 Gabriel A. Zayas
Gabriel A. Zayas Raluca G. Mateescu
Raluca G. Mateescu- Department of Animal Sciences, University of Florida, Gainesville, FL, United States
Introduction: The composite breed Brangus combines the resilience and adaptability of the Bos t. indicus breed Brahman with the superior meat quality and fertility traits of the Bos t. taurus breed Angus. Its diverse genetics not only enables optimal production performance but also adaptability to hot and humid environmental conditions. From a research perspective, this makes Brangus an ideal model for identifying genomic signatures that reveal the effects of both artificial selection and natural adaptation. The aim of this study was to detect genomic signatures of selection by analyzing changes in breed origin of allele (BOA) frequencies across the genome.
Methods: Using a multi-breed Angus and Brahman herd (n = 4,516) as a reference, population structure was measured via principal component analysis and admixture analysis in two commercial Brangus herds (n = 4,720). BOA was estimated in these herds using LAMP-LD, followed by a signature of selection analysis utilizing a median-based Z-score approach and Fst analyses to detect genomic regions under selection.
Results: The analysis revealed a genome-wide increase in Angus ancestry in both Brangus populations (71.46%, 68.7%), reflecting intense selection for traits associated with this lineage. BOA-based intra- and inter-population analyses identified significant shifts in Brahman and Angus ancestry across the genome, indicating potential selection for breed-specific genetics. Key genomic regions were identified on chromosomes 6, 8, 10, 12, 14, 17, 18, 19, 27, and 28, and were linked to traits such as fertility, growth, heat tolerance, carcass characteristics, and meat quality.
Discussion: Expected genes showing signatures of selection included MC1R, responsible for black coat color, and PLAG1, integral to growth, fertility, and carcass traits, underscoring the effectiveness of this methodology. Novel genes under selection, such as CCNB2 (critical for fertility), MTCH2 (associated with meat quality and fertility) and PRLR (associated with coat length and heat tolerance), were also identified. These findings provide deeper insights into the genetic mechanisms driving adaptation and production performance in cattle and offer valuable information for strategic breeding practices aimed at optimizing the strengths of both parental breeds.
1 Introduction
Globally, around 65% of beef cattle are raised in tropical or subtropical climates, regions characterized by hot and humid environments (Burrow, 2012). In the United States, approximately 45% of beef cattle are located in southern and southeastern states, which feature similar subtropical conditions (Cooke et al., 2020). These hot and humid conditions are sub-optimal for cattle, especially for taurine (Bos t. taurus) beef breeds, whose growth rate and reproductive performance decreases in these harsh environments (Burrow, 2015). A prevalent approach used by producers in these areas involves crossbreeding commercial taurine breeds with zebu (Bos t. indicus) breeds. Zebu cattle are well adapted to these subtropical climates, and the new composites outperform the parental breeds in these environments (Burrow, 2015). A notable example of such a composite breed is Brangus, a 5/8 (62.5%) Angus and 3/8 (37.5%) Brahman cross. Brangus cattle take advantage of breed complementarity, combining the high-merit carcass traits of the Angus breed with the environmental resilience and adaptability of the Brahman breed. This results in a breed with good carcass quality, which is resilient enough to endure the harsh conditions in tropical and subtropical regions.
Previous research has noted shifts in the expected Angus-Brahman ratios within Brangus cattle, with a tendency of higher Angus percentages (Paim et al., 2020a; Álvarez Cecco et al., 2022; Li et al., 2023). This is often attributed to intense selection for predominantly Angus traits and the integration of UltraBlack cattle. Expected progeny difference (EPD) developed by the International Brangus Breeders Association allows for selection of a variety of traits including growth, carcass quality, meat quality, and reproduction traits. However, while this artificial selection has changed the original genetic population structure of this breed over time, the specific genomic regions under selection and the underlying genetic mechanisms driving these changes are not fully understood.
Prior research identified signatures of selection using genomic data through haplotype methods (ROH, Rsb and XP-EHH) and Fst values (Paim et al., 2020b; Álvarez Cecco et al., 2022; Zayas et al., 2024b). However, these studies often fall short of directly highlighting regions of selection based on Angus and Brahman ancestry deviations, instead reverting to assigning Angus and Brahman ancestries to regions identified post hoc. While certain studies have mapped deviations in Brahman and Angus ancestry across the genome, they have not consistently applied robust tests to identify these deviations, relying predominantly on threshold alterations (Li et al., 2023). Furthermore, no research has harnessed the breed of origin of alleles (BOA) technique to detect shifts in allele combinations (e.g., AA = Homozygous Angus, AB = Heterozygous, BB = Homozygous Brahman). By identifying BOA across the genome, BOA can be treated as a pseudo-genotype to ascertain genomic variations suggestive of selection. This methodology gains credibility from earlier research that emphasized selection favoring Brahman ancestry in the BoLA region on BTA23 in Argentinian Brangus – the genomic location of the major histocompatibility complex encompassing genes central to the adaptive immune response (Goszczynski et al., 2018). BOA’s robustness has already been demonstrated in tracking the introgression of the SLICK1 allele from Senepol into Holstein cattle (Zayas et al., 2024a) and in identifying regions linked to carcass weight, marbling, and reproductive traits in composite breeds (Warburton et al., 2023; Zayas et al., 2024c). These findings underscore BOA’s reliability and highlight its potential for identifying selection signatures in Brangus cattle.
The benefits of detecting shifts in allele combinations stem from the observance of the heterozygotes (AB) which cannot be detected by other methods. Similar to genotypic heterozygosity, under a neutral evolutionary scenario, the expected level of BOA heterozygosity should remain relatively stable. However, selection (either natural or artificial selection) for favorable traits will alter the frequency of the heterozygotes. These changes in BOA heterozygosity in certain genomic regions indicate selective pressures. For example, a decrease of heterozygote BOA alleles in favor of homozygous Angus alleles can be indicative of selection for traits associated with the Angus alleles such as meat quality or growth traits. Conversely, an increase in heterozygosity can be indicative of hybrid vigor (or heterosis), where the presence of diverse alleles from different breeds confers an advantage. This is particularly relevant in traits where a balance of attributes from both breeds is beneficial. The utilization of BOA information can be incorporated to unveil genomic regions under potential selection. Utilizing tests that compare genome wide heterozygosity and nucleotide diversity, BOA methodology can be used to pinpoint regions under selection for either Angus or Brahman lineage.
The aim of this study was to first determine genome-wide breed compositions of commercial Brangus herds utilizing genomic data. A multi-breed Angus and Brahman (MAB) herd was used as a reference population illustrating that the use of a multi-breed herd as a reference can adequately capture the genetic diversity in commercial Brangus herds. The BOA in the commercial Brangus herds and the MAB were then predicted using purebred animals. Using the BOA data, an intra-population median-based Z–score test and inter-population Fst analysis was implemented to identify deviations of Angus and Brahman ancestry for potential regions of selection.
2 Material and methods
2.1 Animals
The research protocol was approved by the University of Florida Institutional Animal Care and Use Committee number 201003744. A total of 4,022 commercial Brangus cattle were used from the Seminole Tribe of Florida. Out of these, 2,965 were replacement heifers used in a separate thermotolerance study from years 2017, 2018, and 2019. The other 1,057 were steers from a nutritional study from years 2017 and 2018. A second commercial Brangus population was composed of 698 replacement heifers from the Williamson Cattle Company (2020–2022). A total of 4,516 animals from the University of Florida’s MAB herd were used as a reference for population structure analysis. This MAB herd contains animals ranging from 100% Angus to 100% Brahman (Elzo et al., 2012; Gobena et al., 2018).
2.2 Genotyping
Genomic DNA was extracted from blood or meat samples using the QIAamp DNA Mini DNA kit (Qiagen, Valencia, CA, United States). The extracted DNA was genotyped with the Bovine GeneSeek Genome Profiler F-250 array (Neogen Corp. – GeneSeek, Lincoln, NE, United States) identifying 221,115 genetic markers. The map of the Bovine GeneSeek Genome Profiler F-250 array was updated from the UMD3.1 assembly to the ARS-UCD1.2 assembly. Only autosomal markers were kept for further analysis.
2.3 Population structure analysis
2.3.1 Selection of unrelated animals and quality control
Close familial relationships and shared recent ancestry can bias interpretation of breed composition and population structure in model-based analyses (Patterson et al., 2006; Conomos et al., 2016). To counteract this bias, a subset of unrelated cattle from the MAB herd were used as the reference when estimating genomic breed composition on the rest of the MAB herd and on the commercial Brangus herds.
Genotypic quality control filtering was implemented at the animal and marker level using the software PLINK1.9 (Chang et al., 2015). At the animal level, filters were set using genotype completion rate (<90%). At the marker level filters were set using minor allele frequency (<1%), genotype call rate (<90%), and Hardy–Weinberg equilibrium deviation (P-value <1 × 10−8). Widespread linkage disequilibrium (LD) can distort estimates of kinship, ancestry and breed composition by inflating the influence of certain genomic regions (Alexander et al., 2009; Conomos et al., 2016). Thus, LD pruning was performed to remove markers in high LD, ensuring the analysis focuses on more independent markers and leading to more accurate estimations of genetic relationships, kinship coefficients, and breed compositions. Markers above a LD threshold of 0.5 r2 were pruned out using a window size of 5,000 kb and step size of 10bp (Turner et al., 2011). After quality control a total of 42,311 single nucleotide polymorphisms (SNP) remained. These 42,311 SNPs were extracted from the commercial Brangus herds. Brangus cattle which had a genotypic call rate below 90% for these 42,311 SNPs were removed. A total of 4,415 cattle from the MAB and 4,675 cattle from the commercial Brangus herds remained for population structure analysis.
Unrelated animals in the MAB population were identified using the “pcairPartition” function from the R package Genesis (Conomos et al., 2016). The matrix is estimated using the KING-robust method which avoids bias due to population structure (Manichaikul et al., 2010). This method identifies individuals that are not overly related to others in the population but also not too different from the rest of the population. To extract pairwise kinship and divergences from the matrix, the function “snpgdsIBDKing” in the R package Genesis was implemented (Conomos et al., 2016). The unrelated animals were those that had pairwise kinship lower than 0.022, but in addition had the most number of pairwise divergences lower than −0.022 (Conomos et al., 2016; Gobena et al., 2018). The thresholds for kinship and divergence were selected to balance the need for individuals who are both genetically representative of the population and sufficiently unrelated to minimize biases in further population structure analyses.
