 Naibedya Dutta†
Naibedya Dutta† Ryo Higuchi-Sanabria
Ryo Higuchi-Sanabria
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Aging, 24 March 2022
Sec. Aging, Metabolism and Redox Biology
Volume 3 - 2022 | https://doi.org/10.3389/fragi.2022.860404
This article is part of the Research TopicModel Organisms in Aging Research: Caenorhabditis elegansView all 13 articles
Organisms are constantly exposed to stress both from the external environment and internally within the cell. To maintain cellular homeostasis under different environmental and physiological conditions, cell have adapted various stress response signaling pathways, such as the heat shock response (HSR), unfolded protein responses of the mitochondria (UPRMT), and the unfolded protein response of the endoplasmic reticulum (UPRER). As cells grow older, all cellular stress responses have been shown to deteriorate, which is a major cause for the physiological consequences of aging and the development of numerous age-associated diseases. In contrast, elevated stress responses are often associated with lifespan extension and amelioration of degenerative diseases in different model organisms, including C. elegans. Activating cellular stress response pathways could be considered as an effective intervention to alleviate the burden of aging by restoring function of essential damage-clearing machinery, including the ubiquitin-proteosome system, chaperones, and autophagy. Here, we provide an overview of newly emerging concepts of these stress response pathways in healthy aging and longevity with a focus on the model organism, C. elegans.
Rejuvenation of human life by reducing the damaging effects of aging is one of the trending focuses of current biological research across the globe. Aging is an obvious physiological condition for all living organisms on this planet. It has been designated as a cumulative impairment of different cellular events, which are mainly associated with the maintenance of cellular homeostasis. This frailty condition, broadly influenced by various stressors or genetic factors, leads to the development of major disease conditions including diabetes, neurodegenerative disorders, cardiovascular disorders, and cancer, all of which together increases the vulnerability to death (Franceschi et al., 2018). In 2014, Carlos López-Otín et al. has characterized nine major factors as the hallmark of aging: genomic instability, telomere attrition, epigenetic alterations, and loss of protein homeostasis (proteostasis) categorized as the primary hallmarks of aging; stem cell exhaustion and altered intercellular communication referred to as integrative hallmarks; and–mitochondrial dysfunction, deregulated nutrient sensing, and cellular senescence categorized as antagonistic hallmarks (López-Otín et al., 2013).
Stress, generated from different endogenous or exogenous sources can disturb homeostasis across cellular compartments, contribute to cellular dysfunction, and impact the aging process (Li et al., 2021). As such, living organisms have acquired different unique cellular signaling pathways through evolution to cope with the constant exposure of different internal or external stresses (Kirkwood and Austad 2000). These responses generally include the activation of a widespread transcriptional program to promote genes important for restoring cellular homeostasis. These include the heat-shock response (HSR) and organelle-specific stress response pathways like the endoplasmic reticulum unfolded protein response (UPRER) and the mitochondrial unfolded protein response (UPRMT), which have been studied in different organisms, including Caenorhabditis elegans (Kenyon 2010; Kourtis and Tavernarakis 2011). Often, it is the breakdown or dysregulation of these important cellular quality control and stress response machineries that are causative of aging. For example, in neurodegenerative disorders like Parkinson’s disease (PD), Alzheimer’s disease (AD), and Huntington’s disease (HD), misfolded protein accumulation caused by compromised activation of stress responses lead to decreased health. In addition, dysfunction of different organelles such as the mitochondria and endoplasmic reticulum under these conditions can also facilitate neurodegenerative diseases and metabolic disorders (Ozcan and Tabas 2012; Hartl 2017; Colla 2019; Misrani et al., 2021).
Using C. elegans as a model system, research over the last few decades has established the importance of the activation of cellular stress response pathways in healthy life. Stimulation of stress response pathways can restore cellular homeostasis and reduce the risk of the manifestation of age-related diseases, so lengthening life span. In this context, short exposure to mild stress can be beneficial to organismal health and longevity. Termed hormesis, early exposure to stress can activate critical cellular stress responses that can mitigate the accumulation of damage at advanced age (Rattan 2004). Many of these pathways, which include the HSR, UPRMT, and UPRER have originally been identified in C. elegans, again highlighting the strength of this model in stress biology. In this review, we sum up our latest understanding on the significance of successful stress response pathways in longevity, distinctly emphasizing the model organism C. elegans.
In 1962 an accidental finding by an Italian scientist Ferruccio Ritossa opens a new window in biological research. He observed a different and unique puffing pattern in the polytene chromosome of the salivary cells of the fruit fly, Drosophila busckii under an elevated temperature condition (Ritossa 1962; Ritossa 1996). That unknown chromosomal puff was later identified as the active transcriptional region for the synthesis of a special group of proteins known as heat shock proteins, which maintain proteostasis in a cell by facilitating protein stabilization, refolding of misfolded protein structure, protein translocation, and degradation of toxic protein aggregates (Tissiéres et al., 1974; Lindquist and Craig 1988; Anckar and Sistonen 2011). These heat shock proteins are conserved across living organisms and considered as the principal functional unit for the HSR. Heat shock proteins are widely grouped into two families based on their molecular weight: First, the large ATP-dependent heat shock proteins of molecular mass between 40 and 105 kDa, including the major chaperone proteins, HSP70, HSP90, etc., and second, the small heat shock proteins of molecular mass between 8 and 25 kDa, which includes HSP27, HSP25, ubiquitin, and others (Jee 2016).
Most protein chaperones share similarities in their function. However, they differ in respect to their cellular localization, substrate specificity, and mechanistic details. The HSP70 family make up the most abundant chaperone proteins present in the cell. The cytosolic HSP70 has several homologs, which are present in different subcellular compartments including the heat shock cognate 70 (HSC70) present in the cytosol along with HSP70 and GRP78/BiP present in the endoplasmic reticulum (Radons 2016). HSP90 is another ATP-dependent chaperone protein that is constitutively expressed in cells and found in different cellular compartments like the cytosol, mitochondria, endoplasmic reticulum, nucleus etc. HSP90 shows the specificity to bind with a large number of misfolded proteins and provide support to refold to their functional state (Schopf et al., 2017). In contrast to the ATP-dependent chaperone proteins, small heat shock proteins function in an ATP-independent manner. These small heat shock proteins such as HSP27 and αβ-crystallin show induced expression in response to stress. During proteotoxic stress, they bind with misfolded client proteins and block further misfolding and/or the formation of misfolded protein aggregates until the clients are delivered to repairing chaperones like HSP70 and HSP40 (Bakthisaran et al., 18542015; Schopf et al., 2017). In addition to protein refolding, many molecular chaperones also participate in proteasomal degradation. For example, chaperone molecules can couple with ubiquitin ligase proteins that facilitate the polyubiquitination of misfolded proteins and prepare them for proteasomal degradation.
Heat shock proteins are synthesized in a large amount immediately after sensing stress, such as elevated temperature, hypoxia, exposure to heavy metals, etc. This universally conserved cellular stress response is orchestrated by a key transcription factor known as heat shock factor 1 or simply HSF1 (Anckar and Sistonen 2011). In invertebrates, the heat shock factor is encoded by a single gene, whereas in vertebrates, four major heat shock factor isoforms, HSF1, HSF2, HSF3, and HSF4 have evolved. In the case of both vertebrates and invertebrates, HSF1 plays the major decisive role in the activation of the HSR while the other isoforms that exist only in vertebrates are less studied and found to show some unique and sometimes tissue-specific functions. Interestingly, while HSF1, HSF2, and HSF4 have been found in all vertebrates, HSF3 was specifically observed in avian species only (Nakai et al., 1995). HSF2 plays an important role in female fertility, spermatogenesis, and early development, whereas HSF4 participates in eye lens development (Schuetz et al., 1991; Nakai et al., 1997).
HSF1 is made of 529 amino acids and its structure consists of four major structural/functional domains: the DNA binding domain (DBD), oligomerization domain, regulatory domain (RD), and transactivation domain (TAD) (Figure 1A). The DBD is present at the amino-terminal end of HSF1 and forms a helix turn helix structure. Through this domain, HSF1 binds with its specific DNA sequence and that association is stabilized by the interaction between the amphipathic helical region and the hydrophobic DNA pocket (Neudegger et al., 2016). Next to the DBD, the alpha-helix rich oligomerization domain is situated through which one HSF1 molecule interacts with another (Peteranderl et al., 1999). That oligomerization domain is further divided into two subdomains HR-A and HR-B. A separate and similar domain HR-C is present between the transactivation domain and the regulatory domain that helps to inhibit random oligomerization of HSF1 (Anckar and Sistonen 2011; Rabindran et al., 1993). The regulatory domain, present alongside the oligomerization domain (between HR-A/HR-B and HR-C) controls the transactivation of HSF1 and is the site for regulatory post-translational modifications, such as phosphorylation, acetylation/deacetylation, and sumoylation (Anckar and Sistonen 2011). Ultimately at the C-terminus of the HSF1 protein is the TAD, which facilitates the transcriptional activation of its target genes. However, this is the most structurally unknown part of the protein to date. In normal growth conditions, monomeric HSF1 exists as an inactive form in the cytoplasm in association with a complex of some regulatory proteins such as HSP70, HSP90, and TRiC. These molecular chaperones act as a negative regulator for HSF1 function (Anckar and Sistonen 2011; Shi et al., 1998; Neef et al., 2014). In response to stress, the monomeric HSF1 is released from its inhibitory complex and trimerizes, allowing for a series of post-translational modifications which help in its nuclear translocation and conversion to an active DNA binding component (Figure 1B). Those post-translational modifications include phosphorylation, sumoylation, acetylation, and deacetylation, which are vital to the activation-attenuation cycle of HSF1 (Anckar and Sistonen 2011; Dai 2018). HSF1 binds to a conserved pentameric sequence motif termed the heat shock element or HSE (5′nGAAn3′) at the promoter of its target genes, which include the heat shock proteins, autophagy, actin, and innate immunity (Anckar and Sistonen 2011).
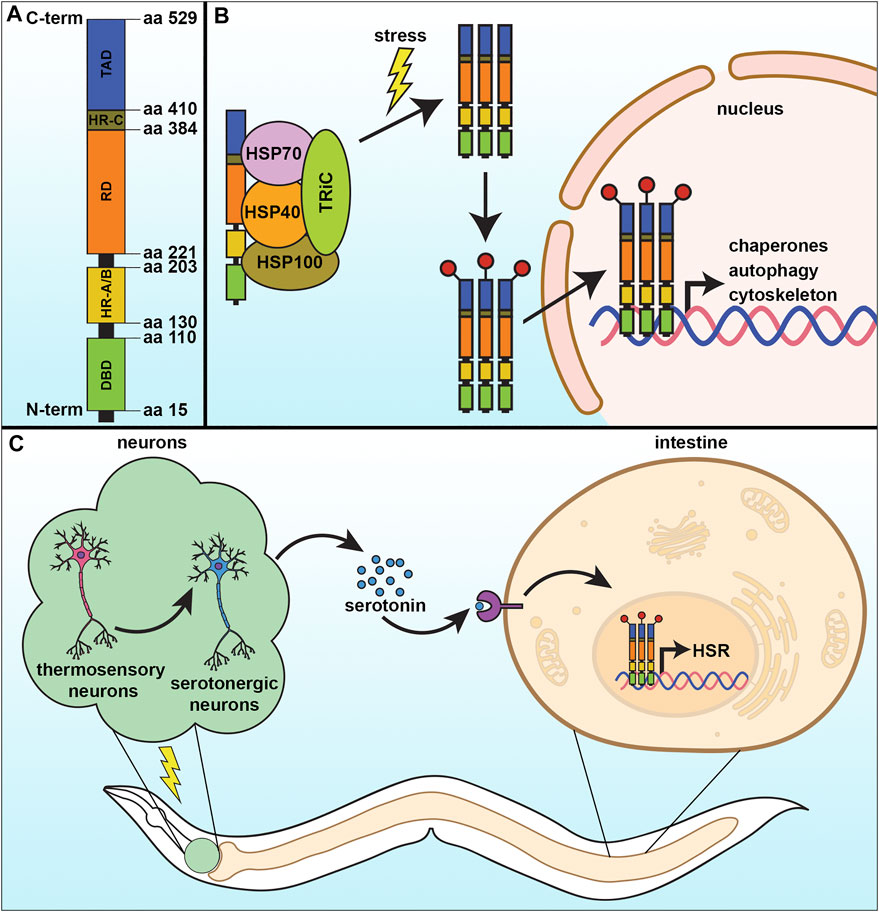
FIGURE 1. The cytosolic heat-shock response. (A) The heat-shock response (HSR) is regulated by the transcription factor, HSF-1, which contains four major structural domains: the DNA-binding domain (DBD), regulatory domain (RD), trans-activating domain (TAD), and the oligomerization domain divided into HR-A and HR-B. (B) Under basal conditions, HSF-1 is bound by regulatory proteins that prevent its activation. Upon stress, these proteins are released from HSF-1 as they are titrated away to serve as molecular chaperones to damaged proteins, allowing HSF-1 to trimerize and serve as a transcription factor to activate genes important to mitigate damage and increase survival. (C) The HSR can be communicated in a non-autonomous manner, whereby thermosensory and serotonergic neurons can communicate to peripheral tissue, including the intestine, to activate a systemic HSR and promote organismal health and lifespan.
As a critical player in cellular stress response, HSF1 function can have a direct impact on physiological health. However, like most quality control mechanisms, HSF1 activity, and the HSR deteriorate during the aging process. Specifically, the HSR in C. elegans exhibits decreased transcriptional output early in adulthood (Labbadia and Morimoto 2015). Shockingly, there is a precipitous drop in the induction of HSR genes (including hsp-70 and hsp-16.11) in response to thermal stress within a narrow 4-h window that coincides with the start of egg-laying. Interestingly, this collapse of the HSR was not due to the decreased binding ability of HSF-1, but rather due to profound changes to the chromatin landscape due to the overt decline in expression of the histone demethylase jmjd-3.1. This decreased functional output of the HSR has direct physiological ramifications, as animals exhibit an age-dependent decrease in thermotolerance and an increase in protein aggregation load (Morley et al., 2002; Ben-Zvi et al., 2009). The repression of HSR can be compensated by increased expression of hsf-1 to diminish the burden of proteotoxicity at advanced age to improve lifespan (Morley and Morimoto 2004; Ben-Zvi et al., 2009; Shemesh et al., 2013). Specifically, overexpression of hsf-1 can significantly extend lifespan (Hsu et al., 2003; Baird et al., 2014), whereas downregulation shortens lifespan and results in premature aging phenotypes (Garigan et al., 2002). Knockdown of the downstream targets of HSF-1, including the heat shock proteins, HSP70 and HSP90 also decreases lifespan (Morley and Morimoto 2004), whereas long-lived mutants including daf-2 and age-1 show increased expression of these chaperones (Hsu et al., 2003; McElwee et al., 2003; Murphy et al., 2003), providing further evidence that HSF-1-mediated stress resilience is important for longevity. In fact, longevity can actually be predicted by the expression of a single target of HSF-1, hsp-16.2 (Mendenhall et al., 2012). Importantly, HSF-1 has a beneficial effect on multiple tissue types, as overexpression solely in neurons, the body wall muscle, or intestinal cells was sufficient to promote lifespan extension (Morley and Morimoto 2004). Finally, hsf-1 has a temporal requirement in lifespan extension, whereby its expression during development impacts lifespan, whereas overexpression later in adulthood has no effect (Volovik et al., 2012).
Beyond just an ectopic expression of hsf-1, activation of the HSR through exposure to low levels of thermal stress have also been shown to impact longevity (Butov et al., 2001). Termed hormesis, this described the phenomenon whereby low-grade exposure to stress can be beneficial to organismal health by activating critical stress response pathways. While these beneficial effects of hormesis were primarily ascribed to heatshock proteins and DAF-16 (insulin growth factor) signaling (Cypser and Johnson 2002; Hsu et al., 2003; Morley and Morimoto 2004; Rattan 2005), it is becoming increasingly clear that heatshock proteins are not the only effector molecules. Based on several genome-scale studies, it has been recognized that HSF1 has a much broader functional spectrum that can control numerous molecular events associated with cellular quality control pathways, including ubiquitin-mediated proteasomal degradation, autophagy, and the maintenance of organelles including mitochondria, ER, and the actin cytoskeleton (Barna et al., 2018). In fact, hypomorphic mutations of hsf-1 and perturbations of some HSR chaperones had no impact on thermotolerance (McColl et al., 2010; Kourtis et al., 2012). One study even found that overexpression of hsf-1 with a truncation in the C-terminal transactivating domain dramatically extended lifespan, despite having no impact on the induction of heat-shock proteins (Baird et al., 2014). Instead, this variant of HSF-1 increased the expression of pat-10, a troponin-like calcium-binding protein that promoted actin cytoskeletal maintenance and function. This increase in cytoskeletal integrity at advanced age directly impacted organismal health and promoted longevity. Similar to the induction of heat-shock proteins (Morley and Morimoto 2004), HSF-1’s beneficial impact on the cytoskeleton independently affected multiple cell types to promote lifespan (Higuchi-Sanabria et al., 2018a).
