 Ermioni S. Arvanitaki
Ermioni S. Arvanitaki Kalliopi Stratigi
Kalliopi Stratigi George A. Garinis
George A. Garinis
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Aging, 07 September 2022
Sec. Genetics, Genomics and Epigenomics of Aging
Volume 3 - 2022 | https://doi.org/10.3389/fragi.2022.973781
Persistent DNA lesions build up with aging triggering inflammation, the body’s first line of immune defense strategy against foreign pathogens and irritants. Once established, DNA damage-driven inflammation takes on a momentum of its own, due to the amplification and feedback loops of the immune system leading to cellular malfunction, tissue degenerative changes and metabolic complications. Here, we discuss the use of murine models with inborn defects in genome maintenance and the DNA damage response for understanding how irreparable DNA lesions are functionally linked to innate immune signaling highlighting their relevance for developing novel therapeutic strategies against the premature onset of aging-associated diseases.
The growing list of syndromes associated with inborn defects in DNA repair and phenotypes resembling accelerated ageing points to genomic damage as a major culprit in the ageing process (Hoeijmakers, 1994). The gradual accumulation of persistent DNA lesions triggers the activation of potent inflammatory responses that are causal to age-related degeneration, metabolic abnormalities and cancer. Indeed, genome maintenance pathways and immune responses are highly conserved and tightly linked biological processes, supporting the notion that cells have encountered genotoxins and foreign pathogens nearly ab initio. For instance, induction of the SOS response, an inducible pathway governing DNA repair in bacteria affects their adaptation to antimicrobial tolerance, resistance, and virulence (Zgur-Bertok, 2013). In prokaryotes, the CRISPR-Cas system is also functionally linked to antiviral immunity and DNA repair (Babu et al., 2011). An efficient adaptive immune system in mammals requires that the non-homologous end-joining (NHEJ) pathway repairs the DNA strand breaks (DSBs) that occur during V(D)J recombination in developing T lymphocytes, ensuring the vast diversity of receptors necessary for overcoming pathogenic insults. (Boboila et al., 2012). Likewise, in B lymphocytes, an error-prone DNA repair mechanism is required to introduce a large number of nucleotide substitutions into a small area of the genome, in a process called somatic hypermutation of immunoglobulin variable genes, which produces variant antibodies with increased affinity for cognate antigens (Di Noia and Neuberger, 2007). Besides the functional contribution of DNA repair factors in the development and maturation of adaptive immunity, the distinct types of cells and tissues in mammals require that genome maintenance, DDR and innate immune responses, the organism’s natural response to harmful genotoxins, are tightly coordinated and timely mobilized [reviewed in (Ioannidou et al., 2016; Stratigi et al., 2017; Chatzidoukaki et al., 2020)].
There is ample evidence to suggest that genome instability drives aging and that DNA damage and DDR activation triggers a wide range of age-related phenotypes, including the activation of cell- and non-cell autonomous signaling pathways; the latter leads to the secretion of pro-inflammatory factors and the gradual infiltration of immune cells at sites of tissue damage with advancing age. DNA damage activates cytoplasmic NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells), an inducible transcription factor that translocates into the nucleus and functions as a pivotal mediator of inflammatory responses by binding to the DNA consensus sequence of target genes (Hayden and Ghosh, 2008). The interferon regulatory factors (IRFs) make up another family of immune-related transcription factors that become activated upon exposure to genotoxins. Induction of IRF-1 protein by either ionizing radiation or etoposide occurs through an increase in IRF-1 protein levels and an increase in the half-life of the IRF-1 protein; the response requires a functional DDR as cells defective in the DNA damage sensor ATM (ataxia telangiectasia, mutated) failed to increase IRF-1 in response to genotoxic stress (Pamment et al., 2002). IRF-3 and 5 provide further evidence that genome maintenance and the DDR are linked to the interferon signaling; IRF-3 is an in vivo target of DNA-PK (DNA-dependent protein kinase) (Karpova et al., 2002) that functions in the repair of DNA breaks by NHEJ and V(D)J recombination (Lieber et al., 2003) whereas IRF-5 is a direct transcriptional target of p53 upon exposure of cells to genotoxic insults (Mori et al., 2002). NKG2D also known as CD159 (Cluster of Differentiation 159) is an activating receptor that is predominantly expressed on the surface of cytotoxic immune cells. The presence of DNA lesions or stalled DNA replication forks triggers the upregulation of NKG2D ligands in mouse or human cells in an ATM-, ATR (Ataxia telangiectasia and Rad3 related)- or Chk1-dependent manner, allowing the immune system to detect and selectively remove damaged cells (Gasser et al., 2005; Gasser and Raulet, 2006; Champsaur and Lanier, 2010). DNAM-1 (DNAX Accessory Molecule-1) is a 65 kDa transmembrane glycoprotein that is constitutively expressed in the majority of T cells, NK cells, and macrophages (Shibuya et al., 1996). Low doses of chemotherapeutic genotoxins trigger the expression of DNAM-1 ligands in multiple myeloma cells in an ATM/ATR-dependent manner (Soriani et al., 2009). Likewise, ICAM-1 (Intercellular Adhesion Molecule 1), a transmembrane glycoprotein that promotes the adhesion of leucocytes to inflamed vascular endothelium, is also induced in response to ionizing irradiation in a p53-dependnent manner (Gaugler et al., 1997). Furthermore, in humans, the expression of all TLR (Toll-Like Receptor) genes, TLR1 to TLR10, in blood lymphocytes and alveolar macrophages is induced by DNA damage with considerable inter-individual variability (Menendez et al., 2011). UV-irradiated keratinocytes form large cytoplasmic complexes, called “inflammasomes” to trigger the maturation, activation and secretion of pro-inflammatory cytokines (Faustin and Reed, 2008; Schroder and Tschopp, 2010).
DNA damage-induced inflammation can be both beneficial and detrimental for organismal survival. In higher organisms, the inflammatory response has evolved as an acute defense mechanism to eliminate the harmful irritant and allow the body to heal. With prolonged stimuli, however, as it is when DNA damage gradually accrues over time in cells, it leads to the activation of persistent DDR-mediated pro-inflammatory signals leading to systemic chronic inflammation, tissue degeneration and malfunction with old age. Cellular senescence is a great example of how irreparable DNA lesions underlie the persistent activation of innate immune signaling (Rodier et al., 2009; Campisi and d'Adda di Fagagna, 2007; Fumagalli and d'Adda di Fagagna, 2009). Senescent cells with chromatin hallmarks of irreparable DSBs secrete a wide range of senescence-associated secretory phenotype (SASP) factors, including inflammatory cytokines and chemokines as well as growth factors and extracellular matrix remodeling enzymes (Coppe et al., 2008; Ohanna et al., 2011; Acosta et al., 2013; Malaquin et al., 2013). SASP factors impinge on cell-fate decisions in neighboring cells or facilitate angiogenesis and promote the growth, invasion, and metastasis of tumor cells (Grivennikov et al., 2010). In support, older individuals show an increase in systemic inflammation as evidenced by the elevated levels of pro-inflammatory cytokines e.g., IL-6, clotting factors and acute phase reactants (Ferrucci et al., 1999; Cohen et al., 2003; Cavanagh et al., 2012; Shaw et al., 2013). Thus, there is an immense need to establish reliable in vivo animal models for understanding how DNA damage-driven inflammatory responses lead to some of the most challenging degenerative disorders of our time, as well as to develop rationalized therapeutic intervention strategies against chronic inflammation with aging.
