 Nicole Scarberry1†
Nicole Scarberry1† Zachary L. McAdams2,3†
Zachary L. McAdams2,3† Derek Benson4
Derek Benson4 Jason Herrick4
Jason Herrick4 Brandon Christopher Moore5,6
Brandon Christopher Moore5,6 Aaron C. Ericsson2,3*
Aaron C. Ericsson2,3*- 1College of Veterinary Medicine (CVM), University of Missouri (MU), Columbia, MO, United States
- 2Molecular Pathogenesis and Therapeutics Program, University of Missouri, Columbia, MO, United States
- 3University of Missouri Metagenomics Center, Department of Veterinary Pathobiology, College of Veterinary Medicine, University of Missouri, Columbia, MO, United States
- 4Amphibian Conservation Center, Omaha’s Henry Doorly Zoo and Aquarium, Omaha, NE, United States
- 5Department of Biomedical Sciences, University of Missouri, Columbia, MO, United States
- 6School of Health Sciences, Stephens College, Columbia, MO, United States
Introduction: At one time thought to be extinct in the wild, the Wyoming toad (Anaxyrus baxteri) is one of the most critically endangered North American amphibian species. Despite approximately 20 years of ex situ breeding and reintroduction programs, this species remains functionally extinct in the wild. There is concern among those working in these programs that individuals bred in captivity fail to develop a microbiome that withstands the stressors of their native habitat following release. In related species, the skin microbiome has been shown to have a defensive function against common pathogens affecting these animals. However, the early-life microbiome of developing tadpoles in this species remains unknown and, therefore, this defensive function is unexplored in the Wyoming toad.
Methods: This study employed 16S rRNA amplicon sequencing to document the baseline microbiome of tadpole and adult Wyoming toads bred for release at Omaha’s Henry Doorly Zoo and Aquarium (Omaha, Nebraska, USA). We characterized the development of the mucosal microbiome in tadpoles and the mucosal and cloacal microbiome of adult toads.
Results: Our results revealed significant differences between tadpole and adult microbiomes, as well as significant sex-dependent differences within the adult Wyoming toads, in terms of richness and composition.
Discussion: These findings have identified the baseline microbiome of this endangered species and variables significantly influencing its composition in captivity. Ongoing studies of the only extant wild population are expected to identify taxa not found in captive toads, and potentially help 100 design husbandry modifications to maximize survivability following reintroduction to the wild.
1 Introduction
Amphibian species serve critical roles in aquatic and terrestrial ecosystems including their capacity in the food web, potential to serve as biological indicators of environmental degradation, and contributions to nutrient cycling (Vitt et al., 1990; Polasik et al., 2016). Thus, special efforts to protect amphibian species and ensure their survival are warranted. At one time thought to be extinct in the wild, the Wyoming toad (Anaxyrus baxteri) is one of the most critically endangered North America amphibian species (Vincent and Abbott, 2015). The Wyoming toad was first listed as an endangered species under the 1983 amendment to the Endangered Species Act (Lewis et al., 1985) but was thought to be extinct until a small population was discovered in Mortenson Lake in Albany County, Wyoming in 1987. Ten Wyoming toads were brought to the Cheyenne Mountain Zoo in 1989 before the species was declared extinct in the wild in 1991, however, an ex situ breeding program was established in 1993 and attempts at reintroduction to their native range began in 1995 (Lewis et al., 1985; Vincent and Abbott, 2015). These efforts to reintroduce the Wyoming toad into the wild have been challenging. For example, despite approximately 10,000 toads/tadpoles being released between 1995 and 2000, only 62 were observed in the wild (USGS, 2001).
A potential factor hindering Wyoming toad repopulation efforts is Batrachochytrium dendrobatidis (Bd) infection, a dominant cause of rapid amphibian decline worldwide (Polasik et al., 2016; Jiménez et al., 2019). The amphibian skin microbiome may play a role in resistance to Bd infection by modulating the host’s innate immune system and promoting colonization resistance (Barnhart et al., 2017; Jiménez et al., 2019). Additionally, the environment in which individuals mature impacts the development of the host’s microbiome in wild species (Comizzoli et al., 2021). Given that Wyoming toads bred for reintroduction are raised in facilities with high biosecurity standards, there is concern that captive-bred individuals fail to develop the proper skin microbiota populations required to withstand the context-specific stresses following release into native Wyoming habitats. In other words, these individuals may be “too sterile” to survive in the wild. The establishment of a context-appropriate, beneficial skin microbiome for these toads to develop may improve the success of captive-bred species reintroduction conservation efforts (West et al., 2019; Dallas and Warne, 2023).
Little is known about the skin microbiome of adult Wyoming toads, let alone during the early stages of development. To address this gap in the field, the U.S. Fish and Wildlife Service (USFWS) and United States Geological Survey (USGS) support ongoing projects assessing the in situ Wyoming toad microbiome (Walters and Chalfoun, 2023). Here, we aimed to complement the ongoing efforts by the USFWS and USGS by characterizing the microbiome of the captive Wyoming toad breeding population (both adults and tadpoles) at Omaha’s Henry Doorly Zoo and Aquarium (OHDZA; Omaha, Nebraska). These data provide a robust characterization of the host-associated microbiome of the Wyoming toad throughout its development and will serve as the basis for future microbiome-based efforts to improve the success of future reintroduction efforts.
2 Materials and methods
2.1 Toads
An Association of Zoos and Aquariums (AZA) Research Proposal form and protocols were submitted and approved by the OHDZA Animal Care and Use Committee in April 2022. As the animals were owned by the USFWS, a Wyoming Toad Research Proposal was also submitted to the Wyoming Toad Recovery Team and approved by vote at their annual spring meeting (2022). Federal Permitting held under OHDZA allowed for housing and research on this species, in accordance with the Endangered Species Act.
Samples were collected from immature (tadpole) and adult (approximately 1-4 years old) Wyoming toads maintained at OHDZA. Toads are part of a captive colony housed in two isolation rooms (Room 2 and Room 8) located in OHDZA’s Amphibian Conservation Area. The room numbers are specific to the OHDZA, thus we maintained these labels for posterity in future analyses. All adult breeder toads were group-housed in condos (n = 2 toads/condo) that allowed access to a constant filtered water bath as well as terrestrial substrate located in Room 2. As sampling was performed during the Wyoming toad breeding season, females received had received subcutaneous gonadotropin-releasing hormone (2×1mg doses). Tadpoles were also housed in condos connected to the same filtered water system as in Room 2, which has never housed any other amphibian species except for the Wyoming toad. Additional tadpoles were housed in isolation Room 8, which was an empty isolation room prior to the successful spawning events of the 2022 breeding season. Tadpoles were house at a density of 20-2,225 tadpoles/enclosure. All water was from a filtered system in accordance with species regulations, to ensure 0ppm ammonia, <0.05ppm nitrite, <10ppm nitrate, and 68-75°F temperature.