HSF-1 has also been shown to function both alongside and as a regulator of autophagy. It can bind directly to the promoter of the autophagy component, ATG7, and induce its expression, which is independent of the canonical function of HSF1 (Desai et al., 2013). Furthermore, it has been also reported that casein kinase 1 phosphorylates the essential autophagy receptor SQSTM1/p62 to lift up the selective autophagic clearance in an HSF1 dependent manner (Watanabe et al., 2017). Induction and activation of the SQSTM1/p62 receptor is sufficient to induce autophagy in distinct tissues and for longevity (Kumsta et al., 2019). A recent study in C. elegans has explored the induction of HSF-1 by hormetic heat stress exposure, which can clear the accumulation of PolyQ aggregates and contributes to enhanced stress resistance and extended lifespan (Kumsta et al., 2017). Similarly, overexpression of hsf-1 is sufficient to induce autophagy and induction of autophagy is required for the beneficial effects of HSF-1. However, a conflicting study found that HSF-1 can actually prevent autophagy by promoting HSP70 expression, which can inhibit starvation and rapamycin-induced autophagy (Dokladny et al., 2013). These seemingly contradicting results may be due to differences in thermal stress conditions. For example, it is entirely possible that under conditions of acute stress, HSF-1 can coordinate heat-shock proteins and chaperones to mitigate damage by protein remodeling. In contrast, under chronic stress when damage exceeds the repair capacity of chaperones, autophagy is essential. Regardless, it is clear that a proper balance between HSR and autophagy is essential for proper maintenance of homeostasis. Indeed, HSR and autophagy can be delicately balanced and coordinated under specific environmental stress and metabolic cues via the homeodomain interacting protein kinase, HPK-1 (Das et al., 2017). Specifically, HPK-1 sits at the center of a dual proteostatic network whereby it is essential for the induction of autophagy gene expression in response to dietary restriction and inactivation of mTOR, but also inhibits sumoylation of HSF-1 to promote its transcriptional activity upon thermal stress.
Studies about the cross-talk between the proteostatic network and immune response of an organism revealed that long-lived C. elegans mutants showed higher resistance to bacterial pathogens along with increased longevity (Garsin et al., 2003). In fact, one of the primary causes of death in C. elelgans is due to pathogenic invasion (Zhao et al., 2017), suggesting that factors that there may be heavy overlap between factors that increase lifespan and immunity. Thus, it is not surprising that HSF-1 has also been implicated in immune response. Specifically in C. elegans, HSP90 and the small heat shock proteins are important for the development of immunity. Interestingly, this HSF-1-mediated defense response did not require p38 MAPK, but recruits the DAF16 pathway for the development of multi-pathogen resistance (Singh and Aballay 2006). Similar to autophagy, this beneficial effect of the HSR is not limited to hsf-1 overexpression and hormetic heat-shock can stimulate the immune response to promote resistance to pathogen exposure (Prithika et al., 2016). In higher eukaryotes with more complex immune systems, the function of induced heat shock proteins is not only limited to ameliorating inflammatory damage, but also encourage the production of anti-inflammatory cytokines (van Eden et al., 2005). HSF1 has been found as a transcriptional activator for the expression of interleukin 10 (IL10) which inhibits the bacterial lipopolysaccharide-mediated production of pro-inflammatory cytokines like TNFα, IL-12, IL-1b, IFNg (Zhang et al., 2012). Furthermore, the inflammatory stress-dependent and the HSF1 mediated induction of heat shock proteins plays an essential role in preparing peptides for proper antigen presentation by CD8+ T lymphocytes (Binder and Srivastava 2005).
In metazoans, the HSR can also be activated non-autonomously irrespective of the presence of thermal stress. It has been reported in rats for the first time by Fawcett et al. that HSF1 activation can be governed by the nervous system in the absence of environmental stress. That study revealed that controlled stress conditions release adrenocorticotropin from the pituitary gland of Wistar rats that facilitate HSF1 trimerization and increase its DNA binding activity, which consequently induces HSP70 expression in adrenal tissue (Fawcett et al., 1994). Later, a similar mechanism involving the nervous system in proteostatic regulation has been observed in C. elegans, where the HSR of somatic cells was found to be controlled in a cell non-autonomous manner by two thermosensory AFD neurons, GCY-3, and TTX-3 (Figure 1C) (Prahlad et al., 2008a). Optogenetic stimulation of the AFD neurons communicates with two serotonergic neurons ADF and NSM that releases serotonin, which was sufficient to activate HSF1 in another cell. In the receiving cell, the serotonin receptor SER-1 drove increased synthesis of chaperone proteins. They firmly established that activation of serotonergic neurons is sufficient to reduce a load of misfolded protein accumulation in C. elegans (Tatum et al., 2015). To survive in nature an organism needs to have the ability to react rapidly to environmental challenges. Thus, it is not surprising that organisms with a developed nervous system have neurons which can sense stress and relay a signal to other tissue. Indeed, neurons can also sense an olfactory experience to prime the function of HSF1 to provide a rapid and stronger response to subsequent exposure to any proteotoxic agents (Ooi and Prahlad 2017). Stimulation of the olfactory neurons by the odorants produced by the toxic bacterium P. aeruginosa can enhance the pathogen-avoiding ability in C. elegans in an HSF-1 and serotonin-signaling dependent manner.
Importantly, one study found that overexpression of hsf-1 in neurons was sufficient to drive increased HSR and DAF-16/FOXO activity in peripheral tissue (Douglas et al., 2015). This combined activation of HSF-1 and DAF-16 targets was sufficient to increase thermotolerance and lifespan, which was similar to the increase in lifespan found in animals with reduced insulin signaling (Son et al., 2018). In addition, neuronal hsf-1 was sufficient to drive protection of the actin cytoskeleton in multiple cell types during aging, including the muscle, hypodermis, and intestine, which was equally important for the increased longevity of these animals. This is curious considering the previously identified involvement of integrins in actin function (Tharp et al., 2021) and HSF-1-mediated depletion of integrin-linked kinase in increased stress resistance and longevity in C. elegans (Kumsta et al., 2014). These data beg the question of whether non-autonomous HSF-1 signaling could potentially utilize similar integrin signaling mechanisms to drive longevity. Finally, neuronal hsf-1 signaling also directly impacts fat metabolism and results in extensive fat remodeling. Specifically, neuronal hsf-1 results in decreased expression of the fat desaturases fat-6/fat-7, while activating the expression of catabolic lysosomal lipases, which shifts the fatty acid composition of the plasma membrane to a more saturated state. This change in fat mimics those downstream of exposure to thermal stress, and is sufficient to drive long-term survival of animals at elevated temperature (Chauve et al., 2021).
Beyond nonautonomous signaling from neurons to periphery, a recent study has found that serotonin can actually transmit HSF-1 signaling to future progeny (Das et al., 2020). Serotonin, released from the maternal neurons can ensure higher longevity and stress resilience in their future offspring through HSF-1 activation in germ cells. Specifically, serotonin signaling promotes protein kinase A (PKA)-dependent modification of HSF-1, increasing the occupancy of RNA Pol II and HSF1 at the promoter of various protective genes, including molecular chaperones which are the targets of HSF1 in response to even minimum heat stress. In addition, HSF-1 promotes the recruitment of a chromatin remodeler FACT (Facilitates Chromatin Transcription) to alter histone dynamics to initiate transcription. This mechanistic pathway is also conserved in mammalian cells. Overall, these studies highlight the numerous pathways HSF-1 modulates to impact organismal health and lifespan.
Beyond its clear role in regulating the HSR, HSF-1 has now been ascribed to several other equally important processes, including autophagy, immune response, and maintenance of the cytoskeleton. Considering its numerous functions, one intriguing question is how this single transcription factor can coordinate such diverse processes, and whether exposure to specific types of stressors can titrate HSF-1 to promoters of appropriate genes. For example, upon deletion of its C-terminal domain, HSF-1 can no longer induce heat-shock proteins, but strongly upregulates genes involved in cytoskeletal maintenance (Baird et al., 2014). It is possible that the C-terminal domain contains an important residue that allows for coordination with other transcriptional regulators recruits HSF-1 to heat-shock proteins, and loss of this domain causes increased accumulation of HSF-1 to cytosolic targets. Still to be understood is the identity of these potential cofactors that would allow HSF-1 to be targeted to specific gene loci.
Another intriguing question is how all the functional roles of HSF-1 are coordinated. To date, separate studies have shown that the induction of chaperones (Morley and Morimoto 2004), cytoskeletal function (Higuchi-Sanabria et al., 2018b), and autophagy (Kumsta et al., 2017) are all necessary for the beneficial effects of HSF-1 activation, such that perturbing any individually can completely abrogate HSF-1-mediated longevity. And while studies with the C-terminal deletion of HSF-1 showed that cytoskeletal regulation can be separated from chaperone induction (Baird et al., 2014), whether the beneficial effects of this HSF-1 variant requires autophagy has yet to be identified. Importantly, all these processes have been shown to be induced upon exposure to heat-shock, suggesting that the majority of HSF-1 targets are simultaneously induced, rather than being separable. Thus, are these seemingly divergent responses actually distinct mechanisms, or all parts of the same pathway? Indeed, all of these pathways do converge into a similar goal: increased proteostasis through protein folding by chaperones, clearance of damaged proteins through autophagy, and even the actin cytoskeleton has clear implications in protein homeostasis (Gross and Kinzy 2007).
Mitochondrial fitness and function are inarguably significant to cell viability, due to their numerous functions, including energy production, regulating apoptotic and necrotic cell death, storing calcium and amino acids, lipid oxidation, and heat production. Mitochondrial dysfunction is linked to aging and several age-related diseases, including Alzheimer’s disease, Parkinson’s, and metabolic syndrome (Higuchi-Sanabria et al., 2018c; Moehle et al., 2019). This means that proper functioning of mitochondria is essential for a healthy physiological state. However, almost counterintuitively, a growing number of studies have shown that perturbations to mitochondrial function can actually lead to lifespan extension (Dillin et al., 2002; Liu et al., 2005; Owusu-Ansah et al., 2013; Yee et al., 2014). The primary reason for these seemingly contradictory observations is that impairment of mitochondria triggers the unfolded protein response of the mitochondria (UPRMT), which results in beneficial effects on organismal health (Durieux et al., 2011).
Considering the importance of mitochondria to cellular health, it is not surprising that cells have adapted many quality control mechanisms to protect mitochondrial function, including the UPRMT. UPRMT is robustly activated by a variety of sources of mitochondrial stress: stoichiometric imbalance between proteins coded in the nuclear or mitochondrial genome (Couvillion et al., 2016; Houtkooper et al., 2013; Molenaars et al., 2020), impaired electron transport chain (ETC) function (Khalimonchuk et al., 2007; Zara et al., 2007; He et al., 2018), mitochondrial protein aggregation (Hallberg et al., 1993; Dubaquié et al., 1998), defects in mitochondrial import (Rolland et al., 2019; Xin et al., 2020), loss of mitochondrial membrane potential (Tharp et al., 2021), or disrupting mitochondrial translation (Nolden et al., 2005) and DNA replication (Kenny and Germain 2017). This process is best understood in C. elegans, where UPRMT is regulated by several transcriptional regulators (Figure 2A). One specific transcription factor, ATFS-1, serves as a signal between the mitochondria and the nucleus. ATFS-1 contains both a nuclear localization sequence and a mitochondrial signal sequence and is preferentially imported into the mitochondria where it is subsequently degraded by mitochondrial Lon proteases. When mitochondrial import is compromised, such as under conditions of stress, ATFS-1 import is reduced and can accumulate in the nucleus to activate UPRMT (Nargund et al., 2012). Several other transcriptional regulators work either in concert with, or independent of, ATFS-1 to regulate UPRMT, including the transcription factor DVE-1 and its ubiquitin-like cofactor UBL-5 (Benedetti et al., 2006; Haynes et al., 2007). Nuclear localization and transcriptional activation by DVE-1 is further promoted by the histone methyltransferase MET-2 and its cofactor LIN-65, which also induce epigenetic changes required for transmission of mitochondrial stress signaling between cells and through generations (Tian et al., 2016). Finally, histone demethylases JMJD-1.2 and JMJD-3.1 remodel chromatin to facilitate access to promoters of numerous UPRMT target genes (Merkwirth et al., 2016).
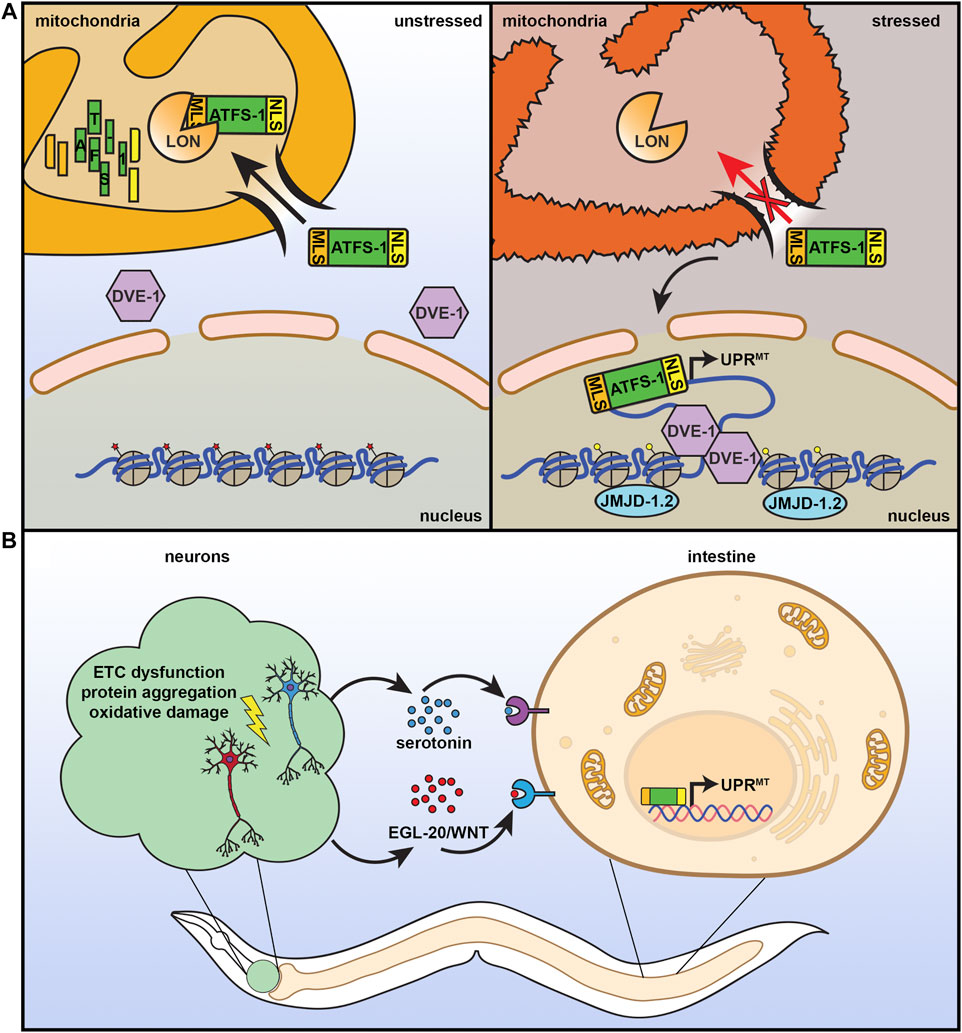
FIGURE 2. The mitochondrial unfolded protein response. (A) ATFS-1 is a unique protein that can serve as a sensor for mitochondrial health and fitness. It contains both a nuclear localization signal (NLS) and a mitochondrial localization signal (MLS). Under basal, unstressed conditions, ATFS-1 is imported into the mitochondria where it is degraded by LON protease. Under conditions of mitochondrial stress or damage, mitochondrial import decreases, allowing ATFS-1 to instead accumulate in the mitochondria where it can activate UPRMT with additional transcriptional regulators including DVE-1 and chromatin regulator JMJD-1.2. (B) Similar to the HSR, UPRMT can also be communicated in a nonautonomous manner. Neurons that experience mitochondrial stress can signal to the peripheral tissue, including the intestine, through serotonin and WNT signaling to result in systemic activation of UPRMT, increased stress resilience, and increased lifespan.
At the genetic level, UPRMT is characterized by a coordinated activation of genes essential for restoring mitochondrial fitness and function. Mitochondrial chaperones, such as Hsp70 and Hsp60/10 can refold misfolded proteins (Castro et al., 2018) and proteases, such as LONP and ClpP can degrade misfolded proteins (Wang et al., 1993) to alleviate proteotoxic stress. Importantly, proteins involved in mitochondrial import are also upregulated to ensure proper import of essential protein homeostatic machinery (Xin et al., 2020). UPRMT targets are not only limited to protein homeostasis, as genes involved in immunity (Pellegrino et al., 2014), autophagy (Haeussler et al., 2020), and xenobiotic stress (Nargund et al., 2012; Nargund et al., 2015) are also induced. Moreover, direct applications of stress to different mitochondrial processes with drugs results in vast differences in transcriptional response (Quirós et al., 2017), thus highlighting the complexities of mitochondrial stress signaling and regulation (refer to (Bar-Ziv et al., 2020a) for a more thorough review). Here, we focus primarily on the impact of UPRMT on aging in the model organism, C. elegans.
The UPRMT was first used to describe a transcriptional response whereby loss of the mitochondrial genome led to increased expression of mitochondrial chaperones. This process was originally identified in mammalian cells and was named after its presumed effects on mitochondrial protein folding (Martinus et al., 1996). Further studies identified additional targets for UPRMT activation, which include ROS detoxification, mitochondrial import, and glycolysis (Zhao et al., 2002). Since then, it has been heavily studied in various model systems, most notably in C. elegans, as a beneficial response to stress. In this model coined “mitohormesis”, exposure to low grade stress to the mitochondria results in the induction of an adaptive program that can benefit both lifespan and healthspan (Ristow and Zarse 2010). In C. elegans, the phenomenon of mitohormesis was first identified in a large-scale RNAi screen of genes found on chromosome I. In this study, RNAi knockdown of three components of the mitochondrial ETC, mitochondrial ATP synthase (atp-1), NADH/ubiquinone oxidoreductase (nuo-2), and cytochrome c reductase (cco-1/cox-5b) all resulted in reduced body size and extended lifespan (Dillin et al., 2002). A similar RNAi screen of nearly all the genes on chromosomes I and II consistently found that knockdown of a large number of mitochondrial genes extended lifespan, including cytochrome C oxidase VIIc (cox-7c), mitochondrial ribosomal protein L47 (mrpl-47), and the mitochondrial solute carrier family 25 member 32 (slc-25A32) (Lee et al., 2003). This study also performed a classical forward genetic screen and identified a mitochondrial leucyl-tRNA synthetase (lrs-2), which similarly extends lifespan when disrupted. Since then, multiple perturbations to mitochondrial function have demonstrated a positive impact on lifespan: mitochondrial ribosomal protein knockdown (Houtkooper et al., 2013), perturbing mitochondrial dynamics in favor of mitochondrial fusion (Chaudhari and Kipreos 2017), mitochondrial genome impairment (Tsang and Lemire 2002), and many others reviewed in (Munkácsy and Rea 2014).