2.3.2 Principal component analysis
A principal component analysis (PCA) was conducted using the “pcair” function from the R package Genesis, which allows for a subset of unrelated samples to be used as a reference (Conomos et al., 2016). The unrelated reference controls for any confounding effects due to familial relationships and recent shared ancestry. PCA was first performed on the genotype data for the unrelated subset of the MAB population by applying eigen decomposition on the covariance matrix of the standardized genotype. Principal components for the rest of the population were then obtained by projecting the unrelated reference standardized genotype data onto the SNP weight matrix calculated for unrelated set in the previous step (Conomos et al., 2016). Overall structuring of the genetic variation was visualized in a scatterplot of the two top principal components using ggplot2 (Wickham, 2009) in R (R Core Team, 2023).
2.3.3 ADMIXTURE analysis
The ADMIXTURE v1.3 software was used to obtain both individual cattle breed composition and breed marker allele frequencies (Alexander et al., 2009). ADMIXTURE uses a maximum likelihood model to divide individuals into subgroups using genomic data (Alexander et al., 2009). The number of subgroups (K) is specified beforehand or can be inferred using a cross-validation procedure. Subgroup memberships were considered as breed membership, using a K of 2 representing the Angus and Brahman origins of the herd. Pedigree information was used to identify the breed associated with each subgroup. The K value of 2 was also confirmed using the cross-validation procedure.
Certain assumptions need to be met when using the maximum likelihood model implemented in ADMIXTURE. These include a relative low level of LD between markers, a sample set with adequate representation of all parental breeds and a sample set composed of only unrelated animals (Alexander et al., 2009). Quality control and LD pruning were performed to combat LD in the sample population to meet the LD assumption. To meet the second assumption, the unrelated subset was used as the reference when inferring genomic breed composition in the related set. This method accounts for the confounding effect of the known and cryptic familial relationships (Shringarpure et al., 2016). To implement this in ADMIXTURE, an unsupervised model on the unrelated MAB subset is ran and the breed allele frequencies (F matrix) from this run is then projected onto the rest of the populations. The F matrix from the unrelated MAB subset was then projected onto the entire MAB herd and the commercial Brangus populations to estimate breed portion of Angus and Brahman in this multi-generational Brangus herds.
2.4 LAMP-LD and breed of origin analysis
Local Ancestry in adMixed Populations (LAMP-LD) was used to infer percentages of local ancestry of the entire MAB herd and both Brangus herds (Baran et al., 2012). LAMP-LD uses hidden Markov models of haplotype diversity of the ancestral/purebred populations within a window-based framework to trace the origin of alleles in the admixed population (Baran et al., 2012). Angus (n=123) and Brahman (n=406) cattle with ≥99% genomic breed composition from the ADMIXTUE were used to represent the purebred populations. Only markers with an allele frequency difference of ≥5% between purebred population were used, to ensure sufficient differentiation between groups.
PLINK was used to conduct quality control before LAMP-LD analysis (Chang et al., 2015). At the animal level, filters included genotype completion rate (<90%). At the marker level filters were applied for minor allele frequency (MAF; <1%), and genotype call rate (<99%). Ultimately 93,751 SNP, all the purebred cattle and 6261 Brangus remained after quality control. The resulting local ancestry results from LAMP-LD where then used to infer the breed origin of alleles (BOA). The BOA of the resulting 93,751 SNPs were then converted into a pseudo-genotype format using in-house scripts with 0 representing homozygous Angus (AA), 1 representing the heterozygote state (AB/BA) and 2 representing homozygous Brahman (BB). The pipeline for these analyses is available at https://github.com/gzayasPR/BOA_Estimation (Zayas et al., 2024c), which provides further details on data processing and management (Tange, 2011; Knaus and Grünwald, 2017; R Core Team, 2023; Barrett et al., 2024; Wickham et al., 2024). Descriptive genome-wide statistics were generated on this new BOA file using PLINK (Chang et al., 2015). Genome-wide breed composition ratios for each population was estimated by calculating the ratio of BOA using PLINK (Chang et al., 2015) software. These estimations were done separately in the Seminole Tribe of Florida Brangus, the Williamson Cattle Company Brangus, and sub-section of the MAB herd containing cattle ranging from 60–80% Angus.
2.5 Identifying signatures of selection
To identify signatures of selection, two methods were employed to detect shifts BOA heterozygotes in the two commercial Brangus herds. The first method is an intra-population analysis that compares the ratios of heterozygous BOA at a locus to the rest of the loci on that particular chromosome. The method employed was a median-based Z-score using the following formula:
Where is the BOA heterozygosity at , is the median BOA heterozygosity of the chromosome of testing . is the median absolute deviation of the chromosome of the testing . This is an intra-population test attempting to identify genomic regions that have large increases or decreases in BOA heterozygosity. The median-based Z-score is typically used to identify outliers within a given dataset, and this application of the test attempts to identify regions whose BOA heterozygous ratio is an “outlier” compared to the rest of chromosome. These “outliers” can be suggestive of regions that have strong selection pressures. The resulting Z-scores were used to infer significance using an FDR of 0.05. Consecutive significant markers were merged into regions to perform a QTL enrichment analysis using GALLO (Fonseca et al., 2020) and gene literature search. Results were plotted in R using the ggplot2 package.
The second method is an inter population analysis that compares the changes/differences between heterozygous BOA across different Brangus populations. To this end, the Fst statistic was estimated using BOA, finding the pairwise differences between the Seminole Tribe of Florida Brangus herd, Williamson Cattle Company Brangus herd and a specific subset of the MAB herd. This subset consists of cattle with an Angus composition ranging between 60–80% and serves essentially as a reference or control group for the Brangus populations. Wright’s Fst estimates (Weir and Cockerham, 1984) were performed using PLINK (Chang et al., 2015). Regions with Fst values above 0.15 were then investigated by performing a QTL and gene literature search. Results were plotted in R using the CMplot package (Yin et al., 2021).
3 Results
3.1 Population structure and breed composition analyses
The “pcairPartition” function identified a total of 255 MAB unrelated and representative animals from the initial 4,415 animals in the MAB population. Figure 1 shows the clustering of the MAB and the two commercial Brangus herds based on the first and second principal components. Figure 1A illustrates the dispersion of the unrelated MAB subset (represented by black stars) along both PC1 and PC2. This level of dispersion indicates that the unrelated subset in the MAB covers the entire population equally and forms an adequate reference. PC1 explained 33.45% of the genetic variation in the entire population and separates the two founder breeds estimating the Angus and Brahman breed fractions. Positive PC1 values are indicative of higher Angus composition while negative PC1 values indicate higher Brahman percentages.
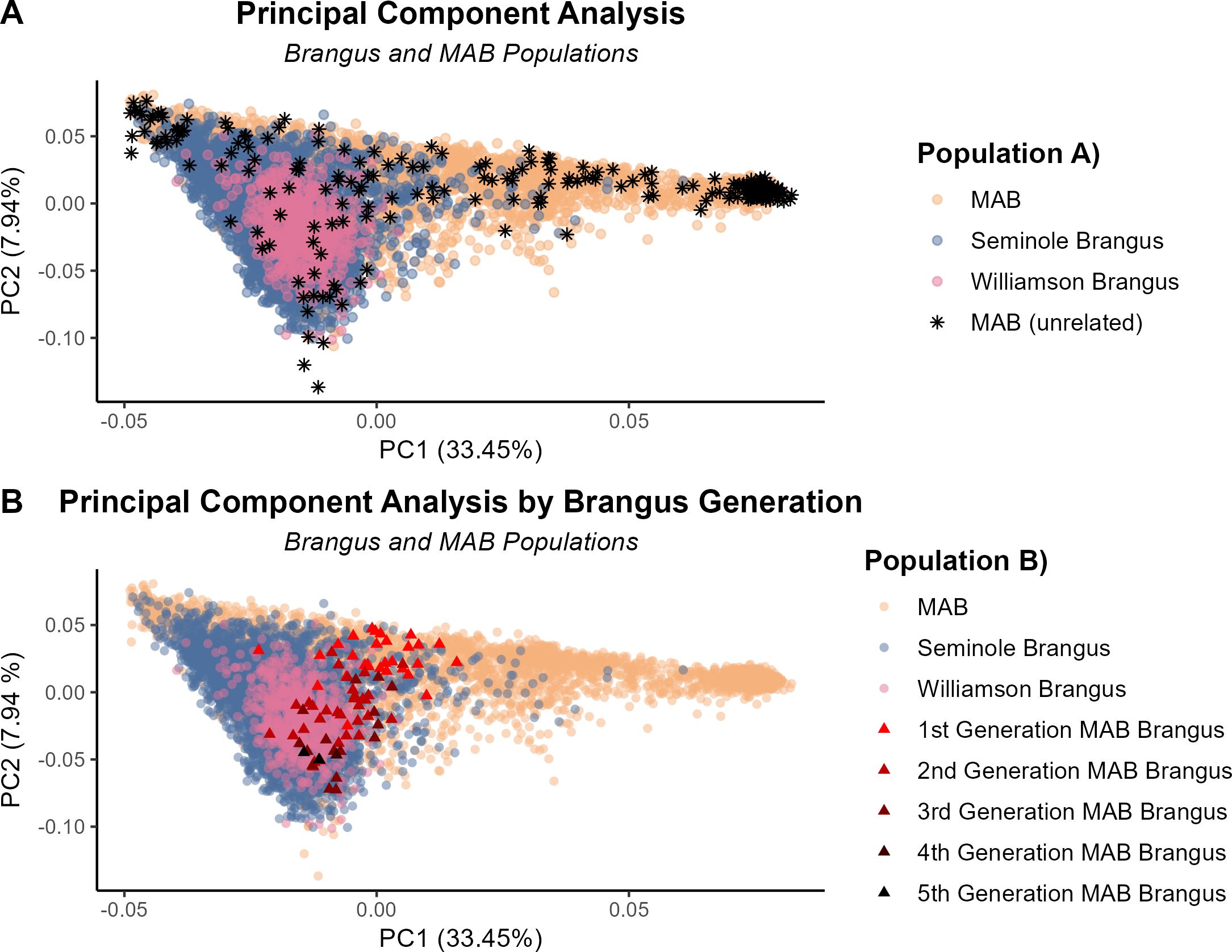
Figure 1. Commercial Brangus and MAB herds distribution along the first principal component (PC1) and second principal component (PC2). (A) The 255 unrelated subset of the MAB herd (black stars), the rest of the MAB herd (orange), the Williamson Brangus in pink and the Seminole Brangus in dark blue. (B) The MAB Brangus separated by generation denoted by red triangles, the rest of the MAB herd (orange), the Williamson Brangus in pink and the Seminole Brangus in dark blue.
