 Marta Weinstock
Marta Weinstock- Institute for Drug Research, School of Pharmacy, Faculty of Medicine, The Hebrew University, Jerusalem, Israel
Alzheimer’s disease (AD) is the most common form of dementia. Mutations in genes and precursors of β amyloid (Aβ) are found in the familial form of the disease. This led to the evaluation of seven monoclonal antibodies against Aβ in subjects with AD, two of which were approved for use by the FDA. They caused only a small improvement in cognitive function, probably because they were given to those with much more prevalent sporadic forms of dementia. They also have potentially serious adverse effects. Oxidative stress and elevated pro-inflammatory cytokines are present in all subjects with AD and are well correlated with the degree of memory impairment. Drugs that affect these processes include TNFα blocking antibodies and MAPK p38 inhibitors that reduce cognitive impairment when given for other inflammatory conditions. However, their adverse effects and inability to penetrate the brain preclude their use for dementia. Rosiglitazone is used to treat diabetes, a risk factor for AD, but failed in a clinical trial because it was given to subjects that already had dementia. Ladostigil reduces oxidative stress and suppresses the release of pro-inflammatory cytokines from activated microglia without blocking their effects. Chronic oral administration to aging rats prevented the decline in memory and suppressed overexpression of genes adversely affecting synaptic function in relevant brain regions. In a phase 2 trial, ladostigil reduced the decline in short-term memory and in whole brain and hippocampal volumes in human subjects with mild cognitive impairment and had no more adverse effects than placebo.
Introduction
Dementia is one of the leading causes of morbidity and mortality worldwide that exerts an immense, negative effect on the quality of life of the subjects, their families and on health care systems. Globally, the total number of people with this condition is expected to reach almost 153 million in 2050 (Li et al., 2022). Alzheimer’s disease (AD), the most common cause of dementia (Hyman et al., 2012), is a progressive neurodegenerative disorder, characterized by the degeneration of cholinergic neurons in the nucleus basalis, and the presence of extracellular plaques beta amyloid (Aβ) and intracellular neurofibrillary tangles composed of phosphorylated tau. AD presents with an impairment in early episodic memory, followed by a gradual and progressive deterioration in cognition and behavior. The characteristic features of the familial form (FAD) were originally described by Alois Alzheimer in 1906. It has an age of onset between 30 and 60 years and is associated with autosomal dominant inheritance of mutations in the amyloid precursor protein, presenilin 1 (PSEN1) or PSEN2 genes (Jagust, 2018). Either PSEN1 or PSEN2 can be the catalytic subunit of γ-secretase, that generates Aβ from amyloid precursor protein. Aβ has been implicated in synaptic dysfunction, disruption of neural connectivity and neuronal death in a brain region-specific manner (Lacalle-Aurioles and Iturria-Medina, 2023).
In FAD, Aβ-containing plaques appear at least 20 years before any signs of memory impairment (Bateman et al., 2012). While prevention of Aβ formation could provide a treatment option for FAD if started early enough, it represents only about 1% of subjects with AD (Hippius and Neundorfer, 2003). The rest have the sporadic form of AD (SAD), with an age of onset of more than 65 years. Their brains also have Aβ-containing plaques, but so do those of healthy, older people with no overt signs of dementia (Rodrigue et al., 2012). Since no correlation was found between the number of Aβ plaques and the degree of cognitive impairment in individuals with SAD (Bennett et al., 2012), the original hypothesis was changed and soluble oligomers of Aβ proposed as the cause of neurodegeneration (Shankar et al., 2008).
Mild cognitive impairment
Petersen originally described mild cognitive impairment in Petersen (2004) and applies to the earliest symptomatic stage of cognitive impairment in which either a single or several cognitive domains are compromised to a mild extent, but functional ability is preserved. However, no differences were found in MCI in subjects with or without evidence of Aβ after applying several different psychological different tests (Insel et al., 2018).
Acetylcholinesterase inhibitors
Acetylcholinesterase (AChE)-positive neurons project diffusely to the cortex from the cholinergic nucleus basalis magnocellularis of Meynert, modulating cortical processing and responses to new and relevant stimuli. The findings that in AD there is a correlation between loss of neurons projecting from the nucleus basalis and the decline in mental status (Whitehouse et al., 1982) led to introduction of AChE inhibitors to preserve acetylcholine levels. Tacrine was the first AChE inhibitor used for the treatment of AD but was withdrawn because of hepatotoxicity (Watkins et al., 1994). Of several others, the two AChE inhibitors most frequently prescribed are donepezil and rivastigmine.
Donepezil, is a selective AChE inhibitor approved in 1996 for mild to moderate AD when administered at a daily dose of 5 mg during the first month of treatment and increased up to 10 mg. The result of a meta-analysis of 5 studies in a total of 1,130 participants showed that 10 mg was associated with a better outcome for cognitive function than placebo after 26 weeks, Alzheimer’s Disease Assessment Scale-Cognitive (ADAS-Cog, range 0 to 70) was −2.67 points. The incidence of adverse effects, insomnia, diarrhea, vomiting, anorexia, and muscle cramps was higher at 12 weeks, but declined by 26 weeks (Birks and Harvey, 2018).