An intriguing phenomenon in C. elegans is that there is a specific temporal requirement during development to enact the positive benefits of mitohormesis. That is, animals exposed to RNAi knockdown of ETC components during development exhibited lifespan extension, even if normal levels of ETC component expression were restored at adulthood (Li et al., 2021). Conversely, reducing levels of ETC components in adulthood had no impact on lifespan. These data suggested that a long-lasting signature exists to elicit lifespan extension following mitochondrial dysfunction. This specific signal is initiated during the L3/L4 larval stage of development (Dillin et al., 2002; Rea et al., 2007). Interestingly, this phenomenon also had a dose-dependent effect. Titrating the RNAi of five genes encoding mitochondrial proteins atp-3, nuo-2, isp-1, cco-1, and frh-1 showed that there was a consistent three-phase lifespan response: low levels of knockdown had no effect, but as expression reduced, lifespan extension lengthened until at the highest level of knockdown, lifespan actually shortened for some conditions (Rea et al., 2007).
The most obvious response to mitochondrial stress is the massive and persistent restructuring of gene expression, which is a hallmark of UPRMT activation. In C. elegans, this process is mediated by the converted effort of the transcriptional regulators ATFS-1, DVE-1, and UBL-5 (Benedetti et al., 2006; Haynes et al., 2007; Nargund et al., 2012). However, the existence of a long-term hormesis suggested that a more permanent signature must exist upon exposure to mitochondrial stress. One study identified the methyltransferase MET-2 and a nuclear cofactor LIN-65 are required for UPRMT activation and lifespan extension found in animals with exposure to mitochondrial stress (cco-1 knockdown or exposure to aggregation prone polyQ described further below) (Tian et al., 2016). Specifically, 1,264 of the 1,312 genes differentially expressed under cco-1 knockdown were dependent on the functional activity of either MET-2 or LIN-65. During mitochondrial stress, MET-2 produces H3K9me1/2. Nuclear localization of LIN-65 then deposits H3K9me2 subunits onto the chromatin, titrating DVE-1 to loose regions of the chromatin and allowing for sustained ATFS-1 localization at these loci. Thus, loss of any of these factors under stress could result in decreased UPRMT activity and loss of lifespan extension.
A complementary study identified another chromatin modifier as a critical regulator of UPRMT-induced longevity (Merkwirth et al., 2016). This study performed a screen to identify genes required for the lifespan extension found in cco-1 knockdown animals and identified the gene encoding the histone demethylase, jmjd-1.2. Knockdown of jmjd-1.2 decreased lifespan and suppressed cco-1 induced lifespan extension, similar to met-2 and lin-65. Importantly, overexpression of jmjd-1.2 was sufficient to induce UPRMT and promote longevity. This was the first study that showed that hyperactivation of UPRMT in the absence of stress was sufficient to extend lifespan. Because JMJD-1.2 functions downstream of mitochondrial stress, its overexpression bypasses the need to cause damage to the mitochondria to induce UPRMT.
An unaddressed conundrum is that the targets of UPRMT include a large number of chaperones and other protein processing components that must be imported into the mitochondria to serve their function to restore organellar homeostasis. However, the dogma for UPRMT activation is that decreased mitochondrial membrane potential drives the mitochondria-to-nuclear signaling by preferential localization of ATFS-1 to the nucleus due to failed entry in the mitochondria. How then do proteins get imported into the mitochondria when membrane potential is compromised? In an exciting piece of work, this paradox was addressed by finding that the induction of UPRMT can actually increase mitochondrial import, despite a loss of membrane potential (Xin et al., 2020). The efficiency of import is increased by upregulating components of the mitochondrial import machinery, including timm-17, timm-23, tomm-20, tomm-22, and tomm-40. In addition, because ATFS-1 has a weak MTS, it continues to fail to enter the mitochondria and can continue to drive UPRMT induction until mitochondrial function is fully restored.
Interestingly, lifespan was never reportedly increased in response to knockdown of complex II subunits (Ichimiya et al., 2002; Rea et al., 2007; Kuang and Ebert 2012). In addition, a screen for novel regulators of UPRMT identified 19 genes that when knocked down induce UPRMT but showed no correlation for lifespan extension. In fact, 6 out of the 19 RNAi conditions actually decreased lifespan, while 3 showed no significant effect (Bennett et al., 2014). Notably, all the genes that decreased lifespan were important for mitochondrial import, which a previous study has implicated as necessary for any beneficial effects of UPRMT (Xin et al., 2020). Thus, it is entirely possible that when mitochondrial import is reduced, there is robust activation of UPRMT without a beneficial impact on organismal health, which would likely require import of newly synthesized mitochondrial protein homeostasis machinery. However, a striking finding in the study was that the lifespan extension found in several conditions of mitochondrial stress induction (RNAi knockdown of transaldolase tald-1, mitochondrial transmembrane protein letm-1, cco-1, and isp-1) were all independent of atfs-1. Moreover, constitutive activation of atfs-1 did not improve lifespan, but instead shortened it, despite measurable activation of UPRMT (Bennett et al., 2014), although it is unclear whether atfs-1 activation may cause unexpected and detrimental consequences to mitochondrial quality (Shpilka et al., 2021). In another study, loss of atfs-1 during adulthood did not affect lifespan extension of clk-1, isp-1, or nuo-6 loss of function animals (Wu et al., 2018), consistent with previous findings that the beneficial impact of mitochondrial stress may be important only during development (Durieux et al., 2011). However, deletion of atfs-1 during development resulted in defects in growth in clk-1 and isp-1 loss of function animals, although it did suppress lifespan of nuo-6 mutants. Overall, these conflicting data argued against the longstanding model that activation of UPRMT improves longevity.
Taken together, it is clear and consistent across numerous studies that mitohormesis is evident: exposure to mitochondrial dysfunction can indeed activate a protective pathway that can promote lifespan extension. However, whether this protective pathway can be fully ascribed to the currently defined model of UPRMT is still up for debate. The response to mitochondrial stress in human cells is complex and context-dependent, as diverse drugs that target different components of the mitochondria show little overlap in transcriptional response (Quirós et al., 2017). Similarly, it is likely that activation of UPRMT in C. elegans is equally context dependent, and a broader definition of UPRMT beyond induction of a small subset of chaperones (e.g., hsp-6 and hsp-60) is necessary. Perhaps a more thorough investigation of specific UPRMT targets downstream of multiple transcriptional regulators–and not just ATFS-1–is essential to understand the nuances between seemingly “UPRMT” dependent and independent mechanisms of lifespan extension downstream of mitochondrial dysfunction. For example, many other pathways including activation of hypoxia inducible factor HIF-1 (Kourtis et al., 2012), homeobox protein CEH-24 (Walter et al., 2011), AMP-activated protein kinase AAK-2 (Curtis et al., 2006), and the p53 homolog CEP-1 (Ventura et al., 2009); and the integrated stress response (ISR) critical for responding to mitochondrial stress in higher mammals (reviewed in (Anderson and Haynes 2020)) have all been implicated in longevity in response to mitochondrial stress in C. elegans. In addition, there are also gene-diet interactions that alter the impact of UPRMT on longevity (Amin et al., 2020). Thus, in its current state, it is clear that a single mechanism cannot describe all the mitochondrial longevity paradigms and more expansive studies are required to better understand this complex phenomenon.
Under conditions of stress, multicellular organisms must coordinate a systemic response across diverse cell types, all with their own unique energetic and metabolic demands. Mitochondrial number, activity, protein composition, morphology, and mtDNA contents can all vary across different tissues (Leary et al., 1998), and thus responses to mitochondrial stress are unique in each cell type. In C. elegans, exposure to mitochondrial stress specifically in the intestine or neurons results in a significant increase in lifespan, whereas exposure of muscle cells to stress had the opposite effect and shortened lifespan (Durieux et al., 2011), highlighting the significance of context-specific benefits of UPRMT activation. Importantly, applying mitochondrial stress to neurons was sufficient to induce systemic activation of UPRMT due to a neuron-to-periphery cell non-autonomous response, which was critical for lifespan extension (Figure 2B) (Leary et al., 1998). This early paradigm of non-autonomous UPRMT involved a “Mjolnor hammer” approach where perturbation of ETC function via cco-1 knockdown caused severe mitochondrial stress in neurons. However, since then, more physiologically relevant stressors confirmed these findings where overexpression of polyQ40 (and larger polyQ repeats) (Berendzen et al., 2016) or expression of ROS-producing KillerRed fluorescent protein (Shao et al., 2016) specifically in neurons robustly induced UPRMT in peripheral cells. Interestingly, overexpression of amyloid ß associated with Alzheimer’s disease or mutant TDP-43 associated with amyotrophic lateral sclerosis in neurons failed to induce peripheral UPRMT activation, suggesting that this neuronal stress signaling paradigm was also context specific (Shao et al., 2016). Importantly, neurons can transmit UPRMT signals to the periphery in the absence of stress, as overexpression of jmjd-1.2 solely in neurons was also able to induce this non-autonomous UPRMT signal and was sufficient to promote longevity (Merkwirth et al., 2016).
The phenomenon whereby neurons can transmit stress signals to the rest of the body is not unique to UPRMT, and has also been described for UPRER (Taylor and Dillin 2013) and the HSR (Prahlad et al., 2008b; Higuchi-Sanabria et al., 2018b) (see individual sections), although the mechanisms driving these responses seem to be distinct. While UPRMT (Berendzen et al., 2016), UPRER (Higuchi-Sanabria et al., 2020), and HSR (Tatum et al., 2015) all seem to utilize serotonergic circuits, Wnt signaling was also identified to be critical for transmitting the UPRMT signal (Zhang et al., 2018). Specifically, knockdown of mig-14, dpy-23, and mig-1, critical components in receiving and internalizing Wnt signals completely abolished UPRMT activation downstream of neuronal polyQ40 expression. Most importantly, overexpression of the gene encoding the secreted Wnt ligand egl-20 specifically in neurons was sufficient to drive systemic UPRMT activation and extend lifespan. Collectively, these findings have led to the conclusion that there exist not one, but two secreted mitokines from neurons that drive mitochondrial stress signaling, serotonin and Wnt. Perhaps most intriguing was the finding that this induced systemic UPRMT downstream of neuronal mitochondrial perturbations were transmitted to offspring over multiple generations in C. elegans. Specifically, neuronal expression of polyQ40 resulted in a transgenerational induction of UPRMT that was observed more than 50 generations out upon loss of the neuronal polyQ40 transgene. This transmission of UPRMT to offspring was through a Wnt-dependent elevation of mtDNA levels across generations (Zhang et al., 2021).
The capacity of cells to transmit mitochondrial stress signals to other cells is not a unique feature of neurons. In fact, germline-specific loss of the cytochrome c ortholog, cyc-2.1 initiates a non-autonomous response that activates UPRMT and AMPK in the intestine, which results in a robust lifespan extension. Importantly, this lifespan extension was dependent on ATFS-1, which drives DRP-1-mediated mitochondrial fragmentation (Lan et al., 2019). Decreased protein homeostasis in the germline–defined in this context as increased aggregation of PGL-1, an RNA-binding protein involved in P granule formation in the germline–also resulted in a non-autonomous activation of UPRMT in the soma, providing evidence that this germline-to-soma transmission of UPRMT could be a general response to germline stress. Increased PGL-1 aggregation resulted in a significant decrease in mitochondrial protein levels in the germline, which resulted in a Wnt-dependent transmission of mitochondrial stress signals to the soma. Specifically, PGL-1 aggregation in the germline resulted in an EGL-20 (Wnt ligand) and MIG-1 (Wnt receptor)-dependent mitochondrial fragmentation and UPRMT induction in the soma (Calculli et al., 2021). However, it is still unclear whether this somatic UPRMT induction is beneficial to organismal health and lifespan.
Perhaps the most controversial point of UPRMT on aging is whether activation of UPRMT directly correlates with aging. As described above, knockdown of complex II genes never exhibited lifespan extension, while other methods to induce UPRMT have inconsistent impacts on lifespan (Bennett et al., 2014). The most obvious hypothesis is that there are varying degrees of mitochondrial stress, whereby only those conditions that induce sufficient mitochondrial stress to activate UPRMT without causing irreversible damage can promote hormesis and extend lifespan. In contrast, causing too much damage would be detrimental, regardless of whether UPRMT is activated or not. Another plausible explanation is that the methods to measure UPRMT activation are not sufficient: most studies rely on artificial transcriptional reporters, such as the overexpression of the hsp-6p::GFP reporter. As an overexpressed system, it is possible that hsp-6p::GFP exaggerates the intensity of UPRMT activation, which is clear in some reports where western blots and qPCR show markedly lower hsp-6 induction than the reporter (Berendzen et al., 2016). Thus, it is possible that a more thorough investigation of UPRMT activation (e.g., survey of more gene targets, measurements of nuclear localization of UPRMT regulators like DVE-1::GFP, measurements of chromatin compaction, etc.) is essential for understanding the true impact of UPRMT on longevity.
Additionally, it would be of great interest to determine whether other cell types can initiate non-autonomous mitochondrial stress signatures. In flies, mitochondrial perturbations through dysfunction of complex I specifically in muscle results also results in impairment of mitochondrial function in the body fat (Song et al., 2017). Moreover, in mice, muscle-specific deletion of a critical autophagy gene Atf7 results in increased secretion of Fgf21, which increased resistance of these animals to diet-induced obesity (Kim et al., 2013). Importantly, Fgf21 serves as an endocrine signal to elicit ATF3-, ATF-4, and ATF-5 dependent ISR and UPRMT (Forsström et al., 2019). Thus, it is entirely possible that other cell types in C. elegans also have the capacity to promote systemic UPRMT through non-autonomous signaling.
Finally, while we survey the impact of UPRMT on longevity in C. elegans, it is of critical importance to put into perspective the translatability of these findings to mammalian systems. One report argued that ATF5, the predicted mammalian homologue of ATFS-1, is critical for mitochondrial quality control (Fiorese et al., 2016). However, large-scale -omics-based approaches showed that the major responses to various drugs that target mitochondrial processes involved ATF4, which activates the ISR (Quirós et al., 2017). Furthermore, studies of human cells in more physiologically similar matrices (softer, 400 Pa hydrogels compared to the ∼3 GPa of polystyrene) highlighted the major involvement of HSF1 and NRF2 in mitochondrial homeostasis in cancer cells (Tharp et al., 2021). Thus, it is still unclear how UPRMT is regulated in mammalian systems, how ISR, UPRMT, HSF1, and NRF2 either simultaneously or independently coordinate aspects of mitochondrial homeostasis, and ultimately, what impact–if any–these mechanisms have on mammalian longevity.
The endoplasmic reticulum (ER) is a complex multi-faceted organelle. While maintaining a contiguous membrane with the nucleus, the ER functions in protein folding, lipid synthesis, lipid droplet formation, and calcium storage (Fagone and Jackowski 2009; Benham 2012; Pol et al., 2014; Raffaello et al., 2016). The ER must also coordinate these responsibilities with other cellular organelles to traffic transmembrane proteins, lipids, and calcium. Therefore, a functional ER is essential to maintaining a healthy cellular status. Perturbations in ER protein quality control have been implicated in age-related diseases like Alzheimer’s disease and Parkinson’s disease (Hartl 2017), while dysregulation of lipid synthesis has been associated with diabetes and cardiovascular disease (Chaurasia and Summers 2015; Sletten et al., 2018). Activation of the unfolded protein response of the ER (UPRER), a transcriptional program activated upon exposure to ER stress, is often correlated with the development of these diseases as well as cancer (Clarke et al., 2014; Zhang et al., 2017). Interestingly, studies have also shown that genetic activation of the most conserved branch of the UPRER can instead result in health benefits and an increased lifespan (Taylor and Dillin 2013; Frakes et al., 2020). These opposing phenotypes highlight the complexity and importance of understanding the role that the UPRER plays in disease and aging.
Similar to the mammalian ER, the C. elegans UPRER is composed of three signaling branches, each branch signaling from a unique transmembrane sensor (Figure 3A) (Shen et al., 2001). Initially discovered through a genetic screen for mutants that failed to induce an Unfolded Protein Response Element (UPRE) reporter in yeast, the most conserved of the these UPRER sensors is inositol-requiring enzyme 1 (ire-1) (Cox et al., 1993; Aragón et al., 2009). The IRE-1 protein is composed of an N-terminal ER luminal domain that is linked by a single-pass transmembrane domain to its cytosolic portion. The cytosolic side of IRE-1 contains both a kinase domain and an RNAse domain (Adams et al., 2019). In unstressed conditions, IRE-1 is bound to the chaperone HSP-4 (HSP70/BiP) at the luminal domain, which maintains IRE-1 as a monomer (Amin-Wetzel et al., 2017). Upon unfolded protein stress, HSP-4 is titrated away from IRE-1, freeing the luminal domain to directly bind unfolded proteins and dimerize (Zhou et al., 2006). Dimerized IRE-1 undergoes autophosphorylation, which activates its RNAse domain (Prischi et al., 2014), resulting in non-canonical splicing of the xbp-1 mRNA to allow synthesis of the active transcription factor, XBP-1s (Calfon et al., 2002). XBP-1s can then enter the nucleus and induce expression of genes aimed at mitigating the stress on the ER, including those involved in protein quality control, secretion, and lipid metabolism (Shen et al., 2005).
