 Cong He
Cong He Baojiang Chen
Baojiang Chen Hecai Yang
Hecai Yang Xiaoqing Zhou3*
Xiaoqing Zhou3*
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Aging Neurosci., 27 March 2025
Sec. Alzheimer's Disease and Related Dementias
Volume 17 - 2025 | https://doi.org/10.3389/fnagi.2025.1554398
Alzheimer’s disease (AD) is a widespread neurodegenerative disorder and one of the major challenges for public health. Despite extensive research, the role of microglia in AD remains complex and dual. The aim of this review is to summarize the most recent advances in research regarding the dual role of microglia in AD concerning both immunomodulation and pathological progression by considering mechanisms of activation of microglia, effects on Aβ clearance, tau pathology, and impacts due to genetic variations on microglial functions. Among these findings are the dual role of microglia, the status of activation for M1 and M2 phenotypes, and the crucial role that genetic variants like TREM2 have in modulating the response of microglia. This review describes how modulation of the microglial signaling pathway might be exploited therapeutically for AD treatment and underlines the relevance of a personalized medicine approach.
Alzheimer’s disease (AD) is a neurodegenerative disease and the most common one; an estimated 6.9 million Americans aged 65 and older live with AD (GBD 2021 US Burden of Disease Collaborators, 2024; Salazar-Londono et al., 2024). If no medical breakthroughs occur in prevention or treatment, the number of people living with AD could double to 13.8 million by 2060. It is estimated that the overall cost of health care, long-term care, and hospice care for people with dementia aged 65 years and older is $360 billion. Health insurance per capita costs for people aged 65 years and older with AD or other dementias is almost three times higher than for people without the disease, and the Medicaid cost is over 22 times higher (Alzheimer’s Association, 2024; Li W. et al., 2024). The challenge of AD is a major public health issue that must be urgently addressed by the public health community around the world. This has turned AD into a global public health challenge.
In recent years, there has been increasing emphasis on the crucial role of microglia in the central nervous system (CNS) during AD pathogenesis with the development of neuroimmunology. As the major immune cells in the CNS, microglia are responsible for maintaining neural environment homeostasis, scavenging pathological proteins, and regulating neuronal function (Is et al., 2024; Jacquet et al., 2024). Recent studies have shown that microglia exert a double role in AD-on one hand, they exert a protective function through immunomodulation and Aβ plaque scavenging; on the other hand, over-activated microglia may trigger chronic inflammatory responses that may further aggravate neuronal damage and pathological development (Onuska, 2020; Long et al., 2022). The mechanism of this dual role has not been fully clarified but is closely related to the regulated expression of multiple AD risk genes carried by microglia, such as TREM2, CD33, etc. (Malik et al., 2015; Liu et al., 2020). Recent advances in genomics and proteomics have further revealed the complex signaling changes of microglia in the development of AD pathology. Large-scale proteomic analyses unraveled early energy metabolic changes associated with the activation of microglia in the brain tissue and cerebrospinal fluid of AD patients and summarized the findings as glial biological patterns underlining the centrality of microglia in AD pathology (Hemonnot et al., 2019; Leng and Edison, 2021; Chen et al., 2023; Zhou X. et al., 2024).
The present review aims to summarize the recent research progress regarding the dual roles of microglia in AD focusing on the correlation between immune modulation and pathological progression. From such a focus, integration of the involved mechanisms into the involved signaling pathways may be able to elucidate the pleiotropic roles microglia may play in AD pathogenesis and therefore provide a new theoretical principle or even potentially research toward a final therapeutic strategy.
As the main immune sensors of the CNS, microglia have highly dynamic morphological and functional properties. In the resting state, microglia assume an astrocytic morphology with abundant dendrites, whose dynamically extending and contracting dendrites are believed to be constantly monitoring the surrounding environment (Pallares-Moratalla and Bergers, 2024). Microglia express a variety of receptors on their surfaces, capable of sensing signaling molecules generated by neuronal activity. Abnormal signals or pathological stimuli rapidly transform microglia from the resting to the activated state. Morphology changes as they migrate to the site of injury and release a variety of cytokines and chemokines (Woodburn et al., 2021). Microglia control neuronal functions through the regulation of cerebral vascular tone, hence local cerebral blood flow and neurovascular coupling, which ensures neurons receive adequate oxygen and nutrition. Besides, microglia cooperate with astrocytes to modulate the neurotransmitter levels so as to prevent excitotoxicity by preventing neurotransmitter overloading (Cai et al., 2024). The expression of different types of cytokines and chemokines produced by microglia maintains the immune responses in the CNS, which guarantees immune homeostasis (Sadeghdoust et al., 2024). The permeability of the blood–brain barrier controlled by microglia was interactively realized by its interaction with endothelial cells within the blood–brain barrier. 20 Similarly, the blood–brain barrier’s permeability controlled by microglia interactively influences maintaining the internal environmental stability of the brain (Weng et al., 2024).
Synaptic pruning is a process necessary in neural development in which the activity of neural networks is sustained by eliminating aberrant or redundant synaptic connections (Hao et al., 2024). Complement proteins C1q and C3, which are surface expressed on microglia, can tag synapses for elimination via phagocytosis by microglia. Microglia also mold the synapse itself through partial phagocytosis or synaptic stripping (Cornell et al., 2022). It is one of the main mechanisms by which neural networks stabilize and become plastic. Microglia also secrete a multitude of neurotrophic factors involved in neuronal survival and regeneration (Gaire, 2022). Microglia have two phenotypes depending on the nature of inflammation: M1 (proinflammatory phenotype) and M2 (anti-inflammatory phenotype), which protect neurons from damage by secreting different cytokines that modulate the local inflammatory environment (Du et al., 2017). Microglia, upon activation or ischemia and subsequent brain injury, migrate to the injured site within a short time by phagocytosing dead cells and debris. Microglia are rapidly activated in response to brain injury or ischemia and migrate into the injured area to restore tissue and regenerate by removing dead cells and cellular debris. Microglia play a role in the regulation of energy metabolism by influencing glycolysis and oxidative phosphorylation that support normal neuronal function (Zhang G. et al., 2024).
Biomarkers, imaging markers, and genetic markers are of crucial importance for the early diagnosis of AD. In biomarkers, low cerebrospinal fluid Aβ42, elevated total tau protein, and phosphorylated tau protein are typical markers for AD, and the latter two alterations occur in the prodromal stage of the disease and may be correlated with the severity of the disease (Andersson et al., 2025). Blood concentrations of neurofilament light chain are extremely elevated in early AD and correlate strongly with the severity of the disease and are also an early marker of diagnosis (Andersson et al., 2020). Exosomal miRNAs also exhibit an altered level of expression, the most notable of which is miR-15. Exosomal miRNAs are also abnormally expressed at early AD, and overexpression of miR-155 increases neuroinflammation and miR-124 inhibits inflammation and neuronal damage (Li Y. B. et al., 2024). Of the imaging markers, brain Aβ plaque and tau pathology positron emission tomography scanning and PET scanning using radiotracers reveal specific Aβ plaque binding and detect Aβ deposits at an early stage. Magnetic resonance imaging, however, reflects structural brain change, which is highly associated with cognitive decline (Mantzavinos and Alexiou, 2017). APOE ε4 allele is one of the most powerful gene risk factors of AD among the genetic markers, and allele carriers are greatly prone to early diagnosis (Di Battista et al., 2016). Gene mutations in TREM2 have been implicated in AD risk, and its absence in microglial cells disables Aβ clearance and neurologic inflammatory response (Heneka, 2023). CD33 gene polymorphism rs3865444 is associated with decreased AD risk and can be utilized in early diagnosis in the regulation of microglial Aβ clearance (Walker et al., 2015). Detection and verification of such markers are an excellent tool for early diagnosis of AD and enable early treatment and intervention of the disease (Li Y. B. et al., 2024).
Aβ has been reported to bind the TLR receptor on microglial surfaces and convert microglia to the M1-type phenotype (Thakur et al., 2023). These cytokines not only support neuroinflammation but also induce neuronal apoptosis. Aberrant activation of microglia also results in disruption of the blood–brain barrier, promoting increased peripheral immune cell infiltration (Cai et al., 2022; Versele et al., 2022; Sarb et al., 2024; van den Brink et al., 2024). Activation of microglia has been shown to trigger signaling cascades such as NF-κB and JNK via TNF-α specific binding to the receptors TNF-R1 and TNF-R2 (Wang D. et al., 2021; Wheeler et al., 2024). These signaling pathways increase the expression of other inflammatory cytokines that participate in pathological mechanisms such as induction of inflammation, apoptosis, and production of Aβ precursor protein and tau protein.
Under physiological conditions, microglia may phagocytose and maintain the homeostasis of Aβ in the brain. Microglial dysfunction contributes to a decreased ability to phagocytose Aβ and results in the accumulation of this protein in amyloid plaques within the AD brain. The released toxic substances by dysfunctional microglia include all kinds of oxygen radicals, nitrogen radicals, and glutamate (Salminen, 2024). It influences the stability and function of neural networks through modulating synaptic functions such as synaptic plasticity and synaptic density, leading to cognitive decline (da Fonseca et al., 2014; Wright et al., 2024). Abnormal activation of microglia promotes the infiltration of T cells, and through secretion of cytokines and direct cell contact driving hyperphosphorylation of tau proteins and the formation of neurofibrillary tangles (Tucsek et al., 2014; Sharallah et al., 2024), this exacerbates neurodegenerative pathology, putting extra emphasis on the multilevel pathological role of microglia in AD.
Peripheral immune cells can cross the central nervous system (CNS) through the blood–brain barrier (BBB) and communicate with microglia to regulate neuroinflammatory and neurodegenerative processes. In AD, Aβ plaque and tau protein tangle accumulation result in disruption of the BBB to make it permeable and provide an easy access to the CNS for peripheral immune cells (Zhang Q. et al., 2024). Microglia and astrocytes are activated simultaneously to secrete chemokines to invite peripheral immune cells into the CNS. Monocytes infiltrate the CNS and become macrophages that can phagocytose Aβ plaques but are hyper-activated to produce pro-inflammatory cytokines (Sun et al., 2024). B cells produce antibodies against Aβ and help clear Aβ plaques through the ADCC pathway. Peripheral immune cells and microglia exhibit competitive and synergistic interactions, for instance, macrophage and microglia can be synergistic while phagocytosing and degrading Aβ, yet the expression of TREM2 in macrophages can repress the action of TREM2 of microglia to affect the efficiency of Aβ clearance (Sharma et al., 2024). Various clinical trials indicated that fluctuations in blood monocyte count and T-cell subset are associated with cognitive impairment of AD patients, the therapeutic mechanism depending on peripheral immune cells, for instance, inhibition of the release of chemokines and modulation of the activity of T-cells, will be an innovative direction to treat AD in the future (Daraban et al., 2024).
The functional state of microglia, the principal immune cells of the CNS, is far more complex than the traditional M1 and M2 phenotype polarization. Current studies have shown that the state of microglia in AD can comprise a wide variety of different phenotypes that play different roles in different stages of the disease and microenvironments (Baligacs et al., 2024). Aside from the classic M1 and M2 phenotypes, studies have characterized conditions such as disease-associated microglia (DAM) and reactive microglia (RAM) that have specific functional and molecular profiles in AD pathology (Lana and Giovannini, 2025).
M1 microglia are activated when stimulated by proinflammatory factors such as interferon-γ and lipopolysaccharide, and release proinflammatory factors such as TNF-α, IL-6, IL-1β and inducible nitric oxide synthase. Excessive secretion of these factors aggravates neuroinflammatory reaction and toxic injury of neurons, and promotes the accumulation of Aβ and the hyperphosphorylation of tau protein (Darwish et al., 2023; Bhardwaj et al., 2024). In contrast, M2 microglia are activated by anti-inflammatory factors such as interleukin −4 (IL-4) and interleukin −13 (IL-13), and secrete anti-inflammatory and neurotrophic factors such as interleukin −10 (IL-10), transforming growth factor β (TGF-β), BDNF and glial-derived neurotrophic factor (GDNF) (Mirarchi et al., 2024; Table 1).