Rivastigmine is a slowly reversible inhibitor of AChE and Butyrylcholinesterase (BuChE). BuChE is distributed in neurons, glia and endothelial cells and BuChE-positive neurons that project to the frontal cortex may play a role in attention and executive function (Bullock and Lane, 2007). While AChE declines in AD, with a loss of cholinergic neurons, BuChE activity progressively increases as the severity of dementia advances. Rivastigmine interacts preferentially with the G1 form of the enzyme found in high levels in the brains of patients with AD. At doses of 6 to 12 mg/day, rivastigmine produced a significant dose related improvement of ≥4-points in the ADAS-cog subscale at 26 weeks after treatment compared to placebo. Adverse effects were nausea and vomiting that were reduced by dose titration (Spencer and Noble, 1998). Rivastigmine is metabolized by its target enzyme and not by CRP450 in the liver. Thus, it can safely be given with the variety of medications used to treat elderly subjects. Since rivastigmine causes less inhibition of AChE in the striatum than in the cortex and hippocampus, it can also be administered to patients with dementia and Parkinson’s disease. None of the AChE inhibitors affect the underlying causes of cholinergic neuron deterioration, thus their effect is only seen as long as acetylcholine is still being released.
N-methyl-d-aspartate receptor antagonists: memantine
Glutamate is the major excitatory neurotransmitter in the brain that acts on several synaptic receptors including N-methyl-d-aspartate (NMDAR). This receptor plays a fundamental role in synaptic plasticity, and the underlying molecular mechanisms of learning and memory. Activation of NMDARs is also important for the survival of neurons apoptosis (Collingridge and Singer, 1990). However, activation of extra-synaptic NMDARs by the spillover of glutamate from astrocytes or presynaptic terminals results in prolonged Ca2+ influx into the postsynaptic neuron, loss of synaptic function and neuronal cell death. This correlates with the decline in cognition and the development of dementia (Hardingham and Bading, 2010).
The observation that memantine was able to suppress signaling through these extra-synaptic receptors resulted in its approval by the FDA for the treatment of AD. The results of a large number of placebo controlled clinical trials of memantine, summarized in two mega analyses indicate that memantine improves cognitive function, AD-associated behavioral disturbances and activities of daily living in subjects with moderate to severe dementia. However, these effect sizes were small (SMD = −0.09 to −0.27) but adverse effects were mild (Matsunaga et al., 2015). There was no clinically significant benefit from the addition of AChE inhibitors like donepezil (McShane et al., 2019).
Secretase inhibitors
During the last decade, the pharmaceutical industry has concentrated its efforts to affect the processes leading to neurodegeneration by developing drugs to decrease Aβ. γ-secretase is a multi-subunit protease that was identified as responsible for the generation of Aβ, and thus considered a prime therapeutic target in AD (Shoji et al., 1992). This led to the development of γ-secretase inhibitors like semagacestat to inhibit the formation of Aβ. However, a phase 3 trial in patients with mild to moderate AD was prematurely stopped because the drug actually worsened several measures of cognitive function (Doody et al., 2013). Like other γ-secretase inhibitors, avagacestat and tarenflurbil (Penninkilampi et al., 2016), semagacestat caused serious adverse effects, including cancer, skin related disorders, hypersensitivity reactions, increase in infections and renal failure (Henley et al., 2014).
β-secretase inhibitors also prevent formation of Aβ from amyloid precursor protein and their adverse effects are less serious than those of γ-secretase inhibitors. However, verubecestat, atabaques and lanabecestat (all worsened cognitive function in subjects with mild–moderate AD) (Egan et al., 2018; Novak et al., 2020; Wessels et al., 2020). Verubecestat also increased the rate of decline of hippocampal volume, compared to placebo.
Antibodies against Aβ
Seven injectable, monoclonal antibodies (Abs) have been prepared to remove Aβ fibrils or plaques (Alves et al., 2023). Positron emission tomography scans for Aβ confirmed its removal by the Abs but the effect on memory decline in patients with mild to moderate degrees of AD of aducanumab, solenazumab and bapineuzumab, which bind to Aβ plaques, was barely different from that of placebo (Mullard, 2019; Mahase, 2021; Mahase, 2022). Lecanemab binds to soluble Aβ protofibrils while donanemab binds to insoluble, modified, N-terminal truncated form of β-amyloid present only in amyloid plaques. They were approved by the FDA after two large placebo-controlled trials in more than 800 subjects. The Clinical Dementia Rating Sum of Boxes is a continuous measure of dementia severity and ranges from 0 to 18 and is suitable for the earliest stages of AD. The difference in this measure between lecanemab and placebo, the primary end point in the trial, was only 0.45 points (van Dyck et al., 2023) and for donanemab, 0.7 points (Sims et al., 2023).
Like all the Abs developed so far, lecanemab and donanemab cause various degrees of magnetic resonance imaging-detectable amyloid-related imaging abnormalities, such as cerebral edema or hemorrhage in addition to ventricular enlargement ranging from 23 to 57% (Alves et al., 2023). Recently, the FDA has approved injection once a month of donanemab after a new trial but the company that who developed it warned of potential adverse in some subjects that include headache, dizziness, nausea, difficulty walking, confusion, vision changes and seizures.
Regulatory agencies other than the FDA have conceded that any cognitive improvement by these agents is well below a minimally, clinically important effect (Sperling et al., 2023). Cerebral bleeding proscribes their use in patients on anti-coagulants and those with the ApoE4 gene, a strong risk factor for AD (Mahley, 2016). Furthermore, their high cost, coupled with the need for administration by intravenous injection, once or twice a month, make them unavailable to most subjects with AD. More consideration should be given to other causes of neurodegeneration in order to develop safer, cheaper treatments to prevent the development of AD, by giving them orally to subjects with MCI, the earliest prodromal stage of AD, mild cognitive impairment.
