 Katherine D. Arias1
Katherine D. Arias1 Amadou Traoré
Amadou Traoré Félix Goyache
Félix Goyache- 1Área de Genética y Reproducción Animal, Servicio Regional de Investigación y Desarrollo Agroalimentario del Principado de Asturias (SERIDA)-Deva, Gijón, Spain
- 2Laboratoire de Biologie et Santé Animales, Institut de l’Environnement et des Recherches Agricoles (INERA), Ouagadougou, Burkina Faso
Methods: Up to 237 individuals belonging to 10 different taurine (Bos taurus), zebu (B. indicus), and taurine × zebu sanga cattle populations sampled in Benin, Burkina Faso, and Niger were typed using the BovineHD BeadChip of Illumina to ascertain the patterns and importance of heterozygosity-rich regions (HRRs) in West African cattle. To account for among-population gene flow, individuals were further classified into three groups (Qtaurine, Qzebu, and admixed) according to maximum likelihood estimates of individual ancestries () obtained using the program Admixture v1.23.
Results and discussion: The 967 HRRs identified on 27 out of 29 bovine chromosomes in 231 cattle individuals were further summarized into 103 HRR islands, covering 40.7 Mb of the bovine genome. Only HRR islands identified in at least 10% of the animals typed or 10% of the individuals classified according to cattle type or admixture class were considered relevant to characterize the West African cattle genomic background. Only one and three HRR islands were considered taurine- and zebu-specific, respectively. Most (14) relevant HRR islands identified were present in cattle individuals despite the cattle type or admixture class into which they were classified (waHRR), suggesting that HRR can give advantages for adaptation to harsh environments. A total of 202 potential candidate genes were identified on the 14 waHRR islands. Most of them belonged to gene families coding zinc finger, protocadherin, adhesion G protein-coupled receptors, solute carrier, and arachidonate lipoxygenase proteins, involved in immune response, with a putative role in adaptation. Furthermore, waHRRs were also enriched on 18 and 11 different genes coding olfactory receptors and pregnancy-associated glycoproteins, respectively, giving additional support to the suggested importance of the HRR islands identified for the adaptive ability of West African cattle. Our research identified new genomic areas that can be targeted for further research in cattle adaptive ability.
Introduction
The seminal work by Williams et al. (2016) introduced the concept of heterozygosity-rich regions (HRRs), also known as runs of heterozygosity (ROHet), as short genomic areas in which polymorphic loci clustered non-randomly, probably due to balancing selection. Williams et al. (2016) illustrated the importance of HRR for conservation purposes, in the highly endangered and homozygous Chillingham cattle. Following this goal, HRR length has been recently shown as a promising parameter to assess increases in molecular inbreeding (Arias et al., 2023). However, the interest in HRR exceeds conservation purposes, and they have been approached in non-endangered cattle populations (Ferenčaković et al., 2017; Peripolli et al., 2020; Biscarini et al., 2020; Mulim et al., 2022). Further analyses have been carried out in other livestock species such as horses (dos Santos et al., 2021), sheep (Selli et al., 2021; Tsartsianidou et al., 2021), pigs (Chen et al., 2022; Ruan et al., 2022; Bordonaro et al., 2023), and goat (Li et al., 2022; Chessari et al., 2024).
Despite the increasing interest in characterizing HRRs in livestock species, their importance is far from being correctly ascertained. In this initial moment of the study of HRR, most works focused on a) the comparison of the distribution patterns between HRR and the well-described runs of homozygosity (Biscarini et al., 2020; Selli et al., 2021; Chen et al., 2022; Li et al., 2022); b) the ability of different methodologies (sliding windows or consecutive approach) to capture HRR, with the advantage for the consecutive approach (Marras et al., 2018; Biscarini et al., 2020; dos Santos et al., 2021); and c) the importance of SNP array density to identify HRR: the lower the density of the panel, the lower the number and the bigger the length in identifying HRR (Mulim et al., 2022).
The selective advantage of heterozygous genotypes showing overdominance causes balancing selection. However, the proportion of loci in which polymorphism is conserved by heterozygote advantage is predictably low (Samuels et al., 2016). This fits well with HRRs as major representatives of the variability in the genome. Across species, HRRs are smaller and less frequent than ROH in the genome (Biscarini et al., 2020; Tsartsianidou et al., 2021; Chen et al., 2022; Arias et al., 2023). Furthermore, a heterozygous advantage is expected to have important adaptive functions (Hedrick, 2012) and to be informative on the evolutionary history of populations (Samuels et al., 2016) due to the potential beneficial effects of HRRs (Williams et al., 2016; Biscarini et al., 2020; Mulim et al., 2022; Chessari et al., 2024).
A major aspect to be ascertained is to which extent HRRs are shared among populations within species. This may be related to the fact that lower-density panels result in fewer identified HRRs, which tend to be of greater length. Williams et al. (2016) found that the HRR patterns identified in the endangered Chillingham cattle were not observable in other cattle breeds (Holstein-Friesian, Chianina, and Romagnola), and therefore, HRRs were suggested to be particular for each population. Mulim et al. (2022), analyzing 16 different worldwide cattle populations, also found that the distribution and patterns of HRRs were specific to each population. This scenario has been described in other livestock species as well (Selli et al., 2021; Ruan et al., 2022; Bordonaro et al., 2023). Therefore, HRRs could reflect the effect of divergent selection and inform on the structure and breeding history of livestock populations.
West Africa is a geographical region including diverse environmental areas in which different cattle types are bred. In the arid Sahel area, the trypanosusceptible humped zebu (Bos indicus) cattle are managed due to their ability to adapt to dry environments with high temperatures, their resistance to tick infestation, and their large body size compared to taurine cattle (Mattioli et al., 2000; Mwai et al., 2015). In turn, trypanotolerant small-sized taurine (B. taurus) cattle, resulting from a unique process of natural adaptation to animal diseases of biogeographic zones south of the Sahel, namely, trypanosomiasis, are managed in the humid-hot Guinean environmental area of West Africa (Mattioli et al., 2000; Mwai et al., 2015). Both cattle types have very different origins and history (Chen et al., 2010; Pérez-Pardal et al., 2010, Pérez-Pardal et al., 2018) and large morphological differences (Traoré et al., 2015, Traoré et al., 2016), and their differences in genomic features have been studied before (Goyache et al., 2021, Goyache et al., 2022). Furthermore, in a breeding scenario in which unsupervised mating is the rule, admixture between taurine and zebu cattle is frequently observed at different degrees (Álvarez et al., 2014; Traoré et al., 2015; Goyache et al., 2021). This admixture process, carried out for centuries and even millennia, led to the development of the so-called sanga cattle, stable taurine × zebu admixed cattle populations, with intermediate performance (Berthier et al., 2015; Mwai et al., 2015).
This research aims at the identification of HRR islands in a sample of West African taurine, sanga, and zebu cattle, describing their distribution patterns across the bovine genome and performing enrichment and functional annotation analyses to identify sets of candidate genes that can give new insights on the importance of HRRs in the species.
Materials and methods
Samples, genotypes, and quality control
A total of 237 cattle genotypes previously analyzed by ( Goyache et al, 2021, Goyache et al, 2022) were available. A complete description of the dataset is given in Supplementary Table S1 and summarized in Table 1. The taurine subset (106 samples) included 33 Lagunaire cattle sampled in Benin (province of Mono), 48 N’Dama cattle sampled in the Comoé Province of Burkina Faso (N’DamaBF), and 25 N’Dama cattle bred in the province of Bas-Congo of Congo (N’DamaCo) although they originated in Guinea (Álvarez et al., 2014). The zebu subset (89 samples) included 21 zebu Bororo cattle sampled in the provinces of Tahoua and Maradi of Niger, 12 zebu Peul sampled in the Alibori Province of Benin (ZPeulBe), and 56 zebu Peul cattle sampled in the Dori Province of Burkina Faso (ZPeulBF). The sanga dataset included a total of 42 samples consisting of 28 Borgou cattle of Benin; 6 Kuri cattle of the Niger shores of Lake Chad; 2 Lobi cattle, the Burkina Faso representative of the Baoulé breed; and 6 Lagune × zebu crosses with different degrees of zebu admixture obtained in the Zou Province of Benin. The morphological differences and breeding scenarios of the sampled populations have been previously described (Álvarez et al., 2014; Traoré et al., 2015, Traoré et al., 2016; Grema et al., 2017; Moussa et al., 2017).