PC2 reveals information of the Brangus ancestry and estimates the distance of a particular Brangus animal from a first-generation Brangus animal. This can be seen in Figure 1B where the red triangles represent Brangus cattle from different generations in the MAB herd, and the darker the shade of red the further the animal is from the initial Brangus formation. These commercial Brangus cattle are multi-generational Brangus. as expected, they cluster alongside the multi-generational MAB Brangus cattle on PC2.
For the breed composition analysis, the F-matrix of the 255 unrelated MAB animals was used as the reference for the projected run on the commercial Brangus herds (Figure 2). The Seminole Brangus cattle had on average 71.65% Angus genetics, with a standard deviation of 7.33%. The Williamson Brangus cattle had less Angus genetics averaging around 68.70% Angus, with a standard deviation of 3.19%. There was a significant positive and strong correlation between the breed composition results from ADMIXTURE and PC1 estimates (R >.99). Both ADMIXTURE and PCA indicate both Seminole and Williamson Brangus were more Angus than the expected 5/8.
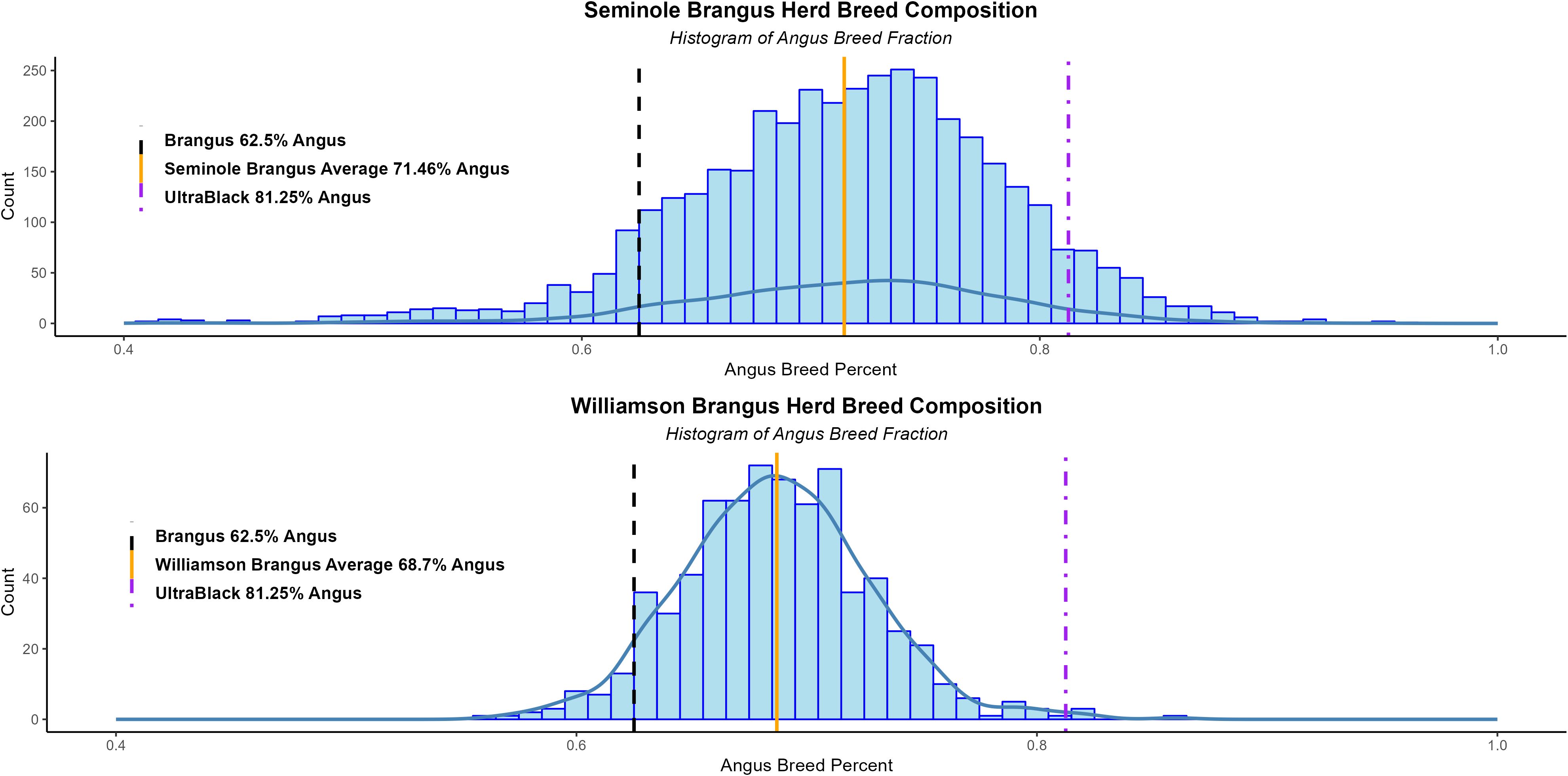
Figure 2. Histogram showing the Seminole and Williamson Brangus breed composition, estimated using ADMXITURE. The x-axis represents the Angus breed percentage, the black dotted line is the expected 5/8 (62.5%) Angus breed percentage. The orange line represents the population’s average Angus breed composition of 71.65%.
3.2 Breed of origin analysis
Descriptive statistics for the homozygous Angus, heterozygous, and homozygous Brahman BOA genotypes observed in the commercial Brangus herds are presented in Table 1. It is important to highlight that the average Angus and Brahman breed composition detailed in this section differs slightly from those derived from the ADMIXTURE analysis. The primary reason for this discrepancy is the methodology employed in each analysis. The aim of the ADMIXTURE analysis was to obtain overall estimation of the breed composition for each individual animal. Thus, LD-pruning is performed to reduce the influence of closely spaced markers, which can result in biased breed composition estimates. By pruning we ensure a more accurate and unbiased representation of the genetic makeup of the individual by focusing only on independent markers. In contrast, the BOA analysis capitalizes on patterns within LD to allocate ancestry. Given that the functional SNP chip used in this study contains many markers in proximity to one another, this approach can often lead to the overestimation of specific ancestries when determining breed composition. However, the correlation between the estimates for genomic breed composition based on ADMIXTURE and BOA was a significant, positive and strong (R > 0.99) as seen in Supplementary Figure 2.
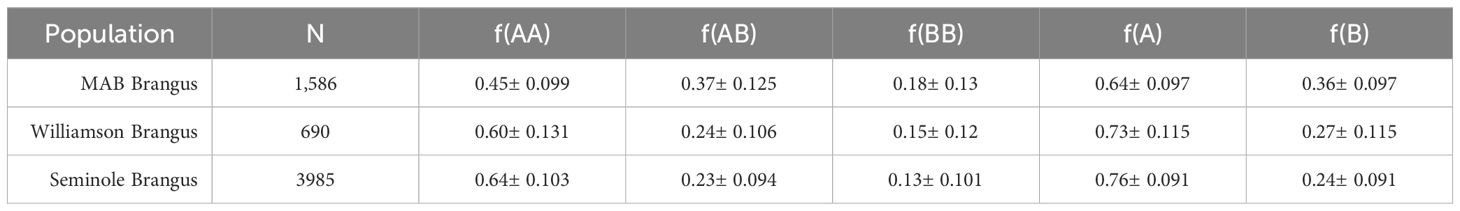
Table 1. Summary statistics for breed of origin of allele in three Brangus populations.
3.3 Signatures of selection analysis
3.3.1 Median-based Z-score signatures of selection analysis
Utilizing the median-based Z-score, a total of 14 regions in the Williamson Brangus herd and 15 regions in the Seminole Brangus herd were significant after accounting for multiple testing as seen in Figure 3. A total of six regions on BTA 14, 17, 18, 19, 20 and 28 were significant in both Brangus populations. Table 2 shows detailed information on these significant signature of selection regions, including the chromosome start and end position, percent Angus and Brahman and significance levels. Overall, as expected, these regions experience a reduction or increase in heterozygote BOA genotypes. Of these regions, four regions in the Seminole herd and four regions in the Williamson herd had an increase in Angus ancestry with f(A) > 0.8, indicating strong selection for Angus ancestry. Supplementary Table 1 shows the results from the QTL enrichment analysis for the Seminole herd and Supplementary Table 2 shows the results from the QTL enrichment analysis for the Williamson herd.