Other causes of neurodegeneration
Neuronal damage leading to cognitive impairment can be produced by a combination of environmental and genetic factors. These include a high body mass index, diabetes, insulin resistance, hypertension and dyslipidemia disorders (Verdile et al., 2015). Cognitive impairment can occur in diabetic subjects with chronic inflammation through disruption of the blood brain barrier and entry of macrophages (Varatharaj and Galea, 2017). The APOE gene encodes a protein involved in lipid and cholesterol binding and transport. The APOE 4 allele is an important risk factor for SAD (Strittmatter et al., 1993) and subjects carrying two copies of this gene have a 15-fold likelihood of developing SAD, and at an earlier than those lacking the gene (Altmann et al., 2014; Mahley, 2016).
Both FAD and SAD have abnormalities in the brain circulation (Cortes-Canteli and Iadecola, 2020), together with mitochondrial dysfunction (de la Monte and Tong, 2014; Grimm et al., 2016), which results in the formation of reactive oxygen species causing damage to lipids and proteins (Cai and Tammineni, 2017). Cognitive status was found to be negatively correlated with the level of oxidation of lipids and proteins in the frontal cortex of subjects with SAD (Ansari and Scheff, 2010). This was accompanied by activation of astrocytes and microglia and the release of pro-inflammatory cytokines, particularly TNFα (Fischer and Maier, 2015) and IL-1β (Griffin and Mrak, 2002). The cytokines also produce reactive oxygen and nitrogen species further exacerbating neurodegeneration (Smith et al., 2012). Oxidative stress (Butterfield, 2021) and elevated pro-inflammatory cytokines (Romero-Sevilla et al., 2022) are present in the brains of subjects with MCI.
Role of activated microglia and astrocytes in neuronal damage
Damage-inducing ligands released from injured cells act on receptors in microglia to produce an intracellular signaling cascade via adaptor proteins, like Myeloid differentiation primary response 88 and Interleukin-1 receptor (IL-1R) associated kinase. These activate a series of mitogen-activated protein kinases (MAPKs), extracellular signal-regulated kinase, c-Jun N-terminal kinase, p38 MAPK and the NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) inflammasome (Blevins et al., 2022; Holbrook et al., 2021), through dual phosphorylation on Thr and Tyr residues within a conserved Thr-X-Tyr motif (Cargnello and Roux, 2011). Among these kinases, the p38 MAPK pathway is considered as the pivotal regulator of inflammation (Rivera-Cervantes et al., 2015). IL-1β is also released through the activation of NLRP3. It increases nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) (Kelly et al., 2003) that promotes cytokine secretion after its translocation to the nucleus (Barnes and Karin, 1997). NF-κB is strongly associated with age in mice and humans (Abbasi et al., 2015) and is also increased in the brains of subjects with neurodegenerative diseases (Granic et al., 2009; Kempuraj et al., 2016).
MAP kinases control the expression and release of inflammatory genes and cytokines in microglia and other immune cells by transcription factors like early growth response protein (EGR1) (Friedle et al., 2011). p38 MAPK activity is elevated in neurons and glial cells in the hippocampus and cortex in patients with AD (Hensley et al., 1999), together with MAPK kinase 6, an upstream activator of p38 MAPK (Zhu et al., 2001). p38 MAPK stimulates pro-apoptotic signaling pathways (Kheiri et al., 2018) and promotes excitotoxicity (Rivera-Cervantes et al., 2015). The expression of p38α MAPK in neurons is associated with the formation of Aβ, inflammation, and tau-induced synaptic dysfunction (Gee et al., 2020).
TNF alpha induced protein 3 (TNFAIP3) is a potent regulator of ubiquitin dependent signals and of immune homeostasis. It is expressed in microglia and other cell types in which it acts as a negative feedback regulator of inflammation (Malynn and Ma, 2019). By controlling ubiquitination of intracellular regulating proteins like Inhibitor of IκB kinase gamma, TNFAIP3 decreases the release of NF-κB from IκBα and its subsequent translocation to the nucleus. It also suppresses proteins like TRAF6, thereby reducing activation both of MAPKs and the NLRP3 inflammasome (Voet et al., 2018) (Figure 1).