Owing to their congenital defects in genome maintenance, DNA repair-deficient animals are valid models to test for the physiological relevance of DNA damage-driven inflammation in premature disease onset during normal and accelerated aging (Supplementary Table S1) (van de Ven et al., 2007; Schumacher et al., 2008a; Schumacher et al., 2008b; Garinis, 2008; Garinis et al., 2008; Garinis et al., 2009; Schumacher et al., 2009; Karakasilioti et al., 2013; Karakasilioti and Garinis, 2014; Ioannidou et al., 2016; Chatzidoukaki et al., 2020). Mice with engineered mutations in NER genes reliably mimic most of the pleiotropic and heterogeneous pathological symptoms seen in NER syndromes (Hasty et al., 2003; Niedernhofer et al., 2006; van der Pluijm et al., 2007; Schumacher et al., 2008a; Schumacher et al., 2008b; Garinis, 2008; Rieckher et al., 2021). ERCC1-XPF is a heterodimeric, structure-specific endonuclease complex required for lesion excision in nucleotide excision repair (NER) (Kamileri et al., 2012; Apostolou et al., 2019) that plays an analogous role in the repair of highly cytotoxic DNA inter-strand crosslinks (ICLs) (Niedernhofer et al., 2004). The Ercc1−/− and Ercc1-/Δ7 mouse models of the human progeroid syndrome XFE (Niedernhofer et al., 2006; Gregg et al., 2012; Schermer et al., 2013; Pieren et al., 2021) provide compelling evidence that the persistent activation of DNA damage-driven innate immune responses lead to tissue-degenerative alterations. For instance, aP2-Ercc1F/- mice that carry the NER defect only in the white adipose tissue, rapidly accumulate DNA damage in adipocytes and manifest a chronic auto-inflammatory response leading to severe fat depletion and metabolic abnormalities (Karakasilioti et al., 2013). In aP2-Ercc1F/- mice, the fat depots manifest hallmarks of persistent DDR together with the transcriptional de-repression of pro-inflammatory factors, the infiltration of activated macrophages as well as the release of DAMPs known to initiate and perpetuate immune responses (Karakasilioti et al., 2013). In line with the contributing role of DNA damage-driven inflammation in the premature onset of age-related diseases, pharmacologic inhibition of NF-κB was shown to delay several pathological symptoms in progeroid ERCC1-defective mice (Tilstra et al., 2012). When the ERCC1 defect is restricted in macrophages in Lys2-Ercc1F/- mice, the tissue-infiltrating Ercc1−/− macrophages release extracellular vesicles whose cargo is targeted to diverse recipient cells leading to systemic metabolic reprogramming associated with enhanced cellular glucose uptake, increased oxygen consumption, chronic inflammation and overt pathology (Goulielmaki et al., 2020). More recently, the gradual accumulation of persistent DNA damage in hematopoietic cells of Vav-iCre+/-; Ercc1F/- mice led to an accelerated aging of the immune system associated with the progressive reduction of lymphocytes (Yousefzadeh et al., 2021); in turn, the aged immune system of Vav-iCre+/-; Ercc1F/- mice led to senescence and loss of tissue homeostasis in non-lymphoid organs. R-loops are nucleic acid structures composed of an RNA–DNA hybrid and a displaced single-stranded DNA that may spontaneously lead to DNA breaks (Skourti-Stathaki et al., 2011; Skourti-Stathaki and Proudfoot, 2014; Goulielmaki et al., 2021). In progeroid Ercc1−/− and naturally aged pancreata, DNA damage triggers the formation of R-loops leading to the release and build-up of single-stranded (ss)DNA fragments in the cytoplasm of cells stimulating a viral-like immune response (Chatzidoukaki et al., 2021). Owing to the great similarity of phenotypic features between Ercc1−/− and Xpg−/− animals mimicking the progeroid Cockayne syndrome (Barnhoorn et al., 2014), it is attractive to speculate that the latter animals would respond similarly in terms of the improper activation of the immune system. Csbm/m mice carrying an inborn defect in CSB involved in transcription-coupled (TC) NER present with lung inflammation and thrombogenic responses upon exposure to ozone, a well-established toxic environmental factor (Kooter et al., 2008). Interestingly, enhanced inflammation has also been noticed in Xpa−/− (Xeroderma Pigmentosum) mice that are defective in both the TC-NER and the global genome repair sub-pathways of NER (Miyauchi-Hashimoto et al., 1996). Loss of myelin and Purkinje cell death in Csa−/−/Xpa−/− mice were accompanied by microglia and astrocyte activation and vascular inflammation in the brain, mirroring the neuronal dysfunction observed in XP patients (Kajitani et al., 2021). Similar to aP2-Ercc1F/- mice, increased cellular senescence in the adipose tissue of 4-weeks old pol η−/− (DNA polymerase η) mice carrying a defect in lesion bypass polymerase was accompanied by the senescence-associated secretion of pro-inflammatory cytokines IL-6 and TNF-α (tumor necrosis factor α) (Chen et al., 2015). Besides DNA lesions associated with NER or post-replication repair machinery, inflammation is also triggered by irreparable modifications to DNA bases, including oxidation, alkylation and deamination. For instance, mice deficient in MBD4, a glycosylase involved in the detection and repair of deamination of methyl-cytosines, manifest greater colon inflammation and tissue injury leading to an increase in tumor burden (Yu et al., 2016). In line, alkyladenine DNA glycosylase and two members from the AlkB family of DNA repair enzymes (ALKBH2, and ALKBH3) are shown to protect against tissue injury and tumorigenesis in an inducible mouse model of inflammation-driven colon cancer (Meira et al., 2008; Calvo et al., 2012). 8-Oxoguanine glycosylase (OGG1) is the most prominent DNA glycosylase that is responsible for the removal of 8-OH-dG, an oxidative DNA lesion that is frequently formed in the DNA of aerobic organisms (Dianov et al., 1998). Importantly, OGG1–deficient macrophages are more apoptotic and inflammatory compared to wild type controls. Moreover, a defect in OGG1 leads to an increase in inflammasome activity and the premature onset of atherosclerosis in mice that are deficient for low-density lipoprotein receptor (Tumurkhuu et al., 2016). Naturally occurring RNA-DNA hybrids are frequently formed on promoters or termination regions when a nascent RNA molecule is hybridized with the DNA template before the two strands of the DNA duplex reanneal, leaving the non-template DNA single-stranded (Skourti-Stathaki et al., 2011; Skourti-Stathaki and Proudfoot, 2014). Persistent R-loops expose long stretches of ssDNA, leading to the spontaneous formation of DSBs or to transcription-associated mutagenesis (Muers, 2011; Wimberly et al., 2013; Costantino and Koshland, 2015). Partial loss‐of‐function biallelic mutations in RNase H2 genes, an endogenous endoribonuclease that cleaves the RNA strand in RNA-DNA duplexes, are the major cause of the autoinflammatory disorder Aicardi-Goutières syndrome (Crow and Manel, 2015). Likewise, a hypomorphic RNase H2 mouse model for Aicardi-Goutières syndrome associates with the induction of interferon-stimulated gene transcripts, a response that is dependent on the cGAS‐STING nucleic acid‐sensing pathway (Mackenzie et al., 2016). Mice with