 Shuangshuang Liu1,2
Shuangshuang Liu1,2 Jingxiao Fu
Jingxiao Fu Meiai Zhao
Meiai Zhao- 1Key Lab of Plant Biotechnology in Universities of Shandong Province, College of Life Sciences, Qingdao Agricultural University, Qingdao, China
- 2Key Laboratory of Qingdao Major Crop Germplasm Resource Innovation and Application, Qingdao, China
- 3College of Agronomy, Qingdao Agricultural University, Qingdao, China
Stalk rot causes significant yield loss in maize worldwide. The breeding of resistant variety is a potential way to effectively control the disease. This study aims to dissect resistance genes for maize stalk rot caused by Fusarium graminearum using an integrated gene mapping method. A diversity panel of 165 inbred lines and an F2 population from the hybridization of CDMA66 and Huangzao 4 were used as materials. The 165 inbred lines were clustered into four subgroups, of which tropical materials are in the majority. Through combining disease severity index (DSI) and single nucleotide polymorphisms (SNPs) of Maize 50K chip and 20K, genome-wide association analysis (GWAS) revealed 34 SNPs that were significantly associated with stalk rot in maize (P < 0.001). These SNPs were distributed on chromosomes 1, 3, 4, 6, 8, and 10, of which the loci on chromosomes 4 and 8 were confirmed by the mapped QTLs for stalk rot. Finally, candidate genes were identified including GRMZM2G082709 which encoded NAC domain-containing protein and thioredoxin reductase (GRMZM5G841142). However, LRR receptor-like serine/threonine-protein kinase (GRMZM2G080041) was down-regulated after inoculation. These SNPs and candidate genes identified would provide essential information for resistant gene cloning and molecular breeding of anti-stalk rot variety in maize.
Introduction
Stalk rot is a destructive disease in maize and occurs worldwide. It has been prevalent in China for decades and causes extensive economic losses (Song et al., 2015). The genetic mechanism of maize stalk rot is complex. It is caused by one or more fungal pathogen(s). The causative agents, including Fusarium graminearum, Fusarium verticillioides, Fusarium moniliforme, Fusarium proliferatum, are soil-borne fungus, indicating it is difficult to control stalk rot. Once stalk rot occurs, manual harvest has to be conducted due to the lodging of plants. Thus, it is necessary to understand the genetic basis of stalk rot and to breed resistant varieties.
A genome-wide association study (GWAS) has been proven to be an effective method in identifying the genes, alleles, or heliotypes associated with agronomic traits. Moreover, many quantitative trait loci (QTL) or genomic regions that confer resistance to important maize diseases have been detected by the method (Gowda et al., 2015). Exploring the genome-wide association analysis between SNPs and phenotypes of a desired trait allows the identification of site for quantitative traits (Brachi et al., 2011).
The stalk rot resistance in maize is controlled by polygene and the additive genetic effects among the genes (Wang et al., 2017). Till now, a number of quantitative trait loci (QTLs) associated with maize stalk rot have been identified. For example, Pè et al. (1993) used an F2:3 population from B87 and 33-16 to detect the QTLs for stalk rot and found 5 QTLs that were located on chromosomes 1, 3, 4, 5, and 10, respectively. Yang et al. (2010) detected a major QTL-qRfg1 with a back cross population from 1145 and Y331. The QTL was located on Chromosome 10 and explained that 36.3% of the phenotype. A gene ZmCCT was further cloned from this region and the epigenetic regulation mechanism was studied (Wang C. et al., 2017). Zhang et al. (2012) narrowed the physical distance of a micro QTL-qRfg2 on bins 1.09/10–300 kb. Ye et al. (2019) cloned a pathogenic gene ZmAuxRP1 at a minor QTL-qRfg2 site and proved that the gene can promote the biosynthesis of indole-3-acetic acid (IAA). Ma et al. (2017) used a RIL population from H127R and C7-2 to map QTLs for maize stalk rot and explored a major QTL-qRfg3 on Chromosome 3, explaining 10.7–19.4% of the phenotype variation. Jung et al. (1994) found a resistant gene Rcg1 on Chromosome 4 for anthracnose stalk rot. The gene encodes NBS-LRR protein, which plays an important role in the pathogen recognition (Frey, 2005; Abad et al., 2006). Chen et al. (2017) identified a resistant QTL (QTL-Rgsr8.1) associated with maize Gibberella stalk rot and located it between 164.69 Mb and 166.72 Mb on Chromosome 8 by the whole-genome resequencing-based approach. Further, two candidate-resistant genes were found in this region. One was responsed to auxin and the other encoded a disease resistance protein. At present, many QTLs (genes) have been reported, but no consistent QTL (gene) is able to be used in the molecular breeding of stalk rot in maize.
In this study, based on 165 maize inbred lines, we performed genetic diversity analysis, GWAS, and linkage analysis and performed phenotypic analysis through the inoculation of stalk rot pathogen on maize plants in the field. Finally, we obtained the candidate resistant genes of stalk rot, which is of great significance for further breeding of maize stalk rot resistant varieties.
Materials and Methods
Materials and Phenotyping
A set of 165 maize inbred lines (Supplementary Table 1), including resources from the International Maize and Wheat Improvement Center (CIMMYT) and China, were used as materials in this study. These inbred lines were collected or bred by the Maize Molecular Breeding Team, Qingdao Agriculture University, China.
The trials were conducted under artificial inoculation using the injection method. Fusarium graminearum were cultured on petri dish including PDA agar medium in the dark at 26°C. A week before inoculation, mung beans were put into boiling water for 10 min; after filtering with gauze, mung bean medium were transferred into flasks for sterilization at 121°C for 30 min. 5–6 Fusarium graminearum clumps with a diameter of about 1 cm were inoculated into 100 ml of mung bean medium. The clumps were shaken at 180 rpm for 5–7 days at 26°C. When conidia were produced, they were filtered and ready for field inoculation. Aseptic conditions were maintained during all the preparation steps. At the tassel emergence stage, 1 mL of inoculum was injected into the second internode of the plant using a sterile syringe and needle after making a 2 mm hole, and the inoculation sites were sealed with tape for preventing the contamination. The control plants were mock-inoculated with sterilized water. The pathogen, kindly provided by Pathologist Professor Yan in Qingdao Agricultural University, was derived from a single isolate. Randomized complete block with three repetitions were adopted in the experiment. Each inbred line was planted in a row, 20 plants per row. The row length was 4 m, the adjacent row spacing was 0.6 m, and the plants within a row were 0.2 m apart from each other.
Plants showing typical stalk rot symptoms at the last milk stages were scored. There are 20 plants scored in each row. The infected plants initially appear brown at lower internodes and stalks, and then wilt, lodge, or even die. After being scored, every investigated plant was longitudinally cut into pieces to assess the mycelia. Disease incidence had six grades: 1, 2, 3, 4, 5, and 6. Grades 1–3 were regarded as resistant materials and 4–6 were regarded as susceptible materials (Yang et al., 2010). The grade values were then converted into the disease severity index (DSI) with following formula: DSI (%) = [sum (class frequency × score of rating class)] / [(total number of plants) × (maximal disease index)] × 100 (Chiang et al., 2017). We conducted experiments in Laiyang in 2017 and 2019, using different plots, and the environment in these 2 years was quite different. Therefore, we treat it as two environments. In order to eliminate the influence of environmental effects and genetic effects and improve the accuracy of the results, we used the lme4 package of R software (https://www.r-project.org) to fit the BLUE (best linear unbiased estimates) value. The mixed linear model was used for correcting these phenotypes.
Genetic Diversity Analysis
A total of 5,585 SNP markers were obtained by comparing maize 50K genotypes and 20K genotypes to the same version of maize B73 reference genome. These SNP sites were then filtered with parameters of missing rate >20%, heterozygosity >20%, and minor allele frequency (MAF) <0.05. At last, 4,666 SNP markers were chosen for GWAS analysis. Linkage disequilibrium (LD) analysis was performed for these SNP markers among 165 inbred lines by Tassel 5.2.3 (Bradbury et al., 2007). The population structure analysis of these inbred lines was conducted by Structure V. 2.3.4 (Pritchard et al., 2000; Evanno et al., 2005), and the principal component analysis (PCA) and genotype analysis were performed by GAPIT (Lipka et al., 2012).