Tadpoles were allowed to mature in Rooms 2 and 8 before being sent to various conservation sites in the Laramie Water Basin, Wyoming for release later in the summer. A small portion of these tadpoles were kept back from release to be used as new breeding stock. Each room was kept in isolation and as a separate environment as to best fit the needs of the species housed there, with handlers changing personal protective equipment (PPE) before entering each room and a work-flow order such that Wyoming toads were handled first by keeper staff throughout the day due to their conservation importance and susceptibility to disease as to not contaminate them with other species housed within the Amphibian Conservation Area. Protocol PPE includes lab coats, nitrile gloves, and rubber boots within the bio secure isolation room.
2.2 Sample collection
Mucosal and cloacal swabs samples were collected from 24 adult toads (n = 12 toads/sex) for a total of 48 samples. Samples were collected from adults using sterile cotton-tipped swabs (Puritan #25-3316-U, Guilford, Maine) using a protocol specific to these animals. Each animal was maintained in a holding condo for the duration of sampling. Prior to any swabbing, they were rinsed front and back with approximately 100 mL of sterile water. Applying uniform pressure each time, the toad was swabbed while rotating the swab back and forth for 10 complete back-and-forth strokes in each of the following areas (in the order listed) for a total of 60 strokes per toad: along the right side of the abdomen, along the left side of the abdomen, along the ventral surface of the right hind leg, along the ventral surface of the left hind leg, along the spine (central dorsal surface) and along the mouth. Swabs were then broken off into sterile 1.5 mL Eppendorf tubes and stored on ice for transportation from the zoo to the lab (approximately 6 hours). Samples were then stored at -20°C until processing.
Cloacal swabs were collected from the adults following mucosal swabbing. Excess skin was pulled away from the cloacal vent opening with the handler’s free hand while the other hand introduced the sterile cotton swab through the cloacal vent three times. The swab tips were again broken off into sterile vials and the samples transported back to the laboratory on ice and stored at -20°C until processing.
Tadpole samples were collected from groups of five (time 1) or three (time 2-3) animals due to their small surface area at three different timepoints. Tadpoles at times 1, 2, and 3 were at Gosner stages 25, 26-35, and 41-46, respectively. Samples were collected from the same colony and living condo by swabbing the tadpoles with the same sterile cotton-tipped swab so as to yield a readable DNA sample while not stripping the animal of all their necessary mucosa. Total sample sizes from each colony were n = 16 (Colony A, Room 2), n = 6 (Colony F, Room 2), n = 8 (Colony O, Room 8), and n = 16 (Colony P, Room 8). Animals were rinsed in a sterile water bath in group sizes as stated above, then moved to a transfer cup to have the water drained off as best as possible. Maintaining even pressure, the animals in each group were sampled with 10 complete back-and-forth strokes using a sterile swab. The swab tips were again broken off into sterile vials and the samples transported back to the laboratory on ice and stored at -20°C until processing.
Longitudinal samples were collected from both tadpole and adult samples. Time points 1 and 2 were collected 15 days apart and time points 2 and 3 were collected 24 days apart from one another.
Biosecurity protocols put in place by the Wyoming Toad SSP and OHDZA’s Amphibian Conservation Area were followed for all Wyoming toad and tadpole handling, manipulating, and sampling. Each round of samples taken from within the Wyoming toad room required new PPE to be worn to ensure no/low pathogen transfer from external sources. Toads were handled one at a time, PIT tags verified for correct identities, and placed back into holding condos as soon as the swabs were complete. Tadpoles were removed from their main holding tanks to smaller volumes of water to facilitate sample collection. While in these smaller volumes of water, temperature remained constant and oxygen was provided from a remote, filtered oxygen source. Tadpole isolation ceased after one hour to ensure water quality parameters remained stable. Care was taken to minimize animal stress by collecting swabs as quickly as possible and closely monitoring the animals after swabbing to ensure their overall welfare remained of the highest quality. Any liquid or solid waste material was thoroughly disinfected with 70% ethanol and/or oxygenated powder bleach mixtures for 12 hours of contact time. Biological samples were stored appropriately and kept separate from any outside influences.
2.3 DNA extraction
DNA was extracted using QIAamp PowerFecal Pro DNA extraction kits (Qiagen) according to the manufacturer instructions with the exception that DNA was eluted in 60 µL of EB buffer (Qiagen). DNA yields were quantified via fluorometry (Qubit 2.0, Invitrogen, Carlsbad, CA) using quant-iT BR dsDNA reagent kits (Invitrogen). Due to the low biomass, all DNA was used for 16S rRNA library preparation.
2.4 16S rRNA library preparation and sequencing
Library preparation and sequencing were performed at the University of Missouri (MU) Genomics Technology Core. Bacterial 16S rRNA amplicons were constructed via amplification of the V4 region of the 16S rRNA gene with universal primers (U515F/806R) previously developed against the V4 region, flanked by Illumina standard adapter sequences (Caporaso et al., 2011; Walters et al., 2011). PCR was performed as 50 µL reactions containing 100 ng metagenomic DNA, dual-indexed forward and reverse primers (0.2 µM each), dNTPs (200 µM each), and Phusion high-fidelity DNA polymerase (1U, Thermo Fisher). Amplification parameters were 98°C(3 min) + [98°C(15 sec) + 50°C(30 sec) + 72°C(30 sec)] × 30 cycles + 72°C(7 min). Amplicon pools were combined, mixed, and then purified by addition of Axygen Axyprep MagPCR clean-up beads to an equal volume of 50 µL of amplicons and incubated for 15 minutes at room temperature. Products were washed multiple times with 80% ethanol and the pellet was resuspended in 32.5 µL EB buffer (Qiagen), incubated for two minutes at room temperature, and then placed on the magnetic stand for five minutes. The final amplicon pool was evaluated using an Advanced Analytical Fragment Analyzer automated electrophoresis system, quantified using quant-iT HS dsDNA reagent kits, and diluted according to the Illumina standard protocol for sequencing as 2×250 bp paired-end reads on the MiSeq instrument.
2.5 Bioinformatics
16S rRNA sequences were processed using Quantitative Insights Into Microbial Ecology 2 (QIIME2) v2021.8 (Bolyen et al., 2019). Illumina adapters and primers were trimmed from forward and reverse reads with cutadapt (Martin, 2011). Reads were then truncated to 150 base pairs, then denoised into unique amplicon sequence variants (ASVs) using DADA2 (Callahan et al., 2016). Unique sequences were then assigned taxonomy using an a sklearn algorithm and the QIIME2-provided 99% non-redundant SILVA v138 reference database (Quast et al., 2013) trimmed to the 515F/806R (Caporaso et al., 2011) region of the 16S rRNA gene.
Following sequence processing, further analyses were performed using R v.4.2.2 (Team, 2022). Alpha diversity metrics (Chao-1 and Simpson Indices) were determined using the microbiome (Lahti and Shetty, 2017) and vegan (Oksanen et al., 2014) libraries. Differences in beta diversity were visualized with principal coordinate analysis (PCoA) using Bray-Curtis (weighted) distances. Briefly, a distance matrix was generated with the vegdist function from the vegan library using a quarter-root transformed feature table. PCoAs were performed using the ape (Paradis and Schliep, 2019) library with a Cailliez correction (Cailliez, 1983). The cladogram was generated using Graphlan v1.1.4 (Asnicar et al., 2015).
