 Yushu Gu
Yushu Gu Miaomiao Liu
Miaomiao Liu Ronald J. Quinn
Ronald J. Quinn
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Anal. Sci. , 10 October 2022
Sec. Omics
Volume 2 - 2022 | https://doi.org/10.3389/frans.2022.1014017
This article is part of the Research Topic Emerging Structural Proteomics Methodologies View all 8 articles
Understanding molecular level interactions between the metabolome and proteome, two of the most important classes of molecules in biology, will generate deeper insight into the function of metabolites (natural products) which have a central role in interactions with therapeutic targets. Drug discovery in today’s pharmaceutical environment is driven by high-throughput screening of large chemical libraries. It is now 10 years since we published a paper on the development of natural product fraction libraries with control of LogP properties. We have now turned our attention to using pure natural product libraries to address the timeframe issues associated with isolation and characterization of the active constituent(s). Native mass spectrometry can be used as a robust platform for identifying the interactions between natural products and their protein targets. The recent development of Collision-Induced Affinity Selection mass spectrometry, a technique using capture of ligand-protein complexes followed by collision induced dissociation to identify library hits followed by direct ligand-protein confirmation in native mass spectrometry also enables screening of a greater proportion of human proteins. We will review native mass spectrometry-based approaches to use natural product extracts, pre-fractionated natural product libraries and pure natural product libraries for screening against molecular targets. We will also discuss some of the other mass-spectrometry based applications that have been implicated in natural product drug discovery.
Natural products (NPs) have historically been an indispensable source for the development of therapeutic agents. There are various types of analytical methods for the discovery of NPs. Traditionally, the discovery of new bioactive NPs begins with extraction from biological samples of plants, microorganisms and marine organisms, fractionation to isolate the bioactive compounds, and screening on therapeutic targets (Koehn and Carter, 2005). Although proven successful, this method requires considerable time and effort, and can be difficult due to the complexity of extracts. Drug discovery in today’s pharmaceutical environment has shifted to high-throughput screening of large chemical libraries. Understanding molecular level interactions between the metabolome and proteome, two of the most important classes of molecules in biology, will generate deeper insight into the function of metabolites (natural products) which have a central role in interactions with therapeutic targets. It is now 10 years since the paper published on the development of natural product fraction libraries with control of physicochemical parameter such as partition coefficient (LogP) properties (Camp et al., 2012). We have now turned our attention to using pure natural product libraries to address the timeframe issues associated with isolation and characterisation of the active constituent(s). This review will highlight approaches to use natural product extracts, pre-fractionated libraries, and pure natural product libraries for screening against molecular targets using high-throughput mass spectrometry (MS) approaches. Amongst the many established techniques for natural product screening in drug discovery, native mass spectrometry (native MS) has emerged as a robust platform for identifying the interactions between NPs and their protein targets (Elnaas et al., 2020; Nguyen et al., 2021). The recent development of Collision-Induced Affinity Selection MS (CIAS-MS), a technique using capture of ligand-protein complexes, collision induced activation in the collision cell to identify library hits followed by direct ligand-protein confirmation in native MS also enable screening of a greater proportion of human proteins (Mak et al., 2022). Table 1 summarises our studies covered in this review of using native MS and CIAS-MS in natural product screening.

TABLE 1. Utilization of native MS and CIAS-MS in natural product screening.
Soft ionization approaches allow the study of protein-ligand complexes with the advantage of direct observation of protein-ligand binding without disruption of non-covalent interactions (Chen et al., 2019a). Electrospray ionization mass spectrometry (ESI–MS) and matrix assisted laser desorption ionization mass spectrometry (MALDI-MS) are two soft ionization methods with the ability to detect protein-ligand interactions in classes of compounds that were previously not achievable by MS, particularly in identifying active compounds in medicinal plant extracts (Tanaka, 2003; Kumar, 2017; Chen et al., 2019a).
In ESI-MS, the sample is pumped into a conductive capillary with a high voltage. Charged droplets of analytes are ejected from the capillary, and due to the high potential difference, charged droplets with the same polarity surface charge as the charge on the needle are sprayed through the sampling interface. Solvent evaporation occurs as the charged droplets travel across the needle tip to the cone, resulting in shrinking of the droplets until they reach the “Rayleigh limit”—the point where the surface tension fails to maintain the charge. The Coulombic explosion, that then takes place, breaks the droplets into smaller droplets to produce gas phase analyte molecules that can be detected by MS detectors (Figure 1). Electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry (ESI-FTICR-MS) is a robust and label-free approach with the advantage of direct observation of protein-ligand binding under non-denaturing conditions (Elnaas et al., 2020). Consequently, the biological functionality of the analyte molecules can be well reflected in the gas phase.
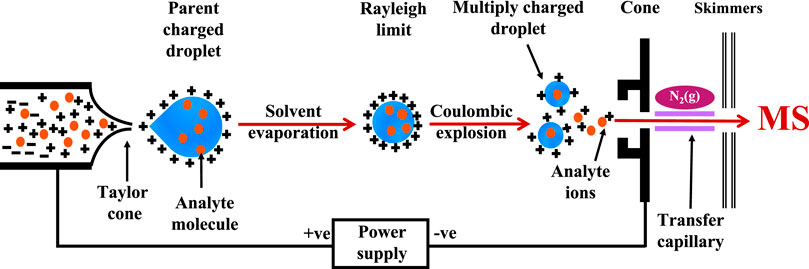
FIGURE 1. Detection of ligand-protein complex: General arrangement of ESI-MS and detection of high molecular weight ligand-protein complex.
Approaches to detecting small molecules involve dissociation of ligand-protein complexes has been achieved using MALDI-MS and affinity capture (also known as affinity selection) methods. This allows the detection of the ligand rather than the direct observation of protein-ligand complexes. MALDI-MS is a soft ionization that involves depositing the analyte molecules with a matrix solution on a metal surface. It embeds the analyte molecule (typically at a low micromolar concentration) into the matrix after drying. A laser producing a brief burst of ions is utilized to irradiate the analyte molecules, forcing them into the gas phase without decomposition (Keller and Li, 2001). Figure 2A illustrates the schematic workflow of a typical MALDI-MS, where the desorption of analyte molecules embedded in the matrix is induced by the laser. The analyte molecules are ionized by attachments of protons and carried into the gas phase by the matrix absorbing the energy (Bizzini and Greub, 2010). The time-of-flight differences between the desorbed molecules can then be measured by MS. However, due to the strong acidic matrix used during sample preparation and the negative impact it has on non-covalent complexes, MALDI-MS is applied less frequently for the study of biological macromolecules and small molecule binding compared to ESI-MS (Chen et al., 2019a). A typical affinity capture method involves incubating compound mixtures with a protein target, the separation of the bound/unbound ligands followed by dissociation of the bound ligand from the protein-ligand complex for detection in a mass spectrometer (Figure 2B).
