) is lower than our maximum result equal to 0.503 eV. Except for the HCO case where the two values coincide, CBS values are also lower than the CCSD(T) ones obtained by Ferrero et al. (Ferrero et al., 2020a), but it should be kept in mind that these authors limited the calculation to only two water molecules in addition to the adsorbate.
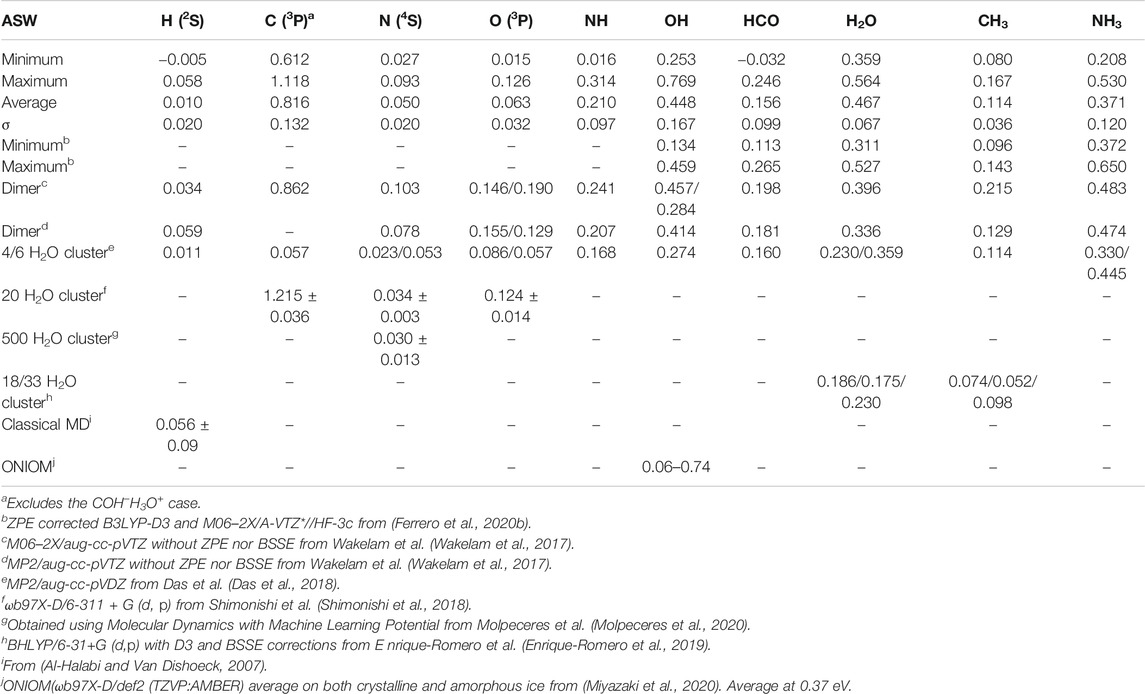
TABLE 5. Calculated BE’s at CBS/DLPNO-CCSD(T):PM6// ONIOM(ωB97X-D/6-31+G**:PM6) including ZPE for amorphous structures.
Table 5 shows the summary of the results obtained for amorphous ices at the CBS/DLPNO-CCSD(T):PM6// ΟΝΙΟΜ(ωB97X-D/6-31+G**:PM6) including scaled ZPE correction. This table also displays when available previous theoretical determinations of the BE’s. A graphical comparison with the crystalline case can be seen in Figure 11. Figure 9A compares the corrected BE0 to the uncorrected BEe DFT results. If we except the special case of hydrogen atom, the correlation coefficient is always larger than 0.97. For the three other atoms, the ZPE correction is in fact very small (a few %). For the molecules, the correction is between 11 and 20 %, except for CH3 where it is 32% as in the crystalline case. Figure 9B plots the comparison between the CBS/DLPNO-CCSD(T) and the DFT calculations. The dispersion of the values is much larger than in the crystalline case, except for the carbon atom where a covalent bond is created. However, it is clear that the CBS results are considerably lower than the DFT ones.
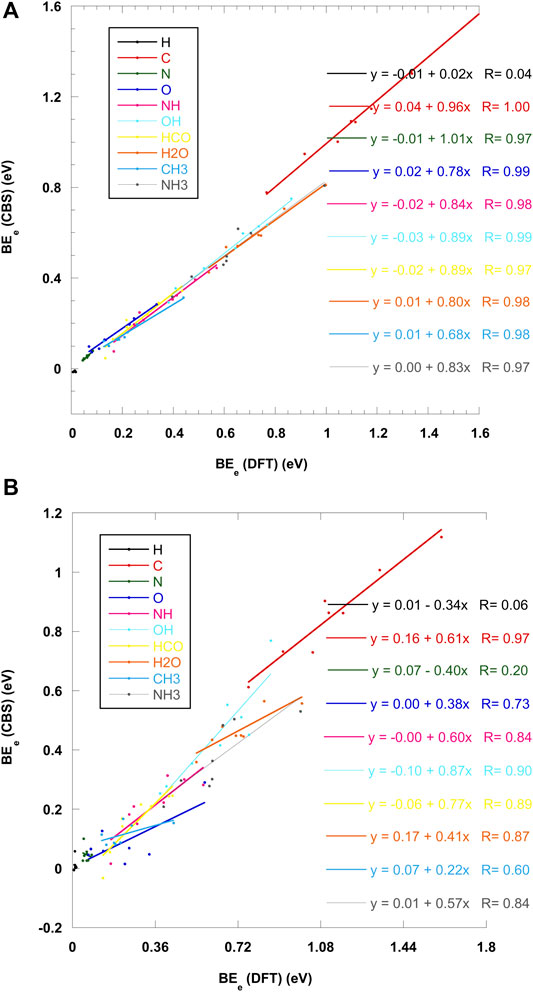
FIGURE 9. Effect of scaled ZPE on ONIOM(ωB97X-D/6-31+G**:PM6) BE’s for amorphous ices (A) Effect of CBS single point on ZPE-corrected BE’s for amorphous ices (B).
