 Madison T. Henniger1
Madison T. Henniger1 Troy N. Rowan1,2,3Jonathan E. Beever1,2,3
Troy N. Rowan1,2,3Jonathan E. Beever1,2,3 Pierre-Yves Mulon2
Pierre-Yves Mulon2 Joe S. Smith2
Joe S. Smith2 Brynn H. Voy1
Brynn H. Voy1 Jim E. Wells4
Jim E. Wells4 Larry A. Kuehn4
Larry A. Kuehn4 Phillip R. Myer1*
Phillip R. Myer1*- 1Department of Animal Science, University of Tennessee, Knoxville, TN, United States
- 2Department of Large Animal Clinical Sciences, University of Tennessee, Knoxville, TN, United States
- 3Genomics Center for the Advancement of Agriculture, University of Tennessee Institute of Agriculture, Knoxville, TN, United States
- 4USDA-US Meat Animal Research Center, Clay Center, NE, United States
The rumen microbiome provides approximately 70% of the required energy for the host by converting low-quality feedstuffs into usable energy for ruminants. The energy produced by the microorganisms is subsequently absorbed through the rumen epithelium and used towards growth and energy maintenance. There is evidence that ruminal epimural microbes directly interact with the rumen epithelium, acting as an intermediary communicator between the rumen liquid fraction and the host. Epimural microbiota have been demonstrated to be distinct from the ruminal liquid microbiome and perform unique roles within the rumen environment. However, methods to sample epimural communities from the rumen wall are limited and typically invasive, requiring animal fistulation or harvesting. To characterize the epimural communities present on the rumen wall, a novel and minimally-invasive surgical method was developed to swab the epithelium of the ventral sac of the rumen. The objective of this study was to validate this sampling method by comparing epimural and liquid fraction bacterial communities. During a 70-day feeding trial, Angus steers (n = 45) were sampled on day 35 using the novel surgery method and tubed on day 70 to sample rumen liquid content. Genomic DNA was used to generate amplicon libraries of the V4 region of the 16S rRNA gene. There were no differences between alpha diversity indices when comparing rumen versus epimural bacterial communities (P > 0.05). The Bray-Curtis dissimilarity was used to ordinate ASV counts, and then tested for differences between rumen and epimural communities using a PERMANOVA with 999 permutations (P < 0.05). Differential abundances of bacterial communities were tested using ANCOM-BC and MaAsLin2, where significance was determined by Q < 0.05 and overlap between both analysis methods. Within the 91 taxa that differed in abundance, 451 ASVs were found to be different between sample types (Q < 0.05). Unique ASVs associated with Prevotella, Succinivibrio, family-level Eubacterium, and family-level Succinivibrio were in greater abundance for the rumen epithelial-associated bacterial communities (Q < 0.05). The results demonstrate that the novel method of sampling from the rumen wall can capture differences between epimural and ruminal fluid bacterial communities, thus facilitating studies investigating the interactions between epimural bacteria with the host.
1 Introduction
The rumen epithelium plays a critical role in the uptake of short chain fatty acids, otherwise known as volatile fatty acids (VFAs), which provide approximately 70% of required energy to the ruminant host (Bergman, 1990). These VFAs are the primary fermentation product from the rumen microbiome, which consists of bacterial, archaeal, protozoal, and fungal communities. Due to the unique fermentative capabilities of the rumen, the rumen microbiome has been associated with the host phenotypes of feed efficiency and methane production (Roehe et al., 2016; Shabat et al., 2016; Li et al., 2017; Li et al., 2022; Andersen et al., 2023). Thus, the rumen microbiome plays a critical role in supplying energy for the host, which in turn alters production-relevant parameters.
The rumen microbiome has been characterized by three main types of ruminal microbes, primarily based on digesta type and tissues that the microbial communities are adherent to within the rumen. The common categories consist of the fiber-adherent, planktonic (liquid-associated), and epimural (papillae-associated) microbial communities. The fiber-adherent bacteria represent the majority of the ruminal bacterial biomass, approximately 70% (Millen et al., 2016), and serve the main role in digestion (McAllister et al., 1994; Larue et al., 2005). Planktonic bacterial communities constitute approximately 30% (Millen et al., 2016) of the bacterial biomass, synergistically working with solid-adherent bacteria for nutrient breakdown (McAllister et al., 1994). Overall, the differences in function and diversity between the fiber-adherent and planktonic fractions of the rumen have been well-studied across species (Larue et al., 2005; Cho et al., 2006; Mao et al., 2015; De Mulder et al., 2016; Liu et al., 2016; Schären et al., 2017). Epimural microbes, while making up only 1% of the total biomass (Czerkawski, 1986; Millen et al., 2016), directly interact with the rumen epithelium, potentially modulating host-microbiome interactions.
Early studies have identified that bacterial communities that adhere to the rumen wall are distinctly different from the liquid and solid fractions (Cheng and Wallace, 1979; McCowan et al., 1980; Sadet et al., 2007). Diet is the primary driver of planktonic and fiber-adherent communities (Tajima et al., 2001); however, the degree to which diet impacts epimural microbiota is unknown. There is evidence that diet can alter the epimural microbiome, especially in diets that induce acidosis (Sadet-Bourgeteau et al., 2010; Chen et al., 2011; Liu et al., 2013; Liu et al., 2015; Petri et al., 2020). Regardless of dietary impact, epimural microbes have been primarily implicated across ruminant species for their distinct functions in the rumen and their influence on oxygen scavenging (Cheng et al., 1979), urea hydrolysis (Cheng and Wallace, 1979), nutrient absorption (Mao et al., 2015; Zhao et al., 2017), and recycling of epithelial tissues (Dinsdale et al., 1980). Thus, epimural microbial communities may play significant roles in modulating health status and feed efficiency by protecting the ruminal epithelium and also in an intermediary role in nutrient uptake.
Many studies have focused on characterizing the planktonic and fiber-adherent communities, which include associations with health status (Khafipour et al., 2009; Zhang et al., 2022), production-relevant traits such as feed efficiency (Jami et al., 2014; Myer et al., 2015), and methane production (Wallace et al., 2019; Martínez-Álvaro et al., 2022). Yet only within the last decade have researchers begun to deeply investigate the abundance and functions of the epimural communities across ruminant species. This includes characterizing the epimural microbiome (Mao et al., 2015; De Mulder et al., 2016; Sbardellati et al., 2020) and examining the influence of ruminal acidosis challenges on the epimural microbiome (Penner et al., 2009; Petri et al., 2013; Wetzels et al., 2017; Li et al., 2019b). However, identifying the epimural microbiome across species and studies can be challenging due to technological differences, data processing, and analysis (Anderson et al., 2021; Pacífico et al., 2021). Further, the ruminal wall is difficult to access, thus resulting in many of these studies using small animal numbers.
Current methods to sample epimural microbiota from the rumen wall typically utilize animal fistulation or harvesting. To improve bovine rumen microbiome studies and recognize the limitations of terminal animal research, novel methods must be developed to effectively and comprehensively sample epimural microorganisms. Therefore, the objectives of this project were to assess an alternative, minimally-invasive technique for sampling microbiota from the rumen wall and validate the method’s ability to capture differences between the rumen liquid and epimural bacterial communities.
2 Materials and methods
2.1 Animal ethics statement
This experiment was approved by the University of Tennessee Animal Care and Use Committee.
2.2 Experimental plan
To test and validate the method to collect epimural microbiota, 45 Angus steers were enrolled in a 70-day feeding trial. During this period, weight and feed intake was measured to determine health status as a response to the surgery method. Steers were kept on the same diet throughout the study, being a total mixed ration (TMR) of sudex (sorghum-sudangrass hybrid) and a growing Co-Op diet (Tennessee Farmer’s Co). The TMR consisted of 14.72% crude protein and 66.19% TDN on a dry-matter basis.
2.3 Surgery method for rumen wall sampling
On day 35 of the 70-day trial, steers were restrained in a chute and a 2-in × 2-in square of hair coat was clipped using a #40 blade ventrally to the lumbar transverse processes and caudally to the last rib in the left paralumbar fossa. The skin was surgically prepared using alternate swabbing of povidone-iodine scrub and isopropyl alcohol. The area was locally anesthetized with subcutaneous and intramuscular infiltration of 10 mL of 2% lidocaine. Six to ten minutes following lidocaine administration, a 1.5-cm incision was made in the center of the prepped area using a #10 scalpel blade. A 10-mm diameter trocar-cannula unit was then inserted through the body wall until penetration of the lumen of the dorsal sac of the rumen. A swab was introduced through the cannula and oriented towards the rumen wall, arching and swabbing against the wall several times. The swab was removed and immediately placed into 1 mL of tris-EDTA buffer in a 15 mL conical tube and flash-frozen in liquid nitrogen. The trocar was retrieved from the body wall and the incision was closed by USP #2 suture material. Samples were stored at -80°C until further analysis. The surgery method for rumen wall sampling is depicted in Figure 1.
