 Karel Bezstarosti†
Karel Bezstarosti† Jeroen A. A. Demmers
Jeroen A. A. Demmers- Proteomics Center, Erasmus University Medical Center, Rotterdam, Netherlands
The reliable, accurate and quantitative targeted detection of proteins is a key technology in molecular and cell biology and molecular diagnostics. The current golden standard for targeted protein detection in complex mixtures such as complete cell lysates or body fluids is immunoblotting, a technology that was developed in the late 1970s and has not undergone major changes since. Although widespread, this methodology suffers from several disadvantages, such as the inability to detect low-abundant proteins or specific posttranslational modifications, the requirement for highly specific antibodies, the lack of quantitative power and the often-tedious practical procedures. Mass spectrometry (MS) based targeted protein detection is an alternative technology that could circumvent these caveats. Here, we compare immunoblotting with targeted protein mass spectrometry using a parallel reaction monitoring (PRM) regime on the Orbitrap mass spectrometer. We show that PRM based MS has superior sensitivity and quantitative accuracy over immunoblotting. The limit of detection for proteolytic peptides of a purified target protein was found to be in the mid- to low-attomole range and approximately one order of magnitude higher when embedded in a complex biological matrix. The incorporation of synthetic heavy isotope labeled (AQUA) peptides as internal calibrants into the PRM workflow allows for even higher accuracy for both the relative and absolute quantitation of tryptic target peptides. In conclusion, PRM is a versatile and sensitive technology, which can overcome the shortcomings of immunoblotting. We argue that PRM based MS could become the method of choice for the targeted detection of proteins in complex cellular matrices or body fluids and may eventually replace standard methods such as Western blotting and ELISA in biomedical research and in the clinic.
Introduction
In molecular biology, biomedical research and clinical diagnostics, the current golden standard of protein detection in complex mixtures such as cell lysates and body fluids is immunoblotting (or: Western blotting). In immunoblotting, a protein sample is first loaded onto a poly-acrylamide electrophoresis (PAGE) gel to separate proteins. Subsequently, proteins are transferred from the gel to a nitrocellulose or polyvinylidene difluoride (PVDF) solid support membrane using an electric field oriented perpendicular to the surface of the gel, causing proteins to move out of the gel and onto the membrane. Specific (primary) antibodies directed against the target protein of interest then bind to the protein population on the membrane surface and these antibodies are subsequently recognized by other (secondary) antibodies. These secondary antibodies are then visualized through staining, immunofluorescence, or other methods (Towbin et al., 1979; Burnette, 1981; Smith, 1994; Westermeier, 2014). While the basic principles of the technique remain the same since its inception in 1979, there have been advancements in terms of sensitivity, quantification methods, and automation. However, the fundamental steps involved in Western blotting, including protein separation by electrophoresis, transfer to a membrane, antibody binding, and detection, have remained relatively constant.
Though generally considered a robust and inexpensive technique, there are several caveats related to immunoblotting. The specificity of the technique heavily depends on the quality of the antibody that is used. The secondary and tertiary structure of proteins can prevent the binding of antibodies to the relevant epitopes under both denaturing and native conditions, as can the occurrence of post-translational modifications (PTMs). Furthermore, cross-reactivity may cause the antibody to target another, non-related protein than the protein of interest, which happens to have similar gel retention properties (Ghosh et al., 2014). A broad specificity test of commercial antibodies showed that cross reaction with other proteins than the target protein is quite common, with more than 75% of the approximately 6,000 antibodies tested demonstrating a cross reaction or no appreciable binding to its intended target (Weller, 2016). Finally, while the running costs for Western blotting are relatively low, the costs for even limited amounts of antibody can be substantial (typically several 100 s of euros/dollars). Finally, Western blotting is a low-throughput technology: even though several samples may be run in parallel on one SDS-PAGE gel, the number of bands that can be visualised are still limited if different target proteins have similar retention properties (Ghosh et al., 2014).
A more fundamental drawback of Western blotting is the lack of quantitative character. Even when targeting relatively highly abundant cellular proteins, it remains difficult to determine relative quantities of the target protein even in a single experiment (Hammond et al., 2013). While it was recently shown that a newly developed Western blotting-based method can give accurate estimates of protein levels (Deng and Zi, 2022), the requirement of a concentration gradient may impair high-throughput analyses. Furthermore, it assumes that a concentration gradient actually displays a linear relationship in immunoblotting staining intensities, which may not necessarily be true over several orders of magnitude considering previous results [e.g., (Hammond et al., 2013),].
Alternative technologies for the targeted detection of specific proteins may overcome caveats such as the inability to detect low-abundant proteins, specific post-translational modifications and the need for highly specific antibodies, and may thus present superior performance. Here, we focus on mass spectrometry-based methods.
Highly specific methods to detect proteins using mass spectrometry in a targeted fashion are selected reaction monitoring (SRM), multiple reaction monitoring (MRM) or parallel reaction monitoring (PRM). This technology allows for highly selective and accurate proteolytic peptide analysis, since (combinations of) these peptides are assumed to have unique amino acid sequences specific to a particular target protein. SRM was originally developed on triple-quadrupole mass spectrometers, where the first quadrupole is employed as a mass filter that filters the targeted precursor ion, which is subsequently fragmented in the second quadrupole that acts as the collision chamber. The peptide fragment ions are subsequently mass analyzed in the third quadrupole. In SRM, the fragment ions are measured sequentially in distinct scans [reviewed in (Kulyyassov et al., 2021)]. While SRM is monitoring only a single fixed mass window, MRM scans rapidly over multiple narrow mass windows and thus acquires traces of multiple fragment ion masses in parallel (Chambers et al., 2014). So, MRM is the application of SRM to multiple product ions from one or more precursor ions. If the third quadrupole is replaced by a time-of-flight (TOF) or an orbitrap detector that has both a higher mass resolution and higher mass accuracy (Makarov, 2000), all fragment ions can be analyzed simultaneously and with high mass accuracy. Hence, this variant is called parallel reaction monitoring (PRM) (Peterson et al., 2012; Shi et al., 2016).
