 Yu Mao1
Yu Mao1 Lifang Yang2*
Lifang Yang2*
- 1Department of Cardiovascular Surgery, Xijing Hospital, Air Force Medical University, Xi’an, China
- 2Department of Anesthesiology, Xi’an Children Hospital, Xi’an, China
The core intent of enhanced recovery after surgery (ERAS) is to alleviate the perioperative stress response and postoperative complications through perioperative multimodal analgesia and intensive surgery. Previous studies have demonstrated that multiple molecular mechanisms function in improving ischemic stroke, reversing the process of ischemia reperfusion, relieving intracerebral hemorrhage and traumatic brain injury. Meanwhile, electroacupuncture (EA) has been validated with regard to its efficacy and safety in perioperative treatment, which has an important impact on rehabilitation research and ERAS development. Currently, studies have showed that EA provides a new approach for cerebrovascular protection by activating various molecular mechanisms. In this review, we display the recent progress of main molecular mechanisms of EA in ERAS from the perspectives of protecting cerebrovascular functions, in order to provide guidance for the clinical treatment.
1. Introduction
Enhanced recovery after surgery (ERAS) was originally proposed by Kehlet and Wilmore in 2001 (1), aiming to reduce the perioperative stress response. ERAS minimizes the perioperative stress response, reduces patient pain and promotes early recovery of organ function through various medical modifications, thus prompting patients to rapidly return to a preoperative state (2). Furthermore, traditional Chinese medicine (TCM) has preliminary practice and experience with ERAS. According to TCM, acupoints are the specific parts of the meridians where qi and blood gather and come in and out of the body surface during acupuncture (3). Electroacupuncture (EA) is performed on the basis of traditional acupuncture, and is a combination of acupuncture and electric stimulation technology (4). EA can improve neuromuscular activity, analgesia, and immune regulation and protect the function of organs (5). In addition, EA pretreatment has been proposed as a novel method to protect cerebrovascular function in the regulation of various molecular mechanisms (6). In this review, we reviewed the evidence for the role of main molecular mechanisms of EA in the cerebrovascular protection of ERAS.
2. Promotion of cerebrovascular ischemia reperfusion injury and ischemic stroke
Stroke is the second leading cause of death worldwide and a main cause of disability, thereby endangering human health (7). The most effective treatments are thrombolysis and interventions, but both treatments have a limited time window. Therefore, there is an urgent need for an alternative treatment strategy for cerebrovascular ischemia reperfusion injury (CIRI), which may promote the recovery of neurological function and reduce the burden of stroke through multidirectional and comprehensive treatment with multiple targets. A number of studies have been conducted to examine acupoints most commonly used to treat ischemic stroke. The most commonly used acupoints for ischemic stroke include Baihui (GV20), ST36, Quchi (LI11), Shuigou (GV26), and Hegu (LI4) (8). Here, we investigated the evidence of autophagy and related molecular mechanisms of EA in the ischemic stroke of ERAS (Table 1).
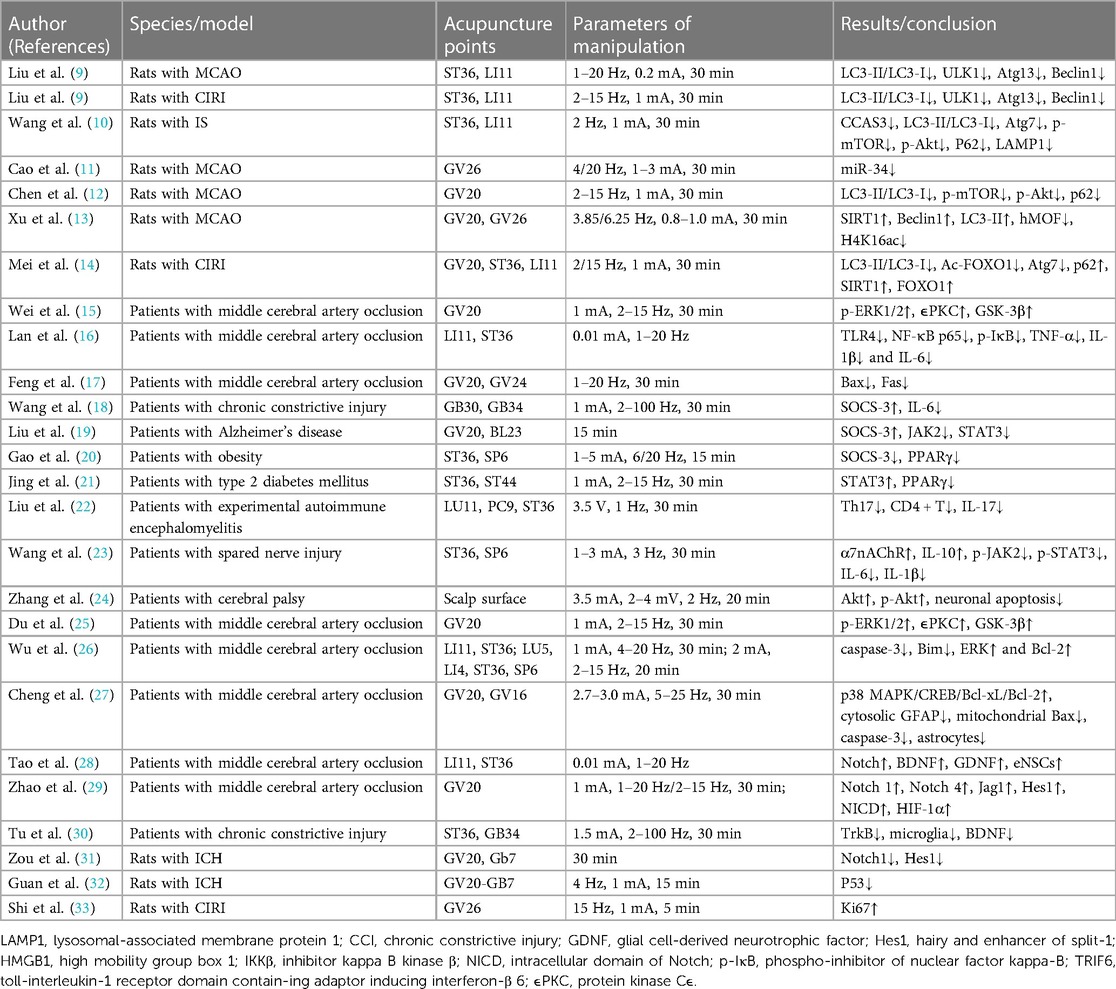
Table 1. Research information of electroacupuncture on cerebrovascular diseases during enhanced recovery after surgery period.
2.1. Autophagy
Autophagy is a highly conserved lysosomal-dependent process that may be in-volved in maintaining cell homeostasis. Autophagy may sequester aged proteins and misfolded molecules for degradation by nonspecifically targeting protein aggregates. Several studies have demonstrated that a variety of interventions may ameliorate CIRI by modulating autophagy. Therefore, autophagy plays a crucial role in the regulation of cerebral ischemia.