2.6 Statistics
Univariate data (reported as mean ± SE) were first tested for normality using the Shapiro-Wilk method, followed by the appropriate parametric or non-parametric test. Whenever possible and practical, multi-factor tests (e.g., two-way analysis of variance, ANOVA) were used. When applicable to data generated tadpoles, nested two-factor analyses were performed using the following modeling: response variable ~ room/colony. Differences in multivariate data were tested using permutational multivariate ANOVA (PERMANOVA) and visualized using PCoAs. PERMANOVAs were performed with 9,999 permutations. Both PERMANOVA and PCoA were performed using weighted (Bray-Curtis) distances. Differential abundance testing was performed using analysis of composition of microbiomes with bias correction 2 (ANCOM-BC2) with a significance threshold of a Benjamani-Hochberg-corrected p < 0.05 (Benjamini and Hochberg, 1995; Lin and Peddada, 2020). Structural zeroes (i.e., taxa present in ≥ 1 group and absent in in ≥ 1 group) were also reported. Pairwise ANCOM-BC2 was utilized as necessary. Microbial community analysis was performed using the open-source R statistical software v4.2.2 (Team, 2022).
3 Results
3.1 Sequencing data pass quality control
All but two samples resulted in usable data, with a total of 8,945,498 high-quality sequence reads, and a mean of 95,165 ± 7,297 sequence reads per sample. Samples from adults returned greater sequence read counts than samples from tadpoles (mean ± SE = 129,198 ± 10,886 in adults, 59,652 ± 6,391 in tadpoles, p < 0.001, Mann-Whitney rank sum test). Testing for group differences in sequencing depth separately within adults and tadpoles, separately, failed to detect any significant differences, although there was a trend toward greater sequence read counts in samples from male adults compared to female adults (Supplementary Figure 1A, p = 0.086, F = 3.0, two-way ANOVA) as well as a trend towards greater reads in mucosal samples compared to cloacal samples (p = 0.095, F = 2.9, two-way ANOVA). No difference in read counts were observed between tadpole colonies (Supplementary Figure 1B).
3.2 Amplicon sequence variant coverage
Denoising paired-end sequences into amplicon sequence variants (ASVs) recovered an average of 62,095 ± 5,824 features per sample ranging from 140 to 229,727 features per sample. As expected from the aforementioned increase in sequencing coverage, more ASVs were detected in adult samples compared to tadpole samples (mean ± SE; 90,908 ± 7,827 in adults, 32,030 ± 6,093 in tadpoles, p < 0.001, Mann-Whitney rank sum test). No significant differences in ASV counts were detected between groups within each age. Given the nature of low biomass samples (i.e., swabs) yielding low feature counts, we estimated the achieved sampling of each community using Good’s coverage. Each sample yielded a Good’s coverage of greater than 99.9%, thus the full, unrarefied feature table (ASV counts per sample) was used in all further analyses.
3.3 Adult and tadpole Wyoming toad microbiomes differ in composition
An initial survey of the entire dataset revealed significant age-associated differences in richness (Chao-1 Index, p < 0.001, Mann-Whitney rank sum test), although these were difficult to interpret in the context of the aforementioned difference in sequencing depth. Within adults, mucosal samples exhibited increased community richness relative to cloacal samples (Figure 1A, p = 0.011, F = 7.0, two-way ANOVA). No sex-dependent effects on sample richness were observed (p = 0.325, F = 0.99, two-way ANOVA). Samples collected from adult males exhibited increased diversity (Simpson Index) relative to females with no effect of sample type (Figure 1B, Sex: p = 0.016, F = 6.3; Sample Type: p = 0.59, F = 0.29, two-way ANOVA). Within tadpole samples, significant differences in community richness were observed between colonies (Room×Colony: p = 0.005, F = 6.1, nested two-factor ANOVA). Within Room 2, samples collected from Colony F exhibited increased richness compared to Colony A (Figure 1C, p = 0.033, Tukey post hoc). No significant differences in alpha diversity were detected between rooms or colonies (Figure 1D).
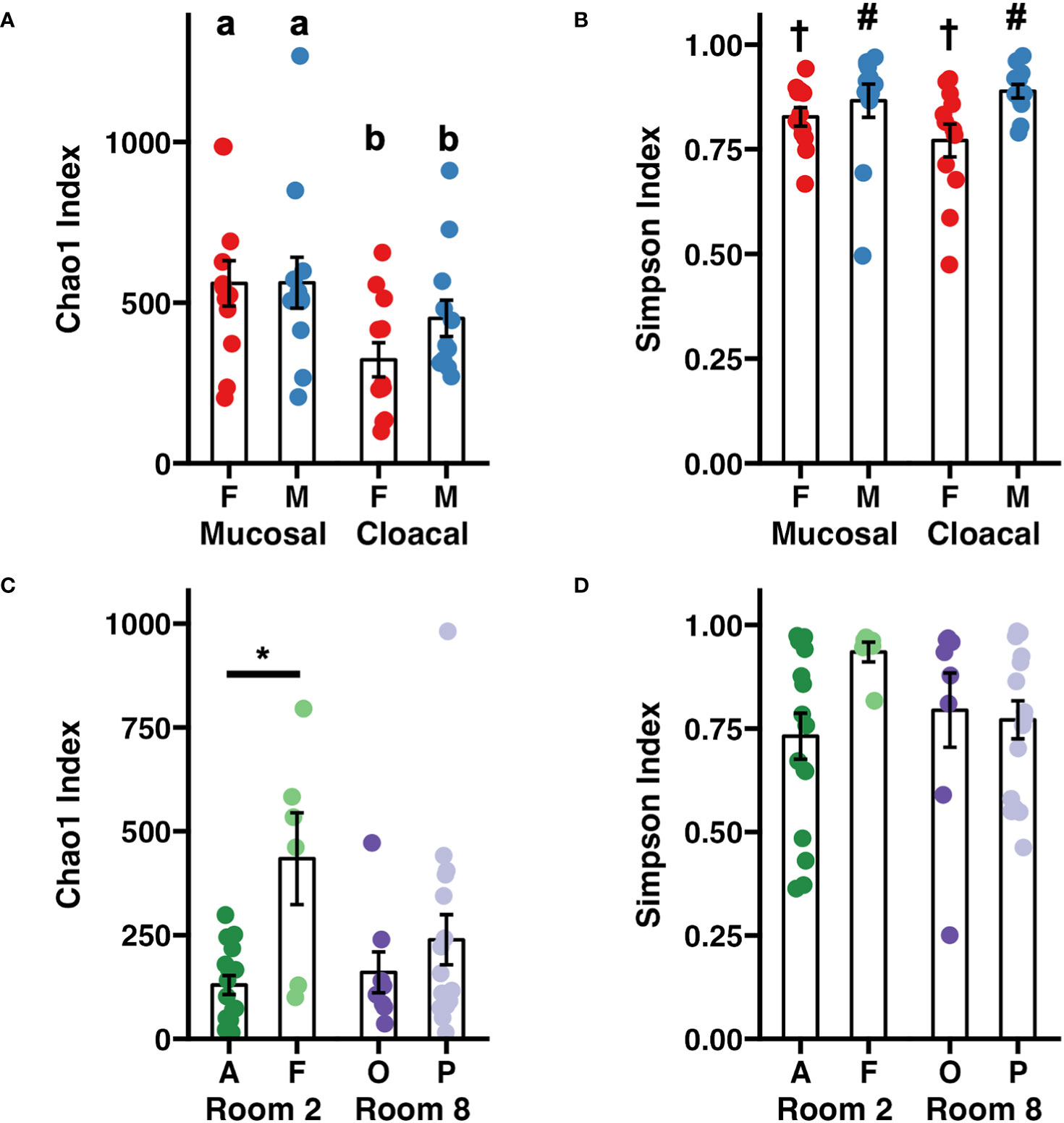
Figure 1 Dot plots depicting adult Wyoming toad (A) richness and (B) diversity and tadpole (C) richness and (D) diversity. Differences between adult samples were assessed using a two-way ANOVA. Differing letters represent sex-dependent differences of p < 0.05. Differing symbols († and #) represent sample type- dependent differences of p < 0.05. Differences between tadpole samples were assessed using a nested two-factor ANOVA. Pairwise differences between tadpole colonies were assessed using a Tukey post hoc test. *p < 0.05. Error bars depict mean ± SE.