It should be noticed that, as in the A4 site of crystalline ice, one of the amorphous clusters produced a H3O+COH− complex when approached by a carbon atom in its triplet ground state (see Figure 10A). The charges of this product were confirmed via a NBO analysis (Figure 10B). The H3O+COH− structure lies about 1.1 eV below the dissociated case, which is the largest calculated value. For the seven other amorphous structures, the final product was the pyramidal triplet state of the COH2 complex already found in crystalline ice.
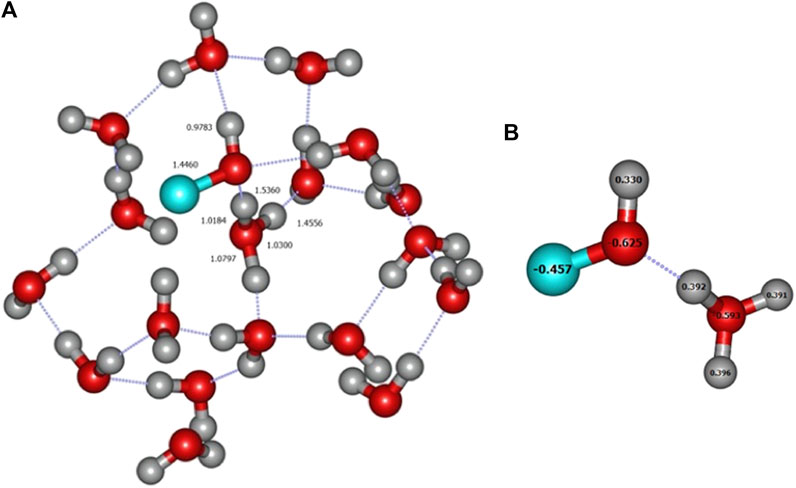
FIGURE 10. (A) Formation of the H3O+COH− complex in an amorphous environment. Distances are given in Angströms. (B) NBO charges from ONIOM(ωB97X-D/6-31+G**:PM6) calculation.
According to Table 5, our best estimate for the binding energy of H is 0.010 ± 0.020 eV. However, the maximum value is 0.058 eV. They are close to the crystalline case (Table 4). Previous theoretical works, either using quantum methods on clusters [(Wakelam et al., 2017; Das et al., 2018)] or classical calculations (Al-Halabi and Van Dishoeck, 2007) also give very dispersed values. One can only conclude that while on average the BE is very small for H, some local configuration of ASW may lead to larger values as large as 0.058 eV.
Concerning the carbon atom, the low value obtained by Das et al. (Das et al., 2018), i.e., only 0.057 eV is easily explained by the fact that the C atom is weakly bound to a hydrogen atom instead of an oxygen [see Figure 3A in supplementary material in (Das et al., 2018)]. Our average value of 0.816 ± 0.132 eV is 33% lower than average of 1.215 ± 0.036 eV obtained at the ωb97X-D/6-311 + G(d, p) level by Shimonishi et al. (Shimonishi et al., 2018). The differences come from the lack of Low level layer, the absence of ZPE and CBS single point correction which impact the BE’s. Their smaller uncertainty is possibly due to a different method to select the adsorption trial geometries and average the largest BE’s.
The binding energy of nitrogen atom on ASW has been measured experimentally by Minissale et al. (Minissale et al., 2016) to be
Minissale et al. also measured the BE for oxygen atom:
To the best of our knowledge, there is no experimental determination of the BE of the NH radical on ice surfaces. As shown in Table 5, only calculations on water clusters are available. Our best estimate of 0.210 ± 0.097 eV is slightly higher than the 0.168 eV value obtained on a water tetramer by Das et al. (Das et al., 2018) but close to the dimer result (0.207 eV) from Wakelam et al. (Wakelam et al., 2017).
Similarly, for the OH radical, no experimental value has been published yet. For example, for OH, Garrod and Herbst (Garrod and Herbst, 2006) assumed that it is half the value of H2O, i.e., 0.246 eV. Dulieu et al. (Dulieu et al., 2013) obtained 0.396 eV but on silicate dust surfaces. Therefore, as for other molecules, only theoretical values are available, most of them obtained with water clusters (Table 5). The only exception is the recent work by Ferrero et al. (Ferrero et al., 2020b), where the BE of OH is calculated to be between 0.134 and 0.459 eV (on 5 amorphous structures). Our maximum value of 0.769 eV is higher, and could be explained by the existence of three hydrogen bonds between OH and the neighboring water molecules. Our average best estimate is thus 0.448 ± 0.167 eV.
As for HCO, again no experimental value is available. The value of Garrod and Herbst (Garrod and Herbst, 2006) of 0.138 eV was deduced by addition and subtraction of energies from other molecules. In Table 5, our slightly negative value is also due to the large CBS/DLPNO-CCSD(T) correction, the first positive value being in fact 0.055 eV, which is lower than the minimum value of Ferrero et al. at 0.116 eV. On the other hand, our maximum values are very similar: 0.246 eV in the present work and 0.265 eV in Ferrero et al.