2.1.1. The mammalian target of rapamycin (mTOR) pathway
mTOR is a protein with serine/threonine kinase activity that orchestrates anabolic and catabolic processes to maintain essential homeostasis. mTORC1 is the main regulator of autophagy, which may promote anabolism and inhibit catabolic processes by inhibiting autophagy. Autophagosome formation is regulated by the mTORC1 pathway and LC3-II (34). LC3-I is localized in the autophagosome membrane, and the number of autophagosomes is closely related to the ratio of LC3-II/LC3-I (35). Furthermore, the activation of autophagy in postischemic neurons occurs through the activation of Beclin1 and LC3 in the periinfarct cortex (35). Liu et al. (9) demonstrated that EA pretreatment may downregulate the LC3-II/LC3-I ratio and reduce the number of autolysosomes, lysosomes, and autophagosomes in ischemic paracortical areas. mTOR regulates autophagy in CIRI through two major pathways: the Akt-mTOR pathway and the adenosine 5′-monophosphate-activated protein kinase (AMPK)-mTOR pathway. The Akt-mTOR pathway is regulated by the phosphatidylinositol 3-kinase (PI3K) enzyme, and its downregulation may induce autophagy (36). Conversely, the increased adenosine monophosphate (AMP)/adenosine triphosphate (ATP) ratio and calcium influx caused by ischemic stress might enhance AMPK activity (37). Wang et al. (10) showed that the protective effect of EA at Quchi (LI11) and ST36 was related to the inhibition of neuronal autophagy and the activation of the PI3K-Akt signaling pathway. In this study, EA pretreatment after IS inhibited the expression levels of ischemia-activated LC3-II/LC3-I and Atg7 and increased the expression levels of ischemia-inhibited PI3K and phosphorylation of mTOR and Akt (10). In addition, since mTORC1 inactivates autophagy-inducing proteins [ULK1/2 and autophagy-related gene 13 (Atg13)], the reduction in mTORC1 activity may enhance the activation of ULK1, thereby promoting autophagic flux (38). Moreover, EA also decreased the expression of autophagosome membrane markers, such as ULK1 and Atg13. Liu et al. (9) showed that autophagy is mediated through the mTORC1-ULK complex-Beclin1 pathway, and EA may prevent IS by inhibiting autophagy. They demonstrated that stimulation of LI11 and ST36 improved neurological deficits and reduced infarct volume. After EA stimulation, the levels of LC3 II/I, ULK1, Atg13 and Beclin1 decreased, while the expression of mTORC1 increased in the peripheral cortex (9).
2.1.2. The hypoxia-inducible factor 1 (HIF-1) pathway and ROS
HIF-1 is a transcription factor that regulates adaptive responses to hypoxia. It has become a major focus of neuroscience research because it regulates postischemic pathological processes such as apoptosis, energy metabolism, and gene transcription. Recent studies have shown that HIF-1 is involved in the regulation of autophagy after IS (39). Activated HIF-1 promotes the expression of BINP under ischemic conditions. BCL2/adenovirus E1B 19 kd-interacting protein (BNIP)3/BNIP3l competes with Beclin1 and dissociates it from the Beclin1/Bcl-2 complex, thereby stimulating Beclin1 to participate in autophagosome formation (40). Lu et al. (41) demonstrated that hypoxic preconditioning (HPC) activates autophagy through the HIF-1/BNIP3/Beclin1 signaling pathway, which indicates that HIF-1 is involved in the activation of autophagy and is a potential therapeutic target. Furthermore, HIF-1 ex-pression is also involved in the upregulation of mitophagy by inhibiting the mTOR signaling pathway, although whether this effect is regulated by BNIP3 needs to be further verified (42). In addition, HIF-1 may activate autophagy by regulating the expression of p53 (43). Oxidative stress after CIRI leads to excessive reactive oxygen species (ROS) accumulation, which is related to the regulation of autophagy (44). ROS mediate autophagy mainly through intracellular transcriptional regulation (45). Elevated ROS increases p53 levels, which activates two autophagy modulators, Tp53-induced glycolysis and apoptosis regulator (TIGAR), and DNA damage-regulated autophagy modulator (DRAM) (46, 47). ROS also enhanced the transcription of Nrf2. Nrf2 promotes the expression of the autophagy-related protein P62 to mediate autophagy (48). ROS stimulate forkhead box O3 (FOXO3) expression by activating LC3 and BNIP3 to regulate autophagy (49). On the other hand, Atg4, which is responsible for autophagosome membrane elongation, has also been shown to be inhibited by ROS, thereby supporting autophagosome formation (50). Atg4 is involved in the formation of LC3-I by exposing the C-terminal residue of LC3, and LC3-I then com-bines with phosphate ethanolamine (PE) to form LC3-II. In addition, Atg4 is involved in LC3-II cleavage. Atg4 protease activity was inhibited by ROS oxidation. Inhibition of Atg4 promotes autophagy by reducing LC3-II cleavage (44, 51, 52).
2.1.3. Extra signaling pathways
Micro ribonucleic acid (MiRNA)s are small endogenous noncoding ribonucleic acid (RNA)s that control messenger RNA (mRNA) degradation and/or regulate protein translation by interacting with specific sites located in the 3′ or 5′-untranslated regions of target mRNAs. Studies have shown that the Wnt pathway may play an important role in EA pretreatment in CIRI. Cao et al. (11) showed that EA attenuated CIRI by inhibiting autophagy through activation of the miR-34/Wnt pathway. The results showed that miR-34 was significantly increased in the middle cerebral artery occlusion (MCAO) group, while EA pretreatment decreased the expression of miR-34. Wnt1 is a target of miR-34, so EA pretreatment after MCAO also alleviated autophagy in the MCAO group. Similarly, Chen et al. (12) found that EA pretreatment inhibited autophagy by inhibiting glycosynthase kinase (GSK)3β in the Wnt pathway, thereby inducing tolerance to CIRI. In addition, Xu et al. (13) showed that EA inhibited the H4K16ac process, promoted autophagy and attenuated CIRI. These findings suggest that modulation of histone H4 lysine 16 acetylation-mediated autophagy may be a key mechanism for EA in CIRI (13). Mei et al. (14) proposed that the neuroprotective mechanism of EA may be realized through the regulation of autophagy by the silent mating type information regulation 2 homolog-1 (SIRT1)-recombinant forkhead box protein O1 (FOXO1) signaling pathway. The experimental results showed that EA pretreatment decreased the LC3-II/LC3-I ratio, the levels of Ac-FoxO1 and Atg7, and the interaction between AcFOXO1 and Atg7 but in-creased the levels of P62, SIRT1 and FOXO1 (14). Wang et al. (53) found that EA could ameliorate mitochondrial damage induced by nitro/oxidative stress and reduce the ac-cumulation of damaged mitochondria through Pink1/Parkin-mediated mitophagy clearance, thereby protecting cells from neuronal injury in CIRI (53). In conclusion, the mechanism of autophagy in the treatment of CIRI and ischemic stroke (IS) has not been clarified, and further studies are urgently needed.
2.2. Related main molecular mechanisms
The pathological mechanism of CI/R is complex, and energy metabolism disorder, excitatory amino acid toxicity, inflammatory response and apoptosis are all involved in the pathophysiological process (54). In addition to autophagy, the cytokine-mediated inflammatory response plays a crucial role in pathological damage in CI/R (55). Therefore, early control of glial cell activation, reduction in leukocyte infiltration and release of inflammatory mediators and inhibition of the inflammatory response are essential to reduce the occurrence of stroke. In particular, the inflammatory cascade following acute cerebral infarction (ACI) is accompanied by the activation of many inflammatory cells and the release of inflammatory mediators, and signaling pathways are critical to these inflammatory cells. Therefore, it is of great significance to study the effect of EA on the inflammatory signaling pathway after ACI. Some studies have shown that the signaling pathways of the inflammatory response after ACI mainly include Toll-IL-1 receptor (TLR)4/ nuclear factor kappa-B (NF-κB), Janus kinase (JAK)2/signal transducer and activator of transcription (STAT)3, phosphatidylinositol-3-hydroxykinase (PI3K)/Akt, extracellular regulated protein kinases (ERK)/Jun N-terminal kinase (JNK)/p38, Notch and brain-derived neurotrophic factor (BDNF)/tyrosine kinase receptor B (TrkB) (Figure 1). In addition, the brain protective mechanism of EA may be related to CE1R (15, 56).