Potentially, targeted mass spectrometry could overcome the caveats described above for immunoassays. In a standard bottom-up proteomics experiment, proteins are first digested into peptides prior to analysis. Hence, multiple proteolytic peptides per target protein can be selected for targeted detection, whereas antibodies generally recognize only one epitope of a protein. In addition, isoform specific peptide sequences or post-translationally modified peptides can be targeted. Since mass spectrometric detection does not require the use of an antibody, the amino acid sequence information is usually sufficient to select the peptide precursors of interest. Peptide fragmentation patterns can be experimentally determined or predicted using AI Based algorithms such as PROSIT (Gessulat et al., 2019) to select the optimal set of fragment ions. Although in many studies targeted mass spectrometry and immunoblotting techniques for protein detection and relative quantitation have been applied and compared (Bluemlein and Ralser, 2011; Lau et al., 2011; Martínez-Márquez et al., 2013; Tsuchiya et al., 2013; Yang et al., 2013; 2014; Prasad and Unadkat, 2014; Parsons and Heazlewood, 2015; Sowers et al., 2015; Jayasena et al., 2016; Thorsen et al., 2023), a systematic assessment of the limits of detection, both for pure proteins and of proteins in complex biological matrices, has not been done. Also, many comparative studies it highlights the latest, sensitive Orbitrap technology for targeted detection as opposed to more widespread and discussed triple quadrupole mass spectrometry.
Here, we set out to make a head-to-head comparison between immunoblotting and PRM in a systematic manner, using a commercially available target protein – antibody combination (glyceraldehyde-3-phosphate dehydrogenase, GAPDH), both in pure form and spiked into a complex biological matrix. First, we determined the limit of detection (LOD) of immunoblotting using a dilution series. Subsequently, we digested GAPDH using trypsin and performed PRM on a selection of GAPDH proteolytic peptides. Peptides could be detected in the low femtogram (i.e., low- to mid-attomole range), presenting over five orders of magnitude higher sensitivity over immunoblotting. Also, the dose-response relationship was linear over a wide concentration range, spanning several orders of magnitude. Next, target peptides were absolutely quantified using known quantities of synthesized heavy isotope labelled (AQUA) peptides as internal calibrants. Furthermore, we show that using PRM, GAPDH proteolytic peptides could be detected and relatively quantified in a complex biological matrix (i.e., a yeast complete lysate). Finally, we present several suggestions for improvement of sample preparation protocols for targeted proteomics, which could even further enhance the sensitivity and quality of such measurements.
Materials and methods
Reagents
Rabbit-GAPDH was purchased from Sigma (G5262-1VL). Saccharomyces cerevisiae extract was purchased from Promega (V6941). Heavy isotope labeled VIPELGNK (13C6, 15N2-Lys) peptide was custom made by Thermo Scientific.
Immunoblotting
Lysates were diluted into 4X Laemmli buffer, boiled and sonicated for 5 min. Lysates were then separated on 10% SDS-PAGE acrylamide gel and transferred to 0.2 µm PVDF (Merck Milipore). Membranes were blocked in 5% skim milk in PBS-0.1% Tween for 1 h, incubated overnight for primary antibodies, and incubated for 1.5 h for the secondary antibody. Bands were visualized using ECL imaging on the Amersham Imager. HRP-Goat-α-Mouse (Sigma, cat # AP308P, used in 1:5,000 dilution) and HRP-Goat-α-Rabbit (Sigma, cat # AP307P, used in 1:5,000 dilution) were used as a secondary antibody to allow for ECL visualization. Primary antibodies used were Mouse-α-GAPDH (Abcam, cat # ab8245, used in 1:5,000 dilution) and Rabbit-α-Drosophila GAPDH (homemade, 1:500).
Mass spectrometry sample preparation
Samples were prepared as described earlier (van der Wal et al., 2018). For absolute peptide quantitation, synthesized heavy stable isotope (ThermoFisher Scientific) labeled VIPELGNK peptide was added to the sample in known quantities before tryptic digestion. A stock solution of 20 fmol/μL was used to spike in 500 pg of the labeled VIPELGNK peptide into a yeast background.
Targeted mass spectrometry
A tryptic digest of purified GAPDH was analyzed using a data dependent acquisition (DDA) regime to select the appropriate proteotypic peptides for PRM. Proteolytic peptides were analyzed by nanoflow liquid chromatography tandem mass spectrometry (nLC-MS/MS) on an EASY-nLC coupled to an Orbitrap Lumos Tribrid mass spectrometer (ThermoFisher Scientific), operating in positive mode and running Tune version 3.3. Peptides were trapped on a 2 cm × 100 μm Pepmap C18 column (Thermo Fisher 164,564) and then separated on an in-house packed 25 cm × 75 μm capillary column with 1.9 μm Reprosil-Pur C18 beads (Dr Maisch) at a flow rate of 250 nL/min, using a linear gradient of 0%–32% acetonitrile in 0.1% formic acid during 90 min. The eluate was directly sprayed into the electrospray ionization (ESI) source of the mass spectrometer. Spectra were acquired in continuum mode in the Orbitrap at 120,000 resolution for MS1 and 30,000 resolution for MS2 in profile mode and with standard AGC target settings. Fragmentation of the peptides was performed by HCD.
For targeted proteomics, a parallel reaction monitoring regime (PRM) was used to select for a set of previously defined peptides on an Orbitrap Tribrid Fusion Lumos mass spectrometer (ThermoFisher Scientific) operating in positive mode. Precursors were selected in the quadrupole with an isolation width of 0.7 m/z and fragmented with HCD using 30% collision energy (CE). MS1 and MS2 spectra were recorded in the orbitrap at 30,000 resolution in profile mode and with standard AGC target settings. The injection time mode was set to dynamic with a minimum of 9 points across the peak. The sequence of sampling was blanks first and then in order of increasing peptide input amounts to avoid any contamination of previous samples.
Data analysis
Analysis of PRM data was performed using Skyline (v 22) (MacLean et al., 2010) and in-house software. Regression analysis was performed using Graphpad Prism (v 5.02).
Data deposition
All raw mass spectrometry data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD050407.
Results
Parallel reaction monitoring (PRM) is a sensitive and specific mass spectrometry technology to target proteolytic peptides of interest in a sample. PRM differs from regular bottom-up data dependent acquisition (DDA) mass spectrometry (Figure 1) in a number of ways. In DDA MS, MS1 spectra are recorded and based on the relative mass spectral intensities of peptide precursors, several are selected for fragmentation. Typically, the TopN most intense precursor peaks are selected in each duty cycle (consisting of MS1 + x MS2 spectra), with a dynamic exclusion time that prevents multiple selection events of the same precursor. This exclusion time ensures the sampling of the highest number of precursor peptides and, thus, the deepest coverage of the proteome. The peptide fragments are subsequently analyzed in the Orbitrap at high mass accuracy and high resolution. Next, fragmentation spectra are matched with in silico generated spectra of tryptic peptides resulting in identification of the peptide. Based on the MS1 and/or MS2 spectral intensities, semi-quantitative analysis of peptides is possible. There is a stochastic element in the DDA protocol, resulting in lower abundant peptides being eluted and detected in the MS1 spectrum, but not selected for fragmentation. These peptides will therefore not be identified in the database matching procedure.