GWAS Analysis
Based on the phenotype and genotype of 165 inbred lines, GWAS was performed to screen the significant SNP markers among the population by Fixed and random model Circulating Probability Unification (FarmCPU) (Liu, 2015). The FarmCPU model alternately uses a fixed-effects model and a random-effects model to control the confounding problem of the model, thereby better controlling the false-positive and false-negative problems, thus greatly improving the statistical power (Liu et al., 2016). The significance of the association between SNP markers and DSI was determined with a less stringent threshold of –log10(P)>3.5, similar to the approach adapted in the references (Phan et al., 2018).
QTL Mapping in a Genetic Linked Population
A stalk rot-resistant inbred line CDMA66 (♀) was hybridized with a stalk rot susceptible inbred line Huangzao4 (♂); the generated F1 and F2 and their parental lines were planted in Laiyang, Shandong, for stalk rot evaluation in the summer of 2018. At the silking stage, 350 well-grown F2 plants were inoculated by Fusarium graminearum by the injection method. The genomic DNA of these inoculated plants was extracted using cetyltrimethylammonium ammonium bromide (CTAB) method (Chen and Ronald, 1999). At the mature stage, the disease grade of each plant was investigated according to the method in section Materials and Phenotyping, and then the DSI was calculated.
The polymorphism markers between CDMA66 and Huangzao4 used for genetic linkage map construction were screened from 985 SSRs that covered 10 maize chromosomes. The linkage map was constructed using the software IciMapping V3.2 (Wang J. L et al., 2012). Inclusive Composite Interval Mapping (ICIM) (Li et al., 2008) was used for QTL mapping.
Candidate Gene Mining and Analysis
With the genomic sequence of the Maize B73 RefGen_v3 as a reference genome, candidate genes were chosen based on the SNP loci that were significantly associated with stalk rot (Schnable et al., 2009). The chosen candidate genes were confirmed at MaizeGDB (https://www.maizegdb.org/). The SNP loci that were significantly associated with stalk rot (P < 0.001) were annotated at MaizeGDB4 (https://www.maizegdb.org/) and NCBI5 (https://www.ncbi.nlm.nih.gov/).
Gene Expression Analysis
Two candidate genes' expression levels in CDMA66 and Huangzao4 were further analyzed using qRT-PCR at time points of 12, 24, 48, and 72 h after inoculation. Total RNA was extracted using an RNAiso kit (TaKaRa, Japan) according to the manufacturer's instructions. Single-strand cDNA was synthesized with a reverse transcription kit (TaKaRa, Japan). Quantitative RT-PCR assays were performed with SYBR Premix Ex Taq (Accurate Biology, China) on a T100™ Thermal Cycler (BIO-RAD). The primers used for qRT-PCR are listed in Supplementary Table 2. The temperature cycling conditions were 94°C for 30 s, then 40 cycles of 95°C for 10 s and 55°C for 10 s and 72°C for 30 s. With Actin1 as an inference gene, the relative expression of interest genes was determined using the 2−ΔΔCt method (Zhang et al., 2017). The qRT-PCR experiment was independently repeated at least twice, with each sample run in triplicate, and a representative data set was shown.
Results
Phenotype Statistics
The DSI information of stalk rot of 165 maize inbred lines evaluated in 2017 and 2019 is presented in Table 1 and Figure 1. The BLUE (best linear unbiased estimates) value is the corrected phenotype value. The DSI values were within the range of 0.14–1 in 2017 and within 0.05–1 in 2019. The absolute values of kurtosis and skewness of these indexes were both <1. That is, the phenotype data were suitable for QTL mapping.
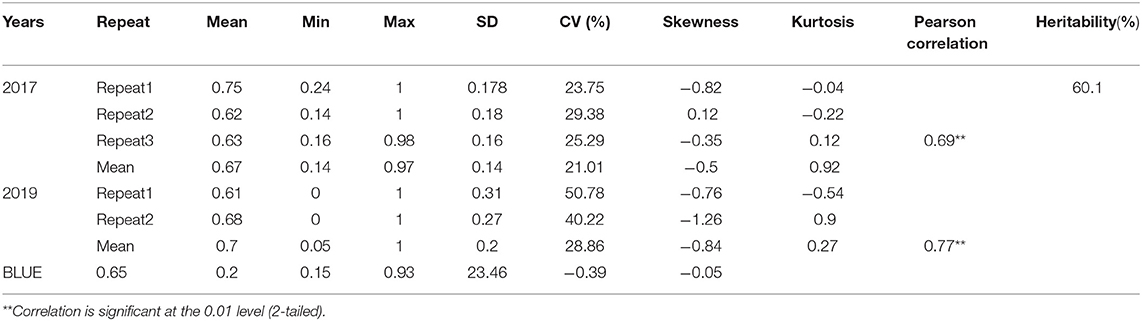
Table 1. Statistical summary of phenotypes of 165 maize inbred lines.
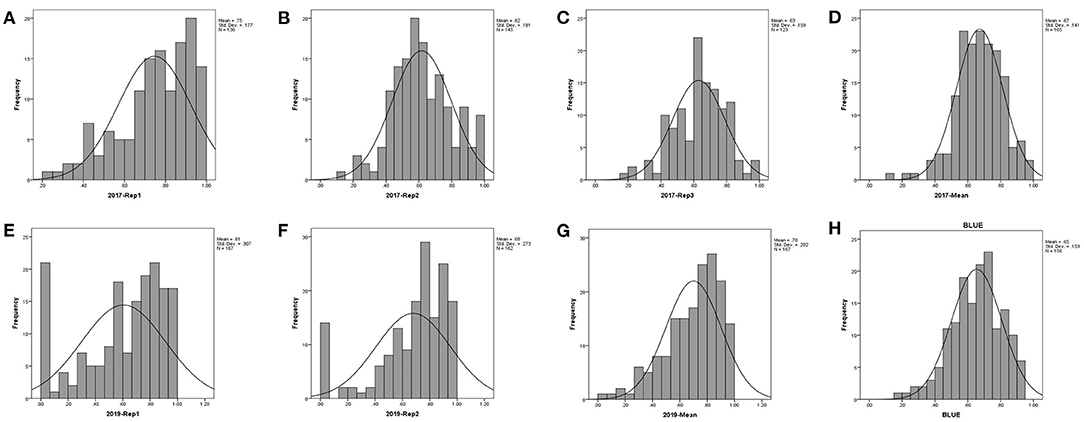
Figure 1. Histogram of the stalk rot disease index for 165 maize inbred lines. (A–D) repeat 1, repeat 2, repeat 3, and mean in 2017; (E–G) repeat 1, repeat 2, and mean in 2019, respectively; (H) is the BLUE value in 2 years).
Genetic Diversity Analysis
We established a correlation map to estimate the genome-wide LD levels of 4,666 SNPs among the 165 inbred lines. LD decayed differently in the 10 chromosomes of maize, with chromosomes 2 and 10 having the most and least decay rates, respectively (Figure 2A). When the cutoff value of r2 was set to 0.14, the averaged LD decay distance of 165 inbred lines was approximately 300 kb. Structure software was used to calculate the K value. ΔK reached its maximum at K = 2 and got its secondary maximum value at K = 4 (Figure 2B). When K = 2, the inbred lines were grouped into temperate and tropical groups and the tropical materials are in the majority. When K = 4, the materials were divided into four subgroups, namely, Lvdahonggu, Tangsipingtou, tropical material group, and P group (Figure 2C). The population grouping of maize inbred lines were analyzed using the principal component analysis (PCA) method, and they were well-separated by the first principal component (PC1), the second principal component (PC2), and the third principal component (PC3) (Figure 2D). PC1, PC2, and PC3 explained 26.2, 9.6, and 2.4% of the total variation, respectively, and the cumulative value was 38.2%.