In contrast to most other species studied in the present work, the adsorption of water on ice has been the subject of numerous experimental works [see Table 1 in (Yocum et al., 2019) for a recent review]. Sandford and Allamandola (Sandford and Allamandola, 1988) proposed 0.415 ± 0.001 and 0.437 ± 0.001 eV, Fraser et al. (Fraser et al., 2001) 0.497 ± 0.005 eV, Haynes et al. (Haynes et al., 1992) 0.516 ± 0.009 eV, Bolina et al. (Bolina et al., 2005) 0.414 ± 0.008 eV, Ulbricht et al. (Ulbricht et al., 2006) 0.477 ± 0.031 eV. Although the experiments differ in the details (for example the substrate), they agree to a value between 0.4–0.5 eV. This is in good agreement with our best estimate of 0.467 ± 0.067 eV (Table 5).
CH3 being a radical as well, experimental determinations seem inexistent. A recent DFT study by Enrique-Romero et al. (Enrique-Romero et al., 2019) on water clusters give values below 0.1 eV (Table 5). Our own results are slightly higher with 0.114 ± 0.036 eV. Our minimum and maximum values are also similar to those of Ferrero et al. (Ferrero et al., 2020b).
Finally, for NH3, the recommended value of 0.477 eV (Hama and Watanabe, 2013) is estimated from the experimental TPD experiments from Collings et al. (Collings et al., 2004). As can be seen in Table 4, this value is close to the water clusters calculation [(Wakelam et al., 2017; Das et al., 2018)] and lies within the interval of our best estimate: 0.371 ± 0.120 eV. The DFT//HF-3c results of Ferrero et al. (Ferrero et al., 2020b), favor larger values between 0.372 and 0.650 eV.
Figure 11. A gives finally a graphical representation of the CBS BEs obtained on the 16 adsorption sites, 8 crystalline and 8 amorphous structures. It shows that BE’s can vary significantly for a given adsorbate or a given substrate, depending strongly on the local environment. Excepting the reactive carbon atom case, the highest value should correspond to the lowest lying equilibrium geometry. However, in most cases, several configurations have very similar BE’s, both for crystalline and amorphous cases, these values being indicative of a configuration that may exist on any ice sample. It is worth mentioning that the statistics could be improved by considering a larger number of initial configurations, but it has to be reminded that for each geometry, a full QM/MM optimization and single point CBS/DLPNO-CCSD(T) correction are carried out, these calculations being expensive, more especially when convergence is difficult to achieve. Figure 11B compares the average BE’s and their standard deviation for both surfaces. In most cases, the amorphous average is above the crystalline value, but the trend is not universal and does not allow to conclude without ambiguity. It should be recalled that the considered amorphous ice surface is non-porous, addition of pores or cavities could lead to more marked differences with respect to the crystalline face. However, such a surface would be difficult to handle with the proposed methodology because of the large cluster size it would require. The comparison of our calculated BE’s with the values that have been reported in UMIST (McElroy et al., 2013) or KIDA (Wakelam et al., 2012) databases indicate that the values appearing in the databases are for most cases contained in the BE’s range we obtain on both surfaces. For OH, HCO, and C, the data implemented in KIDA have a better agreement with our theoretical values than the UMIST ones. This comparison further supports our methodology that can be used to provide BE’s for species remaining undocumented, such as radicals or atoms or even larger molecules, thus improving the chemical networks implemented in the astrochemical models.
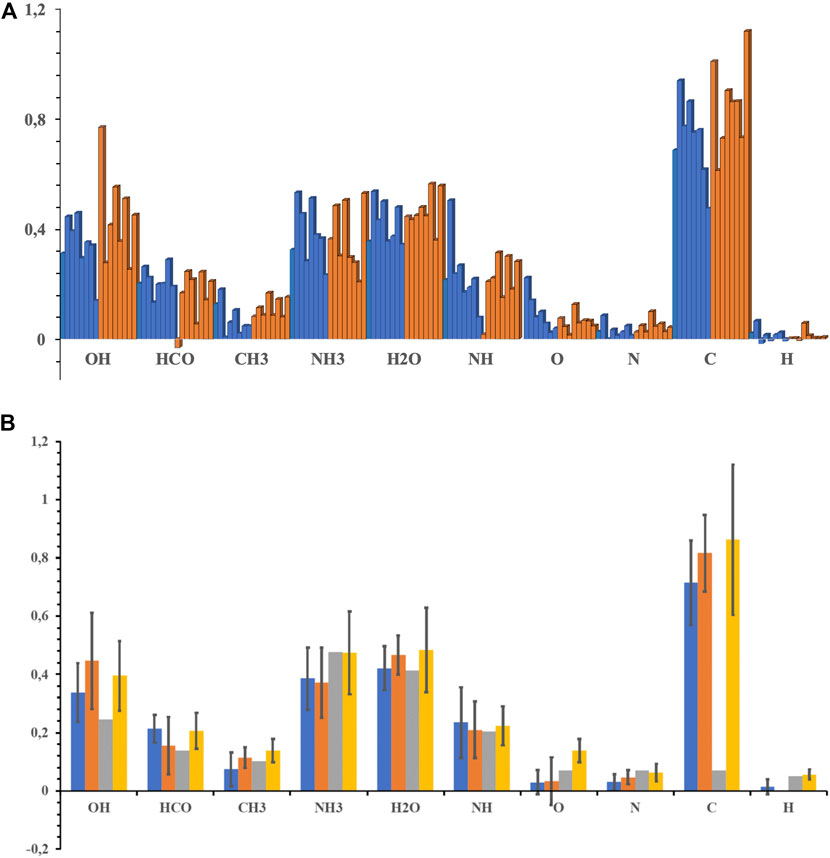
FIGURE 11. (A) Summary of CBS BE (in eV) distributions for crystalline (blue) and amorphous (orange) ices (B) Comparison of calculated average BE’s (with standard deviation) between crystalline (blue) and amorphous (orange) ice. Values, when available, taken from UMIST (McElroy et al., 2013) and KIDA (Wakelam et al., 2012) databases are also represented in grey and yellow, respectively.