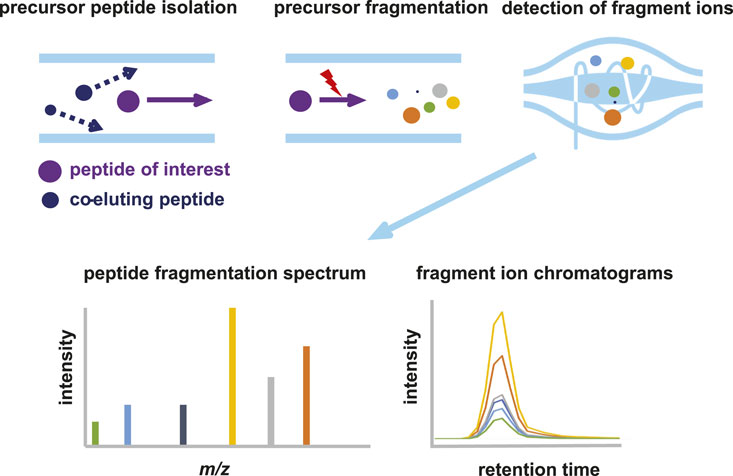
Figure 1. Schematic representation of parallel reaction monitoring (PRM) mass spectrometry. In PRM, the quadrupole is used to select and isolate ions of a predetermined m/z value. Precursor ions are then fragmented by CID or HCD to generate fragment ions, which are analyzed at high resolution and mass accuracy in the orbitrap. Quantitation is achieved by measuring the abundances (peak area under the curve or intensity) of the fragment ions derived from the target peptide. The summed abundances are then used to relatively quantify the peptide in different samples.
In PRM, the mass spectrometer is configured in such a way that only a priori selected peptide precursors are isolated and fragmented. All available time can thus be allocated to the fragmentation and analysis of a limited selection of peptides, resulting in enhanced detection sensitivity. The first quadrupole in the Orbitrap act merely as a filter and allows only predetermined ions to pass, which are selected based on their highly accurate m/z and retention time (RT) window. All precursor ions that fulfill the selection criteria are continuously selected and fragmented, as long as the corresponding peptide elutes from the LC column. The selection of peptides of interest and their properties (i.e., m/z and RT) is based either on recorded spectra in earlier experiments, peptide libraries or AI based prediction models such as PROSIT (Gessulat et al., 2019). Fragmentation of the precursor leads to several fragment ions (called “transitions”), which have both highly specific m/z values and relative spectral intensities. Even using relatively short LC gradients, several 100 s proteolytic peptides can be targeted in a single LC-MS run.
We set out to compare the sensitivity and specificity of immunoblotting versus PRM. Rabbit glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was chosen as the target protein of interest, since both the purified protein and highly specific antibodies were commercially available. First, we estimated the LOD for SDS-PAGE and Western blotting in a dilution series ranging from 100 ng to 1 pg. Using colloidal Coomassie blue (CBB) staining (Figure 2A) the visual LOD was about 100 ng of protein input, which is in accordance with the CBB manufacturer’s indication that it can be used to visualize quantities of protein in the ng range. Using silver staining, the band representing GAPDH at 50 ng input material was still visible by the eye, while lower amounts were below the visual detection limit ((Figure 2B).
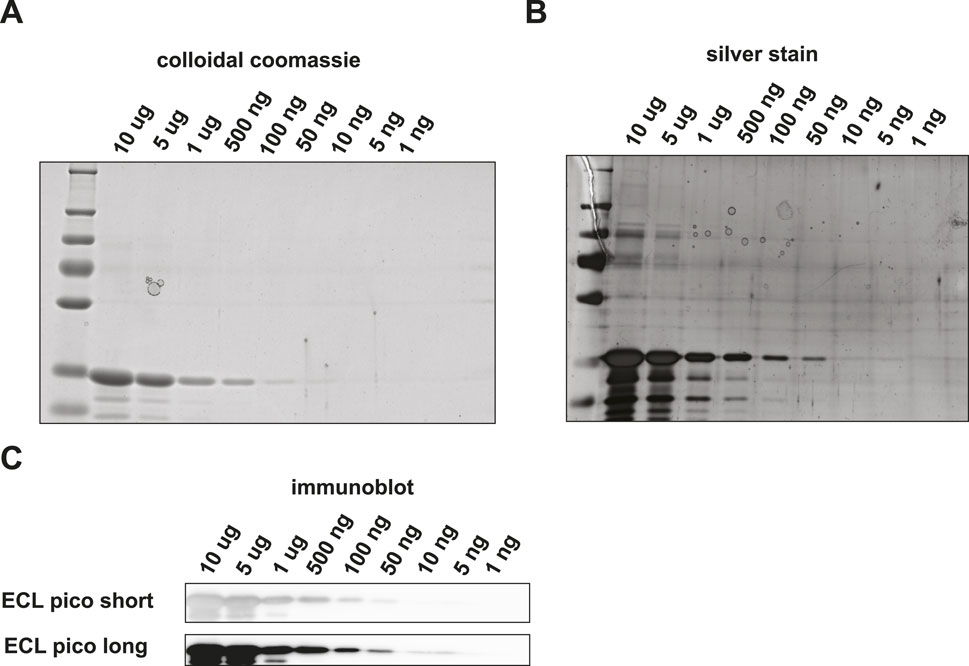
Figure 2. Detection of GAPDH loaded onto an SDS-PAGE gel and subsequent visualization using (A) colloidal coomassie, (B) silver stain (C) or Western blot followed by HRP-antibody binding and ECL Pico visualization.
We then performed Western blotting with HRP-antibody conjugates on a similar dilution series. We used enhanced chemiluminescence (ECL) pico solution to simulate acquisition. With ECL pico, input amounts of down to 10 ng could still be detected (Figure 2C). While a band is visible in the 5 ng input amount long exposure channel, the intensity of the band does not correspond with the expected linear decrease in staining intensity and is thus most likely a background signal. We were not able to visualize protein amounts in the sub-nanogram range. Verification with another protein – antibody pair, i.e., GST tagged Drosophila GAPDH and an in-house generated antibody against Drosophila GAPDH, did not result in the detection of quantities in the sub-nanogram range either (Supplementary Figure S1).