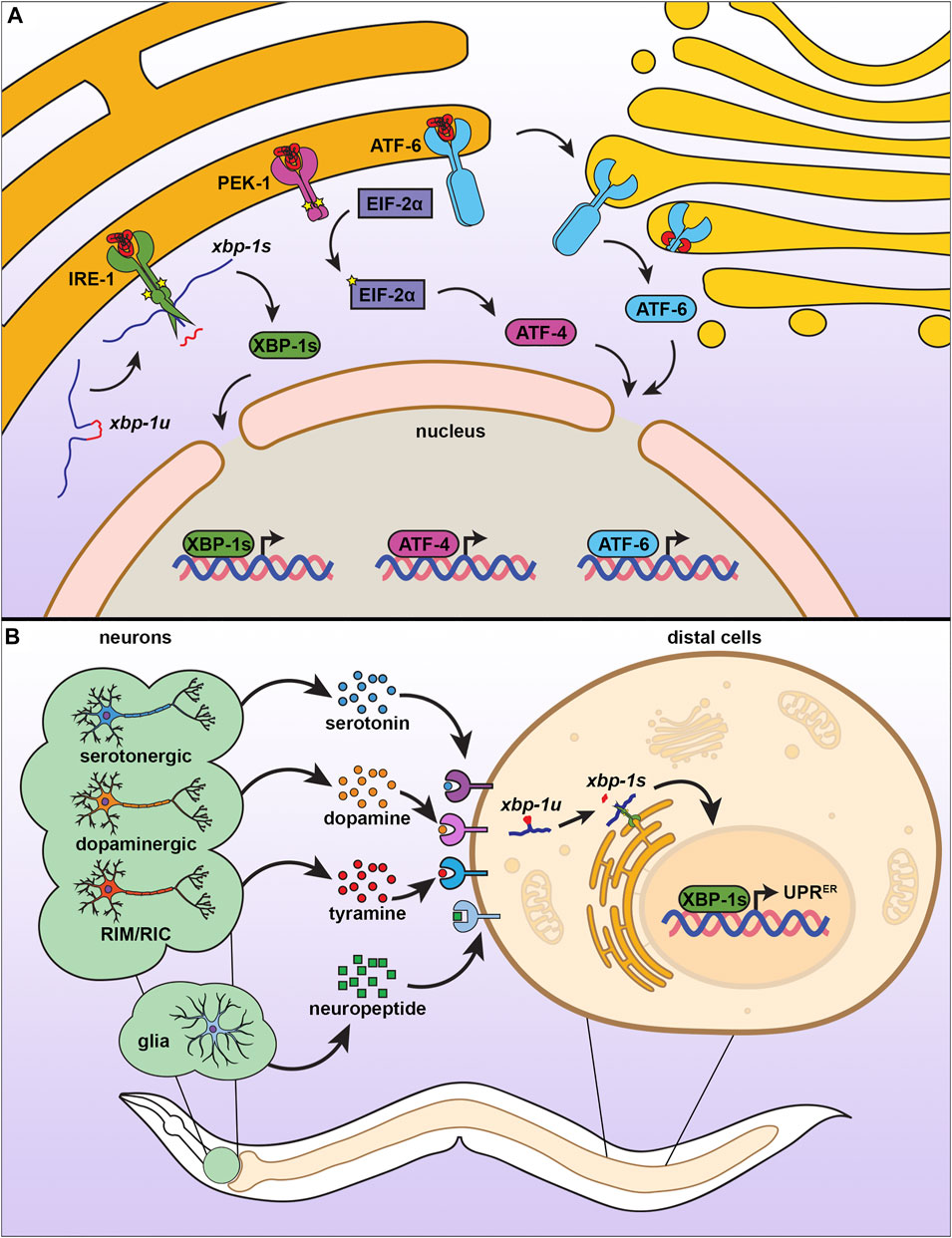
FIGURE 3. The endoplasmic reticulum unfolded protein response. (A) There are three branches of UPRER, each consisting of an ER transmembrane protein with different mechanisms of action. When IRE-1 senses misfolded proteins, it homodimerizes, autophosphorylates, and promotes splicing of xbp-1u to xbp-1s, which can then be translated into the transcription factor XBP-1s, which activates UPRER. Similar to IRE-1, PEK-1 undergoes oligomerization, which induces eIF2α phosphorylation and activation of ATF4 to inhibit global translation. ATF6 is activated by proteolytic cleavage under ER stress, which causes translocation to the Golgi for further processing, resulting in a transcriptionally active ATF6 that promotes UPRER. (B) The UPRER is communicated in a nonautonomous manner through multiple neuronal subtypes and through glial cells. Serotonergic, dopaminergic, and RIM/RIC neurons signal to peripheral cells using specific neurotransmitters, whereas glial cells communicate through neuropeptide signaling. While the exact identify of receptors involved have not yet been fully characterized, distal cells respond by promoting IRE-1-dependent splicing of xbp-1s to induce the UPRER.
In addition to the induction of ER homeostasis genes through activating xbp-1s, IRE-1 also plays a functional role in the degradation of mRNAs in a process known as Regulated Ire1 Dependent Decay (RIDD) (Tam et al., 2014). Targets of RIDD generally include proteins with signal peptides and transmembrane domains, or secretory proteins whose decreased translation is expected to reduce the protein folding burden of the ER (Hollien and Weissman 2006; Lee et al., 2011; Tsuru et al., 2013). Mammalian IRE-1 can also interact with TNF Receptor Associated Factor 2 (TRAF2) and activate the c-Jun N-terminal kinase (JNK) pathway (Urano et al., 2000). Activation of JNK signaling can promote cell death, while inhibiting downstream activation of JNK can promote cell survival under ER stress (Nishitoh et al., 2002; Verma and Datta 2010).
The UPRER sensors of other two branches of the UPRER are the protein kinase R (PKR)-like endoplasmic reticulum kinase (pek-1) and the activating transcription factor 6 (atf-6). Similar to IRE-1, both PEK-1 and ATF-6 contain a luminal domain that binds unfolded proteins, a transmembrane domain, and a cytosolic domain. However, they differ in the functions of the cytosolic domain (Schröder and Kaufman 2005). While PEK-1 also contains a kinase domain on its cytosolic side, it does not possess RNAse function (Cui et al., 2011). Instead, activation of PEK-1 results in the phosphorylation of the alpha subunit of eukaryotic initiation factor (eif-2A) to reduce global translation, but favors translation of the transcription factor ATF-4 to induce expression of genes to aid in mitigation of the ER stress (Han et al., 2013; Ma and Hendershot 2003; B’chir et al., 2013). The final branch of the UPRER has an entirely different mechanism of action and involves the cytosolic domain of the ATF-6 protein, which contains a leucine zipper family transcription factor (Schröder and Kaufman 2005). Upon the accumulation of unfolded proteins, the luminal domain of ATF-6 loses its association with ER-resident chaperones, which results in its translocation to the Golgi (Stauffer et al., 2020) where it is further processed (Haze et al., 1999; Ye et al., 2000). The ATF-6 transcription factor then enters the nucleus to promote expression of UPRER target genes (Shoulders et al., 2013; Stauffer et al., 2020).
Like in most organisms, the IRE-1 branch of the UPRER is the most heavily studied branch in C. elegans. The IRE-1 branch is primarily responsible for the induction of genes encoding ER resident chaperones, including hsp-4 and hsp-3 (Shen et al., 2001). As such, the transcriptional reporter for hsp-4 has become one of the most common and robust methods of monitoring UPRER activation within the nematode (Calfon et al., 2002; Bar-Ziv et al., 2020b). However, it is becoming increasingly clear that chaperone induction and protein homeostasis are not the only targets of UPRER, as other critical processes are also induced including immune response, lysosomal function and autophagy, and lipid homeostasis. The UPRER also plays an important role in development in C. elegans and the simultaneous loss of multiple branches results in developmental arrest of animals (Shen et al., 2005). Importantly, the IRE-1 branch of UPRER is critical for the exit from cellular quiescence (Roux et al., 2016). Newly hatched nematodes that enter L1 arrest due to the absence of food can exit this quiescent state to resume normal development and a normal lifespan upon the introduction of food. Larvae that maintain the L1 arrest for extended periods require ire-1 to recover from this state and resume development. The mRNA degradation function of IRE-1 has also been shown to regulate cellular fate decisions in C. elegans. Activation of IRE-1 within tumorous germline cells stimulates differentiation that halts progression of the tumor growth (Levi-Ferber et al., 2015; Levi-Ferber et al., 2021). This function is also conserved in mammals, drosophila, and yeast, where the UPRER has also been found to be required for normal development of various cell types (Reimold et al., 2001; Jung et al., 2016; Xu et al., 2016; Hillary and FitzGerald 2018; Kroeger et al., 2018).
Dysregulation of the UPRER is a common feature in many diseases, including neurodegeneration, metabolic disease, and cancer, and has been shown to decline in function during aging. In C. elegans, the capacity to induce IRE-1 and XBP-1 mediated UPRER fundamentally declines during aging, which results in increased sensitivity to ER stress (Taylor and Dillin 2013). A similar phenomenon is seen in aged mice where expression of ER quality control genes show marked decline in the brain (Naidoo et al., 2008; Nuss et al., 2008). The loss of UPRER function during aging can lead to the accumulation of damaged and aggregated proteins, which results in the physiological consequences of aging including increased proteotoxicity and cell death (Estébanez et al., 2018).
Although the UPRER is an adaptive and beneficial mechanism involved in clearing and mitigating damage under conditions of stress, sustained and unresolved UPRER can result in the activation of apoptosis. As such, chronic and irreversible UPRER is actually a hallmark for several disease, including neurodegeneration. Unresolved ER stress can activate pro-apoptotic machinery, including the C/EBP homologous protein CHOP, which downregulates anti-apoptotic factors, like B-cell lymphoma 2 (BCL2) (Chauve et al., 2021). Under certain conditions, ER stress can also promote MAPK signaling, including ERK1/2, which can be a pro-cancer signal, which makes inhibiting UPRER a potential therapeutic intervention for these cancers (Jiang et al., 2007; Beck et al., 2013). In addition, activation of the UPRER can also promote an inflammatory signaling cascade, including cytokine release, which can be correlated with common inflammatory disease including diabetes, atherosclerosis, and inflammatory bowel disease (Zhang and Zhang 2012; Coleman and Haller 2019). These studies suggest a potentially negative role for UPRER in organismal health and disease pathology. Indeed, studies in C. elegans have also revealed a potentially negative role for the UPRER, whereby whole-organism overexpression of xbp-1s is not beneficial to longevity and muscle-specific overexpression actually reduces lifespan (Taylor and Dillin 2013). Thus, while it is clear that the connection between UPRER and longevity are complex, myriad studies in C. elegans are in agreement that UPRER in the nervous system can be highly beneficial to organismal health. Thus, we focus on how UPRER function can impact the aging process in C. elegans, which primarily involves non-autonomous communication of ER stress signals through neural cells (Figure 3B).
Multicellular organisms coordinate systemic changes for development and metabolism through secretion of factors from the nervous system (e.g., neurotransmitter, hormones). Indeed, perceived ER stress in neural cells results in secretion of signals to orchestrate an organismal response improve ER homeostasis, ER stress resilience, and longevity. Specifically, overexpression of the spliced xbp-1s transcription factor within neurons or amphid sheath glia of C. elegans results in activation of the UPRER not only within the neurons and glia themselves, but also within distal intestinal cells (Taylor and Dillin 2013; Frakes et al., 2020). This non-autonomous induction of the UPRER results in increased stress resilience and longevity. Interestingly, the mechanisms whereby neurons and glia coordinate this organism-wide response differ: non-autonomous signaling through neurons is dependent on the release of small clear vesicles (SCVs) through UNC-13, such that loss of unc-13 results in elimination of the beneficial effects of neuronal xbp-1s overexpression (Taylor and Dillin 2013); in contrast, non-autonomous signaling through glia are dependent on release of neuropeptides through the release of dense core vesicles mediated by UNC-31 and neuropeptide processing by EGL-3 (Frakes et al., 2020). The distinction between the two origins of the UPRER signal is further highlighted by the partially additive increase in lifespan that is observed when xbp-1s is overexpressed in both types of neural cells. As neurons and glia have a well-established relationship, it would be likely that both paradigms share some form of communication or mechanism to extend lifespan, though current research has not yet clarified whether direct communication between neurons and glia exist in this paradigm.
In its original study, the beneficial effects of neuronal UPRER were ascribed to the upregulation of chaperones, which resulted in increased protein homeostasis and ER stress resilience. Since then, further studies found that neuronal UPRER also improved lipid metabolism. Lipid staining and lipidomic analysis revealed a decrease in neutral lipid stores, while concurrently showing an increase in monounsaturated fatty acids, including oleic acid (Imanikia et al., 2019a). In fact, supplementation of oleic acid was sufficient to increase the lifespan of wild-type and xbp-1 deficient animals but did not further extend the lifespan of xbp-1s overexpressing animals, providing further evidence that the beneficial effects of neuronal xbp-1s could be at least partially ascribed to changes in lipid profiles. Interestingly, oleic acid supplementation also provided protection against the effects of proteotoxic polyQ40 and α-Aß proteins, suggesting that improving lipid metabolism in the ER could also impact protein homeostasis, though the mechanism whereby this happens is still unexplored (Imanikia et al., 2019a). Importantly, this study ascribed the primary mechanism of lipid remodeling to the desaturases FAT-6 and FAT-7, which metabolized neutral lipid stores into oleic acid to promote organismal health.
The contribution of lipid metabolism to the beneficial effects of non-autonomous UPRER phenotypes was further indicated by the expansion of lysosomes and increased lipophagy found in these animals (Imanikia et al., 2019b; Daniele et al., 2020a). Neuronal xbp-1s overexpression resulted in a significant expansion of lysosomes and increased expression of lysosomal lipases. Changes to lysosomal activity were essential for the beneficial effects of neuronal xbp-1s as knockdown of the C. elegans homolog to mammalian TFEB, hlh-30, was sufficient to suppress the lifespan extension of these animals (Imanikia et al., 2019b). This is in direct agreement with another study that found that HPL-2, a chromatin modifying protein, promotes autophagy to increase ER stress resilience (Kozlowski et al., 2014). Further, transcriptional profiling of worms deficient in phosphatidylcholine (PC) synthesis–which causes ER stress through lipid dysregulation–also induced autophagy in an IRE-1/XB-1-dependent manner (Koh et al., 2018). This is highly similar to a process previously described in yeast, where inhibition of PC biosynthesis activates microlipophagy downstream of UPRER (Vevea et al., 2015). Indeed, in C. elegans, promoting lipophagy by overexpression of ehbp-1, a core component of the conserved RME-1/RAB-10/EHBP-1 lipophagy complex, was sufficient to drive lipid remodeling and lifespan extension (Daniele et al., 2020a). Importantly, this beneficial effect of lipophagy downstream of neuronal UPRER was independent from its canonical role in protein homeostasis through chaperones.
A subsequent study found that the chaperone induction and altered lipid metabolism downstream of non-autonomous UPRER signaling was in part due to signaling from distinct subsets of neurons (Higuchi-Sanabria et al., 2020). Overexpression of xbp-1s within dopaminergic neurons was sufficient to drive EHBP-1 regulated lipid metabolism, while overexpression in serotonergic neurons induced expression of ER chaperones to promote protein homeostasis. Both sources of non-autonomous UPRER signals independently promoted organismal health and lifespan, and their effects were observed to be additive, suggesting an intricate model whereby neurons can differ in their capacity to elicit a peripheral response through unique stress signals. Indeed, a separate genetic screen to identify neurotransmitters involved in neuronal UPRER signals failed to identify dopamine or serotonin signaling, and instead found that tyramine synthesis was essential for transmitting UPRER from neurons to the intestine (Özbey et al., 2020). Overexpression of xbp-1s within the tyraminergic neurons was sufficient to induce the UPRER within the intestine and stimulate changes to the animal’s feeding behavior and brood size. While dopaminergic, serotonergic, and tyraminergic neurons have now been shown to contribute to the non-autonomous UPRER, whether these neurons directly communicate with each other or whether other subtypes or specific neurons may also contribute to–or potentially hinder–the longevity phenotypes of the non-autonomous UPRER remains to be known.
Neuronal transmission of an ER stress response is not limited to the genetic models observed in C. elegans. When Xbp1s is overexpressed in POMC neurons of mice, a similar non-autonomous activation of UPRER exists in peripheral cells and beneficially impacts metabolic physiology (e.g., improved glucose homeostasis, increased insulin sensitivity, and protection against high-fat diet induced obesity) (Williams et al., 2014). Importantly, a similar process can occur with naturally occurring stimuli whereby sensory perception of food can result in activation of POMC neurons to activate UPRER within the liver to prepare for incoming nourishment (Brandt et al., 2018). This induction resulted in increased lipid synthesis and remodeling of the ER, likely to prime the liver for its roles in processing the nutrients. However, how this nonautonomous signaling impacts aging or longevity has not been confirmed, still making a conclusive correlation between UPRER activity and lifespan difficult.
As aptly named, the historical function ascribed to the UPRER is to promote protein homeostasis, though it has become increasingly clear that the UPRER regulates many critical functions outside of protein quality control, including autophagy and lipid homeostasis as described briefly above. One important question is how these functional roles overlap. For example, it isn’t difficult to ascertain how increased lysosomal function can promote autophagy to clear damaged proteins, ultimately resulting in increased protein homeostasis. However, increased lipid metabolism downstream of nonautonomous UPRER signals also resulted in increased clearance of protein aggregates (Imanikia et al., 2019a) and increased resistance to protein misfolding stress (Higuchi-Sanabria et al., 2020). It is entirely possible that improved lipid metabolism simply increases general organismal health, making animals more resilient to all sources of stress or increases the resources available for mitigating damage. Alternatively, it is possible that improved lipid homeostasis can actually have direct impacts on protein homeostasis. For example, increased lipid metabolism can increase secretory capacity of the ER (Daniele et al., 2020b), and it is possible that damaged proteins can be secreted to external environments, such as the pseudocoelom in C.elegans, where it will cause less damage.