In this work we have used a first principles methodology tailored to study adsorption processes of small atoms and molecules on both crystalline and amorphous ices, represented by a cluster of about 150 water molecules. A full quantum ONIOM QMHigh/QMLow scheme was employed, using semi-empirical methods for the surrounding water molecules representative of the ice sample.
Several combinations of functionals, basis sets and semi-empirical parametrizations were tested on the different possible adsorption sites of a perfect hexagonal ice. The ΟΝΙΟΜ(ωB97X-D/6-31+G**:PM6) was chosen as a good compromise between cost and accuracy. Despite the fact that the ωB97X-D functional is range-separated and includes dispersion correction, the energy of the QM part was recalculated using single-point CBS/DLPNO-CCSD(T) calculations. This level of theory should remove possible BSSE effects. Inclusion of ZPE effect is also important, even for atoms.
This protocol was applied to both crystalline and amorphous ices. For the latter, eight snapshots obtained from classical MD calculations were selected. This allows us to propose accurate binding energies for the set of studied species. The dispersion of the BE’s is rather large, reflecting a large diversity in the hydrogen bond network and the possible inclusion of the adsorbate on the surface. In the case of carbon atom, two different pathways were identified, depending on the local environment. One of them leads to the formation of a covalently bonded COH2 molecule while the other one, less frequent, produces a COH−H3O+ complex.
The present method is sufficiently versatile to be applied to other systems, including larger adsorbed molecules and/or, mixed ices. The accuracy can also be further benchmarked by adapting the choice of the functional and basis set. A possible extension would be to use a three-layer ONIOM. The computational cost of the DLPNO-CCSD(T) single point calculations can also be limited by using only the def2-TZVPP basis set, for example. It would also be interesting to increase the number of studied snapshots in order to get more statistically significant values. Finally, reactive processes may also be tractable.
As a conclusion, the present study brings new BEs estimations to be included in astrochemical models. When pure quantum chemistry calculations are performed, a single and unique value is produced that corresponds to the global minimum. With the methodology proposed here, the statistics over different configurations provides a set of values: maximum, minimum, average and standard deviation, BE distribution. The average BE accounts for the diversity of adsorption sites, whereas using the maximum BE implies that adsorption occurs preferentially in the most stable sites. An extensive investigation should be carried out to quantify how sensitive the astrochemical models are with respect to BE variations. This is beyond the scope of the present study but would help to target the BE accuracy and consequently to adjust the theoretical methodology accordingly.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
DD did most of the quantum chemical calculations and wrote parts of the manuscripts. CT did the classical dynamics calculations and participated in the writing. MM helped writing the manuscript.
The authors acknowledge support from the CaPPA project (Chemical and Physical Properties of the Atmosphere) funded by the French National Research Agency (ANR) through the PIA (Programme d’Investissement d’Avenir) under Contract No. ANR-10-LABX-005; the Région Hauts de France and the Ministère de l’Enseignement Supérieur et de la Recherche (CPER Climibio) and the European Fund for Regional Economic Development for their financial support. This work was performed using HPC resources from GENCI-TGCC (Grant No. 2020–A0050801859).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank the PCMI (Programme National de Physique et Chimie du Milieu Interstellaire) for support and the Centre de Ressources Informatiques (CRI) of the Université of Lille for providing computing time. Special thanks to A. Rivero-Santamaria for useful discussions and J. C. Loison and V. Wakelam for their insightful comments.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fspas.2021.645243/full#supplementary-material.
Abascal, J. L., and Vega, C. (2005). A general purpose model for the condensed phases of water: TIP4P/2005. J. Chem. Phys. 123 (23), 234505. doi:10.1063/1.2121687
Al-Halabi, A., and Van Dishoeck, E. F. (2007). Hydrogen adsorption and diffusion on amorphous solid water ice. Mon. Notices Royal Astron. Soc. 382 (4), 1648–1656. doi:10.1111/j.1365-2966.2007.12415.x
Bolina, A. S., Wolff, A. J., and Brown, W. A. (2005). Reflection absorption infrared spectroscopy and temperature-programmed desorption studies of the adsorption and desorption of amorphous and crystalline water on a graphite surface. J. Phys. Chem. B. 109 (35), 16836–16845. doi:10.1021/jp0528111
Buch, V., and Devlin, J. P. (1991). Spectra of dangling OH bonds in amorphous ice: assignment to 2 and 3‐coordinated surface molecules. J. Chem. Phys. 94 (5), 4091. doi:10.1063/1.460638
Burke, D. J., and Brown, W. A. (2010). Ice in space: surface science investigations of the thermal desorption of model interstellar ices on dust grain analogue surfaces. Phys. Chem. Chem. Phys. 12 (23), 5947. doi:10.1039/b917005g
Chai, J. D., and Head-Gordon, M. (2008). Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 10 (44), 6615–6620. doi:10.1039/b810189b
Chung, L. W., Sameera, W. M., Ramozzi, R., Page, A. J., Hatanaka, M., Petrova, G. P., et al. (2015). The ONIOM method and its applications. Chem. Rev. 115 (12), 5678–5796. doi:10.1021/cr5004419
Clark, T., Chandrasekhar, J., Spitznagel, G. N. W., and Schleyer, P. V. R. (1983). Efficient diffuse function-augmented basis sets for anion calculations. III. The 3-21 +G basis set for first-row elements, Li-F. J. Comput. Chem. 4 (3), 294–301. doi:10.1002/jcc.540040303
Collings, M. P., Anderson, M. A., Chen, R., Dever, J. W., Viti, S., Williams, D. A., et al. (2004). A laboratory survey of the thermal desorption of astrophysically relevant molecules. Mon. Notices Royal Astron. Soc. 354 (4), 1133–1140. doi:10.1111/j.1365-2966.2004.08272.x
Cuppen, H. M., Walsh, C., Lamberts, T., Semenov, D., Garrod, R. T., Penteado, E. M., et al. (2017). Grain surface models and data for astrochemistry. Space Sci. Rev. 212 (1–2), 1–58. doi:10.1007/s11214-016-0319-3
Das, A., Sil, M., Gorai, P., Chakrabarti, S. K., and Loison, J. C. (2018). An approach to estimate the binding energy of interstellar species. Astrophys. J. Suppl. Ser. 237 (1), 9. doi:10.3847/1538-4365/aac886
David, V., Grinberg, N., Moldoveanu, S. C., Grinberg, N., and Moldoveanu, S. C. (2017). Long-range molecular interactions involved in the retention mechanisms of liquid chromatography. Adv. Chromatogr. 54, 73–110. 10.1201/9781315116372-4.