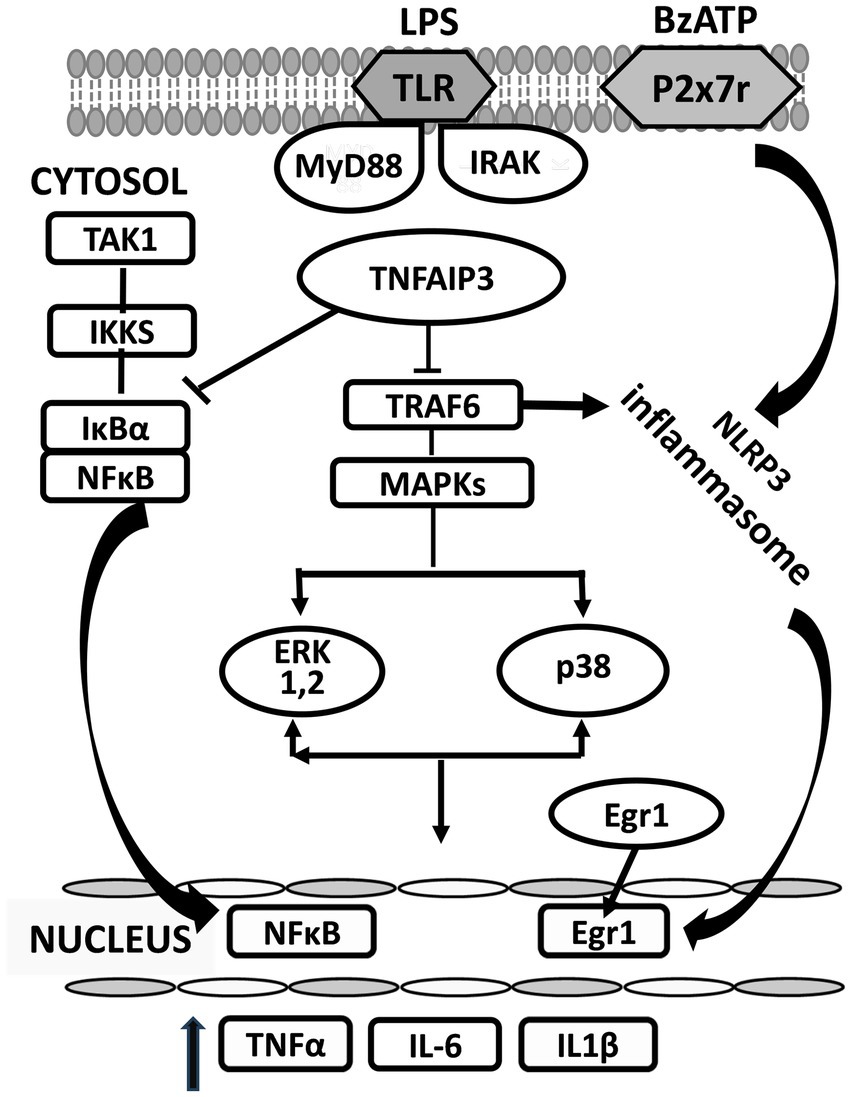
Figure 1. Changes in proteins induced in the cytosol of microglia activated by BzATP and lipopolysaccharide (LPS). LPS (lipopolysaccharide); BzATP (2’-3’-O-(4-benzoyl benzoyl) adenosine 5’-triphosphate); TLR (Toll receptor); MYD88 (myeloid differentiation primary response 88); IRAK (interleukin-1 receptor associated kinase); MAPK (mitogen-activated protein kinase); ERK (extracellular signal-regulated kinase). MyD88 and IRAK are downstream members of the Toll-like receptor (TLR) inflammatory signaling pathway that leads to several functional outputs, including the activation of nuclear factor-kappa B (NFκB), and the release of proinflammatory cytokines. →, activates; ⊥, inhibits.
Treatments that reduce the effect of pro-inflammatory cytokines
TNFα inhibitors
Eternacept, infliximab and adalimumab are three antibodies that bind TNFα and block its actions. They were developed for the treatment of rheumatoid arthritis, ulcerative colitis and Crohn’s disease and can produce a dramatic improvement in these conditions. Although they do not enter the brain, they have been reported to reduce the development of cognitive impairment when given for a period of time to older subjects (Torres-Acosta et al., 2020). This prompted Tobinick et al. (2006) to administer Eternacept by peri spinal injection to a subject with SAD. It produced an immediate and sustained improvement in cognitive function indicating a role for brain TNFα in AD. However, when given to treat peripheral inflammatory conditions TNFα blockers can cause infections, especially reactivation of latent tuberculosis, serious allergic reactions, lymphomas, congestive heart failure and demyelinating disease (Allez et al., 2010; Bongartz et al., 2006). Therefore, there is a need for safer molecules that can prevent oxidative stress and excess cytokine release but do not interfere with their essential activities.
MAPK p38 inhibitors
An excess release of pro-inflammatory cytokines causing organ damage occurs in several other diseases including, cancer (Denny, 2022), cardiovascular disorders (Papaconstantinou, 2019) and lung disease (Breyer et al., 2012). Recognition of the role of p38 MAPK in propagating inflammation led to synthesis and evaluation of p38 MAPK inhibitors for the treatment of such conditions (Bivona et al., 2023). They were shown to alleviate some of them in experimental disease models (Cohen et al., 2009; Marber et al., 2011). However, when assessed in humans they were withdrawn, either because of lack of sufficient efficacy (Damjanov et al., 2009; Watz et al., 2014), or because of adverse effects, like maculo-papular rash, stomatitis and severe headache (Goldman et al., 2018). Moreover, many of these drugs are unable to enter the brain, or avoid exclusion by p-glycoprotein (Lee and Kim, 2017) which has precluded their use in subjects with AD. Neflamapimod, given at a dose of 40 mg twice daily, lowered cerebrospinal fluid biomarkers of synaptic dysfunction but did not improve episodic memory in patients with mild AD. This trial may have failed because the dose was too low or there were too few subjects in the trial (Prins et al., 2021).