Now that we established the baseline detection of GAPDH using SDS-PAGE based immunoblotting methods, we set out to check the detection of GAPDH using PRM. We used a bottom-up mass spectrometry approach to first determine which tryptic peptides display the highest response in a DDA LC-MS analysis. We focused on three proteotypic peptides, i.e., VIPELNGK, TVDGPSGK and QASEGPLK. GAPDH was digested with trypsin in a dilution series from 10 ng down to 10 pg and PRM was performed on the three target peptides. The MS/MS spectrum and extracted fragment ion chromatograms for peptide VIPELGNK are shown in Figures 3A, B; Supplementary Figure S2A, B. Starting from input amounts of 500 fg and lower, the number of detected transitions is reduced, although even at 10 fg one transition could still be clearly observed. In order to determine the limit of detection (LOD) and the dose-response correlation, we performed regression analysis on a dilution series, which revealed an R2 of 0.9960 (Figure 3C). Similar assays for the two other GAPDH tryptic peptides, i.e., TVDGPSGK and QASEGPLK, produced R2 values of 0.9982 and 0.9984, respectively (Supplementary Figures S2C, S2F). Thus, there is a linear relationship between peptide input amount and observed spectral intensities of its specific fragment ions over at least 4 orders of magnitude. Most likely, this linear relationship spans even more order of magnitude, but we did not include higher amounts than 10 ng on column in the dilution series. At this stage, we did not include replicate experiments. Although the individual dilution series were all measured several times, this was done on different LC-MS setups and on different days. Given the variation in the absolute signals because of this, we therefore decided not to treat these as pure replicates.
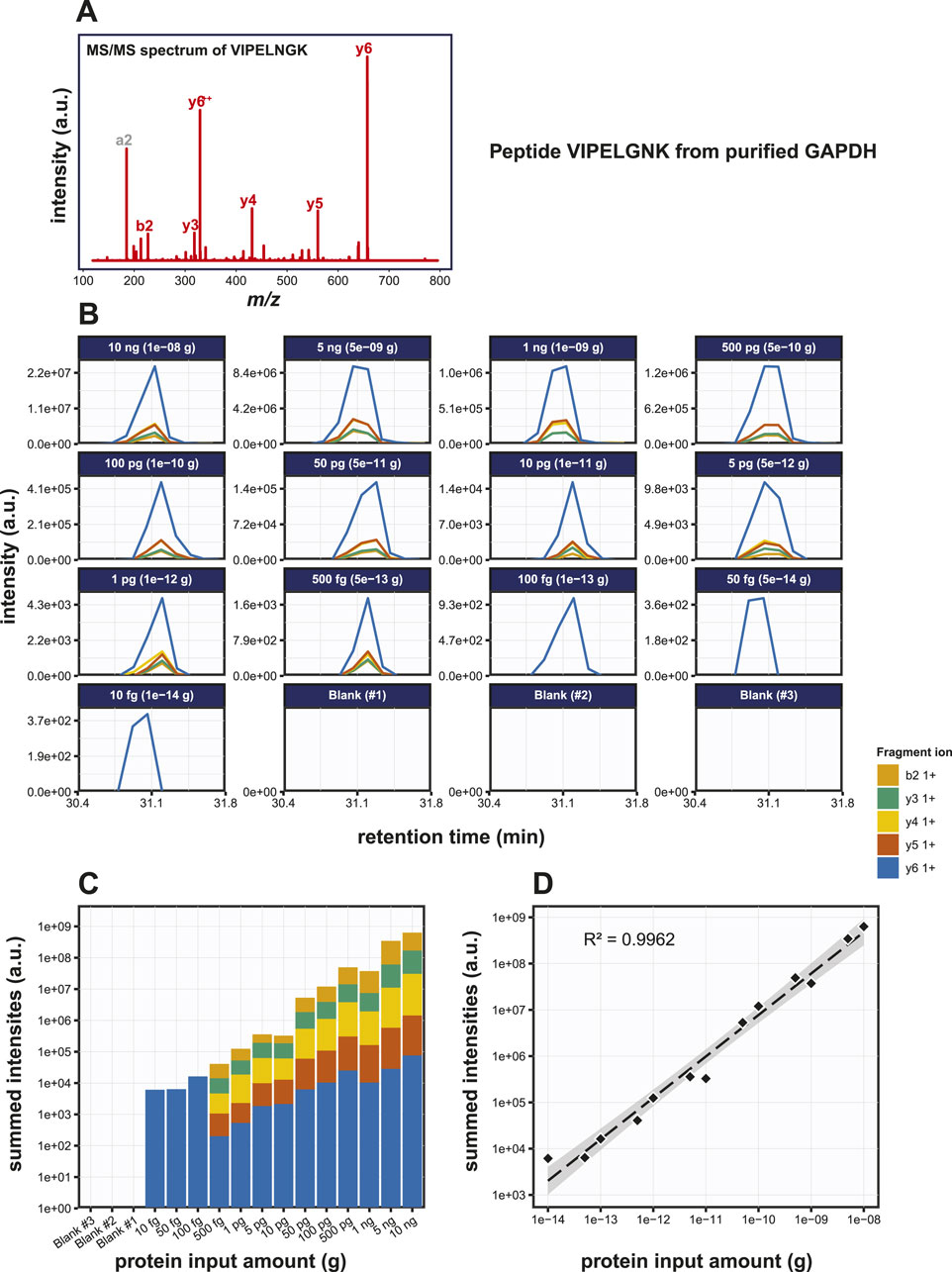
Figure 3. Detection of the GAPDH derived tryptic peptide VIPELGNK using PRM. (A) MS/MS spectrum of the VIPELGNK peptide. (B) Fragment ion chromatograms of VIPELGNK, exported from Skyline. (C) Area under the curve (AUC) plot of the fragment ion chromatograms shown in (B). (D) Regression analysis of the data shown in (C) with the best fit line drawn in striped black.
Although the targeted detection of proteolytic peptides from a pure protein digest is a valid benchmark test to determine the assay sensitivity, the real power of such an assay would be reflected by the capability of detecting target proteins embedded in a complex biological matrix. Therefore, we set out to investigate the sensitivity of this method by targeting the same selection of tryptic peptides mixed into a complex protein matrix. To avoid cross species reactivity issues, we used an S. cerevisiae lysate as the background matrix. The amino acid sequences of the three target peptides under investigation were unique to rabbit GAPDH. First, GAPDH was spiked into 5 µg of a yeast whole cell lysate according to the dilution scheme used before (Figure 4). We then performed Western blotting and subsequent visualization via HRP-conjugated antibodies using two different ECL solutions. No apparent background signal was observed in the Western blotting samples in the MW range where GAPDH was expected. Using ECL pico visualization GAPDH could still be detected at 50 ng total input (Figures 4B, C).