Ditchfield, R., Hehre, W. J., and Pople, J. A. (1971). Self‐consistent molecular‐orbital methods. IX. An extended Gaussian‐type basis for molecular‐orbital studies of organic molecules. J. Chem. Phys. 54, 724–728. doi:10.1063/1.1674902
Dohnálek, Z., Kimmel, G. A., Ayotte, P., Smith, R. S., and Kay, B. D. (2003). The deposition angle-dependent density of amorphous solid water films. J. Chem. Phys. 118 (1), 364. doi:10.1063/1.1525805
Du, S., Francisco, J. S., Schenter, G. K., Iordanov, T. D., Garrett, B. C., Dupuis, M., et al. (2006). The OH radical−H2O molecular interaction potential. J. Chem. Phys. 124, 224318–224415. doi:10.1063/1.2200701
Dulieu, F., Congiu, E., Noble, J., Baouche, S., Chaabouni, H., Moudens, A., et al. (2013). How micron-sized dust particles determine the chemistry of our universe. Sci. Rep. 3 (1), 1338. doi:10.1038/srep01338
Duvernay, F., Rimola, A., Theule, P., Danger, G., Sanchez, T., and Chiavassa, T. (2014). Formaldehyde chemistry in cometary ices: the case of HOCH2OH formation. Phys. Chem. Chem. Phys. 16 (44), 24200–24208. doi:10.1039/c4cp03031a
Ehrenfreund, P., and Charnley, S. B. (2000). Organic molecules in the interstellar medium, comets, and meteorites: a voyage from dark clouds to the early Earth. Annu. Rev. Astron. Astrophys. 38 (1), 427–483. doi:10.1146/annurev.astro.38.1.427
Ehrenfreund, P., Fraser, H. J., Blum, J., Cartwright, J. H. E., Garcia Ruiz, J. M., Hadamcik, E., et al. (2003). Physics and chemistry of icy particles in the universe: answers from microgravity. Planet. Space Sci. 51 (7–8), 473–494. doi:10.1016/s0032-0633(03)00052-7
Enrique-Romero, J., Rimola, A., Ceccarelli, C., Ugliengo, P., Balucani, N., and Skouteris, D. (2019). Reactivity of HCO with CH3 and NH2 on water ice surfaces. a comprehensive accurate quantum chemistry study. ACS Earth Space Chem. 3 (10), 2158–2170. doi:10.1021/acsearthspacechem.9b00156
Ferrero, S., Martínez-Bachs, B., Enrique-Romero, J., Rimola, A., et al. (2020a). “Adsorption of atoms on a crystalline ice surface model: results from periodic ab initio simulations,” in Computational science and its applications–ICCSA 2020. Editors O. Gervasi, B. Murgante, S. Misra, C. Garau, I. Blečić, and D. Taniar (Cham, Switerzland: Springer international publishing), 553–560.
Ferrero, S., Zamirri, L., Ceccarelli, C., Witzel, A., Rimola, A., and Ugliengo, P. (2020b). Binding energies of interstellar molecules on crystalline and amorphous models of water ice by ab-initio calculations. Astrophys. J. 904 (1), 904–911. doi:10.3847/1538-4357/abb953
Fraser, H. J., Collings, M. P., McCoustra, M. R. S., and Williams, D. A. (2001). Thermal desorption of water ice in the interstellar medium. Mon. Notices Royal Astron. Soc. 327 (4), 1165–1172. doi:10.1046/j.1365-8711.2001.04835.x
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2016). Gaussian 16 Rev. B.01. Wallingford, CT: Gaussian Inc.