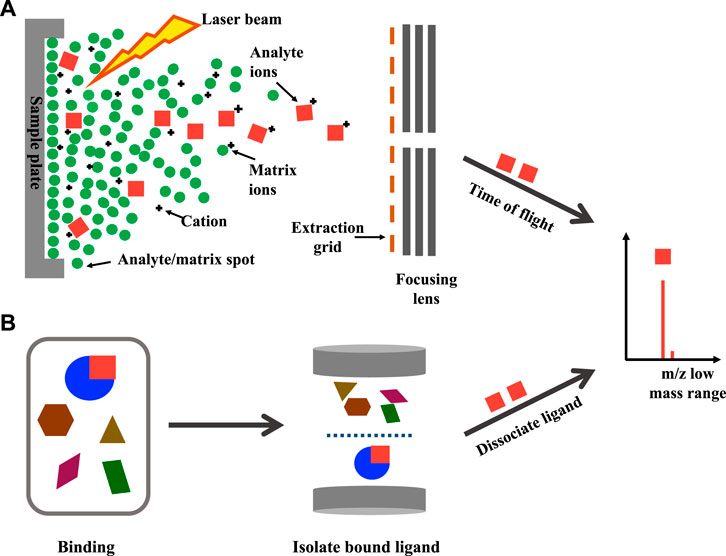
FIGURE 2. Detection of ligand: Schematic workflow of (A) MALDI-MS and (B) affinity capture for the detection of low molecular weight ligand.
After equilibration of a target protein and a mixture of compounds, detection of noncovalent or covalent complexes facilitates the discovery of specific binding with a therapeutic target.
Over a decade ago, we first described a proof-of-principle approach to demonstrate direct screening of NPs using ESI-FTICR-MS (Vu et al., 2008). By incubation of bovine carbonic anhydrase II (bCAII) with spiked plant crude extracts of known specific bCAII inhibitors, noncovalent complexes between bCAII with known ligands were detected by native MS. The desalted extracts and alkaloid-enriched extracts presented no interferences to the formation of the noncovalent complexes. A total number of 85 methanolic plant extracts were then screened and one active extract was detected to contain a ligand (6-(1S-hydroxy-3-methylbutyl)-7-methoxy-2H-chromen-2-one) of bCAII. The molecular weight (MW) of the bound ligand was also identified in this approach. The binding specificity was determined through competition experiments between the active compound and furosemide, a specific ligand of bCAII.
A similar recent study demonstrated a native MS screening strategy using gel filtration coupled with nanoscale ion emitters for the detection of small molecule binders from crude natural product extracts with human carbonic anhydrase I (hCAI) (Nguyen et al., 2021). Potential advancement in throughput by applying SIRIUS, a rapid computational tool for elucidating molecular structures was mentioned (Dührkop et al., 2019; Nguyen et al., 2021).
Native MS has been utilised as part of a PhenoTarget approach, a combination of phenotypic screening to detect cellular active components followed by a target screening approach where the hits are screened against a panel of putative targets (Figure 3). Hit natural product fractions identified by a high throughput phenotypic screening against Mycobacterium tuberculosis H37Rv were then screened by native MS against 37 mycobacterial proteins (Xie et al., 2020). This approach allowed rapid identification of an anti-tuberculosis lead compound polycarpine, an alkaloid interacting with the M. tuberculosis protein Rv1466 (Xie et al., 2020). By using the same approach, altholactone was identified to bind to Rv1466 (Elnaas et al., 2020).
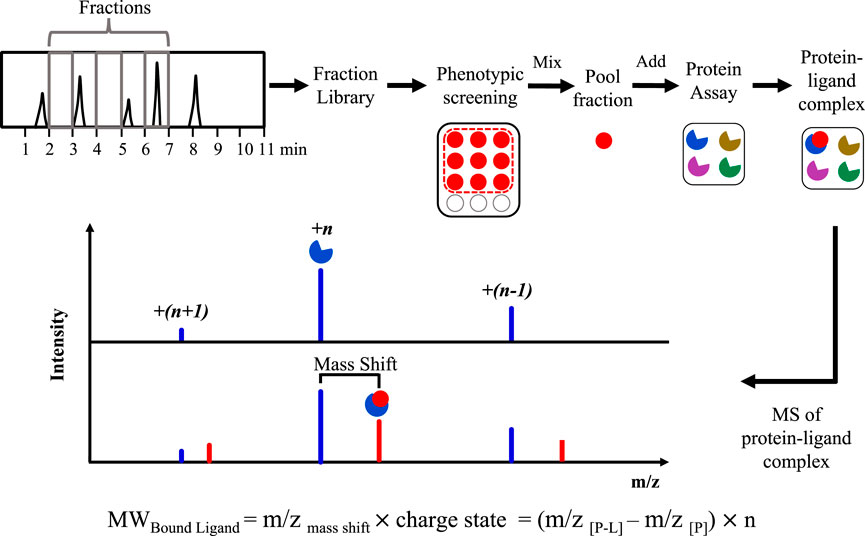
FIGURE 3. Basic strategy of the PhenoTarget screening for identifying lead compounds and target protein.
