 Sam Wouters1,2
Sam Wouters1,2 Ils Pijpers3
Ils Pijpers3 Ninon Vanden Haute1
Ninon Vanden Haute1 Daniel Meston1Lieve Dillen3
Daniel Meston1Lieve Dillen3 Filip Cuyckens2Sebastiaan Eeltink1*
Filip Cuyckens2Sebastiaan Eeltink1*- 1Vrije Universiteit Brussel (VUB), Department of Chemical Engineering, Brussels, Belgium
- 2Janssen R&D, Drug Metabolism and Pharmacokinetics, Beerse, Belgium
- 3Janssen R&D, Bioanalysis Discovery and Development Sciences, Beerse, Belgium
This study reports on the potential of using ion-exchange suppressor technology in liquid chromatography—electrospray ionization mass spectrometry workflows. The aim was to use high salt concentrations to improve separation performance, while overcoming the resulting significant ion suppression during electrospray ionization. As a case study, we apply suppressor technology to the hydrophilic interaction liquid chromatography separation and detection of taurine and glycochenodeoxycholate sulfate, endogenous biomarkers for organic anion transporter protein inhibition. The desired chromatographic selectivity was achieved applying 100 mM ion-pairing agent, while competing ions negatively affecting MS sensitivity were actively removed post-column from the solvent via a charged partially permeable membrane and replaced with protons, resulting in an up to 10-fold increase in detection sensitivity.
Introduction
Ion suppression in electrospray ionization is a major limitation in the techniques applicability, which is also very relevant for hydrophilic interaction liquid chromatography (HILIC) (Mallet et al., 2004; Volmer and Jessome, 2006). HILIC is based on partitioning of analytes between a water-enriched layer of the mobile phase that is immobilized on to a polar stationary phase and the low aqueous/highly polar organic mobile phase (Dejaegher et al., 2008; McCalley, 2017). The presence of buffer salts or ion-pairing agents in the mobile phase are routinely used to tune the retention behavior of charged analytes allowing to improve chromatographic resolution. While HILIC usually enjoys high detector response due to the volatility of the effluent, the increased ionic eluent strength tends to limit MS sensitivity due to charge competition during electrospray ionization (ESI). To address this long-standing limitation, our group recently explored the use of a membrane-based microfluidic chip for selective exchange of trifluoroacetate in HILIC and reversed-phase liquid chromatography (RPLC) mobile phases with formate and propionate prior to MS detection, aiming at greatly reducing adduct formation and significantly improving MS sensitivity in protein and peptide analysis (Wouters et al., 2021). The concept is based on suppressed conductivity detection, commonly used in ion chromatography, where selective post-column exchange of ions from mobile phases is achieved using so-called suppressor or stripper devices, prior to conductivity detection (Wouters et al., 2017; Wouters et al., 2019). In this work, we explore the use of high salt concentration solvent modifiers to tune resolution and subsequently mitigate the MS incompatibility of high molar ammonium acetate applying a post-column mobile-phase modulation approach.
We exemplify from a case study where the concept is applied to the separation of taurine and glycochenodeoxycholate sulfate (GCDCA-S), which are both highly polar, hydrophilic compounds and are therefore incompatible with conventional RPLC. As such, HILIC is better suited to separate these compounds, which are under investigation in the context of drug-drug interactions (DDI). Drug transporter proteins in the liver and kidney play an important role in the uptake and elimination of a wide range of chemicals. The inhibition of these transporters may cause significant DDI, which may increase the risk of adverse events through altered exposure of the combination of drugs (Palleria et al., 2013). Given the growing relevance of polypharmacy, and the increased attention for DDI from the regulatory agencies, the potential for DDI needs to be assessed in drug development as part of the safety program. Traditionally, in vitro DDI evaluations first identify any potential risk followed by clinical DDI studies with probe substrates for the transporter flagged in vitro (Chu et al., 2018). However, an emerging approach has been presented making use of endogenous biomarkers as read-out for in vivo DDI. Several endogenous compounds that can be measured in urine and plasma, have been evaluated as substrates for drug transporters in the liver and kidney (Tsuruya et al., 2016). Previous observation demonstrates that inhibition of two major renal organic anion polypeptide transporters (OAT1 and OAT3) decreases the renal clearance of taurine and GCDCA-S significantly, suggesting these compounds may serve as diagnostic biomarkers of OAT inhibition. While taurine and GCDCA-S are opportune analytes for monitoring OAT activity, they are not without analytical challenges.