The peroxisome proliferator-activated receptor γ agonists: rosiglitazone
The PPARγ receptor acts as a master regulator of adipocyte differentiation and plays an important role in lipid metabolism and glucose homeostasis. Of the various synthetic ligands of this receptor, rosiglitazone has the highest bioavailability and fewest side effects (Lebovitz, 2002). Rosiglitazone can inhibit the release of proinflammatory cytokines TNFα, IL-6 and IL-1β from lipopolysaccharide-activated RAW264.7 cells (Zhou et al., 2021) and the expression of IL-6 and other inflammatory genes in the cortex of mice after controlled cortical impact (Yi et al., 2008). In diabetic subjects, rosiglitazone (4 mg/day) decreased the levels of NF-κB in the nuclei of circulating mononuclear cells (Mohanty et al., 2004), consistent anti-inflammatory effect. It prevented learning and memory deficits induced in rats by a high fat diet by correcting peripheral insulin resistance (Pathan et al., 2008). Thus, it was considered to have the greatest likelihood of reducing the development of SAD when given to diabetic subjects (Akimoto et al., 2020), because it increases their insulin sensitivity and decrease hepatic triglyceride content (Mayerson et al., 2002). In a phase 2 trial of rosiglitazone in patients with mild-to-moderate AD, a beneficial effect was found on cognition in APOE4-ve individuals (Risner et al., 2006). However, in a larger phase 3 trial of 24 weeks duration no difference was found from placebo in the Alzheimer’s disease assessment scale in those with or without an ApoE4 gene with doses of two and eight mg of Rosiglitazone (Gold et al., 2010). This is not surprising if any potential therapeutic effect on cognitive symptoms results from a reduction in the sequelae of diabetes and not from any direct effect of the drug in the brain. It may have succeeded despite its poor brain penetrability if had it been given at the stage of MCI for long enough to enable its effect on peripheral inflammation and insulin resistance to influence a reduction in memory decline. However, the significant levels of cardiotoxicity reported in subjects receiving the drug could outweigh any potential benefits (Nathan, 2007).
In developing a drug for AD, the major focus should be to prevent the pathogenic processes in the brain leading to cognitive impairment and dementia by giving it at the earliest signs of memory loss, amnestic MCI. Patient identification for initiation of treatment and its successful outcome must depend on clinical and biomarker-based measures that can differentiate those that will develop AD within 2–3 years from those who do not. We highlight our experience with ladostigil as a potential candidate.
Ladostigil (SPE 100)
Ladostigil, 6-(N- ethyl, N- methyl carbamyloxy)-N propargyl-1(R)-aminoindan hemitartrate is a small molecule that readily penetrates the brain after oral administration and was originally designed to treat subjects with AD. The carbamate moiety of rivastigmine was introduced into rasagiline, a selective monoamine oxidase B (MAO-B) inhibitor to provide AChE inhibitory activity. It was hypothesized that MAO-B inhibition would reduce oxidative stress resulting from the metabolism of biogenic amines and improve apathy and depression, characteristic of subjects with AD (Weinstock et al., 2000). However, ladostigil was found to be a much weaker inhibitor of AChE and MAO-B enzymes in vitro than either rivastigmine or rasagiline, respectively (Sterling et al., 2002; Weinstock and Groner, 2008). Nevertheless, chronic administration to rats inhibited both enzymes through the formation of active metabolites. Since the results of a phase 2 trial in subjects with AD that received 80 mg, twice daily did not show any added benefit from MAO-B inhibition over rivastigmine alone further development was discontinued.
At a concentration too low to inhibit AChE or MAO-B, ladostigil decreased the nuclear translocation of NF-κB and phosphorylation of extracellular signal-regulated kinase and p38 MAPK and downregulated gene expression of TNFα, IL-6 and IL-1β in microglia activated by lipopolysaccharide (Panarsky et al., 2012). In SH-SY5Y neuroblastoma cells, ladostigil prevented the fall in the mitochondrial potential resulting from oxidative stress by delaying the opening of voltage-dependent anion channels (Maruyama et al., 2003). Oral administration of ladostigil (1 mg/kg/day) for 6 months to 16-month-old rats that did not inhibit either AChE or MAO-B prevented the decline in memory and increase in activation of astrocytes and microglia in selected regions of the hippocampus and other brain areas involved in the control of memory (Weinstock et al., 2013). Ladostigil also decreased the overexpression of genes encoding pro-inflammatory cytokines, regulating calcium homeostasis, ion channels and those adversely affecting synaptic function, while increasing expression of genes providing neurotrophic support in brain regions associated with learning and memory in the aged rats (Linial et al., 2020). Ladostigil had no effect on the astrocyte and microglial immunoreactivity in the same brain regions in young rats.
In search of the mechanism of action, primary cultures of mouse microglia were activated by a combination of 2′-3′-O-(4-benzoyl benzoyl) adenosine 5′-triphosphate, an agonist of P2x7 receptors and lipopolysaccharide. Ladostigil (0.01–1 nM), concentrations compatible with those found in the cerebral cortex of aging rats in which it prevented development of memory deficits, decreased secretion of IL1β and IL-6 proteins by at least 50% (Reichert et al., 2024). RNA sequential analysis performed on activated microglia showed that ladostigil significantly reduced the transition of EGR1 to the nucleus, while increasing levels of TNFaIP3 in the microglia cytoplasm (Reichert et al., 2024). Restoration to normal by ladostigil of the aberrant signaling of these proteins could explain how it reduced the release of pro-inflammatory cytokines and prevented the morphological and inflammatory changes in brain regions of aging rats (Linial et al., 2020; Shoham et al., 2019) (Figure 2).