Finding a ligand for a disease associated protein target is amongst the most crucial aspects of drug discovery. Traditional high-throughput screening (HTS) detects compounds with binding affinities in the low micromolar range. However, molecular complexity, which represents various features of a molecule, such as molecular weight, shape and polarity, has an impact on binding affinity, as the molecular complexity of molecules increases, the chance of maintaining sufficient interactions decreases, leading to suboptimal binding affinity (Hann et al., 2001). To the contrary, smaller fragments that form fewer interactions have a better chance to bind to sites on a greater number of proteins, resulting in higher hit rates (Erlanson et al., 2016). Fragment-based drug discovery (FBDD) aims to identify simpler compounds with lower binding affinity resulting from fewer binding interactions. FBDD has advanced into an alternative approach to HTS lead generation as the smaller fragment sizes and the reduction in unfavourable interactions result in high ligand efficiency (Murray and Rees, 2009). A fragment library of 331 low molecular weight NPs was screened against Plasmodium falciparum 2′-deoxyuridine 5′-triphosphate nucleotidohydrolase (PfdUTPase) using native MS (Vu et al., 2013). Seven securinine-related compounds were found to bind PfdUTPase allosterically, resulting in improving enzyme activity, and inhibiting viability of gametocytes (sexual) and blood (asexual) stage parasites. A larger library of 643 NP-based fragments was employed to investigate 62 potential malaria protein targets using native MS. Ninety-six (96) low molecular weight NPs were identified as binding partners of 32 of the putative malarial targets. Seventy-nine (79) of the fragments showed direct growth inhibition of P. falciparum at concentrations that have the potential to be developed into leads against these protein targets (Vu et al., 2018).
Native MS based natural product library screening was also used to identify small molecule binders of the non-structural protein 9 (Nsp9) of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), a component of the PP1ab polyprotein that is essential for the viral replication of the virus causing the current global pandemic (Littler et al., 2021). Recombinant Nsp9 protein was screened against a NP library of 1,614 compounds by native MS. The MW of the bound ligand was directly calculated by the changes in mass-to-charge ratio (Δm/z) between the unbound protein and the protein–ligand complex ions. An ent-kaurane natural product, oridonin, was identified as a strong binder of Nsp9 with micromolar affinity (Kd = 7.2 μM). Oridonin was found to reduce viral titer following infection of cells with SARS-CoV-2. Subsequently, using native MS, oridonin and its clinical analogue, HAO472, demonstrated significant binding selectivity to SARS-CoV-2 and HCoV-229E Nsp9 over FSCoV-F56 Nsp9 (Liu et al., 2022). Particularly, native MS allowed the observation of direct binding of HAO472 to Nsp9, indicating HAO472 could be active itself, rather than acting entirely as a pro-drug.
Affinity ultrafiltration mass spectrometry (UF-LC/MS) has been used to screen NP libraries, with the ability to detect active compounds from mixtures of unbound inactive components by observing the biological affinities of small molecules and proteins (Chen et al., 2018). Twelve flavonoids were found as potential cyclooxygenase 2 (COX-2) inhibitors in the plant lotus plumule using UF-LC/MS and flavonoid C-glycosides and O-glycosides exhibited similar binding affinity to COX-2 (Chen et al., 2019b). This assay was then used in the screening of matrix metalloproteases-2 (MMP-2) inhibitors from three Chinese traditional medicinal plants, Smilax glabra Roxb., Smilax china L. and Saposhnikovia divaricate (Turcz.) Schischk. (Li et al., 2015). The main components, resveratrol, engelitin and asibinn, identified in extracts of the two Smilax species, and the main component 4′-O-β-d-glucosyl-5-O-methylvisamminol from S. divaricata as well as three other chromones were potent MMP-2 inhibitors (Li et al., 2015). Technologies combining the selectivity of native MS and tandem mass spectrometry and the separation capability of high-performance liquid chromatography (HPLC) have also been developed and advanced in recent years (Ul Haq et al., 2020; Reher et al., 2021; Aron et al., 2022). A two-step native ESI-LC-MS/MS strategy was developed using an online post-column for pH adjustment, an approach commonly used in intact protein MS, coupled to metal infusion to directly detect iron binding siderophores in complex samples such as culture extracts (Aron et al., 2022). With the help of ion identity molecular networking (IIMN), a novel computational tool that integrates correlation analysis into molecular networks to searching for biologically related compounds, this approach can overcome the challenge in identifying relevant metal binding compounds from large datasets (Schmid et al., 2021; Aron et al., 2022). Another example of incorporating native ESI and LC-MS/MS with pH adjustment in natural product screening, is the screening of chymotrypsin binders from crude extracts of cyanobacteria, a prolific source of bioactive natural product (Márquez et al., 1998; Welker et al., 2012; Reher et al., 2021). The results from the experiment revealed 30 cyclodepsipeptides as chymotrypsin binders and led to the identification of a new family of potent protease inhibitors termed ‘rivulariapeptolides’ with the aid of computational tool such as SIRIUS (Reher et al., 2021).
MALDI-MS has been previously employed in the examinations of various NPs. MALDI-TOF MS was used for the identification of flavonol glycosides in almond seedcoats, the identification of polyphenols in various foods, such as grape, cranberry, and pomegranate, and the characterization of hydrolysable tannins from Astronium species (Frison-Norrie and Sporns, 2002; Reed et al., 2005; da Silva et al., 2011). The non-covalent complexes between bovine serum albumin and the tannin, β-1,2,3,4,6-Penta-O-galloyl-d-glucopyranose (PGG), by MALDI-MS gave similar fragmentation patterns of PGG as observed for ESI-MS (Chen and Hagerman, 2004). Intensity-fading MALDI-MS has been applied with less acidic matrices DHAP and THAP to demonstrate it as an alternative approach for the direct MALDI-MS method where observing the intact complexes has proven difficult due to the dissociation of the complexes, to characterize the natural product peptide, melittin binding to calmodulin (Wang et al., 2009). MALDI-TOF-based affinity selection mass spectrometry (AS-MS) has been employed for the high-throughput screening of compound library for human protein tyrosine phosphatase 1B (PTP1B), with two previously published PTP1B inhibitors, an orthosteric active-site binder of PTP1B and an allosteric PTP1B modulator as reference compounds (Wiesmann et al., 2004; Han et al., 2008; Simon et al., 2021). The screening resulted in a final hit rate of 0.31%, including the confirmed binding of both compounds, demonstrating that MALDI-TOF AS-MS was able to detect both orthosteric and allosteric ligands (Simon et al., 2021).
Affinity selection-mass spectrometry (AS-MS), such as size exclusion affinity selection mass spectrometry (AS-MS), pulsed ultrafiltration AS-MS and UF-LC/MS, is a hyphenated MS approach widely used for the identification of a bound ligand (Figure 4A) (Kaur et al., 1997; van Breemen et al., 1997; Chen et al., 2018; Muchiri and van Breemen, 2021). The major drawbacks of this method are the laborious efforts required to remove unbound chemicals, and the possibility of dissociation of the ligand-protein complexes during the separation steps (Liu et al., 2013; Li et al., 2014; Fu et al., 2019).