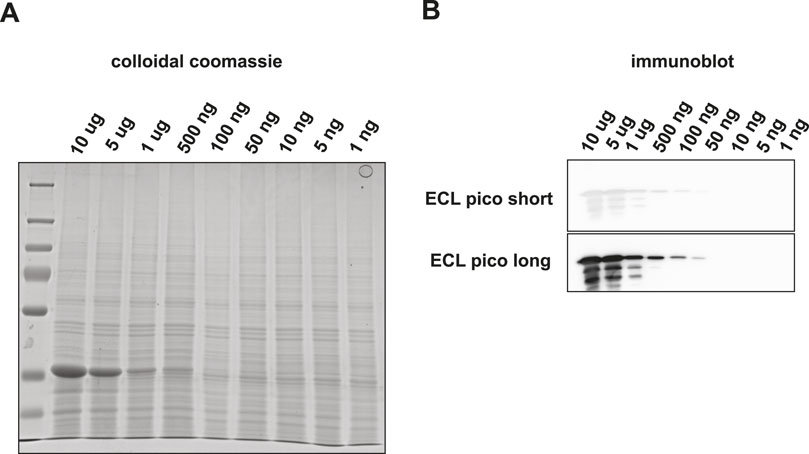
Figure 4. Detection of GAPDH embedded in a 200 ng yeast total lysate background matrix using SDS-PAGE and subsequent visualization by (A) colloidal coomassie staining or by (B) Western blot followed by HRP-antibody binding and ECL Pico visualization.
Next, we performed PRM on digests of various amounts of GAPDH spiked into 200 ng of S. cerevisiae lysate. The same selection of target tryptic peptides as before were used (Figure 5A; Supplementary Figure S4). Highly accurate transitions were detected at input quantities >500 fg GAPDH, but with lower input amounts the background interference masked the specific signals. Transitions were scored based on the dotP value reported by Skyline and the output data were converted to bar graphs (Figure 5B; Supplementary Figures S4B, S4E). Regression analysis for the peptides VIPELNGK, TVDGPSGK and QASEGPLK produced R2 values of the peptides as 0.9986, 0.9980 and 0.9980 respectively (Figure 5C; Supplementary Figures S4C, S4F), while the LODs were 5 pg, 1 pg and 10 pg, respectively. At this stage, we did not include replicate experiments. Although the individual dilution series were all measured several times, this was done on different LC-MS setups and on different days. Given the variation in the absolute signals because of this, we therefore decided not to treat these as pure replicates. In conclusion, the detection of very low amounts of peptide embedded in a complex background matrix was more challenging, and the LODs were roughly one order of magnitude higher than for the purified protein.
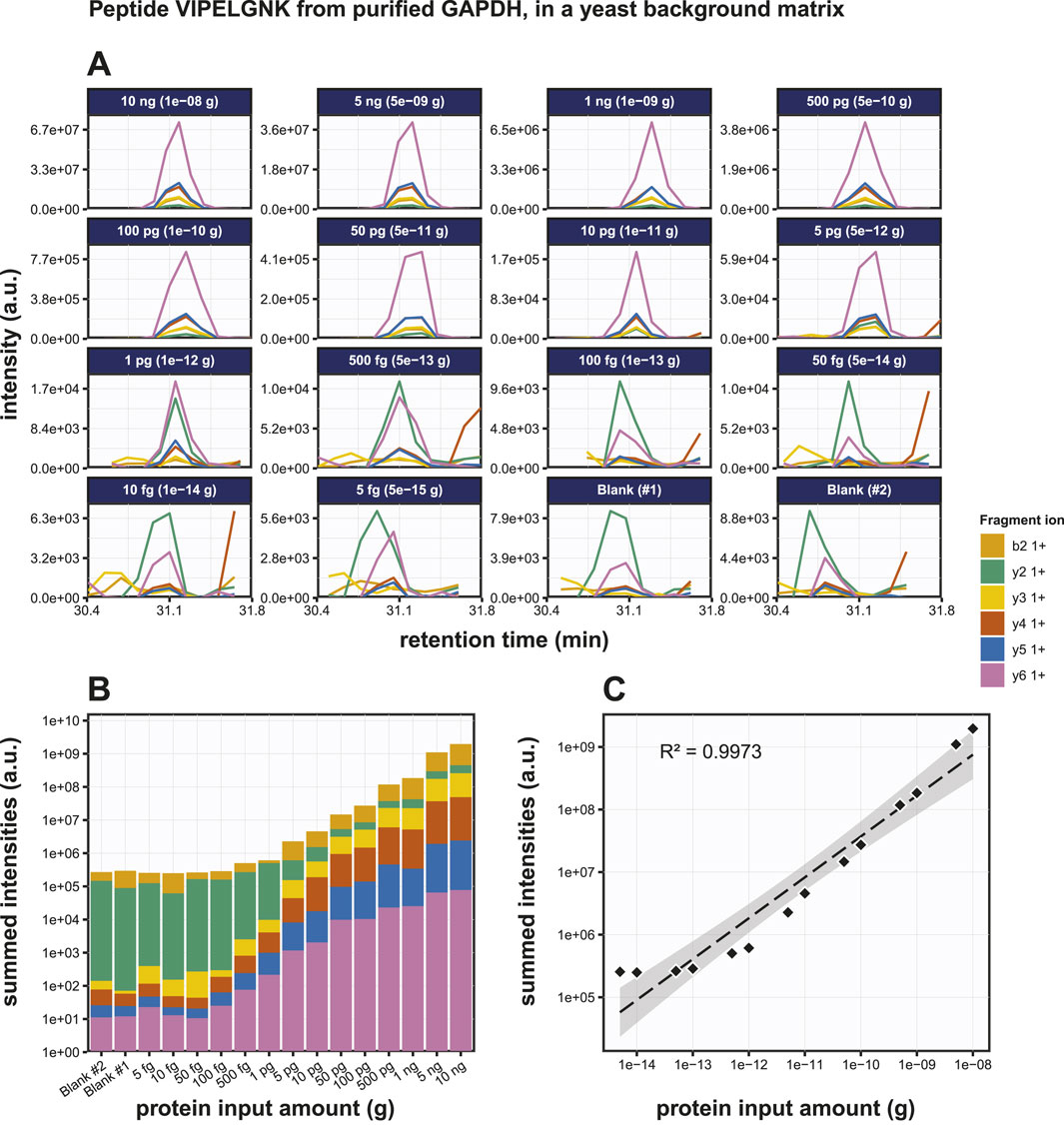
Figure 5. Detection of the GAPDH derived tryptic peptide VIPELGNK in a yeast total lysate background matrix using PRM. (A) Fragment ion chromatograms of peptide VIPELGNK in a yeast background (200 ng per sample), exported from Skyline. (B) AUC bar graphs of the fragment ion chromatograms shown in (B). (C) Regression analysis of the data shown in (B) with the best fit line drawn in in striped black.