Materials and methods
Chemicals and materials
Taurine, acetonitrile (ACN, LC-MS grade), ammonium acetate, ammonium hydroxide, and 10% sulfuric acid were purchased from Sigma-Aldrich (Bornem, Belgium). GCDCA-S, taurine-D4 and GCDC-D4 were obtained at Toronto Research Chemicals (Toronto, Canada). An Acquity UPLC BEH Amide 50 × 2.1 mm i. d. column packed with 1.7 µm particles was purchased from Waters (Milford, MA, United States). A planar dual-membrane chemically-regenerated anion suppressor fitted with sulfonated cation-exchange membranes (ACRS500) was purchased at Thermo Fisher Scientific (Sunnyvale United States).
QC sample preparation and processing
Since taurine and GCDCA-S are endogenously present in plasma (target matrix), Milli-Q water was used as a surrogate matrix for the calibration curve. Combined calibration samples for both analytes were prepared to analyze a range from 200 to 50,000 ng/ml for taurine and 20.0–5,000 ng/ml for GCDCA-S. Combined QC samples were prepared by spiking native pooled K2EDTA human plasma, at a concentration of 20,000 ng/ml for taurine and 2,000 ng/ml for GCDCA-S. These QC samples were serially diluted in pooled K2EDTA plasma. Together with the spiked QC samples, the pooled human plasma used for the preparation of the QC samples, was analyzed for the presence of endogenous concentrations. The nominal values of the QCs were calculated as the sum of the endogenous level plus the spiked amount. To minimize the matrix effect and avoid saturation of the detector of the mass spectrometer, all samples were 10-fold diluted with Milli-Q water before protein precipitation with 10-fold volume of acetonitrile. For taurine, a stable isotope labeled internal standard was available. For GCDCA-S the deuterated analogue of GCDC was used as structural analogue internal standard.
For parallelism evaluation, the endogenous concentration of the blank plasma used for the preparation of the QC samples was determined by interpolation of the response of the QC blank to the calibration curve in surrogate matrix. Secondly, a linear regression with a 1/x2 weighing was performed using the spiked concentration of analyte of the QC sample and the peak area ratios obtained. The endogenous level of the QC was determined by dividing the intercept with the slope of the curve. Parallelism is considered proven if the difference between both values is less than 20.0% and −20%.
LC-MS instrumentation and settings
Separations were performed on a Shimadzu LC30 system (Shimadzu, Kyoto, Japan). Mobile phases consisted of 10 mM or 100 mM aqueous ammonium acetate (A), and ACN as the organic modifier (B). A binary pump was used to deliver a solvent gradient from 95% to 50% B from 0.3 to 3.5 min at a flow rate of 0.6 ml/min. The BEH amide column was thermostatted in a column compartment at 50°C. The suppressor was installed after the column prior to the MS detector and a 12.5 mM sulfuric acid in 10% ACN as regenerant flow was applied at 1.2 ml/min in counter flow using an isocratic pump (Shimadzu). A 10-port, 2-position switching valve (VICI, Schenkon, Switzerland) was used to bypass the suppressor. Detection was performed on an API 4000 triple quadruple MS (AB Sciex, Toronto, Canada) equipped with an electrospray ionization interface. LC-MS/MS was performed in the negative ionization mode at −4.5 keV with single reaction monitoring of the Q1/Q3 transitions at m/z: 452.3/74.0 for GCDC-D4, 528.3/448.3 for GCDCA-S and 124.0/80.0 for taurine and 128.0/80.0 for taurine-D4 while applying declustering potentials of −100/−100/−45 V, collision energies −42/−42/−28 and collision cell exit potentials −25/−25/−5 V, for GCDC-D4/GCDCA-S/taurine (−D4), respectively. Data acquisition and analysis were controlled using Analyst 1.7 software (AB Sciex, Toronto, Canada).