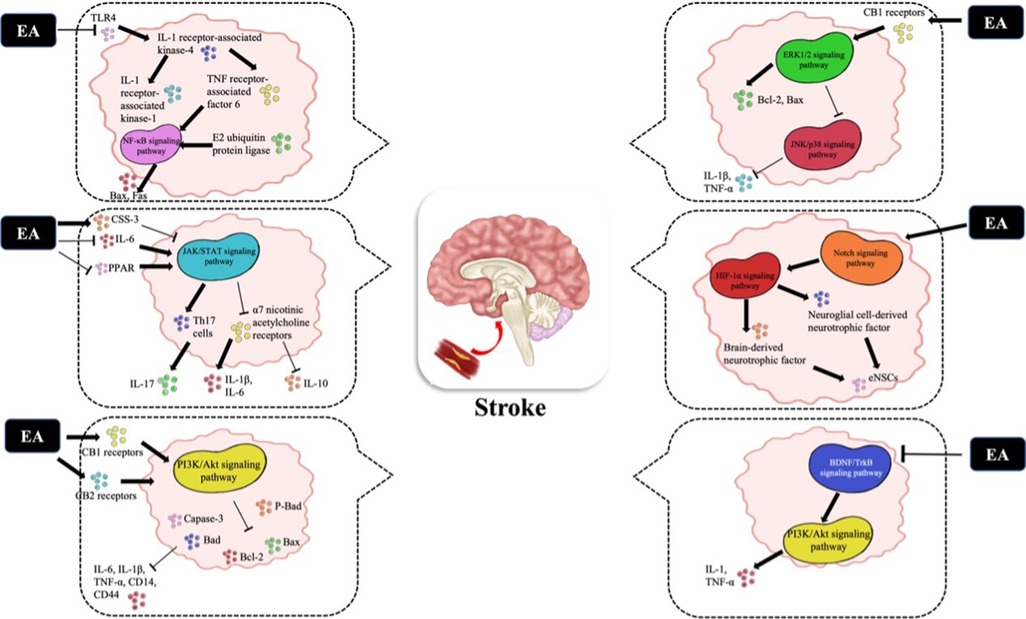
Figure 1. The related molecular mechanisms by which EA promotes CIRI. (I) EA may upregulate the expression of the TLR4/NFκB signaling pathway and decrease the expression of Bax and Fas. (II) After EA stimulation, the JAK2/STAT3 signaling pathway is activated; (i) CSS-3 is upregulat-ed and plays an anti-inflammatory role; (ii) PPARγ expression is downregulated to play an an-ti-inflammatory role; (iii) Th17 cells and the α7 nicotinic acetylcholine receptor are inhibited to reduce brain damage. (III) EA activates the PI3K/Akt signaling pathway to decrease the expres-sion of Bad, p-BAD, Bcl2, Bax and Caspase-3. (IV) The ERK/JNK/P38 signaling pathway is acti-vated by EA to downregulate the expression of Bad, Bax and Caspase-3, which decreases the ex-pression of TNF-α and IL-1β. (V) After EA stimulation, the Notch signaling pathway and down-stream HIF-1α pathway are activated to reverse intracerebroventricular injury. (VI) EA can acti-vate the BDNF/TrkB signaling pathway to activate the downstream P13K/Akt pathway. EA, electropuncture; CI/R, cerebral ischemia/reperfusion; TLR, toll-like receptor; JAK/STAT, Janus ki-nase/signal transduction and transcriptional activator; CSS-3, cytokine signal suppressor protein 3; PPAR, peroxisome proliferator-activated receptor; ERK, extracellular regulated protein kinases; JNK, c-Jun N-terminal kinase; BDNF, brain-derived neurotrophic factor; TrkB, tyrosine kinase receptor B.
2.2.1. TLR4/NF-κB
TLR is a pattern recognition receptor, and the signaling pathway is closely related to the precise regulation of the cell TLR domain adapter, in which TLR4 is closely related to inflammatory gene expression (57). After TLR4 activation, IL-1 receptor-associated kinase-4 activates other members of the IL-1 receptor-associated kinase family, including IL-1 receptor-associated kinase-1 and TNF receptor-associated factor 6. TNF receptor-associated factor 6 is then combined with E2 ubiquitin protein ligase to activate a complex containing transforming growth factor-β (TGF-β) activated kinase 1 and TGF-β-activated kinase 11. This complex activates the mitogen-activated protein kinase (MAPK) and NF-κB pathways (58). The results showed that EA significantly decreased the expression of TLR4, NF-κB and other key signaling molecules in the TLR4/NF-κB signaling pathway after ACI and inhibited the secretion of TNF-α and other inflammatory cytokines. Therefore, it is speculated that EA may mediate the TLR4/NF-κB signaling pathway to exert an anti-inflammatory effect after ACI (16). EA stimulation of GV20 and Shenting (GV24) inhibited the activation of NF-κB signaling and apoptosis of brain cells, decreased the expression of Bax and Fas, and improved learning ability and poststroke recognition ability (17). These studies suggest that EA in ACI can upregulate the expression of the TLR4/NFκB pathway and play a role in nutritional protection and inflammatory response limitation.
2.2.2. JAK/STAT signaling pathway
The JAK/STAT signaling pathway is an important pathway that regulates cellular inflammation and oxidative stress. Activated JAKs catalyze tyrosine phosphorylation, recruit STATs through the SH2 domain, and then increase gene transcription and ex-pression by forming homodimers and binding to corresponding target gene promoters (59). The JAK2/STAT3 pathway is most closely associated with central nervous system (CNS) diseases and is important in the regulation of microglial activation. After CI/R, the expression of JAK2, p-JAK2 and p-STAT3 is increased in the brain, especially in activated microglia and astrocytes, which may aggravate cerebral edema (60, 61). It has been demonstrated that downregulation of JAK2/STAT3 phosphorylation reduces the secretion of TNF-α and IL-1β in lipopolysaccharide-induced microglia (62). After EA stimulation of GV20, IL-1β activates JAK2/STAT3, which promotes phosphorylation of STAT3, levels of glial fibrillary acidic protein (GFAP) and vimentin as well as the release of inflammatory factors and adhesion molecules by binding to IL-1β receptors, resulting in T-cell immune responses (63). Overactivation of the JAK2/STAT3 pathway not only acts as an extramembrane signal of ischemia and hypoxia but also upregulates inflammatory factors and activates neuroglial cells, thus exacerbating the damage to brain tissues. Therefore, identifying downregulated targets for JAK2/STAT3 signaling may be a novel strategy for inhibiting neuroinflammation. Moreover, cytokine signal suppressor protein 3 (CSS-3) belongs to the suppressor of cytokine signaling (SOCS) family of cytokine signal suppressor proteins and is involved in the negative regulation of JAK2/STAT3 signaling. After EA, activation of neuroglial cells was inhibited, and expression of CSS-3 was upregulated, thus providing a negative regulation to JAK2/STAT3 signaling to yield an anti-inflammatory role (18). Another study found that IL-6-induced activation of JAK2/STAT3 signaling increased the expression of superoxide dismutase (SOD)2 target genes and is involved in antioxidant/anti-inflammatory responses (64). However, knocking out or blocking IL-6 signaling exacerbated ischemic injury (65). JAK2/STAT3 mRNA and protein levels were significantly upregulated following EA treatment of chronic cerebral hypoperfusion rats, suggesting that EA may inhibit inflammation by regulating IL-6. Furthermore, EA can regulate the release of microglial inflammatory factors by downregulating the expression of JAK2 and STAT3 in the cerebral cortex JAK2/STAT3 signaling pathway, thereby alleviating chronic inflammatory injury (19, 66). In addition, peroxisome proliferator activated receptor (PPAR) is a ligand-dependent transcription factor in the nuclear hormone receptor family, including three subtypes: PPARα, PPARβ/δ and PPARγ (67). Recent studies have shown that PPARγ is closely related to the JAK2/STAT3 signaling pathway after CI/R. In obese rat models, EA inhibits PPARγ agonist-induced JAK2/STAT3 signal activation, downregulates CSS-3 and PPARγ expression, and increases leptin receptor and STAT3 levels, thereby reducing food intake and weight gain. Therefore, EA stimulation was speculated to have a benign regulatory effect on the overexpression of CSS-3 and PPARγ in obese rat models (20, 21). The effect of EA after CI/R on PPARγ-mediated receptors in the JAK2/STAT3 pathway is still unclear. Further studies are needed to determine how downstream protein and gene expression in the JAK2/STAT3 pathway is involved in pathological brain injury. Last but not least, experimental results showed that CD4+ T lymphocytes differentiated into Th17 cells regulated by JAK2/STAT3 with the induction of IL-6 (68). EA can significantly reduce inflammatory cell infiltration, downregulate CD4+ T lymphocyte expression, improve neurological function scores (69, 70), reduce the expression of Th17 cells and pathological changes in the brain, and effectively inhibit the secretion of the proinflammatory cytokine IL-17 in mice (22, 71). α7 nicotinic acetyl-choline receptors are widely distributed in the CNS. After activation, it regulates the phosphorylation of JAK2/STAT3 signaling pathway proteins and participates in inflammatory responses (72). EA stimulation activates α7 nicotinic acetylcholine receptors by inhibiting JAK2/STAT3 signaling and promotes the balance between the proinflammatory cytokines IL-1β and IL-6 and the anti-inflammatory cytokine IL-10 (23). Therefore, EA may affect various targets in the JAK2/STAT3 signaling pathway to inhibit inflammatory responses and reduce brain damage.