Beyond the beneficial roles of the UPRER, unresolved UPRER signaling can be detrimental. For example, we briefly discussed how chronic UPRER can cause apoptosis in mammalian cells, and overexpression of HAC1s, the S. cerevisiae homolog of xbp-1, can perturb cell cycle progression (Sopko et al., 2006). However, promoting UPRER in C. elegans seems to promote organismal health and extend lifespan, which can be due to the post-mitotic nature of the adult worm, which is not deterred by cell cycle or apoptosis machinery. Indeed, ectopic activation of UPRER has negative consequences in the germline, the few actively dividing cells of the adult worm, whereby neuronal UPRER causes decreased fecundity and brood size (Özbey et al., 2020). However, this differentiation between actively dividing versus post-mitotic cells still does not explain the negative impact of xbp-1s overexpression in muscle cells of C. elegans (Taylor and Dillin 2013), as these cells are also post-mitotic. One plausible explanation is that increased UPRER causes lipid depletion, which may be detrimental to the highly energy-demanding muscle cells.
A final thought that we find important to mention is to understand the multi-faceted response of UPRER. With the increasing number of mechanistic pathways being ascribed to UPRER activation, how then does a cell coordinate these downstream pathways? For example, serotonergic and dopaminergic signals elicit two different responses in peripheral cells: induction of chaperones and lipid regulatory enzymes, respectively. How does one transcription factor, XBP-1s, coordinate two distinct responses? It is possible that other cofactors titrate XBP-1s to its appropriate targets; for example, TFEB/HLH-30 is required for the changes to lysosomal genes downstream of XBP-1s activation, suggesting that interaction of TFEB/XBP-1s is important for lysosomal genes. Still to be identified are what other XBP-1s interactors exist to titrate XBP-1s specifically to other target pathways, including lipid regulation, ERAD, or chaperones.
Perhaps one historically overlooked issue in the field of stress biology is in its often single focus: generally, stress responses are studied with a focus on the single organelle that it impacts. However, it is becoming increasingly clear that activation or perturbation of stress responses elicit pleiotropic changes that alter multiple systems.
The functional state of the mitochondria directly impacts the state of other organelles, and thus it is unsurprising that UPRMT function has ramifications on other cellular processes. For example, ER-mitochondria contact sites (ERMCS) allow for the exchange of proteins, metabolites, ions, and lipids between the organelles and are critical in maintaining cellular homeostasis (Xu et al., 2020). In HeLa cells, mitochondrial stress induced by doxycycline exposure can increase ERMCS (Lopez-Crisosto et al., 2021), potentially through UPRMT activation. Increased ERMCS can also result in creased lifespan in drosophila (Garrido-Maraver et al., 2020), opening up the possibility that UPRMT activation may potentially increase lifespan through effects on ERMCS. One important function of ERMCS is CA2+ homeostasis regulated by ER-to-mitochondria CA2+ transfer. In C. elegans, the UPRER regulator, ATF-6 regulates lifespan through modulation of ER-mitochondria calcium transport. As C. elegans age, ATF-6 activity increases, resulting in activation of UPRER and subsequent increase in the calreticulin, CRT-1. Increased levels of CRT-1 result in aberrant accumulation of CA2+ ions in the ER. Interestingly, perturbations in atf-6 result in decreased CRT-1, calcium efflux through the inositol triphosphate receptor, ITR-1, and increased mitochondrial CA2+ import, resulting in increased metabolic activity of mitochondria and extension of lifespan (Burkewitz et al., 2020).
Considering the close communication between the mitochondria and ER and the impact of the UPRER on mitochondrial health, it would be of great interest to determine the overlapping functions of all stress responses and how each impact the other. For example, induction of the UPRMT has direct impact on the HSR. HSF-1 function declines during the aging process (Trivedi and Jurivich 2020) and the HSR can become dysfunctional as early as the second day of reproductive capacity in C. elegans (Labbadia and Morimoto 2015). However, one study found that low levels of mitochondrial stress through perturbation of a cytochrome C oxidase subunit, cox-6c, can increase functional capacity of the HSR at late age and has a positive impact on stress resilience and longevity. Specifically, activation of both the UPRMT and HSR are both equally responsible for the increase in organismal health and lifespan found in animals exposed to mitochondrial stress (Labbadia et al., 2017). Interestingly, knockdown of hsp-6 (the gene encoding the primary mitochondrial chaperone mtHSP70) also resulted in activation of the HSR. In fact, microarray analysis of animals with hsp-6 knockdown found 187 genes differentially expressed, with 66 being known targets of HSF-1 and DVE-1, highlighting the distinct and important overlap between UPRMT and HSR. Further investigation of these overlapping genes found that many genes were involved in lipid metabolism. Subsequent metabolomics identified that this mitochondria-to-cytosolic stress response (MCSR) was dependent on an overall increase in fatty acid levels. Specifically, increased cardiolipin and inhibition of ceramide synthesis were sufficient to drive MCSR induction. Most importantly, blocking fatty acid oxidation to increase fats using the drug, perhexiline, was sufficient to drive MCSR activation in both worms and human cells (Kim et al., 2016). This study has powerful translational potential, with perhexiline serving as a potential therapeutic intervention for aging and protein aggregation disorders, including Huntington’s, Alzheimer’s, and ALS. Finally, HSF-1 has been shown to directly impact mtDNA gene expression through elevated histone H4 levels. Specifically, knockdown of the conserved heat shock factor binding protein, hsb-1 results in increased H4 levels early in development, which alters chromatin state of mtDNA, decreases expression of mtDNA-encoded genes, reduces mitochondrial respiratory capacity, and promotes lifespan in a UPRMT-dependent manner (Sural et al., 2020).
These studies continued to bridge the functional role of the mitochondria to other organelles, particularly with the surprising finding that HSF-1, the transcriptional regulator originally ascribed to HSR, now being directly influenced by UPRMT. HSF-1 has also been shown to directly impact the actin cytoskeleton (Baird et al., 2014; Higuchi-Sanabria et al., 2018b), and many studies have already highlighted the importance of actin as serving as the scaffold for–and regulating the dynamics of–the mitochondria (Fehrenbacher et al., 2004; Boldogh and Pon 2006; Higuchi et al., 2013; Korobova et al., 2013; Manor et al., 2015). A large-scale cross-organism screen combining the power of CRISPR-Cas9 screening in human cells (Shalem et al., 2014) and RNAi screening in C. elegans identified EPS-8 as a novel link between actin and mitochondrial homeostasis (Moehle et al., 2021). Specifically, knockdown of eps-8 resulted in hyperstabilization of actin through the integrin, PAT-3. This increased stability of actin resulted in fragmentation of mitochondria and a beneficial activation of the UPRMT, such that these animals exhibited increased lifespan. These findings are conserved in human cells where hyperactive integrin signaling can hyperstabilize actin filaments, resulting in increased stress resilience through activation of UPRMT through HSF1 and ATF5 (Tharp et al., 2021). These studies beg the question of what impact–if any–UPRMT and mitochondrial dysfunction can have on the actin cytoskeleton. While one would expect that mitochondrial dysfunction and decreased energetics may negatively influence the actin cytoskeleton, the activation of the HSR and increased function of HSF-1 during mitochondrial stress may actually positively impact actin health and function.
Similar to the overlapping functions of UPRMT and HSR, the UPRER also shows some overlap. In fact, nine stress response genes are induced by both UPRER and HSR (Liu and Chang 2008), including the ER chaperone, HSP-4/BiP (Kohno et al., 1993) and the COPII cargo receptor, Erv29 (Caldwell et al., 2001). Importantly, the activation of the HSR via a constitutively active HSF1 rescued the growth defect of UPRER deficient cells. In addition, induction of the UPRER via heat-shock in mammalian cells can activate canonical XBP1 targets (Heldens et al., 2011). While these studies were not performed in C. elegans, overexpression of xbp-1s in C. elegans did seem to negatively impact the expression of key HSF-1 targets, including hsp-16.2 and hsp-70 (Taylor and Dillin 2013), clearly indicating that UPRER function has at least some indirect impact on HSR. More thorough analysis is certainly necessary to determine the direct ramifications of UPRER on HSR and vice versa during the aging process in C. elegans.
Overall, it is becoming increasingly clear that overlapping functions exist between stress responses and that each response can impact other organelles. For example, UPRMT has direct ramifications on overall health and fitness of an organism–not just on the mitochondria, and not just on the single cell type exposed to mitochondrial stress. The same is true for the UPRER and the HSR, highlighting the critical importance of studying the cross-regulation of stress responses, and moving away from studying these quality control mechanisms in a black box of specificity. One intriguing question is to understand how all quality control machineries are regulated as a whole. For example, under conditions of competing needs, does there exist a preference for a specific stress response? While it is clear that the HSR is a critical response to thermal stress, heat can also damage the mitochondria and ER, so how does the cell prioritize HSR over UPRER/UPRMT activation? Under these more “generalized” stresses, how does the cell preferentially activate one response over the other? Even further, if multiple stress responses are activated, would this have a synergistic effect producing a hyper long-lived animal? Or would there be a detrimental effect when activating too many stress responses? Future studies must continue to focus on the direct and indirect effects of one stress response on all organelles and take into consideration the effects of organelle-to-organelle signaling and even cell-to-cell signaling in the cross communication and cross regulation of stress responses.
ND prepared the introduction and heat-shock response sections, GG prepared the UPRER section and all figures, and RHS prepared the UPRMT and conclusion sections. ND, GG, and RHS reviewed and edited all sections of the manuscript.
GG is supported by T32AG052374 and RHS is supported by R00AG065200 from the National Institute on Aging.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Adams, C. J., Kopp, M. C., Larburu, N., Nowak, P. R., and Ali, M. M. U. (2019). Structure and Molecular Mechanism of ER Stress Signaling by the Unfolded Protein Response Signal Activator IRE1. Front. Mol. Biosci. 6, 1. doi:10.3389/fmolb.2019.00011
Amin, M. R., Mahmud, S. A., Dowgielewicz, J. L., Sapkota, M., and Pellegrino, M. W. (2020). A Novel Gene-Diet Interaction Promotes Organismal Lifespan and Host protection during Infection via the Mitochondrial UPR. Plos Genet. 16, e1009234. doi:10.1371/journal.pgen.1009234
Amin-Wetzel, N., Saunders, R. A., Kamphuis, M. J., Rato, C., Preissler, S., Harding, H. P., et al. (2017). A J-Protein Co-chaperone Recruits BiP to Monomerize IRE1 and Repress the Unfolded Protein Response. Cell 171, 1625e13–1637. doi:10.1016/j.cell.2017.10.040
Anckar, J., and Sistonen, L. (2011). Regulation of HSF1 Function in the Heat Stress Response: Implications in Aging and Disease. Annu. Rev. Biochem. 80, 1089–1115. doi:10.1146/annurev-biochem-060809-095203
Anderson, N. S., and Haynes, C. M. (2020). Folding the Mitochondrial UPR into the Integrated Stress Response. Trends Cell Biol. 30, 428–439. doi:10.1016/j.tcb.2020.03.001
Aragón, T., van Anken, E., Pincus, D., Serafimova, I. M., Korennykh, A. V., Rubio, C. A., et al. (2009). Messenger RNA Targeting to Endoplasmic Reticulum Stress Signalling Sites. Nature 457, 736–740. doi:10.1038/nature07641
Baird, N. A., Douglas, P. M., Simic, M. S., Grant, A. R., Moresco, J. J., Wolff, S. C., et al. (2014). HSF-1-mediated Cytoskeletal Integrity Determines Thermotolerance and Life Span. Science 346, 360–363. doi:10.1126/science.1253168
Bakthisaran, R., Tangirala, R., and Rao, Ch. M. (18542015). Small Heat Shock Proteins: Role in Cellular Functions and Pathology. Biochim. Biophys. Acta 1854, 291–319. doi:10.1016/j.bbapap.2014.12.019
Bar-Ziv, R., Bolas, T., and Dillin, A. (2020). Systemic Effects of Mitochondrial Stress. EMBO Rep. 21, e50094. doi:10.15252/embr.202050094
Bar-Ziv, R., Frakes, A. E., Higuchi-Sanabria, R., Bolas, T., Frankino, P. A., Gildea, H. K., et al. (2020). Measurements of Physiological Stress Responses in C. Elegans. JoVE 159, 1. doi:10.3791/61001
Barna, J., Csermely, P., and Vellai, T. (2018). Roles of Heat Shock Factor 1 beyond the Heat Shock Response. Cell. Mol. Life Sci. 75, 2897–2916. doi:10.1007/s00018-018-2836-6
B’chir, W., Maurin, A.-C., Carraro, V., Averous, J., Jousse, C., Muranishi, Y., et al. (2013). The eIF2α/ATF4 Pathway Is Essential for Stress-Induced Autophagy Gene Expression. Nucleic Acids Res. 41, 7683–7699.