Results and discussion
HILIC-ESI-MS method development and use of post-column modulation
The incorporation of internal standards for quantitative analysis is recommended to adequately control experimental drift and correct for detector response variability. As such, isotopically labelled GCDC-D4 was included in the sample mixture as a structural analogue of GCDCA-S and taurine-D4 as stable isotope labeled internal standard for taurine. In the calibration samples prepared in MilliQ water, GCDC-D4 did not fully compensate for the matrix difference between the surrogate matrix and target matrix when applying 10 mM ammonium acetate. Altering the concentration from 10 mM to 100 mM resulted in an improved matrix effect compensation. Figures 1A,B show a representative chromatogram of the HILIC-ESI-MS analysis while applying 10 mM and 100 mM ammonium acetate to the mobile phase, respectively.
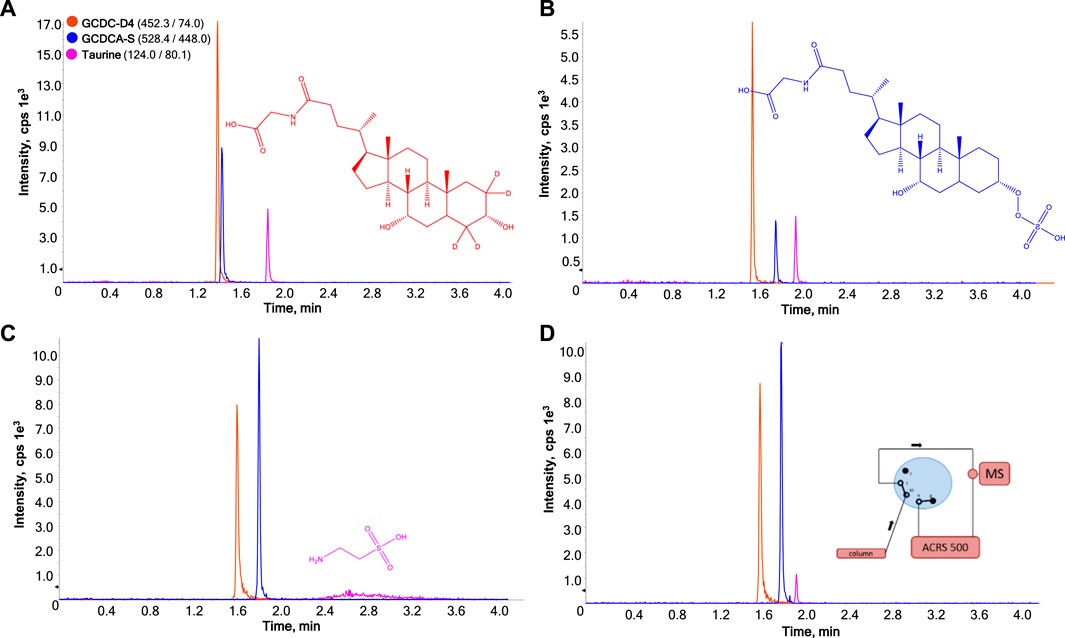
FIGURE 1. Effect of amount of ion pairing agent on HILIC-ESI-MS analysis of GCDC-D4 (1), GCDCA-S (2), and taurine (3) without (A,B) and with (C,D) on-line membrane suppression of ammonium acetate. The ammonium acetate concentrations are 10 mM (A) or 100 mM (B, C, and (D). In (D) GCDC-D4 and GCDCA-S pass though the membrane suppressor prior to MS detection, whereas taurine is directed to the MS bypassing the suppressor by employing a heart-cutting approach programmed at 1.9 min.
While improving resolution, increasing the ammonium acetate content led to a sharp 6.4-fold reduction in MS sensitivity due to ion suppression for GCDCA-S. Taurine was less sensitive to changes in retention but also undergoes substantial (3.2-fold) reduction in MS sensitivity. Figures 1C exemplifies the effect when integrating a cation-exchange membrane suppressor post-column prior to the ESI interface, targeting the depletion of ammonium ions from the HILIC mobile phase. The exchange in the membrane suppressor schematically shown in Figure 2. Positively charged ammonium ions from the effluent flow diffuse through the partially permeable negatively-charged membrane into the regenerant flow and protons from the regenerant flow diffuse into the effluent flow converting ammonium acetate to acetic acid. A dilute solution of a strong acid (sulfuric acid) was applied in the regenerant as a source of protons. The regenerant runs countercurrent with respect to the eluent flow at twice the flowrate to maximize the concentration gradient and thereby promote the complete conversion of ammonium acetate to acetic acid. Details on construction and functioning of this type of suppressor can be found in a recent review by Haddad et al. (Wouters et al., 2017). For details on how to further influence the exchange rate in such concentration-gradient-driven devices, we refer to earlier work (Wouters et al., 2016; Wouters et al., 2021). 10% acetonitrile was added to the regenerant to prevent loss of analytes due to interaction with the polymeric membrane.