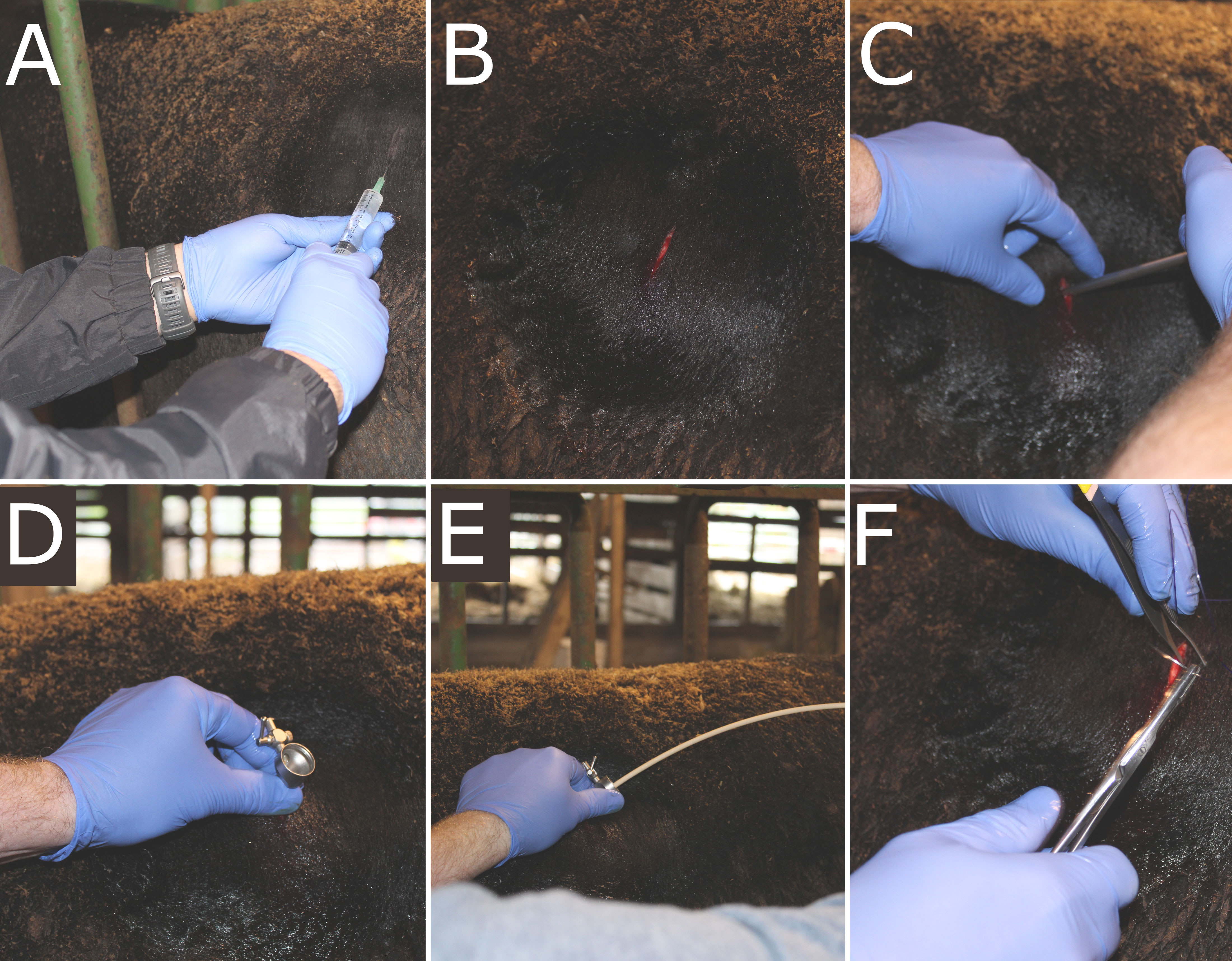
Figure 1 Surgery method for sampling from the rumen wall. (A) Incision site was prepped using alternating povidone iodine scrub and 70% isopropyl alcohol, and lidocaine was administered. (B) A 1.5-cm incision was made, followed by (C) insertion of a 10-mm trocar. (D) Once the trocar was fully inserted into the lumen of the dorsal sac, (E) the swab was introduced and arced to swab against the ventral sac of the rumen wall. Once the swab and trocar were removed, (F) the incision site was closed via USP #2 sutures.
2.4 Surgery scoring
Steers were monitored daily post-operatively for signs of inflammation or discomfort. Two weeks after the surgery date, steers incision sites were measured in height and width of inflammation and swelling. During surgery site scoring, remaining sutures were removed and the presence or absence of sutures at the time of removal was noted. Surgery sites were then scored by veterinarians from the University of Tennessee Large Animal Clinical Sciences and ranged from 0 (no discharge and less than 20 × 20 mm site) to 3 (swelling greater than 60 × 60 mm) (Table 1). Any other indication of swelling or discharge was noted at that time.
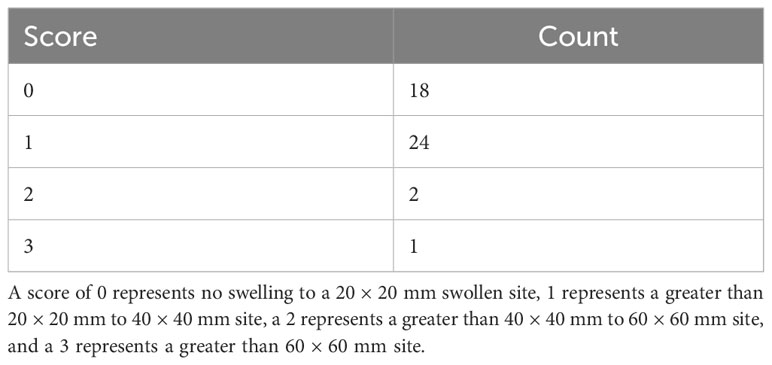
Table 1 Incision scores of steers two weeks after the trocar cannulation surgery to access the rumen wall.
2.5 Sampling of rumen fluid
On day 70 of the 70-day trial, steers were restrained in a chute and an orogastric tubing system was used to obtain rumen fluid. The initial extraction from within the ventral sac of the rumen was discarded to avoid saliva contamination. Following rumen fluid extraction, the tubing system was flushed with 70% ethanol and rinsed with water to reduce between-animal contamination. Approximately 50 mL of rumen liquid was collected per animal and flash-frozen in liquid nitrogen. Samples were stored at -80°C until further analysis.
2.6 Extraction, amplification, and sequencing of bacterial DNA
Genomic DNA was extracted from samples using a previously validated procedure by Yu and Morrison (Yu and Morrison, 2004). In brief, 0.2 g of sample were added to 2 mL bead beating tubes (Zymo Research, Orange, CA, United States) and 1 mL of lysis buffer (500 mM NaCl, 50 mM Tris-HCl pH 8.0, 50 mM EDTA, and 4% sodium dodecyl sulfate [SDS]) was added to tubes. Samples were homogenized at 21 Hz for three minutes and then incubated at 70°C for 15 minutes, with three inversions every five minutes. Samples were centrifuged for 10 minutes at 16,000 × g and 4°C. The supernatant was aspirated and placed into fresh 1.5 mL microcentrifuge tubes, 300 μL of lysis buffer was added to bead-beating tubes, and the steps above were repeated. Following mechanical and chemical lysis of cells, samples underwent isopropanol precipitation of nucleic acids. To each lysate tube, 260 μL of 10 M ammonium acetate was added and incubated on ice for five minutes. Supernatants were transferred to two 1.5 mL microcentrifuge tubes to add one volume of isopropanol. Following, samples were incubated on ice for 30 minutes. Samples were centrifuged for 15 minutes at 16,000 × g at 4°C to spin down the nucleic acid pellet. The supernatant was discarded and the resulting nucleic acid pellet was washed with 200 μL of 70% ethanol. The pellet was air-dried for five minutes at room temperature. Subsequently, 100 μL of Buffer TE was added to dissolve the pellet and aliquots were pooled. The QIAGEN DNeasy Blood and Tissue Kit was used for downstream purification (QIAGEN, Hilden, Germany). Proteinase K and Buffer AL were added to each sample per company protocol and samples were incubated at 70°C for 10 minutes. Following, 200 μL of ethanol was added. Samples were transferred to a QIAamp column and centrifuged for one minute at 16,000 × g. Buffers AW1 and AW2 were added at 500 μL sequentially, with flow through being discarded between each buffer after centrifugation. Columns were then dried at room temperature for one minute. Columns were then placed in a fresh 1.5 mL microcentrifuge tube for final elution. To elute DNA, 70 μL of Buffer AE was added to the column membrane, incubated at room temperature for 2 minutes, and then 30 μL of Buffer AE was added. Samples were centrifuged for one minute at 16,000 × g and 4°C. The quality of DNA was assessed using a 1% TBE agarose gel and quantity was measured using a DeNovix spectrophotometer (DeNovix Inc., Wilmington, DE, United States). Samples were diluted to 10 ng/μL before polymerase chain reaction (PCR) amplification.
Amplicon sequencing of the 16S rRNA region was performed at the University of Tennessee Genomics Core, following their standard operating procedures of a two-step PCR. The V4 region of the 16S rRNA gene was amplified from the extracted DNA using primers 515Fb (GTGYCAGCMGCCGCGGTAA) (Parada et al., 2016) and 806Rb (GGACTACNVGGGTWTCTAAT) (Apprill et al., 2015), modified with adapters for Illumina MiSeq sequencing. The initial PCR consisted of 2 × KAPA HiFi HotStart ReadyMix Taq (Roche, Indianapolis, IN, United States), 1.5 μM each primer, and 2.5 μL template DNA. The reaction consisted of 3 min at 95°C, followed by 25 cycles of 95°C for 30 s, 55°C for 30 s, and 72°C for 30 s, with a final extension at 72°C for 5 min. Successful PCR amplification was confirmed via gel electrophoresis on a 2% agarose gel. The PCR product was then purified using 20 uL of AMPure XP beads (Agencourt, Beverly, MA, United States) and ethanol washes, then eluted in 50 uL of Tris-HCl. Nextera XT indexes (Illumina, Inc., San Diego, CA, United States) were then added to the PCR products using a second, reduced-cycle PCR, such that each sample had a unique combination of forward and reverse indexes. This reduced reaction consisted of 3 min at 95°C, followed by 8 cycles of 95°C for 30 s, 55°C for 30 s, and 72°C for 30 s, with a final extension at 72°C for 5 min. The products were purified again using 56 uL of AMPure XP beads, with ethanol washes and a final elution in 25 uL Tris-HCl. Samples were quantified on a NanoDrop spectrophotometer (Fisher Scientific International, Inc., Hampton, NH, United States) and pooled to approximately equal concentrations. Final product sizes and concentrations were confirmed on an Agilent Bioanalyzer (Santa Clara, CA, United States) using the standard sensitivity kit. The final library was then diluted to 4 pM, and combined with 20% of a 10 pM PhiX library control (Illumina, Inc., San Diego, CA, United States), and run paired-end 250 nucleotides on a v2, 500-cycle flow cell of an Illumina MiSeq sequencer at the University of Tennessee Genomics Core.
2.7 Processing of 16S rRNA gene sequences
Resultant fastq sequences were analyzed in the RStudio environment (R version 3.6.2). Quality was assessed using package ‘fastqcr’ v0.1.2 (Alboukadel, 2019) and sequences were filtered and trimmed using the function filterAndTrim from the package ‘phyloseq’ v1.40.0 (McMurdie and Holmes, 2013). Parameters for filterAndTrim included a truncLen of 240 nucleotides for forward and reverse reads. The trimLeft parameter was used to remove the first 20 nucleotides of reverse reads. Maximum expected errors were 2 for forward reads and 2 for reverse reads. Filtered reads were inspected for an average quality score (Q) of 30 or greater. Reads with less than Q30 were removed. Error rates were learned using the learnErrors function in the ‘dada2’ package v.1.24.0 (Callahan et al., 2016; Prodan et al., 2020). Sequences were then denoised using the dada function, which produced amplicon sequence variants (ASVs) for each sample. Denoised forward and reverse reads were merged using the mergePairs function in ‘dada2’, with a minimum of 12 bp overlap. A sequence table was then created from the merged reads and sequence lengths were checked. Chimeras were removed from data using removeBimeraDenovo with the consensus method. Taxa were assigned to the SILVA 138.1 database with a minimum bootstrap confidence of 80 using assignTaxonomy (Quast et al., 2012; Yilmaz et al., 2013; Glöckner et al., 2017; Henderson et al., 2019). The ASV table, assigned taxa, and metadata were merged into a phyloseq object for downstream analysis. Taxa that were identified as unclassified at any rank were reassigned to their last known taxonomic rank and stored in a separate phyloseq object. Alpha diversity was measured using the observed ASVs, Chao1 metric, and Shannon Diversity Index using the estimate_richness function from the ‘phyloseq’ package. Next, sample counts were transformed and ordinated using Bray-Curtis Dissimilarity to calculate distances for a principal coordinates analysis (PCoA) to assess beta diversity using ‘vegan’ v2.6.2 (Oksanen et al., 2022). The core_members function in the ‘microbiome’ package (v1.18.0) (Lahti et al., 2020) was used to determine within-study core epimural bacteria based on ASVs with greater than 1% detection and 90% prevalence across all epimural samples.