Visualization of beta diversity among the entire dataset using principal coordinate analysis (PCoA) with weighted (Bray-Curtis) distances showed complete separation of samples from adults and tadpoles along the first principal coordinate (Figure 2). One-way PERMANOVA comparing adult and tadpole samples using Bray-Curtis distances confirmed a significant difference in community composition between age groups (p < 0.001, F = 22.98). Five dominant (average relative abundance > 1%) bacterial phlya were shared between adults and tadpoles including Actinobacteriota, Bacillota, Bacteroidota, Cyanobacteria, and Pseudomonadota. Verrucomicrobiota was also dominant in tadpole samples. We then identified differentially abundant phyla between adults and tadpoles using analysis of composition of microbiomes with bias correction (ANCOM-BC2). Of the 35 detected phyla, 12 were differentially abundant including Actinobacteriota and Patescibacteria enriched in adults and WPS-2 and Bdellovibrionota enriched in tadpoles. The phyla Synergistota and Fermentibacterota were only detected in adults whereas Hydrogenedentes, Latescibacterota, Modulibacteria, and Halanaerobiaeota were only detected in tadpoles (Supplementary File 1). Collectively, these data demonstrate large age-dependent differences in alpha and beta diversity as well as unique taxonomic signatures within each group. We next stratified the data by age group to better assess the influence of sex and sample site in the adult toads, and room and colony in the tadpoles.
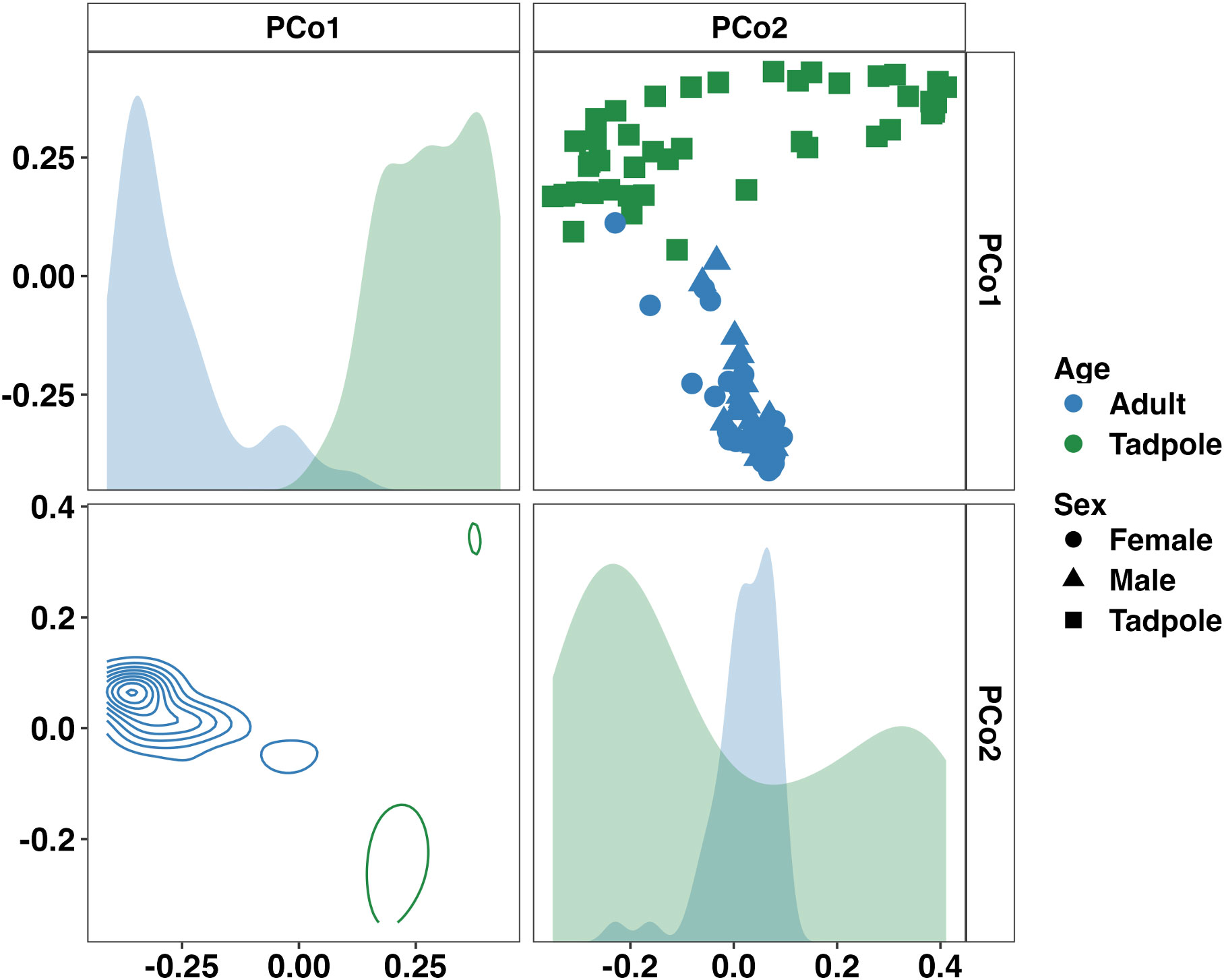
Figure 2 Principal coordinate analysis matrix using weighted Bray-Curtis distances depicting significant differences in community composition between all samples from adult and tadpole Wyoming toads along the first (22.75%) and second (9.06%) principal coordinates. Density plots show distribution of samples along single (diagonal) or multiple axes (lower half). F = 23.0, p < 0.001. One-way PERMANOVA.