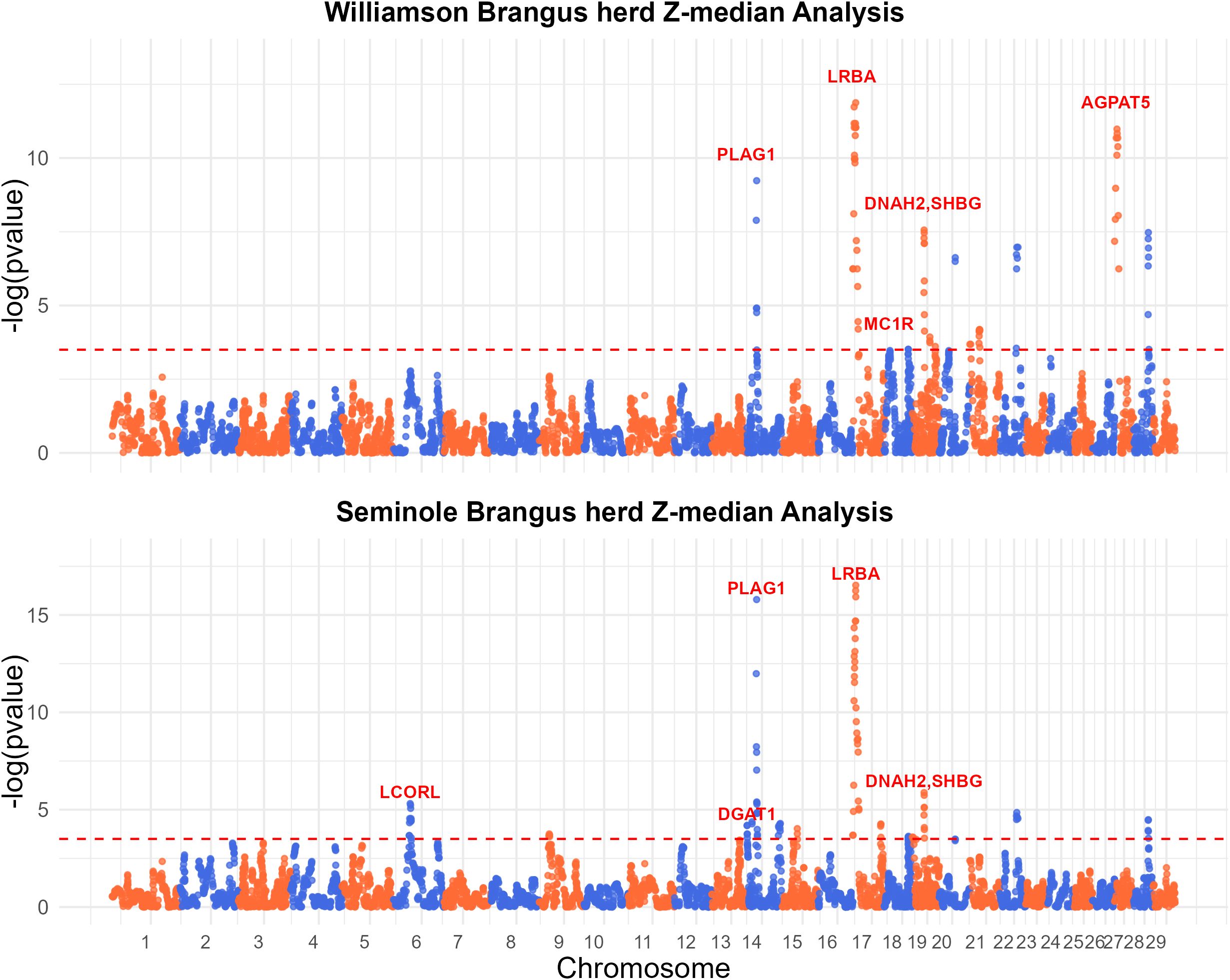
Figure 3. Manhattan plots on single locus median-based Z-score test on BOA heterozygosity in the Williamson Brangus (top) and Seminole Brangus (bottom) populations. The redline represents the significant threshold based on FDR = 0.05.
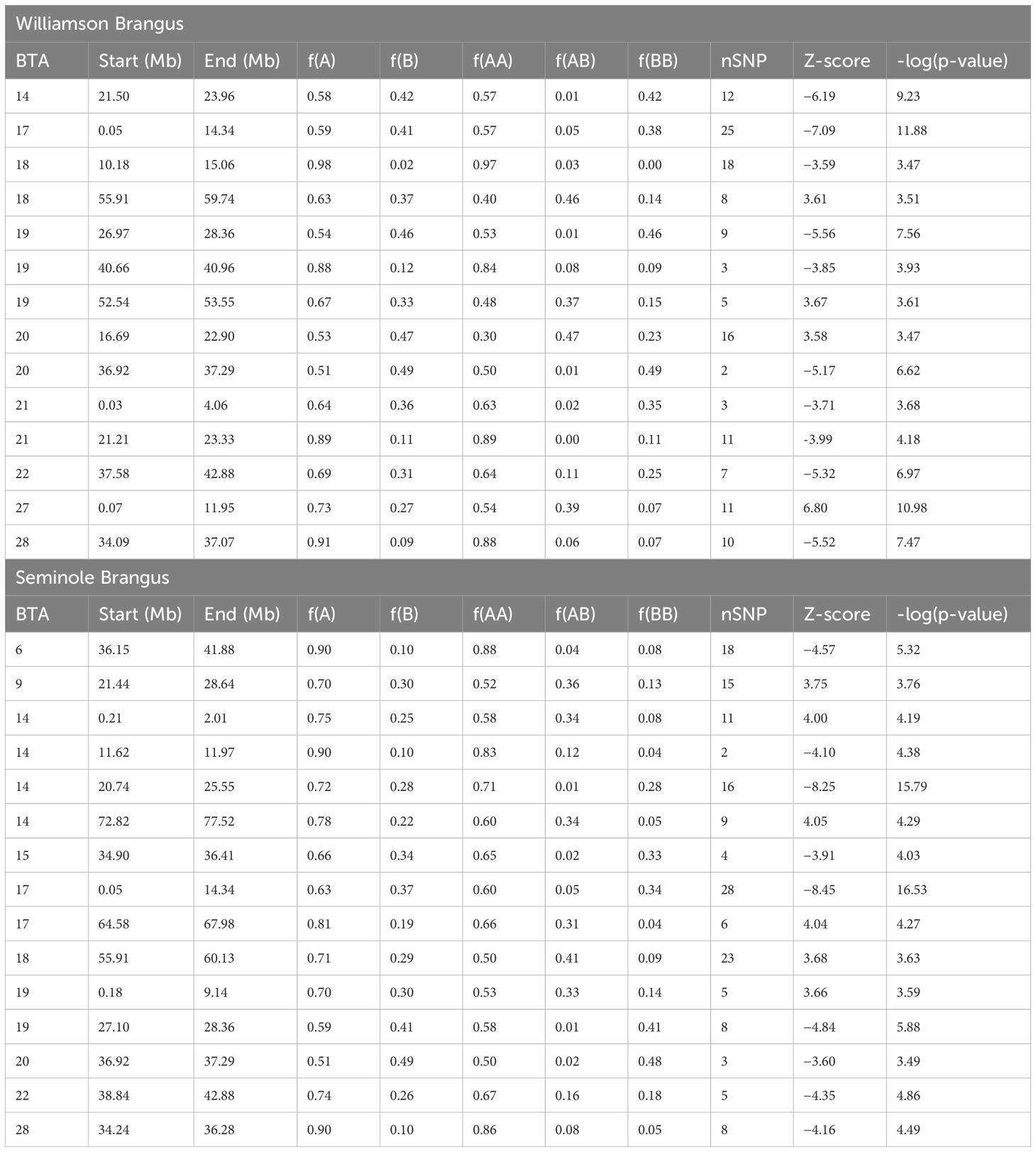
Table 2. Significant signature of selection regions based on the single-locus median-based Z-score test on BOA heterozygosity in the Williamson Brangus (top) and Seminole Brangus (bottom) populations.
3.3.2 Fst signatures of selection analysis
Results from the inter-population Fst tests are presented in Figure 4. A total of 8 regions in the Williamson herd and 4 regions in the Seminole herd surpassed the threshold of 0.15. Of these regions, the BTA10 (73–77 MB) and BTA12 (23.3 – 23.4 MB) regions overlapped between the two populations. Overlapping regions between different herds are important as they might be indicative of key genomic areas that are under similar selective pressures in both herds. Table 3 provides a detailed breakdown of these regions, outlining the chromosome start and end position, percent Angus and Brahman and Fst in the Seminole and Williamson populations. One noteworthy observation from our study is the pronounced prevalence of Angus ancestry in the identified regions, which could indicate these regions could be related to production traits.
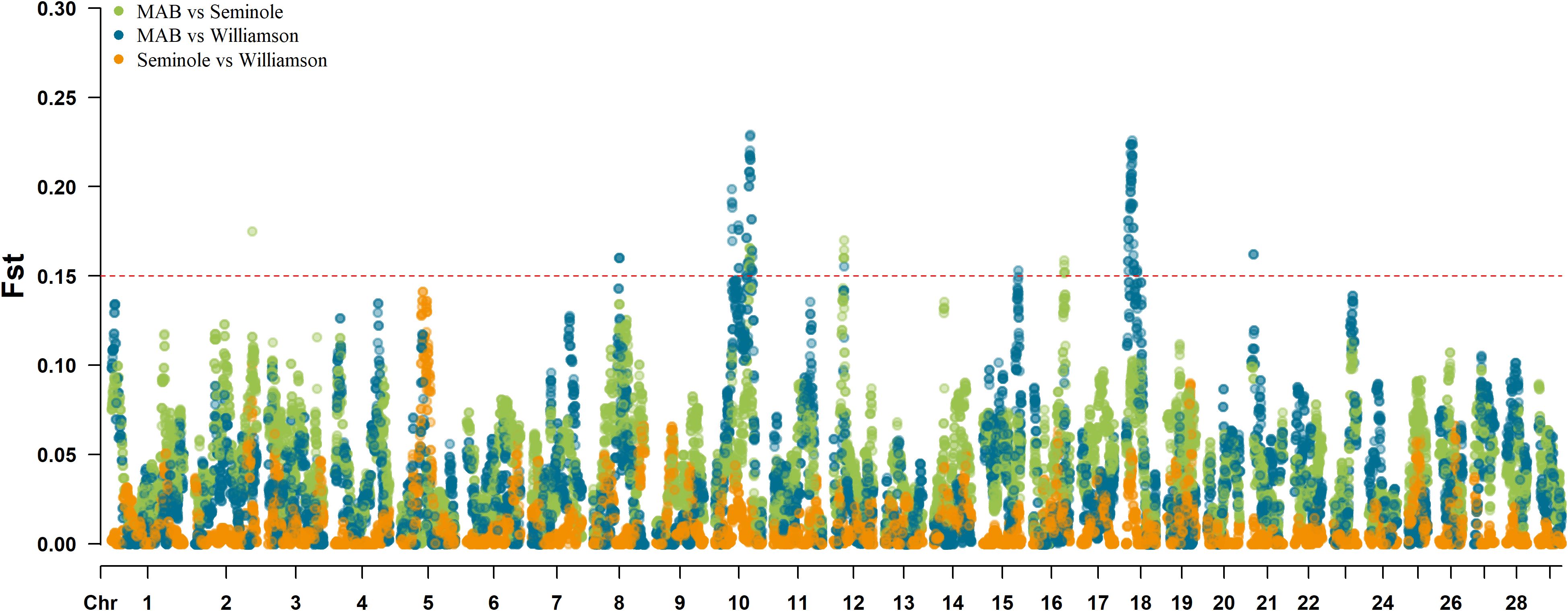
Figure 4. Fst analysis Manhattan plot between MAB Brangus, Seminole Brangus and Williamson Brangus.