intestinal ablation of RNase H2, present with local intestinal inflammation, tissue damage and colorectal cancer (Aden et al., 2019). Werner syndrome is a premature aging disorder caused by mutations in a DNA helicase/exonuclease. Mice lacking the helicase domain of this protein exhibit an increase in serum inflammatory cytokines and metabolic abnormalities (Aumailley et al., 2015) that closely resembles the low grade, age-related inflammatory phenotype seen in Werner syndrome patients (Goto et al., 2012). Loss-of-function mutations in any of the Fanconi anemia (FA) genes, involved in DNA replication and the DDR, lead to neuronal decline, bone marrow failure, cancer, and premature aging (Bogliolo and Surralles, 2015). FANCC is required for antiviral host defenses and for the suppressing of inflammasome activation in mice, by removing damaged mitochondria (Sumpter et al., 2016); however, the mitophagy function of FANCC is independent of its role in DNA damage repair. Besides defects in genome maintenance, mice with mutations in genes coding for kinases involved in DDR manifest prominent inflammatory features. For instance, heterozygous mutant animals for Smg1, coding for a kinase with a known role in nonsense-mediated mRNA decay and the DDR, predisposes mice to higher levels of IL-6, CSF-1 and IL-1β cytokines and chronic inflammation in the lung and kidneys (Roberts et al., 2013). Mice deficient in ataxia telangiectasia mutated gene (ATM), upon exposure to lipopolysaccharide stimulation, associate with a prominent inflammatory phenotype that leads to Purkinje cell death (Yang et al., 2014). Likewise, Atm−/− mice are more sensitive to the deleterious effects of chronic dextran sulfate sodium-induced inflammation with greater upregulation of inflammatory cytokines, and higher percentages of activated CD69+ and CD44+ T-cells in the peripheral blood throughout treatment (Westbrook and Schiestl, 2010). Mice defective in DNA-PK, a nuclear DNA-dependent serine/threonine protein kinase involved in NHEJ and the DDR associate with an activation of cGAS-mediated antiviral innate immunity (Sun et al., 2020); DNA-PK binds DNA in the cytoplasm and stimulates the transcription of type I interferon (IFN), cytokine and chemokine genes in primary fibroblasts and mice (Ferguson et al., 2012). These findings could well explain why most patients with mutations in DNA-PKcs manifest a hyperactivated innate immune response and suffer from autoimmune diseases.
In addition to genetic models with DNA repair/DDR defects, there is a wide range of genotoxins that are known to trigger a pro-inflammatory response in mice. For instance, exposure of the skin to ultraviolet (UV) light leads to the formation of cyclobutane pyrimidine dimers (CPDs) and pyrimidine (6-4) pyrimidone photoproducts (6-4PPs). The UV-induced photolesions interfere with the process of mRNA synthesis and DNA replication (Garinis et al., 2005) triggering skin inflammation and erythema, edema, and hyperplasia. Photolyases are enzymes that are capable of removing the UV-induced CPDs or 6-4PPs upon exposure to visible light (Garinis et al., 2006). However, these enzymes are not present in placental mammals. Using transgenic mice that express a marsupial (Potorous tridactylis) CPD-specific photolyase transgene, either ubiquitously or specifically in the basal keratinocytes of the epidermis, it was shown that light-dependent removal of CPDs (but not 6-4PPs) is the primary cause of the great majority of UV-exposed skin semi-acute responses, including sunburn, apoptosis, hyperplasia as well as non-melanoma skin cancer (Jans et al., 2005; Jans et al., 2006). Murine models of whole thorax or hemithorax irradiation reliably recapitulate the pathogenesis and the clinical symptoms of DNA damage-induced pneumonitis and fibrosis (Wirsdorfer and Jendrossek, 2017). Similar symptoms are also quickly developed in mice exposed to the radiomimetic DNA damaging chemotherapeutic drug Bleomycin (Moeller et al., 2008). Bacterial genotoxins are also known to trigger single- or double strand breaks (DSBs) in target host cells, leading to chronic inflammation that may ultimately facilitate the oncogenic processes. For instance, the enterotoxigenic Bacteroides fragilis secretes B. fragilis toxin, a zinc-dependent metalloprotease that can induce colitis and colorectal cancer in multiple intestinal neoplasia (Min) mice (Housseau and Sears, 2010). Likewise, mice infected with Helicobacter hepaticus associate with the presence of the bacterial genotoxin cytolethal distending toxin, a chronic inflammatory response and the development of hepatic dysplastic nodules (Ge et al., 2007). Regulatory T cells (Tregs) are a subpopulation of T cells that suppress the induction and proliferation of effector T cells. A recent study in Tregs derived from individuals with autoimmune diseases or from an animal model for multiple sclerosis revealed an enhanced DDR signaling in these cells resulting from an increase in mitochondrial oxidative stress and impaired lysosomal function (Alissafi et al., 2020). Thus, as inflammation further inflicts DNA damage upon immune-system cells in the periphery, DNA damage-driven innate immune responses may take on a momentum of their own due to the amplification and feedback loops of the immune system leading to severe degenerative tissue changes over time (Figure 1).
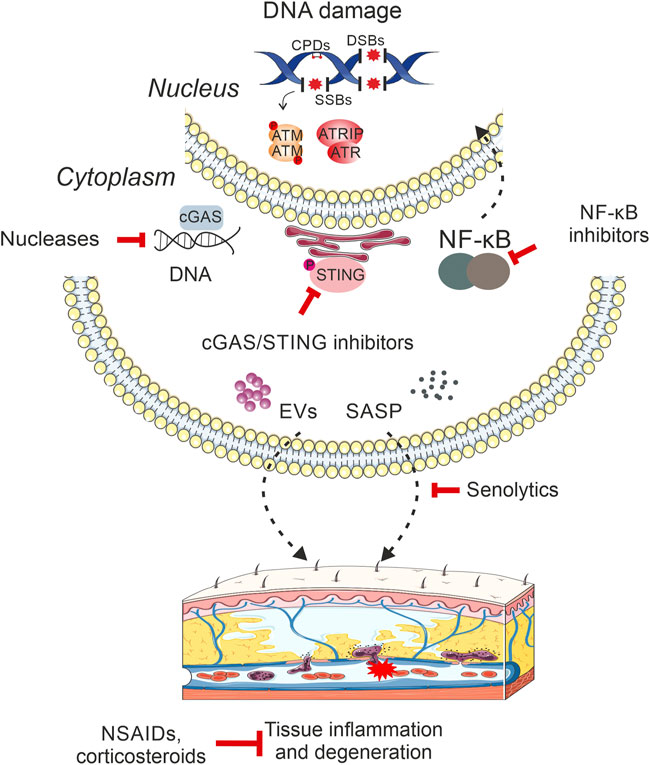
FIGURE 1. DNA damage-driven inflammation drives age-related pathology. DNA damage triggers the activation of innate immune responses, which act as a key mediator of tissue degenerative changes and age-related organismal decline. In particular, the presence of DNA breaks, small base modifications or bulky DNA adducts in the mammalian genome triggers the release of DNA moieties in the cytoplasm, which are predominantly sensed by the cGAS/STING signaling pathway stimulating the type I interferon (IFN) response. DNA damage also triggers the activation of NF-κB in the cytoplasm that translocates into the nucleus to activate the transcription of immune response gene targets. Moreover, the accumulation of irreparable DNA lesions may lead to the release of senescence-associated secretory phenotype (SASP) factors, such as cytokines, soluble growth factors, proteases as well as insoluble extracellular matrix components in the surrounding milieu. Cells with compromised genome integrity are also known to secrete extracellular vesicles that target a wide range of recipient cells leading to metabolic reprogramming and systemic inflammation. Eventually, the multiple links between persistent DNA damage and activation of innate immune responses in mammals lead to chronic inflammation that drives tissue deterioration, malfunction and organismal decline with aging. The recent development of rationalized intervention strategies (nucleases, corticosteroids etc.) against direct products of DNA damage itself i.e., nucleic acids or against inflammatory mediators are expected to profoundly lessen the adverse consequences of degenerative features that manifest with advancing age. Intervention methods targeting DNA damage-induced inflammatory stimuli are depicted in red inhibitory arrows.