3.4 Adult Wyoming toads harbor sex-specific cloacal and mucosal microbiomes
In samples from adults, a sex- and sample type-dependent effect on beta-diversity was observed (Sex: p < 0.001, F = 3.55; Sample Type: p = 0.014, F = 1.92, two-way PERMANOVA). Visualizing these communities using a PCoA revealed a sex-dependent separation along the third principal coordinate (Figure 3). Differential abundance testing using ANCOM-BC2 found only Armatimonadota to be significantly enriched in males (Supplementary Figure 2). Additionally, Fermentibacterota and Fibrobacterota were only detected in males whereas Nitrospirota was only observed in females. Only one, unresolved bacterial taxa was differentially abundant between mucosal and cloacal samples. At the family level, 41 taxa were differentially abundant between males and females (22 and 19 taxa, respectively). Many families within the phylum Pseudomonadota including Alcaligenaceae, Labraceae, and Aeromonadaceae were enriched in males. Deinococcaceae (Phylum Deinococcota), Nitrospiraceae (Phylum Nitrospirota), and multiple families within the phylum Cyanobacteria were enriched in females (Supplementary Figure 2). Only three families were differentially abundant between mucosal and cloacal samples. Mycobacteriaceae and Solirubrobacteraceae (phylum Actinobacteriota) and one unresolved bacteria were enriched in mucosal samples. Additionally, 53 bacterial families were unique to males while 63 were only in females. A full list of differentially abundant taxa at the phylum and family levels in adults is provided in Supplementary File 2. These data demonstrate large sex-dependent differences in the adult Wyoming toad microbiome.
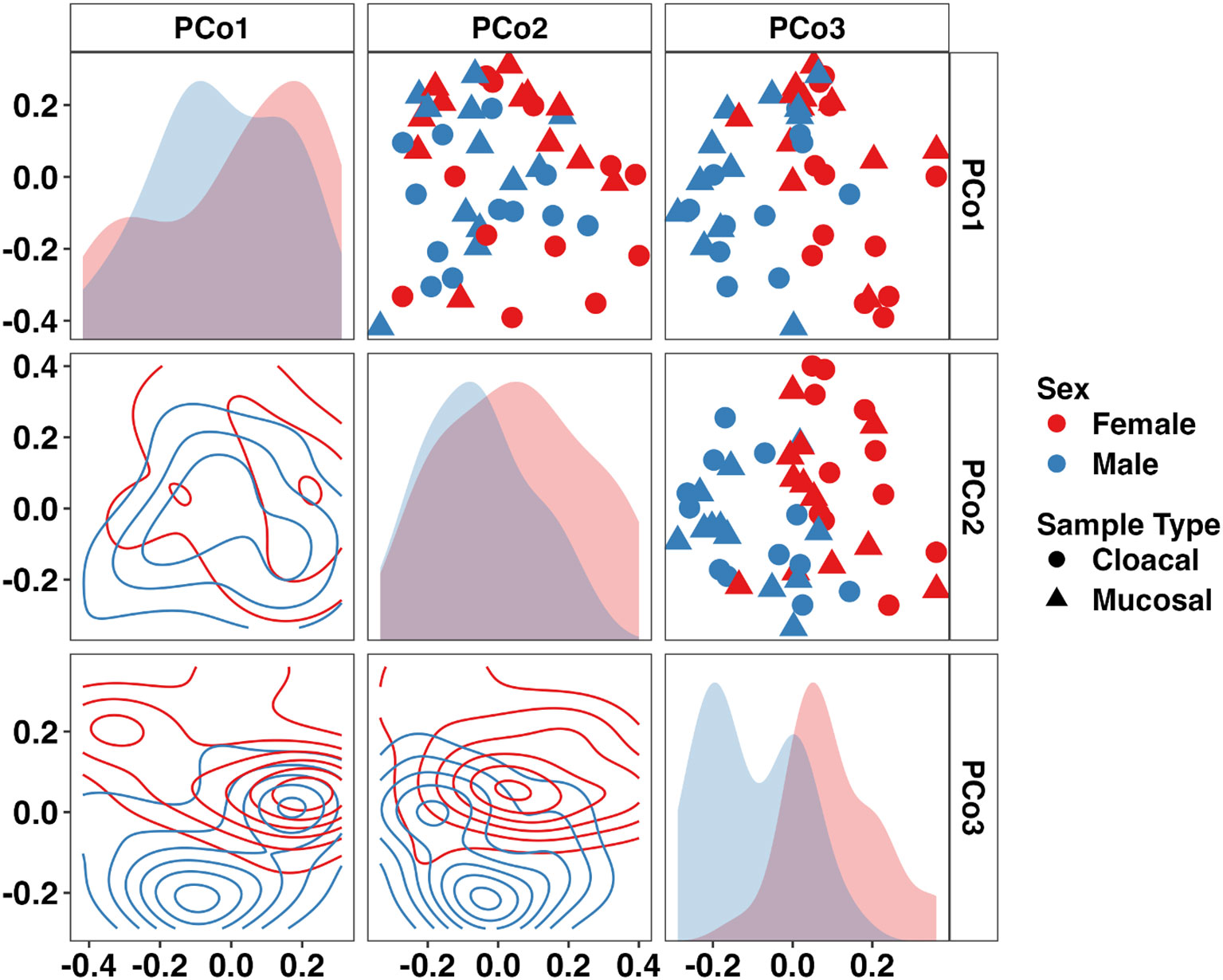
Figure 3 Principal coordinate analysis matrix using weighted Bray-Curtis distances depicting differences in community composition between sex and sample type in adult Wyoming toads along the first (16.31%), second (13.45%), and third (9.85%) principal coordinates. Sex- (p < 0.001, F = 3.6) and sample type-dependent (p = 0.014, F = 1.9) effects on community composition were observed. Density plots show distribution of samples along single (diagonal) or multiple axes (lower half). Two-way PERMANOVA.
3.5 Room-specific husbandry subtly affects the Wyoming toad tadpole microbiome
Focusing on the separate tadpole colonies, we identified significant differences in the weighted microbial composition between colonies but not between rooms (Figure 4, Room: p = 0.053, F = 1.68; Room×Colony: p = 0.021, F = 1.62, nested two-factor PERMANOVA). Pairwise comparisons revealed significant, albeit modest, differences in community composition between Colony A-Colony F (p = 0.044, F = 1.83) and Colony F-Colony P (p = 0.010, F = 2.13). We again applied ANCOM-BC2 at the phylum and family levels to identify differentially abundant taxa within all four colonies. No significant differentially abundant taxa were identified at the phylum or family levels. Twenty of the thirty-three resolved phyla were observed in all four tadpole colonies. Using pairwise ANCOM-BC2 comparisons, five families were identified as enriched, predominantly in Colony F. These families included an uncultured Rhodospirillales and uncultured Alphaproteobacteria (phylum Pseudomonadota), uncultured RBG-13-54-9 (phylum Chloroflexi), Sporichthyaceae (phylum Actinobacteriota), and Pedosphaeraceae (phylum Verrucomicrobiota). A full list of differentially abundant taxa at the phylum and family levels in each tadpole colony is provided in Supplementary File 3.