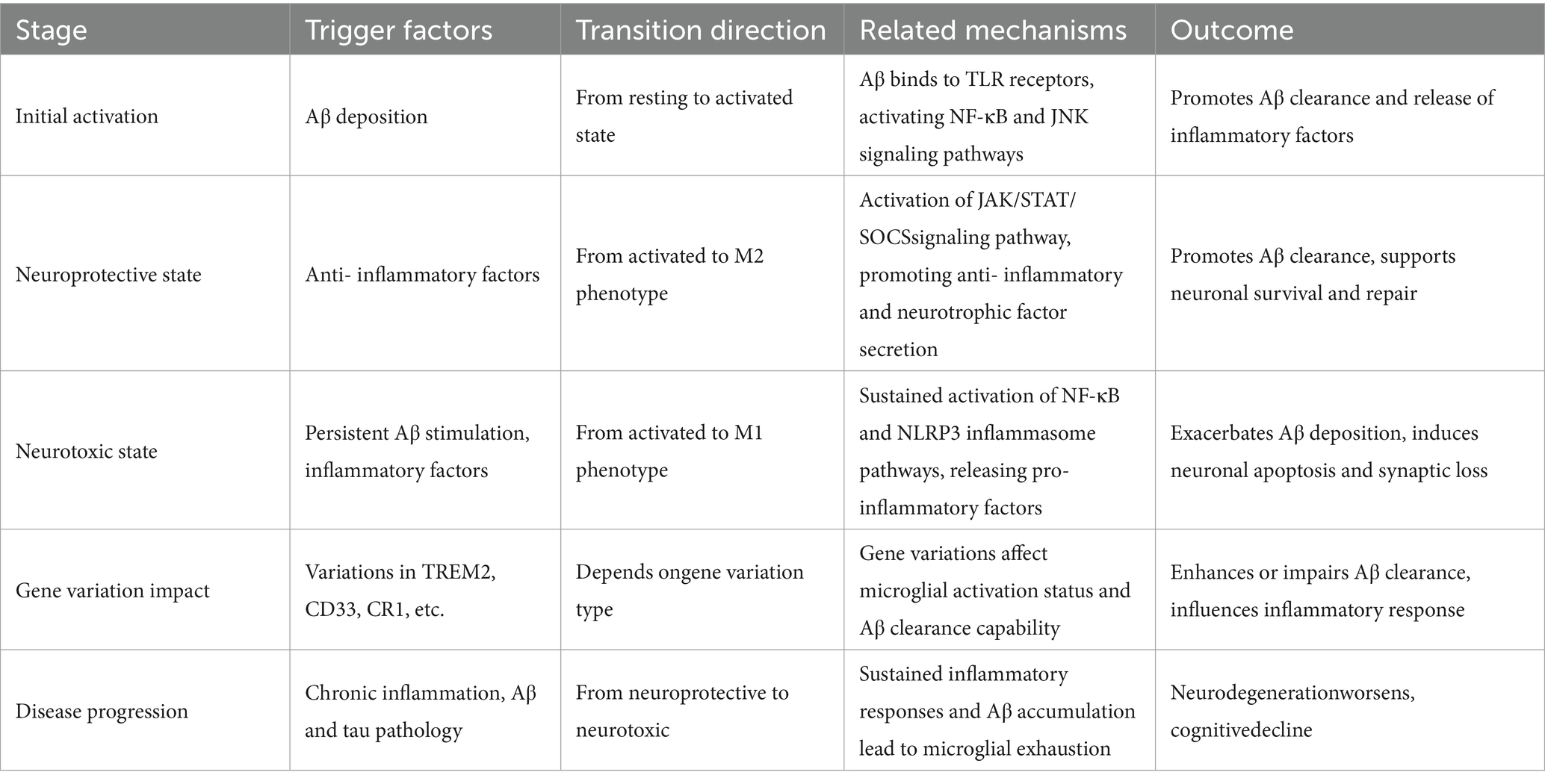
Table 1. The transition mechanisms and related factors of microglia between neuroprotective and neurotoxic states in AD.
Additionally, disease-associated microglia (DAM) have distinct initial AD gene expression patterns and are found surrounding Aβ plaques and clear Aβ as well as modulate tau pathology (Martins-Ferreira et al., 2025). TREM2 variants were significantly associated with AD risk increase and that its physiological function is to allow for DAM formation and thus increased Aβ clearance (Wang H. et al., 2023). Pathology of tau also significantly increases with deficient TREM2 function or microglial deficiency, pointing toward an essential role of DAM in the prevention of tau spreading. The phenotypes of AD in microglia are beyond the simple M1 and M2 phenotypes but more evolved phenotypes such as DAM (Wang et al., 2025). Each state has its own corresponding functions to perform in the different stages of the disease and microenvironment, and in the future, further molecular mechanisms and functional difference among the states would have to be investigated by studies to un-scramble the multi-functional role of microglia in AD (Gao et al., 2023).
Microglial activation is immediately correlated with Aβ plaque development during the early stages of AD. Numerous studies have confirmed that microglia can identify Aβ through multiple pattern recognition receptors (Wu et al., 2024). In addition to mediating the identification of Aβ, these receptors also cause microglia to migrate toward where Aβ deposition occurs and are activated for phagocytosis by triggering downstream signal pathways (Zou et al., 2024). Microglia regulate local inflammation by secreting inflammatory and chemokine factors, which have the potential to affect the clearance and deposition efficacy of Aβ (Streit et al., 2024; Tournier et al., 2024). M2-type-activated microglia execute anti-inflammatory and reparative actions that are beneficial for Aβ clearance. In addition, disease-associated microglia (DAM) accumulate in the vicinity of Aβ plaques and are involved in Aβ clearance modulation and tau pathology. Dysregulated expression of the TREM2 gene, highly associated with increased risk of AD, can drive the formation of DAM to enhance Aβ clearance under physiological conditions. Pathologically stimulated microglia may result in overexuberant inflammatory responses and complement system activation, hence encouraging Aβ deposition and neuronal damage (Madhu et al., 2024; Rodriguez-Vieitez et al., 2024). Investigations in AQP4-deficient mice models have provided evidence that overactivation of microglia is strongly associated with Aβ clearance defect and Aβ deposition. Reducing microglia or inhibiting APOE expression could significantly affect Aβ deposition and clearance (Sethi et al., 2024).
Among the pathologic processes of AD, neuroinflammatory response is widely accepted to be one of the significant mechanisms that are instrumental in disease facilitation. Activated microglia and the cytokines secreted by them have been shown to be one of the major drivers of neuroinflammation in AD, since microglia are the resident immune cells of the CNS (Sian-Hulsmann and Riederer, 2024; Sobue et al., 2024; Yu et al., 2024). The majority of studies indicated that inflammatory cytokine expression, such as IL-1β, TNF-α, and IL-6, was highly upregulated in the brains of AD patients and were secreted predominantly by activated microglia (Shen Y. et al., 2024). In addition, IL-1β, TNF-α, and IL-6 not only increased neuroinflammatory responses but also exhibited direct toxic effects on neurons, inducing neuronal apoptosis and synapse loss (Liu X. H. et al., 2024; Ohm et al., 2024). It was established that Aβ peptide induced higher secretion of cytokines by microglia, supporting the perception that Aβ plays a considerable role in activating microglia (Huang et al., 2024; Kempuraj et al., 2024; Liu D. et al., 2024). This sets a feedback cycle of continuous release of cytokines, establishing a state of chronic neuroinflammation (Botella Lucena and Heneka, 2024; Cafferata et al., 2024; Gasparotto et al., 2024). TLR signaling was established as the regulator of the innate immune response in microglia and astrocytes, further corroborating the key role of cytokines in neuroinflammation (Ana, 2024).
The activation state of microglia is closely related to neurotoxicity in AD. Relevant studies have noted that the activation of microglia is very close to the Aβ deposition, neurofibrillary tangle formation, and neurodegeneration (Yang G. et al., 2023; Yang Y. et al., 2023; Zhou et al., 2023). Different pathological stimuli can influence changes in motility, morphology, phagocytosis, and release of cytokines, chemokine, reactive oxygen species, and prostaglandin metabolites in activated microglia (Maki et al., 2023). These changes allow microglia to turn into a neurotoxic phenotype, which release inflammatory mediators, leading to an enhancement of neuronal damage (Ma et al., 2023). The gene expression profiling has noted characteristic gene expression profiles with microglia that associate with Aβ plaques; moreover, gene expression was reduced with the steady state microglia (Le et al., 2023). This means that the activation of microglia not only correlates with the severity of neurodegeneration but could possibly contribute to disease development due to changes in gene expression (Demuth et al., 2023). More than normal TNF-α and IL-1β release closely relates to neuronal apoptosis and synaptic loss that enhance neurotoxicity (Bivona et al., 2023). In the case of phagocytosis of Aβ, if the microglia get overactivated, it may result in normal neuronal damage.
Microglia, which are central nervous system immune cells, are not only involved in the generation and clearance of Aβ plaques, but also perform the propagation and transmission of tau protein by a myriad of mechanisms (Jiao et al., 2024). Recent research has found that microglia over-secret extracellular vesicles (EVs) containing phosphorylated tau proteins in the vicinity of Aβ plaques, and that EVs transmit tau proteins between neurons (Al-Thani et al., 2024; Fan et al., 2024). This mechanism is responsible for the correlation between Aβ deposition and tau pathology, where the deposition of Aβ plaques activates microglia to secrete more EVs to promote tau protein spread at an accelerated rate (Arber et al., 2024; Fu et al., 2024; Tsering et al., 2025). Studies have shown that when TREM2 function is normal, microglia are able to effectively limit the seeding and spreading of tau protein around Aβ plaques, whereas tau pathology is significantly increased with TREM2 loss of function or microglia depletion (Kloske et al., 2024; Roveta et al., 2024). In the early stages of AD, microglia mainly maintain homeostasis, whereas in the later stages of the disease they are activated and transformed into disease-associated microglia (DAM) (Kanuri and Sirrkay, 2024; Kim et al., 2024).
A different study has shown that TREM2 gene variants strongly associate with AD risk and that TREM2 dysfunction results in microglia’s inability to clear Aβ plaques and tau proteins effectively (Ameli Mojarad and Ameli Mojarad, 2024; Pocock et al., 2024). Aβ plaque promotes phosphorylation and aggregation of tau proteins and neuroprogenitor fiber tangles formation (Chandra and Vassar, 2024). Deposition of Aβ has been shown to increase tau accumulation in studies via mechanisms involving microglia. In a model of microglia depletion, tau pathology near Aβ plaques was significantly increased, suggesting that microglia in general are suppressive of tau pathology (Kaur et al., 2024; Shahidehpour et al., 2024). Microglia over-produce EVs near Aβ plaques, not only facilitating the spreading of tau proteins but also aggravating neuroinflammation. Because microglia have a pivotal role in Aβ and tau pathology, it would be advantageous to diminish Aβ and tau pathology by stimulating TREM2 or augmenting induction of microglia to the DAM phenotype (Bathe et al., 2024; Figure 1).
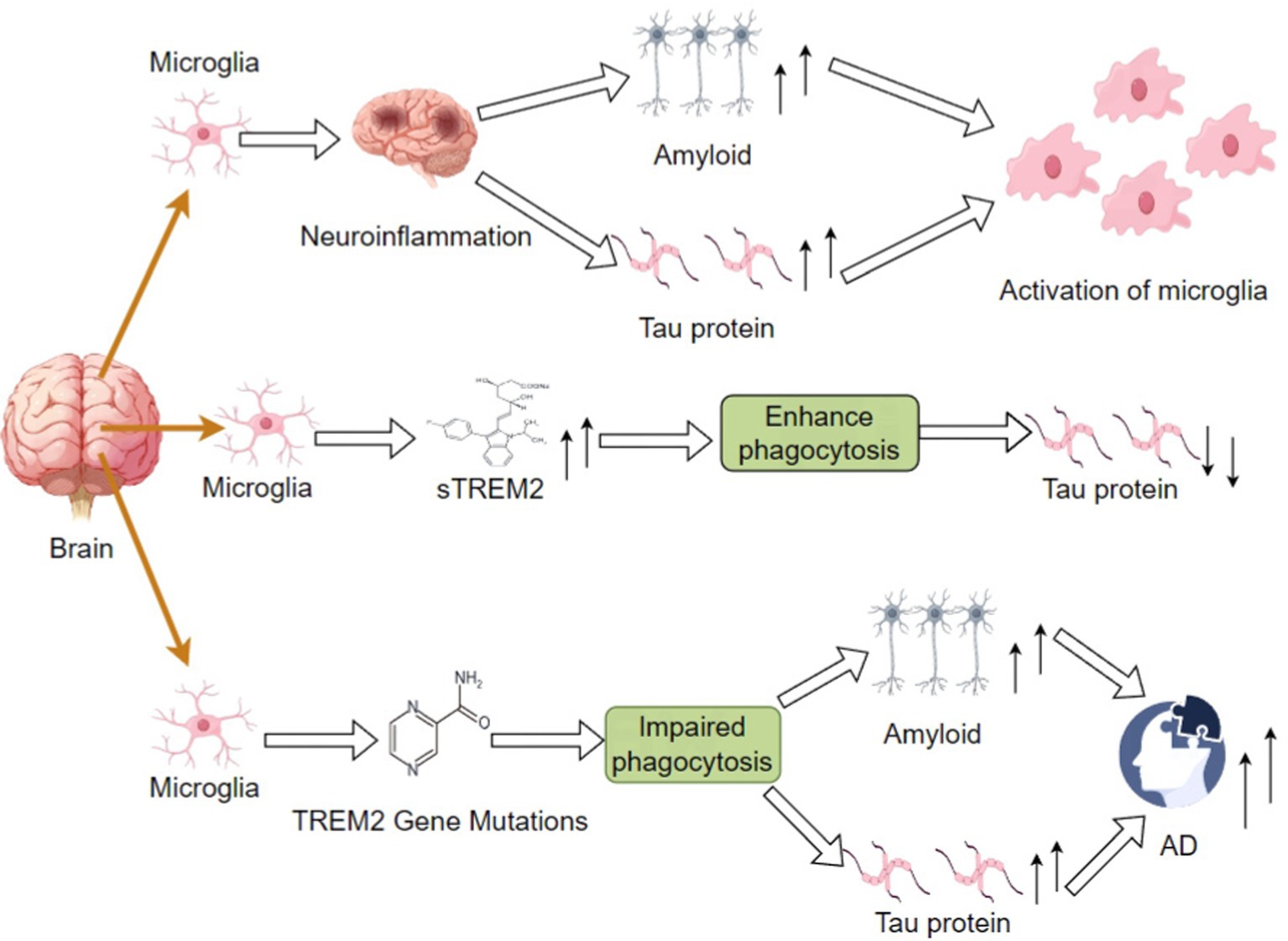
Figure 1. Differential impacts on amyloid and tau protein accumulation in AD via impaired phagocytic function by increased sTREM2 levels or TREM2 mutations; these may outline the mechanisms involved in neuroinflammation and the subsequent course of disease development.