As DNA damage drives inflammation and aging, it is tempting to consider therapeutic strategies that would lessen the inflammatory burden and delay the premature onset of associated tissue degenerative changes with old age. Non-steroidal anti-inflammatory drugs (NSAIDs) are medicines that are widely used to reduce inflammation and relieve symptoms of long-term pain. However, the use of NSAIDs is often associated with gastrointestinal problems, including stomach irritation and reflux and may increase the risk of cardiovascular conditions (Vonkeman and van de Laar, 2010). Corticosteroids present another category of anti-inflammatory drugs that are typically used to treat rheumatologic and chronic inflammatory diseases that associate with aging. However, depending on the dose, type of steroid and length of treatment, corticosteroid drugs may trigger a wide range of side effects, including weight gain, muscle weakness and lower resistance to infection, osteoporosis, diabetes, stomach irritation etc., (Yasir et al., 2022). Moreover, the high prevalence of chronic inflammatory diseases and the pervasive risks associated with the long-term use of available anti-inflammatory drugs requires that a new class of therapeutic regimens becomes available that would avoid the unnecessary use of NSAIDs and corticosteroid drugs. This is particularly relevant for the treatment of chronic inflammatory conditions that require repetitive or continuous treatment regimens. The use of senolytic drugs that can selectively eliminate senescent cells (and by inference also the release of pro-inflammatory SASP factors) or the targeting of the SASP response itself (rather than the senescent cells) may serve as an alternative way to reduce the inflammatory load driven by irreparable DNA damage (Gasek et al., 2021). Importantly, senolytics may not need to be administered continuously to trigger senolysis, thereby minimizing potential harmful side effects. However, the SASP response often entails hundreds of proteins and non-protein signaling factors, whose composition depends on the cell type involved and the mechanism driving senescence (Birch and Gil, 2020). Moreover, in certain cell types, such as in macrophages, the accumulation of age-related biomarkers e.g., lipofuscin and β-galactosidase or the increase in the mRNA levels of senescent-associated factors p16INK4A, p21CIP1 or SASP-associated IL-6 and IL-8 protein levels (Goulielmaki et al., 2020) may only reflect a reversible response of these cells to physiological stimuli (Hall et al., 2017), rather than the premature onset of replicative or stress-induced senescence (Franceschi et al., 2000). For DNA damage-driven inflammatory responses, it is, therefore, valid to consider alternative options that would target the cause rather than the symptoms of inflammation. Owing to the fact that DNA damage triggers the accumulation of cytoplasmic DNA moieties or RNA-DNA hybrids in cells (Chatzidoukaki et al., 2021), there have been recent attempts to develop a new class of therapeutic regimens that may prove valuable in ameliorating the harmful effects of DNA damage-driven inflammation. For instance, cytoplasmic DNA species are rapidly sensed by the enzyme cGAS (cyclic GMP-AMP synthase), which in turn activates the adaptor protein STING (stimulator of interferon genes) on the endoplasmic reticulum. As the cGAS-STING pathway can sense the cytosolic DNA activating the innate immune response, selective small molecules were developed as inhibitors, with the potential to target the cGAS-STING axis in humans. However, inhibition of the cGAS-STING pathway may provoke adverse effects in humans by increasing susceptibility to infection (Decout et al., 2021). Likewise, several immunosuppressant drugs that may be used to combat DNA damage-driven inflammatory responses by inhibiting e.g., JAK/STAT or NF-κB signalling, various TLRs, NADPH oxidase, IL-1β and TNF-α associate with an increased risk of infection, as well as with akinetic mutism, toxic encephalopathy, seizures, tremor and neurobehavioral changes (Fireman et al., 2004). More recently, we reasoned that the removal of cytoplasmic ss DNA moieties or RNA-DNA hybrids from cells would reduce the inflammatory load in damaged cells in vivo. To test this, we used extracellular vesicles (EVs) to deliver recombinant Mung Bean S1 or RNase H1 nucleases to inflamed DNA repair-deficient cells that rapidly accumulate DNA damage or to wild-type cells previously treated with a genotoxin. Interestingly, the nuclease-mediated removal of cytosolic nucleic acids from these cells curbed the Type I IFN response ameliorating the DNA damage-induced phenotype in targeted cells (Chatzidoukaki et al., 2021).
There is much work to be done before we will be able to dissect the functional links between persistent DNA damage and inflammation in vivo. The use of progeroid murine models with tissue-specific defects in genome maintenance will allow us to further delineate the causal contribution of specific cell types to systemic inflammation with old age (Figure 1). In parallel, animal models with tagged DNA repair factors coupled to functional genomics and proteomics strategies may prove valuable for identifying new gene targets or protein partners that could link genome maintenance with innate immune signaling. It will also be essential to identify how an active DDR originating from any alterations in the physicochemical structure of the DNA activates cytoplasmic stress responses and the release of proinflammatory factors in the tissue microenvironment. Likewise, it will be vital to dissect the functional links between DNA damage-driven chronic inflammation and metabolic rewiring with old age. Finally, the recent development of novel therapeutic strategies indicates that, in the long run, it may be more valuable to invest in approaches targeting the DNA damage itself rather than suppressing downstream proinflammatory signals. Such strategies could open new, meaningful avenues towards the development of new rationalized therapeutic interventions against a wide range of adverse pathological outcomes during aging (Tilstra et al., 2012; Karakasilioti et al., 2013).
EA, KS, and GG wrote the manuscript. EA conceived and designed the figure and table of the manuscript. GG contributed to the structure of the review and edited the final manuscript.