2.8 Statistical analyses
Scores from the incision sites were evaluated in RStudio (R version 3.6.2). A histogram was created to visually assess the distribution of scores.
The alpha diversity measurements of observed ASVs, Chao1, and Shannon Diversity Index were visually assessed for normality and then tested using a Shapiro-Wilks test (W), with normality determined at W > 0.90 and P > 0.05. A Kruskal-Wallis H test was then used to test differences between sampling sites. Next, sample counts were transformed from total counts and distances were calculated by the Bray-Curtis Dissimilarity Matrix to calculate beta diversity measurements. Following, dispersion estimates were calculated and then distances were tested by a permutational multivariate analysis of variance (PERMANOVA) with 999 permutations using the adonis function in the package ‘vegan’ (Oksanen et al., 2007). Significance was determined at P < 0.05 for all analyses.
To test for differential abundances of bacterial communities between the rumen wall and the ruminal liquid fraction, the ‘ANCOMBC’ package v1.6.2 (Lin and Peddada, 2020) was used, as recommended by Nearing and others. (Nearing et al., 2022). For ANCOM-BC, the phyloseq object was used with the formula of sample type for the ancombc function, with the fixed effect of sampling site. Further, ASVs with prevalence less than 10% across samples were removed from the analysis. Multiple testing was addressed using Benjamini-Hochberg corrections (Benjamini and Hochberg, 1995). Due to the nature of the samples, there were structural zeroes in the data, where taxa were classified as structural zeroes based on their asymptotic lower bounds. A conservative variance estimator was used for the test statistic based on the assumption that there were large differentially abundant taxa. Significance was determined at Q < 0.05.
Further, the ‘MaAsLin2’ package v1.10.0 (Mallick et al., 2021) was used to test for differential abundances of bacterial communities between the rumen wall and the ruminal liquid fraction. Using the Maaslin2 function, the ASV counts were normalized using the cumulative sum scaling (CSS) method (Paulson et al., 2013). Counts were log-transformed and the linear model was used, as recommended by package developers (Mallick et al., 2021). The statistical analyses used are further supported by Nearing et al. (Nearing et al., 2022). To maintain consistency and quality control, ASVs with a less than 10% prevalence across samples were removed from the analysis. The minimum abundance for each feature was set to 0.0001. Fixed effects included sample type, which was either epimural or liquid samples. Multiple testing was addressed using Benjamini-Hochberg corrections (Benjamini and Hochberg, 1995), with α set to 0.05.
The significantly different features between the ruminal liquid and epimural samples across both ANCOM-BC and MaAsLin2 were used for reporting differential abundance data.
3 Results
3.1 Surgery results
Visual assessment scores of the surgical site two weeks following surgery revealed a right-skewed distribution, with the majority of animals within the 0-1 range (Table 1). Within score ranges, 18 steers had a score of 0, 24 had a score of 1, two had a score of 2, and one had a score of 3. In addition, slight discharge around the incision site was present in four animals, irrespective of incision site score.
3.2 Sequencing results
Libraries were sequenced on an Illumina MiSeq and yielded an average of 157,741 read pairs per sample, with a minimum of 35,498 reads and a maximum of 319,966 reads. After taxonomic assignment, 15,405 taxa were identified out of the 90 samples. Within the rumen wall bacterial communities, 23 phyla and 212 genera were identified. When analyzing the ruminal liquid fraction, 26 phyla and 272 genera were assigned.
3.3 Alpha and beta diversity of bacterial communities
Analysis of the alpha diversity indices of observed ASVs, Chao1, and Shannon Diversity Index did not reveal differences in richness and evenness of ASVs between the rumen wall and rumen liquid bacterial communities (P > 0.05) (Figure 2). However, when assessing distances measured by Bray-Curtis dissimilarity, there were significant differences based on sampling site (P < 0.05) (Figure 3).
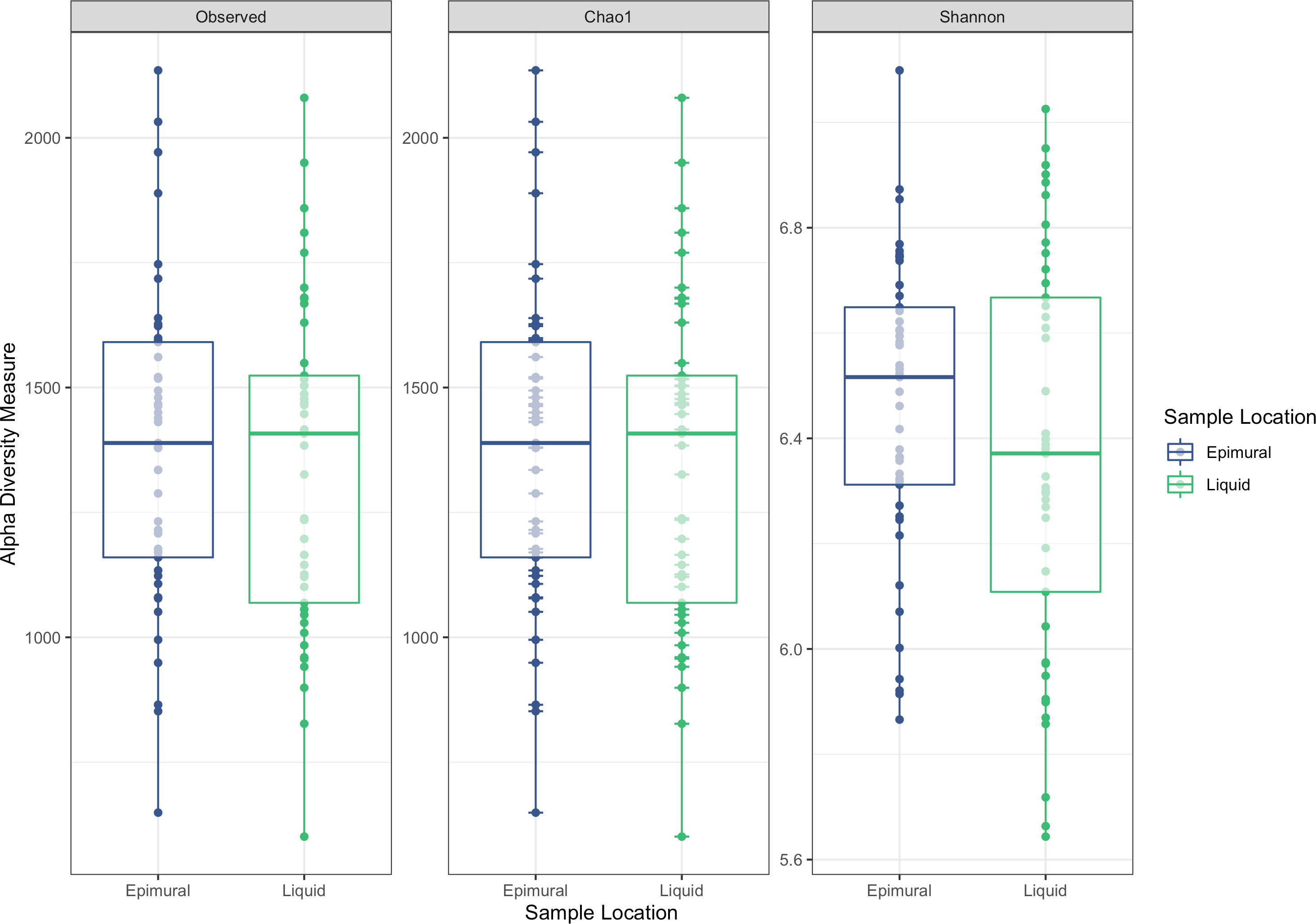
Figure 2 Alpha-diversity measurements between epimural and liquid bacterial communities. Measurements include the observed ASVs, Chao1, and Shannon Diversity Index. Epimural communities are represented in blue and liquid communities are represented in green.
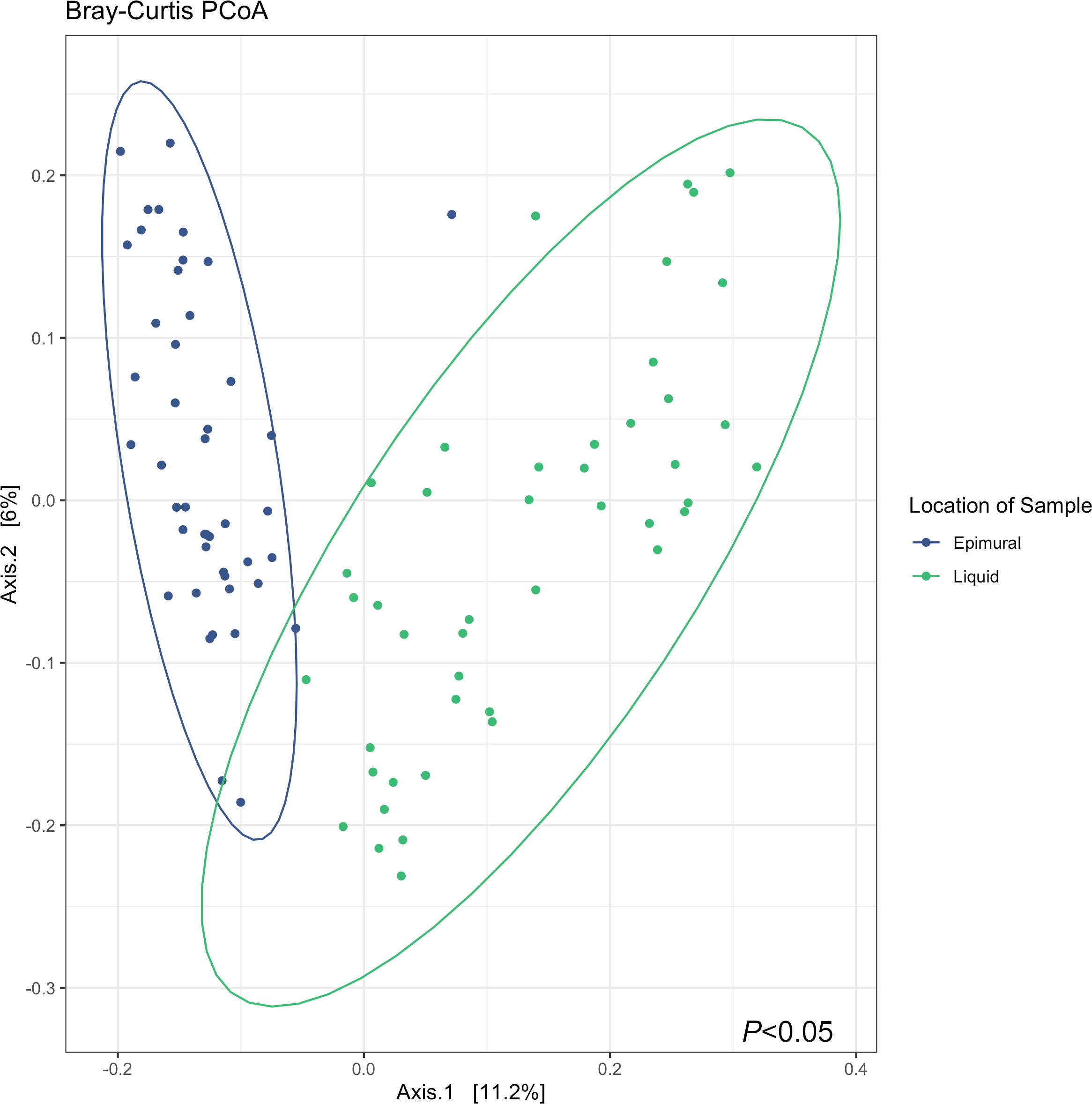
Figure 3 Bray-Curtis principle coordinates analysis (PCoA) describing the dissimilarity between sample locations, where the epimural samples are represented in blue, and the liquid samples are represented in green. Testing differences with a PERMANOVA with 999 permutations revealed differences in between sample diversity (P < 0.05).