Garrod, R., Park, I. H., Caselli, P., and Herbst, E. (2006). Are gas-phase models of interstellar chemistry tenable? the case of methanol. Faraday Discuss. 133, 51. doi:10.1039/b516202e
Garrod, R. T., and Herbst, E. (2006). Formation of methyl formate and other organic species in the warm-up phase of hot molecular cores. Astron. Astrophys. 457 (3), 927–936. doi:10.1051/0004-6361:20065560
Goumans, T. P., and Kästner, J. (2010). Hydrogen-atom tunneling could contribute to H2 formation in space. Angew. Chem. Int. Ed. Engl. 49 (40), 7350–7352. doi:10.1002/anie.201001311
Guedes, R. C., Costa Cabral, B. J., Martinho Simões, J. A., and Cabral do Couto, P. (2003). The hydration of the OH radical: microsolvation modeling and statistical mechanics simulation. J. Chem. Phys. 119 (14), 7344–7355. 10.1063/1.1605939.
Guo, Y., Riplinger, C., Becker, U., Liakos, D. G., Minenkov, Y., Cavallo, L., et al. (2018). Communication: an improved linear scaling perturbative triples correction for the domain based local pair-natural orbital based singles and doubles coupled cluster method [DLPNO-CCSD(T)]. J. Chem. Phys. 148 (1), 011101. doi:10.1063/1.5011798
Hama, T., and Watanabe, N. (2013). Surface processes on interstellar amorphous solid water: adsorption, diffusion, tunneling reactions, and nuclear-spin conversion. Chem. Rev. 113, 8783–8839. doi:10.1021/cr4000978
Hariharan, P. C., and Pople, J. A. (1973). The influence of polarization functions on molecular orbital hydrogenation energies. Theoret. Chim. Acta 28, 213–222. doi:10.1007/bf00533485
Haynes, D. R., Tro, N. J., and George, S. M. (1992). Condensation and evaporation of water on ice surfaces. J. Phys. Chem. 96 (21), 8502–8509. doi:10.1021/j100200a055
He, J., Jing, D., and Vidali, G. (2014). Atomic oxygen diffusion on and desorption from amorphous silicate surfaces. Phys. Chem. Chem. Phys. 16 (8), 3493–3500. doi:10.1039/c3cp54328e
He, J., Shi, J., Hopkins, T., Vidali, G., and Kaufman, M. J. (2015). A new determination of the binding energy of atomic oxygen on dust grain surfaces: experimental results and simulations. Astrophys. J. 801 (2), 120. doi:10.1088/0004-637x/801/2/120
Hehre, W. J., Ditchfield, R., and Pople, J. A. (1972). Self-consistent molecular orbital methods. XII. Further extensions of Gaussian-type basis sets for use in molecular orbital studies of organic molecules. J. Chem. Phys. 56, 2257–2261. doi:10.1063/1.1677527
Hellweg, A., Hättig, C., Höfener, S., and Klopper, W. (2007). Optimized accurate auxiliary basis sets for RI-MP2 and RI-CC2 calculations for the atoms Rb to Rn. Theor. Chem. Acc. 117 (4), 587–597. doi:10.1007/s00214-007-0250-5
Hickson, K. M., Loison, J. C., Nuñez-Reyes, D., and Méreau, R. (2016). Quantum tunneling enhancement of the C + H2O and C + D2O reactions at low temperature. J. Phys. Chem. Lett. 7 (18), 3641–3646. doi:10.1021/acs.jpclett.6b01637
Ioppolo, S., van Boheemen, Y., Cuppen, H. M., van Dishoeck, E. F., and Linnartz, H. (2011). Surface formation of CO2 ice at low temperatures. Mon. Notices Royal Astron. Soc. 413 (3), 2281–2287. doi:10.1111/j.1365-2966.2011.18306.x
Karssemeijer, L. J., Pedersen, A., Jónsson, H., and Cuppen, H. M. (2012). Long-timescale simulations of diffusion in molecular solids. Phys. Chem. Chem. Phys. 14 (31), 10844–10852. doi:10.1039/c2cp41634d
Kimmel, G. A., Dohnálek, Z., Stevenson, K. P., Smith, R. S., and Kay, B. D. (2001). Control of amorphous solid water morphology using molecular beams. II. Ballistic deposition simulations. J. Chem. Phys. 114 (12), 5295–5303. doi:10.1063/1.1350581
Koch, D. M., Toubin, C., Peslherbe, G. H., and Hynes, J. T. (2008). A theoretical study of the formation of the aminoacetonitrile precursor of glycine on icy grain mantles in the interstellar medium. J. Phys. Chem. C 112 (8), 2972–2980. doi:10.1021/jp076221+
Koch, D. M., Toubin, C., Xu, S., Peslherbe, G. H., and Hynes, J. T. (2007). Concerted proton-transfer mechanism and solvation effects in the HNC/HCN isomerization on the surface of icy grain mantles in the interstellar medium. J. Phys. Chem. C 111 (41), 15026–15033. doi:10.1021/jp076220h
Liakos, D. G., Sparta, M., Kesharwani, M. K., Martin, J. M., and Neese, F. (2015). Exploring the accuracy limits of local pair natural orbital coupled-cluster theory. J. Chem. Theory Comput. 11 (4), 1525–1539. doi:10.1021/ct501129s
McElroy, D., Walsh, C., Markwick, A. J., Cordiner, M. A., Smith, K., and Millar, T. J. (2013). The UMIST database for astrochemistry 2012. Astron. Astrophys. 550, 13. doi:10.1051/0004-6361/201220465
Michoulier, E., Noble, J. A., Simon, A., Mascetti, J., and Toubin, C. (2018). Adsorption of PAHs on interstellar ice viewed by classical molecular dynamics. Phys. Chem. Chem. Phys. 20 (13), 8753–8764. doi:10.1039/c8cp00593a
Michoulier, E., Toubin, C., Simon, A., Mascetti, J., Aupetit, C., and Noble, J. A. (2020). Perturbation of the surface of amorphous solid water by the adsorption of polycyclic aromatic hydrocarbons. J. Phys. Chem. C 124 (5), 2994–3001. doi:10.1021/acs.jpcc.9b09499
Minissale, M., Congiu, E., and Dulieu, F. (2016). Direct measurement of desorption and diffusion energies of O and N atoms physisorbed on amorphous surfaces. Astron. Astrophys. 585, A146. doi:10.1051/0004-6361/201526702
Minissale, M., Loison, J-C., Baouche, S., Chaabouni, H., Congiu, E., and Dulieu, F. (2015). Solid-state formation of CO2 via the H2CO + O reaction. Astron. Astrophys. 577, A2. doi:10.1051/0004-6361/201424342
Miyazaki, A., Watanabe, N., Sameera, W. M. C., Nakai, Y., Tsuge, M., Hama, T., et al. (2020). Photostimulated desorption of OH radicals from amorphous solid water: evidence for the interaction of visible light with an OH-ice complex. Phys. Rev. A 102 (5). 052822. 10.1103/PhysRevA.102.052822.