Even though significant progress has been made in understanding the contribution of microglia to Aβ and tau pathology, literature on the link between microglia activation and tau pathology is conflicting, where some studies point toward it predisposing toward tau pathology induction while others suggest it may play a protective role against tau clearance, and these discrepancies may be owing to variations in experimental models, design, and the phase of the disease studied (Baligacs et al., 2024). In order to eliminate these discrepancies, future studies need to focus on the following. To resolve these contradictions, future studies would need to focus on the following aspects: first, normalizing experimental models and using consistent and well-characterized cell lines and animal models to reduce the impact of model variation (Anand et al., 2022); second, conducting longitudinal studies to follow the dynamic evolution of microglia activation and tau pathology with time and determine their functions at different stages of the disease (Wang C. et al., 2022); and third, conducting in-depth mechanistic studies to investigate the relationship between microglia activation and tau pathology (Chen X. et al., 2024). Third, comprehensive mechanistic studies will be conducted to investigate the molecular mechanism of the interaction between microglia activation and tau pathology; fourth, integration of data will be conducted, using meta-analyses and systematic reviews to aggregate data from multiple studies and identify common trends and variations; and fifth, advanced imaging tools will be utilized to visualize spatial and temporal correspondence between microglia activation and tau pathology in vivo, providing more intuitive evidence (Wu et al., 2025).
MicroRNAs are small non-coding RNAs that regulate gene expression by binding to the 3′ untranslated region of target mRNAs, inhibiting their translation, or directly degrading them. miRNAs can regulate key signaling pathways in microglia, affecting the inflammatory response and Aβ clearance of microglia. miRNAs in microglia regulate the inflammatory response and Aβ clearance of microglia by modulating key signaling pathways. miR-155 overexpression promotes microglial polarization to the pro-inflammatory M1 phenotype and NF-κB signaling pathway activation by targeting SHIP1 to promote the release of inflammatory factors (Zhang et al., 2025). The expression of miR-34a is increased in AD patients’ microglial cells, and the targeting of SIRT2 is enhanced in microglia. In microglia of AD patients, miR-34a expression is up-regulated and targets SIRT1 to inhibit its expression, affecting microglia metabolism and inflammatory responses, and thus Aβ clearance and neuroinflammation (Jadhav, 2024). Overexpression of miR-21 targets PTEN to stimulate the PI3K/Akt signaling pathway, promoting inflammatory responses and thus exacerbating neuroinflammation. Unlike miR-155, miR-124 inhibits M1-type polarization and promotes anti-inflammatory M2-type polarization to reduce neuroinflammation and neuronal injury. miR-124 suppresses the secretion of pro-inflammatory mediators and induces autophagy in the inflammation pathogenesis to alleviate neuroinflammation via targeting p62/p38 (Chen Y. et al., 2024).
TREM2, which is highly expressed on the surface of microglia, interacts directly with Aβ through regulation of microglia function (Shi et al., 2024). Several studies have shown that TREM2 recognizes and binds to Aβ, promoting phagocytosis of Aβ from microglia. TREM2 activation enhances Aβ phagocytosis by microglia through many molecular pathways (Serrano-Pozo et al., 2021; Cosma et al., 2023; de Gea et al., 2023). TREM2 activation enhances phagocytosis in microglia by several molecular pathways, thus promoting Aβ clearance (Benitez et al., 2021). TREM2 activation promotes phagocytosis in microglia. TREM2 activation enhances the expression of various genes related to phagocytosis and promotes the phagocytic function of microglia. TREM2 forms complexes with APOE and CLU/APOJ apolipoproteins, regulates metabolic-state regulation of microglia, along with its autophagy function, thus maintaining its phagocytic and survival maintenance (Dvir-Szternfeld et al., 2022). Besides participating in Aβ phagocytosis and its clearance, the role of TREM2 goes to taking part importantly in the modulation of microglia chemotaxis, survival, and proliferation, with the purpose of maintaining their functionality (Gratuze et al., 2021; Murakawa-Hirachi et al., 2021). More recently, the role of a loss of function in TREM2 leading to mTOR pathway impairment in microglia, which has been implicated as having a main function in the autophagic process, apart from a role in cell metabolism. Indeed, lifetime risks of AD for carriers of TREM2 loss-of-function mutations are remarkably increased, rivaling or even surpassing those for carriage of APOE ε4 variants (Burton et al., 2024; Escamilla and Salas-Lucia, 2024; Etxeberria et al., 2024). Such findings underscore the critical role of TREM2 in maintenance of healthy microglia and Aβ clearing.
Loss or impairment of TREM2 function has been associated with a significant reduction of microglia phagocytosis (Giorgio et al., 2024). In the TREM2-deficient 5 × FAD mouse model, there is an increased ratio of lipidated LC3II to nonlipidated LC3I with increased numbers of autophagic vesicles, indicating impaired process of autophagy (Zhong et al., 2024). TREM2 may be considered the central regulator for phagocytosis in microglia. Its activation promotes the phagocytosis and degradation of Aβ by microglia (Zhang W. et al., 2024). Moreover, phagocytic dysfunction has affected the efficiency of Aβ clearance and promoted Aβ accumulation by several pathways. Moreover, it has been shown that the activation of the NLRP3 inflammasome in AD is associated with the deposition of Aβ, which may result from an inflammatory response due to an impaired phagocytosis process in microglia. Microglial-related studies implicate that impaired phagocytosis mediates Aβ production through NF-κB signaling pathways and increases in BACE1 expression, which eventually lead to accumulation in Aβ within brain tissue and hastening of the AD pathological course (Wang et al., 2024; Yan et al., 2024).
Microglia trigger downstream inflammatory signaling cascades by detecting Aβ aggregates through recognition receptors. Aβ42 activates the TRPM2 channels by inducing oxidative stress, resulting in pro-inflammatory cytokine release, and is a positive feedback loop to enhance the inflammation process (Shen Z. et al., 2024). Aβ has the ability to also activate the NLRP3 inflammasomes of microglial cells to cleave caspase-1 and mature IL-1β. This is amplified on microglial inflammation, initiating a negative chronic inflammatory state leading to necrosis to the neurons (Zhang L. et al., 2024). Activation of binding between TREM2 and Aβ initiates proliferation, phagocytosis, and secretion by microglia, modulating their metabolism and viability. Despite being protective, overexposure to Aβ has been shown to lead to microglial dysfunction, reduced clearance, and perpetuation of relentless inflammatory signaling (Wang M. et al., 2022).
Aβ-activated microglia produce proinflammatory cytokines such as TNFα and IL-1β that induce excitotoxicity and neuronal death via the synergistic activation of neuronal TNF receptors and NMDA receptors. This neuroinflammatory cascade response contributes to synaptic dysfunction and neuronal loss in AD (Shi et al., 2022). While microglia phagocytosis is critical in Aβ clearance, sustained Aβ exposure may lead to their functional depletion as evidenced by dendritic shortening, reduction in coverage area, and fragmentation of cellular processes suggestive of loss of surveillance function. Functionally impaired microglia lose the capability of effective Aβ clearance, which consequently accumulates in the brain and promotes AD pathology (Bandyopadhyay, 2021; Wang S. Y. et al., 2021). In cellular models of AD, Aβ-treated microglial cells showed an impairment of autophagy as defined by reduced expression of the autophagy-related protein Beclin-1. Defective autophagy disturbed the degradation capability of microglia for Aβ, resulting in Aβ accumulation and enhancement of inflammatory responses (Katsumoto et al., 2018).
TREM2 is constitutively strongly expressed on microglia in the central nervous system and regulates microglia phagocytosis and signaling through interaction with DAP12 protein. TREM2 directly deoves Aβ oligomers and activates the downstream PI3K/Akt signaling pathway, and facilitates Aβ degradation and phagocytosis by microglia. In the initial stage of AD, TREM2 activation enhances the chemotactic activity of microglia to migrate to the site of Aβ deposition and clear plaques to play a neuroprotective role. TREM2 loss-of-function mutations result in defective phagocytosis of microglia and significant reduction in the effectiveness of Aβ clearance, as well as suppression of secretion of the anti-inflammatory factor IL-10 to further amplify chronic neuroinflammation (Li et al., 2022). TREM2-deficient mice have been shown to be unable to encapsulate Aβ plaques in mouse models, leading to synaptic loss and hyperphosphorylation of tau proteins of neighboring neurons. TREM2 maintains long-term survival of microglia by controlling their metabolism, and its dysfunction may lead to microglia depletion, which could exacerbate Aβ deposition and neurodegeneration (Lin et al., 2024).
CD33 is a salivary acid-binding immunoglobulin-like lectin whose expression level dictates risk for AD by controlling immune checkpoint activity in microglia. rs3865444 CD33 gene polymorphism reduces the expression of intact CD33 but enhances D2-CD33 that lacks exon 2. D2-CD33 enhances microglial cell, via the ITAM signaling pathway, Aβ phagocytosis as well as inflammatory inhibitory factor secretion (Javor et al., 2022). Microglia in subjects with the rs3865444 minor allele are enhanced in Aβ clearance and inhibited in the release of proinflammatory factors. On the other hand, full-length CD33 overexpression inhibits microglia polarization into the M2 subtype and leads to defective Aβ clearance and ongoing neuroinflammation, proving the crucial role of CD33 splicing homeostasis in regulating microglia immune homeostasis (Li et al., 2015).
CR1 is expressed primarily on the surface of microglia and facilitates Aβ coagulation and precipitation by binding to Aβ, thereby facilitating phagocytosis and clearance of Aβ by microglia. CR1 gene polymorphisms are strongly associated with AD susceptibility, affecting the expression level of the CR1 protein and microglial efficiency of Aβ clearance (Sudwarts and Thinakaran, 2023). CR1 gene variants affect the condition of microglial activation and inflammatory reaction, causing bias toward M1-type activation and suppressing their Aβ clearance. CR1 gene variants suppress Aβ clearance through affecting the condition of microglial activation and inflammatory reaction, causing bias toward M1-type microglial activation (Zuroff et al., 2017).
APOE gene polymorphisms tightly control AD pathology by modulating lipid metabolism and inflammatory processes in microglia. APOEε4 has a higher affinity to Aβ than APOE2/3 but, via its defective binding to lipoproteins, causes reduced phagocytosis of Aβ by microglial cells. APOEε4 also induces mitochondrial damage in microglial cells and increases ROS production and NLRP3 inflammatory vesicle activity (Qu et al., 2024). APOEε4 blocks the conversion of microglia into a neuroprotective phenotype, induces the release of proinflammatory extracellular vesicles, and promotes the transneuronal transmission of tau proteins by blocking the TREM2-PI3K/Akt signaling pathway, demonstrating that APOE genes are importantly involved in the pathomechanisms of AD (Narasimhan et al., 2024).
It was identified that PLCG2 is up-regulated selectively in plaque-associated microglia and engaged in the regulation of microglia phagocytosis and inflammation response. These variants bi-directionally control microglia phenotype and function affecting Aβ clearance competency through activating divergent transcriptional programs. These results not only indicate the widespread involvement of PLCG2 in AD pathogenesis, but also infer that it could be a prospective therapeutic target for future AD treatment (Magno et al., 2019; Staley et al., 2024).
The ABI3 gene encodes a transcription factor with a B3 domain and is at the center of microglial immune responses. Some variants, such as p.S209F, which was significantly associated with LOAD risk, may alter the efficiency of Aβ clearance by microglia through their impact on ABI3 function. Such variants lead to impaired activation and phagocytosis of microglia, thus affecting Aβ clearance and exacerbating pathological progression of AD (Ibanez et al., 2022; Smith et al., 2022; Table 2).