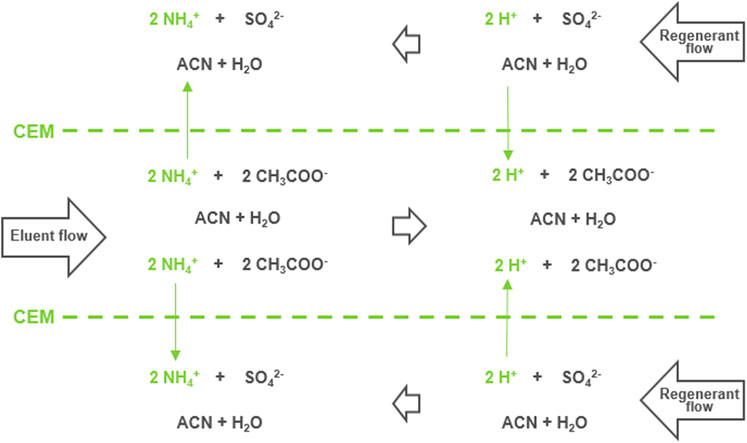
FIGURE 2. Schematic representation of the exchange process taking place in the planar double-sided flow-through membrane suppressor which features a central eluent channel, separated by cation-exchange membranes from a regenerant channel above and below.
Applying 100 mM ammonium acetate and integrating post-column modulation prior to MS led to a substantial increase in the detector response for GCDCA-S, presenting a 6.9-fold increase in peak height and 10.2-fold increase in peak area, despite an increase in peak width by a factor 1.4, which is to be expected with the added dead volume of the suppressor. However, the peak shape of taurine was strongly distorted, likely caused by the zwitterionic character of taurine and consequently, ionic interactions between the positively charged amine on taurine and the negatively-charged membrane. Post-column infusion of 10% v/v ammonium hydroxide between the column eluent and the ion-stripper lead to deprotonation of the carboxylate of taurine, inducing ionic repulsion of taurine from the negatively charged membrane. This led to a strong decrease in tailing factor but also a decrease in the intensity of taurine, again due to competitive ion-suppression in ESI from the additional ammonium ions (result not shown). To overcome the chemical incompatibility of taurine with the ion-stripper a flow diverter valve was integrated, allowing taurine to selectively bypass the ion-stripper, as demonstrated in Figures 1D. This led to a substantial decrease in tailing factor. Limitations would be that such approach is only possible when sufficient retention-time difference is established between both compounds, as is the case in our experiment, and taurine will still suffer to some extent from ion-suppression, as it is still eluting at higher concentrations of ammonium acetate.
Determination of the suitability of the methodologies for quantitative analysis
While taurine and GCDCA-S have been analyzed previously by LC-MS as potential endogenous biomarkers (Peng et al., 2014; Tsuruya et al., 2016; Shen et al., 2017; Horvath et al., 2020), they have not been validated for use in clinical studies. As GCDCA-S and taurine are endogenously present in plasma the calibration curve cannot be made in this matrix, instead water was selected as surrogate matrix. For the optimized method, we determined the robustness, linearity, and precision and confirmed the parallelism between the calibration samples and the authentic analyte in the biological matrix. The accuracy and precision were evaluated with QC samples in six-fold and the dilution integrity shows whether a dilution of the QC samples in water can be used. The criteria for a successful robustness run are: 80.0% ≤ accuracy ≤ 120.0%, 0% ≤ coefficient of variation (CV) ≤ 20.0%. Figure 3 shows the calibration lines for a standard curve in the surrogate matrix as well as for QCs spiked in plasma. Quantitative analysis of taurine and GCDCA-S with the proposed assay could fulfill regulatory acceptance criteria, with taurine determined between 200–50000 ng/ml and GCDCA-S between 20–5,000 ng/ml. For both compounds the accuracy was within criteria (taurine: 95.4%–100.0% and GCDCA-S:100.0%–115.8%) and the %CV was maximum 6.2% for taurine and 3.8% for GCDCA-S. The parallelism between the surrogate matrix and human plasma was -9.0% for taurine and −7.1% for GCDCA-S evaluated as the difference in the endogenous concentrations obtained with surrogate matrix and the target matrix. The method was qualified for the quantification of taurine and GCDCA-S concentrations in plasma originating from clinical studies.