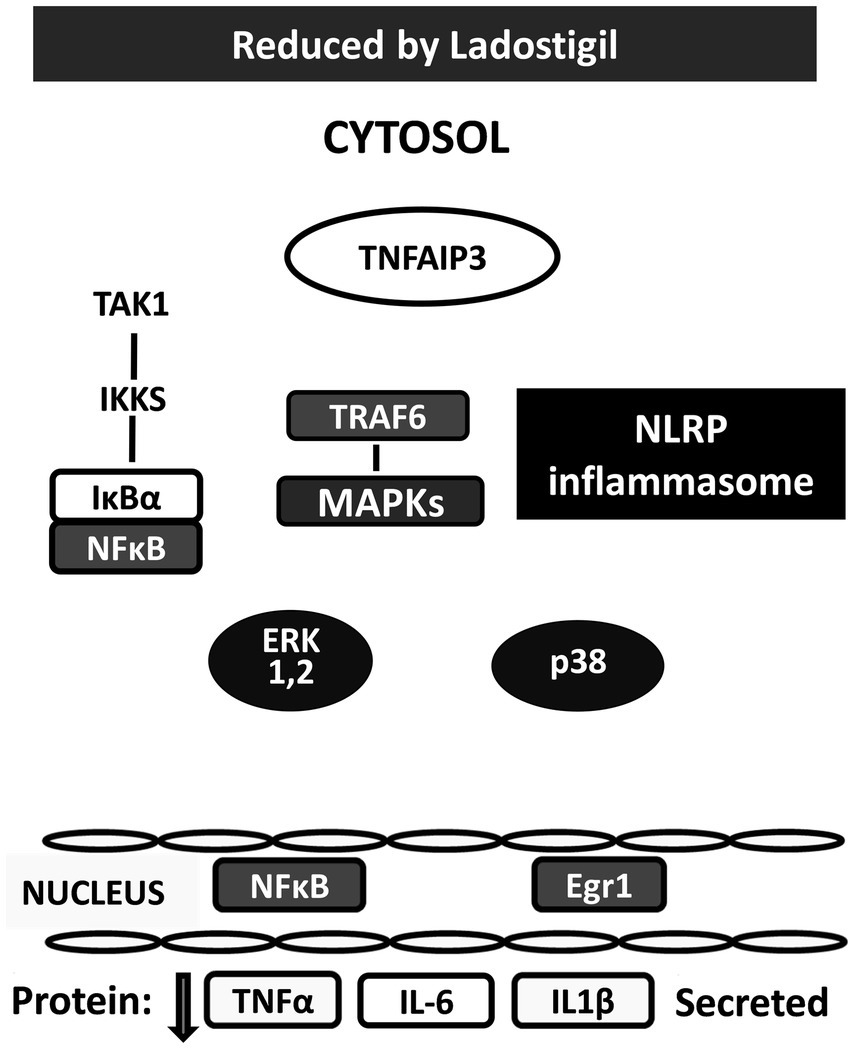
Figure 2. Changes produced by ladostigil in proteins in cytosol and nucleus of microglia activated by BzATP and LPS. Legend as in Figure 1. White colored shapes indicate proteins up-regulated by ladostigil; black colored shapes are proteins downregulated by ladostigil.
In 2015, a phase 2 trial of ladostigil (10 mg/day) was initiated in subjects with MCI. Unlike the trials with Abs against Aβ, the end point then decreed by the European regulatory authorities was per cent of subjects that converted to AD. Based on previous reports that about 15% of patients would progress to dementia per year (Davatzikos et al., 2011), 100 subjects were included in each drug and placebo group. Since about 30% of the subjects in both groups withdrew from the trial in the course of 3 years and the actual conversion rate in those receiving placebo group was only 7% per year, the study was underpowered because there were also subjects insufficiently impaired to convert within 3 years (Schneider et al., 2019). Moreover, once they converted, they were lost to follow up and all measures. The remaining subjects given ladostigil had a significantly lower mini-mental and higher Schelten’s score (a measure of medial temporal lobe atrophy) than those on placebo. Among subjects without an ApoE4 gene, those in the placebo group progressed at twice the rate of ladostigil-treated group (p = 0.028). The age of onset of SAD is younger in ApoE4 carriers (Mahley, 2016). However, nine of them given ladostigil that converted to AD were 6 years older than those receiving placebo and also had a higher CDR-SOB score. When examined in all subjects below 74 years of age, to eliminate this age difference, the rate of decline in the Rey Auditory Verbal Learning Test was significantly lower in those given ladostigil, than in the placebo group (Figure 3).