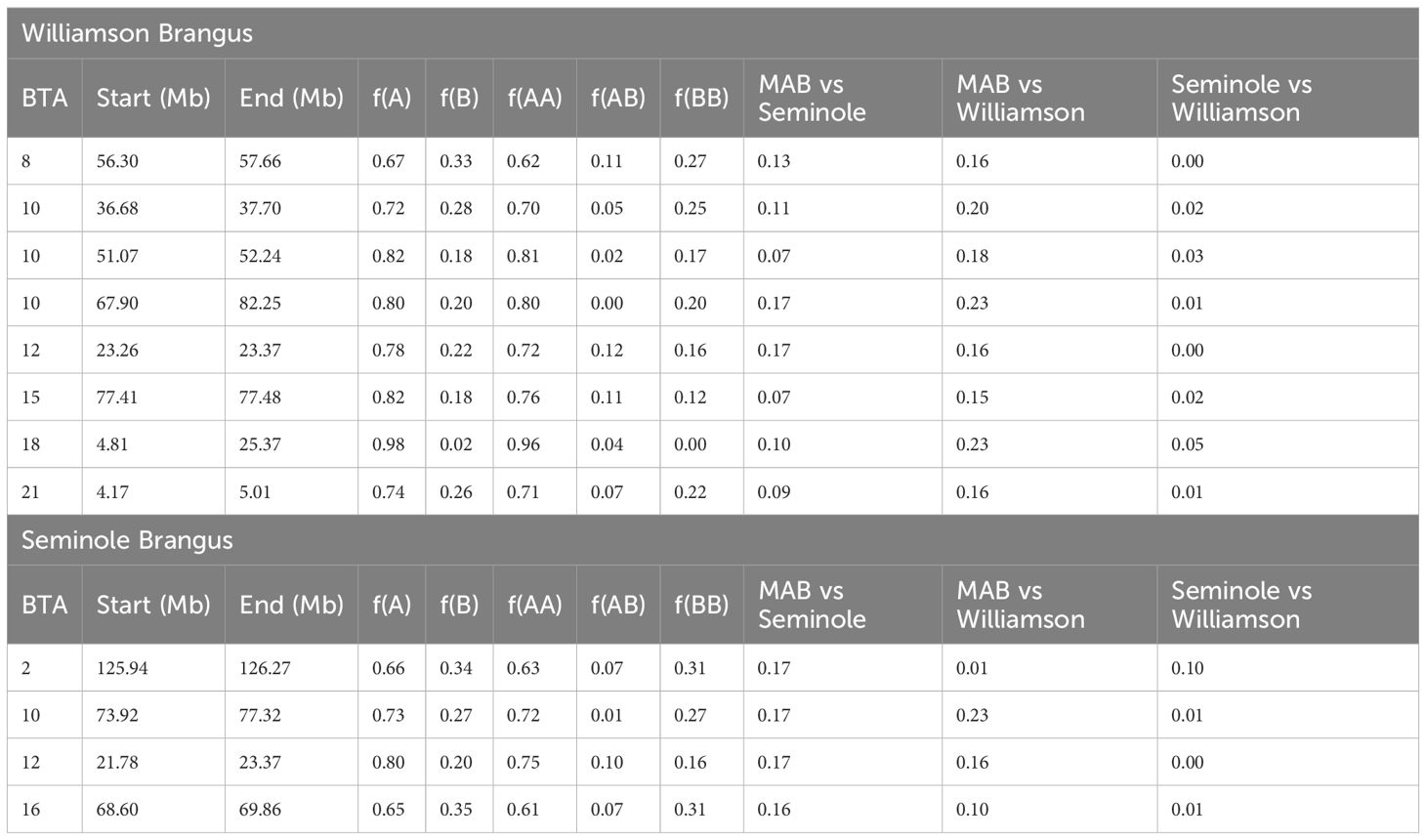
Table 3. Significant regions identified using pairwise Fst estimates in the Williamson Brangus(top) and Seminole Brangus (bottom) using the MAB as a reference population.
4 Discussion
To our knowledge, this study is the first to utilize BOA to identify signatures of selection across the entire genome in a composite cattle breed. The study provides a comprehensive view of the intricate genetic landscape in Brangus cattle, revealing how patterns of selection are shaped by their composite breed structure. By examining the interplay of Angus and Brahman ancestries, we uncover artificial selection and the genetic influences of heterosis and inbreeding on various traits.
4.1 Population structure
The results from the PCA indicate that the MAB herd is a good reference for Brangus and other Angus-Brahman crossbred populations. The first principal component in this analysis can be used to differentiate between Angus and Brahman ancestry. This follows other studies in admixed population, where PC1 separates the two parental breeds (McVean, 2009; Gobena et al., 2018), and worldwide PCA also show differentiation between Bos t. indicus and Bos t. taurus cattle along PC1 (Lewis et al., 2011; Decker et al., 2014). PC2 differentiates between recent and generational Brangus crosses, where recent Brangus crosses have positive PC2 values and generational Brangus crosses have negative PC2 values. These results are similar to those found by Gobena et al. (2018), who performed a similar analysis with the MAB. These results strongly support the conclusion that the MAB herd is a suitable reference population for the Seminole and Williamson commercial Brangus herds for further analysis. The results from the genomic breed composition estimates using ADMIXUTRE and LAMP-LD indicate an increase in Angus ancestry across the genome in both commercial Brangus populations. These results are in agreement with most literature in Brangus cattle, showing that the Angus breed percentage in Brangus is roughly 70% (Paim et al., 2020a; Álvarez Cecco et al., 2022; Li et al., 2023). These results underscore the influence of Angus genetics in shaping the genomic landscape of Brangus cattle.
4.2 Signatures of selection
In this study, a novel approach was employed to identify regions under selection in Brangus cattle, combining BOA methodology with median-based Z-score hypothesis testing. To validate this methodology, significant findings from this method were compared with those previously reported in literature using traditional selection signature methods (Supplementary Table 3). The analysis showed considerable overlap with regions previously identified, underscoring the effectiveness of the current analysis in detecting known selective pressures. Additionally, it uncovered novel regions not previously recognized as regions of selection. These results demonstrate its potential for gaining new insights into selection consequences at the genome level in Brangus and can be applied to other composite breeds.
The advantage of this method stems from the combined use of BOA and median-based Z-score hypothesis testing. Traditional LD-based selection signature methods (XP-Ehh, iHS, ROH) may be less effective in admixed populations like Brangus cattle, where the mixed ancestry leads to rapid breakdown and increased complexity in LD patterns. This complexity can obscure true selection signals. By focusing on BOA rather than generalized population-wide LD patterns, our approach can effectively “correct” for these LD complexities, ultimately providing a clearer picture of the selection processes shaping the genome in composite breeds. Moreover, the use of a median-based Z-score hypothesis testing adds robustness by mitigating the impact of outliers. Regions undergoing selection often encompass large areas with extreme values or highly differentiated values. While mean-based hypothesis testing can be skewed by outliers, a median-based approach offers a more stable measure of central tendency, making it better suited for identifying true deviations due to selection. This stability makes the median-based Z-score approach particularly effective for detecting both strong and subtle selection signals that might otherwise go unnoticed. Using this method, we identified various shifts in ancestry, including increased Angus and Brahman ancestry, as well as changes in BOA heterozygosity.
4.2.1 Increases in Angus ancestry
Our study brought to light several regions with markedly increased Angus ancestry, identified through both Z-median and Fst analyses. The Angus breed is renowned for traits such as meat quality, marbling, growth rate, reproduction, and fertility. In this study, we identified regions with increase Angus ancestry in Brangus linked to key traits such as growth rate, meat quality, carcass quality, fertility, and coat color. Among these, some regions are annotated while others remain unannotated in cattle and have not been previously identified as signatures of selection.
One of the most compelling findings from our study is the identification of the melanocortin 1 receptor (MC1R) gene, located on BTA18 as a signature of selection. The significant region is located on BTA18 from 10.18 - 15.06 MB, showing a significant increase in Angus ancestry. This region is characterized by a near-complete absence of homozygous Brahman ancestry and a very low prevalence of heterozygous BOA. MC1R functions as a key player in the melanocortin pathway, regulating melanogenesis (Wolf Horrell et al., 2016). Specifically, it dictates the type of melanin produced: eumelanin (black/brown pigment) or pheomelanin (yellow/red pigment) (Nasti and Timares, 2015). In cattle, MC1R is widely recognized for controlling the black coat color, a trait heavily selected for in Brangus and Angus-Brahman production in America, primarily because it is one of the qualifying criteria for the premium “Certified Angus beef” label. The contrasting coat color palettes of Brahman (primarily white and grey) and Angus (predominantly black) make it clear that the genetic influence promoting black coat color in Brangus comes largely from the Angus lineage in Brangus. In the Williamson herd, the predominance of Angus ancestry in this region reflects the intensive selection for the black coat color – a trait that meets market preferences and holds significant economic value in the beef industry. Surprisingly this region is rarely identified as a signature of selection in Brangus, despite the strong selection pressure for MC1R. Detecting this selection signal underscores our method’s ability to reveal key genomic areas linked to production traits that may be overlooked by traditional approaches.