The Horizon 2020 ERC Consolidator grant “DeFiNER” (GA 64663); the ERC PoC “Inflacare” (GA 874456); the Horizon 2020 Marie Curie ITN “aDDRess” (GA 812829), and “HealthAge” (GA 812830), ELIDEK grants 631 and 1059; the “Research-Create-Innovate” actions (MIA-RTDI) “Panther”-00852 and “Liquid Pancreas”-00940 supported this work.
For the preparation of the figures, we acknowledge a service to medicine provided by Les Laboratoires. Servier (Servier Medical Art) (https://www.servier.com).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fragi.2022.973781/full#supplementary-material.
Acosta, J. C., Banito, A., Wuestefeld, T., Georgilis, A., Janich, P., Morton, J. P., et al. (2013). A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 15 (8), 978–990. doi:10.1038/ncb2784
Aden, K., Bartsch, K., Dahl, J., Reijns, M. A. M., Esser, D., Sheibani-Tezerji, R., et al. (2019). Epithelial RNase H2 maintains genome integrity and prevents intestinal tumorigenesis in mice. Gastroenterology 156 (1), 145–159. doi:10.1053/j.gastro.2018.09.047
Alissafi, T., Kalafati, L., Lazari, M., Filia, A., Kloukina, I., Manifava, M., et al. (2020). Mitochondrial oxidative damage underlies regulatory T cell defects in autoimmunity. Cell Metab. 32 (4), 591–604. doi:10.1016/j.cmet.2020.07.001
Apostolou, Z., Chatzinikolaou, G., Stratigi, K., and Garinis, G. A. (2019). Nucleotide excision repair and transcription-associated genome instability. Bioessays 41 (4), e1800201. doi:10.1002/bies.201800201
Aumailley, L., Garand, C., Dubois, M. J., Johnson, F. B., Marette, A., and Lebel, M. (2015). Metabolic and phenotypic differences between mice producing a werner syndrome helicase mutant protein and wrn null mice. PLoS One 10 (10), e0140292. doi:10.1371/journal.pone.0140292
Babu, M., Beloglazova, N., Flick, R., Graham, C., Skarina, T., Nocek, B., et al. (2011). A dual function of the CRISPR-Cas system in bacterial antivirus immunity and DNA repair. Mol. Microbiol. 79 (2), 484–502. doi:10.1111/j.1365-2958.2010.07465.x
Barnhoorn, S., Uittenboogaard, L. M., Jaarsma, D., Vermeij, W. P., Tresini, M., Weymaere, M., et al. (2014). Cell-autonomous progeroid changes in conditional mouse models for repair endonuclease XPG deficiency. PLoS Genet. 10 (10), e1004686. doi:10.1371/journal.pgen.1004686
Birch, J., and Gil, J. (2020). Senescence and the SASP: many therapeutic avenues. Genes Dev. 34 (23-24), 1565–1576. doi:10.1101/gad.343129.120
Boboila, C., Alt, F. W., and Schwer, B. (2012). Classical and alternative end-joining pathways for repair of lymphocyte-specific and general DNA double-strand breaks. Adv. Immunol. 116, 1–49. doi:10.1016/B978-0-12-394300-2.00001-6
Bogliolo, M., and Surralles, J. (2015). Fanconi anemia: a model disease for studies on human genetics and advanced therapeutics. Curr. Opin. Genet. Dev. 33, 32–40. doi:10.1016/j.gde.2015.07.002
Calvo, J. A., Meira, L. B., Lee, C. Y. I., Moroski-Erkul, C. A., Abolhassani, N., Taghizadeh, K., et al. (2012). DNA repair is indispensable for survival after acute inflammation. J. Clin. Invest. 122 (7), 2680–2689. doi:10.1172/JCI63338
Campisi, J., and d'Adda di Fagagna, F. (2007). Cellular senescence: when bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 8 (9), 729–740. doi:10.1038/nrm2233
Cavanagh, M. M., Weyand, C. M., and Goronzy, J. J. (2012). Chronic inflammation and aging: DNA damage tips the balance. Curr. Opin. Immunol. 24 (4), 488–493. doi:10.1016/j.coi.2012.04.003
Champsaur, M., and Lanier, L. L. (2010). Effect of NKG2D ligand expression on host immune responses. Immunol. Rev. 235 (1), 267–285. doi:10.1111/j.0105-2896.2010.00893.x
Chatzidoukaki, O., Goulielmaki, E., Schumacher, B., and Garinis, G. A. (2020). DNA damage response and metabolic reprogramming in health and disease. Trends Genet. 36 (10), 777–791. doi:10.1016/j.tig.2020.06.018
Chatzidoukaki, O., Stratigi, K., Goulielmaki, E., Niotis, G., Akalestou-Clocher, A., Gkirtzimanaki, K., et al. (2021). R-loops trigger the release of cytoplasmic ssDNAs leading to chronic inflammation upon DNA damage. Sci. Adv. 7 (47), eabj5769. doi:10.1126/sciadv.abj5769
Chen, Y. W., Harris, R. A., Hatahet, Z., and Chou, K. m. (2015). Ablation of XP-V gene causes adipose tissue senescence and metabolic abnormalities. Proc. Natl. Acad. Sci. U. S. A. 112 (33), E4556–E4564. doi:10.1073/pnas.1506954112
Cohen, H. J., Harris, T., and Pieper, C. F. (2003). Coagulation and activation of inflammatory pathways in the development of functional decline and mortality in the elderly. Am. J. Med. 114 (3), 180–187. doi:10.1016/s0002-9343(02)01484-5
Coppe, J. P., Patil, C. K., Rodier, F., Sun, Y., Munoz, D. P., Goldstein, J., et al. (2008). Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 6 (12), 2853–2868. doi:10.1371/journal.pbio.0060301
Costantino, L., and Koshland, D. (2015). The yin and yang of R-loop biology. Curr. Opin. Cell Biol. 34, 39–45. doi:10.1016/j.ceb.2015.04.008
Crow, Y. J., and Manel, N. (2015). Aicardi-Goutieres syndrome and the type I interferonopathies. Nat. Rev. Immunol. 15 (7), 429–440. doi:10.1038/nri3850
Decout, A., Katz, J. D., Venkatraman, S., and Ablasser, A. (2021). The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol. 21 (9), 548–569. doi:10.1038/s41577-021-00524-z
Di Noia, J. M., and Neuberger, M. S. (2007). Molecular mechanisms of antibody somatic hypermutation. Annu. Rev. Biochem. 76, 1–22. doi:10.1146/annurev.biochem.76.061705.090740
Dianov, G., BisChoff, C., Piotrowski, J., and Bohr, V. A. (1998). Repair pathways for processing of 8-oxoguanine in DNA by mammalian cell extracts. J. Biol. Chem. 273 (50), 33811–33816. doi:10.1074/jbc.273.50.33811
Faustin, B., and Reed, J. C. (2008). Sunburned skin activates inflammasomes. Trends Cell Biol. 18 (1), 4–8. doi:10.1016/j.tcb.2007.10.004