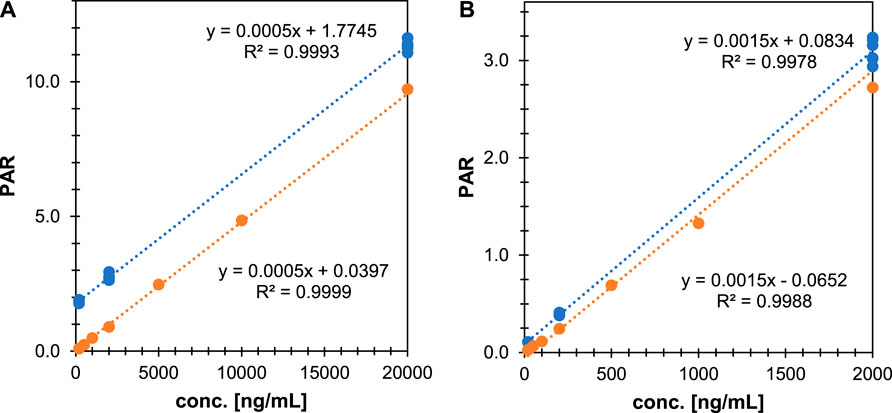
FIGURE 3. Parallelism evaluation of the HILIC-suppressor-ESI-MS with heart-cut of taurine for the evaluation of linearity, precision, and accuracy of (A) taurine and (B) GCDCA-S in QC samples in human plasma (blue circles) as well as surrogate matrix samples (orange circles).
Concluding remarks
Herein, we have provided a proof-of-concept showing the advantages one can expect when implementing suppressor technology for the analysis of taurine and GCDCA-S, diagnostic biomarkers for OAT inhibition. Membrane-suppressor technology can be easily integrated into existing LC-MS workflows and has the potential to provide dramatic increases in analyte detector response in the case MS detrimental mobile phase conditions are used. As such, suppressors may provide a facile methodology to implement in methods applying less “MS-friendly” buffers allowing one to tune selectivity using high concentration ion-pairing agents, without compromising MS detection sensitivity. A current limitation was observed when applying the concept to a zwitterionic compound, where unwanted interactions with the membrane introduced retention in the suppressor. We hypothesize that the alteration of membrane properties would allow suppressors to be tuned to give optimal signal amplification for specific analytes. Additionally, post-column buffering of the pH would provide flexibility when analyzing zwitterionic compounds, such as amino acids, opening several interesting opportunities in proteomic applications as well as for oligonucleotide analysis. The addition of a simple diverter valve allows to switch the application of the suppressor chip between well separated analytes within one chromatogram.
Data availability statement
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
Author contributions
SW, IP, LD, FC, and SE contributed to conception and design of the study. SW, IP, NV, and DM contributed to the methodology, analysis and validation. SW and IP wrote the first draft of the manuscript. SW, IP, LD, FC, and SE contributed to the writing of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
Support of this work by a grants of the Research Foundation Flanders—FWO (grant number: G033018N) and the Flemish Agency of Innovation and Entrepreneurship (IWT.150467) for the “DEBOCS” project are gratefully acknowledged. Luc Sips and Luc Diels are acknowledged for experimental help.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Chu, X., Liao, M., Shen, H., Yoshida, K., Zur, A. A., Arya, V., et al. (2018). Clinical probes and endogenous biomarkers as substrates for transporter drug-drug interaction evaluation: Perspectives from the international transporter consortium. Clin. Pharmacol. Ther. 104, 836–864. doi:10.1002/cpt.1216
Dejaegher, B., Mangelings, D., and Vander Heyden, Y. (2008). Method development for HILIC assays. J. Sep. Sci. 31 (9), 1438–1448. doi:10.1002/jssc.200700680
Horvath, T. D., Haidacher, S. J., Hoch, K. M., Auchtung, J. M., and Haag, A. M. (2020). A high-throughput LC-MS/MS method for the measurement of the bile acid/salt content in microbiome-derived sample sets. Methods 7, 100951. doi:10.1016/j.mex.2020.100951
Mallet, C. R., Lu, Z., and Mazzeo, J. R. (2004). A study of ion suppression effects in electrospray ionization from mobile phase additives and solid-phase extracts. Rapid Commun. Mass Spectrom. 18, 49–58. doi:10.1002/rcm.1276
McCalley, D. V. (2017). Understanding and manipulating the separation in hydrophilic interaction liquid chromatography. J. Chromatogr. A 1523, 49–71. doi:10.1016/j.chroma.2017.06.026
Palleria, C., Di Paolo, A., Giofrè, C., Caglioti, C., Leuzzi, G., Siniscalchi, A., et al. (2013). Pharmacokinetic drug-drug interaction and their implication in clinical management. J. Res. Med. Sci. 18 (7), 601–610.