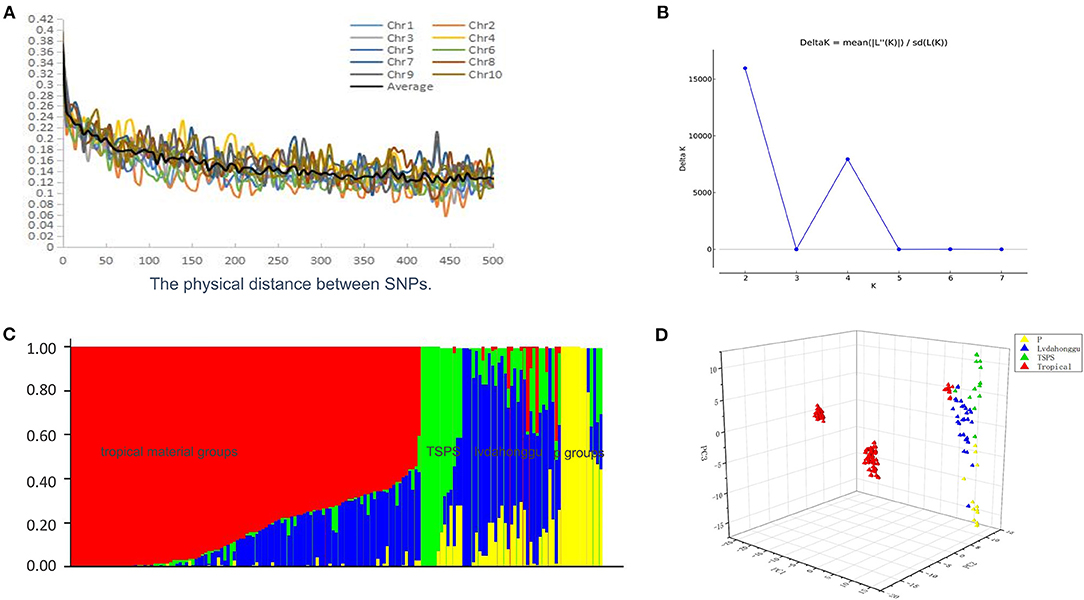
Figure 2. Genetic diversity analysis. (A) Whole-genome LD in the entire panel based on maize inbred lines. (B) ΔK value of maize inbred lines based on 2538 SNPs. (C) Bayes cluster plot of 165 maize inbred lines when K = 4. (D) Principal component analysis based on maize groups.
GWAS Analysis
Filtering the genetic data by quality criteria before GWAS analysis improved the reliability of the results. The filtered genotype showed a low level of the heterozygosity for individuals and markers (Figure 3). The association analysis between selected SNPs and the stalk rot traits was performed using FarmCPU to explore associated loci and allelic variation. The results were shown in Figure 4. Quantile–quantile (Q–Q) plots implied that the population structure and genetic relationship of 165 inbred lines were well-controlled. A total of 19 SNPs that were closely related to stalk rot in maize were detected. These SNPs were distributed on chromosomes 1, 2, 3, 4, 6, 7, 8, and 10 (Table 2). Among them, 3 SNP loci were detected on each of chromosomes 1, 2, and 6; 2 SNP loci were detected on chromosomes 3 and 8; 4 SNP loci were detected on chromosomes 4; and 1 SNP locus was detected on chromosomes 7 and 10.
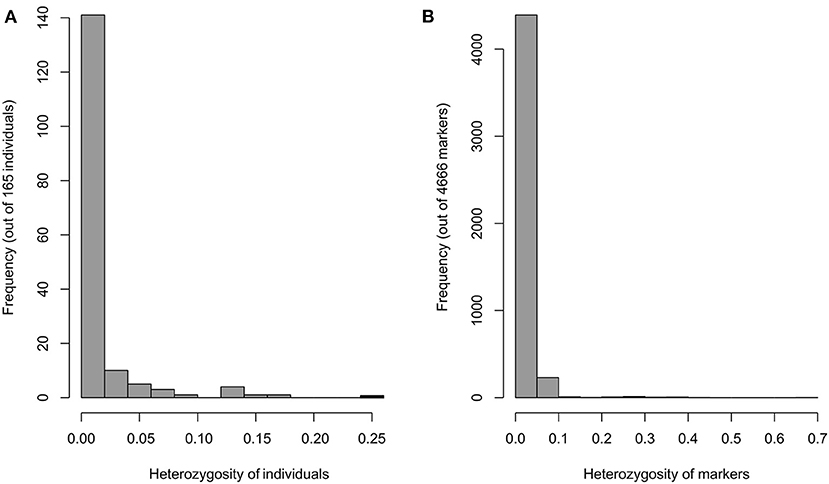
Figure 3. Heterozygosity 4666 SNP markers in the 165 inbred lines. (A) Heterozygosity of individuals. (B) Heterozygosity of markers.
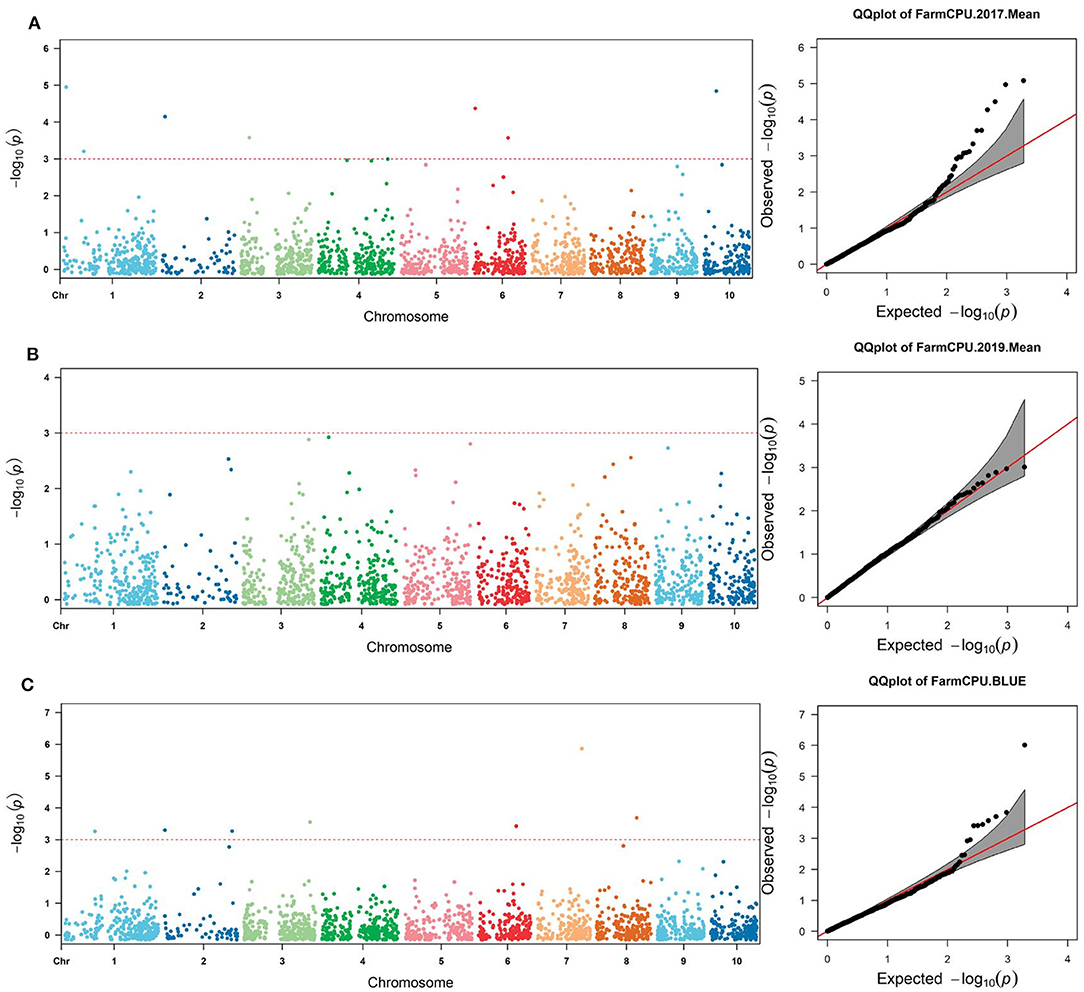
Figure 4. Manhattan plots and (Q–Q) plots of 165 inbred lines resistant to stalk rot [(A) 2017; (B) 2019; (C): BLUE].