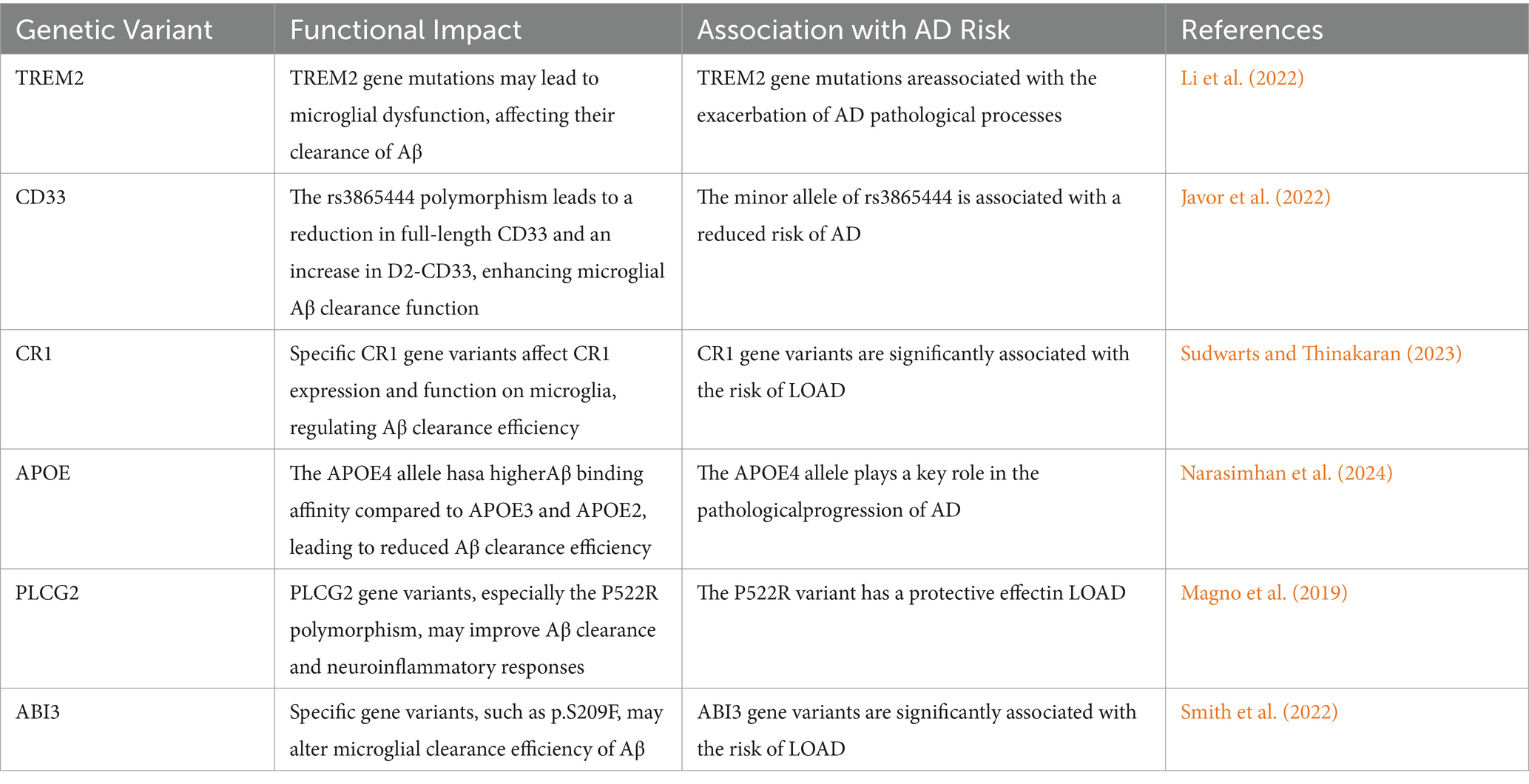
Table 2. Genetic variants and their impact on microglial function and Alzheimer’s disease risk.
At the initial phase of AD, TREM2-PI3K/Akt pathway dominates, and increased Aβ clearance and inhibition of NF-κB can delay pathological development (Hu et al., 2025). At the advanced phase of AD, continuous activation by Aβ leads to TREM2 decrease, decreased PI3K/Akt activity, excessive activation of NF-κB and exuberant release of inflammatory cytokines enhance tau pathology spreading and neuronal loss (Zhang J. et al., 2024). TREM2 mutations lead to the disturbance of PI3K/Akt-NF-κB homeostasis, causing microglial cell dysfunction and exacerbation of AD (Sun et al., 2021).
TREM2 was found to mediate the process in the central nervous system by binding to a series of ligands, such as Aβ oligomers and phospholipids, by transmitting the downstream DAP12/DAP10-SYK-PI3K-AKT signaling cascade to regulate microglia phagocytosis, survival, inflammatory response, and metabolic processes (Lin et al., 2024). TREM2 activation facilitates microglia phagocytosis and enhances Aβ degradation by facilitating the upregulation of phagocytosis-related genes via the PI3K/Akt pathway. TREM2 inhibits nuclear translocation of NF-κB via the PI3K/Akt pathway and reduces secretion of proinflammatory factors to inhibit chronic neuroinflammation. TREM2-PI3K/Akt signaling modulates microglia energy metabolism and maintains their long-term survival and functional homeostasis (Yao et al., 2019).
NF-κB signaling pathway is the key regulator of inflammatory and immune responses of microglia. TREM2, which is one of the highly expressed microglia-restricted receptors, was discovered to exert influence on the production of inflammatory factors by inducing downstream NF-κB signaling upon association with DAP12. Activation of TREM2 inhibits NF-κB-induced production of inflammatory factors to mitigate neuroinflammation. These observations demonstrate an immunomodulatory function of TREM2 in AD and a critical role for the NF-κB pathway in microglial activation. TREM2-activated PI3K/Akt pathway inhibits excessive inflammation by phosphorylating IκB kinase, blocking IκBα degradation and inhibiting NF-κB nuclear translocation (Shi and Huang, 2023).
Being a cytoplasmic multiprotein complex, NLRP3 inflammasome senses endogenous danger signals and pathological protein aggregation. Activation of NLRP3 inflammasome further promotes the maturation and secretion of proinflammatory cytokines IL-1β and IL-18, which further enhance neuroinflammatory responses and drive neurodegenerative processes. Aβ deposition directly activates the NLRP3 inflammatory vesicles in microglia, enhancing the secretion of IL-1β and further exaggeration of neuroinflammation and neuronal injury with positive feedback (Hanslik and Ulland, 2020; Zhang Z. et al., 2024).
Microglia are implicated in diverse neurobiological processes by activating the PI3K/Akt signaling pathway in AD pathology. Experiments demonstrate that activation of the PI3K/Akt pathway suppresses the release of pro-inflammatory mediators and enhances the release of anti-inflammatory mediators that regulate AD-associated neuroinflammation. Activation of the PI3K/Akt pathway suppresses GSK3β activity in microglia and diminishes Tau protein phosphorylation and neurofibrillary tangle formation (Long et al., 2021). PI3K/Akt promotes M2-type polarization of microglia and inhibits M1-type polarization and immune microenvironment balance. PI3K/Akt regulates the glycolysis and oxidative phosphorylation of microglia through mTORC1, which modulates their clearance function and inflammatory phenotype. TREM2 significantly enhanced microglial cell neuroinflammation by activating the PI3K/Akt/GSK3β pathway, significantly enhancing spatial cognition in the APP/PS1 mouse model. These investigations represent the critical function of the PI3K/Akt pathway in regulating AD pathology and promise therapeutic targets (Chu et al., 2021).
Aβ acts on TLR4 on the microglial cell membrane to initiate the MAPK signal pathway, which phosphorylates heterogeneous downstream substrates like transcription factors and cytoskeletal proteins and modulates the synthesis of proinflammatory cytokines and chemokines. The MAPK signal pathway induces polarization of the microglial cells into the M1-type, augmenting the neuroinflammatory microenvironment of the brain and being harmful to the survival and functions of the neurons. MAPK signaling pathway also regulates polarization of microglia to the M2 form, which holds anti-inflammatory as well as neuroprotective function against neuronal damage. Balance in between the two polarized forms, i.e., the M1 or the M2 type, decides outcomes of disease and interruption of MAPK signaling pathway would theoretically result in interruption in balance toward excessively pro-inflammatory track (Wang C. et al., 2023). Confirmatory experiments have also shown that etomidate enhances cognitive function and suppresses oxidative stress and inflammatory response in AD mice brain tissue through regulation of the MAPK/ERK signaling pathway. The results indicate that inhibitors of the MAPK signaling pathway are likely to be potential drugs in the treatment of AD (Zuppe and Reed, 2024; Table 3).
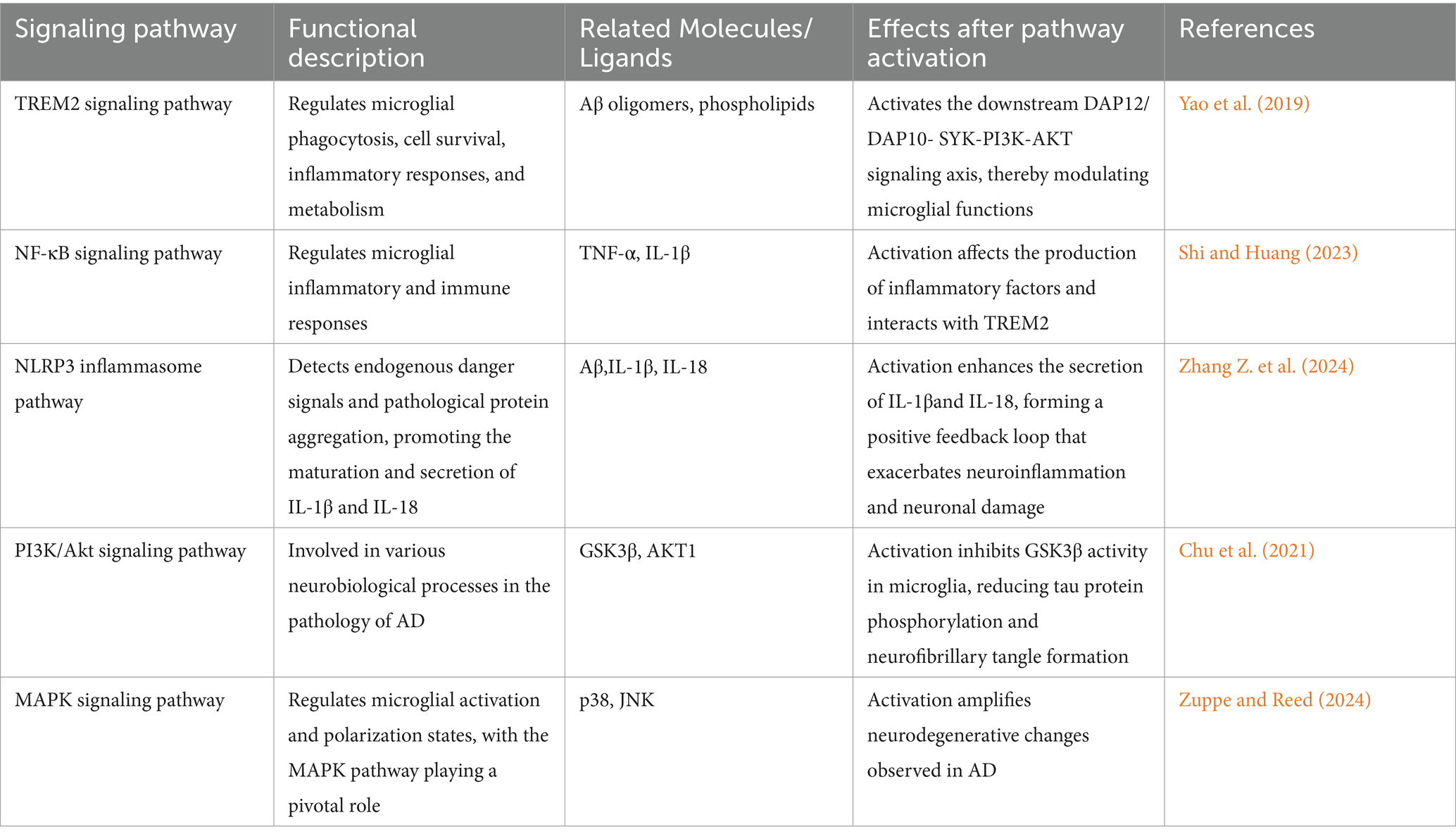
Table 3. Overview of key signaling pathways involved in microglial activation and their roles in Alzheimer’s disease pathology.