3.4 Abundances of epimural bacterial communities
The top phyla present in the epimural bacterial communities were Bacteroidota (46.94%), Firmicutes (39.62%), and Proteobacteria (6.07%), representing greater than 90% of total communities. Verrucomicrobiota (2.43%) and Spirochaetota (1.66%) followed in abundance. Other phyla present represented less than 1% of the relative abundance.
The top ten bacterial genera within the epimural bacterial communities were Prevotella (21.03%), Succiniclasticum (6.99%), Rikenellaceae RC9 gut group (6.03%), Christensenellaceae R-7 group (3.85%), Ruminobacter (3.20%), Ruminococcus (2.71%), Ruminococcaceae NK4A214 group (2.27%), Prevotellaceae UCG-003 (2.09%), Treponema (1.37%), and Prevotellaceae UCG-001 (1.19%). There were 197 genera present at less than 1% of the relative abundance. Unassigned reads to the genus level represented 28.49% of the relative abundance. All bacterial communities identified are present in Supplementary Table 1.
3.5 Abundances of liquid-associated bacterial communities
The top phyla in the ruminal liquid bacterial communities, representing greater than 90% of the total relative abundance, were Bacteroidota (52.17%), Firmicutes (34.96%), and Proteobacteria (4.65%). Other phyla present at greater than 1% abundance were Verrucomicrobiota (3.05%), Spirochaetota (1.47%), and Patescibacteria (1.12%).
The top ten bacterial genera within the planktonic communities were Prevotella (26.26%), Rikenellaceae RC9 gut group (5.54%), Succiniclasticum (4.49%), Christensenellaceae R-7 group (2.62%), Ruminobacter (2.47%), Prevotellaceae UCG-003 (2.10%), Ruminococcus (1.78%), Prevotellaceae UCG-001 (1.46%), Ruminococcaceae NK4A214 group (1.36%), and Treponema (1.17%). There were 259 genera present at less than 1% of the relative abundance. Unassigned reads to the genus level represented 32.02% of the relative abundance. All bacterial communities identified are present in Supplementary Table 1.
3.6 Prevalent epimural bacteria
To describe the most prevalent epimural bacterial communities, the communities were based on ASVs with a greater than 1% detection and 90% prevalence among epimural samples. The 99 ASVs filtered based on these criteria were assigned to 23 unique genera (Table 2).
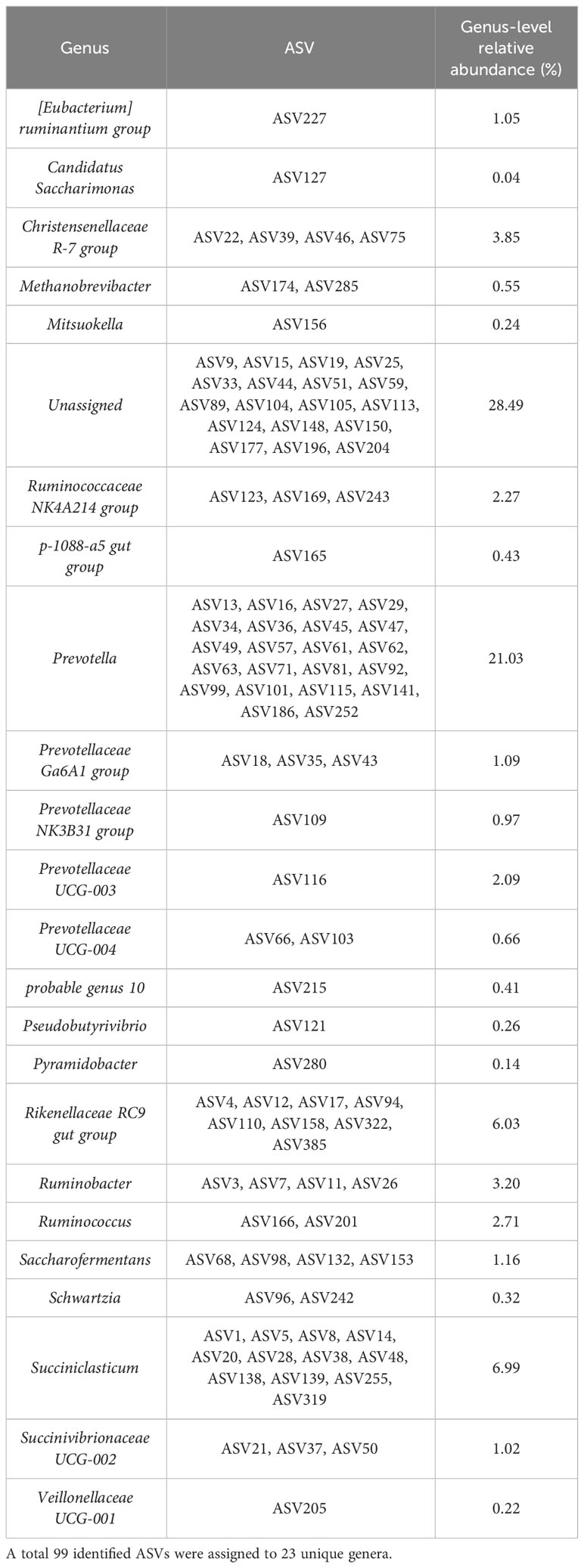
Table 2 The most prevalent bacterial communities present based on a criteria of amplicon sequence variants (ASVs) with a greater than 1% detection and prevalence across all epimural samples.
A full table representing the last known taxonomic assignment of unassigned bacteria at the genus level is located in Supplementary Table 2.
3.7 Differential abundances between epimural and liquid bacteria
After filtering MaAsLin2 and ANCOM-BC results for the overlapping bacterial communities, 451 ASVs significantly differed between the epimural and liquid fractions (Q < 0.05). These ASVs represented 91 taxa, where 62 were identified at the genus level and the remaining 29 were associated with their last-known taxonomic rank. Log2 fold changes were calculated for all significantly different ASVs, shown in Figure 4. Log2 fold changes for all significantly different ASVs, including unclassified genera that were reclassified to their last known taxonomic rank, are present in Supplementary Figure 1. The returned MaAsLin2 coefficients and ANCOM-BC log2 values are available in Supplementary Table 3.
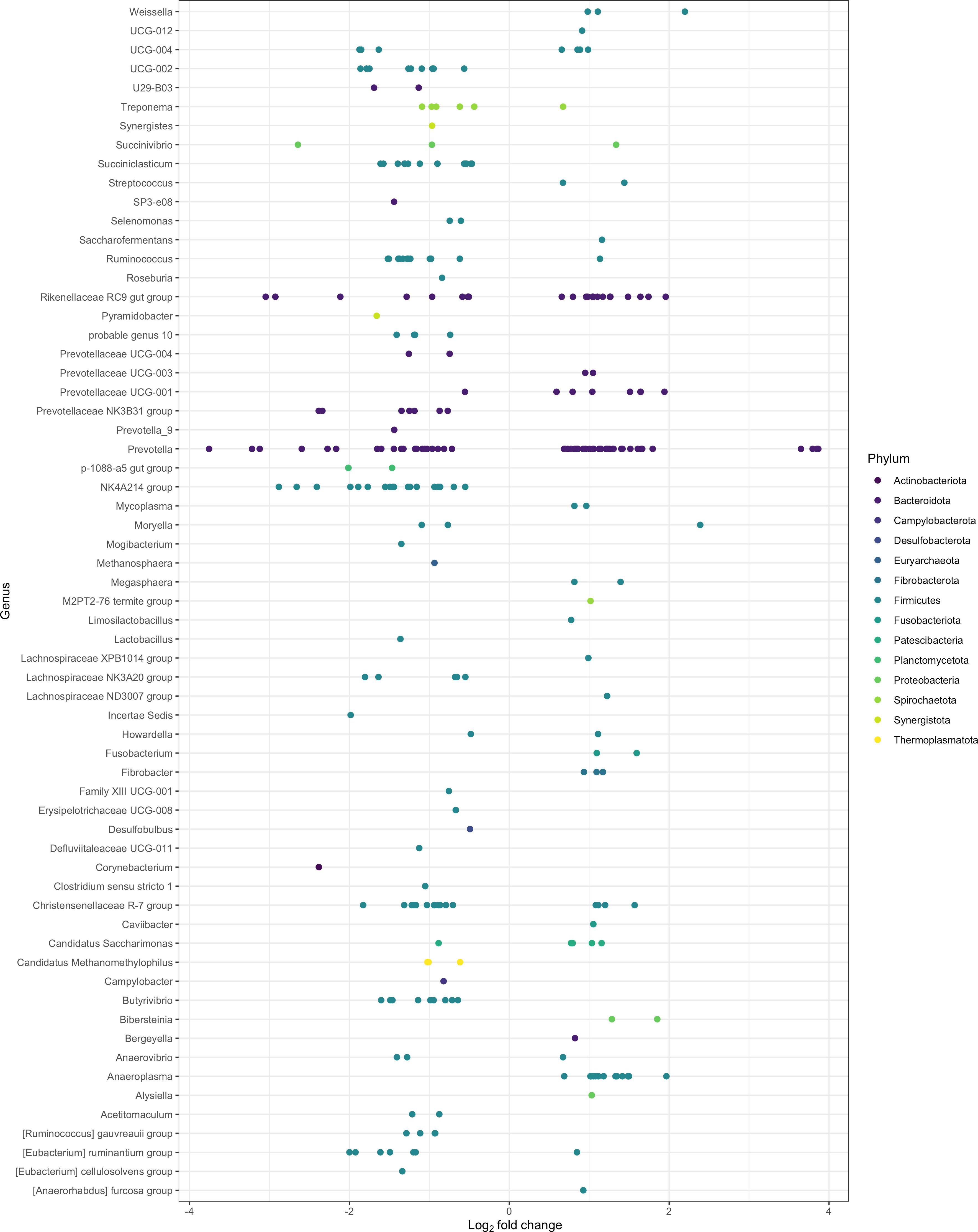
Figure 4 Significantly different log2 fold changes in the abundance of amplicon sequence variants (ASVs) when using epimural as a reference level (P < 0.05; Q < 0.05). A negative log2 fold change represents ASVs more abundant within the epimural microbiome and a positive log2 fold change represents ASVs more abundant within the liquid microbiome. Colors represent different phyla, and rows label the ASVs to the genus level.