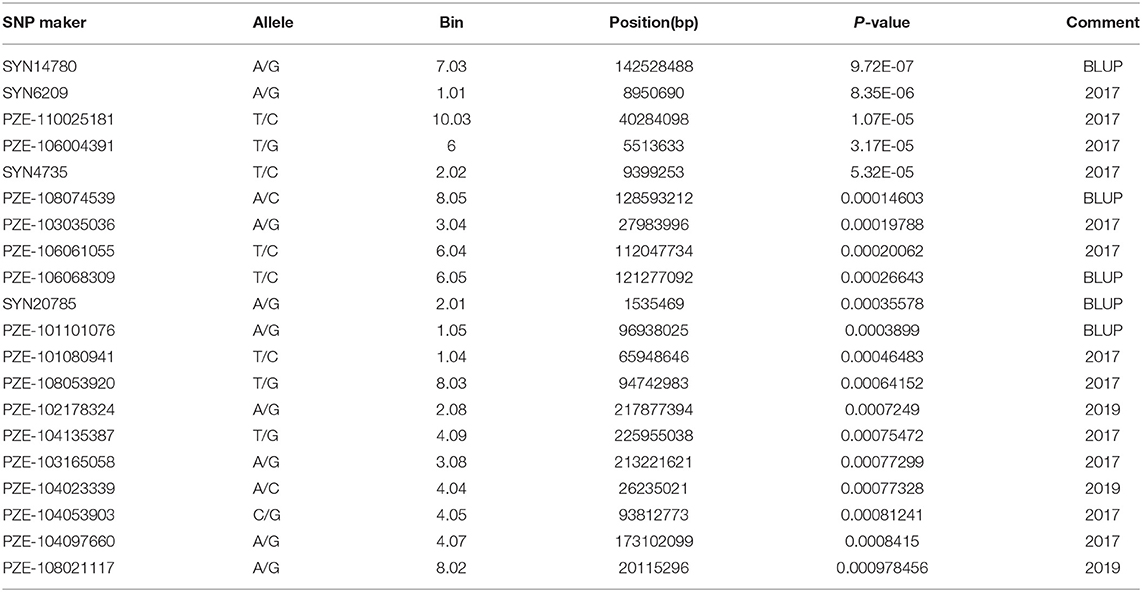
Table 2. The SNPs closely related to stalk rot in maize.
Preliminary QTLs Mapping Using the Linked Population
A total of 428 polymorphism markers between CDMA66 and Huangzao4 were screened out of 985 SSR markers covering 10 chromosomes of maize (Supplementary Figure 1). Taking chromosome 1 polymorphism marker bnlg1556 and some individuals in the F2 population as an example, the genotype analysis was performed, and the amplification results are shown in Supplementary Figure 2. Individuals in the F2 population with the same bands as the parent P1 or P2 are homozygous, and individuals with bands from both parents are heterozygous. The polymorphism markers on chromosomes 9 and 2 showed the highest polymorphism rate (63%) and the lowest polymorphism rate (26%), respectively. After removing the partial segregation markers, 227 SSR markers were remained to construct a genetic linkage map (Figure 5). Using LOD 2.5 as the threshold, three QTLs closely related to disease resistance were mapped on chromosomes 3, 4, and 8 (Table 3, Figure 6A). Comparing the results of QTL mapping and GWAS, two SNP loci (PZE-108021117 and PZE-104097660) were exactly located at QTL intervals at bin 8.02 and bin 4.07. The QTL on Chromosome 4 was mapped between mmc 0371 and umc 1808, had a LOD value of 2.59 and a contribution rate of 5.49%, and showed an additive effect of 0.3916. The QTL on Chromosome 8 was spanned by marker bnlg 2235 and umc1530, having the LOD value of 4.96 (Figure 6B), explaining 10.62% of phenotypic variation, and showing additive and dominant effects of 0.3262 and 0.6065, respectively.
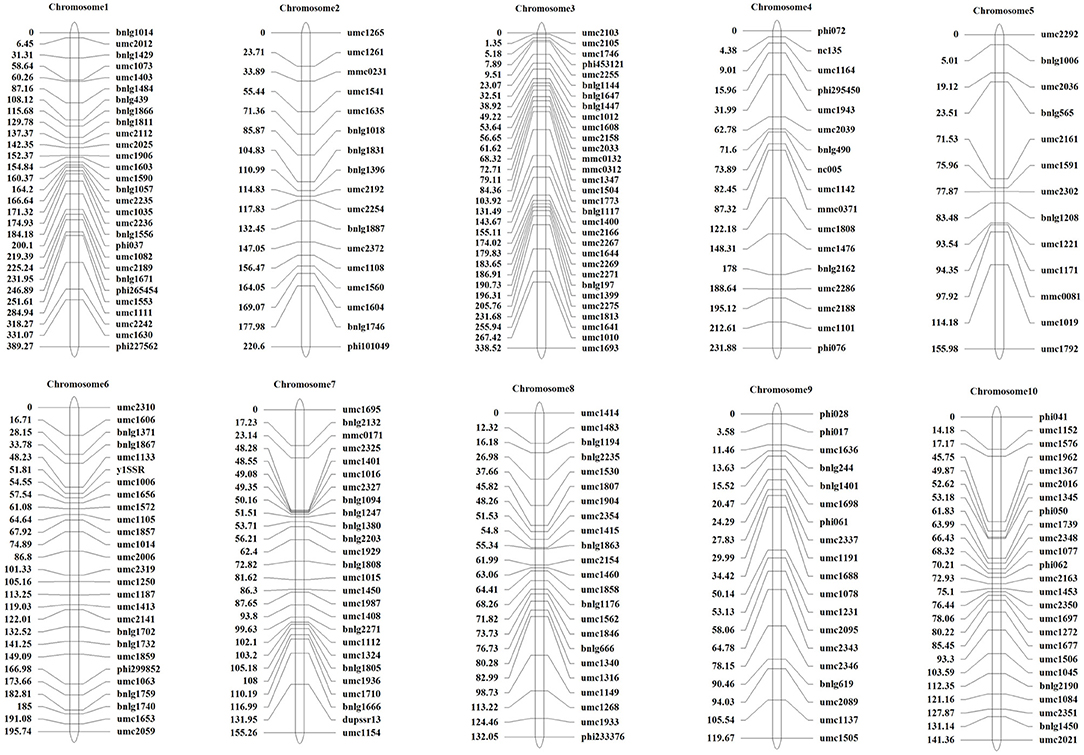
Figure 5. SSR marker linkage map of 10 chromosomes in maize.
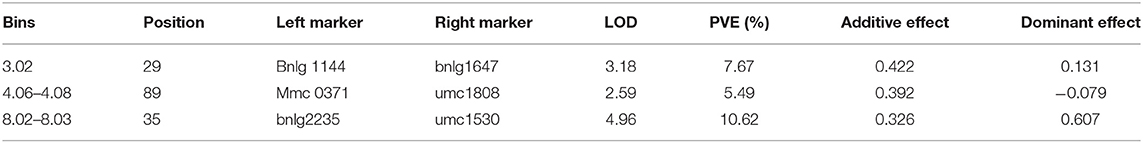
Table 3. QTLs for maize stalk rot.