Gene therapy technology also shows much potential in AD treatment studies. By utilizing gene editing techniques like CRISPR/Cas9, it is possible to directly regulate the expression level of genes associated with AD in an effort to enhance microglia function. From studies, microglia-induced neuroinflammatory responses could be significantly diminished by regulating TREM2 and CD33 genes. Small molecule drugs also regulate the inflammatory response of microglia (Garcia-Gonzalez et al., 2024). The P2X7 receptor inhibitors and the NLRP3 inflammatory vesicle inhibitors have been shown to inhibit the inflammatory response of microglia. They inhibit the release of inflammatory mediators and eliminate the threat of damage and death in neurons by controlling the signaling pathways involved selectively (Doshi et al., 2024). Their activities provide new hope for pharmacologic treatment of AD, and show promising activity against microglia-induced neuroinflammation.
Though microglia-directed therapeutic interventions for AD have shown promise, there are still numerous areas where clinical translation is problematic. Drug delivery effectiveness is limited by the blood–brain barrier, and although nanocarriers, targeted ultrasound, and other platforms are used to enhance penetration, their safety, stability, and scale-up remain open questions, and dynamic change in expression of microglial cell-specific receptors reduces targeting efficacy (Liu et al., 2022). Inadequate targeting and functional/molecular marker overlap between microglial cells and peripheral macrophages may lead to drug Misuse of the peripheral immune system to cause systemic inflammation or immunosuppression (Baumgartner et al., 2025). Clinical trial failures are not unusual, and several clinical trials for neuroinflammatory disorders have failed to meet expected endpoints. Experiments have shown that although the anti-Aβ monoclonal antibody Aducanumab activates microglia to clear plaques, its contentious efficacy and side effects such as cerebral edema highlight the importance of balancing immune activation and neuroprotection (Niazi, 2024). Drugs that block NLRP3 inflammatory vesicles reduce tau pathology in animal models but have limited effectiveness in humans due to the activation of compensatory pathway activity or off-targeting (Zhou L. et al., 2024).
The second main challenge is the functional changes in microglia during aging. Microglia are morphologically, phenotypically, and functionally modified in brain aging, and such changes may impact their response to therapeutic intervention. With age, microglia experience alterations including diminished proliferative and phagocytic function, as well as increased basal inflammatory activity, and these alterations can modulate the activity of microglia in order to maintain neuronal homeostasis (Antignano et al., 2023). As age progresses, increased blood–brain barrier permeability can impact drug delivery and distribution to microglia. Microglial heterogeneity of the aging brain also makes it challenging to develop microglia-specific treatments, as different subpopulations of microglia may respond differently to treatment. Therefore, IAG-dependent microglial changes should be considered when creating and evaluating treatments in AD (Stamataki et al., 2024).
During the last decade, microglial activation has been a pivotal driving force of AD pathogenesis, and therapeutic intervention in engaged signaling pathways has emerged as a novel therapeutic approach. Gene therapy and small-molecule drugs have been shown to possess the capacity to reduce inflammation and neuroprotection, but their clinical utility remains handicapped by challenges of targeting efficacy, pharmacokinetics, and long-term safety. Future and ongoing studies should examine deeper the microglia signaling pathways and design new drugs that guarantee safety, efficacy, and person-specific adaptability of the new treatment. We should further utilize single-cell sequencing technology to intensively investigate the various subpopulations of microglia in AD and their multiple functional differences under different phases of the disease; design nanocarriers or targeted ultrasound technology capable of crossing the blood–brain barrier for improving drug-delivery efficacy; study in particular the roles of peripheral immune cells in AD and design therapy with the goal to target microglia and peripheral immune cells simultaneously; and intensively investigate the role of miRNA in microglia activation and neuroinflammation. Cell activation and neuroinflammation, and devise miRNA-based therapeutic strategies; adopt more adaptive clinical trials, and utilize biomarkers for early diagnosis and evaluation of therapeutic effects; explore the combined modality of gene therapy and small molecule drugs, and explore the application of gene editing technology in microglia; perform longitudinal studies of dynamic changes of microglia at different stages of AD and explore their role in early diagnosis and control of disease; perform longitudinal studies of dynamic changes of microglia at different stages of AD and explore their role in early diagnosis and control of disease. Referring to their potential roles in early detection and monitoring of the disease. In these specific research directions, future studies may have a clearer understanding of microglia’s multi-faceted mechanisms in AD and establish a firm foundation for more effective therapeutic strategies.
CH: Conceptualization, Data curation, Writing – original draft. BC: Validation, Writing – review & editing. HY: Supervision, Writing – review & editing. XZ: Funding acquisition, Writing – review & editing.
The author(s) declare that financial support was received for the research and/or publication of this article. This work was partially supported by the Shenzhen Municipal Science and Technology Innovation Commission (JCYJ20210324122210028), awarded to Xiaoqing Zhou.
We thank all the authors for their contribution to the article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors declare that no Gen AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Al-Thani, N. A., Zinck, D., Stewart, G. S., and Costello, D. A. (2024). Modulation of urea transport attenuates TLR2-mediated microglial activation and upregulates microglial metabolism in vitro. Meta 14:634. doi: 10.3390/metabo14110634
Alzheimer’s Association (2024). 2024 Alzheimer’s disease facts and figures. Alzheimers Dement. 20, 3708–3821. doi: 10.1002/alz.13809
Ameli Mojarad, M., and Ameli Mojarad, M. (2024). The neuroinflammatory role of microglia in Alzheimer’s disease and their associated therapeutic targets. CNS Neurosci. Ther. 30:e14856. doi: 10.1111/cns.14856
Ana, B. (2024). Aged-related changes in microglia and neurodegenerative diseases: exploring the connection. Biomedicines 12:1737. doi: 10.3390/biomedicines12081737
Anand, C., Maia, P. D., Torok, J., Mezias, C., and Raj, A. (2022). The effects of microglia on tauopathy progression can be quantified using Nexopathy in silico (Nexis) models. Sci. Rep. 12:21170. doi: 10.1038/s41598-022-25131-3
Andersson, E., Janelidze, S., Lampinen, B., Nilsson, M., Leuzy, A., Stomrud, E., et al. (2020). Blood and cerebrospinal fluid neurofilament light differentially detect neurodegeneration in early Alzheimer’s disease. Neurobiol. Aging 95, 143–153. doi: 10.1016/j.neurobiolaging.2020.07.018
Andersson, E., Lindblom, N., Janelidze, S., Salvado, G., Gkanatsiou, E., Soderberg, L., et al. (2025). Soluble cerebral Abeta protofibrils link Abeta plaque pathology to changes in CSF Abeta (42)/Abeta (40) ratios, neurofilament light and tau in Alzheimer’s disease model mice. Nat Aging. doi: 10.1038/s43587-025-00810-8
Antignano, I., Liu, Y., Offermann, N., and Capasso, M. (2023). Aging microglia. Cell. Mol. Life Sci. 80:126. doi: 10.1007/s00018-023-04775-y
Arber, C., Casey, J. M., Crawford, S., Rambarack, N., Yaman, U., Wiethoff, S., et al. (2024). Microglia contribute to the production of the amyloidogenic ABri peptide in familial British dementia. Acta Neuropathol. 148:65. doi: 10.1007/s00401-024-02820-z
Baligacs, N., Albertini, G., Borrie, S. C., Serneels, L., Pridans, C., Balusu, S., et al. (2024). Homeostatic microglia initially seed and activated microglia later reshape amyloid plaques in Alzheimer’s disease. Nat. Commun. 15:10634. doi: 10.1038/s41467-024-54779-w
Bandyopadhyay, S. (2021). Role of neuron and glia in Alzheimer’s disease and associated vascular dysfunction. Front. Aging Neurosci. 13:653334. doi: 10.3389/fnagi.2021.653334
Bathe, T., Hery, G. P., Villareal, J. A. B., Phillips, J. L., Cohen, E. M., Sharma, R. V., et al. (2024). Disease and brain region specific immune response profiles in neurodegenerative diseases with pure and mixed protein pathologies. Acta Neuropathol. Commun. 12:54. doi: 10.1186/s40478-024-01770-7
Baumgartner, A., Robinson, M., Ertekin-Taner, N., Golde, T. E., Jaydev, S., Huang, S., et al. (2025). Fokker-Planck diffusion maps of microglial transcriptomes reveal radial differentiation into substates associated with Alzheimer’s pathology. Commun Biol 8:279. doi: 10.1038/s42003-025-07594-y
Benitez, D. P., Jiang, S., Wood, J., Wang, R., Hall, C. M., Peerboom, C., et al. (2021). Knock-in models related to Alzheimer’s disease: synaptic transmission, plaques and the role of microglia. Mol. Neurodegener. 16:47. doi: 10.1186/s13024-021-00457-0
Bhardwaj, V., Kumari, S., Dhapola, R., Sharma, P., Beura, S. K., Singh, S. K., et al. (2024). Shedding light on microglial dysregulation in Alzheimer’s disease: exploring molecular mechanisms and therapeutic avenues. Inflammopharmacology 33, 679–702. doi: 10.1007/s10787-024-01598-6
Bivona, G., Iemmolo, M., and Ghersi, G. (2023). CX3CL1 pathway as a molecular target for treatment strategies in Alzheimer’s disease. Int. J. Mol. Sci. 24:8230. doi: 10.3390/ijms24098230
Botella Lucena, P., and Heneka, M. T. (2024). Inflammatory aspects of Alzheimer’s disease. Acta Neuropathol. 148:31. doi: 10.1007/s00401-024-02790-2
Burton, E. A., Argenziano, M., Cook, K., Ridler, M., Lu, S., Su, C., et al. (2024). Variant-to-function mapping of late-onset Alzheimer’s disease GWAS signals in human microglial cell models implicates RTFDC1 at the CASS4 locus. bio Rxiv. doi: 10.1101/2024.08.22.609230
Cafferata, E. A., Ramanauskaite, A., Cuypers, A., Obreja, K., Dohle, E., Ghanaati, S., et al. (2024). Experimental peri-implantitis induces neuroinflammation: an exploratory study in rats. BMC Oral Health 24:1238. doi: 10.1186/s12903-024-04995-z
Cai, L., Fan, Q., Pang, R., Chen, C., Zhang, Y., Xie, H., et al. (2024). Microglia programmed cell death in neurodegenerative diseases and CNS injury. Apoptosis 30, 446–465. doi: 10.1007/s10495-024-02041-5
Cai, Y., Liu, J., Wang, B., Sun, M., and Yang, H. (2022). Microglia in the Neuroinflammatory pathogenesis of Alzheimer’s disease and related therapeutic targets. Front. Immunol. 13:856376. doi: 10.3389/fimmu.2022.856376