Beck, D., Niessner, H., Smalley, K. S., Flaherty, K., Paraiso, K. H., Busch, C., et al. (2013). Vemurafenib Potently Induces Endoplasmic Reticulum Stress-Mediated Apoptosis in BRAFV600E Melanoma Cells. Sci. Signal. 6, ra7. doi:10.1126/scisignal.2003057
Ben-Zvi, A., Miller, E. A., and Morimoto, R. I. (2009). Collapse of Proteostasis Represents an Early Molecular Event in Caenorhabditis elegans Aging. Proc. Natl. Acad. Sci. 106, 14914–14919. doi:10.1073/pnas.0902882106
Benedetti, C., Haynes, C. M., Yang, Y., Harding, H. P., and Ron, D. (2006). Ubiquitin-like Protein 5 Positively Regulates Chaperone Gene Expression in the Mitochondrial Unfolded Protein Response. Genetics 174, 229–239. doi:10.1534/genetics.106.061580
Benham, A. M. (2012). Protein Secretion and the Endoplasmic Reticulum. Cold Spring Harbor Perspect. Biol. 4, a012872. doi:10.1101/cshperspect.a012872
Bennett, C. F., Vander Wende, H., Simko, M., Klum, S., Barfield, S., Choi, H., et al. (2014). Activation of the Mitochondrial Unfolded Protein Response Does Not Predict Longevity in Caenorhabditis elegans. Nat. Commun. 5, 3483. doi:10.1038/ncomms4483
Berendzen, K. M., Durieux, J., Shao, L.-W., Tian, Y., Kim, H.-E., Wolff, S., et al. (2016). Neuroendocrine Coordination of Mitochondrial Stress Signaling and Proteostasis. Cell 166, 1553–1563. doi:10.1016/j.cell.2016.08.042
Binder, R. J., and Srivastava, P. K. (2005). Peptides Chaperoned by Heat-Shock Proteins Are a Necessary and Sufficient Source of Antigen in the Cross-Priming of CD8+ T Cells. Nat. Immunol. 6, 593–599. doi:10.1038/ni1201
Boldogh, I. R., and Pon, L. A. (2006). Interactions of Mitochondria with the Actin Cytoskeleton. Biochim. Biophys. Acta (Bba) - Mol. Cell Res. 1763, 450–462. doi:10.1016/j.bbamcr.2006.02.014
Brandt, C., Nolte, H., Henschke, S., Engström Ruud, L., Awazawa, M., Morgan, D. A., et al. (2018). Food Perception Primes Hepatic ER Homeostasis via Melanocortin-dependent Control of mTOR Activation. Cell 175, 1321–1335. e20. doi:10.1016/j.cell.2018.10.015
Burkewitz, K., Feng, G., Dutta, S., Kelley, C. A., Steinbaugh, M., Cram, E. J., et al. (2020). Atf-6 Regulates Lifespan through ER-Mitochondrial Calcium Homeostasis. Cell Rep. 32, 108125. doi:10.1016/j.celrep.2020.108125
Butov, A., Johnson, T., Cypser, J., Sannikov, I., Volkov, M., Sehl, M., et al. (2001). Hormesis and Debilitation Effects in Stress Experiments Using the Nematode Worm Caenorhabditis elegans: the Model of Balance between Cell Damage and HSP Levels. Exp. Gerontol. 37, 57–66. doi:10.1016/s0531-5565(01)00161-9
Calculli, G., Lee, H. J., Shen, K., Pham, U., Herholz, M., Trifunovic, A., et al. (2021). Systemic Regulation of Mitochondria by Germline Proteostasis Prevents Protein Aggregation in the Soma of C. elegans. Sci. Adv. 7, eabg3012. doi:10.1126/sciadv.abg3012
Caldwell, S. R., Hill, K. J., and Cooper, A. A. (2001). Degradation of Endoplasmic Reticulum (ER) Quality Control Substrates Requires Transport between the ER and Golgi. J. Biol. Chem. 276, 23296–23303. doi:10.1074/jbc.m102962200
Calfon, M., Zeng, H., Urano, F., Till, J. H., Hubbard, S. R., Harding, H. P., et al. (2002). IRE1 Couples Endoplasmic Reticulum Load to Secretory Capacity by Processing the XBP-1 mRNA. Nature 415, 92–96. doi:10.1038/415092a
Castro, J. P., Wardelmann, K., Grune, T., and Kleinridders, A. (2018). Mitochondrial Chaperones in the Brain: Safeguarding Brain Health and Metabolism? Front. Endocrinol. 9, 196. doi:10.3389/fendo.2018.00196
Chaudhari, S. N., and Kipreos, E. T. (2017). Increased Mitochondrial Fusion Allows the Survival of Older Animals in Diverse C. elegans Longevity Pathways. Nat. Commun. 8, 182. doi:10.1038/s41467-017-00274-4
Chaurasia, B., and Summers, S. A. (2015). Ceramides - Lipotoxic Inducers of Metabolic Disorders. Trends Endocrinol. Metab. 26, 538–550. doi:10.1016/j.tem.2015.07.006
Chauve, L., Hodge, F., Murdoch, S., Masoudzadeh, F., Mann, H.-J., Lopez-Clavijo, A. F., et al. (2021). Neuronal HSF-1 Coordinates the Propagation of Fat Desaturation across Tissues to Enable Adaptation to High Temperatures in C. elegans. Plos Biol. 19, e3001431. doi:10.1371/journal.pbio.3001431
Clarke, H. J., Chambers, J. E., Liniker, E., and Marciniak, S. J. (2014). Endoplasmic Reticulum Stress in Malignancy. Cancer Cell 25, 563–573. doi:10.1016/j.ccr.2014.03.015
Coleman, O. I., and Haller, D. (2019). ER Stress and the UPR in Shaping Intestinal Tissue Homeostasis and Immunity. Front. Immunol. 10, 1. doi:10.3389/fimmu.2019.02825
Colla, E. (2019). Linking the Endoplasmic Reticulum to Parkinson's Disease and Alpha-Synucleinopathy. Front. Neurosci. 13. doi:10.3389/fnins.2019.00560
Couvillion, M. T., Soto, I. C., Shipkovenska, G., and Churchman, L. S. (2016). Synchronized Mitochondrial and Cytosolic Translation Programs. Nature 533, 499–503. doi:10.1038/nature18015
Cox, J. S., Shamu, C. E., and Walter, P. (1993). Transcriptional Induction of Genes Encoding Endoplasmic Reticulum Resident Proteins Requires a Transmembrane Protein Kinase. Cell 73, 1197–1206. doi:10.1016/0092-8674(93)90648-a
Cui, W., Li, J., Ron, D., and Sha, B. (2011). The Structure of the PERK Kinase Domain Suggests the Mechanism for its Activation. Acta Crystallogr. D Biol. Cryst. 67, 423–428. doi:10.1107/s0907444911006445
Curtis, R., O'Connor, G., and DiStefano, P. S. (2006). Aging Networks inCaenorhabditis Elegans: AMP-Activated Protein Kinase (Aak-2) Links Multiple Aging and Metabolism Pathways. Aging Cell 5, 119–126. doi:10.1111/j.1474-9726.2006.00205.x
Cypser, J. R., and Johnson, T. E. (2002). Multiple Stressors in Caenorhabditis elegans Induce Stress Hormesis and Extended Longevity. Journals Gerontol. Ser. A: Biol. Sci. Med. Sci. 57, B109–B114. doi:10.1093/gerona/57.3.b109
Dai, C. (2018). The Heat-Shock, or HSF1-Mediated Proteotoxic Stress, Response in Cancer: from Proteomic Stability to Oncogenesis. Phil. Trans. R. Soc. B 373, 20160525. doi:10.1098/rstb.2016.0525
Daniele, J. R., Higuchi-Sanabria, R., Durieux, J., Monshietehadi, S., Ramachandran, V., Tronnes, S. U., et al. (2020). UPRER Promotes Lipophagy Independent of Chaperones to Extend Life Span. Sci. Adv. 6, eaaz1441. doi:10.1126/sciadv.aaz1441
Daniele, J. R., Higuchi-Sanabria, R., Durieux, J., Monshietehadi, S., Ramachandran, V., Tronnes, S. U., et al. (2020). UPR ER Promotes Lipophagy Independent of Chaperones to Extend Life Span. Sci. Adv. 6, 1. doi:10.1126/sciadv.aaz1441
Das, R., Melo, J. A., Thondamal, M., Morton, E. A., Cornwell, A. B., Crick, B., et al. (2017). The Homeodomain-Interacting Protein Kinase HPK-1 Preserves Protein Homeostasis and Longevity through Master Regulatory Control of the HSF-1 Chaperone Network and TORC1-Restricted Autophagy in Caenorhabditis elegans. Plos Genet. 13, e1007038. doi:10.1371/journal.pgen.1007038
Das, S., Ooi, F. K., Cruz Corchado, J., Fuller, L. C., Weiner, J. A., and Prahlad, V. (2020). Serotonin Signaling by Maternal Neurons upon Stress Ensures Progeny Survival. Elife 9, e55246. doi:10.7554/eLife.55246
Desai, S., Liu, Z., Yao, J., Patel, N., Chen, J., Wu, Y., et al. (2013). Heat Shock Factor 1 (HSF1) Controls Chemoresistance and Autophagy through Transcriptional Regulation of Autophagy-Related Protein 7 (ATG7). J. Biol. Chem. 288, 9165–9176. doi:10.1074/jbc.m112.422071
Dillin, A., Hsu, A.-L., Arantes-Oliveira, N., Lehrer-Graiwer, J., Hsin, H., Fraser, A. G., et al. (2002). Rates of Behavior and Aging Specified by Mitochondrial Function during Development. Science 298, 2398–2401. doi:10.1126/science.1077780
Dokladny, K., Zuhl, M. N., Mandell, M., Bhattacharya, D., Schneider, S., Deretic, V., et al. (2013). Regulatory Coordination between Two Major Intracellular Homeostatic Systems. J. Biol. Chem. 288, 14959–14972. doi:10.1074/jbc.m113.462408
Douglas, P. M., Baird, N. A., Simic, M. S., Uhlein, S., McCormick, M. A., Wolff, S. C., et al. (2015). Heterotypic Signals from Neural HSF-1 Separate Thermotolerance from Longevity. Cell Rep. 12, 1196–1204. doi:10.1016/j.celrep.2015.07.026
Dubaquié, Y., Looser, R., Fünfschilling, U., Jenö, P., and Rospert, S. (1998). Identification of Invivo Substrates of the Yeast Mitochondrial Chaperonins Reveals Overlapping but Non-identical Requirement for Hsp60 and Hsp10. EMBO J. 17, 5868–5876. doi:10.1093/emboj/17.20.5868
Durieux, J., Wolff, S., and Dillin, A. (2011). The Cell-Non-Autonomous Nature of Electron Transport Chain-Mediated Longevity. Cell 144, 79–91. doi:10.1016/j.cell.2010.12.016
Estébanez, B., de Paz, J. A., Cuevas, M. J., and González-Gallego, J. (2018). Endoplasmic Reticulum Unfolded Protein Response, Aging and Exercise: An Update. Front. Physiol. 9. doi:10.3389/fphys.2018.01744
Fagone, P., and Jackowski, S. (2009). Membrane Phospholipid Synthesis and Endoplasmic Reticulum Function. J. Lipid Res. 50, S311–S316. doi:10.1194/jlr.r800049-jlr200
Fawcett, T. W., Sylvester, S. L., Sarge, K. D., Morimoto, R. I., and Holbrook, N. J. (1994). Effects of Neurohormonal Stress and Aging on the Activation of Mammalian Heat Shock Factor 1. J. Biol. Chem. 269, 32272–32278. doi:10.1016/s0021-9258(18)31631-4
Fehrenbacher, K. L., Yang, H.-C., Gay, A. C., Huckaba, T. M., and Pon, L. A. (2004). Live Cell Imaging of Mitochondrial Movement along Actin Cables in Budding Yeast. Curr. Biol. 14, 1996–2004. doi:10.1016/j.cub.2004.11.004
Fiorese, C. J., Schulz, A. M., Lin, Y.-F., Rosin, N., Pellegrino, M. W., and Haynes, C. M. (2016). The Transcription Factor ATF5 Mediates a Mammalian Mitochondrial UPR. Curr. Biol. 26, 2037–2043. doi:10.1016/j.cub.2016.06.002
Forsström, S., Jackson, C. B., Carroll, C. J., Kuronen, M., Pirinen, E., Pradhan, S., et al. (2019). Fibroblast Growth Factor 21 Drives Dynamics of Local and Systemic Stress Responses in Mitochondrial Myopathy with mtDNA Deletions. Cell Metab. 30, 1040–1054. e7. doi:10.1016/j.cmet.2019.08.019
Frakes, A. E., Metcalf, M. G., Tronnes, S. U., Bar-Ziv, R., Durieux, J., Gildea, H. K., et al. (2020). Four Glial Cells Regulate ER Stress Resistance and Longevity via Neuropeptide Signaling in C. elegans. Science 367, 436–440. doi:10.1126/science.aaz6896
Franceschi, C., Garagnani, P., Morsiani, C., Conte, M., Santoro, A., Grignolio, A., et al. (2018). The Continuum of Aging and Age-Related Diseases: Common Mechanisms but Different Rates. Front. Med. 5, 61. doi:10.3389/fmed.2018.00061
Garigan, D., Hsu, A.-L., Fraser, A. G., Kamath, R. S., Ahringer, J., and Kenyon, C. (2002). Genetic Analysis of Tissue Aging in Caenorhabditis elegans: a Role for Heat-Shock Factor and Bacterial Proliferation. Genetics 161, 1101–1112. doi:10.1093/genetics/161.3.1101
Garrido-Maraver, J., Loh, S. H. Y., and Martins, L. M. (2020). Forcing Contacts between Mitochondria and the Endoplasmic Reticulum Extends Lifespan in a Drosophila Model of Alzheimer's Disease. Biol. Open 9, bio047530. doi:10.1242/bio.047530
Garsin, D. A., Villanueva, J. M., Begun, J., Kim, D. H., Sifri, C. D., Calderwood, S. B., et al. (2003). Long-lived C. elegans Daf-2 Mutants Are Resistant to Bacterial Pathogens. Science 300, 1921. doi:10.1126/science.1080147
Gross, S. R., and Kinzy, T. G. (2007). Improper Organization of the Actin Cytoskeleton Affects Protein Synthesis at Initiation. Mol. Cell Biol 27, 1974–1989. doi:10.1128/mcb.00832-06
Haeussler, S., Köhler, F., Witting, M., Premm, M. F., Rolland, S. G., Fischer, C., et al. (2020). Autophagy Compensates for Defects in Mitochondrial Dynamics. Plos Genet. 16, e1008638. doi:10.1371/journal.pgen.1008638
Hallberg, E. M., Shu, Y., and Hallberg, R. L. (1993). Loss of Mitochondrial Hsp60 Function: Nonequivalent Effects on Matrix-Targeted and Intermembrane-Targeted Proteins. Mol. Cel. Biol. 13, 3050–3057. doi:10.1128/mcb.13.5.3050
Han, J., Back, S. H., Hur, J., Lin, Y.-H., Gildersleeve, R., Shan, J., et al. (2013). ER-stress-induced Transcriptional Regulation Increases Protein Synthesis Leading to Cell Death. Nat. Cell Biol 15, 481–490. doi:10.1038/ncb2738
Hartl, F. U. (2017). Protein Misfolding Diseases. Annu. Rev. Biochem. 86, 21–26. doi:10.1146/annurev-biochem-061516-044518
Haynes, C. M., Petrova, K., Benedetti, C., Yang, Y., and Ron, D. (2007). ClpP Mediates Activation of a Mitochondrial Unfolded Protein Response in C. elegans. Dev. Cell 13, 467–480. doi:10.1016/j.devcel.2007.07.016
Haze, K., Yoshida, H., Yanagi, H., Yura, T., and Mori, K. (1999). Mammalian Transcription Factor ATF6 Is Synthesized as a Transmembrane Protein and Activated by Proteolysis in Response to Endoplasmic Reticulum Stress. MBoC 10, 3787–3799. doi:10.1091/mbc.10.11.3787
He, J., Ford, H. C., Carroll, J., Douglas, C., Gonzales, E., Ding, S., et al. (2018). Assembly of the Membrane Domain of ATP Synthase in Human Mitochondria. Proc. Natl. Acad. Sci. USA 115, 2988–2993. doi:10.1073/pnas.1722086115
Heldens, L., Hensen, S. M. M., Onnekink, C., van Genesen, S. T., Dirks, R. P., and Lubsen, N. H. (2011). An Atypical Unfolded Protein Response in Heat Shocked Cells. PLoS ONE 6, e23512. doi:10.1371/journal.pone.0023512
Higuchi, R., Vevea, J. D., Swayne, T. C., Chojnowski, R., Hill, V., Boldogh, I. R., et al. (2013). Actin Dynamics Affect Mitochondrial Quality Control and Aging in Budding Yeast. Curr. Biol. 23, 2417–2422. doi:10.1016/j.cub.2013.10.022
Higuchi-Sanabria, R., Durieux, J., Kelet, N., Homentcovschi, S., de Los Rios Rogers, M., Monshietehadi, S., et al. (2020). Divergent Nodes of Non-autonomous UPRER Signaling through Serotonergic and Dopaminergic Neurons. Cell Rep. 33, 108489. doi:10.1016/j.celrep.2020.108489
Higuchi-Sanabria, R., Frankino, P. A., Paul, J. W., Tronnes, S. U., and Dillin, A. (2018). A Futile Battle? Protein Quality Control and the Stress of Aging. Dev. Cell 44, 139–163. doi:10.1016/j.devcel.2017.12.020
Higuchi-Sanabria, R., Paul, J. W., Durieux, J., Benitez, C., Frankino, P. A., Tronnes, S. U., et al. (2018). Spatial Regulation of the Actin Cytoskeleton by HSF-1 during Aging. MBoC 29, 2522–2527. doi:10.1091/mbc.e18-06-0362
Higuchi-Sanabria, R., Paul, J. W., Durieux, J., Benitez, C., Frankino, P. A., Tronnes, S. U., et al. (2018). Spatial Regulation of the Actin Cytoskeleton by HSF-1 during Aging. MBoC 29, 2522–2527. doi:10.1091/mbc.e18-06-0362
Hillary, R. F., and FitzGerald, U. (2018). A Lifetime of Stress: ATF6 in Development and Homeostasis. J. Biomed. Sci. 25, 48. doi:10.1186/s12929-018-0453-1
Hollien, J., and Weissman, J. S. (2006). Decay of Endoplasmic Reticulum-Localized mRNAs during the Unfolded Protein Response. Science 313, 104–107. doi:10.1126/science.1129631
Houtkooper, R. H., Mouchiroud, L., Ryu, D., Moullan, N., Katsyuba, E., Knott, G., et al. (2013). Mitonuclear Protein Imbalance as a Conserved Longevity Mechanism. Nature 497, 451–457. doi:10.1038/nature12188
Hsu, A.-L., Murphy, C. T., and Kenyon, C. (2003). Regulation of Aging and Age-Related Disease by DAF-16 and Heat-Shock Factor. Science 300, 1142–1145. doi:10.1126/science.1083701
Ichimiya, H., Huet, R. G., Hartman, P., Amino, H., Kita, K., and Ishii, N. (2002). Complex II Inactivation Is Lethal in the Nematode Caenorhabditis elegans. Mitochondrion 2, 191–198. doi:10.1016/s1567-7249(02)00069-7
Imanikia, S., Özbey, N. P., Krueger, C., Casanueva, M. O., and Taylor, R. C. (2019). Neuronal XBP-1 Activates Intestinal Lysosomes to Improve Proteostasis in C. elegans. Curr. Biol. 29, 2322–2338. e7. doi:10.1016/j.cub.2019.06.031