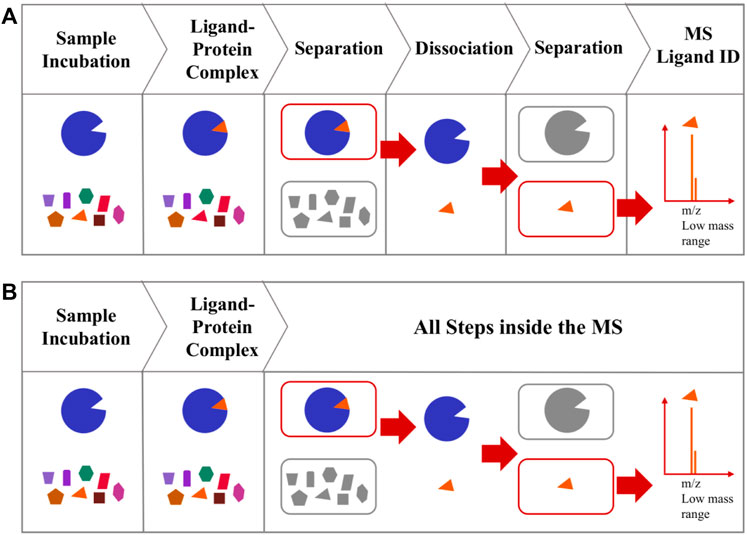
FIGURE 4. (A) A typical AS-MS. AS-MS start with incubation to allow protein-ligand complex formation and the separation of the protein-ligand complexes from the unbound compounds, followed by dissociating the bound ligand from the protein-ligand complex and an additional separation step to capture the low MW ligand, then injecting the ligand into MS for analysis (B) Workflow of collision induced affinity selection MS (CIAS-MS). Direct injection of sample mixture with all following steps completed inside the MS.
The requirement for prior removal of unbound molecules in such general AS-MS methods, has been achieved using CIAS-MS (Mak et al., 2022). CIAS-MS utilizes electrospray ionization to preserve protein-ligand complexes in their native state. Unlike the typical AS-MS, CIAS-MS does not require multiple external preparation steps prior to MS screening, as all steps are performed inside the mass spectrometer (Figure 4B). In CIAS-MS, the samples are directly injected into the ESI sources for ionization, with the quadrupole used for mass selection, allowing the protein-ligand complexes to be trapped and moved to the collision cell for collision induced dissociation (CID), that dissociates the bound ligand from the protein-ligand complex. The dissociated protein and ligand are transferred to the ion guide with a low time-of-flight which permits only the small-sized ligand to be transferred into the mass analyzer for detection.
CIAS-MS was applied to the Nsp9 protein of SARS-CoV-2 incubated with a pool of nine compounds, including the known ligand, oridonin (Littler et al., 2021). The major signal detected in the MS screening, corresponded to oridonin with a sodium adduct (Figure 5A). A minor signal that corresponds to compound 6 was also detected (Figure 5A) and binding between this compound and Nsp9 was confirmed by native MS. CIAS-MS could identify bound ligands from pooled compound libraries and identify multiple bindings in a single screening. CIAS-MS was also applied to detect binding ligands of Nsp9 in Rabdosia rubescens extracts, a rich natural source of oridonin (Ding et al., 2016). The major signals detected were consistent with oridonin plus sodium, potassium and manganese adduct ions, respectively (Figure 5B).
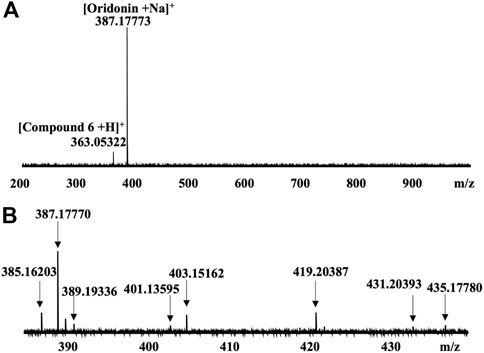
FIGURE 5. (A) CIAS-MS of nsp9 incubated with a pool of nine compounds; (B) CIAS-MS of nsp9 mixed with Rabdosia rubescens extract with major signals of oridonin with adduct ions observed: 387.17 m/z ([M + Na]+), 403.15 m/z ([M + K]+) and 419.20 m/z ([M + Mn]+). Adapted from (Mak et al., 2022)
Despites extensive effort made in facilitating the use of NPs for treating disease, target identification remains a major challenge in the drug discovery pipeline. There are many established label-free biochemical screening techniques utilizing physical interactions of proteins with the ability to study the molecular mechanism of efficacy applicable to human diseases, with some compatible with NPs (Schenone et al., 2013). One example is pulse proteolysis (PP) which studies the change in protein thermodynamic stability upon interaction with a ligand by selectively digesting the unfolded proteins, and calculating the proportion of the folded proteins compared to the whole folded and unfolded protein mixture (Park and Marqusee, 2005; Schlebach et al., 2011). Another example is the stability of proteins from rates of oxidation (SPROX) technique which takes advantage of the chemical denaturant dependence of the hydrogen peroxide-mediated oxidation in proteins to study the thermodynamic properties of the proteins (Strickland et al., 2013). This strategy was applied in the investigation of the protein targets of resveratrol, a bioactive natural product (Dearmond et al., 2011). Drug affinity responsive target stability (DARTS) is another strategy which relies on the decreased protease susceptibility of the target protein upon ligand binding (Lomenick et al., 2011). Previous studies have demonstrated its success in the target identification of natural products such as the molecular target for didemnin B and resveratrol (Lomenick et al., 2011). These techniques have their own advantage over the traditional affinity approaches; however, most are not appliable on all proteins, with technical challenges in experimental designs for measuring the conformational stability on a proteome-wide scale. Additionally, these approaches typically rely on ligands with high affinity, which can bind efficiently on the target proteins.
Native MS can be used for target identification, allowing detection of noncovalent protein-ligand binding between a bioactive small molecule and its target protein in a protein mixture (Liu et al., 2021). The screening of a mixture of five malarial proteins, adenosine deaminase from P. vivax PvADA (protein 1), PfdUTPase (protein 2), thioredoxin PfTrx (protein 3), ubiquitin conjugated enzyme (P. falciparum) (protein 4) and serine/threonine protein kinase PvNEK4 (protein 5), revealed that parthenolide, a natural product from the medicinal herb Feverfew bound to PfTrx (Figure 6) (Vu et al., 2018). The effects of ammonium acetate concentrations, pH conditions and MS instrument conditions with different skimmer, CID and ISCID setup on signal intensities of protein-ligand interactions were optimised for protein detection. This experiment demonstrated the potential of native MS in target identification.