Chandra, S., and Vassar, R. (2024). The role of the gut microbiome in the regulation of astrocytes in Alzheimer’s disease. Neurotherapeutics 21:e00425. doi: 10.1016/j.neurot.2024.e00425
Chen, X., Firulyova, M., Manis, M., Herz, J., Smirnov, I., Aladyeva, E., et al. (2024). Author correction: microglia-mediated T cell infiltration drives neurodegeneration in tauopathy. Nature 634:E15. doi: 10.1038/s41586-024-08200-7
Chen, Y., Mateski, J., Gerace, L., Wheeler, J., Burl, J., Prakash, B., et al. (2024). Non-coding RNAs and neuroinflammation: implications for neurological disorders. Exp. Biol. Med. (Maywood) 249:10120. doi: 10.3389/ebm.2024.10120
Chen, Z., Yuan, Z., Yang, S., Zhu, Y., Xue, M., Zhang, J., et al. (2023). Brain energy metabolism: astrocytes in neurodegenerative diseases. CNS Neurosci. Ther. 29, 24–36. doi: 10.1111/cns.13982
Chu, E., Mychasiuk, R., Hibbs, M. L., and Semple, B. D. (2021). Dysregulated phosphoinositide 3-kinase signaling in microglia: shaping chronic neuroinflammation. J. Neuroinflammation 18:276. doi: 10.1186/s12974-021-02325-6
Cornell, J., Salinas, S., Huang, H. Y., and Zhou, M. (2022). Microglia regulation of synaptic plasticity and learning and memory. Neural Regen. Res. 17, 705–716. doi: 10.4103/1673-5374.322423
Cosma, N. C., Eren, N., Usekes, B., Gerike, S., Heuser, I., Peters, O., et al. (2023). Acute and chronic macrophage differentiation modulates TREM2 in a personalized Alzheimer’s patient-derived assay. Cell. Mol. Neurobiol. 43, 3047–3060. doi: 10.1007/s10571-023-01351-7
da Fonseca, A. C., Matias, D., Garcia, C., Amaral, R., Geraldo, L. H., Freitas, C., et al. (2014). The impact of microglial activation on blood-brain barrier in brain diseases. Front. Cell. Neurosci. 8:362. doi: 10.3389/fncel.2014.00362
Daraban, B. S., Popa, A. S., and Stan, M. S. (2024). Latest perspectives on Alzheimer’s disease treatment: the role of blood-brain barrier and antioxidant-based drug delivery systems. Molecules 29:4056. doi: 10.3390/molecules29174056
Darwish, S. F., Elbadry, A. M. M., Elbokhomy, A. S., Salama, G. A., and Salama, R. M. (2023). The dual face of microglia (M1/M2) as a potential target in the protective effect of nutraceuticals against neurodegenerative diseases. Front Aging 4:1231706. doi: 10.3389/fragi.2023.1231706
de Gea, P., Benkeder, S., Bouvet, P., Aimard, M., Chounlamountri, N., Honnorat, J., et al. (2023). VEGF controls microglial phagocytic response to amyloid-beta. Front. Cell. Neurosci. 17:1264402. doi: 10.3389/fncel.2023.1264402
Demuth, H., Hosseini, S., Dusedeau, H. P., Dunay, I. R., Korte, M., and Zagrebelsky, M. (2023). Deletion of p 75 (NTR) rescues the synaptic but not the inflammatory status in the brain of a mouse model for Alzheimer’s disease. Front. Mol. Neurosci. 16:1163087. doi: 10.3389/fnmol.2023.1163087
Di Battista, A. M., Heinsinger, N. M., and Rebeck, G. W. (2016). Alzheimer’s disease genetic risk factor APOE-epsilon4 also affects Normal brain function. Curr. Alzheimer Res. 13, 1200–1207. doi: 10.2174/1567205013666160401115127
Doshi, V., Joshi, G., Sharma, S., and Choudhary, D. (2024). Gene therapy: an alternative to treat Alzheimer’s disease. Naunyn Schmiedeberg’s Arch. Pharmacol. 397, 3675–3693. doi: 10.1007/s00210-023-02873-z
Du, L., Zhang, Y., Chen, Y., Zhu, J., Yang, Y., and Zhang, H. L. (2017). Role of microglia in neurological disorders and their potentials as a therapeutic target. Mol. Neurobiol. 54, 7567–7584. doi: 10.1007/s12035-016-0245-0
Dvir-Szternfeld, R., Castellani, G., Arad, M., Cahalon, L., Colaiuta, S. P., Keren-Shaul, H., et al. (2022). Alzheimer’s disease modification mediated by bone marrow-derived macrophages via a TREM2-independent pathway in mouse model of amyloidosis. Nat Aging 2, 60–73. doi: 10.1038/s43587-021-00149-w
Escamilla, S., and Salas-Lucia, F. (2024). Thyroid hormone and Alzheimer disease: bridging epidemiology to mechanism. Endocrinology 165:bqae124. doi: 10.1210/endocr/bqae124
Etxeberria, A., Shen, Y. A., Vito, S., Silverman, S. M., Imperio, J., Lalehzadeh, G., et al. (2024). Neutral or detrimental effects of TREM2 agonist antibodies in preclinical models of Alzheimer’s disease and multiple sclerosis. J. Neurosci. 44:e2347232024. doi: 10.1523/JNEUROSCI.2347-23.2024
Fan, X., Chen, H., He, W., and Zhang, J. (2024). Emerging microglial biology highlights potential therapeutic targets for Alzheimer’s disease. Ageing Res. Rev. 101:102471. doi: 10.1016/j.arr.2024.102471
Fu, X. X., Huang, Z. H., Wang, S. Y., Qi, J. W., Luo, Z. J., Yan, E., et al. (2024). Knockdown of TREML2 alleviates neuropathological hallmarks and cognitive deficiency in a model of sporadic Alzheimer’s disease. J. Inflamm. Res. 17, 10471–10478. doi: 10.2147/JIR.S489474
Gaire, B. P. (2022). Microglia as the critical regulators of neuroprotection and functional recovery in cerebral ischemia. Cell. Mol. Neurobiol. 42, 2505–2525. doi: 10.1007/s10571-021-01145-9
Gao, C., Jiang, J., Tan, Y., and Chen, S. (2023). Microglia in neurodegenerative diseases: mechanism and potential therapeutic targets. Signal Transduct. Target. Ther. 8:359. doi: 10.1038/s41392-023-01588-0
Garcia-Gonzalez, N., Goncalves-Sanchez, J., Gomez-Nieto, R., Goncalves-Estella, J. M., and Lopez, D. E. (2024). Advances and challenges in gene therapy for neurodegenerative diseases: a systematic review. Int. J. Mol. Sci. 25:12485. doi: 10.3390/ijms252312485
Gasparotto, J., Somensi, N., Girardi, C. S., Bittencourt, R. R., de Oliveira, L. M., Hoefel, L. P., et al. (2024). Is it all the RAGE? Defining the role of the receptor for advanced glycation end products in Parkinson’s disease. J. Neurochem. 168, 1608–1624. doi: 10.1111/jnc.15890
GBD 2021 US Burden of Disease Collaborators (2024). The burden of diseases, injuries, and risk factors by state in the USA, 1990-2021: a systematic analysis for the global burden of disease study 2021. Lancet 404, 2314–2340. doi: 10.1016/S0140-6736(24)01446-6
Giorgio, J., Jonson, C., Wang, Y., Yokoyama, J. S., Wang, J., Jagust, W., et al. (2024). Variable and interactive effects of sex, APOE epsilon4 and TREM2 on the deposition of tau in entorhinal and neocortical regions. Res Sq. doi: 10.21203/rs.3.rs-4804430/v1
Gratuze, M., Chen, Y., Parhizkar, S., Jain, N., Strickland, M. R., Serrano, J. R., et al. (2021). Activated microglia mitigate Abeta-associated tau seeding and spreading. J. Exp. Med. 218:e20210542. doi: 10.1084/jem.20210542
Hanslik, K. L., and Ulland, T. K. (2020). The role of microglia and the Nlrp3 Inflammasome in Alzheimer’s disease. Front. Neurol. 11:570711. doi: 10.3389/fneur.2020.570711
Hao, W., Ma, Q., Wang, L., Yuan, N., Gan, H., He, L., et al. (2024). Gut dysbiosis induces the development of depression-like behavior through abnormal synapse pruning in microglia-mediated by complement C3. Microbiome 12:34. doi: 10.1186/s40168-024-01756-6
Hemonnot, A. L., Hua, J., Ulmann, L., and Hirbec, H. (2019). Microglia in Alzheimer disease: well-known targets and new opportunities. Front. Aging Neurosci. 11:233. doi: 10.3389/fnagi.2019.00233
Heneka, M. T. (2023). ApoE4 makes microglia trem(2)bling. Neuron 111, 142–144. doi: 10.1016/j.neuron.2022.12.032
Hu, L., Liu, J., Peng, J., Li, X., Huang, Z., Zhang, C., et al. (2025). TREM2 alleviates Neuroinflammation by maintaining cellular metabolic homeostasis and Mitophagy activity during early inflammation. Diseases 13:60. doi: 10.3390/diseases13020060
Huang, Q., Wang, Y., Chen, S., and Liang, F. (2024). Glycometabolic reprogramming of microglia in neurodegenerative diseases: insights from Neuroinflammation. Aging Dis. 15, 01–1175. doi: 10.14336/AD.2023.0807
Ibanez, K. R., McFarland, K. N., Phillips, J., Allen, M., Lessard, C. B., Zobel, L., et al. (2022). Deletion of Abi3/Gngt2 influences age-progressive amyloid beta and tau pathologies in distinctive ways. Alzheimers Res. Ther. 14:104. doi: 10.1186/s13195-022-01044-1
Is, O., Min, Y., Wang, X., Oatman, S. R., Abraham Daniel, A., and Ertekin-Taner, N. (2024). Multi layered omics approaches reveal glia specific alterations in Alzheimer’s disease: a systematic review and future prospects. Glia 73, 539–573. doi: 10.1002/glia.24652
Jacquet, R. G., Gonzalez Ibanez, F., Picard, K., Funes, L., Khakpour, M., Gouras, G. K., et al. (2024). Microglia degrade Alzheimer’s amyloid-beta deposits extracellularly via digestive exophagy. Cell Rep. 43:115052. doi: 10.1016/j.celrep.2024.115052
Jadhav, S. P. (2024). MicroRNAs in microglia: deciphering their role in neurodegenerative diseases. Front. Cell. Neurosci. 18:1391537. doi: 10.3389/fncel.2024.1391537
Javor, J., Bucova, M., Durmanova, V., Radosinska, D., Parnicka, Z., Cierny, D., et al. (2022). Alzheimer’s disease risk variant rs3865444 in the CD33 gene: a possible role in susceptibility to multiple sclerosis. Life 12:1094. doi: 10.3390/life12071094
Jiao, J. J., Hu, Y., Cui, Y. J., Tuo, C. M., Wang, Y. X., Li, X. Y., et al. (2024). Anisomycin alleviates cognitive impairments and pathological features in 3xTg-AD mice. Neuropharmacology 261:110159. doi: 10.1016/j.neuropharm.2024.110159
Kanuri, S. H., and Sirrkay, P. J. (2024). Profiling of microglial-originated microvesicles to unearthing their lurking potential as potent foreseeable biomarkers for the diagnosis of Alzheimer’s disease: a systematic review. Brain Circ 10, 193–204. doi: 10.4103/bc.bc_113_23
Katsumoto, A., Takeuchi, H., Takahashi, K., and Tanaka, F. (2018). Microglia in Alzheimer’s disease: risk factors and inflammation. Front. Neurol. 9:978. doi: 10.3389/fneur.2018.00978
Kaur, S. K. M., Sharma, A., Giridharan, V. V., and Dandekar, M. P. (2024). Brain resident microglia in Alzheimer’s disease: foe or friends. Inflammopharmacology 32, 2781–2800. doi: 10.1007/s10787-024-01550-8
Kempuraj, D., Dourvetakis, K. D., Cohen, J., Valladares, D. S., Joshi, R. S., Kothuru, S. P., et al. (2024). Neurovascular unit, neuroinflammation and neurodegeneration markers in brain disorders. Front. Cell. Neurosci. 18:1491952. doi: 10.3389/fncel.2024.1491952
Kim, Y., Lim, J., and Oh, J. (2024). Taming neuroinflammation in Alzheimer’s disease: the protective role of phytochemicals through the gut-brain axis. Biomed. Pharmacother. 178:117277. doi: 10.1016/j.biopha.2024.117277
Kloske, C. M., Mahinrad, S., Barnum, C. J., Batista, A. F., Bradshaw, E. M., Butts, B., et al. (2024). Advancements in immunity and dementia research: highlights from the 2023 AAIC advancements: immunity conference. Alzheimers Dement. 21:e14291. doi: 10.1002/alz.14291
Lana, D., and Giovannini, M. G. (2025). Special issue: recent advances in microglia research. Int. J. Mol. Sci. 26:507. doi: 10.3390/ijms26020507
Le, L., Feidler, A. M., Li, H., Kara-Pabani, K., Lamantia, C., O’Banion, M. K., et al. (2023). Noradrenergic signaling controls Alzheimer’s disease pathology via activation of microglial beta 2 adrenergic receptors. bio Rxiv. doi: 10.1101/2023.12.01.569564
Leng, F., and Edison, P. (2021). Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat. Rev. Neurol. 17, 157–172. doi: 10.1038/s41582-020-00435-y
Li, Y. B., Fu, Q., Guo, M., Du, Y., Chen, Y., and Cheng, Y. (2024). MicroRNAs: pioneering regulators in Alzheimer’s disease pathogenesis, diagnosis, and therapy. Transl. Psychiatry 14:367. doi: 10.1038/s41398-024-03075-8