Ferguson, B. J., Mansur, D. S., Peters, N. E., Ren, H., and Smith, G. L. (2012). DNA-PK is a DNA sensor for IRF-3-dependent innate immunity. Elife 1, e00047. doi:10.7554/eLife.00047
Ferrucci, L., Harris, T. B., Guralnik, J. M., Tracy, R. P., Corti, M. C., Cohen, H. J., et al. (1999). Serum IL-6 level and the development of disability in older persons. J. Am. Geriatr. Soc. 47 (6), 639–646. doi:10.1111/j.1532-5415.1999.tb01583.x
Fireman, M., DiMartini, A. F., Armstrong, S. C., and Cozza, K. L. (2004). Immunosuppressants. Psychosomatics 45 (4), 354–360. doi:10.1176/appi.psy.45.4.354
Franceschi, C., BonafeM., , ValenSin, S., OlivieriF., , De LucaM., , Ottaviani, E., et al. (2000). Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 908, 244–254. doi:10.1111/j.1749-6632.2000.tb06651.x
Fumagalli, M., and d'Adda di Fagagna, F. (2009). SASPense and DDRama in cancer and ageing. Nat. Cell Biol. 11 (8), 921–923. doi:10.1038/ncb0809-921
Garinis, G. A., Mitchell, J. R., Moorhouse, M. J., Hanada, K., de Waard, H., Vandeputte, D., et al. (2005). Transcriptome analysis reveals cyclobutane pyrimidine dimers as a major source of UV-induced DNA breaks. EMBO J. 24 (22), 3952–3962. doi:10.1038/sj.emboj.7600849
Garinis, G. A., Jans, J., and van der Horst, G. T. (2006). Photolyases: capturing the light to battle skin cancer. Future Oncol. 2 (2), 191–199. doi:10.2217/14796694.2.2.191
Garinis, G. A., van der Horst, G. T. J., Vijg, J., and Hoeijmakers, J. H. J. (2008). DNA damage and ageing: new-age ideas for an age-old problem. Nat. Cell Biol. 10 (11), 1241–1247. doi:10.1038/ncb1108-1241
Garinis, G. A., Uittenboogaard, L. M., Stachelscheid, H., Fousteri, M., van Ijcken, W., Breit, T. M., et al. (2009). Persistent transcription-blocking DNA lesions trigger somatic growth attenuation associated with longevity. Nat. Cell Biol. 11 (5), 604–615. doi:10.1038/ncb1866
Garinis, G. A. (2008). Nucleotide excision repair deficiencies and the somatotropic axis in aging. Hormones 7 (1), 9–16. doi:10.14310/horm.2002.1111032
Gasek, N. S., Kuchel, G. A., Kirkland, J. L., and Xu, M. (2021). Strategies for targeting senescent cells in human disease. Nat. Aging 1 (10), 870–879. doi:10.1038/s43587-021-00121-8
Gasser, S., and Raulet, D. H. (2006). The DNA damage response arouses the immune system. Cancer Res. 66 (8), 3959–3962. doi:10.1158/0008-5472.CAN-05-4603
Gasser, S., Orsulic, S., Brown, E. J., and Raulet, D. H. (2005). The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature 436 (7054), 1186–1190. doi:10.1038/nature03884
Gaugler, M. H., Squiban, C., vAn der Meeren, A., Bertho, J. M., VandaMMeM., , and Mouthon, M. A. (1997). Late and persistent up-regulation of intercellular adhesion molecule-1 (ICAM-1) expression by ionizing radiation in human endothelial cells in vitro. Int. J. Radiat. Biol. 72 (2), 201–209. doi:10.1080/095530097143428
Ge, Z., Rogers, A. B., Feng, Y., Lee, A., Xu, S., Taylor, N. S., et al. (2007). Bacterial cytolethal distending toxin promotes the development of dysplasia in a model of microbially induced hepatocarcinogenesis. Cell. Microbiol. 9 (8), 2070–2080. doi:10.1111/j.1462-5822.2007.00939.x
Goto, M., Sugimoto, K., Hayashi, S., Ogino, T., Sugimoto, M., Furuichi, Y., et al. (2012). Aging-associated inflammation in healthy Japanese individuals and patients with Werner syndrome. Exp. Gerontol. 47 (12), 936–939. doi:10.1016/j.exger.2012.08.010
Goulielmaki, E., Ioannidou, A., Tsekrekou, M., Stratigi, K., Poutakidou, I. K., Gkirtzimanaki, K., et al. (2020). Tissue-infiltrating macrophages mediate an exosome-based metabolic reprogramming upon DNA damage. Nat. Commun. 11 (1), 42. doi:10.1038/s41467-019-13894-9
Goulielmaki, E., Tsekrekou, M., Batsiotos, N., Ascensao-Ferreira, M., Ledaki, E., Stratigi, K., et al. (2021). The splicing factor XAB2 interacts with ERCC1-XPF and XPG for R-loop processing. Nat. Commun. 12 (1), 3153. doi:10.1038/s41467-021-23505-1
Gregg, S. Q., Gutierrez, V., Robinson, A. R., Woodell, T., Nakao, A., Ross, M. A., et al. (2012). A mouse model of accelerated liver aging caused by a defect in DNA repair. Hepatology 55 (2), 609–621. doi:10.1002/hep.24713
Grivennikov, S. I., Greten, F. R., and Karin, M. (2010). Immunity, inflammation, and cancer. Cell 140 (6), 883–899. doi:10.1016/j.cell.2010.01.025
Hall, B. M., Balan, V., Gleiberman, A. S., Strom, E., Krasnov, P., Virtuoso, L. P., et al. (2017). p16(Ink4a) and senescence-associated beta-galactosidase can be induced in macrophages as part of a reversible response to physiological stimuli. Aging (Albany NY) 9 (8), 1867–1884. doi:10.18632/aging.101268
Hasty, P., Campisi, J., Hoeijmakers, J., van Steeg, H., and Vijg, J. (2003). Aging and genome maintenance: lessons from the mouse? Science 299 (5611), 1355–1359. doi:10.1126/science.1079161
Hayden, M. S., and Ghosh, S. (2008). Shared principles in NF-kappaB signaling. Cell 132 (3), 344–362. doi:10.1016/j.cell.2008.01.020
Hoeijmakers, J. H. (1994). Human nucleotide excision repair syndromes: molecular clues to unexpected intricacies. Eur. J. Cancer 30A (13), 1912–1921. doi:10.1016/0959-8049(94)00381-e
Housseau, F., and Sears, C. L. (2010). Enterotoxigenic Bacteroides fragilis (ETBF)-mediated colitis in min (Apc+/-) mice: a human commensal-based murine model of colon carcinogenesis. Cell Cycle 9 (1), 3–5. doi:10.4161/cc.9.1.10352
Ioannidou, A., Goulielmaki, E., and Garinis, G. A. (2016). DNA damage: From chronic inflammation to age-related deterioration. Front. Genet. 7, 187. doi:10.3389/fgene.2016.00187
Jans, J., Schul, W., Sert, Y. G., Rijksen, Y., Rebel, H., Eker, A. P. M., et al. (2005). Powerful skin cancer protection by a CPD-photolyase transgene. Curr. Biol. 15 (2), 105–115. doi:10.1016/j.cub.2005.01.001