2.2.3. PI3K/Akt
The PI3K/Akt signaling pathway plays a role in regulating cell survival, proliferation, metabolism and cancer progression (73). Cannabinoid receptor (CB) 1 receptor mediates activation of the PI3K/Akt pathway and increases phosphorylation of glycogen synthase kinase 3β. Therefore, the activation of the PI3K/Akt signaling pathway may be related to neuroprotective properties (74). Increased expression of micro-glia/macrophages and the inflammatory cytokines IL-6, IL-1β, TNF-α, CD14, CD44 and inducible nitric oxide synthase were observed after cerebral ischemia (75). CI/R sig-nificantly increased the expression of Bad, p-BAD, Bcl-2 and Bax and the number of Caspase-3-positive cells in the PI3K/Akt signaling pathway. Similar to the CB1 receptor, the CB2 receptor also shows neuroprotective functions by activating the PI3K/Akt pathway in the MAPK cascade (76). After EA treatment, the expression levels of PI3K and p-Akt were significantly increased, while the expression levels of Bad, p-BAD, Bcl2, Bax and Caspase-3 were significantly decreased, indicating that EA may interfere with the expression of downstream apoptotic genes through the PI3K/Akt signaling pathway. Zhang et al. (24) combined TCM with modern medical theory, and scalp acupuncture was used to stimulate the corresponding motor cortex projection area (GV20, etc.), and it was found that this therapy can inhibit the PI3K/Akt signaling pathway, reduce hip-pocampal neuron apoptosis, and promote functional recovery. The effect of EA on the PI3K/Akt signaling pathway may be mediated by CB receptors, which downregulate the expression of inflammatory cells.
2.2.4. ERK/JNK/p38
The MAPK family is an important signal regulating enzyme that connects cell membrane surface receptors and regulatory genes. Cellular responses to neuroprotection may involve the CB1 receptor and associated signal transduction elements, including ERK 1/2, JNK and p38, which are phosphorylated to activate and promote transcription factors to regulate cell survival, differentiation and apoptosis (77). It was reported that p38 mitogen-activated protein kinase (p38 MAPK) signaling not only promotes reactive astrocyte injury but also exacerbates progressive infarction in CI/R (78). Activation of the MAPK/ERK pathway promotes proliferation and differentiation of hippocampal endogenous neural stem cells (eNSCs) in rats and reduces neuronal apoptosis after CI (79). Another study found that activation of the MAPK/ERK signaling pathway exacerbates brain damage after CI/R, and this mechanism may be associated with activation of JNK/p38 (80, 81). The protective effects of EA may be associated with activation of the ERK1/2 pathway by the CB1 receptor, promotion of endogenous protein kinase Cε (εPKC)-mediated anti-apoptosis, and enhancement of glycogen synthase kinase 3β (15, 25). Liu et al. (82) demonstrated by dUTP gap terminal labeling and transmission electron microscopy that EA stimulation of GV20 and GV24 inhibited JNK and P38 activation, enhanced ERK1/2 activation and upregulated Bcl-2/Bax signaling pathway protein expression to promote apoptosis clearance. Studies have shown that EA may induce the ERK/JNK/P38 pathway to inhibit the upregulation of caspase-3, reverse the inhibition of ERK and Bcl-2, and exert an-ti-apoptotic and neuroprotective effects (26, 83). In addition, the ERK/p38 signaling pathway activates adenosine monophosphate response element binding protein after EA stimulation, which is a kind of selective nuclear transcription factor that regulates gene expression and nuclear translocation, is involved in cell survival, neurogenesis and neural plasticity, inhibits reactive astrocytes, and reduces TNF-α, IL-1β, and other downstream inflammatory factors (27).
2.2.5. Notch
In mammals, the Notch receptor is a highly conserved membrane-surface receptor that regulates neuronal cell development, including intracellular, transmembrane and extracellular segments. Notch 3, which is closely associated with stroke, was the first Notch gene to be identified. First, Notch 3 mutations are associated with subcortical infarction and autosomal dominant cerebral arteriopathy in leukoencephalopathy syndrome (84). In addition, Notch 1 is involved in microglia-mediated inflammation driven by NF-κB, and the use of γ-secretase inhibitors and Notch 1 antisense transgenic techniques may inhibit cerebral ischemia and reduce microglial toxicity in a synergistic manner (85). It was found that activation of eNSC CI/R in the spinal cord by the Notch signaling pathway induces differentiated astrocytes, amplifies the inflammatory cascade, inhibits eNSC diffusion and inhibits nerve repair. EA stimulation may inhibit the expression of signaling pathways and proinflammatory cytokines, induce the proliferation and differentiation of eNSCs, inhibit the differentiation of eNSCs into as-trocytes and promote the recovery of damaged nerves (86). EA stimulation promoted the proliferation and differentiation of eNSCs into functional neurons and neuroglial cells. Tao et al. (28) found that EA of GV20 promoted the proliferation and differentiation of hippocampal eNSCs by promoting the secretion of brain-derived neurotrophic factor and neuroglial cell-derived neurotrophic factor through the Notch signaling pathway. After I/R, the level of hypoxia-induced factor-1α (HIF-1α) was significantly increased in the EA group. This neuroprotective effect was reversed by intracerebroventricular injection of a γ-secretase inhibitor and HIF-1α antagonist, suggesting that EA ischemic pretreatment may be induced by activating the Notch signaling pathway and downstream HIF-1α pathway (29).
2.2.6. BDNF/TrkB
BDNF is involved in synaptic connections and synaptic plasticity between nerve cells and is a neurotrophic factor for neuronal survival and growth (87). After stroke, activated microglia secrete more BDNF, which works by binding to the high-affinity receptor TrkB. Microglia-mediated BDNF-TrkB signaling pathway interactions play an important role in hyperalgesia after spinal cord injury. Downregulation of BDNF/TrkB signaling pathway protein expression is associated with neuronal apoptosis in the striatum. In the model of CI, inflammation is triggered by activation of microglia and astrocytes and upregulation of the BDNF/TrkB pathway in the hypothalamus and amygdala, accompanied by release of TNF-α. However, the mRNA and protein expression of BDNF and TrkB in the hippo-campus was opposite to that in the hypothalamus and amygdala and decreased after brain injury (88). Activation of the BDNF/TrkB signaling pathway may further promote neutrophil and microglia/macrophage infiltration and secretion of IL-1 and NF-κB (89). Proinflammatory cytokines in the CNS induce depression-like symptoms by activating the BDNF-TrkB pathway in the prefrontal cortex, hippocampus, and nucleus accumbens (90). EA of GV20 induces the BDNF/TrkB signaling pathway and reduces abnormal mechanical pain and hyperalgesia by mediating microglial activity and BDNF expression (30). EA increases serum BDNF secretion, activates the downstream P13K/Akt pathway, and has a neuroprotective effect on apoptosis (91). In addition, the application of the TrkB-specific inhibitor K252a inhibited the recovery of neural functions, suggesting that EA-induced upregulation of BDNF may be related to the activation of the BDNF/TrkB/p-Akt/p-ERK1/2 signaling pathway (91).