Under ideal digestion conditions, 50 fg (1.4 amol) GAPDH should produce 1.4 amol of each tryptic peptide. We determined the trypsin digestion efficiency by spiking a known amount of a heavy isotope labeled variant of peptide VIPELNGK (VIPELNGK(+8)) into the sample to absolutely quantitate the peptide. The heavy isotope labeled lysine residue ensures that it can be distinguished from endogenous, non-labeled VIPELNGK peptide by its higher m/z value, while keeping similar biophysical properties such as electrospray ioinization responsiveness and retention time unchanged. As such, mass spectral intensity ratios can be used for absolute quantitation.
Known amounts of AQUA peptide were spiked into both the purified GAPDH sample and the GAPDH mixed with yeast lysate sample. 500 pg (14 fmol) of GAPDH was digested under standard conditions and then mixed with 10 fmol of VIPELNGK (+8). The resulting mixed sample was then added to 200 ng yeast lysate. The results of the PRM assay on these samples are shown in Figures 6A, B. The overall intensities for the transitions of the endogenous peptide were 2.74 ± 0.05 x lower than those of the heavy peptide (Figure 6B). Precision is generally expressed as coefficient of variation (CV) and the repeatability in a triplicate analysis was very good as evidenced by the low standard deviations and CV values for both the endogenous and the spiked-in AQUA target peptides in both the purified GAPDH sample and the GAPDH mixed in a complex biological matrix sample (Figure 6C). The latter represents a more realistic biological situation and although high levels of contaminating proteins might disturb the detection of the target peptide, these data show that the target peptide can still be detected at very low levels, illustrating the robustness of the methodology. All CV’s were far below 20%, which is considered a good precision in a PRM assay (see, e.g., (Gallien et al., 2015)). The use of an AQUA peptide spike-in allows the absolute quantitation of target peptides. If the digestion efficiency were 100%, the mass spectral intensities for the endogenous peptide are expected to be 1.4 x higher than for the AQUA peptide. In contrast, since the total area fragment signal of the endogenous peptide is 3.84 x lower, this indicates that only 3.65 fmol of the endogenous peptide was present in the sample, suggesting a digestion efficiency of only 27%. From this, we conclude that the genuine LOD is in fact approximately four times lower than was calculated from the initial experiment without the presence of AQUA peptide.
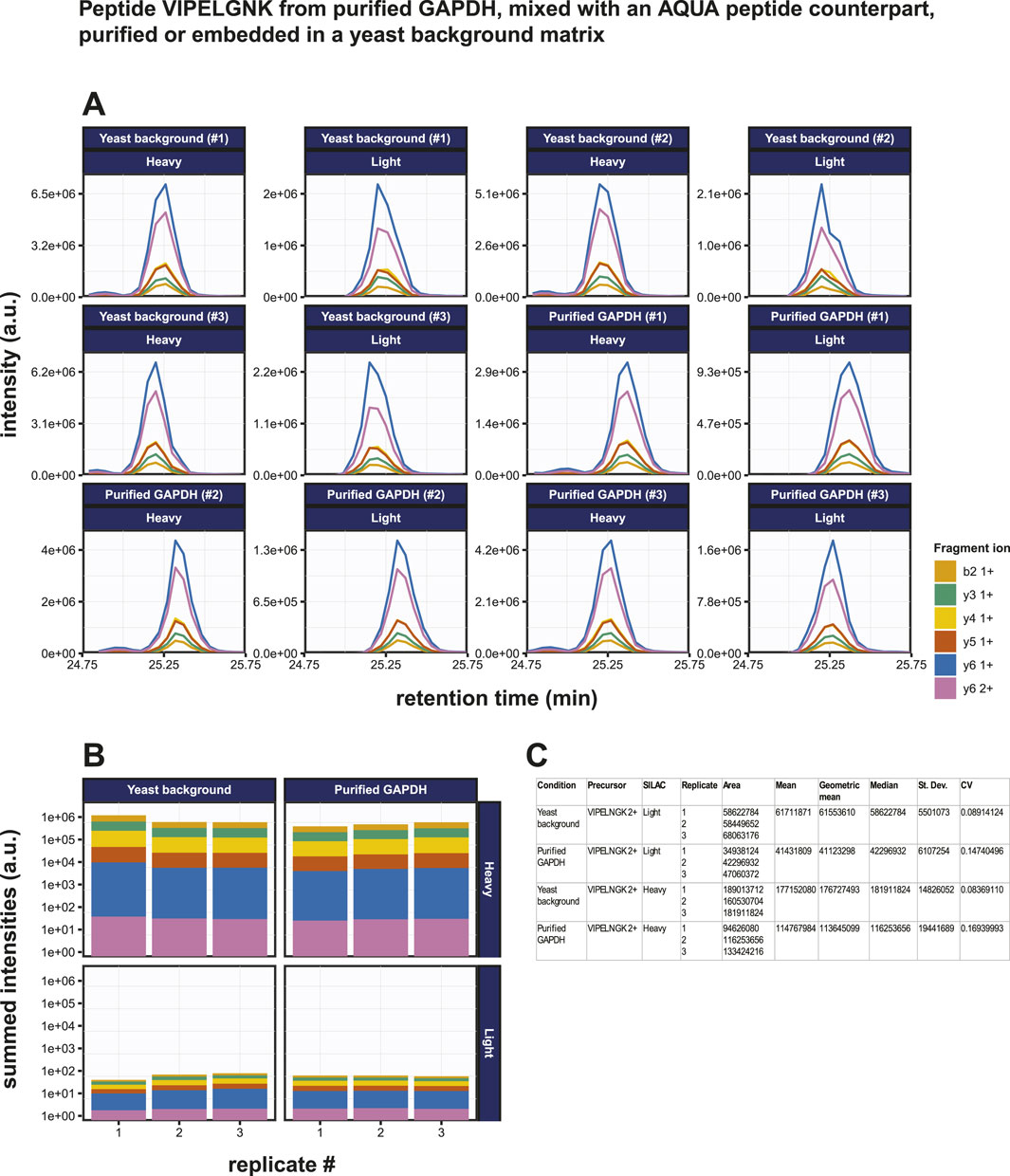
Figure 6. Absolute quantification of the GAPDH derived tryptic peptide VIPELGNK using PRM. (A) Fragment ion chromatograms exported from Skyline of the endogenous peptide VIPELGNK (light) and of the isotopically labeled AQUA peptide VIPELGNK(+8) (heavy). Ten fmol of the AQUA peptide was spiked into the sample. Results from purified GAPDH as well as from GAPDH embedded in a 200 ng yeast total lysate background matrix are shown. (B) AUC bar graphs of light and heavy VIPELGNK peptide based on data in (A). (C) Statistical and variability analysis of data in (B).