The top 10 ASVs in greater abundance in the epimural samples are found in Table 3 (Q < 0.05). Their respective genus-level assignments belonged to Prevotella, Succinivibrio, and Rikenellaceae RC9 gut group. Unassigned ASVs at the genus level were mapped back to their last-known taxonomic rank, belonging to family-level Eubacterium and Prevotella.
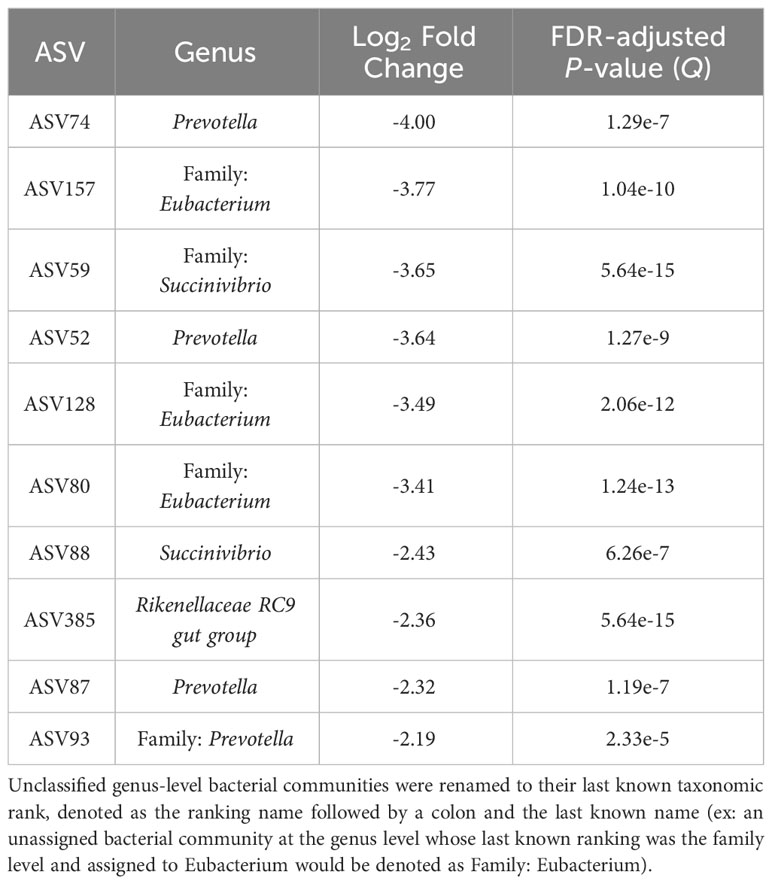
Table 3 The top 10 amplicon sequence variants (ASVs) based on log2 fold changes associated with the epimural bacterial communities present in the rumen.
The top 10 ASVs that were in greater abundance in the planktonic, or liquid-associated, samples are found in Table 4 (Q < 0.05). Their respective genus-level assignments belonged to Prevotella and Weissella. Unassigned ASVs at the genus level were mapped back to their last-known taxonomic rank, belonging to family-level Bacteroidales RF16 group and F082.
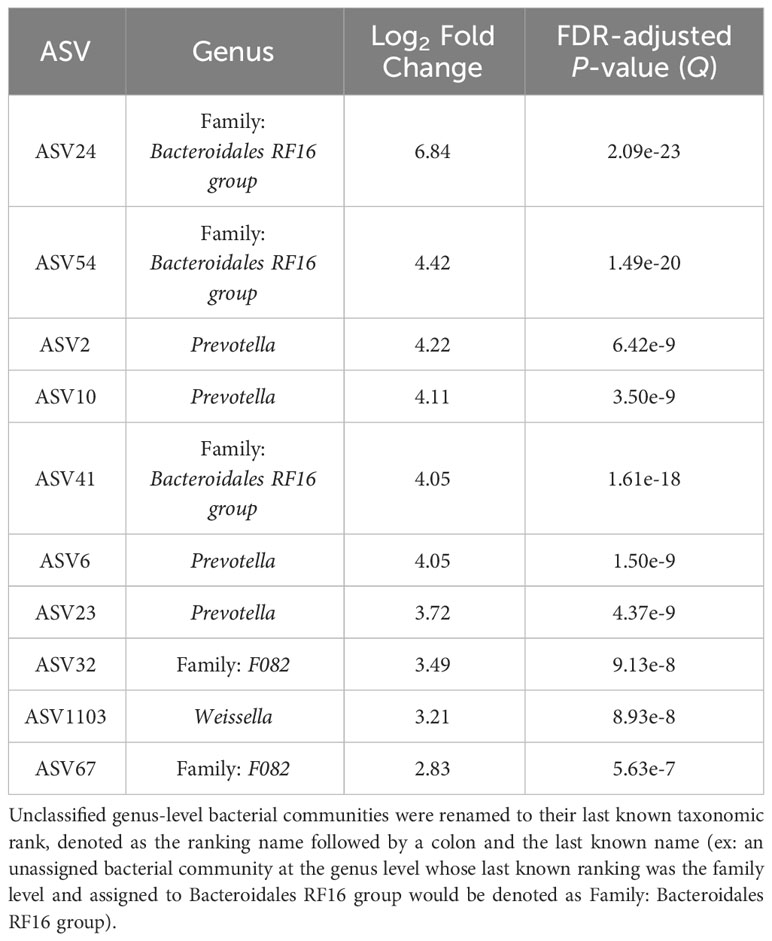
Table 4 The top 10 genera based on log2 fold changes associated with the planktonic bacterial communities present in the rumen.
4 Discussion
Most current approaches for sampling these ruminal epimural communities rely on two main methods: 1) harvesting the animal and collecting rumen papillae that are washed to collect epimural communities or, 2) cannulating the animal to sample papillae or directly swab the rumen wall. Few previous studies have suggested using a trocar for collecting ruminal liquid. However, those methods are not directed toward sampling epimural microbiota and not widely used (Follis and Spillett, 1972; Wilson et al., 1977). Minimizing animal sacrifice and pain remains a key concern for animal welfare in research, providing the impetus for developing a live animal, minimally-invasive method to sample the microbial communities present on the rumen epithelium important. Following surgery, animals were monitored daily for changes in feed intake and health conditions. During the immediate postoperative period, animals showed no decreased appetite or mobility. To determine the procedure’s invasiveness, incision sites were scored based on inflammation and swelling two weeks following the surgery. Few steers had discharge and swelling around the incision site and all steers retained normal mobility; therefore, the method was considered minimally invasive. Due to the limited techniques to sample from the rumen wall, many studies use small animal populations that impact the ability to determine differences within microbial communities. The surgery method allows for a non-invasive, quick technique to sample the rumen wall via swabbing through a trocar while capturing a comprehensive view of the microbial communities present.
While the surgical method allows for a swab to reach the rumen wall of the ventral sac, the swab must be pushed through the fiber mat and the rumen liquid fraction to reach the rumen wall. The cotton swab absorbing some of the liquid content from the rumen results in contamination from the liquid-associated bacterial communities. However, the sampling method did not aim to sample exclusively epimural bacterial communities but rather provide a minimally-invasive method to detect differences between the liquid and the epimural bacteria. The method is best used in a study that will examine the epimural communities in addition to the liquid and/or solid-associated microbial communities to aid in distinguishing the origin. Ultimately, there may be many microbial communities that overlap between epimural and liquid-associated samples using this sampling technique.
Limited research is available regarding the mechanisms of attachment of bacteria to the rumen epithelia. The associated bacterial communities have been previously proposed to adhere via fibrous carbohydrate coats (McCowan et al., 1978; Cheng and Costerton, 1980) or pili (Cheng and Costerton, 1980). Due to the mechanisms of attachment and the sloughing of the rumen epithelia, there is a likelihood that luminal microbiota may also be associated with or nearby epimural communities. As the proximity of these microbial communities may indicate their ability to interact with one another, distinguishing truly epithelial-associated versus planktonic microbes may be difficult. Some studies investigating the epimural microbial communities will rinse the excised rumen papillae with saline (Petri et al., 2013; Wetzels et al., 2017), while others will scrape the rumen epithelium as well (Chen et al., 2011; De Mulder et al., 2016). The swabbing method proposed in the current study may differentially separate adhered microbes from papillae compared to rinsing methods.
The composition and structure of the rumen microbiome has been demonstrated to alter in cases of major changes, such as alteration of diet, management practices, and environment. Steers underwent surgery on day 35 of the study and liquid samples were collected on day 70. At this time, the microbiome should already have reached stability, or near stability, which should result in minimal fluctuations between sampling dates. The time between sampling dates allowed for animals to be closely monitored for changes in health conditions before tubing for rumen fluid. Several studies have investigated the stability of ruminal bacterial communities within the rumen over time, concluding that after adjustment to diet, the most abundant microbial communities remain relatively stable (Clemmons et al., 2019; Snelling et al., 2019). Within the last decade, host genetics have been suggested to play an increasingly important role on the composition of the rumen microbiome (Sasson et al., 2017; Difford et al., 2018; Li et al., 2019a; Wallace et al., 2019; Martínez-Álvaro et al., 2022) and are likely contributing to the stability of the rumen microbiome during this time period. Further, recent studies have identified that host-influenced microbial genes do not have major shifts over the course of a 56-day feeding trial, supporting previous literature findings that the rumen microbiome remains stable over time (Lima et al., 2023). As the diet, management, and environment for the steers enrolled in the current study were maintained consistently, no major perturbations in the rumen microbiome would occur during this time frame. Therefore, differences identified in the rumen bacterial communities within the current study are more likely due to true differences in sample type and not due to temporal changes in the community structure.
In a previous study examining four rumen-cannulated Holstein-Friesian cows, the alpha diversity indices for the epimural bacterial communities indicated an increased variance compared to the liquid and solid-associated bacteria (De Mulder et al., 2016), which was likely due to individual animal differences. Other studies examining the alpha diversity of the epimural samples compared to rumen content samples have found increased diversity in the ruminal epimural bacteria (Abbas et al., 2020). However, this is inconsistent across studies, with others noting no significant differences in alpha diversity between epimural and liquid samples (Zhou et al., 2021). No differences in any alpha diversity metrics (observed ASVs, Chao1, and Shannon Diversity Index) existed between sampling locations within the current study, indicating that the within-sample richness and diversity were similar. There is a high likelihood that the within-sample richness and diversity of ASVs between the ruminal epimural and liquid bacterial communities would be very similar, as they could be distinctly different but equally diverse.