3. Blood–brain barrier and intracerebral hemorrhage
Vascular endothelial cells in the brain are closely connected by various connexins and interact with pericytes and astrocytes to form the blood brain barrier (BBB) system (92). Treatment options for CNS disorders are greatly limited by the BBB. EA may be used to induce an increase in BBB permeability, providing a potential approach to deliver drugs from the systemic circulation to the brain (Table 1). Furthermore, intracerebral hemorrhage (ICH) is a cerebrovascular disease with a high mortality rate and usually results in impaired neurological function, which leads to long-term disability (93). Nontraumatic ICH results from cerebrovascular rupture and subsequent compression, and the pathophysiology is characterized by perihematoma ischemia, edema, apoptosis, and necrosis (94–96). Zou et al. (31) showed that inhibition of the Notch-Hes signaling pathway in the basal ganglia after ICH inhibited neuronal differentiation and maintained neural stem cell proliferation. In addition, Zhang et al. (97) found that EA pretreatment may be achieved by enhancing autophagic clearance of damaged mitochondria. The protective effect of autophagy during the reperfusion phase of IS may be attributed to the inhibition of mitophagy-related mitochondrial clearance and downstream apoptosis, as reflected by the decrease in cyclooxidase IV and translocase outer mitochondrial membrane 20 (TOMM20) and the increase in LC3-II/glyceraldehyde-3-phosphate dehydrogenase (GAPDH) after oxygen-glucose deprivation/reperfusion (97). Guan et al. (32) found that stimulation of GV20 with Qubin (GB7) could enhance mitophagy and thereby alleviate ICH. Therefore, EA (GV20-GB7) treatment may upregulate mitophagy and inhibit apoptosis after ICH.
4. Traumatic brain injury (TBI)
TBI is a common cerebrovascular disease. Due to direct/indirect violence to the head, the brain parenchyma is damaged, causing temporary/permanent loss of consciousness, memory loss, cognitive and neurological dysfunction, and a high disability/mortality rate (98, 99). Studies have demonstrated that EA is effective in the treatment of TBI (100). Moreover, a series of secondary neuroinflammatory reactions and neurological function loss caused by hemiplegia may be alleviated (Table 1). Gu et al. (101) showed that EA may inhibit the overactivation of autophagy in cortical neurons by regulating the activation of AMPK, mTOR and ULKl, downregulating AMPK and upregulating p-mTOR and p-ULK1, thereby improving the symptoms of TBI. Shi et al. (33) reported that EA could significantly improve the blood flow and oxygen uptake capacity in the damaged brain areas, promote the proliferation and differentiation of oligodendrocyte precursor cells, and improve the repair ability of neurons. He et al. (102) showed that p-AMPK induced autophagy and promoted cell apoptosis during TBI by inhibiting the activity of mTOR. Fan et al. (103) confirmed that when cells are in an abnormal state, activated AMPK may inhibit the activity of mTOR, leading to the dissociation of the mTOR-ULK1 complex and thereby activating autophagy.
5. Future directions and conclusions
The brain is the most sensitive organ to hypoxia. Cerebrovascular diseases seriously threaten human health. Above studies have shown that EA pretreatment may alleviate IS and its related symptoms and alleviate CIRI. Meanwhile, EA may relieve the symptoms of ICH and TBI. However, there are still defects in the design of EA-related experiments, and how to improve the design of experiments to avoid bias is also an urgent problem to be solved. Furthermore, EA may be used to treat a variety of diseases, but the disease spectrum is not clear, and the molecular mechanisms need to be further expanded. Additionally, perioperative EA affects the long-term health status of patients after surgery still needs to be further explored. This review mainly focus on the role of EA in the induction of molecular mechanisms in cerebrovascular protection to provide potential methods for further research. In the future, we believe that as the mechanism of EA is further revealed and clarified, the clinical application of EA during perioperative ERAS will be further expanded, and it will have brighter development prospects.
Author contributions
YM: Writing – original draft. LY: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article.
This research was supported by National Natural Science Foundation of China, Award Numbers: 81774415, 82174493; Outstanding Youth Foundation of Shaanxi Province, Award Number: 2021JC-49.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kehlet H, Wilmore DW. Multimodal strategies to improve surgical outcome. Am J Surg. (2002) 183:630–41. doi: 10.1016/s0002-9610(02)00866-8
2. Adamina M, Kehlet H, Tomlinson GA, Senagore AJ, Delaney CP. Enhanced recovery pathways optimize health outcomes and resource utilization: a meta-analysis of randomized controlled trials in colorectal surgery. Surgery. (2011) 149:830–40. doi: 10.1016/j.surg.2010.11.003
3. Zhang J, Xu W. Frequent ventricular extra systole treated by needing neiguan (PC6) plus oral administration of mexiletine: a report of 30 cases. J Tradit Chin Med. (2004) 24:40–1.15119171
4. Huang W, Li X, Wang Y, Yan X, Wu S. Electroacupuncture for women with stress urinary incontinence: protocol for a systematic review and meta-analysis. Medicine (Baltimore). (2017) 96:e9110. doi: 10.1097/MD.0000000000009110
5. Capodice JL, Parkhomenko E, Tran TY, Thai J, Blum KA, Chandhoke RA, et al. A randomized, double-blind, sham-controlled study assessing electroacupuncture for the management of postoperative pain after percutaneous nephrolithotomy. J Endourol. (2019) 33:194–200. doi: 10.1089/end.2018.0665
6. Zhang J, Zhu L, Li H, Tang Q. Electroacupuncture pretreatment as a novel avenue to protect heart against ischemia and reperfusion injury. Evid Based Complement Altern Med. (2020) 2020:9786482. doi: 10.1155/2020/9786482
7. Feigin V, Brainin M. Reducing the burden of stroke: opportunities and mechanisms. Int J Stroke. (2019) 14:761–2. doi: 10.1177/1747493019874718
8. Chavez LM, Huang SS, MacDonald I, Lin JG, Lee YC, Chen YH. Mechanisms of acupuncture therapy in ischemic stroke rehabilitation: a literature review of basic studies. Int J Mol Sci. (2017) 18:2270. doi: 10.3390/ijms18112270
9. Liu W, Shang G, Yang S, Huang J, Xue X, Lin Y, et al. Electroacupuncture protects against ischemic stroke by reducing autophagosome formation and inhibiting autophagy through the mTORC1-ULK1 complex-Beclin1 pathway. Int J Mol Med. (2016) 37:309–18. doi: 10.3892/ijmm.2015.2425
10. Wang MM, Zhang M, Feng YS, Xing Y, Tan ZX, Li WB, et al. Electroacupuncture inhibits neuronal autophagy and apoptosis via the PI3K/AKT pathway following ischemic stroke. Front Cell Neurosci. (2020) 14:134. doi: 10.3389/fncel.2020.00134
11. Cao S, Yang Y, Yu Q, Shu S, Zhou S. Electroacupuncture alleviates ischaemic brain injury by regulating the miRNA-34/wnt/autophagy axis. Brain Res Bull. (2021) 170:155–61. doi: 10.1016/j.brainresbull.2021.02.002
12. Chen C, Yu Q, Xu K, Cai L, Felicia BM, Wang L, et al. Electroacupuncture pretreatment prevents ischemic stroke and inhibits Wnt signaling-mediated autophagy through the regulation of GSK-3β phosphorylation. Brain Res Bull. (2020) 158:90–8. doi: 10.1016/j.brainresbull.2020.03.002
13. Xu SY, Lv HQ, Li WQ, Hong H, Peng YJ, Zhu BM. Electroacupuncture alleviates cerebral ischemia/reperfusion injury in rats by histone H4 lysine 16 acetylation-mediated autophagy. Front Psychiatry. (2020) 11:576539. doi: 10.3389/fpsyt.2020.576539
14. Mei ZG, Huang YG, Feng ZT, Luo YN, Yang SB, Du LP, et al. Electroacupuncture ameliorates cerebral ischemia/reperfusion injury by suppressing autophagy via the SIRT1-FOXO1 signaling pathway. Aging. (2020) 12:13187–205. doi: 10.18632/aging.103420
15. Wei H, Yao X, Yang L, Wang S, Guo F, Zhou H, et al. Glycogen synthase kinase-3β is involved in electroacupuncture pretreatment via the cannabinoid CB1 receptor in ischemic stroke. Mol Neurobiol (2014) 49:326–36. doi: 10.1007/s12035-013-8524-5
16. Lan LAN, Tao J, Chen A,, Xie G,, Huang JIA,, Lin J, et al. Electroacupuncture exerts anti-inflammatory effects in cerebral ischemia-reperfusion injured rats via suppression of the TLR4/NF-κB pathway. Int J Mol Med. (2012) 31:75–80. doi: 10.3892/ijmm.2012.1184
17. Feng X, Yang S, Liu J, Huang JIA, Peng JUN, Lin J, et al. Electroacupuncture ameliorates cognitive impairment through inhibition of NF-κB-mediated neuronal cell apoptosis in cerebral ischemia-reperfusion injured rats. Mol Med Rep. (2013) 7:1516–22. doi: 10.3892/mmr.2013.1392
18. Wang ZF, Yu XM, Gong DG, Wang YQ, Wu CG. Effects of electroacupuncture at “Yanglinquan” and “Huantiao” acupoints on expression of SOCS3 in spinal cord in rats with neuropathic pain. Zhongguo Tengtong Yixue Zazhi. (2015) 21:335–40.