Discussion
Targeted detection of specific proteins in complex matrices is a cornerstone in molecular biology and reliable detection and quantitation of protein levels over a large input range and in a robust manner is of utmost importance. Traditionally, immunoassays have been used since several decades and although several minor improvements have been made since these methodologies still suffer from severe limitations. Here, we investigate the advantages of targeted mass spectrometry based on parallel reaction monitoring (PRM) over immuno-based methods. PRM is presented as a superior alternative for targeted detection of proteins and is able to overcome at least some of the limitations posed by immunoblotting.
One of the major limitations of immunoassays is the dependence on highly specific antibodies targeting the protein of interest. It is not uncommon for an antibody to have off-target effects, i.e., target the wrong epitope and, thus, protein. Such off-target effects obviously hamper a correct interpretation of the results. Targeted mass spectrometry does not require the use of antibodies. The specificity of the methodology is fully embedded in the unique character of the amino acid sequence of the target peptide. Fragmentation of a peptide results in a unique combination of fragment ion m/z values (referred to as transitions) and intensities and often also retention time when LC-MS based methods are used. Even when no experimental peptide fragmentation spectra are available to serve as library spectrum for comparison purposes, in silico digestion in combination with AI based approaches [e.g., (Gessulat et al., 2019; Searle et al., 2020),] can result in reliable transition predictions. In addition, large repositories of high-resolution DDA peptide fragmentation mass spectrometry data contain millions of spectra that can be used as transition libraries for PRM acquisition experiments. To increase the reliability even further, multiple tryptic peptides per protein can be targeted in one assay and multiple proteins (several 10 or even 100 s) can thus be targeted in a single LC-MS run. Importantly, we show here that PRM shows a linear dose-dependent intensity correlation over an extended range of input values that spans at least five orders of magnitude and probably more. In addition, for absolute quantitation of peptides we also show here that heavy isotope labeled AQUA peptides can be used as a spike-in or calibrants. To increase the sensitivity of PRM, prior fractionation of the digested protein sample into multiple peptide fractions by HILIC, high pH reversed phase (RP) or any other fractionation method, could be included in the workflow. We have shown before that this methodology can be used to detect and relatively quantify SARS-CoV-2 Nucleocapsid derived tryptic peptides both in research samples as well as in clinical specimens (van der Wal et al., 2018), illustrating that the technology can indeed be used for diagnostic purposes.
The reliable quantitation of proteins using Western blotting is challenging. Although one advantage of immunoblotting is fractionation of proteins by SDS-PAGE electrophoresis prior to transferring the proteins to the Western blot membrane, differences in protein expression can be only relatively quantified and also the expression levels under different conditions should not differ too much. Large differences in expression levels cannot be accurately quantified because the dynamic range is only limited, while more subtle differences present additional challenges. A twofold increase of protein input amount often does not result in a likewise increase of the staining intensities (Hammond et al., 2013), even though Western blot quantitation is often intensity based. Therefore, the staining intensities on an immunoblot may not accurately represent the absolute protein differences. The staining intensity on an immunoblot depends on several factors, such as the readout method that is used, the concentration of antibody, the absolute amount of protein, the exposure time, etc.
The time investment required to perform SDS-PAGE based protein detection and Western blotting is reasonable low. The whole procedure can be completed in half a day, with some waiting time. The time investment in PRM is about half a day to a day, including sample prep and input of the necessary information for the mass spectrometer to detect the necessary precursor ions. However, in this time frame, a much higher number of proteins can be detected, with negligible extra mass spectrometry time or sample preparation required. Thus, although in speed Western blotting may outperform PRM, this is completely compensated for by the much higher power to multiplex the PRM assay without substantial extra time investment [see, e.g., (Sijm et al., 2022)]. In terms of costs, assuming that a high-resolution mass spectrometer is available, the running costs are relatively low as no expensive antibodies are needed.
In conclusion, in terms of detection sensitivity, specificity and multiplexing targeted proteomics-based methods like PRM are superior over immunoblotting assays. We advocate the use of PRM as an alternative to Western blotting and ELISA assays in molecular biology, biomedical research and clinical and diagnostic assays.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
KB: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing–review and editing. LV: Data curation, Methodology, Project administration, Visualization, Writing–review and editing. WD: Data curation, Formal Analysis, Methodology, Software, Visualization, Writing–review and editing. JD: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/frans.2024.1397810/full#supplementary-material
References
Bluemlein, K., and Ralser, M. (2011). Monitoring protein expression in whole-cell extracts by targeted label- and standard-free LC-MS/MS. Nat. Protoc. 6, 859–869. doi:10.1038/nprot.2011.333
Burnette, W. N. (1981). “Western blotting”: electrophoretic transfer of proteins from sodium dodecyl sulfate--polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein A. Anal. Biochem. 112, 195–203. doi:10.1016/0003-2697(81)90281-5
Chambers, A. G., Percy, A. J., Simon, R., and Borchers, C. H. (2014). MRM for the verification of cancer biomarker proteins: recent applications to human plasma and serum. Expert Rev. Proteomics 11, 137–148. doi:10.1586/14789450.2014.877346
Deng, D., and Zi, Z. (2022). “Absolute quantification of TGF-β signaling proteins using quantitative western blot,” in Methods in molecular biology, 1–12.
Gallien, S., Kim, S. Y., and Domon, B. (2015). Large-scale targeted proteomics using internal standard triggered-parallel reaction monitoring (IS-PRM). Mol. Cell. Proteomics 14, 1630–1644. doi:10.1074/mcp.O114.043968
Gessulat, S., Schmidt, T., Zolg, D. P., Samaras, P., Schnatbaum, K., Zerweck, J., et al. (2019). Prosit: proteome-wide prediction of peptide tandem mass spectra by deep learning. Nat. Methods 16 (16), 509–518. doi:10.1038/s41592-019-0426-7
Ghosh, R., Gilda, J. E., and Gomes, A. V. (2014). The necessity of and strategies for improving confidence in the accuracy of western blots. Expert Rev. Proteomics 11, 549–560. doi:10.1586/14789450.2014.939635
Hammond, M., Kohn, J., Oh, K., and Liu, N. (2013). A method for greater reliability in western blot loading controls: stain-free total protein quantitation. Biorad Bull. 6360.