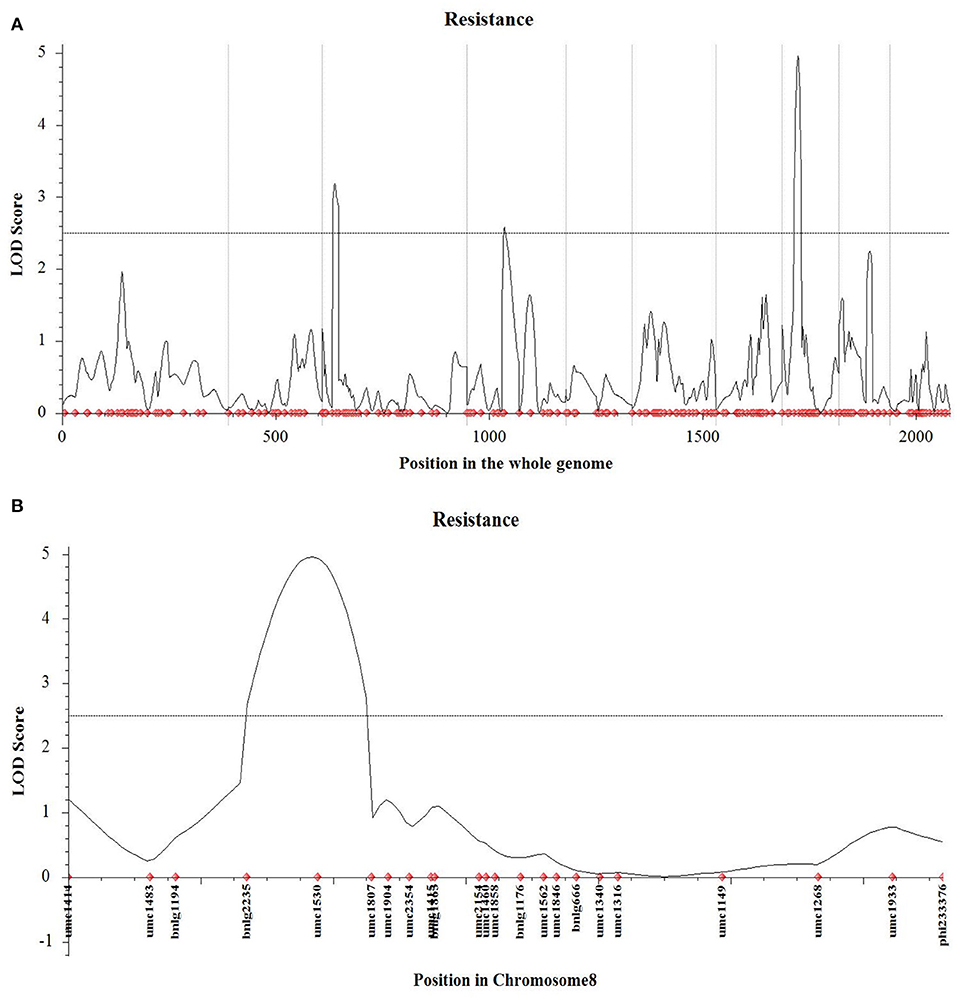
Figure 6. (A) The resistant QTLs for maize stalk rot. (B) The QTL loci on chromosome 8.
Candidate Gene Prediction
Some candidate genes containing the SNPs that were significantly associated with the resistance of stalk rot were selected, and the function was predicted at MaizeGDB4 (https://www.maizegdb.org/) and NCBI5 (https://www.ncbi.nlm.nih.gov/) (Table 4). Gene GRMZM2G010912 was detected by PZE-104097660, but no functions were reported till now. SNP locus PZE-101101076 on maize Chromosome 1 was located within the interval of gene GRMZM2G082709, whose functional annotation is NAC domain-containing protein 86; SNP locus SYN4735 on maize Chromosome 2 was located within the interval of gene GRMZM2G080041, whose functional annotation is LRR receptor-like serine/threonine-protein kinase FLS2; SNP locus PZE-106068309 on maize Chromosome 6 was located within the interval of gene GRMZM5G841142 whose functional annotation is thioredoxin reductase 2; and SNP locus PZE-108053920 on maize Chromosome 8 were located within the interval of gene GRMZM2G006745, whose functional annotation is DRE-binding protein 1c.
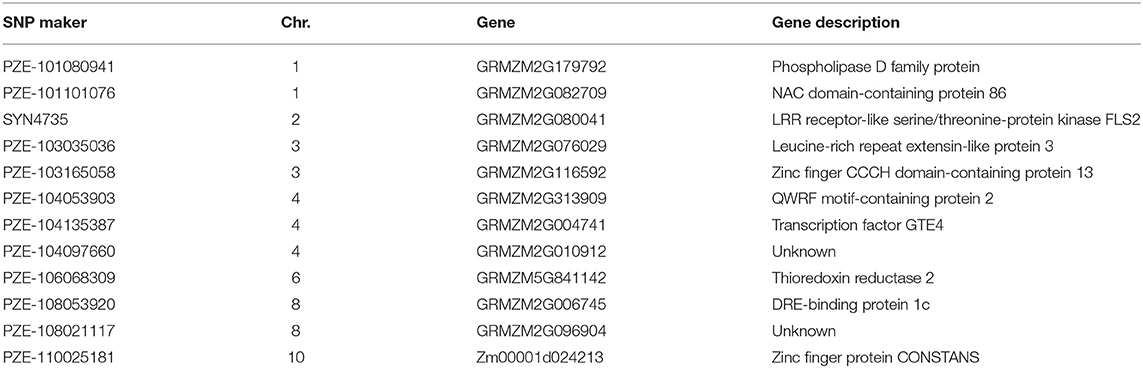
Table 4. Information of candidate genes for resistance to maize stalk rot.
Gene Expression Analysis
The expression levels of some candidate genes were analyzed in the stalk rot-resistant inbred line CDMA66 and susceptible inbred line Huangzao4. After CDMA66 was inoculated by Fusarium graminearum, the expression levels of GRMZM2G082709 and GRMZM5G841142 peaked at 12 and 72 h, respectively, and with the relative expression values of 5.28 and 6.95, respectively. When Huangzao4 was inoculated, the expression of GRMZM2G082709 was down-regulated, while GRMZM5G841142 showed an unchanged expression level. In addition, GRMZM2G080041 was up-regulated in Huangzao 4 but down-regulated in CDMA66 after the two inbred lines were inoculated by pathogen. In Huangzao 4, the maximum relative expression value of 4.37 appeared at 12 after inoculation. The gene GRMZM2G006745 changed the expression level in both Huangzao 4 and CDMA66 (Figure 7). The above genes may be involved in the mechanism of maize stalk rot.
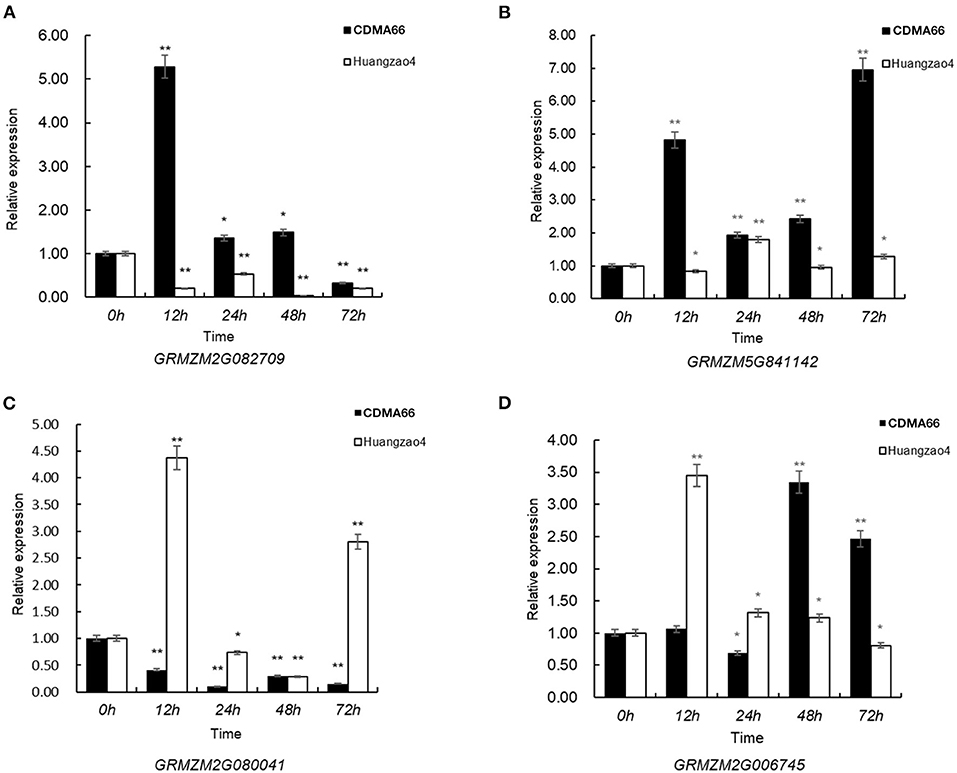
Figure 7. Relative expression of the candidate genes. (A) GRMZM2G082709; (B) GRMZM5G841142; (C) GRMZM2G080041; (D) GRMZM2G006745. **P < 0.01 and *P < 0.05, respectively.