19. Liu J, Du YJ, Zhou QL, Sun GJ. Acupuncture plus moxibustion intervention improves learning-memory ability by suppressing hippocampal JAK2/STAT3 signaling in Alzheimer’s rats. Zhen Ci Yan Jiu. (2019) 44:79–84. doi: 10.13702/j.1000-0607.180456
20. Gao J, Tang CL, Liu RJ, Chen XL, Xie H, Hou YX, et al. Effect of different intensities of electroacupuncture stimulation on expression of SOCS-3 and PPAR-gamma mRNA in adipose tissues of obesity rats. Zhen Ci Yan Jiu. (2013) 38:31–4.23650797
21. Jing X, Ou C, Chen H, Wang T, Xu B, Lu S, et al. Electroacupuncture reduces weight gain induced by rosiglitazone through PPARγ and leptin receptor in CNS. Evid Based Complement Altern Med. (2016) 2016:8098561. doi: 10.1155/2016/8098561
22. Liu Y, Wang H, Wang X, Mu L, Kong Q, Wang D, et al. The mechanism of effective electroacupuncture on T cell response in rats with experimental autoimmune encephalomyelitis. PLoS One. (2013) 8:e51573. doi: 10.1371/journal.pone.0051573
23. Wang Y, Xue M, Xia Y, Jiang Q, Huang Z, Huang C. Electroacupuncture treatment upregulates α7nAChR and inhibits JAK2/STAT3 in dorsal root ganglion of rat with spared nerve injury. J Pain Res. (2019) 12:1947–55. doi: 10.2147/JPR.S203867
24. Zhang H, Gao J, Wang M, Yu X, Lv X, Deng H, et al. Effects of scalp electroacupuncture on the PI3K/Akt signalling pathway and apoptosis of hippocampal neurons in a rat model of cerebral palsy. Acupunct Med. (2018) 36:96–102. doi: 10.1136/acupmed-2016-011335
25. Du J, Wang Q, Hu B, Peng Z, Zhao Y, Ma L, et al. Involvement of ERK 1/2 activation in electroacupuncture pretreatment via cannabinoid CB1 receptor in rats. Brain Res. (2010) 1360:1–7. doi: 10.1016/j.brainres.2010.07.034
26. Wu C, Wang J, Li C, Zhou G, Xu X, Zhang X, et al. Effect of electroacupuncture on cell apoptosis and ERK signal pathway in the hippocampus of adult rats with cerebral ischemia-reperfusion. Evid Based Complement Altern Med. (2015) 2015:414965. doi: 10.1155/2015/414965
27. Cheng CY, Lin JG, Tang NY, Kao ST, Hsieh CL. Electroacupuncture at different frequencies (5hz and 25hz) ameliorates cerebral ischemia-reperfusion injury in rats: possible involvement of p38 MAPK-mediated anti-apoptotic signaling pathways. BMC Complement Altern Med. (2015) 15:241. doi: 10.1186/s12906-015-0752-y
28. Tao J, Chen B, Gao Y, Yang S, Huang J, Jiang X, et al. Electroacupuncture enhances hippocampal NSCs proliferation in cerebral ischemia-reperfusion injured rats via activation of notch signaling pathway. Int J Neurosci. (2014) 124:204–12. doi: 10.3109/00207454.2013.840781
29. Zhao Y, Deng B, Li Y, Zhou L, Yang L, Gou X, et al. Electroacupuncture pretreatment attenuates cerebral ischemic injury via notch pathway-mediated up-regulation of hypoxia inducible factor-1α in rats. Cell Mol Neurobiol. (2015) 35:1093–103. doi: 10.1007/s10571-015-0203-9
30. Tu WZ, Li SS, Jiang X, Qian XR, Yang GH, Gu PP, et al. Effect of electro-acupuncture on the BDNF-TrkB pathway in the spinal cord of CCI rats. Int J Mol Med. (2018) 41:3307–15. doi: 10.3892/ijmm.2018.3563
31. Zou W, Chen QX, Sun XW, Chi QB, Kuang HY, Yu XP, et al. Acupuncture inhibits Notch1 and Hes1 protein expression in the basal ganglia of rats with cerebral hemorrhage. Neural Regen Res. (2015) 10:457–62. doi: 10.4103/1673-5374.153696
32. Guan R, Li Z, Dai X, Zou W, Yu X, Liu H, et al. Electroacupuncture at GV20-GB7 regulates mitophagy to protect against neurological deficits following intracerebral hemorrhage via inhibition of apoptosis. Mol Med Rep. (2021) 24:492. doi: 10.3892/mmr.2021.12131
33. Shi L, Cao HM, Li Y, Xu SX, Zhang Y, Zhang Y, et al. Electroacupuncture improves neurovascular unit reconstruction by promoting collateral circulation and angiogenesis. Neural Regen. Res. (2017) 12:2000–6. doi: 10.4103/1673-5374.221156
34. Xing Y, Zhang M, Li WB, Dong F, Zhang F. Mechanisms involved in the neuroprotection of electroacupuncture therapy for ischemic stroke. Front Neurosci. (2018) 12:929. doi: 10.3389/fnins.2018.00929
35. Li X, Wang MH, Qin C, Fan WH, Tian DS, Liu JL. Fingolimod suppresses neuronal autophagy through the mTOR/p70S6K pathway and alleviates ischemic brain damage in mice. PLoS One. (2017) 12:e0188748. doi: 10.1371/journal.pone.0188748
36. Yudushkin I. Getting the Akt together: guiding intracellular Akt activity by PI3K. Biomolecules. (2019) 9:67. doi: 10.3390/biom9020067
37. Ke R, Xu Q, Li C, Luo L, Huang D. Mechanisms of AMPK in the maintenance of ATP balance during energy metabolism. Cell Biol Int. (2018) 42:384–92. doi: 10.1002/cbin.10915
38. Hardie DG. Why starving cells eat themselves. Science. (2011) 331:410–1. doi: 10.1126/science.1201691
39. Hou Y, Wang J, Feng J. The neuroprotective effects of curcumin are associated with the regulation of the reciprocal function between autophagy and HIF-1α in cerebral ischemia-reperfusion injury. Drug Des Dev Ther. (2019) 13:1135–44. doi: 10.2147/DDDT.S194182
40. Mo Y, Sun YY, Liu KY. Autophagy and inflammation in ischemic stroke. Neural Regen Res. (2020) 15:1388–96. doi: 10.4103/1673-5374.274331
41. Lu N, Li X, Tan R, An J, Cai Z, Hu X, et al. HIF-1α/Beclin1-mediated autophagy is involved in neuroprotection induced by hypoxic preconditioning. J Mol Neurosci. (2018) 66:238–50. doi: 10.1007/s12031-018-1162-7
42. Sun Y, Zhu Y, Zhong X, Chen X, Wang J, Ying G. Crosstalk between autophagy and cerebral ischemia. Front Neurosci. (2019) 12:1022. doi: 10.3389/fnins.2018.01022
43. Hou K, Xu D, Li F, Chen S, Li Y. The progress of neuronal autophagy in cerebral ischemia stroke: mechanisms, roles and research methods. J Neurol Sci. (2019) 400:72–82. doi: 10.1016/j.jns.2019.03.015
44. Huang J, Lam GY, Brumell JH. Autophagy signaling through reactive oxygen species. Antioxid Redox Signal. (2011) 14:2215–31. doi: 10.1089/ars.2010.3554
45. Wang P, Shao BZ, Deng Z, Chen S, Yue Z, Miao CY. Autophagy in ischemic stroke. Prog Neurobiol. (2018) 163–164:98–117. doi: 10.1016/j.pneurobio.2018.01.001
46. Pietrocola F, Izzo V, Niso-Santano M, Vacchelli E, Galluzzi L, Maiuri MC, et al. Regulation of autophagy by stress-responsive transcription factors. Semin Cancer Biol. (2013) 23:310–22. doi: 10.1016/j.semcancer.2013.05.008
47. Crighton D, Wilkinson S, O’Prey J, Syed N, Smith P, Harrison PR, et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. (2006) 126:121–34. doi: 10.1016/j.cell.2006.05.034