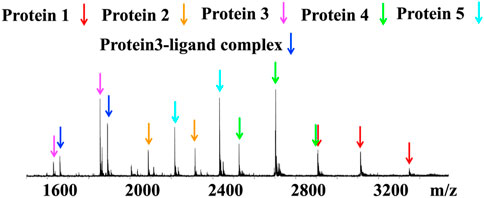
FIGURE 6. Native MS spectra of protein–ligand complex formed between the 5-protein mixture and ligand parthenolide. Blue arrows: unbound PfTrx, magenta arrow: protein–ligand complex. Adapted from (Liu et al., 2021).
Proteolysis targeting chimerics (PROTACs) are heterobifunctional molecules that promote degradation of endogenous protein targets (Potjewyd et al., 2020). A PROTAC contains a motif (usually a small molecule drug or a peptide) that binds the protein target connecting through a chemical linker to another motif that binds an E3 ubiquitin ligase (E3) ligand (Figure 7) (Buckley et al., 2015; Kregel et al., 2020). This connection allows the E3 ubiquitin ligase to ubiquitinylate a specific protein target and promote its degradation via the ubiquitin-proteasome system.
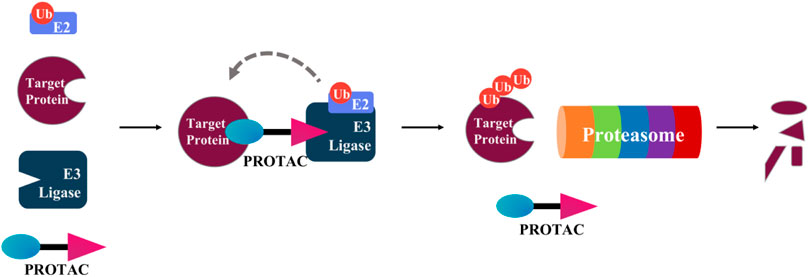
FIGURE 7. Mechanism of PROTAC-induced protein degradation. A PROTAC contains a motif (blue) that binds to the target protein, connected by a linker to another motif (purple) that binds an E3-Ubiquitin ligase. Followed by polyubiquitination of the target protein by an E2 conjugating enzyme, the polyubiquitinated target protein is then degraded by the proteasome.
Natural products have been used in the design of PROTACs (Yang et al., 2022). PROTAC technology has been developed based on ursolic acid (UA), a natural triterpenoid that can be found in herbs and fruits (Qi et al., 2021). Of the six UA PROTACs linked to a thalidomide E3 ligand, the compound connected with a 3-polyoxyether demonstrated in vitro anti-tumour activity by the degradation of the target protein murine double minute-2 protein.
We have previously reported the detection of ternary complexes formed by the PROTAC GNE-987 between Brd4 bromodomains 1 and 2, the drug target for several cancers, and Hippel Lindeau E3-Ubiquitin Ligase using native MS (Lu et al., 2015; Sternicki et al., 2021). The technique allowed the direction observation of ternary complex formed by the target protein, PROTAC and the E3- Ubiquitin ligase with a 1: 1: 1 subunit stoichiometry. Native MS showed larger amounts of ternary complexes formed with Brd4B1 than Brd4B2, which corresponds to the outcome of SPR-based ternary complex half-life measurements that reported Brd4B1-containing complex being more stable than Brd4B2-containing complex (Pillow et al., 2020). This demonstrated the potential of using native MS for direct observation of ternary complexes formed between PROTAC and NPs.
In this review, we have focused on some examples of applying native MS in the field of NP research, such as in identifying the interactions between several NPs and their viral protein targets. Native MS provides great insights into the interactions between the metabolome and the proteome, with the advantage of direct observation of protein-ligand binding. The technique allows the discovery of new bioactive NPs to reveal broader biological information with minimal sample preparation efforts. We have briefly discussed the huge effort for sample separation and dissociation in AS-MS. A new CIAS-MS method for a simpler and more time efficient alternative to previous AS-MS, provides a complementary method to native MS. One cannot help but wonder, what the future holds?
The necessity for complex procedures to purify and isolate proteins is the main limitation for using native MS to study metabolome-proteome interactions, particularly for membrane proteins. There has been progress made in detergents and alternate systems for better stabilizing membrane proteins (Marty et al., 2016; Urner et al., 2020). Reasonable success has been achieved for directly identifying proteins or protein-ligand interactions in crude cell lysate with native MS (Gan et al., 2017; Vimer et al., 2020; Rogawski et al., 2021). Such advancement will continue and elicit discovery of bioactive NPs against proteins that were previously difficult to investigate. Much of the effort in MS has also been shifted towards the development of better MS data automation system, with a wide-scale adoption of standardized workflow for rapid dereplication of NPs awaits (Orton et al., 2018; Park et al., 2020).
There are potential future advances in instruments, sample preparation, throughput, and data analysis automation, for the application of native MS in NP research, and one can envision wider applications of native MS for the development of natural product drug discovery in the upcoming years.
RQ conceptualization; YG, writing–original draft; YG, ML, and RQ, writing–review and editing.