Imanikia, S., Sheng, M., Castro, C., Griffin, J. L., and Taylor, R. C. (2019). XBP-1 Remodels Lipid Metabolism to Extend Longevity. Cell Rep. 28, 581e4–589. doi:10.1016/j.celrep.2019.06.057
Jee, H. (2016). Size Dependent Classification of Heat Shock Proteins: a Mini-Review. J. Exerc. Rehabil. 12, 255–259. doi:10.12965/jer.1632642.321
Jiang, C. C., Chen, L. H., Gillespie, S., Wang, Y. F., Kiejda, K. A., Zhang, X. D., et al. (2007). Inhibition of MEK Sensitizes Human Melanoma Cells to Endoplasmic Reticulum Stress-Induced Apoptosis. Cancer Res. 67, 9750–9761. doi:10.1158/0008-5472.can-07-2047
Jung, K.-W., So, Y.-S., and Bahn, Y.-S. (2016). Unique Roles of the Unfolded Protein Response Pathway in Fungal Development and Differentiation. Sci. Rep. 6, 33413. doi:10.1038/srep33413
Kenny, T. C., and Germain, D. (2017). mtDNA, Metastasis, and the Mitochondrial Unfolded Protein Response (UPRmt). Front. Cell Dev. Biol. 5, 37. doi:10.3389/fcell.2017.00037
Khalimonchuk, O., Bird, A., and Winge, D. R. (2007). Evidence for a Pro-oxidant Intermediate in the Assembly of Cytochrome Oxidase. J. Biol. Chem. 282, 17442–17449. doi:10.1074/jbc.m702379200
Kim, H.-E., Grant, A. R., Simic, M. S., Kohnz, R. A., Nomura, D. K., Durieux, J., et al. (2016). Lipid Biosynthesis Coordinates a Mitochondrial-To-Cytosolic Stress Response. Cell 166, 1539–1552. e16. doi:10.1016/j.cell.2016.08.027
Kim, K. H., Jeong, Y. T., Oh, H., Kim, S. H., Cho, J. M., Kim, Y.-N., et al. (2013). Autophagy Deficiency Leads to protection from Obesity and Insulin Resistance by Inducing Fgf21 as a Mitokine. Nat. Med. 19, 83–92. doi:10.1038/nm.3014
Kirkwood, T. B. L., and Austad, S. N. (2000). Why Do We Age? Nature 408, 233–238. doi:10.1038/35041682
Koh, J. H., Wang, L., Beaudoin-Chabot, C., and Thibault, G. (2018). Lipid Bilayer Stress-Activated IRE-1 Modulates Autophagy during Endoplasmic Reticulum Stress. J. Cel. Sci. 131, 1. doi:10.1242/jcs.217992
Kohno, K., Normington, K., Sambrook, J., Gething, M. J., and Mori, K. (1993). The Promoter Region of the Yeast KAR2 (BiP) Gene Contains a Regulatory Domain that Responds to the Presence of Unfolded Proteins in the Endoplasmic Reticulum. Mol. Cel. Biol. 13, 877–890. doi:10.1128/mcb.13.2.877-890.1993
Korobova, F., Ramabhadran, V., and Higgs, H. N. (2013). An Actin-dependent Step in Mitochondrial Fission Mediated by the ER-Associated Formin INF2. Science 339, 464–467. doi:10.1126/science.1228360
Kourtis, N., Nikoletopoulou, V., and Tavernarakis, N. (2012). Small Heat-Shock Proteins Protect from Heat-Stroke-Associated Neurodegeneration. Nature 490, 213–218. doi:10.1038/nature11417
Kourtis, N., and Tavernarakis, N. (2011). Cellular Stress Response Pathways and Ageing: Intricate Molecular Relationships. EMBO J. 30, 2520–2531. doi:10.1038/emboj.2011.162
Kozlowski, L., Garvis, S., Bedet, C., and Palladino, F. (2014). The Caenorhabditis elegans HP1 Family Protein HPL-2 Maintains ER Homeostasis through the UPR and Hormesis. Proc. Natl. Acad. Sci. 111, 5956–5961. doi:10.1073/pnas.1321698111
Kroeger, H., Grimsey, N., Paxman, R., Chiang, W. C., Plate, L., Jones, Y., et al. (2018). The Unfolded Protein Response Regulator ATF6 Promotes Mesodermal Differentiation. Sci. Signal. 11, eaan5785. doi:10.1126/scisignal.aan5785
Kuang, J., and Ebert, P. R. (2012). The Failure to Extend Lifespan via Disruption of Complex II Is Linked to Preservation of Dynamic Control of Energy Metabolism. Mitochondrion 12, 280–287. doi:10.1016/j.mito.2011.10.003
Kumsta, C., Chang, J. T., Lee, R., Tan, E. P., Yang, Y., Loureiro, R., et al. (2019). The Autophagy Receptor p62/SQST-1 Promotes Proteostasis and Longevity in C. elegans by Inducing Autophagy. Nat. Commun. 10, 5648. doi:10.1038/s41467-019-13540-4
Kumsta, C., Chang, J. T., Schmalz, J., and Hansen, M. (2017). Hormetic Heat Stress and HSF-1 Induce Autophagy to Improve Survival and Proteostasis in C. elegans. Nat. Commun. 8, 14337. doi:10.1038/ncomms14337
Kumsta, C., Ching, T.-T., Nishimura, M., Davis, A. E., Gelino, S., Catan, H. H., et al. (2014). Integrin-linked Kinase Modulates Longevity and Thermotolerance inC. Elegansthrough Neuronal Control of HSF-1. Aging cell 13, 419–430. doi:10.1111/acel.12189
Labbadia, J., Brielmann, R. M., Neto, M. F., Lin, Y.-F., Haynes, C. M., and Morimoto, R. I. (2017). Mitochondrial Stress Restores the Heat Shock Response and Prevents Proteostasis Collapse during Aging. Cell Rep. 21, 1481–1494. doi:10.1016/j.celrep.2017.10.038
Labbadia, J., and Morimoto, R. I. (2015). Repression of the Heat Shock Response Is a Programmed Event at the Onset of Reproduction. Mol. Cel. 59, 639–650. doi:10.1016/j.molcel.2015.06.027
Lan, J., Rollins, J. A., Zang, X., Wu, D., Zou, L., Wang, Z., et al. (2019). Translational Regulation of Non-autonomous Mitochondrial Stress Response Promotes Longevity. Cell Rep. 28, 1050–1062. e6. doi:10.1016/j.celrep.2019.06.078
Leary, S. C., Battersby, B. J., Hansford, R. G., and Moyes, C. D. (1998). Interactions between Bioenergetics and Mitochondrial Biogenesis. Biochim. Biophys. Acta (Bba) - Bioenerg. 1365, 522–530. doi:10.1016/s0005-2728(98)00105-4
Lee, A.-H., Heidtman, K., Hotamisligil, G. S., and Glimcher, L. H. (2011). Dual and Opposing Roles of the Unfolded Protein Response Regulated by IRE1 and XBP1 in Proinsulin Processing and Insulin Secretion. Proc. Natl. Acad. Sci. 108, 8885–8890. doi:10.1073/pnas.1105564108
Lee, S. S., Lee, R. Y. N., Fraser, A. G., Kamath, R. S., Ahringer, J., and Ruvkun, G. (2003). A Systematic RNAi Screen Identifies a Critical Role for Mitochondria in C. elegans Longevity. Nat. Genet. 33, 40–48. doi:10.1038/ng1056
Levi-Ferber, M., Gian, H., Dudkevich, R., and Henis-Korenblit, S. (2015). Transdifferentiation Mediated Tumor Suppression by the Endoplasmic Reticulum Stress Sensor IRE-1 in C. elegans. eLife 4, e08005. doi:10.7554/eLife.08005
Levi-Ferber, M., Shalash, R., Le-Thomas, A., Salzberg, Y., Shurgi, M., Benichou, J. I., et al. (2021). Neuronal Regulated Ire-1-dependent mRNA Decay Controls Germline Differentiation in Caenorhabditis elegans. eLife 10, e65644. doi:10.7554/elife.65644
Li, Z., Zhang, Z., Ren, Y., Wang, Y., Fang, J., Yue, H., et al. (2021). Aging and Age‐related Diseases: from Mechanisms to Therapeutic Strategies. Biogerontology 22, 165–187. doi:10.1007/s10522-021-09910-5
Lindquist, S., and Craig, E. A. (1988). The Heat-Shock Proteins. Annu. Rev. Genet. 22, 631–677. doi:10.1146/annurev.ge.22.120188.003215
Liu, X., Jiang, N., Hughes, B., Bigras, E., Shoubridge, E., and Hekimi, S. (2005). Evolutionary Conservation of the Clk-1-dependent Mechanism of Longevity: Loss of Mclk1 Increases Cellular Fitness and Lifespan in Mice. Genes Dev. 19, 2424–2434. doi:10.1101/gad.1352905
Liu, Y., and Chang, A. (2008). Heat Shock Response Relieves ER Stress. EMBO J. 27, 1049–1059. doi:10.1038/emboj.2008.42
Lopez-Crisosto, C., Díaz-Vegas, A., Castro, P. F., Rothermel, B. A., Bravo-Sagua, R., and Lavandero, S. (2021). Endoplasmic Reticulum−mitochondria Coupling Increases during Doxycycline-Induced Mitochondrial Stress in HeLa Cells. Cell Death Dis 12, 1–12. doi:10.1038/s41419-021-03945-9
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M., and Kroemer, G. (2013). The Hallmarks of Aging. Cell 153, 1194–1217. doi:10.1016/j.cell.2013.05.039
Ma, Y., and Hendershot, L. M. (2003). Delineation of a Negative Feedback Regulatory Loop that Controls Protein Translation during Endoplasmic Reticulum Stress. J. Biol. Chem. 278, 34864–34873. doi:10.1074/jbc.m301107200
Manor, U., Bartholomew, S., Golani, G., Christenson, E., Kozlov, M., Higgs, H., et al. (2015). A Mitochondria-Anchored Isoform of the Actin-Nucleating Spire Protein Regulates Mitochondrial Division. eLife 4, e08828. doi:10.7554/eLife.08828
Martinus, R. D., Garth, G. P., Webster, T. L., Cartwright, P., Naylor, D. J., Høj, P. B., et al. (1996). Selective Induction of Mitochondrial Chaperones in Response to Loss of the Mitochondrial Genome. Eur. J. Biochem. 240, 98–103. doi:10.1111/j.1432-1033.1996.0098h.x
McColl, G., Rogers, A. N., Alavez, S., Hubbard, A. E., Melov, S., Link, C. D., et al. (2010). Insulin-like Signaling Determines Survival during Stress via Posttranscriptional Mechanisms in C. elegans. Cell Metab. 12, 260–272. doi:10.1016/j.cmet.2010.08.004
McElwee, J., Bubb, K., and Thomas, J. H. (2003). Transcriptional Outputs of the Caenorhabditis elegans Forkhead Protein DAF-16. Aging cell 2, 111–121. doi:10.1046/j.1474-9728.2003.00043.x
Mendenhall, A. R., Tedesco, P. M., Taylor, L. D., Lowe, A., Cypser, J. R., and Johnson, T. E. (2012). Expression of a Single-Copy hsp-16.2 Reporter Predicts Life Span. Journals Gerontol. Ser. A: Biol. Sci. Med. Sci. 67, 726–733. doi:10.1093/gerona/glr225
Merkwirth, C., Jovaisaite, V., Durieux, J., Matilainen, O., Jordan, S. D., Quiros, P. M., et al. (2016). Two Conserved Histone Demethylases Regulate Mitochondrial Stress-Induced Longevity. Cell 165, 1209–1223. doi:10.1016/j.cell.2016.04.012
Misrani, A., Tabassum, S., and Yang, L. (2021). Mitochondrial Dysfunction and Oxidative Stress in Alzheimer's Disease. Front. Aging Neurosci. 13, 18. doi:10.3389/fnagi.2021.617588
Moehle, E. A., Higuchi-Sanabria, R., Tsui, C. K., Homentcovschi, S., Tharp, K. M., Zhang, H., et al. (2021). Cross-species Screening Platforms Identify EPS-8 as a Critical Link for Mitochondrial Stress and Actin Stabilization. Sci. Adv. 7, eabj6818. doi:10.1126/sciadv.abj6818
Moehle, E. A., Shen, K., and Dillin, A. (2019). Mitochondrial Proteostasis in the Context of Cellular and Organismal Health and Aging. J. Biol. Chem. 294, 5396–5407. doi:10.1074/jbc.tm117.000893
Molenaars, M., Janssens, G. E., Williams, E. G., Jongejan, A., Lan, J., Rabot, S., et al. (2020). A Conserved Mito-Cytosolic Translational Balance Links Two Longevity Pathways. Cell Metab. 31, 549e7–563. doi:10.1016/j.cmet.2020.01.011
Morley, J. F., Brignull, H. R., Weyers, J. J., and Morimoto, R. I. (2002). The Threshold for Polyglutamine-Expansion Protein Aggregation and Cellular Toxicity Is Dynamic and Influenced by Aging in Caenorhabditis elegans. Proc. Natl. Acad. Sci. 99, 10417–10422. doi:10.1073/pnas.152161099
Morley, J. F., and Morimoto, R. I. (2004). Regulation of Longevity inCaenorhabditis Elegansby Heat Shock Factor and Molecular Chaperones. MBoC 15, 657–664. doi:10.1091/mbc.e03-07-0532
Munkácsy, E., and Rea, S. L. (2014). The Paradox of Mitochondrial Dysfunction and Extended Longevity. Exp. Gerontol. 56, 221–233. doi:10.1016/j.exger.2014.03.016
Murphy, C. T., McCarroll, S. A., Bargmann, C. I., Fraser, A., Kamath, R. S., Ahringer, J., et al. (2003). Genes that Act Downstream of DAF-16 to Influence the Lifespan of Caenorhabditis elegans. Nature 424, 277–283. doi:10.1038/nature01789
Naidoo, N., Ferber, M., Master, M., Zhu, Y., and Pack, A. I. (2008). Aging Impairs the Unfolded Protein Response to Sleep Deprivation and Leads to Proapoptotic Signaling. J. Neurosci. 28, 6539–6548. doi:10.1523/jneurosci.5685-07.2008
Nakai, A., Kawazoe, Y., Tanabe, M., Nagata, K., and Morimoto, R. I. (1995). The DNA-Binding Properties of Two Heat Shock Factors, HSF1 and HSF3, Are Induced in the Avian Erythroblast Cell Line HD6. Mol. Cell Biol 15, 5268–5278. doi:10.1128/mcb.15.10.5268
Nakai, A., Tanabe, M., Kawazoe, Y., Inazawa, J., Morimoto, R. I., and Nagata, K. (1997). HSF4, a New Member of the Human Heat Shock Factor Family Which Lacks Properties of a Transcriptional Activator. Mol. Cell Biol 17, 469–481. doi:10.1128/mcb.17.1.469
Nargund, A. M., Fiorese, C. J., Pellegrino, M. W., Deng, P., and Haynes, C. M. (2015). Mitochondrial and Nuclear Accumulation of the Transcription Factor ATFS-1 Promotes OXPHOS Recovery during the UPRmt. Mol. Cell 58, 123–133. doi:10.1016/j.molcel.2015.02.008
Nargund, A. M., Pellegrino, M. W., Fiorese, C. J., Baker, B. M., and Haynes, C. M. (2012). Mitochondrial Import Efficiency of ATFS-1 Regulates Mitochondrial UPR Activation. Science 337, 587–590. doi:10.1126/science.1223560
Neef, D. W., Jaeger, A. M., Gomez-Pastor, R., Willmund, F., Frydman, J., and Thiele, D. J. (2014). A Direct Regulatory Interaction between Chaperonin TRiC and Stress-Responsive Transcription Factor HSF1. Cell Rep. 9, 955–966. doi:10.1016/j.celrep.2014.09.056
Neudegger, T., Verghese, J., Hayer-Hartl, M., Hartl, F. U., and Bracher, A. (2016). Structure of Human Heat-Shock Transcription Factor 1 in Complex with DNA. Nat. Struct. Mol. Biol. 23, 140–146. doi:10.1038/nsmb.3149
Nishitoh, H., Matsuzawa, A., Tobiume, K., Saegusa, K., Takeda, K., Inoue, K., et al. (2002). ASK1 Is Essential for Endoplasmic Reticulum Stress-Induced Neuronal Cell Death Triggered by Expanded Polyglutamine Repeats. Genes Dev. 16, 1345–1355. doi:10.1101/gad.992302
Nolden, M., Ehses, S., Koppen, M., Bernacchia, A., Rugarli, E. I., and Langer, T. (2005). The M-AAA Protease Defective in Hereditary Spastic Paraplegia Controls Ribosome Assembly in Mitochondria. Cell 123, 277–289. doi:10.1016/j.cell.2005.08.003
Nuss, J. E., Choksi, K. B., DeFord, J. H., and Papaconstantinou, J. (2008). Decreased Enzyme Activities of Chaperones PDI and BiP in Aged Mouse Livers. Biochem. Biophysical Res. Commun. 365, 355–361. doi:10.1016/j.bbrc.2007.10.194
Ooi, F. K., and Prahlad, V. (2017). Olfactory Experience Primes the Heat Shock Transcription Factor HSF-1 to Enhance the Expression of Molecular Chaperones in C. elegans. Sci. Signal. 10, eaan4893. doi:10.1126/scisignal.aan4893
Owusu-Ansah, E., Song, W., and Perrimon, N. (2013). Muscle Mitohormesis Promotes Longevity via Systemic Repression of Insulin Signaling. Cell 155, 699–712. doi:10.1016/j.cell.2013.09.021
Özbey, N. P., Imanikia, S., Krueger, C., Hardege, I., Morud, J., Sheng, M., et al. (2020). Tyramine Acts Downstream of Neuronal XBP-1s to Coordinate Inter-tissue UPRER Activation and Behavior in C. elegans. Dev. Cell 55, 754–770. e6. doi:10.1016/j.devcel.2020.10.024
Ozcan, L., and Tabas, I. (2012). Role of Endoplasmic Reticulum Stress in Metabolic Disease and Other Disorders. Annu. Rev. Med. 63, 317–328. doi:10.1146/annurev-med-043010-144749
Pellegrino, M. W., Nargund, A. M., Kirienko, N. V., Gillis, R., Fiorese, C. J., and Haynes, C. M. (2014). Mitochondrial UPR-Regulated Innate Immunity Provides Resistance to Pathogen Infection. Nature 516, 414–417. doi:10.1038/nature13818
Peteranderl, R., Rabenstein, M., Shin, Y.-K., Liu, C. W., Wemmer, D. E., King, D. S., et al. (1999). Biochemical and Biophysical Characterization of the Trimerization Domain from the Heat Shock Transcription Factor. Biochemistry 38, 3559–3569. doi:10.1021/bi981774j
Pol, A., Gross, S. P., and Parton, R. G. (2014). Biogenesis of the Multifunctional Lipid Droplet: Lipids, Proteins, and Sites. J. Cell Biol 204, 635–646. doi:10.1083/jcb.201311051
Prahlad, V., Cornelius, T., and Morimoto, R. I. (2008). Regulation of the Cellular Heat Shock Response in Caenorhabditis elegans by Thermosensory Neurons. Science 320, 811–814. doi:10.1126/science.1156093
Prahlad, V., Cornelius, T., and Morimoto, R. I. (2008). Regulation of the Cellular Heat Shock Response in Caenorhabditis elegans by Thermosensory Neurons. Science 320, 811–814. doi:10.1126/science.1156093