48. Puissant A, Fenouille N, Auberger P. When autophagy meets cancer through p62/SQSTM1. Am J Cancer Res. (2012) 2:397–413.22860231
49. Mahalingaiah PKS, Singh KP. Chronic oxidative stress increases growth and tumorigenic potential of MCF-7 breast cancer cells. PLoS One. (2014) 9:e87371. doi: 10.1371/journal.pone.0087371
50. Li L, Tan J, Miao Y, Lei P, Zhang Q. ROS And autophagy: interactions and molecular regulatory mechanisms. Cell Mol Neurobiol. (2015) 35:615–21. doi: 10.1007/s10571-015-0166-x
51. Parzych KR, Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal. (2014) 20:460–73. doi: 10.1089/ars.2013.5371
52. Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. (2019) 38:e101812. doi: 10.15252/embj.2019101812
53. Wang H, Chen S, Zhang Y, Xu H, Sun H. Electroacupuncture ameliorates neuronal injury by Pink1/Parkin-mediated mitophagy clearance in cerebral ischemia-reperfusion. Nitric Oxide. (2019) 91:23–34. doi: 10.1016/j.niox.2019.07.004
54. George PM, Steinberg GK. Novel stroke therapeutics: unraveling stroke pathophysiology and its impact on clinical treatments. Neuron. (2015) 87:297–309. doi: 10.1016/j.neuron.2015.05.041
55. Kawabori M, Yenari MA. Inflammatory responses in brain ischemia. Curr Med Chem. (2015) 22:1258–77. doi: 10.2174/0929867322666150209154036
56. Wang Q, Peng Y, Chen S, Gou X, Hu B, Du J, et al. Pretreatment with electroacupuncture induces rapid tolerance to focal cerebral ischemia through regulation of endocannabinoid system. Stroke. (2009) 40:2157–64. doi: 10.1161/strokeaha.108.541490
57. Chen L, Yu J. Modulation of toll-like receptor signaling in innate immunity by natural products. Int Immunopharmacol. (2016) 37:65–70. doi: 10.1016/j.intimp.2016.02.005
58. Hanamsagar R, Hanke ML, Kielian T. Toll-like receptor (TLR) and inflammasome actions in the central nervous system. Trends Immunol. (2012) 33:333–42. doi: 10.1016/j.it.2012.03.001
59. Chen XM, Yu YH, Wang L, Zhao XY, Li JR. Effect of the JAK2/STAT3 signaling pathway on nerve cell apoptosis in rats with white matter injury. Eur Rev Med Pharmacol Sci. (2019) 23:321–7. doi: 10.26355/eurrev_201901_16779
60. Gong P, Zhang Z, Zou Y, Tian Q, Han S, Xu Z, et al. Tetramethylpyrazine attenuates blood-brain barrier disruption in ischemia/reperfusion injury through the JAK/STAT signaling pathway. Eur J Pharmacol. (2019) 854:289–97. doi: 10.1016/j.ejphar.2019.04.028
61. Porro C, Cianciulli A, Trotta T, Lofrumento DD, Panaro MA. Curcumin regulates anti-inflammatory responses by JAK/STAT/SOCS signaling pathway in BV-2 microglial cells. Biology (Basel). (2019) 8:51. doi: 10.3390/biology8030051
62. Shrivastava K, Llovera G, Recasens M, Chertoff M, Giménez-Llort L, Gonzalez B, et al. Temporal expression of cytokines and signal transducer and activator of transcription factor 3 activation after neonatal hypoxia/ischemia in mice. Dev Neurosci. (2013) 35:212–25. doi: 10.1159/000348432
63. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. (2017) 541:481–7. doi: 10.1038/nature21029
64. Jung JE, Kim GS, Chan PH. Neuroprotection by interleukin-6 is mediated by signal transducer and activator of transcription 3 and antioxidative signaling in ischemic stroke. Stroke. (2011) 42:3574–9. doi: 10.1161/STROKEAHA.111.626648
65. Yamashita T, Sawamoto K, Suzuki S, Suzuki N, Adachi K, Kawase T, et al. Blockade of interleukin-6 signaling aggravates ischemic cerebral damage in mice: possible involvement of Stat3 activation in the protection of neurons. J Neurochem. (2005) 94:459–68. doi: 10.1111/j.1471-4159.2005.03227.x
66. Chiba T, Yamada M, Aiso S. Targeting the JAK2/STAT3 axis in Alzheimer’s disease. Expert Opin Ther Targets. (2009) 13:1155–67. doi: 10.1517/14728220903213426
67. Michalik L, Desvergne B, Wahli W. Peroxisome-proliferator-activated receptors and cancers: complex stories. Nat Rev Cancer. (2004) 4:61–70. doi: 10.1038/nrc1254
68. You Z, Timilshina M, Jeong BS, Chang JH. BJ-2266 ameliorates experimental autoimmune encephalomyelitis through down-regulation of the JAK/STAT signaling pathway. Eur J Immunol. (2017) 47:1488–500. doi: 10.1002/eji.201646860
69. Liu YM, Liu XJ, Bai SS, Mu LL, Kong QF, Sun B, et al. The effect of electroacupuncture on T cell responses in rats with experimental autoimmune encephalitis. J Neuroimmunol. (2010) 220:25–33. doi: 10.1016/j.jneuroim.2009.12.005
70. Kim KY, Yang WJ, Shin TK, Jeong HJ, Kim HM. Impact of acupuncture by using life-energy (qi) oriental needle on the paralysis of rats with experimental autoimmune encephalomyelitis. Am J Chin Med. (2012) 40:769–78. doi: 10.1142/s0192415/12500577
71. Lee MJ, Jang M, Choi J, Lee G, Min HJ, Chung WS, et al. Bee venom acupuncture alleviates experimental autoimmune encephalomyelitis by upregulating regulatory T cells and suppressing Th1 and Th17 responses. Mol Neurobiol. (2016) 53:1419–45. doi: 10.1007/s12035-014-9012-2
72. Yang YH, Li DL, Bi XY, Sun L, Yu XJ, Fang HL, et al. Acetylcholine inhibits LPS-induced MMP-9 production and cell migration via the a7 nAChR-JAK2/STAT3 pathway in RAW264.7 cells. Cell Physiol Biochem. (2015) 36:2025–38. doi: 10.1159/000430170
73. Fruman DA, Meyers RE, Cantley LC. Phosphoinositide kinases. Annu Rev Biochem. (1998) 67:481–507. doi: 10.1146/annurev.biochem.67.1.481
74. Ozaita A, Puighermanal E, Maldonado R. Regulation of PI3K/Akt/GSK-3 pathway by cannabinoids in the brain. J Neurochem. (2007) 102:1105–14. doi: 10.1111/j.1471-4159.2007.04642.x
75. Chen J, Wang Z, Zheng Z, Chen Y, Khor S, Shi K, et al. Neuron and microglia/macrophage-derived FGF10 activate neuronal FGFR2/PI3K/Akt signaling and inhibit microglia/macrophages TLR4/NF-κB-dependent neuroinflammation to improve functional recovery after spinal cord injury. Cell Death Dis. (2017) 8:e3090. doi: 10.1038/cddis.2017.490
76. Fernández-Ruiz J, Pazos MR, García-Arencibia M, Sagredo O, Ramos JA. Role of CB2 receptors in neuroprotective effects of cannabinoids. Mol Cell Endocrinol. (2008) 286:S91–6. doi: 10.1016/j.mce.2008.01.001
77. van der Stelt M, Di Marzo V. Cannabinoid receptors and their role in neuroprotection. NeuroMolecular Med. (2005) 7:37–50. doi: 10.1385/nmm:7:1-2:037