Past studies have found that Firmicutes, Bacteroidetes, and Proteobacteria are the most abundant phyla in the epimural communities. However, these studies disagree on whether Firmicutes or Bacteroidetes are in the greatest abundance (Chen et al., 2011; Petri et al., 2013; De Mulder et al., 2016; Sbardellati et al., 2020; Zhou et al., 2021). In this study, Firmicutes was in greater abundance in the epimural bacteria compared to the liquid fraction. Overall, Bacteroidetes was the dominant phylum for both sampling locations. Due to diet influencing both the liquid and epimural microbiota (Petri et al., 2020), shifts between Bacteroidetes and Firmicutes are expected among different studies. Additionally, primers targeting different regions of the 16S rRNA gene differentially preference specific bacterial communities via amplification bias, resulting in some studies discovering greater levels of one microbe over another (Klindworth et al., 2012). Overall, this study captures the main phyla present in other epimural community studies.
Previous studies assessing the composition of the epimural bacterial or archaeal communities have recently been combined into a few meta-analyses. A meta-analysis aiming to identify core epimural microbial communities, defined as operational taxonomic units (OTUs) present in over 90% of samples, found 11 genera representing the epithelial bacterial communities (Pacífico et al., 2021). These 11 genera consisted of Campylobacter, Ruminococcaceae NK4A214 group, Desulfobulbus, Christensenellaceae R-7 group, Comamonas, uncultured Neisseriaceae, Succiniclasticum, Ruminococcaceae UCG-014 and UCG-010, Lachnospiraceae UCG-010, and Defluvitaleaceae UCG-011. A separate meta-analysis assessing the core epimural bacterial communities across 17 16S rRNA gene datasets, defined as a community present in greater than or equal to 80% of samples, found 147 core OTUs from closed-reference clustering within cattle species across dairy and beef breeds (Anderson et al., 2021). When narrowing the criteria for a core microbiome to bacterial communities present in 90% of cattle samples, there were 63 OTUs associated with the epithelium (Anderson et al., 2021). The 63 OTUs corresponded to 24 genus-level assigned taxa, where at least 4 OTUs were assigned to Butyrvibrio, Christensenellaceae R7 group, Desulfobulbus, and Fretibacterium. Based on the above literature, the predominant bacterial communities present on the rumen epithelium have been well-described and therefore can be compared to the current study.
Based on existing literature regarding the core epimural bacterial communities, the swabbing method employed in the current study captures previously-identified core epimural bacterial communities. When the most prevalent, considered as “core” within the current study, ASVs were aggregated to the genus level, many ASVs were assigned to Succiniclasticum, Rikenellaceae RC9 gut group, Christensenellaceae R7 group, Ruminobacter, Ruminococcus, Ruminococcaceae NK4A214 group, and Prevotella. There are several considerations to make when using a core microbiome analysis, such as sample size, prevalence criteria, and the difficulty in determining a true “core” microbiome across studies (Neu et al., 2021). While core bacterial communities are defined within the current study, the epimural bacteria identified are used strictly for comparison to validate that the microbiota present are reported in previous literature. Several genera that were in greater abundance in the epimural communities in comparison to the rumen fluid communities were also considered a core bacteria within the study. Several Succiniclasticum, Ruminococcus, and Ruminococcaceae NK4A214 group were in significantly greater abundance in the epimural samples.
When assigning ASVs to the genus level, the majority of genera belonged to either the epimural or rumen samples, with very few having genera represented in both locations. There were 20 distinct genera that were present in rumen samples; in comparison, 33 genera were identified exclusively in the epimural samples. While there were 9 genera that had ASVs in both sample locations, these possibly indicate different species or common genera between the two locations. As there were distinct genera associated with sample location, there is evidence that the sampling method is not solely contaminated by planktonic bacteria. The number of differentially abundant communities between sampling location indicates that the minimally-invasive swabbing method provides information on the epithelium-associated microbes independent from the liquid-associated bacteria. These findings help validate the sampling ability of the surgery method.
Although not typically considered a core bacteria on the rumen wall, Prevotella and several Prevotellaceae met this study’s criteria as core bacterial communities at the genus and family level. The bacteria are well-characterized in literature for their presence in rumen fluid samples (Stevenson and Weimer, 2007; Myer et al., 2015), which may indicate the potential degree of contamination via the swabbing method. However, other studies that examine epimural bacteria have found Prevotellaceae in small to moderate abundances, suggesting that these luminal communities may be closely associated with the rumen wall and thus appear in epimural samples (Sbardellati et al., 2020). The ASVs assigned to Prevotella within our study separate into two groups depending on the sample location. When examining the log2 fold change between the epimural and rumen liquid samples, the observed Prevotella are presumably serving different functions based on locations. Some caution should be employed when using ASVs to represent different species, as there is the possibility that genomes are artificially split using strict ASV thresholds (Schloss, 2021); however, there is also evidence that ASVs can capture a higher resolution of variation within an environment (Callahan et al., 2016; Callahan et al., 2017). Therefore, these ASVs assigned to Prevotella are possibly different species present depending on the sample locations.
Many ASVs identified as significantly different between the epimural and rumen fluid were unassigned at the genus level. Unassigned reads at the genus level are not uncommon in short-read amplicon studies (Henderson et al., 2019). Due to the difficulty in culturing ruminal bacteria (Creevey et al., 2014), many bacteria are unrepresented in databases, negatively impacting taxonomic resolution. One benefit of using ASVs is that they are not generally constrained to taxonomic classification. The unassigned ASVs could indicate bacterial communities that are not yet represented in databases. There are several attempts within the field of rumen microbiology to improve gaps within databases (Henderson et al., 2019), such as making rumen-specific databases (Seedorf et al., 2014; Seshadri et al., 2018) or improving culturing techniques (Kenters et al., 2011; Nyonyo et al., 2013). As these improve, more information will be available regarding unknown rumen species that are performing unique and important functions within the rumen.
Due to the proximity of the epimural microbiota to the rumen wall and their capacity to act as intermediary communicators to the ruminant host and rumen liquid fraction, there are opportunities to utilize the surgery method to elucidate functionality of the ruminal epimural communities. Studies have started to define the transcriptome of the microbial communities present at the rumen wall (Wang et al., 2017; Mann et al., 2018; Tan et al., 2021) and ruminal epithelial tissue (Kong et al., 2016; Wang et al., 2017; Elolimy et al., 2018). This has offered opportunities to better understand the functions that epimural bacterial and archaeal communities may perform. Studies examining the epimural microbiome have found that individual animal variation contributes to differences in abundance (Sbardellati et al., 2020), which may be explained by individual host control. These microbial communities could be modulating host-microbiome interactions. The non-invasive surgery method proposed within this work will allow for the investigation of mechanisms between the host and epimural microbial communities.
5 Conclusions
The non-invasive surgery method used within the current study captured epimural microbiota while also improving epimural sampling methods in the context of animal welfare in research. There is an opportunity within the field of rumen microbiology to use this minimally-invasive method to explore the microbiome on the rumen wall. In addition, this method can allow for examination of the cross-talk occurring between the rumen environment and the host, mediated by the epimural communities, through different -omics technologies.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA918692.
Ethics statement
The animal study was approved by University of Tennessee Animal Care and Use Committee. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
MH: Data curation, Formal Analysis, Investigation, Methodology, Project administration, Visualization, Writing – original draft, Writing – review and editing. TR: Investigation, Methodology, Supervision, Writing – original draft, Writing – review and editing. JB: Investigation, Methodology, Supervision, Writing – original draft, Writing – review and editing. PM: Methodology, Writing – review and editing. JS: Methodology, Writing – review and editing. BV: Investigation, Methodology, Writing – original draft, Writing – review and editing. JW: Investigation, Methodology, Writing – original draft, Writing – review and editing. LK: Investigation, Methodology, Writing – original draft, Writing – review and editing. PM: Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Supervision, Writing – original draft, Writing – review and editing.
Funding
The authors declare financial support was received for the research, authorship, and/or publication of this article. This work is supported by the Agriculture and Food Research Initiative grant no. 2020-67015-30832 from the USDA National Institute of Food and Agriculture.
Acknowledgments
The authors would like to thank the staff at the University of Tennessee’s Plateau AgResearch and Education Center and the UTIA Genomics Center for the Advancement of Agriculture for their assistance with the project.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Author disclaimer
Mention of a trade name, proprietary product, or specific equipment does not constitute a guarantee or warranty by the USDA and does not imply approval to the exclusion of other products that may be suitable. USDA is an equal opportunity provider and employer.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fanim.2023.1270550/full#supplementary-material
Supplementary Table 1 | Relative abundances and taxonomic assignments for all bacterial communities between the rumen liquid and epimural fractions.
Supplementary Table 2 | The most prevalent epimural amplicon sequence variants based on a prevalence greater than 90% and detection greater than 1% across all epimural samples.
Supplementary Table 3 | Results from the overlap of MaAsLin2 and ANCOMBC for differential abundance analyses, including coefficients, amplicon sequence variant numbers, taxonomic assignments, standard errors, p-values, and FDR-adjusted p-values.
Supplementary Figure 1 | Significantly different log2 fold changes in the abundance of amplicon sequence variants (ASVs) when using epimural as a reference level (P < 0.05; FDR < 0.05). A negative log2 fold change represents ASVs more abundant within the epimural microbiome and a positive log2 fold change represents ASVs more abundant within the liquid microbiome. Colors represent different phyla, and rows label the ASVs to the genus level. The ASVs that were unassigned at the genus level are renamed to their last known taxonomic assignment, represented by the taxonomic rank followed by an underscore with the taxonomic assignment.
References
Abbas W., Keel B. N., Kachman S. D., Fernando S. C., Wells J. E., Hales K. E., et al. (2020). Rumen epithelial transcriptome and microbiome profiles of rumen epithelium and contents of beef cattle with and without liver abscesses. J. Anim. Sci. 98 (12), 1–13. doi: 10.1093/jas/skaa359
Andersen T. O., Altshuler I., Vera-Ponce de León A., Walter J. M., McGovern E., Keogh K., et al. (2023). Metabolic influence of core ciliates within the rumen microbiome. ISME J. 17 (7), 1128–1140. doi: 10.1038/s41396-023-01407-y
Anderson C. J., Koester L. R., Schmitz-Esser S. (2021). Rumen epithelial communities share a core bacterial microbiota: A meta-analysis of 16S rRNA gene illumina miSeq sequencing datasets. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.625400
Apprill A., McNally S., Parsons R., Weber L. (2015). Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microbial. Ecol. 75 (2), 129–137. doi: 10.3354/ame01753
Benjamini Y., Hochberg Y. (1995). Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B. 57, 1, 289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
Bergman E. (1990). Energy contributions of volatile fatty acids from the gastrointestinal tract in various species. Physiol. Rev. 70, 2, 567–590. doi: 10.1152/physrev.1990.70.2.567
Callahan B. J., McMurdie P. J., Holmes S. P. (2017). Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 11 (12), 2639–2643. doi: 10.1038/ismej.2017.119
Callahan B. J., McMurdie P. J., Rosen M. J., Han A. W., Johnson A. J. A., Holmes S. P. (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13 (7), 581–583. doi: 10.1038/nmeth.3869
Chen Y., Penner G. B., Li M., Oba M., Guan L. L. (2011). Changes in bacterial diversity associated with epithelial tissue in the beef cow rumen during the transition to a high-grain diet. Appl. Environ. Microbiol. 77 (16), 5770–5781. doi: 10.1128/AEM.00375-11
Cheng K.-J., Costerton J. (1980). “Adherent rumen bacteria—their role in the digestion of plant material, urea and epithelial cells,” in Digestive physiology and metabolism in ruminants (Netherlands: Springer), 227–250.