Peng, C., Tian, J., Lv, M., Huang, Y., Tian, Y., and Zhang, Z. (2014). Development and validation of a sensitive LC-MS-MS method for the simultaneous determination of multicomponent contents in artificial calculus bovis. J. Chromatogr. Sci. 52 (2), 128–136. doi:10.1093/chromsci/bms256
Shen, H., Chen, W., Drexler, D. M., Mandlekar, S., Holenarsipur, V. K., Shields, E. E., et al. (2017). Comparative evaluation of plasma bile acids, dehydroepiandrosterone sulfate, hexadecanedioate, and tetradecanedioate with coproporphyrins I and III as markers of OATP inhibition in healthy subjects. Drug Metab. Dispos. 45 (8), 908–919. doi:10.1124/dmd.117.075531
Tsuruya, Y., Kato, K., Sano, Y., Imamura, Y., Maeda, K., Kumagai, Y., et al. (2016). Investigation of endogenous compounds applicable to drug-drug interaction studies involving the renal organic anion transporters, OAT1 and OAT3, in humans. Drug Metab. Dispos. 44 (12), 1925–1933. doi:10.1124/dmd.116.071472
Volmer, D. A., and Jessome, L. L. (2006). Ion suppression: A major concern in mass spectrometry. LC GC N. Am. 24, 498–510.
Wouters, S., Bruggink, C., Agroskin, Y., Pohl, C., and Eeltink, S. (2016). Microfluidic membrane suppressor module design and evaluation for capillary ion chromatography. J. Chromatogr. A 1484, 26–33. doi:10.1016/j.chroma.2016.12.078
Wouters, S., Dores-Sousa, J. L., Liu, Y., Pohl, C. A., and Eeltink, S. (2019). Ultra-high-pressure ion chromatography with suppressed conductivity detection at 70 MPa using columns packed with 2.5 μm anion-exchange particles. Anal. Chem. 91 (21), 13824–13830. doi:10.1021/acs.analchem.9b03283
Wouters, S., Eeltink, S., Haselberg, R., Somsen, G. W., and Gargano, A. F. G. (2021). Microfluidic ion stripper for removal of trifluoroacetic acid from mobile phases used in HILIC-MS of intact proteins. Anal. Bioanal. Chem. 413 (17), 4379–4386. doi:10.1007/s00216-021-03414-4
Keywords: ion suppression, biomarker analysis, organic anion transporter proteins, MS sensitivity, ion exchange suppressor
Citation: Wouters S, Pijpers I, Vanden Haute N, Meston D, Dillen L, Cuyckens F and Eeltink S (2022) Making high salt concentrations for optimal chromatography compatible with electrospray ionization mass spectrometry using an ion exchange membrane suppressor: Analysis of biomarkers for transporter protein inhibition as a case study. Front. Anal. Sci. 2:1002935. doi: 10.3389/frans.2022.1002935
Received: 25 July 2022; Accepted: 15 August 2022;
Published: 12 September 2022.
Edited by:
Marianne Fillet, University of Liège, BelgiumReviewed by:
Roman Reminek, Institute of Analytical Chemistry (ASCR), CzechiaLeandro Augusto Calixto, Federal University of São Paulo, Brazil
Copyright © 2022 Wouters, Pijpers, Vanden Haute, Meston, Dillen, Cuyckens and Eeltink. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sebastiaan Eeltink, c2ViYXN0aWFhbi5lZWx0aW5rQHZ1Yi5iZQ==