Another key region identified in this study is the NCAPG-LCORL locus on BTA6. This locus is a candidate gene and has been extensively associated with a range of production traits including feed intake, feed gain, meat quality and carcass quality in beef cattle (Lindholm-Perry et al., 2011, 2013). Our QTL enrichment analysis corroborates these associations, revealing enrichment for average daily gain, carcass weight, ribeye area and lean meat yield (Supplementary Table 2). This genomic region has been previously identified as a signature of selection in Brangus, particularly in studies comparing genetic differences between Brangus and Brahman cattle (Álvarez Cecco et al., 2022). This signature of selection reveals selection for beneficial Angus haplotypes in this region due to artificial selection for production traits.
A signature of selection on BTA15 overlaps the MTCH2 gene, a gene which has garnered significant attention in porcine studies. Research indicates that MTCH2 promotes adipogenesis of preadipocytes in muscle and is overexpressed in pigs with higher intra-muscular fat/marbling (Jiang et al., 2019). Supporting this notion, the region housing MTCH2 was also found significant for meat tenderness in Angus cattle (Leal-Gutiérrez et al., 2020). This connection underscores the gene’s potential influences in both marbling and tenderness, which relate to overall better meat quality.
The previous three regions illustrate the identification of known genes related to production traits, originating from the Angus breed. However, several novel signatures of selection were identified using our methodology. Firstly, we identified a significant peak on BTA28 in both Brangus populations overlapping the ZMIZ1 gene. ZMIZ1 plays a crucial role in the development and function of mature β-cells, particularly in relation to mass expansion during high-fat dietary conditions, as demonstrated by Alghamdi et al. (2022). Alghamdi et al. (2022) knocked out ZMIZ1 in mice resulting in impaired insulin secretion, leading to development of severe diabetes and obesity. We speculate the ZMIZ1 gene may contribute to the rapid growth rate characterizing the Angus breed, thus selection for production has led to an increase in Angus haplotypes in this region. Secondly, the region BTA10 (51.07–52.24 MB) encompasses the gene CCNB2 which encodes for Cyclin B2, which is vital for cell cycle progression. CCNB2 is linked to embryonic competence in cattle, implying a potential role in fertility (Mourot et al., 2006). In mice, research has further underlined CCNB2 importance in reproductive processes, where CCNB2 depletion led to ovulation of immature oocytes, premature ovarian failure, and compromised female fecundity (Daldello et al., 2019). The increase in Angus ancestry in this region points to a selection bias for Angus haplotypes, suggesting that Angus genetics in this region may carry advantageous material for fertility and embryonic development. Overall, the identified regions highlight the targeted artificial selection for advantageous production traits, many of which are derived from Angus ancestry.
4.2.2 Increases in Brahman ancestry
Several regions with pronounced increases in Brahman ancestry were identified in this study, highlighting the unique contributions of Brahman genetics to various desirable traits in cattle. Brahman are known for their thermotolerance, disease resistance, longevity and maternal instincts.
A significant region on BTA20 in both populations is positioned downstream of the PRLR gene, which is linked to the slick-hair phenotype in cattle, a significant marker for thermotolerance (Sosa et al., 2021, 2022). Previous analysis on the Seminole population identified a significant marker near this region for hair length (Sarlo Davila et al., 2020). The increase in Brahman homozygotes in this region may indicate selection for Brahman haplotypes which confer slicker coats leading to increased thermotolerance. This finding emphasizes the importance of Brahman genetics in adapting to heat stress conditions, a critical trait for cattle in warmer climates.
In the Seminole herd, another noteworthy region on BTA2 (125–126 MB) encompasses the WTDC1 gene. This gene is involved in regulating fat-related gene transcription (Bedhane et al., 2019) and has been associated with fatty acid traits in Nellore (de Lemos et al., 2018; Berton et al., 2022). A single region in the Seminole on BTA16 overlaps PROX1 which has been shown to be upregulated in Nellore cattle with high ribeye area (Santos Silva et al., 2020).
Delving deeper into the genetic landscape of both peaks on BTA2 and BTA16, an intriguing pattern emerges in the MAB herd. This specific region is densely populated with Brahman BOA in the MAB Brangus, suggesting certain attributes from the Bos t. indicus lineage that might be advantageous or preferential for this populations potentially the meat quality benefits mentioned before. In contrast, the Seminole herd predominantly showcases Angus BOA throughout its genome. However, closer examination reveals a relative enrichment of Brahman BOA around the regions of WTDC1 and PROX1 genes. This enrichment indicates a nuanced selection strategy: while there’s an overarching preference for the meat quality and other desirable traits associated with the Angus lineage, the specific region near WTDC1 and PROX1 favors Brahman genetics. This is likely due to the advantages these Brahman genes confer, particularly in terms of meat quality.
4.2.3 Changes in BOA heterozygosity
The study also identified changes in BOA heterozygosity that did not seemingly favor either Angus or Brahman haplotypes. These regions are of particular interest since an increase in BOA heterozygosity can be indicative of heterosis, whilst decreases in BOA heterozygosity can indicate beneficial inbreeding. The changes in BOA heterozygotes highlights the complexity of selection and the potential for optimizing traits through a balanced approach that leverages both homozygosity and heterozygosity.
A key observation across both Brangus populations was a marked decrease in BOA heterozygosity near the pleiomorphic adenoma (PLAG1) gene on BTA14. PLAG1 is a known candidate gene associated with various production, carcass and reproductive traits (Fortes et al., 2012; Ali et al., 2015; Seabury et al., 2017; Zhang et al., 2020). Bouwman et al. (2018) in their meta-analysis also highlighted PLAG1’s role in cattle stature, with tall breeds showing near-fixed haplotypes and shorter breeds predominantly carrying alleles favoring increased height. This region’s importance is further supported by our QTL enrichment analysis (Supplementary Tables 1, 2) and aligns with findings from other studies identifying this region as a selection signature in Brangus (Paim et al., 2020b; Álvarez Cecco et al., 2022; Li et al., 2023). In a previous study this region was identified as a run of homozygosity (ROH) island in the Seminole Brangus population (Zayas et al., 2024b). Additionally, this region has been associated with Bos t. taurus introgression into Brahman during the formation of Brahman, likely due to strong selection for production traits (Koufariotis et al., 2018). These findings underscore the importance of PLAG1 in shaping Brangus genetics, particularly through selection pressures favoring traits that enhance productivity.
A large region on BTA17 showed a decrease in BOA heterozygosity in both Brangus populations, and the QTL enrichment analysis showed enrichment for reproduction traits such as maturity rate, maternal stillbirth, maternal calving ease, and daughter pregnancy rate (Supplementary Tables 1, 2). At the peak of this region is the LPS Responsive Beige-Like Anchor Protein (LRBA) gene, which has been associated to a variety of reproduction traits in Holstein (Cole et al., 2011). Though this region has not being previously identified as a signature of selection in other Brangus populations, it is has been identified as a signature of selection in Holstein, Hanwoo, and N’Dama cattle using Tajima’s D, XP-CLR, and XP-EHH population statistical methods (Taye et al., 2017). The region on BTA19: 26.97–28.36 MB which was found significant in both herds, overlapped the sex hormone binding globulin (SHBG) and Dynein Axonemal Heavy Chain 2 (DNAH2) genes. SHBG has previously been linked to prenatal fatalities in dairy cattle, suggesting its role in reproductive success and overall herd health (Fritz et al., 2013). The DNAH2 gene on the other hand is important in male reproductive biology. DNAH2 is a candidate gene associated with male infertility and sperm flagella abnormalities in various species (Fang et al., 2019; Li et al., 2019; Hwang et al., 2021). This region has also been suggested to overlap the scurs locus in cattle (Asai et al., 2004). Scurs is a condition where small bony growths develop in the same area as horns but are not firmly attached to the skull (Gehrke et al., 2020). The inheritance pattern of scurs is not fully understood, however there is evidence to suggest that the condition is only apparent when the locus is in the heterozygous state (Gehrke et al., 2020). Selection against the scurs phenotype may explain the decrease BOA heterozygosity seen in this region. However, another possibility is that these regions which experience a decrease in BOA heterozygosity are also linked to reproduction.
In the Williamson Brangus herd, a significant region on BTA27 (0.07–11.95 MB) demonstrated a marked increase in BOA heterozygosity. At the peak of this region is the Tracheal antimicrobial peptide (TAP) gene, TAP is a member of the β-defensin family and is expressed in the ciliated epithelium of the bovine trachea (Diamond et al., 2000). This gene is upregulated upon exposure to bacteria or bacterial lipopolysaccharide, showing its importance in innate immunity and disease resistance (Diamond et al., 2000). The genomic region on BTA18 (55.91–59.74 MB), found to be significant in both Brangus populations, was notably enriched for traits linked to reproductive success (Supplementary Tables 1, 2) shows a marked increase in BOA heterozygosity. Thus, this region’s genetic makeup could be contributing to these advantageous traits in the Brangus breed. A prominent candidate gene in this region is the Cytosolic Thiouridylase Subunit 1 (CTU1) gene, which has been significantly associated to calving ability in beef and dairy cattle (Purfield et al., 2015; Xiang et al., 2017). The results of our QTL enrichment analysis (Supplementary Table 2) also indicate a significant enrichment for length of productive life, and calving ability. Further enforcing this region’s importance in reproductive success, indicating that the increased BOA heterozygosity in this area may be an adaptive advantage, enhancing traits critical for effective breeding and longevity in cattle.
The changes in BOA heterozygosity observed in our study reflect the complex dynamics of selection in cattle genetics. Decreased heterozygosity, or increased homozygosity, often indicates strong selection pressures for specific alleles that confer advantageous traits. These regions can reveal critical genes influencing production, reproduction, and health, as seen with the PLAG1, LRBA, SHBG, and DNAH2 genes. Conversely, increased heterozygosity suggests a selective advantage for maintaining genetic diversity. This can enhance traits through heterosis, where crossbreeding leads to superior traits such as reproductive efficiency and longevity, as demonstrated by the reproductive performance of the CTU1 gene and the importance in innate immunity of the TAP gene.
Understanding these patterns provides valuable insights into the selection strategies and genetic foundations that shape the desirable traits in Brangus cattle. It highlights the importance of both homozygosity and heterozygosity in breeding programs aimed at optimizing cattle performance and health.