Jayasena, T., Poljak, A., Braidy, N., Zhong, L., Rowlands, B., Muenchhoff, J., et al. (2016). Application of targeted mass spectrometry for the quantification of sirtuins in the central nervous system. Sci. Rep. 6, 35391. doi:10.1038/srep35391
Kulyyassov, A., Fresnais, M., and Longuespée, R. (2021). Targeted liquid chromatography-tandem mass spectrometry analysis of proteins: basic principles, applications, and perspectives. Proteomics 21, 2100153. doi:10.1002/PMIC.202100153
Lau, T. Y. K., Collins, B. C., Stone, P., Tang, N., Gallagher, W. M., and Pennington, S. R. (2011). Absolute quantification of toxicological biomarkers by multiple reaction monitoring. Methods Mol. Biol. 691, 417–427. doi:10.1007/978-1-60761-849-2_25
MacLean, B., Tomazela, D. M., Shulman, N., Chambers, M., Finney, G. L., Frewen, B., et al. (2010). Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968. doi:10.1093/bioinformatics/btq054
Makarov, A. (2000). Electrostatic axially harmonic orbital trapping: a high-performance technique of mass analysis. Anal. Chem. 72, 1156–1162. doi:10.1021/AC991131P
Martínez-Márquez, A., Morante-Carriel, J., Sellés-Marchart, S., Martínez-Esteso, M. J., Pineda-Lucas, J. L., Luque, I., et al. (2013). Development and validation of MRM methods to quantify protein isoforms of polyphenol oxidase in loquat fruits. J. Proteome Res. 12, 5709–5722. doi:10.1021/pr4006712
Parsons, H. T., and Heazlewood, J. L. (2015). Beyond the Western front: targeted proteomics and organelle abundance profiling. Front. Plant Sci. 6, 301. doi:10.3389/fpls.2015.00301
Peterson, A. C., Russell, J. D., Bailey, D. J., Westphall, M. S., and Coon, J. J. (2012). Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Mol. and Cell. Proteomics 11, 1475–1488. doi:10.1074/MCP.O112.020131
Prasad, B., and Unadkat, J. D. (2014). Optimized approaches for quantification of drug transporters in tissues and cells by MRM proteomics. AAPS J. 16, 634–648. doi:10.1208/s12248-014-9602-y
Searle, B. C., Swearingen, K. E., Barnes, C. A., Schmidt, T., Gessulat, S., Küster, B., et al. (2020). Generating high quality libraries for DIA MS with empirically corrected peptide predictions. Nat. Commun. 11, 1548–1610. doi:10.1038/s41467-020-15346-1
Shi, T., Song, E., Nie, S., Rodland, K. D., Liu, T., Qian, W. J., et al. (2016). Advances in targeted proteomics and applications to biomedical research. Proteomics 16, 2160–2182. doi:10.1002/pmic.201500449
Sijm, A., Atlasi, Y., Knaap, J. A. V. D., Meer, J. W. V. D., Chalkley, G. E., Bezstarosti, K., et al. (2022). USP7 regulates the ncPRC1 Polycomb axis to stimulate genomic H2AK119ub1 deposition uncoupled from H3K27me3. Sci. Adv. 2. doi:10.1126/sciadv.abq7598
Smith, B. J. (1994). “SDS polyacrylamide gel electrophoresis of proteins,” in Basic protein and peptide protocols (New Jersey: Humana Press), 23–34. doi:10.1385/0-89603-268-X:23
Sowers, J. L., Mirfattah, B., Xu, P., Tang, H., Park, I. Y., Walker, C., et al. (2015). Quantification of histone modifications by parallel-reaction monitoring: a method validation. Anal. Chem. 87, 10006–10014. doi:10.1021/acs.analchem.5b02615
Thorsen, A. S. F., Riber, L. P. S., Rasmussen, L. M., and Overgaard, M. (2023). A targeted multiplex mass spectrometry method for quantitation of abundant matrix and cellular proteins in formalin-fixed paraffin embedded arterial tissue. J. Proteomics 272, 104775. doi:10.1016/j.jprot.2022.104775
Towbin, H., Staehelin, T., and Gordon, J. (1979). Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl. Acad. Sci. U. S. A. 76, 4350–4354. doi:10.1073/PNAS.76.9.4350
Tsuchiya, H., Tanaka, K., and Saeki, Y. (2013). The parallel reaction monitoring method contributes to a highly sensitive polyubiquitin chain quantification. Biochem. Biophys. Res. Commun. 436, 223–229. doi:10.1016/j.bbrc.2013.05.080
van der Wal, L., Bezstarosti, K., Sap, K. A., Dekkers, D. H. W., Rijkers, E., Mientjes, E., et al. (2018). Improvement of ubiquitylation site detection by Orbitrap mass spectrometry. J. Proteomics 172, 49–56. doi:10.1016/j.jprot.2017.10.014
Weller, M. G. (2016). Quality issues of research antibodies. Anal. Chem. Insights 2016, 21–27. doi:10.4137/Aci.s31614
Westermeier, R. (2014). Looking at proteins from two dimensions: a review on five decades of 2D electrophoresis. Arch. Physiol. Biochem. 120, 168–172. doi:10.3109/13813455.2014.945188
Yang, T., Xu, F., Xu, J., Fang, D., Yu, Y., and Chen, Y. (2013). Comparison of liquid chromatography-tandem mass spectrometry-based targeted proteomics and conventional analytical methods for the determination of P-glycoprotein in human breast cancer cells. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 936, 18–24. doi:10.1016/j.jchromb.2013.07.023
Keywords: parallel reaction monitoring (PRM), targeted mass spectrometry, immunoblotting, limit of detection, GAPDH
Citation: Bezstarosti K, Van der Wal L, Doff WAS and Demmers JAA (2024) Parallel reaction monitoring targeted mass spectrometry as a fast and sensitive alternative to antibody-based protein detection. Front. Anal. Sci. 4:1397810. doi: 10.3389/frans.2024.1397810
Received: 08 March 2024; Accepted: 14 August 2024;
Published: 10 September 2024.
Edited by:
Elizabeth Neumann, University of California, Davis, United StatesReviewed by:
Mowei Zhou, Zhejiang University, ChinaRoman Řemínek, Institute of Analytical Chemistry (ASCR), Czechia
Copyright © 2024 Bezstarosti, Van der Wal, Doff and Demmers. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jeroen A. A. Demmers, ai5kZW1tZXJzQGVyYXNtdXNtYy5ubA==
†These authors have contributed equally to this work