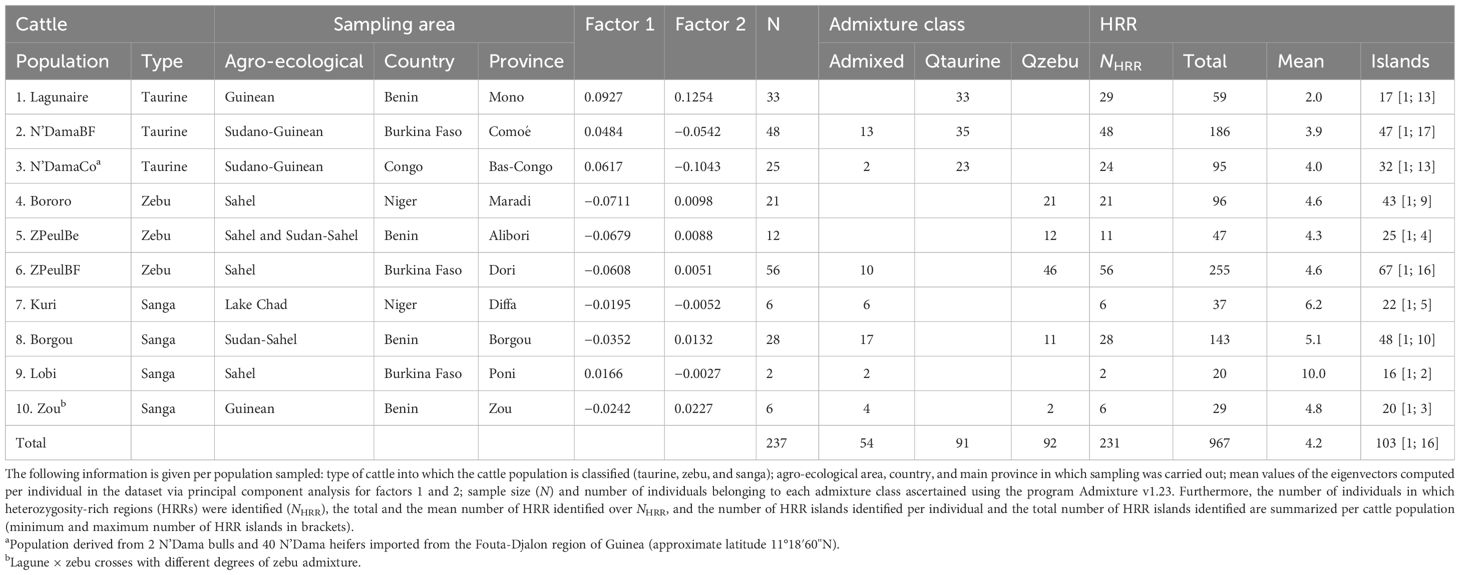
Table 1. Description of sampling.
Genotypes were obtained using the BovineHD BeadChip of Illumina (Illumina Inc., San Diego, CA, USA; 777,962 SNPs) following standard protocols. The software GenomeStudio (Illumina Inc., San Diego, CA) was used to generate standard.ped and.map files. The program PLINK v1.9 (Chang et al., 2015) was used to perform sample and marker-based quality control measures. A GenCall score cutoff of 0.15 and an average sample call rate of 99% were fitted as thresholds. All unmapped SNPs, those mapping to sexual chromosomes, and SNPs with a genotyping rate lower than 90% were removed. Although the genotypes used in (Goyache et al, 2021, Goyache et al, 2022) were edited using a minor allele frequency of 0.05 and departures from Hardy–Weinberg proportions for p ≤ 0.001 as additional quality thresholds, in the current research, SNP data were edited following the recommendations by Arias et al. (2022) for populations in which pedigrees are not available, fitting FIS >0.2 as a threshold to avoid the presence of null and partially null alleles due to technical issues. Compared with classical editing strategies using minor allele frequency and deviation from Hardy–Weinberg proportions, this strategy has been proven to be useful in removing genotyping errors while keeping putatively informative loci and dataset size (Arias et al., 2022, Arias et al., 2024). Therefore, unlike Goyache et al. (2021, 2022), who used 543,595 SNPs, a total of 714,299 SNPs located on the 29 bovine autosomes passed the quality control measures and were used for further analyses.
Population structuring analyses
The program PLINK v1.9 (Chang et al., 2015) was used to carry out principal component analysis (PCA). Eigenvectors computed for each individual were used to construct dispersion plots using the library ggplot2 version 3.5.1 of R version 4.1.2.
To ascertain the degree of admixture of the genotypes under study, clustering analysis was carried out using the program Admixture v1.23 (Alexander et al., 2009; Alexander and Lange, 2011) forcing K = 2, with K representing the number of clusters given in the dataset. The program Admixture calculates maximum likelihood estimates of individual ancestries () based on the data provided by multiple loci. Forcing K = 2 assumes that data include individuals belonging to two parental populations (here zebu and taurine cattle) and others admixed. Individuals belonging to parental populations were defined as those with ranging from 0 to 0.1 (Qzebu admixture class) or from 0.8 to 1.0 (Qtaurine admixture class), while admixed (admixed class) individuals were those with ranging from 0.1 to 0.8. Individual values were used to construct a boxplot by cattle population using the library ggplot2 version 3.5.1 of R version 4.1.2.
HRR identification and selection of HRR islands
Identification of HRRs in each individual was performed using the consecutive runs approach implemented in the package detectRuns version 0.9.6 of R version 4.1.2 (Biscarini et al., 2019). In consistency with Biscarini et al. (2020), parameters for the detection of HRRs were fitted as follows: i) 15 as the minimum number of consecutive SNPs included in an HRR, ii) 250 kb as the minimum length of an HRR, iii) 1,000 kb as the maximum gap between consecutive homozygous SNPs, iv) 2 as the maximum number of SNPs with a missing genotype, and v) 3 as the maximum number of homozygous genotypes allowed in an HRR.
Using the intersectBed function of the BedTools version 2.30.0 software (Quinlan and Hall, 2010), HRRs identified at the individual level were summarized into HRR islands. Similar to Mulim et al. (2022), only HRR islands being present in at least 10% of the animals typed or 10% of the individuals belonging to any of the classes fitted were considered relevant for the characterization of the genomic background of the cattle studied. More precisely, a) those HRRs identified in at least 23 individuals belonging to at least one population within both the taurine and the zebu cattle types and included into both the Qtaurine and the Qzebu admixture classes were considered relevant at the West African cattle level (waHRR); b) those HRRs identified in at least 10 individuals belonging to at least two different cattle populations within the taurine cattle type or in at least one taurine and one sanga cattle populations and not identified in the Qzebu admixture class were considered relevant at the taurine cattle level (tHRR); and c) those HRRs identified in at least nine individuals belonging to at least two different cattle populations within the zebu cattle type or in at least one zebu and one sanga cattle populations and not identified in the Qtaurine admixture class were considered relevant at the zebu cattle level (zHRR).
The distribution of the HRR islands identified was illustrated using the RIdeogram package version 0.2.2 of R version 4.1.2 (Hao et al., 2020).
Enrichment and functional annotation analyses
The waHRR, tHRR, and zHRR islands selected were subject to enrichment analyses using the BioMart tool (Kinsella et al., 2011). Protein-coding genes found within these HRR islands were retrieved from the Ensembl Genes 111 database based on the bovine UMD 3.1 reference genome.
Results
Population structure analyses
Figure 1 illustrates the between-populations relationships ascertained via PCA. On the X-axis, factor 1 (explaining 20.6% of the total variance) separates West African taurine and zebu cattle breeds, whereas factor 2 (on the Y-axis; 8.6% of the variance) separates the taurine Lagunaire cattle from the two N’Dama populations analyzed. Sanga cattle tended to position closer to the zebu cattle breeds.