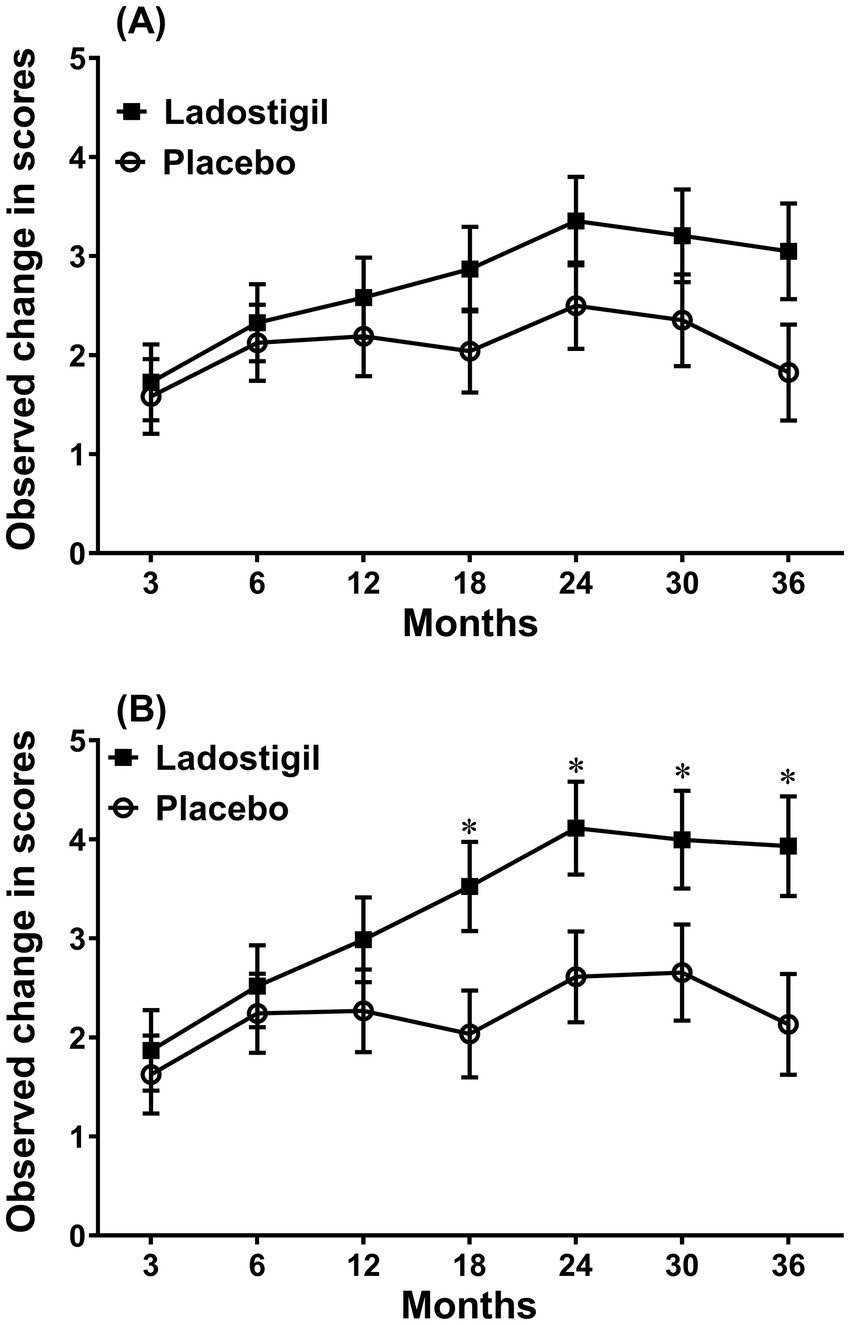
Figure 3. Comparison of the change in score of Rey’s verbal learning delayed recall in subjects that received ladostigil or placebo. (A) All subjects. (B) Subjects less than 74 years of age. Significant difference between groups * p < 0.05. Taken from data in Schneider et al. (2019).
Unlike monoclonal Abs against Aβ, ladostigil significantly reduced the decline in whole brain and hippocampal volumes. Furthermore, adverse effects were mild and did not differ from those in the placebo group. A new study must consider more recent findings that not all aspects of memory decline are faster in those with MCI than in healthy elderly subjects and use appropriate tests to detect this (Grande et al., 2018). They should also include more accurate measures of magnetic resonance imaging in relevant brain regions and other biomarkers of inflammation and oxidative stress appropriate for MCI subjects.
Discussion
Alzheimer’s disease is a complex disorder with several risk factors of which the greatest is advancing age. A minority of subjects have genetic abnormalities in the processing of amyloid precursor protein and generation of Aβ, that occur 20 years before appearance of symptoms. The more prevalent SAD is associated with the presence of the ApoE4 gene and with other pathologies including metabolic syndrome, insulin resistance and cardiovascular disease. The recognition that mitochondrial dysfunction and oxidative stress accompanied by excess glial activation correlate better with neuronal damage and memory loss than the presence of Aβ in subjects with SAD has led to the development and evaluation of other medications to address these factors. However, so far, either they were unable to reduce deterioration in cognitive function or their development was discontinued because of serious adverse effects. To be more effective in preventing loss of memory the drugs should be given at the stage of MCI and appropriate tests applied rather than those used in previous studies developed for subjects with AD. Their cost should not prevent them from being readily available to all who need them, and they should be administered orally, reach the brain, and be devoid of adverse effects.
Author contributions
MW: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
Aβ, β amyloid; Abs, Antibodies; AChE, Acetylcholinesterase; AD, Alzheimer’s disease; ADAS-Cog, Alzheimer’s Disease Assessment Scale-Cognitive; BuChE, Butyrylcholinesterase; EGR1, Early growth response protein1; FAD, Familial form AD; MAO-B, Monoamine oxidase B; MAPK, Mitogen-activated protein kinase; MCI, Mild cognitive impairment; NF-Κb, Nuclear factor kappa-light-chain-enhancer of activated B cells; NMDAR, N-methyl-d-aspartate; PSEN, Presenilin; SAD, Sporadic form of AD; TNFAIP3, TNF alpha induced protein 3.