This work was facilitated by funding from the Australian Research Council Equipment Grant (LE120100170), the Australian Research Council Linkage Project LP120100485 and the Bill and Melinda Gates Foundation grants OPP1008376, OPP1035218, OPP1069415, OPP1174957. This research was supported by the Griffith University (GU) Postgraduate PhD Scholarship, GU International Postgraduate PhD Scholarship, and GU Environmental and Science Top up Scholarship.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aron, A. T., Petras, D., Schmid, R., Gauglitz, J. M., Büttel, I., Antelo, L., et al. (2022). Native mass spectrometry-based metabolomics identifies metal-binding compounds. Nat. Chem. 14 (1), 100–109. doi:10.1038/s41557-021-00803-1
Bizzini, A., and Greub, G. (2010). Matrix-assisted laser desorption ionization time-of-flight mass spectrometry, a revolution in clinical microbial identification. Clin. Microbiol. Infect. 16 (11), 1614–1619. doi:10.1111/j.1469-0691.2010.03311.x
Buckley, D. L., Raina, K., Darricarrere, N., Hines, J., Gustafson, J. L., Smith, I. E., et al. (2015). HaloPROTACS: Use of small molecule PROTACs to induce degradation of HaloTag fusion proteins. ACS Chem. Biol. 10 (8), 1831–1837. doi:10.1021/acschembio.5b00442
Camp, D., Davis, R. A., Campitelli, M., Ebdon, J., and Quinn, R. J. (2012). Drug-like properties: Guiding principles for the design of natural product libraries. J. Nat. Prod. 75 (1), 72–81. doi:10.1021/np200687v
Chen, Y., and Hagerman, A. E. (2004). Characterization of soluble non-covalent complexes between bovine serum albumin and β-1, 2, 3, 4, 6-penta-O-galloyl-d-glucopyranose by MALDI-TOF MS. J. Agric. Food Chem. 52 (12), 4008–4011. doi:10.1021/jf035536t
Chen, G., Wu, J., Li, N., and Guo, M. (2018). Screening for anti-proliferative and anti-inflammatory components from rhamnus davurica pall. Using bio-affinity ultrafiltration with multiple drug targets. Anal. Bioanal. Chem. 410 (15), 3587–3595. doi:10.1007/s00216-018-0953-6
Chen, G., Fan, M., Liu, Y., Sun, B., Liu, M., Wu, J., et al. (2019a). Advances in MS based strategies for probing ligand-target interactions: Focus on soft ionization mass spectrometric techniques. Front. Chem. 7, 703. doi:10.3389/fchem.2019.00703
Chen, G.-L., Fan, M.-X., Wu, J.-L., Li, N., and Guo, M.-Q. (2019b). Antioxidant and anti-inflammatory properties of flavonoids from Lotus plumule. Food Chem. 277, 706–712. doi:10.1016/j.foodchem.2018.11.040
da Silva, V. C., Napolitano, A., Eletto, D., Rodrigues, C. M., Pizza, C., and Vilegas, W. (2011). Characterization of gallotannins from Astronium species by flow injection analysis-electrospray ionization-ion traptandem mass spectrometry and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Eur. J. Mass Spectrom. 17 (4), 365–375. doi:10.1255/ejms.1141
Dearmond, P. D., Xu, Y., Strickland, E. C., Daniels, K. G., and Fitzgerald, M. C. (2011). Thermodynamic analysis of protein-ligand interactions in complex biological mixtures using a shotgun proteomics approach. J. Proteome Res. 10 (11), 4948–4958. doi:10.1021/pr200403c
Ding, Y., Ding, C., Ye, N., Liu, Z., Wold, E. A., Chen, H., et al. (2016). Discovery and development of natural product oridonin-inspired anticancer agents. Eur. J. Med. Chem. 122, 102–117. doi:10.1016/j.ejmech.2016.06.015
Dührkop, K., Fleischauer, M., Ludwig, M., Aksenov, A. A., Melnik, A. V., Meusel, M., et al. (2019). SIRIUS 4 - a rapid tool for turning tandem mass spectra into metabolite structure information. Nat. Methods 16 (4), 299–302. doi:10.1038/s41592-019-0344-8
Elnaas, A. R., Grice, D., Han, J., Feng, Y., Capua, A. D., Mak, T., et al. (2020). Discovery of a natural product that binds to the Mycobacterium tuberculosis protein Rv1466 using native mass spectrometry. Molecules 25 (10), E2384. doi:10.3390/molecules25102384
Erlanson, D. A., Fesik, S. W., Hubbard, R. E., Jahnke, W., and Jhoti, H. (2016). Twenty years on: the impact of fragments on drug discovery. Nat. Rev. Drug Discov. 15 (9), 605–619. doi:10.1038/nrd.2016.109
Frison-Norrie, S., and Sporns, P. (2002). Identification and quantification of flavonol glycosides in almond seedcoats using MALDI-TOF MS. J. Agric. Food Chem. 50 (10), 2782–2787. doi:10.1021/jf0115894
Fu, Y., Luo, J., Qin, J., and Yang, M. (2019). Screening techniques for the identification of bioactive compounds in natural products. J. Pharm. Biomed. Anal. 168, 189–200. doi:10.1016/j.jpba.2019.02.027
Gan, J., Ben-Nissan, G., Arkind, G., Tarnavsky, M., Trudeau, D., Noda Garcia, L., et al. (2017). Native mass spectrometry of recombinant proteins from crude cell lysates. Anal. Chem. 89 (8), 4398–4404. doi:10.1021/acs.analchem.7b00398
Han, Y., Belley, M., Bayly, C. I., Colucci, J., Dufresne, C., Giroux, A., et al. (2008). Discovery of [(3-bromo-7-cyano-2-naphthyl)(difluoro)methyl]phosphonic acid, a potent and orally active small molecule PTP1B inhibitor. Bioorg. Med. Chem. Lett. 18 (11), 3200–3205. doi:10.1016/j.bmcl.2008.04.064
Hann, M. M., Leach, A. R., and Harper, G. (2001). Molecular complexity and its impact on the probability of finding leads for drug discovery. J. Chem. Inf. Comput. Sci. 41 (3), 856–864. doi:10.1021/ci000403i
Kaur, S., McGuire, L., Tang, D., Dollinger, G., and Huebner, V. (1997). Affinity selection and mass spectrometry-based strategies to identify lead compounds in combinatorial libraries. J. Protein Chem. 16 (5), 505–511. doi:10.1023/a:1026369729393
Keller, B. O., and Li, L. (2001). Detection of 25,000 Molecules of Substance P by MALDI-TOF Mass Spectrometry and Investigations into the Fundamental Limits of Detection in MALDI. J. Am. Soc. Mass Spectrom. 12 (9), 1055–1063. doi:10.1016/S1044-0305(01)00288-4