Molpeceres, G., Zaverkin, V., and Kästner, J. (2020). Neural-network assisted study of nitrogen atom dynamics on amorphous solid water–I. adsorption and desorption. Mon. Notices Royal Astron. Soc. 499 (1), 1373–1384. doi:10.1093/mnras/staa2891
Neese, F., and Valeev, E. F. (2011). Revisiting the atomic natural orbital approach for basis sets: robust systematic basis sets for explicitly correlated and conventional correlated ab initio methods? J. Chem. Theory Comput. 7 (1), 33–43. doi:10.1021/ct100396y
Neese, F., Wennmohs, F., Becker, U., and Riplinger, C. (2020). The ORCA quantum chemistry program package. J. Chem. Phys. 152 (22), 224108. doi:10.1063/5.0004608
Neese, F. (2018). Software update: the ORCA program system, version 4.0. WIREs Comput. Mol. Sci. 8 (1), e1327. doi:10.1002/wcms.1327
Noble, J. A., Congiu, E., Dulieu, F., and Fraser, H. J. (2012). Thermal desorption characteristics of CO, O2, and CO2 on non-porous water, crystalline water and silicate surfaces at sub-monolayer and multilayer coverages. Mon. Notices Royal Astron. Soc. 421, 768–779. doi:10.1111/j.1365-2966.2011.20351.x
Noble, J. A., Martin, C., Fraser, H. J., Roubin, P., and Coussan, S. (2014). Unveiling the surface structure of amorphous solid water via selective infrared irradiation of OH stretching modes. J. Phys. Chem. Lett. 5 (5), 826–829. doi:10.1021/jz5000066
Penteado, E. M., Walsh, C., and Cuppen, H. M. (2017). Sensitivity analysis of grain surface chemistry to binding energies of ice species. Astrophys. J. 844 (1), 71. doi:10.3847/1538-4357/aa78f9
Peters, P. S., Duflot, D., Wiesenfeld, L., and Toubin, C. (2013). The H + CO ⇌ HCO reaction studied by ab initio benchmark calculations. J. Chem. Phys. 139 (16), 164310. doi:10.1063/1.4826171
Pirim, C., and Krim, L. (2011). An FTIR study on the catalytic effect of water molecules on the reaction of CO successive hydrogenation at 3K. Chem. Phys. 380 (1–3), 67–76. doi:10.1016/j.chemphys.2010.12.008
Pontoppidan, K. M., Boogert, A. C. A., Fraser, H. J., van Dishoeck, E. F., Blake, G. A., Lahuis, F., et al. (2008). The c2d Spitzer Spectroscopic survey of ices around low‐mass young stellar objects. II. CO2. Astrophys. J. 678 (2), 1005. doi:10.1086/533431
Rimola, A., Taquet, V., Ugliengo, P., Balucani, N., and Ceccarelli, C. (2014). Combined quantum chemical and modeling study of CO hydrogenation on water ice. Astron. Astrophys. 572, A70. doi:10.1051/0004-6361/201424046
Rimola, A., and Ugliengo, P. (2009). The role of defective silica surfaces in exogenous delivery of prebiotic compounds: clues from first principles calculations. Phys. Chem. Chem. Phys. 11 (14), 2497–2506. doi:10.1039/b820577a
Ruaud, M., Wakelam, V., and Hersant, F. (2016). Gas and grain chemical composition in cold cores as predicted by the Nautilus three-phase model. Mon. Notice Royal Astron. Soc. 459 (4), 3756–3767. doi:10.1093/mnras/stw887
Sameera, W. M. C., and Maseras, F. (2018). Expanding the range of force fields available for ONIOM calculations: the SICTWO interface. J. Chem. Inf. Model. 58 (9), 1828–1835. doi:10.1021/acs.jcim.8b00332
Sameera, W. M. C., Senevirathne, B., Andersson, S., Al-lbadi, M., Hidaka, H., Kouchi, A., et al. (2021). CH3O radical binding on hexagonal water ice and amorphous solid water. J. Phys. Chem. A 125 (1), 387–393. doi:10.1021/acs.jpca.0c09111
Sameera, W. M. C., Senevirathne, B., Andersson, S., Maseras, F., and Nyman, G. (2017). ONIOM(QM:AMOEBA09) study on binding energies and binding preference of OH, HCO, and CH3 radicals on hexagonal water ice (Ih). J. Phys. Chem. C 121 (28), 15223–15232. doi:10.1021/acs.jpcc.7b04105
Sandford, S. A., and Allamandola, L. J. (1988). The condensation and vaporization behavior of H2O: CO ices and implications for interstellar grains and cometary activity. Icarus 76 (2), 201–224. doi:10.1016/0019-1035(88)90069-3
Schlegel, H. B., and McDouall, J. J. W. (1991). “Do you have SCF stability and convergence problems? Molecular structure and reactivity,” in Computational advances in organic chemistry. Editors C. Ögretir, and I. G. Csizmadia (Dordrecht: NATO ASI Series), 167–185.