78. Roy Choudhury G, Ryou MG, Poteet E, Wen Y, He R, Sun F, et al. Involvement of p38 MAPK in reactive astrogliosis induced by ischemic stroke. Brain Res. (2014) 1551:45–58. doi: 10.1016/j.brainres.2014.01.013
79. Shioda N, Han F, Fukunaga K. Role of Akt and ERK signaling in the neurogenesis following brain ischemia. Int Rev Neurobiol. (2009) 85:375–87. doi: 10.1016/s0074-7742(09)85026-5
80. Alessandrini A, Namura S, Moskowitz MA, Bonventre JV. MEK1 protein kinase inhibition protects against damage resulting from focal cerebral ischemia. Proc Natl Acad Sci USA. (1999) 96:12866–9. doi: 10.1073/pnas.96.22.12866
81. Yamazaki Y, Arita K, Harada S, Tokuyama S. Activation of c-Jun N-terminal kinase and p38 after cerebral ischemia upregulates cerebral sodium-glucose transporter type 1. J Pharmacol Sci. (2018) 138:240–6. doi: 10.1016/j.jphs.2017.02.016
82. Liu J, Wang Q, Yang S, Huang J, Feng X, Peng J, et al. Electroacupuncture inhibits apoptosis of peri-ischemic regions via modulating p38, extracellular signal-regulated kinase (ERK1/2), and c-Jun N terminal kinases (JNK) in cerebral ischemia-reperfusion-injured rats. Med Sci Monit. (2018) 24:4395–404. doi: 10.12659/MSM.908473
83. Xing Y, Yang SD, Wang MM, Dong F, Feng YS, Zhang F. Electroacupuncture alleviated neuronal apoptosis following ischemic stroke in rats via midkine and ERK/JNK/p38 signaling pathway. J Mol Neurosci. (2018) 66:26–36. doi: 10.1007/s12031-018-1142-y
84. Papakonstantinou E, Bacopoulou F, Brouzas D, Megalooikonomou V, D’Elia D, Bongcam-Rudloff E, et al. NOTCH3 And CADASIL syndrome: a genetic and structural overview. EMBnet J. (2019) 24:e921. doi: 10.14806/ej.24.0.921
85. Wei Z, Chigurupati S, Arumugam TV, Jo DG, Li H, Chan SL. Notch activation enhances the microglia-mediated inflammatory response associated with focal cerebral ischemia. Stroke. (2011) 42:2589–94. doi: 10.1161/strokeaha.111.614834
86. Geng X, Sun T, Li JH, Zhao N, Wang Y, Yu HL. Electroacupuncture in the repair of spinal cord injury: inhibiting the notch signaling pathway and promoting neural stem cell proliferation. Neural Regen Res. (2015) 10:394–403. doi: 10.4103/1673-5374.153687
87. Kramár EA, Chen LY, Lauterborn JC, Simmons DA, Gall CM, Lynch G. BDNF Upregulation rescues synaptic plasticity in middle-aged ovariectomized rats. Neurobiol Aging. (2012) 33:708–19. doi: 10.1016/j.neurobiolaging.2010.06.008
88. de la Tremblaye PB, Benoit SM, Schock S, Plamondon H. CRHR1 exacerbates the glial inflammatory response and alters BDNF/TrkB/pCREB signaling in a rat model of global cerebral ischemia: implications for neuroprotection and cognitive recovery. Prog Neuro-Psychopharmacol Biol Psychiatry. (2017) 79:234–48. doi: 10.1016/j.pnpbp.2017.06.021
89. Jin M, Sheng W, Han L, He Q, Ji X, Liu K. Activation of BDNF-TrkB signaling pathway-regulated brain inflammation in pentylenetetrazole-induced seizures in zebrafish. Fish Shellfish Immunol. (2018) 83:26–36. doi: 10.1016/j.fsi.2018.09.010
90. Zhang JC, Yao W, Hashimoto K. Brain-derived neurotrophic factor (BDNF)-TrkB signaling in inflammation-related depression and potential therapeutic targets. Curr Neuropharmacol. (2016) 14:721–31. doi: 10.2174/1570159/14666160119094646
91. Lin D, De La Pena I, Lin L, Zhou SF, Borlongan CV, Cao C. The neuroprotective role of acupuncture and activation of the BDNF signaling pathway. Int J Mol Sci. (2014) 15:3234–52. doi: 10.3390/ijms15023234
92. Banks WA. From blood–brain barrier to blood–brain interface: new opportunities for CNS drug delivery. Nat Rev Drug Discov. (2016) 15:275–92. doi: 10.1038/nrd.2015.21
93. Caplan LR. Intracerebral haemorrhage. Lancet. (1992) 339:656–8. doi: 10.1016/0140-6736(92)90804-c
94. Keep RF, Hua Y, Xi G. Intracerebral haemorrhage: mechanisms of injury and therapeutic targets. Lancet. Neurol. (2012) 11:720–31. doi: 10.1016/S1474-4422(12)70104-7
95. Nehls DG, Mendelow DA, Graham DI, Sinar EJ, Teasdale GM. Experimental intracerebral hemorrhage: progression of hemodynamic changes after production of a spontaneous mass lesion. Neurosurgery. (1988) 23:439–44. doi: 10.1227/00006123-198810000-00006
96. Belur PK, Chang JJ, He S, Emanuel BA, Mack WJ. Emerging experimental therapies for intracerebral hemorrhage: targeting mechanisms of secondary brain injury. Neurosurg Focus. (2013) 34:E9. doi: 10.3171/2013.2.focus1317
97. Zhang X, Yan H, Yuan Y, Gao J, Shen Z, Cheng Y, et al. Cerebral ischemia-reperfusion-induced autophagy protects against neuronal injury by mitochondrial clearance. Autophagy. (2013) 9:1321–33. doi: 10.4161/auto.25132
98. Assis-Nascimento P, Tsenkina Y, Liebl DJ. Ephb3 signaling induces cortical endothelial cell death and disrupts the blood-brain barrier after traumatic brain injury. Cell Death Dis. (2018) 9:7. doi: 10.1038/s41419-017-0016-5
99. Sitsapesan H, Richards P. The management of traumatic brain injury. Paediatr Child Health. (2013) 23:375–9. doi: 10.1016/j.paed.2013.05.020
100. Li X, Chen C, Yang X, Wang J, Zhao ML, Sun H, et al. Acupuncture improved neurological recovery after traumatic brain injury by activating BDNF/TrkB pathway. Evid Based Complement Altern Med. (2017) 2017:8460145. doi: 10.1155/2017/8460145
101. Gu M, He P, Lyu C, Liu X, Xu Y, Cheng S, et al. Spinosin and 6′-feruloylspinosin protect the heart against acute myocardial ischemia and reperfusion in rats. Mol Med Rep. (2019) 20:4253–61. doi: 10.3892/mmr.2019.10686
102. He X, Zheng Y, Liu S, Shi S, Liu Y, He Y, et al. MiR-146a protects small intestine against ischemia/reperfusion injury by down-regulating TLR4/TRAF6/NF-κB pathway. J Cell Physiol. (2018) 233:2476–88. doi: 10.1002/jcp.26124
Keywords: enhanced recovery after surgery, electroacupuncture, cerebrovascular protection, molecular mechanism, traditional Chinese medicine
Citation: Mao Y and Yang L (2023) Molecular mechanisms of electropuncture in cerebrovascular protection during enhanced recovery after surgery period. Front. Anesthesiol. 2:1335583. doi: 10.3389/fanes.2023.1335583
Received: 9 November 2023; Accepted: 12 December 2023;
Published: 22 December 2023.
Edited by:
Alparslan Turan, Cleveland Clinic, United StatesReviewed by:
Firoozeh Madadi, Shahid Beheshti University of Medical Sciences, IranNishant Kumar, University of Delhi, India
© 2023 Mao and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lifang Yang Mjc4OTE1MzNAcXEuY29t