Koehn, F. E., and Carter, G. T. (2005). The evolving role of natural products in drug discovery. Nat. Rev. Drug Discov. 4 (3), 206–220. doi:10.1038/nrd1657
Kregel, S., Wang, C., Han, X., Xiao, L., Fernandez-Salas, E., Bawa, P., et al. (2020). Androgen receptor degraders overcome common resistance mechanisms developed during prostate cancer treatment. Neoplasia 22 (2), 111–119. doi:10.1016/j.neo.2019.12.003
Kumar, B. R. (2017). Application of HPLC and ESI-MS techniques in the analysis of phenolic acids and flavonoids from green leafy vegetables (GLVs). J. Pharm. Anal. 7 (6), 349–364. doi:10.1016/j.jpha.2017.06.005
Li, D. Q., Zhao, J., Li, S. P., and Zhang, Q. W. (2014). Discovery of xanthine oxidase inhibitors from a complex mixture using an online, restricted-access material coupled with column-switching liquid chromatography with a diode-array detection system. Anal. Bioanal. Chem. 406 (7), 1975–1984. doi:10.1007/s00216-013-7612-8
Li, L., Li, B., Zhang, H., Zhao, A., Han, B., Liu, C., et al. (2015). Ultrafiltration LC-ESI-MSn screening of MMP-2 inhibitors from selected Chinese medicinal herbs Smilax glabra Roxb., Smilax China L. and Saposhnikovia divaricata (turcz.) Schischk as potential functional food ingredients. J. Funct. Foods 15, 389–395. doi:10.1016/j.jff.2015.03.038
Littler, D. R., Liu, M., McAuley, J. L., Lowery, S. A., Illing, P. T., Gully, B. S., et al. (2021). A natural product compound inhibits coronaviral replication in vitro by binding to the conserved Nsp9 SARS-CoV-2 protein. J. Biol. Chem. 297 (6), 101362. doi:10.1016/j.jbc.2021.101362
Liu, Y., Liu, S., and Liu, Z. (2013). Screening and determination of potential xanthine oxidase inhibitors from radix salviae miltiorrhizae using ultrafiltration liquid chromatography-mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 923-924, 48–53. doi:10.1016/j.jchromb.2013.02.009
Liu, M., Van Voorhis, W. C., and Quinn, R. J. (2021). Development of a target identification approach using native mass spectrometry. Sci. Rep. 11 (1), 2387. doi:10.1038/s41598-021-81859-4
Liu, M., Littler, D. R., Rossjohn, J., and Quinn, R. J. (2022). Binding studies of the prodrug HAO472 to SARS-cov-2 Nsp9 and variants. ACS Omega 7 (8), 7327–7332. doi:10.1021/acsomega.1c07186
Lomenick, B., Jung, G., Wohlschlegel, J. A., and Huang, J. (2011). Target identification using drug affinity responsive target stability (DARTS). Curr. Protoc. Chem. Biol. 3 (4), 163–180. doi:10.1002/9780470559277.ch110180
Lu, J., Qian, Y., Altieri, M., Dong, H., Wang, J., Raina, K., et al. (2015). Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chem. Biol. 22 (6), 755–763. doi:10.1016/j.chembiol.2015.05.009
Mak, T., Rossjohn, J., Littler, D. R., Liu, M., and Quinn, R. J. (2022). Collision-induced affinity selection mass spectrometry for identification of ligands. ACS Bio Med. Chem. Au. doi:10.1021/acsbiomedchemau.2c00021
Márquez, B., Verdier-Pinard, P., Hamel, E., and Gerwick, W. H. (1998). Curacin D, an antimitotic agent from the marine cyanobacterium lyngbya majuscula. Phytochemistry 49 (8), 2387–2389. doi:10.1016/S0031-9422(98)00365-3
Marty, M. T., Hoi, K. K., and Robinson, C. V. (2016). Interfacing membrane mimetics with mass spectrometry. Acc. Chem. Res. 49 (11), 2459–2467. doi:10.1021/acs.accounts.6b00379
Muchiri, R. N., and van Breemen, R. B. (2021). Affinity selection–mass spectrometry for the discovery of pharmacologically active compounds from combinatorial libraries and natural products. J. Mass Spectrom. 56 (5), e4647. doi:10.1002/jms.4647
Murray, C. W., and Rees, D. C. (2009). The rise of fragment-based drug discovery. Nat. Chem. 1 (3), 187–192. doi:10.1038/nchem.217
Nguyen, G. T. H., Bennett, J. L., Liu, S., Hancock, S. E., Winter, D. L., Glover, D. J., et al. (2021). Multiplexed screening of thousands of natural products for protein–ligand binding in native mass spectrometry. J. Am. Chem. Soc. 143 (50), 21379–21387. doi:10.1021/jacs.1c10408
Orton, D. J., Tfaily, M. M., Moore, R. J., LaMarche, B. L., Zheng, X., Fillmore, T. L., et al. (2018). A customizable flow injection system for automated, high throughput, and time sensitive ion mobility spectrometry and mass spectrometry measurements. Anal. Chem. 90 (1), 737–744. doi:10.1021/acs.analchem.7b02986
Park, C., and Marqusee, S. (2005). Pulse proteolysis: A simple method for quantitative determination of protein stability and ligand binding. Nat. Methods 2 (3), 207–212. doi:10.1038/nmeth740
Park, H.-M., Winton, V. J., Drader, J. J., Manalili Wheeler, S., Lazar, G. A., Kelleher, N. L., et al. (2020). Novel interface for high-throughput analysis of biotherapeutics by electrospray mass spectrometry. Anal. Chem. 92 (2), 2186–2193. doi:10.1021/acs.analchem.9b04826
Pillow, T. H., Adhikari, P., Blake, R. A., Chen, J., Del Rosario, G., Deshmukh, G., et al. (2020). Antibody conjugation of a chimeric BET degrader enables in vivo activity. ChemMedChem 15 (1), 17–25. doi:10.1002/cmdc.201900497
Potjewyd, F., Turner, A. W., Beri, J., Rectenwald, J. M., Norris-Drouin, J. L., Cholensky, S. H., et al. (2020). Degradation of polycomb repressive complex 2 with an EED-targeted bivalent chemical degrader. Cell Chem. Biol. 27 (1), 47–56.e15. doi:10.1016/j.chembiol.2019.11.006
Qi, Z., Yang, G., Deng, T., Wang, J., Zhou, H., Popov, S. A., et al. (2021). Design and linkage optimization of ursane-thalidomide-based PROTACs and identification of their targeted-degradation properties to MDM2 protein. Bioorg. Chem. 111, 104901. doi:10.1016/j.bioorg.2021.104901