Shimonishi, T., Nakatani, N., Furuya, K., and Hama, T. (2018). Adsorption energies of carbon, nitrogen, and oxygen atoms on the low-temperature amorphous water ice: a systematic estimation from quantum chemistry calculations. Astrophys. J. 855 (1), 27. doi:10.3847/1538-4357/aaaa6a
Stewart, J. J. P. (2007). Optimization of parameters for semiempirical methods V: modification of NDDO approximations and application to 70 elements. J. Mol. Model. 13 (12), 1173–1213. doi:10.1007/s00894-007-0233-4
Taquet, V., Peters, P. S., Kahane, C., Ceccarelli, C., López-Sepulcre, A., Toubin, C., et al. (2013). Water ice deuteration: a tracer of the chemical history of protostars. Astron. Astrophys. 550, A127. 10.1051/0004-6361/201220084
Throssell, K., and Frisch, M. J. (Forthcoming 2018). Evaluation and Improvement of Semi-empirical methods I: PM7R8: a variant of PM7 with numerically stable hydrogen bonding corrections. doi:10.14418/wes01.3.76
Throssell, K. (2017). Evaluating and approximate, improving LCAO-MO theory with restored overlap and bond order bond energy corrections. MS dissertation. Middletown (Connecticut): Wesleyan University.
Ulbricht, H., Zacharia, R., Cindir, N., and Hertel, T. (2006). Thermal desorption of gases and solvents from graphite and carbon nanotube surfaces. Carbon 44 (14), 2931–2942. doi:10.1016/j.carbon.2006.05.040
van Dishoeck, E. F. (1998). What can ISO tell us about gas-grain chemistry?. Faraday Disc. 109, 31–46. doi:10.1039/a800815i
Vega, C., and Abascal, J. L. (2011). Simulating water with rigid non-polarizable models: a general perspective. Phys. Chem. Chem. Phys. 13 (44), 19663–19688. doi:10.1039/c1cp22168j
Wakelam, V., Herbst, E., Loison, J.-C., Smith, I. W. M., Chandrasekaran, V., Pavone, B., et al. (2012). A KInetic database for astrochemistry (KIDA). Astrophys. J., Suppl. Ser. 199 (1), 21. doi:10.1088/0067-0049/199/1/21
Wakelam, V., Loison, J.-C., Mereau, R., and Ruaud, M. (2017). Binding energies: new values and impact on the efficiency of chemical desorption. Mol. Astrophys. 6, 22–35. doi:10.1016/j.molap.2017.01.002
Weigend, F., and Ahlrichs, R. (2005). Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy. Phys. Chem. Chem. Phys. 7 (18), 3297–3305. doi:10.1039/b508541a
Weigend, F. (2008). Hartree-Fock exchange fitting basis sets for H to Rn. J. Comput. Chem. 29 (2), 167–175. doi:10.1002/jcc.20702
Weigend, F., Kattannek, M., and Ahlrichs, R. (2009). Approximated electron repulsion integrals: cholesky decomposition versus resolution of the identity methods. J. Chem. Phys. 130 (16), 164106. doi:10.1063/1.3116103
Weinhold, F., and Carpenter, J. E. (1988). “The natural bond orbital lewis structure concept for molecules, radicals, and radical ions,” in The structure of small molecules and ions. Editors R. Naaman, and Z. Vager (Boston, MA: Springer United States), 227–236.
Whittet, D. C. B., Schutte, W. A., Tielens, A., Boogert, A. C. A., de Graauw, T., Ehrenfreund, P., et al. (1996). An ISO SWS view of interstellar ices: first results. Astron. Astrophys. 315, L357–L360.
Wiberg, K. B. (1968). Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron 24 (3), 1083–1096. doi:10.1016/0040-4020(68)88057-3
Yocum, K. M., Smith, H. H., Todd, E. W., Mora, L., Gerakines, P. A., Milam, S. N., et al. (2019). Millimeter/submillimeter spectroscopic detection of desorbed ices: a new technique in laboratory astrochemistry. J. Phys. Chem. A. 123 (40), 8702–8708. doi:10.1021/acs.jpca.9b04587
Zhao, Y., and Truhlar, D. G. (2008). The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Account. 120 (1), 215–241. doi:10.1007/s00214-007-0310-x
Keywords: interstellar, ices, binding energies, theoretical, amorphous, interstellar medium
Citation: Duflot D, Toubin C and Monnerville M (2021) Theoretical Determination of Binding Energies of Small Molecules on Interstellar Ice Surfaces. Front. Astron. Space Sci. 8:645243. doi: 10.3389/fspas.2021.645243
Received: 22 December 2020; Accepted: 08 February 2021;
Published: 26 March 2021.
Edited by:
Piero Ugliengo, University of Turin, ItalyReviewed by:
Feliu Maseras, Institut Català d’Investigació Química, SpainCopyright © 2021 Duflot, Toubin and Monnerville. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Céline Toubin, Y2VsaW5lLnRvdWJpbkB1bml2LWxpbGxlLmZy; Denis Duflot, ZGVuaXMuZHVmbG90QHVuaXYtbGlsbGUuZnI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.