Prischi, F., Nowak, P. R., Carrara, M., and Ali, M. M. U. (2014). Phosphoregulation of Ire1 RNase Splicing Activity. Nat. Commun. 5, 3554. doi:10.1038/ncomms4554
Prithika, U., Deepa, V., and Balamurugan, K. (2016). External Induction of Heat Shock Stimulates the Immune Response and Longevity of Caenorhabditis elegans towards Pathogen Exposure. Innate Immun. 22, 466–478. doi:10.1177/1753425916654557
Quirós, P. M., Prado, M. A., Zamboni, N., D’Amico, D., Williams, R. W., Finley, D., et al. (2017). Multi-omics Analysis Identifies ATF4 as a Key Regulator of the Mitochondrial Stress Response in Mammals. J. Cell Biol 216, 2027–2045. doi:10.1083/jcb.201702058
Rabindran, S. K., Haroun, R. I., Clos, J., Wisniewski, J., and Wu, C. (1993). Regulation of Heat Shock Factor Trimer Formation: Role of a Conserved Leucine Zipper. Science 259, 230–234. doi:10.1126/science.8421783
Radons, J. (2016). The Human HSP70 Family of Chaperones: where Do We Stand? Cell Stress and Chaperones 21, 379–404. doi:10.1007/s12192-016-0676-6
Raffaello, A., Mammucari, C., Gherardi, G., and Rizzuto, R. (2016). Calcium at the Center of Cell Signaling: Interplay between Endoplasmic Reticulum, Mitochondria, and Lysosomes. Trends Biochem. Sci. 41, 1035–1049. doi:10.1016/j.tibs.2016.09.001
Rattan, S. I. (2004). Aging Intervention, Prevention, and Therapy through Hormesis. J. Gerontol. A. Biol. Sci. Med. Sci. 59, 705–709. doi:10.1093/gerona/59.7.b705
Rattan, S. I. S. (2005). Hormetic Modulation of Aging and Longevity by Mild Heat Stress. Dose-Response 3 (4), dose–response. doi:10.2203/dose-response.003.04.008
Rea, S. L., Ventura, N., and Johnson, T. E. (2007). Relationship between Mitochondrial Electron Transport Chain Dysfunction, Development, and Life Extension in Caenorhabditis elegans. Plos Biol. 5, e259. doi:10.1371/journal.pbio.0050259
Reimold, A. M., Iwakoshi, N. N., Manis, J., Vallabhajosyula, P., Szomolanyi-Tsuda, E., Gravallese, E. M., et al. (2001). Plasma Cell Differentiation Requires the Transcription Factor XBP-1. Nature 412, 300–307. doi:10.1038/35085509
Ristow, M., and Zarse, K. (2010). How Increased Oxidative Stress Promotes Longevity and Metabolic Health: The Concept of Mitochondrial Hormesis (Mitohormesis). Exp. Gerontol. 45, 410–418. doi:10.1016/j.exger.2010.03.014
Ritossa, F. (1962). A New Puffing Pattern Induced by Temperature Shock and DNP in drosophila. Experientia 18, 571–573. doi:10.1007/bf02172188
Ritossa, F. (1996). Discovery of the Heat Shock Response. Cell Stress Chaper 1, 97–98. doi:10.1379/1466-1268(1996)001<0097:dothsr>2.3.co;2
Rolland, S. G., Schneid, S., Schwarz, M., Rackles, E., Fischer, C., Haeussler, S., et al. (2019). Compromised Mitochondrial Protein Import Acts as a Signal for UPRmt. Cell Rep. 28, 1659e5–1669. doi:10.1016/j.celrep.2019.07.049
Roux, A. E., Langhans, K., Huynh, W., and Kenyon, C. (2016). Reversible Age-Related Phenotypes Induced during Larval Quiescence in C. elegans. Cell Metab. 23, 1113–1126. doi:10.1016/j.cmet.2016.05.024
Schopf, F. H., Biebl, M. M., and Buchner, J. (2017). The HSP90 Chaperone Machinery. Nat. Rev. Mol. Cell Biol 18, 345–360. doi:10.1038/nrm.2017.20
Schröder, M., and Kaufman, R. J. (2005). The Mammalian Unfolded Protein Response. Annu. Rev. Biochem. 74, 739–789. doi:10.1146/annurev.biochem.73.011303.074134
Schuetz, T. J., Gallo, G. J., Sheldon, L., Tempst, P., and Kingston, R. E. (1991). Isolation of a cDNA for HSF2: Evidence for Two Heat Shock Factor Genes in Humans. Proc. Natl. Acad. Sci. 88, 6911–6915. doi:10.1073/pnas.88.16.6911
Shalem, O., Sanjana, N. E., Hartenian, E., Shi, X., Scott, D. A., Mikkelsen, T. S., et al. (2014). Genome-scale CRISPR-Cas9 Knockout Screening in Human Cells. Science 343, 84–87. doi:10.1126/science.1247005
Shao, L.-W., Niu, R., and Liu, Y. (2016). Neuropeptide Signals Cell Non-autonomous Mitochondrial Unfolded Protein Response. Cell Res 26, 1182–1196. doi:10.1038/cr.2016.118
Shemesh, N., Shai, N., and Ben-Zvi, A. (2013). Germline Stem Cell Arrest Inhibits the Collapse of Somatic Proteostasis Early inCaenorhabditis Elegansadulthood. Aging cell 12, 814–822. doi:10.1111/acel.12110
Shen, X., Ellis, R. E., Lee, K., Liu, C.-Y., Yang, K., Solomon, A., et al. (2001). Complementary Signaling Pathways Regulate the Unfolded Protein Response and Are Required for C. elegans Development. Cell 107, 893–903. doi:10.1016/s0092-8674(01)00612-2
Shen, X., Ellis, R. E., Sakaki, K., and Kaufman, R. J. (2005). Genetic Interactions Due to Constitutive and Inducible Gene Regulation Mediated by the Unfolded Protein Response in C. elegans. Plos Genet. 1, e37. doi:10.1371/journal.pgen.0010037
Shi, Y., Mosser, D. D., and Morimoto, R. I. (1998). Molecularchaperones as HSF1-specific Transcriptional Repressors. Genes Dev. 12, 654–666. doi:10.1101/gad.12.5.654
Shoulders, M. D., Ryno, L. M., Genereux, J. C., Moresco, J. J., Tu, P. G., Wu, C., et al. (2013). Stress-Independent Activation of XBP1s And/or ATF6 Reveals Three Functionally Diverse ER Proteostasis Environments. Cell Rep. 3, 1279–1292. doi:10.1016/j.celrep.2013.03.024
Shpilka, T., Du, Y., Yang, Q., Melber, A., Uma Naresh, N., Lavelle, J., et al. (2021). UPRmt Scales Mitochondrial Network Expansion with Protein Synthesis via Mitochondrial Import in Caenorhabditis elegans. Nat. Commun. 12, 479. doi:10.1038/s41467-020-20784-y
Singh, V., and Aballay, A. (2006). Heat-shock Transcription Factor (HSF)-1 Pathway Required for Caenorhabditis elegans Immunity. Proc. Natl. Acad. Sci. 103, 13092–13097. doi:10.1073/pnas.0604050103
Sletten, A. C., Peterson, L. R., and Schaffer, J. E. (2018). Manifestations and Mechanisms of Myocardial Lipotoxicity in Obesity. J. Intern. Med. 284, 478–491. doi:10.1111/joim.12728
Son, H. G., Seo, K., Seo, M., Park, S., Ham, S., An, S. W. A., et al. (2018). Prefoldin 6 Mediates Longevity Response from Heat Shock Factor 1 to FOXO in C. elegans. Genes Dev. 32, 1562–1575. doi:10.1101/gad.317362.118
Song, W., Owusu-Ansah, E., Hu, Y., Cheng, D., Ni, X., Zirin, J., et al. (2017). Activin Signaling Mediates Muscle-To-Adipose Communication in a Mitochondria Dysfunction-Associated Obesity Model. Proc. Natl. Acad. Sci. USA 114, 8596–8601. doi:10.1073/pnas.1708037114
Sopko, R., Huang, D., Preston, N., Chua, G., Papp, B., Kafadar, K., et al. (2006). Mapping Pathways and Phenotypes by Systematic Gene Overexpression. Mol. Cell 21, 319–330. doi:10.1016/j.molcel.2005.12.011
Stauffer, W. T., Arrieta, A., Blackwood, E. A., and Glembotski, C. C. (2020). Sledgehammer to Scalpel: Broad Challenges to the Heart and Other Tissues Yield Specific Cellular Responses via Transcriptional Regulation of the ER-Stress Master Regulator ATF6α. Ijms 21, 1134. doi:10.3390/ijms21031134
Sural, S., Liang, C. Y., Wang, F. Y., Ching, T. T., and Hsu, A. L. (2020). HSB-1/HSF-1 Pathway Modulates Histone H4 in Mitochondria to Control mtDNA Transcription and Longevity. Sci. Adv. 6, eaaz4452. doi:10.1126/sciadv.aaz4452
Tam, A. B., Koong, A. C., and Niwa, M. (2014). Ire1 Has Distinct Catalytic Mechanisms for XBP1/HAC1 Splicing and RIDD. Cell Rep. 9, 850–858. doi:10.1016/j.celrep.2014.09.016
Tatum, M. C., Ooi, F. K., Chikka, M. R., Chauve, L., Martinez-Velazquez, L. A., Steinbusch, H. W. M., et al. (2015). Neuronal Serotonin Release Triggers the Heat Shock Response in C. elegans in the Absence of Temperature Increase. Curr. Biol. 25, 163–174. doi:10.1016/j.cub.2014.11.040
Taylor, R. C., and Dillin, A. (2013). XBP-1 Is a Cell-Nonautonomous Regulator of Stress Resistance and Longevity. Cell 153, 1435–1447. doi:10.1016/j.cell.2013.05.042
Tharp, K. M., Higuchi-Sanabria, R., Timblin, G. A., Ford, B., Garzon-Coral, C., Schneider, C., et al. (2021). Adhesion-mediated Mechanosignaling Forces Mitohormesis. Cell Metab 33, 1322–1341. doi:10.1016/j.cmet.2021.04.017
Tian, Y., Garcia, G., Bian, Q., Steffen, K. K., Joe, L., Wolff, S., et al. (2016). Mitochondrial Stress Induces Chromatin Reorganization to Promote Longevity and UPR Mt. Cell 165, 1197–1208. doi:10.1016/j.cell.2016.04.011
Tissiéres, A., Mitchell, H. K., and Tracy, U. M. (1974). Protein Synthesis in Salivary Glands of Drosophila melanogaster: Relation to Chromosome Puffs. J. Mol. Biol. 84, 389–398. doi:10.1016/0022-2836(74)90447-1
Trivedi, R., and Jurivich, D. A. (2020). A Molecular Perspective on Age-dependent Changes to the Heat Shock axis. Exp. Gerontol. 137, 110969. doi:10.1016/j.exger.2020.110969
Tsang, W. Y., and Lemire, B. D. (2002). Mitochondrial Genome Content Is Regulated during Nematode Development. Biochem. Biophysical Res. Commun. 291, 8–16. doi:10.1006/bbrc.2002.6394
Tsuru, A., Fujimoto, N., Takahashi, S., Saito, M., Nakamura, D., Iwano, M., et al. (2013). Negative Feedback by IRE1 Optimizes Mucin Production in Goblet Cells. Proc. Natl. Acad. Sci. 110, 2864–2869. doi:10.1073/pnas.1212484110
Urano, F., Wang, X., Bertolotti, A., Zhang, Y., Chung, P., Harding, H. P., et al. (2000). Coupling of Stress in the ER to Activation of JNK Protein Kinases by Transmembrane Protein Kinase IRE1. Science 287, 664–666. doi:10.1126/science.287.5453.664
van Eden, W., van der Zee, R., and Prakken, B. (2005). Heat-shock Proteins Induce T-Cell Regulation of Chronic Inflammation. Nat. Rev. Immunol. 5, 318–330. doi:10.1038/nri1593
Ventura, N., Rea, S. L., Schiavi, A., Torgovnick, A., Testi, R., and Johnson, T. E. (2009). p53/CEP-1 Increases or Decreases Lifespan, Depending on Level of Mitochondrial Bioenergetic Stress. Aging Cell 8, 380–393. doi:10.1111/j.1474-9726.2009.00482.x
Verma, G., and Datta, M. (2010). IL-1β Induces ER Stress in a JNK Dependent Manner that Determines Cell Death in Human Pancreatic Epithelial MIA PaCa-2 Cells. Apoptosis 15, 864–876. doi:10.1007/s10495-010-0498-4
Vevea, J. D., Garcia, E. J., Chan, R. B., Zhou, B., Schultz, M., Di Paolo, G., et al. (2015). Role for Lipid Droplet Biogenesis and Microlipophagy in Adaptation to Lipid Imbalance in Yeast. Dev. Cell 35, 584–599. doi:10.1016/j.devcel.2015.11.010
Volovik, Y., Maman, M., Dubnikov, T., Bejerano-Sagie, M., Joyce, D., Kapernick, E. A., et al. (2012). Temporal Requirements of Heat Shock Factor-1 for Longevity Assurance. Aging Cell 11, 491–499. doi:10.1111/j.1474-9726.2012.00811.x
Walter, L., Baruah, A., Chang, H.-W., Pace, H. M., and Lee, S. S. (2011). The Homeobox Protein CEH-23 Mediates Prolonged Longevity in Response to Impaired Mitochondrial Electron Transport Chain in C. elegans. Plos Biol. 9, e1001084. doi:10.1371/journal.pbio.1001084
Wang, N., Gottesman, S., Willingham, M. C., Gottesman, M. M., and Maurizi, M. R. (1993). A Human Mitochondrial ATP-dependent Protease that Is Highly Homologous to Bacterial Lon Protease. Pnas 90, 11247–11251. doi:10.1073/pnas.90.23.11247
Watanabe, Y., Tsujimura, A., Taguchi, K., and Tanaka, M. (2017). HSF1 Stress Response Pathway Regulates Autophagy Receptor SQSTM1/p62-Associated Proteostasis. Autophagy 13, 133–148. doi:10.1080/15548627.2016.1248018
Williams, K. W., Liu, T., Kong, X., Fukuda, M., Deng, Y., Berglund, E. D., et al. (2014). Xbp1s in Pomc Neurons Connects ER Stress with Energy Balance and Glucose Homeostasis. Cell Metab. 20, 471–482. doi:10.1016/j.cmet.2014.06.002
Wu, Z., Senchuk, M. M., Dues, D. J., Johnson, B. K., Cooper, J. F., Lew, L., et al. (2018). Mitochondrial Unfolded Protein Response Transcription Factor ATFS-1 Promotes Longevity in a Long-Lived Mitochondrial Mutant through Activation of Stress Response Pathways. BMC Biol. 16, 147. doi:10.1186/s12915-018-0615-3
Xin, N., Durieux, J., Yang, C., Wolff, S., Kim, H.-E., and Dillin, A. (2020). The UPRmt Preserves Mitochondrial Import to Extend Lifespan. New York, NY: Cold Spring Harbor Laboratory. doi:10.1101/2020.07.01.182980
Xu, L., Wang, X., and Tong, C. (2020). Endoplasmic Reticulum-Mitochondria Contact Sites and Neurodegeneration. Front. Cell Dev. Biol. 8, 428. doi:10.3389/fcell.2020.00428
Xu, Z., Chikka, M. R., Xia, H., and Ready, D. F. (2016). Ire1 Supports normal ER Differentiation in Developing Drosophila Photoreceptors. J. Cell Sci 129, 921–929. doi:10.1242/jcs.180406
Ye, J., Rawson, R. B., Komuro, R., Chen, X., Davé, U. P., Prywes, R., et al. (2000). ER Stress Induces Cleavage of Membrane-Bound ATF6 by the Same Proteases that Process SREBPs. Mol. Cell 6, 1355–1364. doi:10.1016/s1097-2765(00)00133-7
Yee, C., Yang, W., and Hekimi, S. (2014). The Intrinsic Apoptosis Pathway Mediates the Pro-longevity Response to Mitochondrial ROS in C. elegans. Cell 157, 897–909. doi:10.1016/j.cell.2014.02.055
Zara, V., Conte, L., and Trumpower, B. L. (2007). Identification and Characterization of Cytochrome Bc1 Subcomplexes in Mitochondria from Yeast with Single and Double Deletions of Genes Encoding Cytochrome Bc1 Subunits. FEBS J. 274, 4526–4539. doi:10.1111/j.1742-4658.2007.05982.x
Zhang, C., Syed, T. W., Liu, R., and Yu, J. (2017). Role of Endoplasmic Reticulum Stress, Autophagy, and Inflammation in Cardiovascular Disease. Front. Cardiovasc. Med. 4, 1. doi:10.3389/fcvm.2017.00029
Zhang, H., Zhang, L., Yu, F., Liu, Y., Liang, Q., Deng, G., et al. (2012). HSF1 Is a Transcriptional Activator of IL-10 Gene Expression in RAW264.7 Macrophages. Inflammation 35, 1558–1566. doi:10.1007/s10753-012-9471-4
Zhang, Q., Wang, Z., Zhang, W., Wen, Q., Li, X., Zhou, J., et al. (2021). The Memory of Neuronal Mitochondrial Stress Is Inherited Transgenerationally via Elevated Mitochondrial DNA Levels. Nat. Cell Biol 23, 870–880. doi:10.1038/s41556-021-00724-8
Zhang, Q., Wu, X., Chen, P., Liu, L., Xin, N., Tian, Y., et al. (2018). The Mitochondrial Unfolded Protein Response Is Mediated Cell-Non-Autonomously by Retromer-dependent Wnt Signaling. Cell 174, 870e17–883. doi:10.1016/j.cell.2018.06.029
Zhang, X., and Zhang, K. (2012). Endoplasmic Reticulum Stress-Associated Lipid Droplet Formation and Type II Diabetes. Biochem. Res. Int. 2012, 1–5. doi:10.1155/2012/247275
Zhao, Q., Wang, J., Levichkin, I. V., Stasinopoulos, S., Ryan, M. T., and Hoogenraad, N. J. (2002). A Mitochondrial Specific Stress Response in Mammalian Cells. EMBO J. 21, 4411–4419. doi:10.1093/emboj/cdf445
Zhao, Y., Gilliat, A. F., Ziehm, M., Turmaine, M., Wang, H., Ezcurra, M., et al. (2017). Two Forms of Death in Ageing Caenorhabditis elegans. Nat. Commun. 8, 15458. doi:10.1038/ncomms15458
Zhou, J., Liu, C. Y., Back, S. H., Clark, R. L., Peisach, D., Xu, Z., et al. (2006). The crystal Structure of Human IRE1 Luminal Domain Reveals a Conserved Dimerization Interface Required for Activation of the Unfolded Protein Response. Proc. Natl. Acad. Sci. 103, 14343–14348. doi:10.1073/pnas.0606480103
Keywords: mitochondria, endoplasmic reticulum, heat-shock, stress, aging, C. elegans
Citation: Dutta N, Garcia G and Higuchi-Sanabria R (2022) Hijacking Cellular Stress Responses to Promote Lifespan. Front. Aging 3:860404. doi: 10.3389/fragi.2022.860404
Received: 22 January 2022; Accepted: 23 February 2022;
Published: 24 March 2022.
Edited by:
Cindy Voisine, Northeastern Illinois University, United StatesReviewed by:
Patricija Van Oosten-Hawle, University of Leeds, United KingdomCopyright © 2022 Dutta, Garcia and Higuchi-Sanabria. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ryo Higuchi-Sanabria, cnlvLnNhbmFicmlhQHVzYy5lZHU=
†These authors have contributed equally to the work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.