Cheng K. J., McCowan R. P., Costerton J. W. (1979). Adherent epithelial bacteria in ruminants and their roles in digestive tract function. Am. J. Clin. Nutr. 32 (1), 139–148. doi: 10.1093/ajcn/32.1.139
Cheng K. J., Wallace R. J. (1979). The mechanism of passage of endogenous urea through the rumen wall and the role of ureolytic epithelial bacteria in the urea flux. Br. J. Nutr. 42 (3), 553–557. doi: 10.1079/BJN19790147
Cho S.-J., Cho K.-M., Shin E.-C., Lim W.-J., Hong S.-Y., Choi B.-R., et al. (2006). 16S rDNA analysis of bacterial diversity in three fractions of cow rumen. J. Microbiol. Biotechnol. 16 (1), 92–101.
Clemmons B. A., Martino C., Schneider L. G., Lefler J., Embree M. M., Myer P. R. (2019). Temporal stability of the ruminal bacterial communities in beef steers. Sci. Rep. 9 (1), 9522. doi: 10.1038/s41598-019-45995-2
Creevey C. J., Kelly W. J., Henderson G., Leahy S. C. (2014). Determining the culturability of the rumen bacterial microbiome. Microbial. Biotechnol. 7 (5), 467–479. doi: 10.1111/1751-7915.12141
Czerkawski J. (1986)Degradation of solid feeds in the rumen: spatial distribution of microbial activity and its consequences. In: Proceedings of 6th International Symposium on Ruminant Physiology (Banff, Canada: Prentice-Hall) (Accessed 10-14 Sep 1984).
De Mulder T., Goossens K., Peiren N., Vandaele L., Haegeman A., De Tender C., et al. (2016). Exploring the methanogen and bacterial communities of rumen environments: solid adherent, fluid and epimural. FEMS Microbiol. Ecol. 93 (3), fiw251. doi: 10.1093/femsec/fiw251
Difford G. F., Plichta D. R., Løvendahl P., Lassen J., Noel S. J., Højberg O., et al. (2018). Host genetics and the rumen microbiome jointly associate with methane emissions in dairy cows. PloS Genet. 14 (10), e1007580. doi: 10.1371/journal.pgen.1007580
Dinsdale D., Cheng K. J., Wallace R. J., Goodlad R. A. (1980). Digestion of epithelial tissue of the rumen wall by adherent bacteria in infused and conventionally fed sheep. Appl. Environ. Microbiol. 39 (5), 1059–1066. doi: 10.1128/aem.39.5.1059-1066.1980
Elolimy A. A., Abdelmegeid M. K., McCann J. C., Shike D. W., Loor J. J. (2018). Residual feed intake in beef cattle and its association with carcass traits, ruminal solid-fraction bacteria, and epithelium gene expression. J. Anim. Sci. Biotechnol. 9 (1), 67. doi: 10.1186/s40104-018-0283-8
Follis T. B., Spillett J. J. (1972). A new method for rumen sampling. J. Wildlife Manage. 36 (4), 1336–1340. doi: 10.2307/3799280
Glöckner F. O., Yilmaz P., Quast C., Gerken J., Beccati A., Ciuprina A., et al. (2017). 25 years of serving the community with ribosomal RNA gene reference databases and tools. J. Biotechnol. 261, 169–176. doi: 10.1016/j.jbiotec.2017.06.1198
Henderson G., Yilmaz P., Kumar S., Forster R. J., Kelly W. J., Leahy S. C., et al. (2019). Improved taxonomic assignment of rumen bacterial 16S rRNA sequences using a revised SILVA taxonomic framework. PeerJ 7, e6496. doi: 10.7717/peerj.6496
Jami E., White B. A., Mizrahi I. (2014). Potential role of the bovine rumen microbiome in modulating milk composition and feed efficiency. PloS One 9 (1), e85423–e85423. doi: 10.1371/journal.pone.0085423
Kenters N., Henderson G., Jeyanathan J., Kittelmann S., Janssen P. H. (2011). Isolation of previously uncultured rumen bacteria by dilution to extinction using a new liquid culture medium. J. Microbiol. Methods 84 (1), 52–60. doi: 10.1016/j.mimet.2010.10.011
Khafipour E., Li S., Plaizier J. C., Krause D. O. (2009). Rumen microbiome composition determined using two nutritional models of subacute ruminal acidosis. Appl. Environ. Microbiol. 75 (22), 7115–7124. doi: 10.1128/AEM.00739-09
Klindworth A., Pruesse E., Schweer T., Peplies J., Quast C., Horn M., et al. (2012). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41 (1), e1–e1. doi: 10.1093/nar/gks808
Kong R. S. G., Liang G., Chen Y., Stothard P., Guan L. L. (2016). Transcriptome profiling of the rumen epithelium of beef cattle differing in residual feed intake. BMC Genomics 17 (1), 592. doi: 10.1186/s12864-016-2935-4
Lahti L., Shetty S., Turgaga N., Leung E., Gilmore R., Salojärvi J., et al. (2020). “Tools for microbiome analyses in R. Microbiome package version v1.18.0,” in Bioconductor.
Larue R., Yu Z., Parisi V. A., Egan A. R., Morrison M. (2005). Novel microbial diversity adherent to plant biomass in the herbivore gastrointestinal tract, as revealed by ribosomal intergenic spacer analysis and rrs gene sequencing. Environ. Microbiol. 7 (4), 530–543. doi: 10.1111/j.1462-2920.2005.00721.x
Li W., Gelsinger S., Edwards A., Riehle C., Koch D. (2019b). Transcriptome analysis of rumen epithelium and meta-transcriptome analysis of rumen epimural microbial community in young calves with feed induced acidosis. Sci. Rep. 9 (1), 1–13. doi: 10.1038/s41598-019-40375-2
Li F., Guan L. L., McBain A. J. (2017). Metatranscriptomic profiling reveals linkages between the active rumen microbiome and feed efficiency in beef cattle. Appl. Environ. Microbiol. 83 (9), e00061–e00017. doi: 10.1128/AEM.00061-17
Li F., Li C., Chen Y., Liu J., Zhang C., Irving B., et al. (2019a). Host genetics influence the rumen microbiota and heritable rumen microbial features associate with feed efficiency in cattle. Microbiome 7 (1), 92. doi: 10.1186/s40168-019-0699-1
Li Z., Wang X., Zhang Y., Yu Z., Zhang T., Dai X., et al. (2022). Genomic insights into the phylogeny and biomass-degrading enzymes of rumen ciliates. ISME J. 16 (12), 2775–2787. doi: 10.1038/s41396-022-01306-8
Lima J., Martínez-Álvaro M., Mattock J., Auffret M. D., Duthie C. A., Cleveland M. A., et al. (2023). “Host-genomically influenced ruminal microbial genes are temporally stable during the finishing phase in beef cattle,” in Proceedings of 12th World Congress on Genetics Applied to Livestock Production (WCGALP) Technical and species orientated innovations in animal breeding, and contribution of genetics to solving societal challenges. (Wageningen Academic Publishers) p. 2616–2619.
Lin H., Peddada S. D. (2020). Analysis of compositions of microbiomes with bias correction. Nat. Commun. 11 (1), 3514. doi: 10.1038/s41467-020-17041-7
Liu J.-h., Bian G.-r., Zhu W.-y., Mao S.-y. (2015). High-grain feeding causes strong shifts in ruminal epithelial bacterial community and expression of Toll-like receptor genes in goats. Front. Microbiol. 6. doi: 10.3389/fmicb.2015.00167
Liu J.-h., Xu T.-t., Liu Y.-j., Zhu W.-y., Mao S.-y. (2013). A high-grain diet causes massive disruption of ruminal epithelial tight junctions in goats. Am. J. Physiol-Regulatory Integr. Comp. Physiol. 305 (3), R232–R241. doi: 10.1152/ajpregu.00068.2013
Liu J.-h., Zhang M.-l., Zhang R.-y., Zhu W.-y., Mao S.-y. (2016). Comparative studies of the composition of bacterial microbiota associated with the ruminal content, ruminal epithelium and in the faeces of lactating dairy cows. Microbial. Biotechnol. 9 (2), 257–268. doi: 10.1111/1751-7915.12345
Mallick H., Rahnavard A., McIver L. J., Ma S., Zhang Y., Nguyen L. H., et al. (2021). Multivariable association discovery in population-scale meta-omics studies. PloS Comput. Biol. 17 (11), e1009442. doi: 10.1371/journal.pcbi.1009442
Mann E., Wetzels S. U., Wagner M., Zebeli Q., Schmitz-Esser S. (2018). Metatranscriptome sequencing reveals insights into the gene expression and functional potential of rumen wall bacteria. Front. Microbiol. 9 (43). doi: 10.3389/fmicb.2018.00043
Mao S., Zhang M., Liu J., Zhu W. (2015). Characterising the bacterial microbiota across the gastrointestinal tracts of dairy cattle: membership and potential function. Sci. Rep. 5 (1), 16116. doi: 10.1038/srep16116
Martínez-Álvaro M., Auffret M. D., Duthie C.-A., Dewhurst R. J., Cleveland M. A., Watson M., et al. (2022). Bovine host genome acts on rumen microbiome function linked to methane emissions. Commun. Biol. 5 (1), 350. doi: 10.1038/s42003-022-03293-0
McAllister T. A., Bae H. D., Jones G. A., Cheng K.-J. (1994). Microbial attachment and feed digestion in the rumen. J. Anim. Sci. 72 (11), 3004–3018. doi: 10.2527/1994.72113004x
McCowan R. P., Cheng K. J., Bailey C. B., Costerton J. W. (1978). Adhesion of bacteria to epithelial cell surfaces within the reticulo-rumen of cattle. Appl. Environ. Microbiol. 35 (1), 149–155. doi: 10.1128/aem.35.1.149-155.1978
McCowan R. P., Cheng K. J., Costerton J. W. (1980). Adherent bacterial populations on the bovine rumen wall: distribution patterns of adherent bacteria. Appl. Environ. Microbiol. 39 (1), 233–241. doi: 10.1128/aem.39.1.233-241.1980
McMurdie P. J., Holmes S. (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PloS One 8 (4), e61217. doi: 10.1371/journal.pone.0061217
Millen D. D., Arrigoni M. D. B., Pacheco R. D. L. (2016). Rumenology. (Berlin/Heidelberg, Germany: Springer).