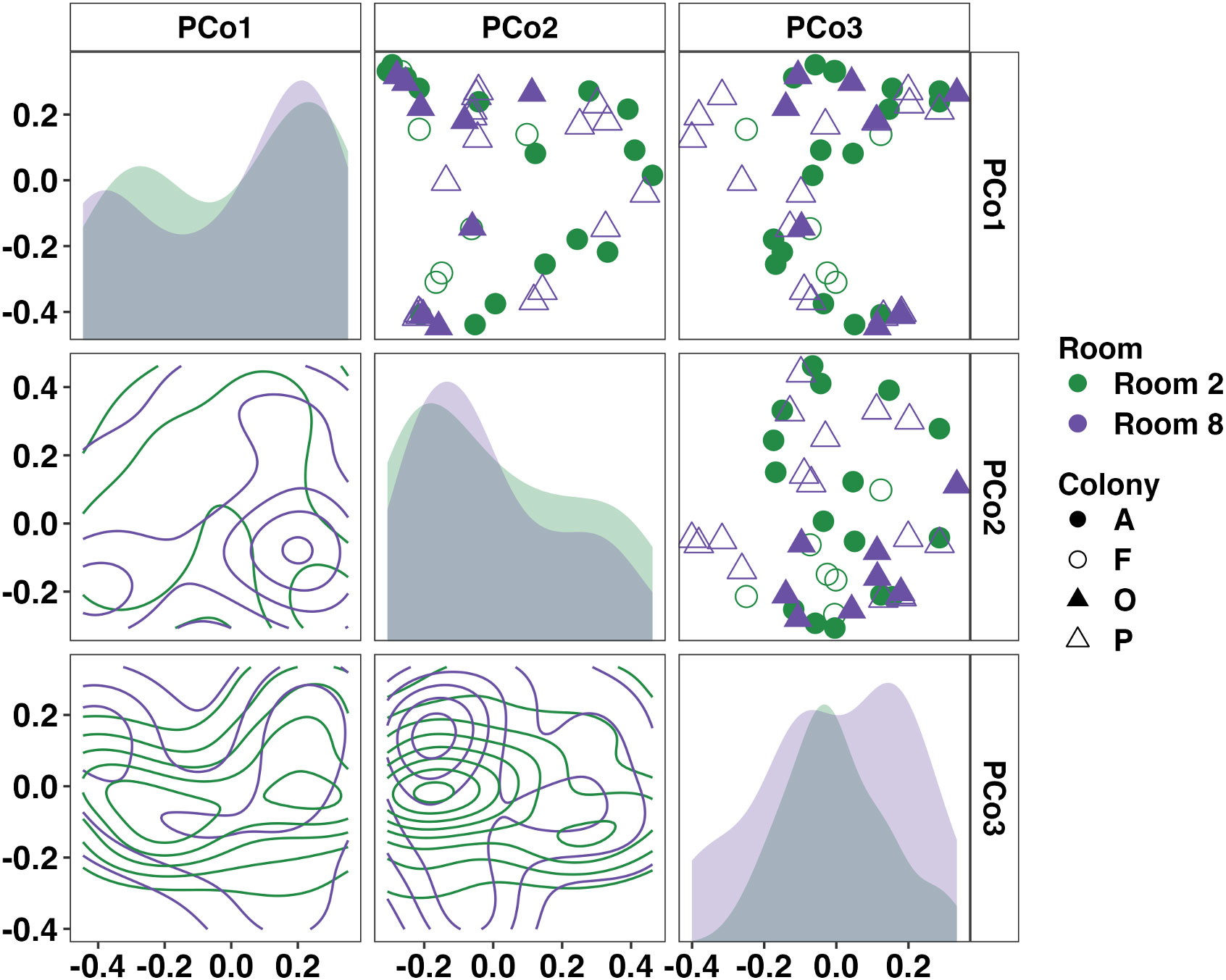
Figure 4 Principal coordinate analysis matrix using weighted Bray-Curtis distances depicting differences in community composition between Wyoming tadpole colonies along the first (19.41%), second (13.43%), and third (7.89%) principal coordinates. Significant colony-dependent differences were observed. Room: p = 0.053, F = 1.7; Room×Colony: p = 0.021, F = 1.6. Density plots show distribution of samples along single (diagonal) or multiple axes (lower half). Nested two-factor PERMANOVA.
3.6 Longitudinal analysis of adult and tadpole Wyoming toads
To characterize the stability of the Wyoming toad microbiome, samples were collected at three separate time points for all adults and 2-3 separate time points for tadpole colonies (Supplementary Figure 3). Given the modest difference in beta diversity between sample types in adults, we combined cloacal and mucosal samples for longitudinal analysis in adults. Two-way PERMANOVA analysis of adult samples revealed no significant effect of time on microbial composition (Sex: p < 0.001, F = 3.6; Time Point: p = 0.123, F = 1.3). Pairwise comparison of every adult sample across time using Bray-Curtis distances supported this as no patterns of dissimilarity were revealed (Figure 5A). When assessing tadpoles, two-way PERMANOVA analysis revealed significant differences in community composition between colonies (p < 0.001, F = 2.1), across time (p < 0.001, F = 5.0), and an interaction of the two (p = 0.001, F = 1.7), however, visualization of the pairwise Bray-Curtis distances revealed high dissimilarity between all samples with no discernable patterns (Figure 5B).
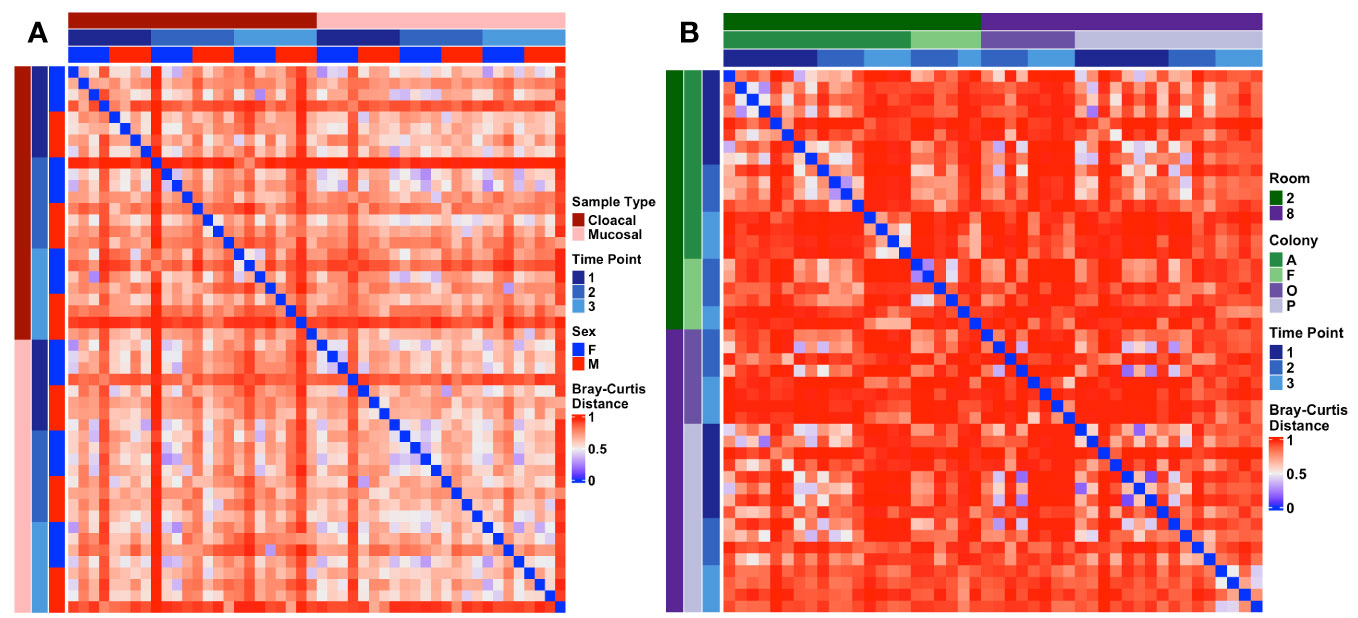
Figure 5 Heatmaps depicting longitudinal Bray-Curtis distances within (A) adult and (B) tadpole samples. Metadata describing sample type and timepoint are displayed as colored bars along the left and top of each heatmap.