Discussion
GWAS and genetic linkage map have been widely used to mine the resistance genes of crops. With the method, Li et al. (2015) detected two major resistance-related QTLs (09HR and 03HR) and 31 SNP loci associated with head smut in maize. Moreover, four and one of the 31 identified SNPs were located within the interval of 09HR and 03HR, respectively. Chen et al. (2016) identified 45 SNP loci that were closely related to Fusarium ear rot (FER) resistance in maize by the GWAS method, eight of which were located within FER QTLs' intervals through integrating the results of GWAS and QTL mapping. Xiao et al. (2019) detected two QTLs (qRBSDVD5 and qRBSDVD6) resistance to RBSDVD on chromosomes 5 and 6 and further detected an associated locus in the interval of qRBSDVD6 by GWAS. In our study, we used the GWAS and linkage mapping methods to explore the resistance genes of maize stalk rot and found 3 QTL loci and 34 SNP loci that were significantly related to maize stalk rot. The two SNP loci, PZE-108021117 and PZE-104097660, were located at the QTL intervals on bin 8.02 and bin 4.07, respectively. Previously, some QTLs for maize stalk rot have been reported to be located on bin 4.03–4.08 (Pè et al., 1993; Jung et al., 1994; Dong and Wang, 2001), and a QTL locus associated with stalk rot was on bin 8.06–8.08 (Chen et al., 2017). Furthermore, Yang et al. (2010) found two QTLs (qRfg1 and qRfg2) for stalk rot and narrowed the physical distance through fine mapping. The qRfg1 locus was located in a ~500-kb interval on Chromosome 10 and flanked by SSR334 and SSR58. The qRfg2 locus was located in a ~300-kb interval on Chromosome 1 and had markers SSRZ68 and SSRZ93 on both sides (Zhang et al., 2012). For the previous studies, most materials used for genome-wide association analysis and genetic linkage map construction are temperate germplasms, while in our study, both temperate and tropical materials were included in order to find new resistance sites of maize stalk rot.
GWAS is a powerful tool to identify marker variants that are significantly associated with complex traits (Pritchard et al., 2000). It is a powerful approach for exploring the molecular basis of phenotypic variations and has been widely used for functional gene discovery in plants (Li et al., 2016). In our study, we performed GWAS to detect functional genes associated with maize stalk rot. With the significant associated SNPs obtained from GWAS, NAC family gene was detected after comparing with the reference genome. NAC transcription factors are a large plant-specific transcription factor (TF) family and the NAC domain protein functions as homo- or hetero-dimer forms (Nakashima et al., 2012). Most NAC proteins have conserved DNA-binding domains that contain approximately 150 amino acids at the N-terminus. These domains are divided into five subdomains (A–E). The subdomains A, C, and D are highly conserved, while B and E are variable and involved in many unique functions of NAC proteins (Olsen et al., 2005). So we speculate that the functions of the maize NAC domain protein are mainly the functions that the B and E subdomains are involved in.
The NAC transcription factor family plays a key role in multiple biological processes, such as plant growth, development and stress responses (Wang et al., 2019). Harrington et al. (2019) found that the NAM-A1 NAC domain was required for protein binding in wheat and the mutation of the NAC domain would lead to the delayed peduncle and flag leaf senescence. Nogueira et al. (2005) found Saccharum sp. NACs (SsNAC23) was significantly associated with cold and water stresses in sugarcane. The Arabidopsis thaliana NAC proteins ANAC019 and ANAC055 might function as transcription activators to regulate the expression of Jasmonic acid (JA) and then regulate plant defense responses against herbivore attack, pathogen infection, and mechanical damages (Bu et al., 2008). Lv et al. (2016) found the overexpression of a novel NAC domain-containing transcription factor gene (AaNAC1) enhanced the content of artemisinin and increases tolerance to drought and Botrytis cinerea in Artemisia annua. Sun et al. (2013) found that the two rice genes ONAC122 and ONAC131 played an important role in the resistance to rice blast fungus. Lin et al. (2018) found that the maize NAC transcription factor ZmNAM played an important role in regulating the growth and development of root in maize. Zhang et al. (2014) found that NAC transcription factors participated in the response process of maize to drought stress.
We also found a gene related to thioredoxin reductase. Thioredoxin is a small protein that catalyzes the redox of disulfide bonds. It plays an important role by regulating the redox state of cells. In plants, the thioredoxin system is particularly complex, and it participates in plant metabolism, transcription and translation regulation, signal transduction, and plant resistance. The function of thioredoxin in plants is regulated by the dimer thioredoxin reductase (NTR) (Charles et al., 2000). Thioredoxin reductase (TrxR) is a dimer flavinase, a member of the pyridine nucleotide disulfide reductase family, and is widely expressed in cells of all levels of organisms from prokaryotes to humans (Holmgren, 2000). A thioredoxin gene, AtTrxh5, in Arabidopsis thaliana plays a role in the stress response of Arabidopsis to the Victorian oat blight (Avena sativa) disease (Sweat and Wolpert, 2007). Laloi et al. (2004) found that cytosolic thioredoxin AtTRXh5 may have potential significance in response to pathogens and oxidative stresses.
Besides, our research team examined one gene related to DRE-binding protein in Chromosome 8 and LRR receptor-like serine/threonine-protein kinase in Chromosome 2. At present, dehydration-responsive element-binding protein (DRE-binding protein) is the most thoroughly studied stress-related transcription factor (Wei et al., 2013). DRE-binding protein transcription factors are involved in regulating the expression of abiotic stress-mediated gene in plants. Sharma et al. (2019) found that apple transcription factor, MdDREB76, conferred salt and drought tolerance in transgenic tobacco by activating the expression of stress-responsive genes. Guo et al. (2018) found that the overexpression of peanut AhDREB1 gene was effective in enhancing salt tolerance in peanuts. Plant receptor-like protein kinases (RLKs) are one of the larger gene families in plants. They have a special protein structure and play an important role in signal transmission. They can sense signal of growth, development and external environmental stress (Lally et al., 2001). Many reports have proved that most of the disease resistance of plants belongs to the LRR-RLK subfamily; for example, the bacterial blight resistance gene Xa21 was cloned from rice and belonged to the LRR subtype, which prevents pathogens from attacking rice (Song et al., 1995). The FLS2 gene of A. thaliana can encode LRR-type RLKs, which can play an important role in plant disease resistance and pathogen identification (Robatzek et al., 2006). These results suggest that multiple mechanisms might be involved in conditioning maize stalk rot resistance.
Conclusion
Genetic diversity analysis and GWAS about maize stalk rot were performed in a panel of 165 maize inbred lines. These materials were clustered into four subgroups. Thirty-four SNP loci and three QTLs that significantly associated with maize stalk rot were detected using FarmCPU. These three QTLs are located on chromosomes 3, 4, and 8, respectively. Among them, two SNP loci, PZE-108021117 and PZE-104097660, were located within QTL interval. Moreover, two genes (GRMZM2G096904 and GRMZM2G010912) which contain the two SNP loci respectively were chosen as candidate resistance genes for stalk rot in maize, but no more information was reported. We also found some other candidate genes such as NAC domain-containing protein 86 and DRE-binding protein that may be related to maize stalk rot. How these candidate genes are involved in the pathogenesis of maize stalk rot needs further verification. Our results would be helpful in discovering the mechanism of stalk rot resistance, and to breed resistant varieties of maize.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author Contributions
XS and MZ conceived and designed the study. MZ, SL, XS, JF, and ZS performed the experiments, phenotyped the populations, and field work. SL and JF contributed to genotype and phenotype data analysis. SL drafted the manuscript. All authors read and approved the final manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (31371636), The Innovation Team in Maize Modern Agricultural System of Shandong Province, China (SDAIT-02-01), Key R&D Project of Shandong Province, China (2016GNC110018), and Applied Basic Research Project of Qingdao, China (14-2-4-13-jch).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fagro.2020.590374/full#supplementary-material
Supplementary Table 1. The list of 165 maize inbred lines.