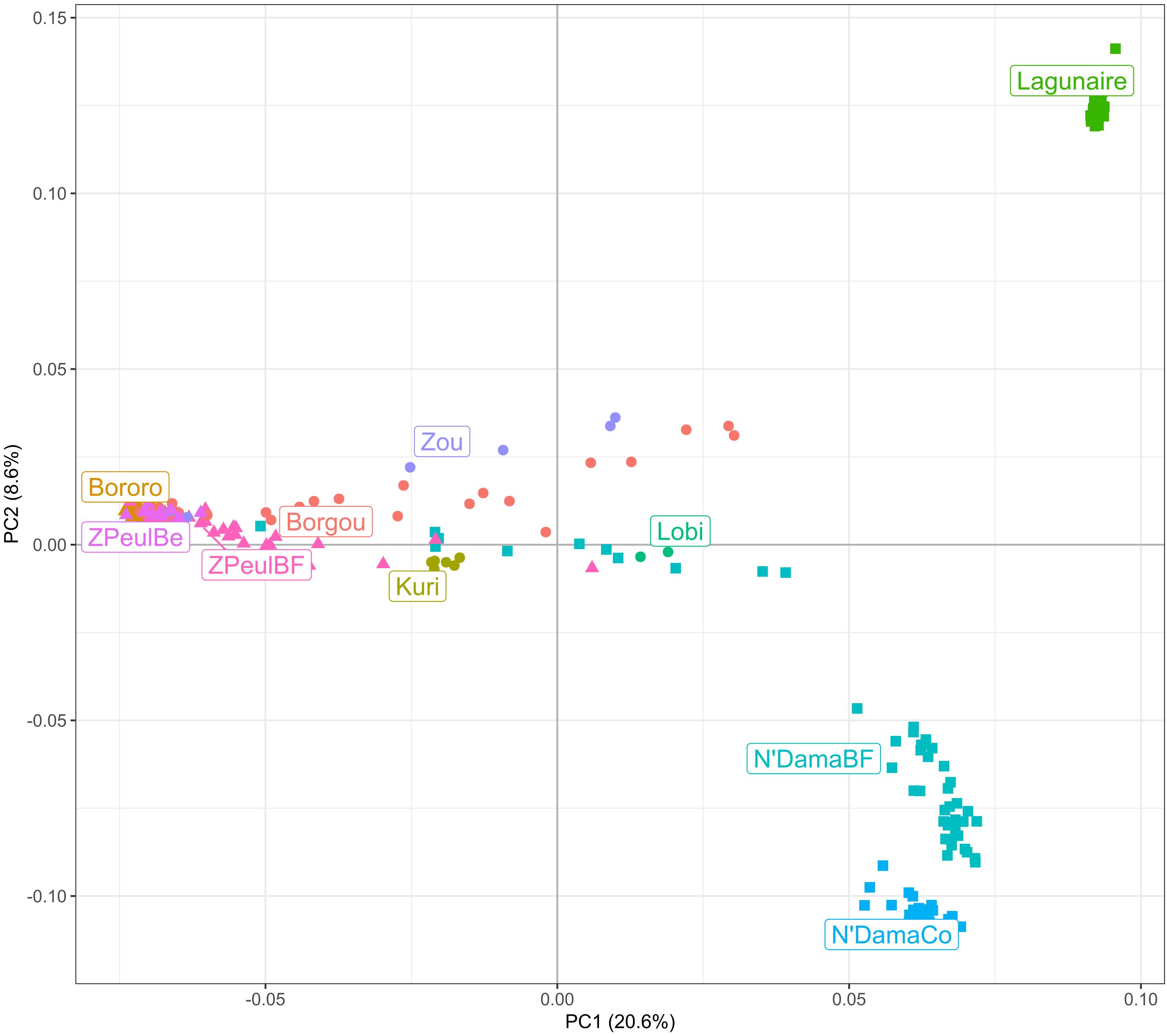
Figure 1. Dispersion plot constructed by cattle breed using the eigenvalues computed for the two first factors identified via PCA (see Supplementary Table S1; Table 1). Mean values for factors 1 (on the X-axis) and 2 (on the Y-axis) explained 20.6% and 8.6% of the total variance, respectively. Zebu cattle breeds are in triangles, sanga cattle breeds in circles, and taurine cattle breeds in squares.
Figure 2 illustrates the dispersion, within cattle populations, of the admixture coefficients estimated using the program Admixture v1.23 (see Table 1; Supplementary Table S1). In the extremes of the distribution, both the taurine Lagunaire and the zebu Bororo populations appeared as genetically uniform. Most zebu individuals had values ranging from 0 to 0.1. Although the N’DamaBF individuals showed large admixture variation (with a significant number of outliers), most N’Dama individuals had values between 0.8 and 0.9. Sanga genotypes tended to be nearer to the zebu individuals, with a non-negligible number of Borgou and Zou genotypes having values lower than 0.1. Individuals with values ≤0.1 were assigned to the Qzebu admixture class (92 individuals), those with values ≥0.8 were assigned to the Qtaurine admixture class (91 individuals), and those with ranging from 0.1 to 0.8 were classified as “admixed” (54 individuals).
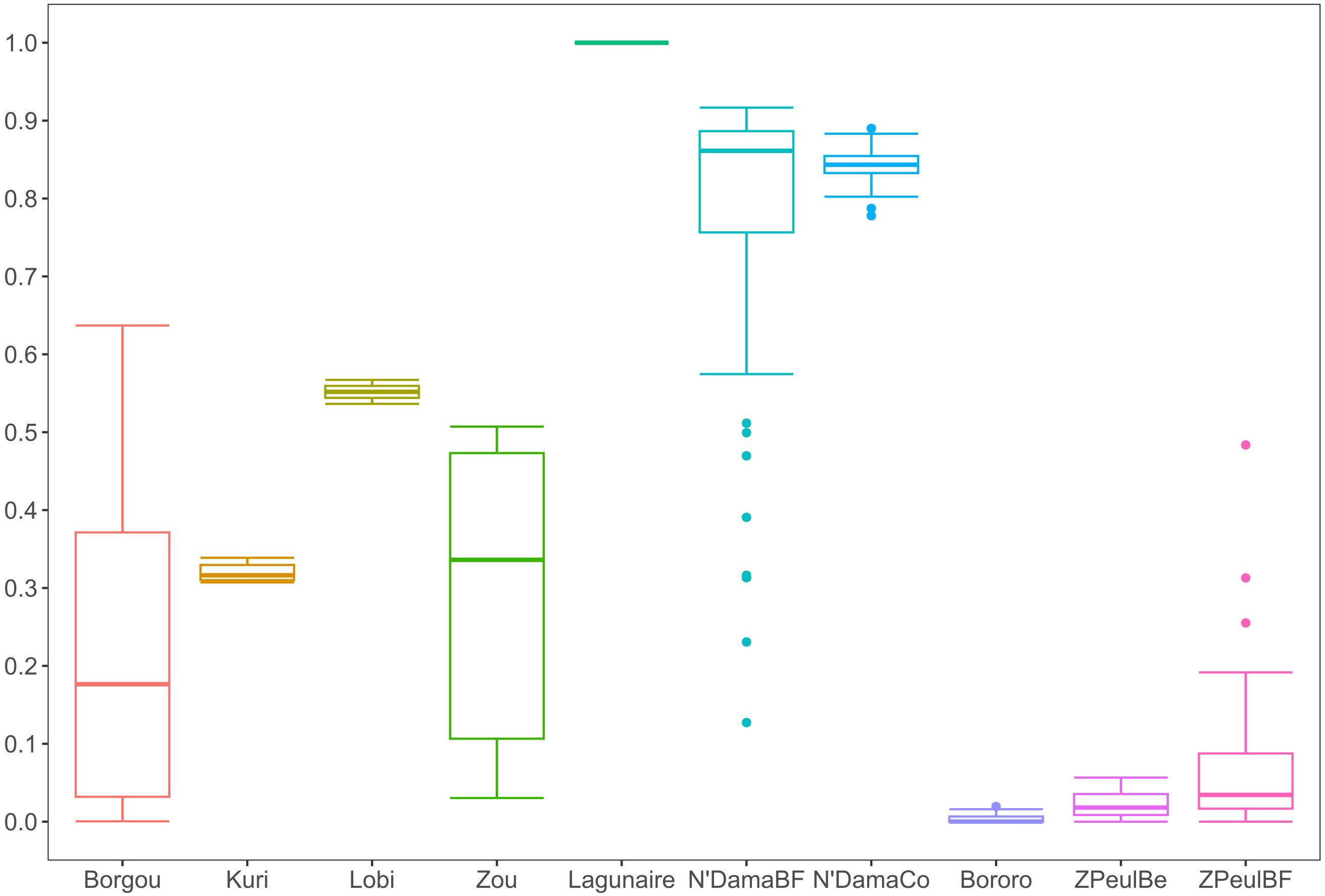
Figure 2. Boxplot illustrating the within-breed variation in individual admixture coefficients, The boxes represent the ranges between the interquartile ranges (between the second and the third quartiles), the lines within the boxes indicate the median values, and the whiskers illustrate the non-outlier lower and upper bounds ( ± 1.5-fold of the interquartile range). Circles illustrate the outlier values.