Jans, J., Garinis, G. A., Schul, W., van Oudenaren, A., Moorhouse, M., Smid, M., et al. (2006). Differential role of basal keratinocytes in UV-induced immunosuppression and skin cancer. Mol. Cell. Biol. 26 (22), 8515–8526. doi:10.1128/MCB.00807-06
Kajitani, G. S., Brace, L., Trevino-Villarreal, J. H., Trocha, K., MacArthur, M. R., Vose, S., et al. (2021). Neurovascular dysfunction and neuroinflammation in a Cockayne syndrome mouse model. Aging (Albany NY) 13 (19), 22710–22731. doi:10.18632/aging.203617
Kamileri, I., Karakasilioti, I., and Garinis, G. A. (2012). Nucleotide excision repair: new tricks with old bricks. Trends Genet. 28 (11), 566–573. doi:10.1016/j.tig.2012.06.004
Karakasilioti, I., and Garinis, G. A. (2014). Tissue-specific aging: a tale of functional asymmetry. Aging (Albany NY) 6 (1), 7–8. doi:10.18632/aging.100635
Karakasilioti, I., Kamileri, I., Chatzinikolaou, G., Kosteas, T., Vergadi, E., Robinson, A. R., et al. (2013). DNA damage triggers a chronic autoinflammatory response, leading to fat depletion in NER progeria. Cell Metab. 18 (3), 403–415. doi:10.1016/j.cmet.2013.08.011
Karpova, A. Y., Trost, M., Murray, J. M., Cantley, L. C., and Howley, P. M. (2002). Interferon regulatory factor-3 is an in vivo target of DNA-PK. Proc. Natl. Acad. Sci. U. S. A. 99 (5), 2818–2823. doi:10.1073/pnas.052713899
Kooter, I. M., Frederix, K., Spronk, H. M. H., Boere, A. J. F., Leseman, D. L. A. C., van Steeg, H., et al. (2008). Lung inflammation and thrombogenic responses in a time course study of Csb mice exposed to ozone. J. Appl. Toxicol. 28 (6), 779–787. doi:10.1002/jat.1339
Lieber, M. R., Ma, Y., Pannicke, U., and Schwarz, K. (2003). Mechanism and regulation of human non-homologous DNA end-joining. Nat. Rev. Mol. Cell Biol. 4 (9), 712–720. doi:10.1038/nrm1202
Mackenzie, K. J., Carroll, P., Lettice, L., Tarnauskaite, Z., Reddy, K., Dix, F., et al. (2016). Ribonuclease H2 mutations induce a cGAS/STING-dependent innate immune response. EMBO J. 35 (8), 831–844. doi:10.15252/embj.201593339
Malaquin, N., Vercamer, C., Bouali, F., Martien, S., Deruy, E., Wernert, N., et al. (2013). Senescent fibroblasts enhance early skin carcinogenic events via a paracrine MMP-PAR-1 axis. PLoS One 8 (5), e63607. doi:10.1371/journal.pone.0063607
Meira, L. B., Bugni, J. M., Green, S. L., Lee, C. W., Pang, B., Borenshtein, D., et al. (2008). DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. J. Clin. Invest. 118 (7), 2516–2525. doi:10.1172/JCI35073
Menendez, D., Shatz, M., Azzam, K., Garantziotis, S., Fessler, M. B., and Resnick, M. A. (2011). The Toll-like receptor gene family is integrated into human DNA damage and p53 networks. PLoS Genet. 7 (3), e1001360. doi:10.1371/journal.pgen.1001360
Miyauchi-Hashimoto, H., Tanaka, K., and Horio, T. (1996). Enhanced inflammation and immunosuppression by ultraviolet radiation in xeroderma pigmentosum group A (XPA) model mice. J. Invest. Dermatol. 107 (3), 343–348. doi:10.1111/1523-1747.ep12363295
Moeller, A., Ask, K., Warburton, D., Gauldie, J., and Kolb, M. (2008). The bleomycin animal model: a useful tool to investigate treatment options for idiopathic pulmonary fibrosis? Int. J. Biochem. Cell Biol. 40 (3), 362–382. doi:10.1016/j.biocel.2007.08.011
Mori, T., Anazawa, Y., Iiizumi, M., Fukuda, S., Nakamura, Y., and Arakawa, H. (2002). Identification of the interferon regulatory factor 5 gene (IRF-5) as a direct target for p53. Oncogene 21 (18), 2914–2918. doi:10.1038/sj.onc.1205459
Muers, M. (2011). Mutation: the perils of transcription. Nat. Rev. Genet. 12 (3), 156. doi:10.1038/nrg2960
Niedernhofer, L. J., Odijk, H., Budzowska, M., van Drunen, E., Maas, A., Theil, A. F., et al. (2004). The structure-specific endonuclease Ercc1-Xpf is required to resolve DNA interstrand cross-link-induced double-strand breaks. Mol. Cell. Biol. 24 (13), 5776–5787. doi:10.1128/MCB.24.13.5776-5787.2004
Niedernhofer, L. J., Garinis, G. A., Raams, A., Lalai, A. S., Robinson, A. R., Appeldoorn, E., et al. (2006). A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature 444 (7122), 1038–1043. doi:10.1038/nature05456
Ohanna, M., Giuliano, S., Bonet, C., Imbert, V., Hofman, V., Zangari, J., et al. (2011). Senescent cells develop a PARP-1 and nuclear factor-{kappa}B-associated secretome (PNAS). Genes Dev. 25 (12), 1245–1261. doi:10.1101/gad.625811
Pamment, J., Ramsay, E., Kelleher, M., Dornan, D., and Ball, K. L. (2002). Regulation of the IRF-1 tumour modifier during the response to genotoxic stress involves an ATM-dependent signalling pathway. Oncogene 21 (51), 7776–7785. doi:10.1038/sj.onc.1205981
Pieren, D. K. J., Smits, N. A. M., Imholz, S., Nagarajah, B., van Oostrom, C. T., Brandt, R. M. C., et al. (2021). Compromised DNA repair promotes the accumulation of regulatory T cells with an aging-related phenotype and responsiveness. Front. Aging 2, 667193. doi:10.3389/fragi.2021.667193
Rieckher, M., Garinis, G. A., and Schumacher, B. (2021). Molecular pathology of rare progeroid diseases. Trends Mol. Med. 27, 907–922. doi:10.1016/j.molmed.2021.06.011
Roberts, T. L., Ho, U., Luff, J., Lee, C. S., Apte, S. H., MacDonald, K. P. A., et al. (2013). Smg1 haploinsufficiency predisposes to tumor formation and inflammation. Proc. Natl. Acad. Sci. U. S. A. 110 (4), E285–E294. doi:10.1073/pnas.1215696110
Rodier, F., Coppe, J. P., Patil, C. K., Hoeijmakers, W. A. M., Munoz, D. P., Raza, S. R., et al. (2009). Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 11 (8), 973–979. doi:10.1038/ncb1909
Schermer, B., Bartels, V., Frommolt, P., Habermann, B., Braun, F., Schultze, J. L., et al. (2013). Transcriptional profiling reveals progeroid Ercc1(-/Δ) mice as a model system for glomerular aging. BMC Genomics 14, 559. doi:10.1186/1471-2164-14-559