4 Discussion
Here we have provided a thorough characterization of the host-associated microbiome of the critically endangered Wyoming toad across stages of development ranging from tadpole to adult. These animals were a part of breeding colonies included in OHDZA’s ongoing efforts to repopulate the Wyoming toad in its native habitat - the Laramie Basin. The microbiomes of adult breeding animals differed in richness, alpha-diversity, and composition from that of tadpole colonies, suggesting a dynamic development of the mucosal microbiome in this amphibian species. Interestingly, we observed novel sex-specific differences in the mucosal and cloacal microbiome of the adult breeding toads. These sex-specific differences may be attributable to the fact that animals were sampled during the Wyoming toad breeding season and females had been receiving an artificial hormone therapy – two factors known to influence the cloacal microbiome of avians (Escallón et al., 2019). While this hormone therapy was administered subcutaneously, future investigations to determine the biological relevance of this therapy on the female mucosal and cloacal microbiome of captive Wyoming toad populations are required.
The utility of our data from captive Wyoming toads is twofold: 1) the conservation of this critically endangered species, and 2) the characterization of the microbial diversity of amphibian host-associated microbiomes throughout development. The immediate use for these data will be their comparison to the microbiome of wild toads surviving at the Laramie Basin release sites. This approach of comparing captive-bred toads to their wild counterparts may support the hypothesis that captive-bred toads do not have a comparable microbiome (e.g., lower richness/diversity, absent taxa) to wild animals leaving them vulnerable to the context-specific challenges of the wild including the environment and temperature (Kueneman et al., 2019). If such differences between captive and wild populations are identified, then these data will guide husbandry practices to maximize the potential for toads to develop a dynamic, complex microbiome ultimately increasing the odds of survival in the wild. Such practices may include the introduction of relevant probiotics or environmental exposures acquired directly from the Laramie Basin.
The timing of such interventions may also be of relevance when considering the developmental stage at which the toads are reintroduced into the wild. Determining at which point in the immature toad’s development a microbiome-based intervention is most effective at improving the odds of adult survival in the wild is critical for future repopulation efforts. This is particularly relevant when considering the development of the host immune system as establishing robust mucosal immunity may improve resistance against many amphibian pathogens including Bd. A recent study evaluating the genetic diversity of Toll-like receptors (TLR) and major histocompatibility complex (MHC) loci in three captive male Wyoming toads found reduced diversity in various TLR and MHCII loci (Carlson et al., 2022). This limited genetic diversity is expected given the small size of breeding colonies of this endangered species; however, it raises concern for susceptibility to various infections for those reintroduced into the wild. Boosting the mucosal immunity (e.g., secreted immunoglobulins) via the microbiome may provide the enhanced immunity required to overcome the lack of genetic diversity in certain immune-related genes.
A limitation of the current study was that the size of developing Wyoming toad tadpoles prevented us from sampling individual subjects without stripping them of their necessary mucosa. Pooling multiple tadpoles allowed us to achieve a necessary sample for DNA extraction and sequencing but prevented us from tracking the development of the skin-associated microbiome at the individual level. Our data, however, provide novel insight into the development of the Wyoming toad tadpole microbiome at a colony and population level. The dynamic morphological changes a toad undergoes between the tadpole and adulthood were complemented by equally dynamic shifts in host-associated microbial composition (Figure 5, Supplementary Figure 3). Our data reveal that, at population level, the microbiome of Wyoming toad tadpoles is distinct from their adult counterparts.
Our goal was to document the host-associated microbiome development of the captive Wyoming toad, and we have provided a comprehensive characterization of this species from the tadpole stage through adulthood. These efforts complement similar ongoing investigations of wild populations in Laramie Basin area. The novel comparison of datasets generated from captive-bred amphibians and their wild counterparts will be of great value to the field as the awareness of microbiome-based efforts to improve repopulation success of the Wyoming toad in the wild is increasing. Ultimately, the goal is to increase the indigenous population of the Wyoming toad in its native land, however, the data presented here will advance repopulation efforts of other amphibian species serving critical roles in their native ecosystems.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: BioProject, PRJNA943179.
Ethics statement
The animal study was approved by OHDZA Animal Care and Use Committee. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
NS: Data curation, Formal analysis, Methodology, Writing – original draft, Writing – review & editing. ZM: Data curation, Formal analysis, Visualization, Writing – original draft, Writing – review & editing. DB: Conceptualization, Data curation, Methodology, Writing – original draft, Writing – review & editing. JH: Conceptualization, Data curation, Methodology, Writing – original draft, Writing – review & editing. BCM: Conceptualization, Data curation, Methodology, Writing – original draft, Writing – review & editing. AE: Conceptualization, Data curation, Formal analysis, Methodology, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. We thank the University of Missouri College of Veterinary Medicine’s Veterinary Research Scholars Program (VRSP) for program support and mentorship. Stipend support for NS was provided by the Morris Animal Foundation through the Veterinary Student Scholars grant.
Acknowledgments
We would like to thank the staff at Omaha’s Henry Doorly Zoo and Aquarium’s Amphibian Conservation center for their support of this project, including J. Krebs. We would also like to thank the keeper staff at the Amphibian Conservation Center for their role in sample collection and ensuring animal husbandry and welfare needs were met.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/famrs.2023.1329897/full#supplementary-material
Supplementary Figure 1 | Forward read counts recovered from 16S rRNA sequencing of (A) adult and (B) tadpole Wyoming toad samples. Differences between adult samples were assessed using a two-way ANOVA. Sex: p = 0.086, F = 3.1; Sample Type: p = 0.095, F = 2.9. Differences between tadpole samples were assessed using a nested two-factor ANOVA. Room: p = 0.364, F = 0.84; Room×Colony: p = 0.172, F = 1.8.