Li, R. Y., Qin, Q., Yang, H. C., Wang, Y. Y., Mi, Y. X., Yin, Y. S., et al. (2022). TREM2 in the pathogenesis of AD: a lipid metabolism regulator and potential metabolic therapeutic target. Mol. Neurodegener. 17:40. doi: 10.1186/s13024-022-00542-y
Li, X., Shen, N., Zhang, S., Liu, J., Jiang, Q., Liao, M., et al. (2015). CD33 rs3865444 polymorphism contributes to Alzheimer’s disease susceptibility in Chinese, European, and north American populations. Mol. Neurobiol. 52, 414–421. doi: 10.1007/s12035-014-8880-9
Li, W., Wang, X., Sun, L., Yue, L., and Xiao, S. (2024). Correlation between the APOE epsilon4 genotype, lifestyle habits, and cognitive deficits in Chinese adults over 60: a cross-sectional analysis in China. Front. Public Health 12:1417499. doi: 10.3389/fpubh.2024.1417499
Lin, M., Yu, J. X., Zhang, W. X., Lao, F. X., and Huang, H. C. (2024). Roles of TREM2 in the pathological mechanism and the therapeutic strategies of Alzheimer’s disease. J. Prev Alzheimers Dis. 11, 1682–1695. doi: 10.14283/jpad.2024.164
Liu, Y., Cheng, X., Li, H., Hui, S., Zhang, Z., Xiao, Y., et al. (2022). Non-coding RNAs as novel regulators of neuroinflammation in Alzheimer’s disease. Front. Immunol. 13:908076. doi: 10.3389/fimmu.2022.908076
Liu, D., Hsueh, S. C., Tweedie, D., Price, N., Glotfelty, E., Lecca, D., et al. (2024). Chronic inflammation with microglia senescence at basal forebrain: impact on cholinergic deficit in Alzheimer’s brain haemodynamics. Brain Commun 6:fcae204. doi: 10.1093/braincomms/fcae204
Liu, X. H., Liu, X. T., Wu, Y., Li, S. A., Ren, K. D., Cheng, M., et al. (2024). Broadening horizons: exploring the Cathepsin family as therapeutic targets for Alzheimer’s disease. Aging Dis. :01. doi: 10.14336/AD.2024.0456
Liu, W., Taso, O., Wang, R., Bayram, S., Graham, A. C., Garcia-Reitboeck, P., et al. (2020). Trem 2 promotes anti-inflammatory responses in microglia and is suppressed under pro-inflammatory conditions. Hum. Mol. Genet. 29, 3224–3248. doi: 10.1093/hmg/ddaa209
Long, H. Z., Cheng, Y., Zhou, Z. W., Luo, H. Y., Wen, D. D., and Gao, L. C. (2021). PI3K/AKT signal pathway: a target of natural products in the prevention and treatment of Alzheimer’s disease and Parkinson’s disease. Front. Pharmacol. 12:648636. doi: 10.3389/fphar.2021.648636
Long, H. Z., Zhou, Z. W., Cheng, Y., Luo, H. Y., Li, F. J., Xu, S. G., et al. (2022). The role of microglia in Alzheimer’s disease from the perspective of immune inflammation and Iron metabolism. Front. Aging Neurosci. 14:888989. doi: 10.3389/fnagi.2022.888989
Ma, Y., Wang, W., Liu, S., Qiao, X., Xing, Y., Zhou, Q., et al. (2023). Epigenetic regulation of neuroinflammation in Alzheimer’s disease. Cells 13:79. doi: 10.3390/cells13010079
Madhu, L. N., Kodali, M., Upadhya, R., Rao, S., Somayaji, Y., Attaluri, S., et al. (2024). Extracellular vesicles from human-induced pluripotent stem cell-derived neural stem cells alleviate proinflammatory cascades within disease-associated microglia in Alzheimer’s disease. J Extracell Vesicles 13:e12519. doi: 10.1002/jev2.12519
Magno, L., Lessard, C. B., Martins, M., Lang, V., Cruz, P., Asi, Y., et al. (2019). Alzheimer’s disease phospholipase C-gamma-2 (PLCG2) protective variant is a functional hypermorph. Alzheimers Res. Ther. 11:16. doi: 10.1186/s13195-019-0469-0
Maki, T., Sawahata, M., Uta, D., Irie, K., and Kume, T. (2023). Chronic treatment with Abeta 42 with a toxic conformer and LPS induces inflammatory responses in BV-2 microglia with dysregulation of hypoxia-inducible factor expression. Biol. Pharm. Bull. 46, 359–363. doi: 10.1248/bpb.b22-00855
Malik, M., Parikh, I., Vasquez, J. B., Smith, C., Tai, L., Bu, G., et al. (2015). Genetics ignite focus on microglial inflammation in Alzheimer’s disease. Mol. Neurodegener. 10:52. doi: 10.1186/s13024-015-0048-1
Mantzavinos, V., and Alexiou, A. (2017). Biomarkers for Alzheimer’s disease diagnosis. Curr. Alzheimer Res. 14, 1149–1154. doi: 10.2174/1567205014666170203125942
Martins-Ferreira, R., Calafell-Segura, J., Leal, B., Rodriguez-Ubreva, J., Martinez-Saez, E., Mereu, E., et al. (2025). The human microglia atlas (HuMicA) unravels changes in disease-associated microglia subsets across neurodegenerative conditions. Nat. Commun. 16:739. doi: 10.1038/s41467-025-56124-1
Mirarchi, A., Albi, E., and Arcuri, C. (2024). Microglia signatures: a cause or consequence of microglia-related brain disorders? Int. J. Mol. Sci. 25:10951. doi: 10.3390/ijms252010951
Murakawa-Hirachi, T., Mizoguchi, Y., Ohgidani, M., Haraguchi, Y., and Monji, A. (2021). Effect of memantine, an anti-Alzheimer’s drug, on rodent microglial cells in vitro. Sci. Rep. 11:6151. doi: 10.1038/s41598-021-85625-4
Narasimhan, S., Holtzman, D. M., Apostolova, L. G., Cruchaga, C., Masters, C. L., Hardy, J., et al. (2024). Apolipoprotein E in Alzheimer’s disease trajectories and the next-generation clinical care pathway. Nat. Neurosci. 27, 1236–1252. doi: 10.1038/s41593-024-01669-5
Niazi, S. K. (2024). Safety concerns in neurological clinical trials: a challenge that the FDA must resolve. Biomedicines 12:2918. doi: 10.3390/biomedicines12122918
Ohm, M., Hosseini, S., Lonnemann, N., He, W., More, T., Goldmann, O., et al. (2024). The potential therapeutic role of itaconate and mesaconate on the detrimental effects of LPS-induced neuroinflammation in the brain. J. Neuroinflammation 21:207. doi: 10.1186/s12974-024-03188-3
Onuska, K. M. (2020). The dual role of microglia in the progression of Alzheimer’s disease. J. Neurosci. 40, 1608–1610. doi: 10.1523/JNEUROSCI.2594-19.2020
Pallares-Moratalla, C., and Bergers, G. (2024). The ins and outs of microglial cells in brain health and disease. Front. Immunol. 15:1305087. doi: 10.3389/fimmu.2024.1305087
Pocock, J., Vasilopoulou, F., Svensson, E., and Cosker, K. (2024). Microglia and TREM2. Neuropharmacology 257:110020. doi: 10.1016/j.neuropharm.2024.110020
Qu, L., Xu, S., Lan, Z., Fang, S., Xu, Y., and Zhu, X. (2024). Apolipoprotein E in Alzheimer’s disease: focus on synaptic function and therapeutic strategy. Mol. Neurobiol. 62, 3040–3052. doi: 10.1007/s12035-024-04449-1
Rodriguez-Vieitez, E., Kumar, A., Malarte, M. L., Ioannou, K., Rocha, F. M., and Chiotis, K. (2024). Imaging Neuroinflammation: quantification of Astrocytosis in a multitracer PET approach. Methods Mol. Biol. 2785, 195–218. doi: 10.1007/978-1-0716-3774-6_13
Roveta, F., Bonino, L., Piella, E. M., Rainero, I., and Rubino, E. (2024). Neuroinflammatory biomarkers in Alzheimer’s disease: from pathophysiology to clinical implications. Int. J. Mol. Sci. 25:11941. doi: 10.3390/ijms252211941
Sadeghdoust, M., Das, A., and Kaushik, D. K. (2024). Fueling neurodegeneration: metabolic insights into microglia functions. J. Neuroinflammation 21:300. doi: 10.1186/s12974-024-03296-0
Salazar-Londono, S., Silva-Buritica, C., Herrera-Velez, L., and Rosselli, D. (2024). Using routinely collected health data to estimate the prevalence of Alzheimer’s disease and potentially modifiable risk factors in Colombia. Int. J. Geriatr. Psychiatry 39:e70029. doi: 10.1002/gps.70029
Salminen, A. (2024). The role of inhibitory immune checkpoint receptors in the pathogenesis of Alzheimer’s disease. J. Mol. Med. 103, 1–19. doi: 10.1007/s00109-024-02504-x
Sarb, O. F., Sarb, A. D., Iacobescu, M., Vlad, I. M., Milaciu, M. V., Ciurmarnean, L., et al. (2024). From gut to brain: uncovering potential serum biomarkers connecting inflammatory bowel diseases to neurodegenerative diseases. Int. J. Mol. Sci. 25:5676. doi: 10.3390/ijms25115676
Serrano-Pozo, A., Li, Z., Noori, A., Nguyen, H. N., Mezlini, A., Li, L., et al. (2021). Effect of APOE alleles on the glial transcriptome in normal aging and Alzheimer’s disease. Nat Aging 1, 919–931. doi: 10.1038/s43587-021-00123-6
Sethi, P., Bhaskar, R., Singh, K. K., Gupta, S., Han, S. S., Avinash, D., et al. (2024). Exploring advancements in early detection of Alzheimer’s disease with molecular assays and animal models. Ageing Res. Rev. 100:102411. doi: 10.1016/j.arr.2024.102411
Shahidehpour, R. K., Nelson, P. T., Katsumata, Y., and Bachstetter, A. D. (2024). Exploring the link between dystrophic microglia and the spread of Alzheimer’s neuropathology. Brain 148, 89–101. doi: 10.1093/brain/awae258
Sharallah, O. A., Poddar, N. K., and Alwadan, O. A. (2024). Delineation of the role of G6PD in Alzheimer’s disease and potential enhancement through microfluidic and nanoparticle approaches. Ageing Res. Rev. 99:102394. doi: 10.1016/j.arr.2024.102394
Sharma, M., Pal, P., and Gupta, S. K. (2024). Deciphering the role of mi RNAs in Alzheimer’s disease: predictive targeting and pathway modulation – a systematic review. Ageing Res. Rev. 101:102483. doi: 10.1016/j.arr.2024.102483
Shen, Y., Liu, F., and Zhang, M. (2024). Therapeutic potential of plant-derived natural compounds in Alzheimer’s disease: targeting microglia-mediated neuroinflammation. Biomed. Pharmacother. 178:117235. doi: 10.1016/j.biopha.2024.117235
Shen, Z., Yang, X., Lan, Y., and Chen, G. (2024). The neuro-inflammatory microenvironment: an important regulator of stem cell survival in Alzheimer’s disease. J. Alzheimers Dis. 98, 741–754. doi: 10.3233/JAD-231159
Shi, K., Chen, L., Chen, L., Tan, A., Xie, G., Long, Q., et al. (2022). Epimedii folium and Curculiginis Rhizoma ameliorate lipopolysaccharides-induced cognitive impairment by regulating the TREM2 signaling pathway. J. Ethnopharmacol. 284:114766. doi: 10.1016/j.jep.2021.114766
Shi, Q., Gutierrez, R. A., and Bhat, M. A. (2024). Microglia, Trem 2, and neurodegeneration. Neuroscientist. doi: 10.1177/10738584241254118
Shi, J., and Huang, S. (2023). Comparative insight into microglia/macrophages-associated pathways in glioblastoma and Alzheimer’s disease. Int. J. Mol. Sci. 25:16. doi: 10.3390/ijms25010016
Sian-Hulsmann, J., and Riederer, P. (2024). Virus-induced brain pathology and the neuroinflammation-inflammation continuum: the neurochemists view. J. Neural Transm. 131, 1429–1453. doi: 10.1007/s00702-023-02723-5
Smith, D. C., Karahan, H., Wijeratne, H. R. S., Al-Amin, M., McCord, B., Moon, Y., et al. (2022). Deletion of the Alzheimer’s disease risk gene Abi3 locus results in obesity and systemic metabolic disruption in mice. Front. Aging Neurosci. 14:1035572. doi: 10.3389/fnagi.2022.1035572
Sobue, A., Komine, O., Endo, F., Kakimi, C., Miyoshi, Y., Kawade, N., et al. (2024). Microglial cannabinoid receptor type II stimulation improves cognitive impairment and neuroinflammation in Alzheimer’s disease mice by controlling astrocyte activation. Cell Death Dis. 15:858. doi: 10.1038/s41419-024-07249-6
Staley, H. A., Jernigan, J. E., Bolen, M. L., Titus, A. M., Neighbarger, N., Cole, C., et al. (2024). Alzheimer’s disease-associated protective variant Plcg 2-P522R modulates peripheral macrophage function in a sex-dimorphic manner. J. Neuroinflammation 21:280. doi: 10.1186/s12974-024-03271-9
Stamataki, M., Rissiek, B., Magnus, T., and Korbelin, J. (2024). Microglia targeting by adeno-associated viral vectors. Front. Immunol. 15:1425892. doi: 10.3389/fimmu.2024.1425892