Figure 3 illustrates the consistency between the classification of cattle into cattle types (sanga, taurine, and zebu) and admixture classes (admixed, Qtaurine, and Qzebu). All cattle type classes had some degree of admixture. Up to 70% (29) of the individuals classified as sanga cattle belonged to the admixed class, whereas the other sanga cattle (13) were included in the Qzebu admixture class. Up to 86% (91) of the taurine individuals were classified into the Qtaurine admixture class, whereas the others (15) were considered admixed. Up to 89% (79) of the zebu individuals were classified into the Qzebu admixture class, whereas the remaining 10 zebu individuals were considered admixed.
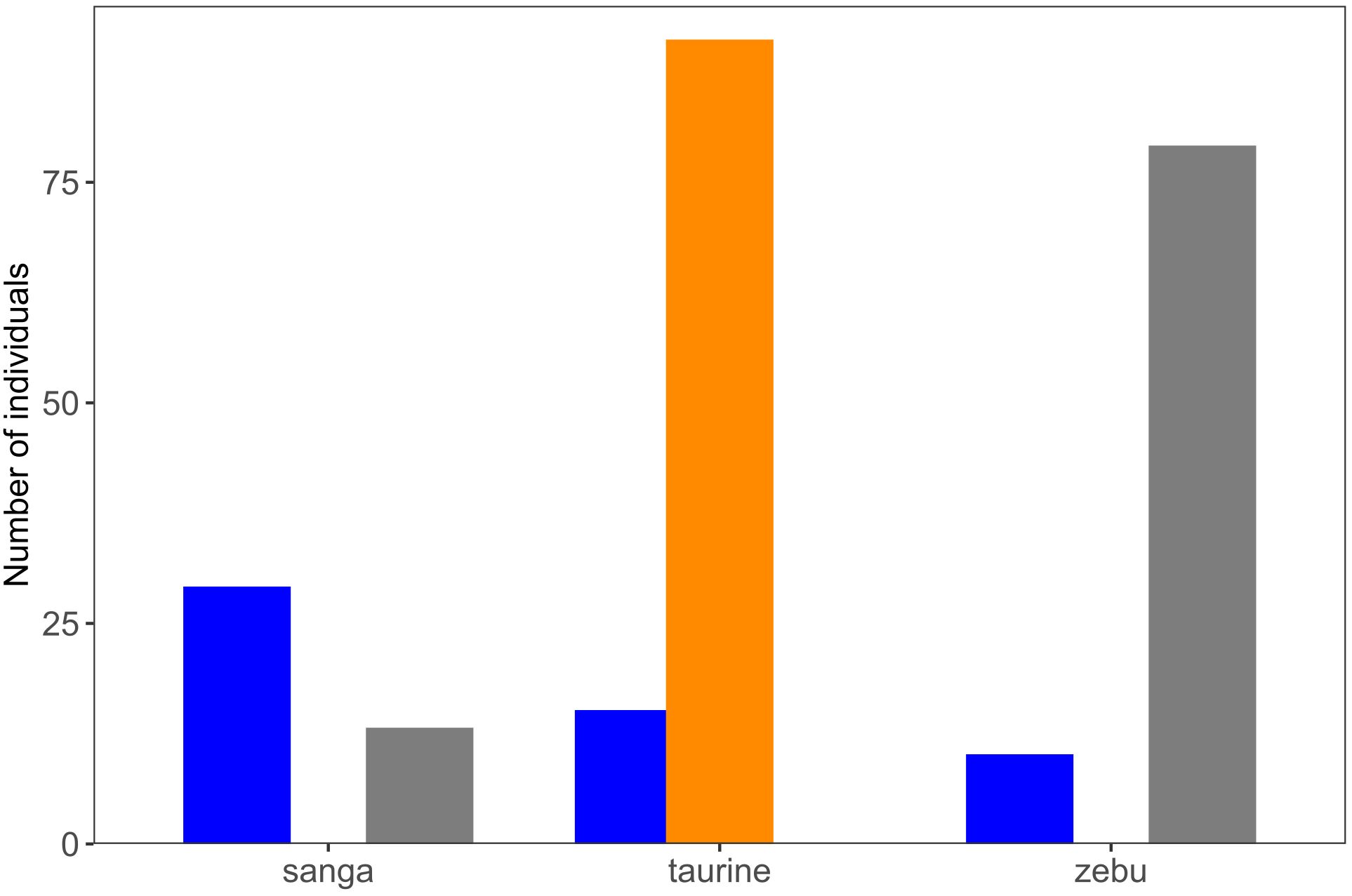
Figure 3. Number of individuals classified as sanga (42), taurine (106), and zebu (89) cattle classified into three different admixture classes: admixed (in blue), Qtaurine (in orange), and Qzebu (in gray). The total number of individuals classified as admixed, Qtaurine, and Qzebu are 54, 91, and 92, respectively.
Identification of HRR and HRR islands
A total of 967 HRRs were identified on 27 out of 29 bovine chromosomes (BTA) in 231 of the cattle analyzed (Table 1; Supplementary Table S2). The number of HRRs identified per individual varied from 1 to 12 (4.2 HRRs per individual, on average). No HRRs were identified in four Lagunaire, one N’DamaCo, and one ZPeulBe cattle. No HRRs were identified in either BTA25 or BTA28. Up to 34% (330) of the HRRs were identified in three bovine chromosomes only: BTA5 (113), BTA7 (97), and BTA18 (120). Up to 35% (340) and 41% (398) of the HRRs were identified in taurine and zebu cattle individuals, respectively. The individuals assigned to the Qzebu admixture class carried the majority of the HRRs identified (412; 43%), whereas those assigned to the Qtaurine class carried 27% (260) only. Most HRRs identified (568; 59%) were smaller than 0.5 Mb, with 189 HRRs (20%) being bigger than 1 Mb (maximum length of 1.57 Mb; Supplementary Table S2).
Using BedTools v2.30.0, the 967 HRRs identified at the individual level were summarized into 103 HRR islands, covering 40.7 Mb of the bovine genome. Supplementary Table S3 gives a full description of the HRR islands identified together with their frequency per cattle breed, cattle type, and admixture class. The location of the HRR islands identified across bovine autosomes is illustrated in Figure 4.