Myer P. R., Smith T. P. L., Wells J. E., Kuehn L. A., Freetly H. C. (2015). Rumen microbiome from steers differing in feed efficiency. PloS One 10 (6), e0129174–e0129174. doi: 10.1371/journal.pone.0129174
Nearing J. T., Douglas G. M., Hayes M. G., MacDonald J., Desai D. K., Allward N., et al. (2022). Microbiome differential abundance methods produce different results across 38 datasets. Nat. Commun. 13 (1), 342. doi: 10.1038/s41467-022-28034-z
Neu A. T., Allen E. E., Roy K. (2021). Defining and quantifying the core microbiome: Challenges and prospects. Proc. Natl. Acad. Sci. 118 (51), e2104429118. doi: 10.1073/pnas.2104429118
Nyonyo T., Shinkai T., Tajima A., Mitsumori M. (2013). Effect of media composition, including gelling agents, on isolation of previously uncultured rumen bacteria. Lett. Appl. Microbiol. 56 (1), 63–70. doi: 10.1111/lam.12019
Oksanen J., Kindt R., Legendre P., O’Hara B., Stevens M. H. H., Oksanen M. J., et al. (2007). The vegan package, Vol. 10. 719.
Oksanen J., Simpson G., Blanchet F., Kindt R., Legendre P., Minchin P., et al. (2022). vegan: Community Ecology Package. R package version 2.6.2.
Pacífico C., Petri R. M., Ricci S., Mickdam E., Wetzels S. U., Neubauer V., et al. (2021). Unveiling the bovine epimural microbiota composition and putative function. Microorganisms 9 (2), 342. doi: 10.3390/microorganisms9020342
Parada A. E., Needham D. M., Fuhrman J. A. (2016). Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 18 (5), 1403–1414. doi: 10.1111/1462-2920.13023
Paulson J. N., Stine O. C., Bravo H. C., Pop M. (2013). Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 10 (12), 1200–1202. doi: 10.1038/nmeth.2658
Penner G. B., Aschenbach J., Gäbel G., Rackwitz R., Oba M. (2009). Epithelial capacity for apical uptake of short chain fatty acids is a key determinant for intraruminal pH and the susceptibility to subacute ruminal acidosis in sheep. J. Nutr. 139 (9), 1714–1720. doi: 10.3945/jn.109.108506
Petri R. M., Neubauer V., Humer E., Kröger I., Reisinger N., Zebeli Q. (2020). Feed additives differentially impact the epimural microbiota and host epithelial gene expression of the bovine rumen fed diets rich in concentrates. Front. Microbiol. 11. doi: 10.3389/fmicb.2020.00119
Petri R. M., Schwaiger T., Penner G. B., Beauchemin K. A., Forster R. J., McKinnon J. J., et al. (2013). Changes in the rumen epimural bacterial diversity of beef cattle as affected by diet and induced ruminal acidosis. Appl. Environ. Microbiol. 79 (12), 3744–3755. doi: 10.1128/AEM.03983-12
Prodan A., Tremaroli V., Brolin H., Zwinderman A. H., Nieuwdorp M., Levin E. (2020). Comparing bioinformatic pipelines for microbial 16S rRNA amplicon sequencing. PloS One 15 (1), e0227434. doi: 10.1371/journal.pone.0227434
Quast C., Pruesse E., Yilmaz P., Gerken J., Schweer T., Yarza P., et al. (2012). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41 (D1), D590–D596. doi: 10.1093/nar/gks1219
Roehe R., Dewhurst R. J., Duthie C.-A., Rooke J. A., McKain N., Ross D. W., et al. (2016). Bovine host genetic variation influences rumen microbial methane production with best selection criterion for low methane emitting and efficiently feed converting hosts based on metagenomic gene abundance. PloS Genet. 12 (2), e1005846. doi: 10.1371/journal.pgen.1005846
Sadet S., Martin C., Meunier B., Morgavi D. P. (2007). PCR-DGGE analysis reveals a distinct diversity in the bacterial population attached to the rumen epithelium. animal 1 (7), 939–944. doi: 10.1017/S1751731107000304
Sadet-Bourgeteau S., Martin C., Morgavi D. P. (2010). Bacterial diversity dynamics in rumen epithelium of wethers fed forage and mixed concentrate forage diets. Veterinary Microbiol. 146 (1), 98–104. doi: 10.1016/j.vetmic.2010.04.029
Sasson G., Ben-Shabat S. K., Seroussi E., Doron-Faigenboim A., Shterzer N., Yaacoby S., et al. (2017). Heritable bovine rumen bacteria are phylogenetically related and correlated with the cow’s capacity to harvest energy from its feed. mBio 8 (4), e00703–e00717. doi: 10.1128/mBio.00703-17
Sbardellati D. L., Fischer A., Cox M. S., Li W., Kalscheur K. F., Suen G. (2020). The bovine epimural microbiota displays compositional and structural heterogeneity across different ruminal locations. J. Dairy Sci. 103 (4), 3636–3647. doi: 10.3168/jds.2019-17649
Schären M., Kiri K., Riede S., Gardener M., Meyer U., Hummel J., et al. (2017). Alterations in the rumen liquid-, particle- and epithelium-associated microbiota of dairy cows during the transition from a silage- and concentrate-based ration to pasture in spring. Front. Microbiol. 8. doi: 10.3389/fmicb.2017.00744
Schloss P. D. (2021). Amplicon sequence variants artificially split bacterial genomes into separate clusters. mSphere 6 (4), e00191–e00121. doi: 10.1128/mSphere.00191-21
Seedorf H., Kittelmann S., Henderson G., Janssen P. H. (2014). RIM-DB: a taxonomic framework for community structure analysis of methanogenic archaea from the rumen and other intestinal environments. PeerJ 2, e494. doi: 10.7717/peerj.494
Seshadri R., Leahy S. C., Attwood G. T., Teh K. H., Lambie S. C., Cookson A. L., et al. (2018). Cultivation and sequencing of rumen microbiome members from the Hungate1000 Collection. Nat. Biotechnol. 36 (4), 359–367. doi: 10.1038/nbt.4110
Shabat S. K. B., Sasson G., Doron-Faigenboim A., Durman T., Yaacoby S., Berg Miller M. E., et al. (2016). Specific microbiome-dependent mechanisms underlie the energy harvest efficiency of ruminants. ISME J. 10 (12), 2958–2972. doi: 10.1038/ismej.2016.62
Snelling T. J., Auffret M. D., Duthie C.-A., Stewart R. D., Watson M., Dewhurst R. J., et al. (2019). Temporal stability of the rumen microbiota in beef cattle, and response to diet and supplements. Anim. Microbiome 1 (1), 16. doi: 10.1186/s42523-019-0018-y
Stevenson D. M., Weimer P. J. (2007). Dominance of Prevotella and low abundance of classical ruminal bacterial species in the bovine rumen revealed by relative quantification real-time PCR. Appl. Microbiol. Biotechnol. 75 (1), 165–174. doi: 10.1007/s00253-006-0802-y
Tajima K., Aminov R. I., Nagamine T., Matsui H., Nakamura M., Benno Y. (2001). Diet-dependent shifts in the bacterial population of the rumen revealed with real-time PCR. Appl. Environ. Microbiol. 67 (6), 2766–2774. doi: 10.1128/AEM.67.6.2766-2774.2001
Tan R. S. G., Zhou M., Li F., Guan L. L. (2021). Identifying active rumen epithelial associated bacteria and archaea in beef cattle divergent in feed efficiency using total RNA-seq. Curr. Res. Microbial. Sci. 2, 100064. doi: 10.1016/j.crmicr.2021.100064
Wallace R. J., Sasson G., Garnsworthy P. C., Tapio I., Gregson E., Bani P., et al. (2019). A heritable subset of the core rumen microbiome dictates dairy cow productivity and emissions. Sci. Adv. 5 (7), eaav8391. doi: 10.1126/sciadv.aav8391
Wang B., Wang D., Wu X., Cai J., Liu M., Huang X., et al. (2017). Effects of dietary physical or nutritional factors on morphology of rumen papillae and transcriptome changes in lactating dairy cows based on three different forage-based diets. BMC Genomics 18 (1), 353. doi: 10.1186/s12864-017-3726-2
Wetzels S. U., Mann E., Pourazad P., Qumar M., Pinior B., Metzler-Zebeli B. U., et al. (2017). Epimural bacterial community structure in the rumen of Holstein cows with different responses to a long-term subacute ruminal acidosis diet challenge. J. Dairy Sci. 100 (3), 1829–1844. doi: 10.3168/jds.2016-11620
Wilson D. E., Hirst S. M., Ellis R. P. (1977). Determination of feeding preferences in wild ruminants from trocar samples. J. Wildlife Manage. 41 (1), 70–75. doi: 10.2307/3800092
Yilmaz P., Parfrey L. W., Yarza P., Gerken J., Pruesse E., Quast C., et al. (2013). The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 42 (D1), D643–D648. doi: 10.1093/nar/gkt1209
Yu Z., Morrison M. (2004). Improved extraction of PCR-quality community DNA from digesta and fecal samples. Biotechniques 36, 5, 808–812. doi: 10.2144/04365st04
Zhang T., Mu Y., Zhang R., Xue Y., Guo C., Qi W., et al. (2022). Responsive changes of rumen microbiome and metabolome in dairy cows with different susceptibility to subacute ruminal acidosis. Anim. Nutr. 8, 331–340. doi: 10.1016/j.aninu.2021.10.009
Zhao K., Chen Y. H., Penner G. B., Oba M., Guan L. L. (2017). Transcriptome analysis of ruminal epithelia revealed potential regulatory mechanisms involved in host adaptation to gradual high fermentable dietary transition in beef cattle. BMC Genomics 18 (1), 976. doi: 10.1186/s12864-017-4317-y
Keywords: rumen, epithelium, bacteria, epimural, beef
Citation: Henniger MT, Rowan TN, Beever JE, Mulon P-Y, Smith JS, Voy BH, Wells JE, Kuehn LA and Myer PR (2023) Validation of a minimally-invasive method for sampling epithelial-associated microorganisms on the rumen wall. Front. Anim. Sci. 4:1270550. doi: 10.3389/fanim.2023.1270550
Received: 31 July 2023; Accepted: 29 September 2023;
Published: 13 October 2023.
Edited by:
Todd Riley Callaway, University of Georgia, United StatesReviewed by:
Zongjun Li, Northwest A&F University, ChinaTansol Park, Chung-Ang University, Republic of Korea
Copyright © 2023 Henniger, Rowan, Beever, Mulon, Smith, Voy, Wells, Kuehn and Myer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Phillip R. Myer, cG15ZXJAdXRrLmVkdQ==