Supplementary Table 2. qRT-PCR primers.
Supplementary Figure 1. Polymorphic SRR primers for PCR results of partial (1, umc2177; 2, umc1546; 3, umc1545; 4, umc1241; 5, umc1642; 6, umc1695; 7, umc1378; 8, bnlg2132; 9, umc1672; 10, umc1695; 11, mmc0171; 12, phi057; 13, phi112; 14, umc1270; there are polymorphic markers in black box).
Supplementary Figure 2. The amplified bands of marker bnlg 1556 in parents and F2 population(P1, parent 1; P2, parent 2; 1–38 are F2 individuals).
References
Abad, L., Wolters, P., and Stucker, D. (2006). “Advances in anthracnose stalk rot resistance,” in Fifth National IPM Symposium (St. Louis, MO). City.
Brachi, B., Morris, G. P., and Borevitz, J. O. (2011). Genome-wide association studies in plants: the missing heritability is in the field. Genome Biol. 12, 232–239. doi: 10.1186/gb-2011-12-10-232
Bradbury, P. J., Zhang, Z., Kroon, D. E., Casstevens, T. M., Ramdoss, Y., and Buckler, E. S. (2007). TASSEL: software f or association mapping of complex traits in diverse samples. Bioinformatics. 23, 2633–2635. doi: 10.1093/bioinformatics/btm308
Bu, Q. Y., Jiang, H. L., Li, C. B., Zhai, Q. Z., Zhang, J., Wu, X. Y., et al. (2008). Role of the Arabidopsis thaliana NAC transcription factors ANAC019 and ANAC055 in regulating jasmonic acid-signaled defense responses. Cell Res. 018, 756−767. doi: 10.1038/cr.2008.53
Charles, H., Williams, Jr. L. Jr., and David, Arscott. (2000). Thioredoxin reductase. J. Biochem. 267: 6110−6117. doi: 10.1046/j.1432-1327.2000.01702.x
Chen, D. H., and Ronald, P. (1999). A rapid DNA minipreparation method suitable for AFLP and other PCR applications. Plant Mol. Biol. Rep. 17, 53–57.
Chen, J. F., Shrestha, R., Ding, J. Q., Zheng, H. J., Mu, C. H., Wu, J. Y., et al. (2016). Genome-wide association study and QTL mapping reveal genomic loci associated with Fusarium ear rot resistance in tropical maize germplasm. Genesgenetics. 6, 3803–3815. doi: 10.1534/g3.116.034561
Chen, Q., Song, J., Du, W. P., Xu, L. Y., Jiang, Y., Zhang, J., et al. (2017). Identification, mapping, and molecular marker development for rgsr8.1: a new quantitative trait locus conferring resistance to Gibberella stalk rot in maize (Zea mays L.). Plant Science. 8, 1355–1364. doi: 10.3389/fpls.2017.01355
Chiang, K. S., Liu, H. I., and Bock, C. H. (2017). A discussion on disease severity index values. Part I: warning on inherent errors and suggestions to maximise accuracy. Ann. als of Appl. ied Biology. 171, 139–154. doi: 10.1111/aab.12362
Dong, W. B., and Wang, G. Y. (2001). RAPD correction and identification of RAPD markers linked to resistance gene of maize against corn stalk rot caused by Pythium inflatum. J. of Agric. Biotechnology. 2, 131–139.
Evanno, G., Regnaut, S., and Goudet, J. (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14, 2611−2620. doi: 10.1111/j.1365-294X.2005.02553.x
Frey, T. J. (2005). Fine Mapping, Cloning, Verification, and Fitness Evaluation of a QTL, Rcg1, which Confers Resistance to Colletotrichum graminicola in Maize. Newark: University of Delaware.
Gowda, M., Das, B., Makumbi, D., Babu, R., Semagn, K., Mahuku, G., et al. (2015). Genome-wide association and genomic prediction of resistance to maize lethal necrosis disease in tropical maize germplasm. Theor. Appl. Genet. 128, 1957–1968. doi: 10.1007/s00122-015-2559-0
Guo, R. J., Li, W., Liu, F. Z., Zhang, K., and Wan, S. Y. (2018). Salt-tolerance analysis of T2 transgenic peanut plants of AhDREB1. Shand Agric. Sci. 50, 72–78. doi: 10.14083/j.issn.1001-4942.2018.06.012
Harrington, S. A., Overend, L. E., Cobo, N., Borrill, P., and Uauy, C. (2019). Conserved residues in the wheat (Triticum aestivum) NAM-A1 NAC domain are required for protein binding and when mutated lead to delayed peduncle and flag leaf senescence. BMC Plant Biol. 19, 407–422. doi: 10.1186/s12870-019-2022-5
Holmgren, A. (2000). Redox regulation by thioredoxin and thioredoxin reductase. Biofactors 11, 63–64. doi: 10.1002/biof.5520110117
Jung, M., Weldekidan, T., Schaff, D., Paterson, A., Tingey, S., and Hawk, J. (1994). Generation-means analysis and quantitative trait locus mapping of anthracnose stalk rot genes in maize. Theor. Appl. Genet. 89, 413–418. doi: 10.1007/BF00225375
Lally, D., Ingmire, P., Tong, H. Y., and He, Z. H. (2001). Antisense expression of a cell wall-associated protein kinase, WAK4, inhibits cell elongation and alters morphology. Plant Cell 13, 1317–1332. doi: 10.1105/tpc.13.6.1317
Laloi, C., Mestres-Ortega, D., Marco, Y., Meyer, Y., and Reichheld, J. (2004). The Arabidopsis cytosolic thioredoxin h5 gene induction by oxidative stress and its W-Box-Mediated response to pathogen elicitor. Plant Physiol. 134, 1006–1016. doi: 10.1104/pp.103.035782
Li, H. H., Ribaut, J. M., Li, Z. L, and Wang, J. K. (2008). Inclusive composite interval mapping (ICIM) for digenic epistasis of quantitative traits in biparental populations. Theor. Appl. Genet. 116, 243–260. doi: 10.1007/s00122-007-0663-5
Li, X. P., Zhou, Z. J., Ding, J. Q., Wu, Y. B., Zhou, B., Wang, R. X., et al. (2016). Combined linkage and association mapping reveals QTL and candidate genes for plant and ear height in maize. Front. Plant Sci. 7, 833–843. doi: 10.3389/fpls.2016.00833
Li, Y. X., Wu, X., Jaqueth, J., Zhang, D. F., Cui, D. H., Li, C. H., et al. (2015). The identification of two head smut resistance-related QTL in maize by the joint approach of linkage mapping and association analysis. PLoS ONE. 10:e0145549. doi: 10.1371/journal.pone.0145549
Lin, J., Yu, Y. T., Ma, S. M., and Hu, J. G. (2018). Cloning and expression analysis of transcription factor gene ZmNAM in maize(Zea mays L.). Acta Agric. Nuclear Sci. 5, 864–874. doi: 10.11869/j.issn.100-8551.2018.05.0864
Lipka, A. E., Tian, F., Wang, Q. S., Peiffer, J., Li, M., and Bradbury, P. J. (2012). GAPIT: genome association and prediction integrated tool. Bioinformatics 28, 2397–2399. doi: 10.1093/bioinformatics/bts444
Liu, X. H., Huang, M., Fan, B., Buckler, E. S., and Zhang, Z. W. (2016). Iterative usage of fixed and random effect models for powerful and efficient genome-wide association studies. PLoS Genet. 12:e1005767. doi: 10.1371/journal.pgen.1005767
Liu, X. L. (2015). Development of an iterative usage of fixed effect and random effect models for powerful and efficient genome-wide association studies (Master's thesis). Huazhong Agricultural University, Wuhan, China.