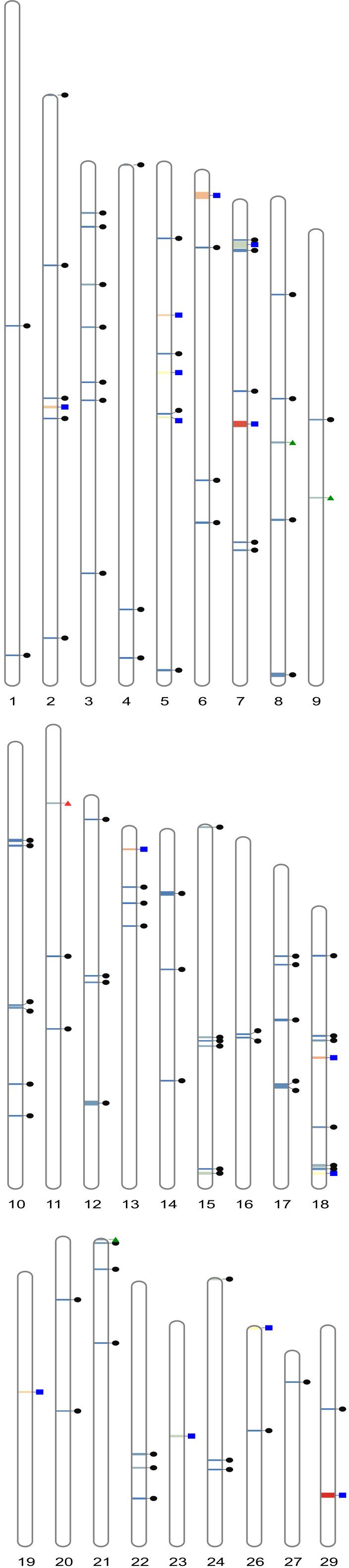
Figure 4. Ideogram illustrating, per bovine autosome, the distribution of the HRR islands identified. HRR islands identified in one individual, or one cattle population only, are in black circles, taurine-specific and zebu-specific HRR islands are in red and green triangles, respectively, and waHRR islands are in blue squares.
Four HRR islands (HRR26 on BTA6, HRR31 and HRR34 on BTA7, and HRR103 on BTA29) were bigger than 1 Mb and the other 13 HRR islands had lengths between 0.5 and 1 Mb (Figure 4; Supplementary Table S3). Thirty out of the 103 HRR islands (29%) were identified in one individual only, 18 of them of Qzebu ancestry. Six additional HRRs (HRR5, HRR16, HRR27, HRR46, HRR49, and HRR73), identified in two or four individuals, were present in one cattle population only.
Only one HRR island (HRR50 on BTA11), identified in 13 Lagunaire and one sanga Zou individuals sampled in Benin, was classified as tHRR. Three HRR islands (HRR39 on BTA8, HRR43 on BTA9, and HRR88 on BTA21), identified in individuals (from 10 to 18) belonging to at least two different zebu and two different sanga cattle breeds, were classified as zHRR. tHRR and zHRR islands were short, with lengths from 258.6 kb (HRR43) to 382.3 kb (HRR39).
Up to 14 HRR islands (HRR6 on BTA2; HRR20, HRR22, and HRR24 on BTA5; HRR26 on BTA6; HRR31 and HRR34 on BTA7; HRR57 on BTA13; HRR80 and HRR84 on BTA18; HRR85 on BTA19; HRR95 on BTA23; HRR99 on BTA26; HRR103 on BTA29) were considered relevant for the characterization of the West African cattle genomic background (waHRR; Supplementary Table S3). waHRRs were identified in at least six cattle breeds belonging to the three cattle types analyzed. The most frequent HRR island (HRR 103) was identified in 64 individuals belonging to the 10 cattle breeds available. Although 6 out of the 14 waHRRs identified were shorter than 0.5 Mb, waHRRs tended to be bigger [mean length of 720.1 kb (minimum = 278.8 kb; maximum = 1,568.8 kb)], with 4 waHRRs having lengths more than 1Mb.
Identification of functional candidate genes
Gene annotation enrichment analysis identified a total of 202 potential candidate genes on the 14 waHRR islands (Supplementary Table S4), 10 on the three zHRR islands, and 1 candidate gene in the only tHRR identified (Supplementary Table S5). A noticeable proportion of these potential candidate genes were not characterized yet: 51 (25%), 5 (50%), and 1 (100%) out of those identified on waHRR, zHRR, and tHRR islands, respectively.
The candidate genes located on zHRR islands were enriched on immunoglobulin-coding genes (such as IGHM) on BTA21 and the sperm acrosome-associated 1 gene (SPACA1) on BTA9. The candidate genes located on waHRR were enriched on genes related to immune and inflammatory response, mainly located on BTA7 and BT18. The set of genes involved in immune response included eight genes coding both alpha and beta protocadherin protein subfamilies (e.g., PCDHAC2 and PCDHGB4), eight genes coding zinc finger (ZNF) proteins, three genes coding adhesion G protein-coupled receptors (e.g., ADGRE3, LOC100299045, and LOC100300051), three genes coding solute carrier family proteins (SLC4A9, LOC538810, and SLC35A4), and three genes coding arachidonate lipoxygenases (ALOX15B, ALOX12B, and ALOXE3) forming a cluster of 65 kb on BTA19.
Furthermore, waHRRs were also enriched on genes coding olfactory receptors (18 different genes on BTA5 and BTA7) and pregnancy-associated glycoproteins (11 PAG family genes forming a cluster on BTA29) spanning a chromosomal area of 1 Mb (from position 38,750,566 to position 39,736,545; Supplementary Table S4).
Discussion
The current analysis adds insights to the previously described genomic structuring of the cattle population analyzed (Goyache et al., 2021, Goyache et al, 2022). The depicted scenario is consistent with a high degree of admixture between zebu and taurine cattle. Admixture is clear in sanga cattle, which tend to be genetically nearer to the zebu cattle rather than to taurine cattle, but also to the taurine N’DamaBF cattle (Figure 1). The N’Dama cattle of Burkina Faso analyzed were previously shown to have a high proportion of zebu admixture (Álvarez et al., 2014), which is reflected in the high number of outliers identified via Admixture analysis (Figure 2). Although the N’DamaBF cattle were sampled in the tsetse-challenged Mangodara area of the country, their householders belonged to the Fulani ethnic group, which is predominant in the Sahel area. This makes it more probable that they would engage in exchanges and subsequent admixture with Sahelian zebu cattle. In any case, although the N’DamaCo cattle were derived from a small number of N’Dama individuals from Guinea and have been managed separately in Congo for decades (Álvarez et al., 2014), the genomic backgrounds of the two N’Dama cattle populations analyzed are still consistent (Figure 1).
In general, the scenario analyzed reinforces the fact that the classification of African cattle into breeds tends to be arbitrary, mainly following geographical or stockbreeders’ ethnic criteria (Traoré et al., 2015), making it necessary to perform additional admixture analyses to ensure that the assignment of genomic features to a given genomic background is not biased (Álvarez et al., 2014).
HRR patterns
The identification of HRRs in the individuals typed followed the expected patterns: they were few (approximately four HRRs per individual) and small. Although various previous analyses suggested that the majority of HRRs have 500 to 1,000 kb length (Biscarini et al., 2020; dos Santos et al., 2021; Tsartsianidou et al., 2021), more than half of HRRs identified in West African cattle were smaller than 500 kb. In contrast to most previous reports, which used medium-density SNP arrays, our data were obtained using a high-density chip leading to a more correct assessment of the length of the HRRs identified (Selli et al., 2021; Mulim et al., 2022). After merging the HRRs identified into HRR islands, this pattern even increases with 83.5% of the HRR islands being smaller than 500 kb and 12.6% only ranging from 500 to 1000 kb. The merging procedure did not lead to an enlargement of the HRR bounds, suggesting that the HRRs identified across breeds and cattle types were highly stable.
In this respect, despite the strong structuring of the population analyzed, including cattle breeds of different origins and spreading areas (Pérez-Pardal et al., 2018; Goyache et al., 2021, Goyache et al, 2022), 44 HRR islands (43%) were identified in individuals belonging to both taurine and zebu cattle breeds. Furthermore, 37 HRR islands (36%) were identified in individuals classified into the Qzebu and Qtaurine admixture classes, respectively. The number of either taurine- or zebu-specific HRRs was very few, and the tHRR identified, only shared by one sanga Zou individual sampled in Benin, can more likely be considered private to the Benin’s Lagunaire cattle. Since HRR distribution patterns have been suggested to be population-specific (Selli et al., 2021; Ruan et al., 2022; Mulim et al., 2022; Bordonaro et al., 2023), the West African cattle scenario departs from expectations. Li et al. (2022) identified one HRR hotspot in goats worldwide only. In any case, our results are not biased due to HRR length. The 14 waHRR islands included the four largest (>1 Mb) identified, as well as six smaller islands (HRR20, HRR24, HRR57, HRR80, HRR85, and HRR95) below 0.5 Mb (Supplementary Table S3). However, the HRR islands identified do not coincide either with the 26 population-specific HRRs previously reported in six different European taurine and Asian zebu cattle populations (see Table 3 of Mulim et al., 2022) or with the three HRRs identified in the Maremmana semi-feral cattle breed (see Table 3 of Biscarini et al., 2020).