5 Conclusion
This study leverages genomic data to establish the MAB herd as a suitable reference population for examining population structure and breed composition in commercial Brangus herds. Our findings revealed higher-than-expected Angus breed fractions (62.5%), with averages of 71.65% and 68.70% in the two commercial Brangus herds. This reflects a strong selection preference for Angus genetics, likely due to their association with desirable production and meat quality traits. Our novel methodology enabled us to pinpoint specific genomic regions under selection for increased Angus ancestry. Notably, we observed increased Angus ancestry overlapping the MC1R gene on BTA18, which controls black coat color, and the NCAPG-LCORL locus on BTA6, a candidate gene associated with growth and carcass traits. We also identified a notable decrease in heterozygosity around PLAG1, a gene integral to growth, fertility, and carcass traits. Other regions associated with meat quality, production, and fertility showed an increase in Angus ancestry, while regions related to fertility, immune response, and thermotolerance displayed either increased Brahman origin or shifts in BOA heterozygosity. Understanding these selected regions can enhance insights into genetic pathways related to adaptation and production, supporting cattle production in subtropical and tropical climates.
Data availability statement
Publicly available datasets were analyzed in this study. This data can be found here: European Variation Archive (EVA), accession numbers PRJEB60100 and PRJEB75981.
Ethics statement
The animal study was approved by University of Florida Institutional Animal Care and Use Committee number 201003744. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
GZ: Data curation, Formal analysis, Methodology, Writing – original draft, Writing – review & editing. RM: Data curation, Formal analysis, Funding acquisition, Resources, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was funded by UF Agricultural Experimental Station Hatch FLA-ANS-005548 and USDA-NIFA Grant 2017-67007-26143. GZ is a USDA National Needs Fellow supported by USDA NIFA grant 2019-38420-28977 and Town Creek Farms.
Acknowledgments
Authors thank the owners and staff of the Seminole Tribe of Florida and the Williamson Cattle Company for their participation in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fanim.2024.1450639/full#supplementary-material
References
Alexander D. H., Novembre J., Lange K. (2009). Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664. doi: 10.1101/gr.094052.109
Alghamdi T. A., Krentz N. A. J., Smith N., Spigelman A. F., Rajesh V., Jha A., et al. (2022). Zmiz1 is required for mature β-cell function and mass expansion upon high fat feeding. Mol. Metab. 66, 101621. doi: 10.1016/j.molmet.2022.101621
Ali A. A., Khatkar M. S., Kadarmideen H. N., Thomson P. C. (2015). Additive and epistatic genome-wide association for growth and ultrasound scan measures of carcass-related traits in Brahman cattle. J. Anim. Breed. Genet. 132, 187–197. doi: 10.1111/jbg.12147
Álvarez Cecco P., Rogberg Muñoz A., Balbi M., Bonamy M., Munilla S., Forneris N. S., et al. (2022). Genome-wide scan for signatures of selection in the Brangus cattle genome. J. Anim. Breed. Genet. 139, 679–694. doi: 10.1111/jbg.12733
Asai M., Berryere T. G., Schmutz S. M. (2004). The scurs locus in cattle maps to bovine chromosome 19. Anim. Genet. 35, 34–39. doi: 10.1111/j.1365-2052.2003.01079.x
Baran Y., Pasaniuc B., Sankararaman S., Torgerson D. G., Gignoux C., Eng C., et al. (2012). Fast and accurate inference of local ancestry in latino populations. Bioinformatics 28, 1359–1367. doi: 10.1093/bioinformatics/bts144
Barrett T., Dowle M., Srinivasan A., Gorecki J., Chirico M., Hocking T., et al. (2024).data.table: extension of `data.frame. Available online at: https://CRAN.R-project.org/package=data.table (Accessed March 02, 2024).
Bedhane M., van der Werf J., Gondro C., Duijvesteijn N., Lim D., Park B., et al. (2019). Genome-wide association study of meat quality traits in hanwoo beef cattle using imputed whole-genome sequence data. Front. Genet. 10. Available at: https://www.frontiersin.org/articles/10.3389/fgene.2019.01235 (Accessed October 5, 2023).
Berton M. P., de Lemos M. V. A., Stafuzza N. B., Simielli Fonseca L. F., Silva D. B., dos S., et al. (2022). Integration analyses of structural variations and differential gene expression associated with beef fatty acid profile in Nellore cattle. Anim. Genet. 53, 570–582. doi: 10.1111/age.13242
Bouwman A. C., Daetwyler H. D., Chamberlain A. J., Ponce C. H., Sargolzaei M., Schenkel F. S., et al. (2018). Meta-analysis of genome-wide association studies for cattle stature identifies common genes that regulate body size in mammals. Nat. Genet. 50, 362–367. doi: 10.1038/s41588-018-0056-5
Burrow H. M. (2012). Importance of adaptation and genotype × environment interactions in tropical beef breeding systems. Animal 6, 729–740. doi: 10.1017/S175173111200002X
Burrow H. M. (2015). Genetic aspects of cattle adaptation in the tropics. Genet. cattle, 571–597. doi: 10.1079/9781780642215.0571
Chang C. C., Chow C. C., Tellier L. C. A. M., Vattikuti S., Purcell S. M., Lee J. J. (2015). Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience 4, 1–16. doi: 10.1186/s13742-015-0047-8
Cole J. B., Wiggans G. R., Ma L., Sonstegard T. S., Lawlor T. J., Crooker B. A., et al. (2011). Genome-wide association analysis of thirty one production, health, reproduction and body conformation traits in contemporary U.S. Holstein cows. BMC Genomics 12, 408. doi: 10.1186/1471-2164-12-408
Conomos M. P., Reiner A. P., Weir B. S., Thornton T. A. (2016). Model-free estimation of recent genetic relatedness. Am. J. Hum. Genet. 98, 127–148. doi: 10.1016/j.ajhg.2015.11.022
Cooke R. F., Daigle C. L., Moriel P., Smith S. B., Tedeschi L. O., Vendramini J. M. B. (2020). Cattle adapted to tropical and subtropical environments: social, nutritional, and carcass quality considerations. J. Anim. Sci. 98, skaa014. doi: 10.1093/jas/skaa014
Daldello E. M., Luong X. G., Yang C.-R., Kuhn J., Conti M. (2019). Cyclin B2 is required for progression through meiosis in mouse oocytes. Development 146 (8). doi: 10.1242/dev.172734
Decker J. E., McKay S. D., Rolf M. M., Kim J., Molina Alcalá A., Sonstegard T. S., et al. (2014). Worldwide patterns of ancestry, divergence, and admixture in domesticated cattle. PloS Genet. 10, e1004254. doi: 10.1371/journal.pgen.1004254
de Lemos M. V. A., Peripolli E., Berton M. P., Feitosa F. L. B., Olivieri B. F., Stafuzza N. B., et al. (2018). Association study between copy number variation and beef fatty acid profile of Nellore cattle. J. Appl. Genet. 59, 203–223. doi: 10.1007/s13353-018-0436-7
Diamond G., Kaiser V., Rhodes J., Russell J. P., Bevins C. L. (2000). Transcriptional regulation of β-defensin gene expression in tracheal epithelial cells. Infect. Immun. 68, 113–119. doi: 10.1128/IAI.68.1.113-119.2000
Elzo M. A., Johnson D. D., Wasdin J. G., Driver J. D. (2012). Carcass and meat palatability breed differences and heterosis effects in an Angus–Brahman multibreed population. Meat Sci. 90, 87–92. doi: 10.1016/j.meatsci.2011.06.010
Fang L., Jiang J., Li B., Zhou Y., Freebern E., Vanraden P. M., et al. (2019). Genetic and epigenetic architecture of paternal origin contribute to gestation length in cattle. Commun. Biol. 2, 1–11. doi: 10.1038/s42003-019-0341-6
Fritz S., Capitan A., Djari A., Rodriguez S. C., Barbat A., Baur A., et al. (2013). Detection of haplotypes associated with prenatal death in dairy cattle and identification of deleterious mutations in GART, SHBG and SLC37A2. PloS One 8, e65550. doi: 10.1371/journal.pone.0065550
Fonseca P. A. S., Suárez-Vega A., Marras G., Cánovas Á. (2020). GALLO: An R package for genomic annotation and integration of multiple data sources in livestock for positional candidate loci. Gigascience 9, giaa149. doi: 10.1093/gigascience/giaa149
Fortes M. R. S., Reverter A., Hawken R. J., Bolormaa S., Lehnert S. A. (2012). Candidate genes associated with testicular development, sperm quality, and hormone levels of inhibin, luteinizing hormone, and insulin-like growth factor 1 in brahman bulls1. Biol. Reprod. 87, 1–8. doi: 10.1095/biolreprod.112.101089
Gehrke L. J., Capitan A., Scheper C., König S., Upadhyay M., Heidrich K., et al. (2020). Are scurs in heterozygous polled (Pp) cattle a complex quantitative trait? Genet. Selection Evol. 52, 6. doi: 10.1186/s12711-020-0525-z
Gobena M., Elzo M. A., Mateescu R. G. (2018). Population structure and genomic breed composition in an angus–brahman crossbred cattle population. Front. Genet. 9. doi: 10.3389/fgene.2018.00090
Goszczynski D. E., Corbi-Botto C. M., Durand H. M., Rogberg-Muñoz A., Munilla S., Peral-Garcia P., et al. (2018). Evidence of positive selection towards Zebuine haplotypes in the BoLA region of Brangus cattle. Animal 12, 215–223. doi: 10.1017/S1751731117001380
Hwang J. Y., Nawaz S., Choi J., Wang H., Hussain S., Nawaz M., et al. (2021). Genetic defects in DNAH2 underlie male infertility with multiple morphological abnormalities of the sperm flagella in humans and mice. Front. Cell Dev. Biol. 9. doi: 10.3389/fcell.2021.662903
Jiang Q., Sun B., Liu Q., Cai M., Wu R., Wang F., et al. (2019). MTCH2 promotes adipogenesis in intramuscular preadipocytes via an m6A-YTHDF1–dependent mechanism. FASEB J. 33, 2971–2981. doi: 10.1096/fj.201801393RRR
Knaus B. J., Grünwald N. J. (2017). VCFR: a package to manipulate and visualize variant call format data in R. Mol. Ecol. Resour. 17, 44–53. doi: 10.1111/1755-0998.12549
Koufariotis L., Hayes B. J., Kelly M., Burns B. M., Lyons R., Stothard P., et al. (2018). Sequencing the mosaic genome of Brahman cattle identifies historic and recent introgression including polled. Sci. Rep. 8, 17761. doi: 10.1038/s41598-018-35698-5
Leal-Gutiérrez J. D., Rezende F. M., Reecy J. M., Kramer L. M., Peñagaricano F., Mateescu R. G. (2020). Whole genome sequence data provides novel insights into the genetic architecture of meat quality traits in beef. Front. Genet. 11. doi: 10.3389/fgene.2020.538640
Lewis J., Abas Z., Dadousis C., Lykidis D., Paschou P., Drineas P. (2011). Tracing cattle breeds with principal components analysis ancestry informative SNPs. PLoS ONE. 6 (4), e18007. doi: 10.1371/journal.pone.0018007
Li Y., Sha Y., Wang X., Ding L., Liu W., Ji Z., et al. (2019). DNAH2 is a novel candidate gene associated with multiple morphological abnormalities of the sperm flagella. Clin. Genet. 95, 590–600. doi: 10.1111/cge.13525
Li Z., He J., Yang F., Yin S., Gao Z., Chen W., et al. (2023). A look under the hood of genomic-estimated breed compositions for brangus cattle: What have we learned? Front. Genet. 14. Available at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1080279 (Accessed May 21, 2024).