Schroder, K., and Tschopp, J. (2010). The inflammasomes. Cell 140 (6), 821–832. doi:10.1016/j.cell.2010.01.040
Schumacher, B., van der Pluijm, I., Moorhouse, M. J., Kosteas, T., Robinson, A. R., Suh, Y., et al. (2008). Delayed and accelerated aging share common longevity assurance mechanisms. PLoS Genet. 4 (8), e1000161. doi:10.1371/journal.pgen.1000161
Schumacher, B., Garinis, G. A., and Hoeijmakers, J. H. (2008). Age to survive: DNA damage and aging. Trends Genet. 24 (2), 77–85. doi:10.1016/j.tig.2007.11.004
Schumacher, B., Hoeijmakers, J. H., and Garinis, G. A. (2009). Sealing the gap between nuclear DNA damage and longevity. Mol. Cell. Endocrinol. 299 (1), 112–117. doi:10.1016/j.mce.2008.10.031
Shaw, A. C., Goldstein, D. R., and Montgomery, R. R. (2013). Age-dependent dysregulation of innate immunity. Nat. Rev. Immunol. 13 (12), 875–887. doi:10.1038/nri3547
Shibuya, A., Hannum, C., Yssel, H., Franz-Bacon, K., and McClanahan, T. (1996). DNAM-1, a novel adhesion molecule involved in the cytolytic function of T lymphocytes. Immunity 4 (6), 573–581. doi:10.1016/s1074-7613(00)70060-4
Skourti-Stathaki, K., and Proudfoot, N. J. (2014). A double-edged sword: R loops as threats to genome integrity and powerful regulators of gene expression. Genes Dev. 28 (13), 1384–1396. doi:10.1101/gad.242990.114
Skourti-Stathaki, K., Proudfoot, N. J., and Gromak, N. (2011). Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol. Cell 42 (6), 794–805. doi:10.1016/j.molcel.2011.04.026
Soriani, A., Zingoni, A., Cerboni, C., Iannitto, M. L., Ricciardi, M. R., Di Gialleonardo, V., et al. (2009). ATM-ATR-dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood 113 (15), 3503–3511. doi:10.1182/blood-2008-08-173914
Stratigi, K., Chatzidoukaki, O., and Garinis, G. A. (2017). DNA damage-induced inflammation and nuclear architecture. Mech. Ageing Dev. 165, 17–26. doi:10.1016/j.mad.2016.09.008
Sumpter, R., Sirasanagandla, S., Fernandez, A. F., Wei, Y., Dong, X., Franco, L., et al. (2016). Fanconi anemia proteins function in mitophagy and immunity. Cell 165 (4), 867–881. doi:10.1016/j.cell.2016.04.006
Sun, X., Liu, T., Zhao, J., Xia, H., Xie, J., Guo, Y., et al. (2020). DNA-PK deficiency potentiates cGAS-mediated antiviral innate immunity. Nat. Commun. 11 (1), 6182. doi:10.1038/s41467-020-19941-0
Tilstra, J. S., Robinson, A. R., Wang, J., Gregg, S. Q., Clauson, C. L., Reay, D. P., et al. (2012). NF-κB inhibition delays DNA damage-induced senescence and aging in mice. J. Clin. Invest. 122 (7), 2601–2612. doi:10.1172/JCI45785
Tumurkhuu, G., Shimada, K., Dagvadorj, J., Crother, T. R., Zhang, W., Luthringer, D., et al. (2016). Ogg1-Dependent DNA repair regulates NLRP3 inflammasome and prevents atherosclerosis. Circ. Res. 119 (6), e76–90. doi:10.1161/CIRCRESAHA.116.308362
van de Ven, M., Andressoo, J. O., Holcomb, V. B., Hasty, P., Suh, Y., van Steeg, H., et al. (2007). Extended longevity mechanisms in short-lived progeroid mice: identification of a preservative stress response associated with successful aging. Mech. Ageing Dev. 128 (1), 58–63. doi:10.1016/j.mad.2006.11.011
van der Pluijm, I., Garinis, G. A., Brandt, R. M. C., Gorgels, T. G. M. F., Wijnhoven, S. W., Diderich, K. E. M., et al. (2007). Impaired genome maintenance suppresses the growth hormone-insulin-like growth factor 1 axis in mice with Cockayne syndrome. PLoS Biol. 5 (1), e2. doi:10.1371/journal.pbio.0050002
Vonkeman, H. E., and van de Laar, M. A. (2010). Nonsteroidal anti-inflammatory drugs: adverse effects and their prevention. Semin. Arthritis Rheum. 39 (4), 294–312. doi:10.1016/j.semarthrit.2008.08.001
Westbrook, A. M., and Schiestl, R. H. (2010). Atm-deficient mice exhibit increased sensitivity to dextran sulfate sodium-induced colitis characterized by elevated DNA damage and persistent immune activation. Cancer Res. 70 (5), 1875–1884. doi:10.1158/0008-5472.CAN-09-2584
Wimberly, H., Shee, C., Thornton, P. C., Sivaramakrishnan, P., Rosenberg, S. M., and Hastings, P. J. (2013). R-loops and nicks initiate DNA breakage and genome instability in non-growing Escherichia coli. Nat. Commun. 4, 2115. doi:10.1038/ncomms3115
Wirsdorfer, F., and Jendrossek, V. (2017). Modeling DNA damage-induced pneumopathy in mice: insight from danger signaling cascades. Radiat. Oncol. 12 (1), 142. doi:10.1186/s13014-017-0865-1
Yang, Y., Hui, C. W., Li, J., and Herrup, K. (2014). The interaction of the atm genotype with inflammation and oxidative stress. PLoS One 9 (1), e85863. doi:10.1371/journal.pone.0085863
Yasir, M., Goyal, A., and Sonthalia, S. (2022). Corticosteroid adverse effects. Treasure Island FL: StatPearls.
Yousefzadeh, M. J., Flores, R. R., Zhu, Y., Schmiechen, Z. C., Brooks, R. W., Trussoni, C. E., et al. (2021). An aged immune system drives senescence and ageing of solid organs. Nature 594 (7861), 100–105. doi:10.1038/s41586-021-03547-7
Yu, A. M., Calvo, J. A., Muthupalani, S., and Samson, L. D. (2016). The Mbd4 DNA glycosylase protects mice from inflammation-driven colon cancer and tissue injury. Oncotarget 7 (19), 28624–28636. doi:10.18632/oncotarget.8721
Keywords: DNA damage, DNA repair, inflammation, aging, diseases
Citation: Arvanitaki ES, Stratigi K and Garinis GA (2022) DNA damage, inflammation and aging: Insights from mice. Front. Aging 3:973781. doi: 10.3389/fragi.2022.973781
Received: 20 June 2022; Accepted: 26 August 2022;
Published: 07 September 2022.
Edited by:
Ingrid Van Der Pluijm, Erasmus MC, NetherlandsReviewed by:
Atsushi Shibata, Gunma University, JapanCopyright © 2022 Arvanitaki, Stratigi and Garinis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: George A. Garinis, Z2FyaW5pc0BpbWJiLmZvcnRoLmdy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.