Considering all of the above, it cannot be excluded that the HRR island patterns identified, no matter the different origins or the different challenging environmental areas in which the individuals sampled are managed, reflect the particular history of West African cattle. Recently, it has been suggested that the distribution of the HRR islands in the goat genome might be mainly related to the breeds’ geography (Chessari et al., 2024). Furthermore, in pigs, Chen et al. (2022) suggested that HRRs shared among local Chinese populations reflect similar adaptive needs.
The present West African cattle result from a very complex history of overlapping processes of spreading and admixture. Although most African cattle carry mitochondrial DNA of taurine origin, most sires in Africa carry zebu Y-chromosomes, probably due to male-mediated introgression of B. indicus into the pre-extant B. taurus African cattle (Hanotte et al., 2000; Bradley and Magee, 2006; Álvarez et al., 2017; Pérez-Pardal et al., 2018). One can hypothesize that HRRs may be an ancient genomic legacy of the local West African taurine cattle. In such a complex historical process of formation, HRRs of taurine origin could have been integrated into the West African zebu and sanga cattle if they provided evolutionary advantages for adaptation to harsh environments. In any case, it is worth noting that, in the analyzed cattle population, the HRR distribution patterns depart from that previously reported for homozygous genomic stretches (Goyache et al., 2021): a non-negligible proportion of ancient autozygous genomic segments subject to positive selection were private to each cattle type.
Biological importance of the candidate genes and annotation clusters identified
Regardless of the livestock species analyzed, reports suggest that HRRs are highly enriched in genes related to immune function and health (Mulim et al., 2022; Bordonaro et al., 2023). However, the importance of the genes shaping immune response goes beyond the maintenance of animal health and may reflect the ability of livestock to adapt to difficult environments (Goyache et al., 2021, Goyache et al, 2022). Previous reports on cattle suggested that HRRs may play a role in local adaptation in rare breeds (Williams et al., 2016; Biscarini et al., 2020). Furthermore, a recent worldwide analysis of HRRs in goats suggested that the patterns identified would reflect both environmental interaction and anthropogenic selection (Chessari et al., 2024). This suggestion fits well with the West African cattle scenario in which the more frequent HRR islands are shared by the different cattle populations, despite their differences in origin, morphology, and harsh environments (forest hot-humid and Sahelian dry) in which they are managed. Together with gene sets primarily related to adaptation, candidate genes identified on waHRR mainly belong to gene families with pleiotropic effects involved in immunity and adaptation.
waHRRs are enriched of genes coding zinc finger proteins, protocadherins, adhesion G protein-coupled receptors, solute carrier proteins, and arachidonate lipoxygenases, involved in immune response: zinc finger proteins regulate the E3 ubiquitin ligase activity and are primarily involved in the innate immune processes due to their role in the regulation of pathogen recognition (Hatakeyama, 2017); cadherins comprise a large family of Ca2+-dependent cell–cell adhesion molecules involved in the adherens junction between epithelial cells exerting various functions related with immunoregulation such as resistance to parasites including Haemonchus contortus (Guo et al., 2016); adhesion G protein-coupled receptors are involved in the development of immune cells and are implicated in the immune regulatory functions of both innate and adaptive immunity (Lin et al., 2017); solute carrier proteins are amino acid transporters affecting T-cell fate decision, which has a central role in immune response (Ren et al., 2017); and finally, arachidonate lipoxygenases are involved in the inflammatory process and homeostasis restoration by catalyzing the oxidation of arachidonic acid into leukotrienes (Serhan et al., 2008).
However, even though immunity plays a major role in the adaptive ability of animals, the gene families described above have been reported to have functions in performance traits. For instance, genes coding zinc finger proteins, as it has been shown for other immunity-related genes (Lindholm-Perry et al., 2020), can be involved in feed efficiency, growth, and performance (Santana et al., 2014); the solute carrier family of genes plays a major role in reproduction, gestation, nutrient absorption, milk synthesis, and secretion (Lázaro et al., 2024); and cadherins are also involved in pregnancy (Cerri et al., 2012).
More importantly, genes involved in the immune response, including members of the gene families described above, have been strongly suggested to have a function in resilience to heat stress (Livernois et al., 2021; Lemal et al., 2023), therefore supporting a putative role in adaptation to the harsh environments of West Africa.
Other candidate genes identified can also be involved in cattle adaptation ability. Pregnancy-associated glycoproteins (PAGs) are encoded by a family of genes with pleiotropic effects (Barbato et al., 2022): in addition to their function in the regulation of gestation and the synthesis of growth factors at the placenta–uterine interface, these genes have a role in local and systemic immunological functions, such as immunomodulation at the maternal–fetal level by binding or sequestering peptides susceptible to recognition by the major histocompatibility complex (MHC), and they exert local immunosuppression that takes place at the beginning of pregnancy. Approximately 10% of the candidate genes identified on waHRRs encode olfactory receptors. This large family of genes has a major function in ecological adaptation (Hughes et al., 2018). In previous reports, it has been shown that copy number variation regions identified on BTA23 in taurine West African cattle are enriched in this family of genes, suggesting that they could play a role in the costly process of adaptation of these cattle to the challenging environment of West Africa (Goyache et al., 2022).
Conclusion
The current research adds to our understanding of the genomic background of West African cattle. Our results point out that HRRs are widely shared among West African cattle populations despite their different origins (B. taurus and B. indicus), degree of admixture, and the dissimilar environments in which they are managed, therefore suggesting that HRRs can give advantages for adaptation to harsh environments. Although waHRRs are mainly enriched on genes involved in immune response, their putative importance for adaptation to harsh environments including resilience to heat stress cannot be neglected. The fact that waHRRs are also enriched on genes belonging to families, such as olfactory receptors and pregnancy-associated glycoproteins, that have been suggested to be involved in adaptation, would also support the role of HRRs in the adaptive ability of West African cattle.
Data availability statement
The data analyzed in this study is subject to the following licenses/restrictions: All data generated during this study are included in this publication and its Supplementary Material. Additional datasets used and analyzed during the study are available upon reasonable request to the corresponding author. Requests to access these datasets should be directed to ZmdveWFjaGVAc2VyaWRhLm9yZw==.
Ethics statement
The requirement of ethical approval was waived by the Ethics Committee for Health Research in Burkina Faso for the studies involving animals because samples were obtained using standard veterinary procedures in full respect with animal welfare and minimizing stress. DNA was obtained from blood and hair root samples collected by veterinary practitioners with permission and in the presence of the owners. Due to conditions of sampling, permission from the Ethics Committee for Health Research in Burkina Faso (Joint Order 2004-147/MS/MESSE of 11 May 2004) was not required. The studies were conducted in accordance with the local legislation and institutional requirements.
Author contributions
KA: Conceptualization, Writing – original draft, Writing – review & editing, Formal analysis, Methodology. IF: Formal analysis, Writing – review & editing, Data curation, Resources. AT: Data curation, Resources, Writing – review & editing. FG: Writing – review & editing, Conceptualization, Funding acquisition, Project administration, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was partially supported by the grant MICINN-FEDER AGL2016-77813-R. CompGen. K.D.A. was funded by AEI-ESF, grant no. PRE2020-092905.
Conflict of interest
The authors declare that this research was conducted with no financial and nonfinancial competing interests.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fanim.2024.1468575/full#supplementary-material
Abbreviations
AC, annotation cluster; BTA, bovine chromosome; HRR, heterozygosity-rich regions; N’DamaBF, N’Dama cattle sampled in the Comoé Province of Burkina Faso; N’DamaCo, N’Dama cattle sampled in the Bas-Congo Province of Congo; Qtaurine, admixture class including individuals with admixture coefficients higher than 0.8; Qzebu, admixture class including individuals with admixture coefficients lower than 0.1; tHRR, HRR considered relevant at the taurine cattle level; waHRR, HRR considered relevant at the West African cattle level; zHRR, HRR considered relevant at the zebu cattle level; ZPeulBe, zebu Peul cattle sampled in the Alibori Province of Benin; ZPeulBF, zebu Peul cattle sampled in the Dori Province of Burkina Faso.