Lindholm-Perry A. K., Kuehn L. A., Oliver W. T., Sexten A. K., Miles J. R., Rempel L. A., et al. (2013). Adipose and muscle tissue gene expression of two genes (NCAPG and LCORL) located in a chromosomal region associated with cattle feed intake and gain. PLoS One 8, e80882. doi: 10.1371/journal.pone.0080882
Lindholm-Perry A. K., Sexten A. K., Kuehn L. A., Smith T. P., King D. A., Shackelford S. D., et al. (2011). Association, effects and validation of polymorphisms within the NCAPG - LCORL locus located on BTA6 with feed intake, gain, meat and carcass traits in beef cattle. BMC Genet. 12, 103. doi: 10.1186/1471-2156-12-103
McVean G. (2009). A genealogical interpretation of principal components analysis. PloS Genet. 5, e1000686. doi: 10.1371/journal.pgen.1000686
Manichaikul A., Mychaleckyj J. C., Rich S. S., Daly K., Sale M., Chen W. M. (2010). Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873. doi: 10.1093/bioinformatics/btq559
Mourot M., Dufort I., Gravel C., Algriany O., Dieleman S., Sirard M.-A. (2006). The influence of follicle size, FSH-enriched maturation medium, and early cleavage on bovine oocyte maternal mRNA levels. Mol. Reprod. Dev. 73, 1367–1379. doi: 10.1002/mrd.20585
Nasti T. H., Timares L. (2015). MC1R, eumelanin and pheomelanin: their role in determining the susceptibility to skin cancer. Photochem. Photobiol. 91, 188–200. doi: 10.1111/php.12335
Paim T., do P., Hay E. H. A., Wilson C., Thomas M. G., Kuehn L. A., et al. (2020a). Dynamics of genomic architecture during composite breed development in cattle. Anim. Genet. 51, 224–234. doi: 10.1111/age.12907
Paim T., do P., Hay E. H. A., Wilson C., Thomas M. G., Kuehn L. A., et al. (2020b). Genomic breed composition of selection signatures in brangus beef cattle. Front. Genet. 11. doi: 10.3389/fgene.2020.00710
Patterson N., Price A. L., Reich D. (2006). Population structure and eigenanalysis. PLoS Genet. 2 (12), e190. doi: 10.1371/journal.pgen.0020190
Purfield D. C., Bradley D. G., Evans R. D., Kearney F. J., Berry D. P. (2015). Genome-wide association study for calving performance using high-density genotypes in dairy and beef cattle. Genet. Sel Evol. 47, 47. doi: 10.1186/s12711-015-0126-4
R Core Team (2023). R: A language and Environment for Statistical Computing (Vienna, Austria: R Foundation for Statistical Computing). Available at: https://www.R-project.org/.
Santos Silva D. B., Fonseca L. F. S., Magalhães A. F. B., Muniz M. M. M., Baldi F., Ferro J. A., et al. (2020). Transcriptome profiling of muscle in Nelore cattle phenotypically divergent for the ribeye muscle area. Genomics 112, 1257–1263. doi: 10.1016/j.ygeno.2019.07.012
Sarlo Davila K. M., Howell A., Nunez A., Orelien A., Roe V., Rodriguez E., et al. (2020). Genome-wide association study identifies variants associated with hair length in Brangus cattle. Anim. Genet. 51, 811–814. doi: 10.1111/age.12970
Seabury C. M., Oldeschulte D. L., Saatchi M., Beever J. E., Decker J. E., Halley Y. A., et al. (2017). Genome-wide association study for feed efficiency and growth traits in U.S. beef cattle. BMC Genomics 18, 386. doi: 10.1186/s12864-017-3754-y
Shringarpure S. S., Bustamante C. D., Lange K., Alexander D. H. (2016). Efficient analysis of large datasets and sex bias with ADMIXTURE. BMC Bioinf. 17, 1–6. doi: 10.1186/s12859-016-1082-x
Sosa F., Carmickle A. T., Jiménez-Cabán E., Ortega M. S., Dikmen S., Negrón-Pérez V., et al. (2021). Inheritance of the SLICK1 allele of PRLR in cattle. Anim. Genet. 52, 887–890. doi: 10.1111/age.13145
Sosa F., Santos J. E. P., Rae D. O., Larson C. C., Macchietto M., Abrahante J. E., et al. (2022). Effects of the SLICK1 mutation in PRLR on regulation of core body temperature and global gene expression in liver in cattle. animal 16, 100523. doi: 10.1016/j.animal.2022.100523
Taye M., Lee W., Jeon S., Yoon J., Dessie T., Hanotte O., et al. (2017). Exploring evidence of positive selection signatures in cattle breeds selected for different traits. Mamm Genome 28, 528–541. doi: 10.1007/s00335-017-9715-6
Turner S., Armstrong L. L., Bradford Y., Carlsony C. S., Crawford D. C., Crenshaw A. T., et al. (2011). Quality control procedures for genome-wide association studies. Curr. Protoc. Hum. Genet. 68, 1.19.1–1.19.18. doi: 10.1002/0471142905.hg0119s68
Warburton C. L., Costilla R., Engle B. N., Moore S. S., Corbet N. J., Fordyce G., et al. (2023). Concurrently mapping quantitative trait loci associations from multiple subspecies within hybrid populations. Heredity 131,, 350–360. doi: 10.1038/s41437-023-00651-4
Weir B. S., Cockerham C. C. (1984). Estimating F-statistics for the analysis of population structure. Evolution 38, 1358–1370. doi: 10.2307/2408641
Wickham H. (2009). ggplot2: Elegant Graphics for Data Analysis (New York, NY: Springer New York). doi: 10.1007/978-0-387-98141-3
Wickham H., Vaughan D., Girlich M. (2024). tidyr: Tidy Messy Data. Available online at: https://CRAN.R-project.org/package=tidyr.
Wolf Horrell E. M., Boulanger M. C., D’Orazio J. A. (2016). Melanocortin 1 receptor: structure, function, and regulation. Front. Genet. 7. Available at: https://www.frontiersin.org/articles/10.3389/fgene.2016.00095 (Accessed October 10, 2023).
Xiang R., MacLeod I. M., Bolormaa S., Goddard M. E. (2017). Genome-wide comparative analyses of correlated and uncorrelated phenotypes identify major pleiotropic variants in dairy cattle. Sci. Rep. 7, 9248. doi: 10.1038/s41598-017-09788-9
Yin L., Zhang H., Tang Z., Xu J., Yin D., Zhang Z., et al. (2021). rMVP: A memory-efficient, visualization-enhanced, and parallel-accelerated tool for genome-wide association study. Genomics Proteomics Bioinf. 19, 619–628. doi: 10.1016/j.gpb.2020.10.007
Zayas G. A., Dikmen S., Mateescu R. G., Hansen P. J. (2024a). Maintaining breed integrity: Successful introgression of the SLICK1 allele into the Holstein breed. J. Heredity, esae057. doi: 10.1093/jhered/esae057
Zayas G. A., Rodriguez E. E., Hernandez A. S., Rezende F. M., Mateescu R. G. (2024b). Exploring genomic inbreeding and selection signatures in a commercial Brangus herd through functional annotation. J. Appl. Genet. 65 (2), 383–394. doi: 10.1007/s13353-024-00859-y
Zayas G. A., Rodriguez E., Hernandez A., Rezende F. M., Mateescu R. G. (2024c). Breed of origin analysis in genome-wide association studies: enhancing SNP-based insights into production traits in a commercial Brangus population. BMC Genomics 25, 654. doi: 10.1186/s12864-024-10465-1
Zhang F., Wang Y., Mukiibi R., Chen L., Vinsky M., Plastow G., et al. (2020). Genetic architecture of quantitative traits in beef cattle revealed by genome wide association studies of imputed whole genome sequence variants: I: feed efficiency and component traits. BMC Genomics 21, 36. doi: 10.1186/s12864-019-6362-1
Keywords: Angus, Brahman, adaptation, composite breeds, artificial selection, BOA
Citation: Zayas GA and Mateescu RG (2025) Genomic signatures of selection in Brangus cattle revealing the genetic foundations of adaptability and production traits using a breed of origin approach. Front. Anim. Sci. 5:1450639. doi: 10.3389/fanim.2024.1450639
Received: 17 June 2024; Accepted: 27 December 2024;
Published: 20 January 2025.
Edited by:
Kristopher Irizarry, Western University of Health Sciences, United StatesReviewed by:
Juliana Petrini, Clinica do Leite Ltda, BrazilMartina Miluchová, Slovak University of Agriculture, Slovakia
Copyright © 2025 Zayas and Mateescu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gabriel A. Zayas, Z3pheWFzOTdAdWZsLmVkdQ==