Supplementary Figure 2 | Family-level cladogram of taxa resolved in adult samples. Rings represent (from innermost out) Phylum-level classification, differentially abundant phyla, and differently abundant families between sexes. DA: Differentially abundant by significance (Benjamini-Hochberg corrected p < 0.05). SZ: Structural zero determined by presence/absence. ANCOM-BC2.
Supplementary Figure 3 | Representative images of Wyoming toad tadpoles at the three collection timepoints. (A) Time Point 1: 1-5 days post hatch. (B) Time Point 2: Midpoint Evaluation (C) Time Point 3: 2 days post metamorphosis.
References
Asnicar F., Weingart G., Tickle T. L., Huttenhower C., Segata N. (2015). Compact graphical representation of phylogenetic data and metadata with GraPhlAn. Peerj 3, e1029. doi: 10.7717/peerj.1029
Barnhart K., Forman M. E., Umile T. P., Kueneman J., McKenzie V., Salinas I., et al. (2017). Identification of bufadienolides from the boreal toad, anaxyrus boreas, active against a fungal pathogen. Microbial Ecol. 74, 990–1000. doi: 10.1007/s00248-017-0997-8
Benjamini Y., Hochberg Y. (1995). Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol 57, 289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
Bolyen E., Rideout J. R., Dillon M. R., Bokulich N. A., Abnet C. C., Al-Ghalith G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. doi: 10.1038/s41587-019-0209-9
Cailliez F. (1983). The analytical solution of the additive constant problem. Psychometrika 48, 305–308. doi: 10.1007/bf02294026
Callahan B. J., McMurdie P. J., Rosen M. J., Han A. W., Johnson A. J., Holmes S. P. (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Caporaso J. G., Lauber C. L., Walters W. A., Berg-Lyons D., Lozupone C. A., Turnbaugh P. J., et al. (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. 108, 4516–4522. doi: 10.1073/pnas.1000080107
Carlson K. B., Wcisel D. J., Ackerman H. D., Romanet J., Christiansen E. F., Niemuth J. N., et al. (2022). Transcriptome annotation reveals minimal immunogenetic diversity among Wyoming toads, Anaxyrus baxteri. Conserv. Genet. 23, 669–681. doi: 10.1007/s10592-022-01444-8
Comizzoli P., Power M. L., Bornbusch S. L., Muletz-Wolz C. R. (2021). Interactions between reproductive biology and microbiomes in wild animal species. Anim. Microbiome 3, 87. doi: 10.1186/s42523-021-00156-7
Dallas J. W., Warne R. W. (2023). Captivity and animal microbiomes: potential roles of microbiota for influencing animal conservation. Microb. Ecol. 85, 820–838. doi: 10.1007/s00248-022-01991-0
Escallón C., Belden L. K., Moore I. T. (2019). The cloacal microbiome changes with the breeding season in a wild bird. Integr. Org Biol. 1, oby009. doi: 10.1093/iob/oby009
Jiménez R. R., Alvarado G., Estrella J., Sommer S. (2019). Moving beyond the host: unraveling the skin microbiome of endangered Costa Rican amphibians. Front. Microbiol. 10. doi: 10.3389/fmicb.2019.02060
Kueneman J. G., Bletz M. C., McKenzie V. J., Becker C. G., Joseph M. B., Abarca J. G., et al. (2019). Community richness of amphibian skin bacteria correlates with bioclimate at the global scale. Nat. Ecol. Evol. 3, 381–389. doi: 10.1038/s41559-019-0798-1
Lahti L., Shetty S. (2017). Tools for microbiome analysis in R. Version 2.1.26. Available at: http://microbiome.github.com/microbiome.
Lewis D. L., Baxter G. T., Johnson K. M., Stone M. D. (1985). Possible Extinction of the Wyoming Toad, Bufo hemiophrys baxteri. J. Herpetology 19, 166–168. doi: 10.2307/1564434
Lin H., Peddada S. D. (2020). Analysis of compositions of microbiomes with bias correction. Nat. Commun. 11, 3514. doi: 10.1038/s41467-020-17041-7
Martin M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet J. 17, 10–12. doi: 10.14806/ej.17.1.200
Oksanen J., Blanchet F. G., Kindt R., Legendre P., Minchin P. R., O’Hara R. B., et al. (2014) Vegan: Community Ecoloy Package. R package version 2.2-0. Available at: https://github.com/vegandevs/vegan.
Paradis E., Schliep K. (2019). ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528. doi: 10.1093/bioinformatics/bty633
Polasik J. S., Murphy M. A., Abbott T., Vincent K. (2016). Factors limiting early life stage survival and growth during endangered Wyoming toad reintroductions. J. Wildl Manage. 80, 540–552. doi: 10.1002/jwmg.1031
Quast C., Pruesse E., Yilmaz P., Gerken J., Schweer T., Yarza P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Team, R. D. C (2022). R: A Language and Environment for Statistical Computing. Available at: http://www.R-project.org.
USGS. (2001). Population and habitat viability assessment for the Wyoming toad (Bufo baxteri): Final workshop report (Apple Valley, MN: Conservation Breeding Specialist Group). Available at: http://pubs.er.usgs.gov/publication/70159722.
Vincent K., Abbott T. (2015). First Revised Recovery Plan for Wyoming Toad. Available at: https://ecos.fws.gov/docs/recovery_plan/First%20Revised%20Recovery%20Plan%20for%20Wyoming%20Toad_07.16.2015.pdf.
Vitt L. J., Caldwell J. P., Wilbur H. M., Smith D. C. (1990). Amphibians as harbingers of decay. BioScience 40, 418–418. doi: 10.1093/bioscience/40.6.418
Walters W. A., Caporaso J. G., Lauber C. L., Berg-Lyons D., Fierer N., Knight R. (2011). PrimerProspector: de novo design and taxonomic analysis of barcoded polymerase chain reaction primers. Bioinformatics 27, 1159–1161. doi: 10.1093/bioinformatics/btr087
Walters A., Chalfoun A. (2023). Research and Monitoring of Wyoming Toad Reintroductions: Linking Survival, Behavior and Genetics to Inform Species Recovery. Available at: https://www1.usgs.gov/coopunits/project/170219780096/awalter8 (Accessed June 5, 2023).
Keywords: Wyoming toad, microbiome, 16S rRNA, adult toad, toad tadpole
Citation: Scarberry N, McAdams ZL, Benson D, Herrick J, Moore BC and Ericsson AC (2024) Longitudinal characterization of the captive adult and tadpole Wyoming toad (Anaxyrus baxteri) microbiome. Front. Amphib. Reptile Sci. 1:1329897. doi: 10.3389/famrs.2023.1329897
Received: 30 October 2023; Accepted: 20 December 2023;
Published: 11 January 2024.
Edited by:
Douglas Woodhams, University of Massachusetts Boston, United StatesReviewed by:
Yan-Fu Qu, Nanjing Normal University, ChinaJason Dallas, Middle Tennessee State University, United States
Copyright © 2024 Scarberry, McAdams, Benson, Herrick, Moore and Ericsson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aaron C. Ericsson, ZXJpY3Nzb25hQG1pc3NvdXJpLmVkdQ==
†These authors have contributed equally to this work