References
Alexander D. H., Lange K. (2011). Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinform. 12, 246. doi: 10.1186/1471-2105-12-246
Alexander D. H., Novembre J., Lange K. (2009). Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664. doi: 10.1101/gr.094052.109
Álvarez I., Pérez-Pardal L., Traoré A., Koudandé D. O., Fernández I., Soudré A., et al. (2017). Differences in genetic structure assessed using Y-chromosome and mitochondrial DNA markers do not shape the contributions to diversity in African sires. J. Anim. Breed Genet. 134, 393–404. doi: 10.1111/jbg.12278
Álvarez I., Traoré A., Fernández I., Cuervo M., Lecomte T., Soudré A., et al. (2014). Assessing introgression of Sahelian zebu genes into native Bos taurus breeds in Burkina Faso. Mol. Biol. Rep. 41, 3745–3754. doi: 10.1007/s11033-014-3239-x
Arias K. D., Álvarez I., Gutiérrez J. P., Fernández I., Menéndez J., Menéndez-Arias N. A., et al. (2022). Understanding Mendelian errors in SNP arrays data using a Gochu Asturcelta pig pedigree: genomic alterations, family size and calling errors. Sci. Rep. 12, 19686. doi: 10.1038/s41598-022-24340-0
Arias K. D., Gutiérrez J. P., Fernández I., Álvarez I., Goyache F. (2023). Approaching autozygosity in a small pedigree of Gochu Asturcelta pigs. Genet. Sel Evol. 55, 74. doi: 10.1186/s12711-023-00846-7
Arias K. D., Lee H., Bozzi R., Álvarez I., Gutiérrez J. P., Fernández I., et al. (2024). Ascertaining the genetic background of the Celtic-Iberian pig strain: a signatures of selection approach. J. Anim. Breed Genet. 141 , 96–96. doi: 10.1111/jbg.12829
Barbato O., Menchetti L., Brecchia G., Barile V. L. (2022). Using pregnancy-associated glycoproteins (PAGs) to improve reproductive management: from dairy cows to other dairy livestock. Animals 12, 2033. doi: 10.3390/ani12162033
Berthier D., Peylhard M., Dayo G. K., Flori L., Sylla S., Bolly S., et al. (2015). A comparison of phenotypic traits related to trypanotolerance in five west african cattle breeds highlights the value of shorthorn taurine breeds. PloS One 10, e0126498. doi: 10.1371/journal.pone.0126498
Biscarini F., Cozzi P., Gaspa G., Marras G. (2019). Package ‘detectRUNS’: detect runs of homozygosity and runs of heterozygosity in diploid genomes. R Package.
Biscarini F., Mastrangelo S., Catillo G., Senczuk G., Ciampolini R. (2020). Insights into genetic diversity, runs of homozygosity and heterozygosity-rich regions in maremmana semi-feral cattle using pedigree and genomic data. Animals 10. doi: 10.3390/ani10122285
Bordonaro S., Chessari G., Mastrangelo S., Senczuk G., Chessa S., Castiglioni B., et al. (2023). Genome-wide population structure, homozygosity, and heterozygosity patterns of Nero Siciliano pig in the framework of Italian and cosmopolitan breeds. Anim. Genet. 54, 591–605. doi: 10.1111/age.13344
Bradley D. G., Magee D. A. (2006). “Genetics and origins of domesticated cattle,” in Documenting domestication. New genetic and archaelogical paradigms. Eds. Zeder M. A., Bradley D. G., Emshwiller B. D., Smith B. D. (Berkeley, California, USA: University of California Press), 317–328. doi: 10.1525/9780520932425-025
Cerri R. L. A., Thompson I. M., Kim I. H., Ealy A. D., Hansen P. J., Staples C. R., et al. (2012). Effects of lactation and pregnancy on gene expression of endometrium of Holstein cows at day 17 of the estrous cycle or pregnancy. J. Dairy Sci. 95, 5657–5675. doi: 10.3168/jds.2011-5114
Chang C. C., Chow C. C., Tellier L. C. A. M., Vattikuti S., Purcell S. M., Lee J. J. (2015). Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience 4, 7. doi: 10.1186/s13742-015-0047-8
Chen S., Lin B.-Z., Baig M., Mitra B., Lopes R. J., Santos A. M., et al. (2010). Zebu cattle are an exclusive legacy of the South Asia neolithic. Mol. Biol. Evol. 27, 1–6. doi: 10.1093/molbev/msp213
Chen Z., Zhang Z., Wang Z., Zhang Z., Wang Q., Pan Y. (2022). Heterozygosity and homozygosity regions affect reproductive success and the loss of reproduction: A case study with litter traits in pigs. Comput. Struct. Biotechnol. J. 20, 4060–4071. doi: 10.1016/j.csbj.2022.07.039
Chessari G., Criscione A., Marletta D., Crepaldi P., Portolano B., Manunza A., et al. (2024). Characterization of heterozygosity-rich regions in Italian and worldwide goat breeds. Sci. Rep. 14, 3. doi: 10.1038/s41598-023-49125-x
dos Santos W. B., Schettini G. P., Fonseca M. G., Pereira G. L., Loyola-Chardulo L. A., Neto M., et al. (2021). Fine-scale estimation of inbreeding rates, runs of homozygosity and genome-wide heterozygosity levels in the Mangalarga MarChador horse breed. J. Anim. Breed Genet. 138, 161–173. doi: 10.1111/jbg.12508
Ferenčaković M., Banadinović M., Mercvajler M., Khayatzadeh N., Mészáros G., Cubric-Curik V., et al. (2017). Mapping of heterozygosity rich regions in Austrian pinzgauer cattle. Acta Agric. Slov. Supp. 5, 41–44.