Lv, Z. Y., Shu Wang, S., Zhang, F. Y., Chen, L. X., Hao, X. L., Pan, Q. F., et al. (2016). Overexpression of a novel NAC domain-containing transcription factor gene (AaNAC1) enhances the content of artemisinin and increases tolerance to drought and botrytis cinerea in artemisia annua. Plant Cell Physiol. 9, 1961–1971. doi: 10.1093/pcp/pcw118
Ma, C. Y., Ma, X. N., Yao, L. S., Liu, Y. J., Du, F. L., Yang, X. H., et al. (2017). qRfg3, a novel quantitative resistance locus against Gibberella stalk rot in maize. Theor. Appl. Genet. 130, 1723–1734. doi: 10.1007/s00122-017-2921-5
Nakashima, K., Takasaki, H., Mizoi, J., Shinozaki, K., and Shinozaki, K. Y. (2012). NAC transcription factors in plant abiotic stress responses. Bba Gene Regul. Mech. 1819, 97–103. doi: 10.1016/j.bbagrm.2011.10.005
Nogueira, F. T. S., Schlögl, P. S., Sandra, R., Camargo, S. R., Fernandez, J. H., DeRosa, V. E. Jr., et al. (2005). SsNAC23, a member of the NAC domain protein family, is associated with cold, herbivory and water stress in sugarcane. Plant Sci. 169, 93–106. doi: 10.1016/j.plantsci.2005.03.008
Olsen, A. N., Ernst, H. A., Leggio, L. L., and Skriver, K. (2005). NAC transcription factors: structurally distinct, functionally diverse. Trends Plant Sci. 10, 79–87. doi: 10.1016/j.tplants.2004.12.010
Pè, M. E., Gianfranceschi, L., Taramino, G., Tarchini, R., Angelini, P., Dani, M., et al. (1993). Mapping quantitative trait loci (QTLs) for resistance to Gibberellazeae infection in maize. Mol. Gen. Genet. 241, 11–16. doi: 10.1007/BF00280195
Phan, H. T. T., Rybak, K., Bertazzoni, S., Furuki, E., Dinglasan, E., Hickey, L. T., Oliver, R. P., et al. (2018). Novel sources of resistance to Septoria nodorum blotch in the Vavilov wheat collection identifed by genome-wide association studies. Theor. Appl. Genet. 131, 1223–1238. doi: 10.1007/s00122-018-3073-y
Pritchard, J. K., Stephens, M., and Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics 155, 945–959.
Robatzek, S., Chinchilla, D., and Boller, T. (2006). Ligand-induced endocytosis of the pattern recognition receptor FLS2 in Arabidopsis. Genes Dev. 20, 537–542. doi: 10.1101/gad.366506
Schnable, P. S., Hsia, A. P., and Guo, L. (2009). Molecular Genetic Approaches to Maize Improvement. Berlin; Heidelberg: Springer.
Sharma, V., Goel, P., Kumar, S., and Singh, A. K. (2019). An apple transcription factor, MdDREB76, confers salt and drought tolerance in transgenic tobacco by activating the expression of stress-responsive genes. Plant Cell Rep. 38, 221–241. doi: 10.1007/s00299-018-2364-8
Song, F. J., Xiao, M. G., Duan, C. X., Li, H. J., Zhu, Z. D., Liu, B. T., et al. (2015). Two genes conferring resistance to Pythium stalk rot in maize inbred line Qi319. Mol. Genet. Genom. 290, 1543–1549. doi: 10.1007/s00438-015-1019-5
Song, W. Y., Wang, G. L., Chen, L. L., Kim, H. S., Pi, L. Y., Hoisten, T., et al. (1995). A receptor kinase-like protein encoded by the rice disease resistance gene, Xa21. Science 270, 1804–1806. doi: 10.1126/science.270.5243.1804
Sun, L. J., Zhang, H. J., Li, D. Y., Huang, L., Hong, Y. B., Ding, X. S., et al. (2013). Functions of rice NAC transcriptional factors, ONAC122 and ONAC131, in defense responses against Magnaporthe grisea. Plant Mol. Biol. 81, 41–56. doi: 10.1007/s11103-012-9981-3
Sweat, T. A., and Wolpert, T. J. (2007). Thioredoxin h5 is required for victorin sensitivity mediated by a CC-NBS-LRR in Arabidopsis. Plant Cell 19, 673–687. doi: 10.1105/tpc.106.047563
Wang, C., Yang, Q., Wang, W. X., Li, Y. P., Guo, Y. L., Zhang, D. F., et al. (2017). A transposon-directed epigenetic change in ZmCCT underlies quantitative resistance to Gibberella stalk rot in maize. New Phytol. 215, 1503–1515. doi: 10.1111/nph.14688
Wang, F., Sun, L. J., Zhao, X. Y., Wang, J. W., and Song, X. S. (2019). Research progresses on plant NAC transcription factors. Biotechnol. Bull. 4, 88–93. doi: 10.13560/j.cnki.biotech.bull.1985.2018-0905
Wang, J. L., Li, H., Zhang, L., and Meng, L. (2012). Users' Manual of QTL IciMapping Version 3.2. Beijing; Mexico: The Quantitative Genetics group; Institute of Crop Science; Chinese Academy of Agricultural Science (CAAS); Genetic Resources Programme, CIMMYT, 208.
Wang, J. P., Liu, Y. W., Sun, G. Z., and Wang, H. B. (2017). Evaluation and validation of molecular markers associated with stalk rot resistance in 159 maize inbred lines. J. Plant Genet. Resour. 18, 754–762. doi: 10.13430/j.cnki.jpgr.2017.04.019
Wei, S. J., Wu, Y. F., Xu, X. J., Gao, F., and Zhou, Y. J. (2013). Cloning and function analysis of ThDREB2B gene encoding a putative DRE-binding transcription factor from Thellungiella halophila. Acta Bot. Boreali-Occidentalia Sin. 2, 215–222.
Xiao, S. Z., Wang, B. X., Liu, Y. Q., Miao, T. H., Zhang, H. L., Wen, P. Z., et al. (2019). Genome-wide association study and linkage analysis on resistance to rice black-streaked dwarf virus disease. Mol. Breed. 39:73. doi: 10.1007/s11032-019-0980-9
Yang, Q., Yin, G. M., Guo, Y. L., Zhang, D. F., Chen, S. J., and Xu, M. L. (2010). A major QTL for resistance to Gibberella stalk rot in maize. Theor. Appl. Genet. 121, 673–687. doi: 10.1007/s00122-010-1339-0
Ye, J. R., Zhong, T., Zhang, D. F., Ma, C. Y., Wang, L. N., Yao, L. S., et al. (2019). The auxin-regulated protein ZmAuxRP1 coordinates the balance between root growth and stalk rot disease resistance in maize. Mol. Plant. 12, 360–373. doi: 10.1016/j.molp.2018.10.005
Zhang, D. F., Liu, Y. J., Guo, Y. L., Yang, Q., Ye, J. R., Chen, S. J., et al. (2012). Fine-mapping of qRfg2, a QTL for resistance to Gibberella stalk rot in maize. Theor. Appl. Genet. 124, 585–596. doi: 10.1007/s00122-011-1731-4
Zhang, J. Y., Chen, F., Li, L., Ma, Y. W., Wang, Z. P., Jiang, L. Y., et al. (2014). Sequence characteristics and expression analysis of 16 NAC transcription factors in maize under water stress. J. Shanxi Agric. Sci. 4, 307–312. doi: 10.3969/j.issn.1002-2481.2014.04.01
Keywords: maize stalk rot, genetic diversity, GWAS, QTL, candidate gene
Citation: Liu S, Fu J, Shang Z, Song X and Zhao M (2021) Combination of Genome-Wide Association Study and QTL Mapping Reveals the Genetic Architecture of Fusarium Stalk Rot in Maize. Front. Agron. 2:590374. doi: 10.3389/fagro.2020.590374
Received: 01 August 2020; Accepted: 30 November 2020;
Published: 03 February 2021.
Edited by:
Geert Jozej Willem Haesaert, Ghent University, BelgiumCopyright © 2021 Liu, Fu, Shang, Song and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Meiai Zhao, bWVpYWkyMDE1QDE2My5jb20=; Xiyun Song, c29uZ3hpeXVuMjAyMEAxNjMuY29t