Reed, J. D., Krueger, C. G., and Vestling, M. M. (2005). MALDI-TOF mass spectrometry of oligomeric food polyphenols. Phytochemistry 66 (18), 2248–2263. doi:10.1016/j.phytochem.2005.05.015
Reher, R., Aron, A. T., Fajtová, P., Liu, C., Ben Shalom, I. Y., Bittremieux, W., et al. (2021). Native metabolomics identifies the rivulariapeptolide family of protease inhibitors. Nat. Commun. 13 (1), 4619. doi:10.1101/2021.09.03.458897
Rogawski, R., Rogel, A., Bloch, I., Gal, M., Horovitz, A., London, N., et al. (2021). Intracellular protein–drug interactions probed by direct mass spectrometry of cell lysates. Angew. Chem. Int. Ed. Engl. 60 (36), 19637–19642. doi:10.1002/anie.202104947
Schenone, M., Dancik, V., Wagner, B. K., and Clemons, P. A. (2013). Target identification and mechanism of action in chemical biology and drug discovery. Nat. Chem. Biol. 9 (4), 232–240. doi:10.1038/nchembio.1199
Schlebach, J. P., Kim, M.-S., Joh, N. H., Bowie, J. U., and Park, C. (2011). Probing membrane protein unfolding with pulse proteolysis. J. Mol. Biol. 406 (4), 545–551. doi:10.1016/j.jmb.2010.12.018
Schmid, R., Petras, D., Nothias, L.-F., Wang, M., Aron, A. T., Jagels, A., et al. (2021). Ion identity molecular networking for mass spectrometry-based metabolomics in the GNPS environment. Nat. Commun. 12 (1), 3832. doi:10.1038/s41467-021-23953-9
Simon, R. P., Winter, M., Kleiner, C., Wehrle, L., Karnath, M., Ries, R., et al. (2021). MALDI-TOF-Based affinity selection mass spectrometry for automated screening of protein-ligand interactions at high throughput. SLAS Discov. 26 (1), 44–57. doi:10.1177/2472555220959266
Sternicki, L. M., Nonomiya, J., Liu, M., Mulvihill, M. M., and Quinn, R. J. (2021). Native mass spectrometry for the study of PROTAC GNE-987-containing ternary complexes. ChemMedChem 16 (14), 2206–2210. doi:10.1002/cmdc.202100113
Strickland, E. C., Geer, M. A., Tran, D. T., Adhikari, J., West, G. M., DeArmond, P. D., et al. (2013). Thermodynamic analysis of protein-ligand binding interactions in complex biological mixtures using the stability of proteins from rates of oxidation. Nat. Protoc. 8 (1), 148–161. doi:10.1038/nprot.2012.146
Tanaka, K. (2003). The origin of macromolecule ionization by laser irradiation (nobel lecture). Angew. Chem. Int. Ed. Engl. 42 (33), 3860–3870. doi:10.1002/anie.200300585
Ul Haq, F., Ali, A., Akhtar, N., Aziz, N., Khan, M. N., Ahmad, M., et al. (2020). A high-throughput method for dereplication and assessment of metabolite distribution in salvia species using LC-MS/MS. J. Adv. Res. 24, 79–90. doi:10.1016/j.jare.2020.02.001
Urner, L. H., Liko, I., Yen, H. Y., Hoi, K. K., Bolla, J. R., Gault, J., et al. (2020). Modular detergents tailor the purification and structural analysis of membrane proteins including G-protein coupled receptors. Nat. Commun. 11 (1), 564. doi:10.1038/s41467-020-14424-8
van Breemen, R. B., Huang, C. R., Nikolic, D., Woodbury, C. P., Zhao, Y. Z., and Venton, D. L. (1997). Pulsed ultrafiltration mass spectrometry: A new method for screening combinatorial libraries. Anal. Chem. 69 (11), 2159–2164. doi:10.1021/ac970132j
Vimer, S., Ben-Nissan, G., and Sharon, M. (2020). Direct characterization of overproduced proteins by native mass spectrometry. Nat. Protoc. 15 (2), 236–265. doi:10.1038/s41596-019-0233-8
Vu, H., Pham, N. B., and Quinn, R. J. (2008). Direct screening of natural product extracts using mass spectrometry. J. Biomol. Screen. 13 (4), 265–275. doi:10.1177/1087057108315739
Vu, H., Roullier, C., Campitelli, M., Trenholme, K. R., Gardiner, D. L., Andrews, K. T., et al. (2013). Plasmodium gametocyte inhibition identified from a natural-product-based fragment library. ACS Chem. Biol. 8 (12), 2654–2659. doi:10.1021/cb400582b
Vu, H., Pedro, L., Mak, T., McCormick, B., Rowley, J., Liu, M., et al. (2018). Fragment-based screening of a natural product library against 62 potential malaria drug targets employing native mass spectrometry. ACS Infect. Dis. 4 (4), 431–444. doi:10.1021/acsinfecdis.7b00197
Wang, Z., Yu, X., Cui, M., Liu, Z., Song, F., and Liu, S. (2009). Investigation of calmodulin-peptide interactions using matrix-assisted laser desorption/ionization mass spectrometry. J. Am. Soc. Mass Spectrom. 20 (4), 576–583. doi:10.1016/j.jasms.2008.11.017
Welker, M., Dittmann, E., and von Döhren, H. (2012). “Chapter two - cyanobacteria as a source of natural products,” in Methods in enzymology. Editor D. A. Hopwood (Academic Press), 23–46.
Wiesmann, C., Barr, K. J., Kung, J., Zhu, J., Erlanson, D. A., Shen, W., et al. (2004). Allosteric inhibition of protein tyrosine phosphatase 1B. Nat. Struct. Mol. Biol. 11 (8), 730–737. doi:10.1038/nsmb803
Xie, Y., Feng, Y., Di Capua, A., Mak, T., Buchko, G. W., Myler, P. J., et al. (2020). A phenotarget approach for identifying an alkaloid interacting with the tuberculosis protein Rv1466. Mar. Drugs 18 (3), E149. doi:10.3390/md18030149
Keywords: native mass spectrometry, natural products, metabolome, proteome, CIAS-MS, drug discovery
Citation: Gu Y, Liu M and Quinn RJ (2022) Metabolite-protein interactions: Native mass spectrometry and collision induced affinity selection mass spectrometry in natural product screening. Front. Anal. Sci. 2:1014017. doi: 10.3389/frans.2022.1014017
Received: 08 August 2022; Accepted: 23 September 2022;
Published: 10 October 2022.
Edited by:
Mowei Zhou, Pacific Northwest National Laboratory (DOE), United StatesReviewed by:
Suk-Joon Hyung, Genentech, Inc., United StatesCopyright © 2022 Gu, Liu and Quinn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ronald J. Quinn, ci5xdWlubkBncmlmZml0aC5lZHUuYXU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.