Streit, W. J., Phan, L., and Bechmann, I. (2024). Ferroptosis and pathogenesis of neuritic plaques in Alzheimer’s disease. Pharmacol. Rev. 77:100005. doi: 10.1124/pharmrev.123.000823
Sudwarts, A., and Thinakaran, G. (2023). Alzheimer’s genes in microglia: a risk worth investigating. Mol. Neurodegener. 18:90. doi: 10.1186/s13024-023-00679-4
Sun, X. Y., Li, L. J., Dong, Q. X., Zhu, J., Huang, Y. R., Hou, S. J., et al. (2021). Rutin prevents tau pathology and neuroinflammation in a mouse model of Alzheimer’s disease. J. Neuroinflammation 18:131. doi: 10.1186/s12974-021-02182-3
Sun, Z., Zhang, X., So, K. F., Jiang, W., and Chiu, K. (2024). Targeting microglia in Alzheimer’s disease: pathogenesis and potential therapeutic strategies. Biomol. Ther. 14:833. doi: 10.3390/biom14070833
Thakur, S., Dhapola, R., Sarma, P., Medhi, B., and Reddy, D. H. (2023). Neuroinflammation in Alzheimer’s disease: current Progress in molecular signaling and therapeutics. Inflammation 46, 1–17. doi: 10.1007/s10753-022-01721-1
Tournier, B. B., Ceyzeriat, K., Badina, A. M., Gloria, Y., Fall, A. B., Amosse, Q., et al. (2024). Impairment of hippocampal astrocyte-mediated striatal dopamine release and locomotion in Alzheimer’s disease. NeuroImage 298:120778. doi: 10.1016/j.neuroimage.2024.120778
Tsering, W., de la Rosa, A., Ruan, I. Y., Philips, J. L., Bathe, T., Villareal, J. A., et al. (2025). Preferential clustering of microglia and astrocytes around neuritic plaques during progression of Alzheimer’s disease neuropathological changes. J. Neurochem. 169:e16275. doi: 10.1111/jnc.16275
Tucsek, Z., Toth, P., Sosnowska, D., Gautam, T., Mitschelen, M., Koller, A., et al. (2014). Obesity in aging exacerbates blood-brain barrier disruption, neuroinflammation, and oxidative stress in the mouse hippocampus: effects on expression of genes involved in beta-amyloid generation and Alzheimer’s disease. J. Gerontol. A Biol. Sci. Med. Sci. 69, 1212–1226. doi: 10.1093/gerona/glt177
van den Brink, H., Voigt, S., Kozberg, M., and van Etten, E. S. (2024). The role of neuroinflammation in cerebral amyloid angiopathy. EBioMedicine 110:105466. doi: 10.1016/j.ebiom.2024.105466
Versele, R., Sevin, E., Gosselet, F., Fenart, L., and Candela, P. (2022). TNF-alpha and IL-1beta modulate blood-brain barrier permeability and decrease amyloid-beta peptide efflux in a human blood-brain barrier model. Int. J. Mol. Sci. 23:10235. doi: 10.3390/ijms231810235
Walker, D. G., Whetzel, A. M., Serrano, G., Sue, L. I., Beach, T. G., and Lue, L. F. (2015). Association of CD33 polymorphism rs3865444 with Alzheimer’s disease pathology and CD33 expression in human cerebral cortex. Neurobiol. Aging 36, 571–582. doi: 10.1016/j.neurobiolaging.2014.09.023
Wang, D., Chen, F., Han, Z., Yin, Z., Ge, X., and Lei, P. (2021). Relationship between amyloid-beta deposition and blood-brain barrier dysfunction in Alzheimer’s disease. Front. Cell. Neurosci. 15:695479. doi: 10.3389/fncel.2021.695479
Wang, C., Fan, L., Khawaja, R. R., Liu, B., Zhan, L., Kodama, L., et al. (2022). Microglial NF-kappaB drives tau spreading and toxicity in a mouse model of tauopathy. Nat. Commun. 13:1969. doi: 10.1038/s41467-022-29552-6
Wang, H., Li, X., Wang, Q., Ma, J., Gao, X., and Wang, M. (2023). TREM2, microglial and ischemic stroke. J. Neuroimmunol. 381:578108. doi: 10.1016/j.jneuroim.2023.578108
Wang, S. Y., Liu, Y., Li, X. M., Algradi, A. M., Jiang, H., Sun, Y. P., et al. (2021). Discovery of active ingredients targeted TREM2 by SPR biosensor-UPLC/MS recognition system, and investigating the mechanism of anti-neuroinflammatory activity on the lignin-amides from Datura metel seeds. Molecules 26:5946. doi: 10.3390/molecules26195946
Wang, L., Ouyang, D., Li, L., Cao, Y., Wang, Y., Gu, N., et al. (2025). TREM2 affects DAM-like cell transformation in the acute phase of TBI in mice by regulating microglial glycolysis. J. Neuroinflammation 22:6. doi: 10.1186/s12974-025-03337-2
Wang, M., Pan, W., Xu, Y., Zhang, J., Wan, J., and Jiang, H. (2022). Microglia-mediated neuroinflammation: a potential target for the treatment of cardiovascular diseases. J. Inflamm. Res. 15, 3083–3094. doi: 10.2147/JIR.S350109
Wang, Y., Ye, M., Ji, Q., Liu, Q., Xu, X., and Zhan, Y. (2024). The longitudinal trajectory of CSF sTREM2: the Alzheimer’s disease neuroimaging initiative. Alzheimers Res. Ther. 16:138. doi: 10.1186/s13195-024-01506-8
Wang, C., Zong, S., Cui, X., Wang, X., Wu, S., Wang, L., et al. (2023). The effects of microglia-associated neuroinflammation on Alzheimer’s disease. Front. Immunol. 14:1117172. doi: 10.3389/fimmu.2023.1117172
Weng, Y., Chen, N., Zhang, R., He, J., Ding, X., Cheng, G., et al. (2024). An integral blood-brain barrier in adulthood relies on microglia-derived PDGFB. Brain Behav. Immun. 115, 705–717. doi: 10.1016/j.bbi.2023.11.023
Wheeler, K. V., Irimia, A., and Braskie, M. N. (2024). Using neuroimaging to study cerebral amyloid Angiopathy and its relationship to Alzheimer’s disease. J. Alzheimers Dis. 97, 1479–1502. doi: 10.3233/JAD-230553
Woodburn, S. C., Bollinger, J. L., and Wohleb, E. S. (2021). The semantics of microglia activation: neuroinflammation, homeostasis, and stress. J. Neuroinflammation 18:258. doi: 10.1186/s12974-021-02309-6
Wright, B., King, S., and Suphioglu, C. (2024). The importance of phosphoinositide 3-kinase in Neuroinflammation. Int. J. Mol. Sci. 25:11638. doi: 10.3390/ijms252111638
Wu, D. P., Wei, Y. S., Hou, L. X., Du, Y. X., Yan, Q. Q., Liu, L. L., et al. (2025). Circular RNA APP contributes to Alzheimer’s disease pathogenesis by modulating microglial polarization via mi R-1906/CLIC1 axis. Alzheimers Res. Ther. 17:44. doi: 10.1186/s13195-025-01698-7
Wu, Z., Yang, S., Fang, X., Shu, Q., and Chen, Q. (2024). Function and mechanism of TREM2 in bacterial infection. PLoS Pathog. 20:e1011895. doi: 10.1371/journal.ppat.1011895
Yan, H., Wang, W., Cui, T., Shao, Y., Li, M., Fang, L., et al. (2024). Advances in the understanding of the correlation between Neuroinflammation and microglia in Alzheimer’s disease. Immunotargets Ther 13, 287–304. doi: 10.2147/ITT.S455881
Yang, Y., Garcia-Cruzado, M., Zeng, H., Camprubi-Ferrer, L., Bahatyrevich-Kharitonik, B., Bachiller, S., et al. (2023). LPS priming before plaque deposition impedes microglial activation and restrains Abeta pathology in the 5xFAD mouse model of Alzheimer’s disease. Brain Behav. Immun. 113, 228–247. doi: 10.1016/j.bbi.2023.07.006
Yang, G., Tong, Y., Wang, X., Zhao, C., Ba, Z., Ahelijiang, R., et al. (2023). Guizhi Fuling capsule relieves memory deficits by inhibition of microglial neuroinflammation through blocking JAK2/STAT3 pathway in presenilin1/2 conditional double knockout mice. Front. Immunol. 14:1185570. doi: 10.3389/fimmu.2023.1185570
Yao, H., Coppola, K., Schweig, J. E., Crawford, F., Mullan, M., and Paris, D. (2019). Distinct signaling pathways regulate TREM2 phagocytic and NFkappaB antagonistic activities. Front. Cell. Neurosci. 13:457. doi: 10.3389/fncel.2019.00457
Yu, Y., Lv, J., Ma, D., Han, Y., Zhang, Y., Wang, S., et al. (2024). Microglial ApoD-induced NLRC4 inflammasome activation promotes Alzheimer’s disease progression. Animal Model Exp Med. doi: 10.1002/ame2.12361
Zhang, Z., Li, M., Li, X., Feng, Z., Luo, G., Wang, Y., et al. (2024). Glutamine metabolism modulates microglial NLRP3 inflammasome activity through mitophagy in Alzheimer’s disease. J. Neuroinflammation 21:261. doi: 10.1186/s12974-024-03254-w
Zhang, W., Liu, T., Li, J., Singh, J., Chan, A., Islam, A., et al. (2024). Decreased extrasynaptic delta-GABA(a) receptors in PNN-associated parvalbumin interneurons correlates with anxiety in APP and tau mouse models of Alzheimer’s disease. Br. J. Pharmacol. 181, 3944–3975. doi: 10.1111/bph.16441
Zhang, Q., Yang, G., Luo, Y., Jiang, L., Chi, H., and Tian, G. (2024). Neuroinflammation in Alzheimer’s disease: insights from peripheral immune cells. Immun. Ageing 21:38. doi: 10.1186/s12979-024-00445-0
Zhang, L., Yao, Q., Hu, J., Qiu, B., Xiao, Y., Zhang, Q., et al. (2024). Hotspots and trends of microglia in Alzheimer’s disease: a bibliometric analysis during 2000-2022. Eur. J. Med. Res. 29:75. doi: 10.1186/s40001-023-01602-9
Zhang, J., Zhang, Y., Liu, L., Zhang, M., Zhang, X., Deng, J., et al. (2025). Chemerin-9 is neuroprotective in APP/PS1 transgenic mice by inhibiting NLRP3 inflammasome and promoting microglial clearance of Abeta. J. Neuroinflammation 22:5. doi: 10.1186/s12974-024-03325-y
Zhang, J., Zhang, Y., Wang, J., Xia, Y., Zhang, J., and Chen, L. (2024). Recent advances in Alzheimer’s disease: mechanisms, clinical trials and new drug development strategies. Signal Transduct. Target. Ther. 9:211. doi: 10.1038/s41392-024-01911-3
Zhang, G., Zhao, A., Zhang, X., Zeng, M., Wei, H., Yan, X., et al. (2024). Glycolytic reprogramming in microglia: a potential therapeutic target for ischemic stroke. Cell. Signal. 124:111466. doi: 10.1016/j.cellsig.2024.111466
Zhong, R., Xu, Y., Williams, J. W., and Li, L. (2024). Loss of TREM2 diminishes CAA despite an overall increase of amyloid load in Tg-SwDI mice. Alzheimers Dement. 20, 7595–7612. doi: 10.1002/alz.14222
Zhou, X., Cao, H., Jiang, Y., Chen, Y., Zhong, H., Fu, W. Y., et al. (2024). Transethnic analysis identifies SORL1 variants and haplotypes protective against Alzheimer’s disease. Alzheimers Dement. 21:e14214. doi: 10.1002/alz.14214
Zhou, R., Chen, S. H., Zhao, Z., Tu, D., Song, S., Wang, Y., et al. (2023). Complement C3 enhances LPS-elicited Neuroinflammation and neurodegeneration via the mac 1/NOX2 pathway. Mol. Neurobiol. 60, 5167–5183. doi: 10.1007/s12035-023-03393-w
Zhou, L., Wang, Y., Xu, Y., Zhang, Y., and Zhu, C. (2024). A comprehensive review of AAV-mediated strategies targeting microglia for therapeutic intervention of neurodegenerative diseases. J. Neuroinflammation 21:232. doi: 10.1186/s12974-024-03232-2
Zou, J., McNair, E., DeCastro, S., Lyons, S. P., Mordant, A., Herring, L. E., et al. (2024). Microglia either promote or restrain TRAIL-mediated excitotoxicity caused by Abeta(1-42) oligomers. J. Neuroinflammation 21:215. doi: 10.1186/s12974-024-03208-2
Zuppe, H., and Reed, E. (2024). Common cytokine receptor gamma chain family cytokines activate MAPK, PI3K, and JAK/STAT pathways in microglia to influence Alzheimer’s disease. Front. Mol. Neurosci. 17:1441691. doi: 10.3389/fnmol.2024.1441691
Keywords: Alzheimer’s disease, microglia, neuroinflammation, amyloid-beta clearance, genetic variants
Citation: He C, Chen B, Yang H and Zhou X (2025) The dual role of microglia in Alzheimer’s disease: from immune regulation to pathological progression. Front. Aging Neurosci. 17:1554398. doi: 10.3389/fnagi.2025.1554398
Edited by:
Tormod Fladby, University of Oslo, NorwayReviewed by:
Yukari Shigemoto-mogami, National Institute of Health Sciences (NIHS), JapanCopyright © 2025 He, Chen, Yang and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoqing Zhou, MjIzMzA5MzAxNUBxcS5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.