Goyache F., Pérez-Pardal L., Fernández I., Traoré A., Menéndez-Arias N. A., Álvarez I. (2021). Ancient autozygous segments subject to positive selection suggest adaptive immune responses in West African cattle. Gene 803, 145899. doi: 10.1016/j.gene.2021.145899
Goyache F., Pérez-Pardal L., Fernández I., Traoré A., Menéndez-Arias N. A., Arias K. D., et al. (2022). Identification and characterization of copy number variations regions in west african taurine cattle. Animals 12. doi: 10.3390/ani12162130
Grema M., Traoré A., Issa M., Hamani M., Abdou M., Fernández I., et al. (2017). Morphological assessment of Niger Kuri cattle using multivariate methods. SA J. Sci. 47, 505. doi: 10.4314/sajas.v47i4.9
Guo Z., González J. F., Hernandez J. N., McNeilly T. N., Corripio-Miyar Y., Frew D., et al. (2016). Possible mechanisms of host resistance to Haemonchus contortus infection in sheep breeds native to the Canary Islands. Sci. Rep. 6, 26200. doi: 10.1038/srep26200
Hanotte O., Tawah C. L., Bradley D. G., Okomo M., Verjee Y., Ochieng J., et al. (2000). Geographic distribution and frequency of a taurine Bos taurus and an indicine Bos indicus Y specific allele amongst sub-saharan African cattle breeds. Mol. Ecol. 9, 387–396. doi: 10.1046/j.1365-294x.2000.00858.x
Hao Z., Lv D., Ge Y., Shi J., Weijers D., Yu G., et al. (2020). RIdeogram: drawing SVG graphics to visualize and map genome-wide data on the idiograms. PeerJ Comput. Sci. 6, e251. doi: 10.7717/peerj-cs.251
Hatakeyama S. (2017). TRIM family proteins: roles in autophagy, immunity, and carcinogenesis. Trends Biochem. Sci. 42, 297–311. doi: 10.1016/j.tibs.2017.01.002
Hedrick P. W. (2012). What is the evidence for heterozygote advantage selection? Trends Ecol. Evol. 27, 698–704. doi: 10.1016/j.tree.2012.08.012
Hughes G. M., Boston E. S. M., Finarelli J. A., Murphy W. J., Higgins D. G., Teeling E. C. (2018). The birth and death of olfactory receptor gene families in mammalian niche adaptation. Mol. Biol. Evol. 35, 1390–1406. doi: 10.1093/molbev/msy028
Kinsella R. J., Kahari A., Haider S., Zamora J., Proctor G., Spudich G., et al. (2011). Ensembl BioMarts: a hub for data retrieval across taxonomic space. Database 2011, bar030–bar030. doi: 10.1093/database/bar030
Lázaro S. F., Tonhati H., Oliveira H. R., Silva A. A., Scalez D. C. B., Nascimento A. V., et al. (2024). Genetic parameters and genome-wide association studies for mozzarella and milk production traits, lactation length, and lactation persistency in Murrah buffaloes. J. Dairy Sci. 107, 992–1021. doi: 10.3168/jds.2023-23284
Lemal P., May K., König S., Schroyen M., Gengler N. (2023). Invited review: From heat stress to disease—Immune response and candidate genes involved in cattle thermotolerance. J. Dairy Sci. 106, 4471–4488. doi: 10.3168/jds.2022-22727
Li G., Tang J., Huang J., Jiang Y., Fan Y., Wang X., et al. (2022). Genome-wide estimates of runs of homozygosity, heterozygosity, and genetic load in two chinese indigenous goat breeds. Front. Genet. 13. doi: 10.3389/fgene.2022.774196
Lin H.-H., Hsiao C.-C., Pabst C., Hébert J., Schöneberg T., Hamann J. (2017). Adhesion GPCRs in regulating immune responses and inflammation. Adv. Immunol. 136, 163–201. doi: 10.1016/bs.ai.2017.05.005
Lindholm-Perry A. K., Freetly H. C., Oliver W. T., Rempel L. A., Keel B. N. (2020). Genes associated with body weight gain and feed intake identified by meta-analysis of the mesenteric fat from crossbred beef steers. PloS One 15, e0227154. doi: 10.1371/journal.pone.0227154
Livernois A. M., Mallard B. A., Cartwright S. L., Cánovas A. (2021). Heat stress and immune response phenotype affect DNA methylation in blood mononuclear cells from Holstein dairy cows. Sci. Rep. 11, 11371. doi: 10.1038/s41598-021-89951-5
Marras G., Wood B. J., Makanjuola B., Malchiodi F., Peeters K., van As P., et al. (2018). “Characterization of runs of homozygosity and heterozygosity-rich regions in a commercial Turkey (Meleagris gallopavo) population,” in Presented at the 11th world congr genet appl to livest prod. 2018(Auckland, New Zealand).
Mattioli R. C., Pandey V. S., Murray M., Fitzpatrick J. L. (2000). Immunogenetic influences on tick resistance in African cattle with particular reference to trypanotolerant N’Dama (Bos taurus) and trypanosusceptible Gobra zebu (Bos indicus) cattle. Acta Tropica 75, 263–277. doi: 10.1016/S0001-706X(00)00063-2
Moussa M. M. A., Issa M., Traoré A., Grema M., Hamani M., Fernández I., et al. (2017). Morphological assessment of the Zebu Bororo (Wodaabé) cattle of Niger in the West African zebu framework. Arch. Anim. Breed. 60, 363–371. doi: 10.5194/aab-60-363-2017
Mulim H. A., Brito L. F., Pinto L. F. B., Ferraz J. B. S., Grigoletto L., Silva M. R., et al. (2022). Characterization of runs of homozygosity, heterozygosity-enriched regions, and population structure in cattle populations selected for different breeding goals. BMC Genomics 23, 209. doi: 10.1186/s12864-022-08384-0
Mwai O., Hanotte O., Kwon Y.-J., Cho S. (2015). African indigenous cattle: unique genetic resources in a rapidly changing world. Asian-australasian. J. Anim. Sci. 28, 911–921. doi: 10.5713/ajas.15.0002R
Pérez-Pardal L., Royo L. J., Beja-Pereira A., Curik I., Traoré A., Fernández I., et al. (2010). Y-specific microsatellites reveal an African subfamily in taurine (Bos taurus) cattle. Anim. Genet. 41, 232–241. doi: 10.1111/j.1365-2052.2009.01988.x
Pérez-Pardal L., Sánchez-Gracia A., Álvarez I., Traoré A., Ferraz J. B. S., Fernández I., et al. (2018). Legacies of domestication, trade and herder mobility shape extant male zebu cattle diversity in South Asia and Africa. Sci. Rep. 8, 18027. doi: 10.1038/s41598-018-36444-7
Peripolli E., Stafuzza N. B., Amorim S. T., de Lemos M. V. A., Grigoletto L., Kluska S., et al. (2020). Genome-wide scan for runs of homozygosity in the composite Montana Tropical® beef cattle. J. Anim. Breed Genet. 137, 155–165. doi: 10.1111/jbg.12428
Quinlan A. R., Hall I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. doi: 10.1093/bioinformatics/btq033
Ren W., Liu G., Yin J., Tan B., Wu G., Bazer F. W., et al. (2017). Amino-acid transporters in T-cell activation and differentiation. Cell Death Dis. 8, e2655. doi: 10.1038/cddis.2016.222
Ruan D., Yang J., Zhuang Z., Ding R., Huang J., Quan J., et al. (2022). Assessment of heterozygosity and genome-wide analysis of heterozygosity regions in two duroc pig populations. Front. Genet. 12. doi: 10.3389/fgene.2021.812456
Samuels D. C., Wang J., Ye F., He J., Levinson R. T., Sheng Q., et al. (2016). Heterozygosity ratio, a robust global genomic measure of autozygosity and its association with height and disease risk. Genetics 204, 893–904. doi: 10.1534/genetics.116.189936
Santana M. H., Utsunomiya Y. T., Neves H. H., Gomes R. C., Garcia J. F., Fukumasu H., et al. (2014). Genome-wide association analysis of feed intake and residual feed intake in Nellore cattle. BMC Genet. 15, 21. doi: 10.1186/1471-2156-15-21
Selli A., Ventura R. V., Fonseca P. A. S., Buzanskas M. E., Andrietta L. T., Balieiro J. C. C., et al. (2021). Detection and visualization of heterozygosity-rich regions and runs of homozygosity in worldwide sheep populations. Animals 11. doi: 10.3390/ani11092696
Serhan C. N., Chiang N., Van Dyke T. E. (2008). Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 8, 349–361. doi: 10.1038/nri2294
Traoré A., Koudandé D. O., Fernández I., Soudré A., Álvarez I., Diarra S., et al. (2016). Multivariate characterization of morphological traits in West African cattlesires. Arch. Anim. Breed 59, 337–344. doi: 10.5194/aab-59-337-2016
Traoré A., Koudandé D. O., Fernández I., Soudré A., Granda V., Álvarez I., et al. (2015). Geographical assessment of body measurements and qualitative traits in West African cattle. Trop. Anim. Health Prod 47, 1505–1513. doi: 10.1007/s11250-015-0891-7
Tsartsianidou V., Sánchez-Molano E., Kapsona V. V., Basdagianni Z., Chatziplis D., Arsenos G., et al. (2021). A comprehensive genome-wide scan detects genomic regions related to local adaptation and climate resilience in Mediterranean domestic sheep. Genet. Sel Evol. 53, 90. doi: 10.1186/s12711-021-00682-7
Keywords: taurine cattle, zebu cattle, heterozygosity-rich region islands, admixture, enrichment analyses, candidate genes
Citation: Arias KD, Fernández I, Traoré A and Goyache F (2024) West African cattle share non-random heterozygosity-rich region islands enriched on adaptation-related genes despite their different origins. Front. Anim. Sci. 5:1468575. doi: 10.3389/fanim.2024.1468575
Received: 22 July 2024; Accepted: 31 October 2024;
Published: 19 November 2024.
Edited by:
Vincenzo Landi, University of Bari Aldo Moro, ItalyReviewed by:
Mario Barbato, University of Messina, ItalyDailu Guan, University of California, Davis, United States
Copyright © 2024 Arias, Fernández, Traoré and Goyache. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Félix Goyache, ZmdveWFjaGVAc2VyaWRhLm9yZw==
†ORCID: Félix Goyache, orcid.org/0000-0002-6872-1045