References
Abbasi, A., Forsberg, K., and Bischof, F. (2015). The role of the ubiquitin-editing enzyme A20 in diseases of the central nervous system and other pathological processes. Front. Mol. Neurosci. 8:21. doi: 10.3389/fnmol.2015.00021
Akimoto, H., Negishi, A., Oshima, S., Wakiyama, H., Okita, M., Horii, N., et al. (2020). Antidiabetic drugs for the risk of Alzheimer disease in patients with type 2 DM using FAERS. Am. J. Alzheimers Dis. Other Demen. 35:1533317519899546. doi: 10.1177/1533317519899546
Allez, M., Karmiris, K., Louis, E., Assche, G. V., Ben-Horin, S., Klein, A., et al. (2010). Report of the ECCO pathogenesis workshop on anti-TNF therapy failures in inflammatory bowel diseases: definitions, frequency and pharmacological aspects. J. Crohns Colitis 4, 355–366. doi: 10.1016/j.crohns.2010.04.004
Altmann, A., Tian, L., Henderson, V. W., and Greicius, M. D. (2014). Alzheimer's disease neuroimaging initiative investigators. Sex modifies the APOE-related risk of developing Alzheimer disease. Ann. Neurol. 75, 563–573. doi: 10.1002/ana.24135
Alves, F., Kalinowski, P., and Ayton, S. (2023). Accelerated brain volume loss caused by anti-beta-amyloid drugs: a systematic review and Meta-analysis. Neurology 100, e2114–e2124. doi: 10.1212/WNL.0000000000207156
Ansari, M. A., and Scheff, S. W. (2010). Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J. Neuropathol. Exp. Neurol. 69, 155–167. doi: 10.1097/NEN.0b013e3181cb5af4
Barnes, P. J., and Karin, M. (1997). Nuclear factor-B: a pivotal transcription factor in chronic inflammatory diseases. N. Engl. J. Med. 336, 1066–1071. doi: 10.1056/NEJM199704103361506
Bateman, R. J., Xiong, C., Benzinger, T. L., Fagan, A. M., Goate, A., Fox, N. C., et al. (2012). Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N. Engl. J. Med. 367, 795–804. doi: 10.1056/NEJMoa1202753
Bennett, D. A., Schneider, J. A., Arvanitakis, Z., and Wilson, R. S. (2012). Overview and findings from the religious orders study. Curr. Alzheimer Res. 9, 628–645. doi: 10.2174/156720512801322573
Birks, J. S., and Harvey, R. J. (2018). Donepezil for dementia due to Alzheimer’s disease. Cochrane Database Syst. Rev. 6:CD001190. doi: 10.1002/14651858
Bivona, G., Iemmolo, M., Agnello, L., Lo Sasso, B., Gambino, C. M., Giglio, R. V., et al. (2023). Microglial activation and priming in Alzheimer's disease: state of the art and future perspectives. Int. J. Mol. Sci. 24:884. doi: 10.3390/ijms24010884
Blevins, H. M., Xu, Y., Biby, S., and Zhang, S. (2022). The NLRP3 Inflammasome pathway: a review of mechanisms and inhibitors for the treatment of inflammatory diseases. Front. Aging Neurosci. 14:879021. doi: 10.3389/fnagi.2022.879021
Bongartz, T., Sutton, A. J., Sweeting, M. J., Buchan, I., Matteson, E. L., and Montori, V. (2006). Anti-TNF antibody therapy in rheumatoid arthritis and the risk of serious infections and malignancies: systematic review and meta-analysis of rare harmful effects in randomized controlled trials. JAMA 295, 2275–2285. doi: 10.1001/jama.295.19.2275
Breyer, J. P., Avritt, T. G., McReynolds, K. M., Dupont, W. D., and Smith, J. R. (2012). Confirmation of the HOXB13 G84E germline mutation in familial prostate cancer. Cancer Epidemiol. Biomarkers Prev. 21, 1348–1353. doi: 10.1158/1055-9965.EPI-12-0495
Bullock, R., and Lane, R. (2007). Executive Dyscontrol in dementia, with emphasis on subcortical pathology and the role of Butyrylcholinesterase. Curr. Alzheimer Res. 4, 277–293. doi: 10.2174/156720507781077313
Butterfield, D. A. (2021). Ubiquitin carboxyl-terminal hydrolase L-1 in brain: focus on its oxidative/nitrosative modification and role in brains of subjects with Alzheimer disease and mild cognitive impairment. Free Radic. Biol. Med. 177, 278–286. doi: 10.1016/j.freeradbiomed.2021.10.036
Cai, Q., and Tammineni, P. (2017). Mitochondrial aspects of synaptic dysfunction in Alzheimer's disease. J. Alzheimers Dis. 57, 1087–1103. doi: 10.3233/JAD-160726
Cargnello, M., and Roux, P. P. (2011). Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 75, 50–83. doi: 10.1128/MMBR.00031-10
Cohen, S. B., Cheng, T. T., Chindalore, V., Damjanov, N., Burgos-Vargas, R., Delora, P., et al. (2009). Evaluation of the efficacy and safety of pamapimod, a p38 MAP kinase inhibitor, in a double-blind, methotrexate-controlled study of patients with active rheumatoid arthritis. Arthritis Rheum. 60, 335–344. doi: 10.1002/art.24266
Collingridge, G. L., and Singer, W. (1990). Excitatory amino acid receptors and synaptic plasticity. Trends Pharmacol. Sci. 11, 290–296. doi: 10.1016/0165-6147(90)90011-V
Cortes-Canteli, M., and Iadecola, C. (2020). Alzheimer's disease and vascular aging: JACC focus seminar. J. Am. Coll. Cardiol. 75, 942–951. doi: 10.1016/j.jacc.2019.10.062
Damjanov, N., Kauffman, R. S., and Spencer-Green, G. T. (2009). Efficacy, pharmacodynamics, and safety of VX-702, a novel p38 MAPK inhibitor, in rheumatoid arthritis: results of two randomized, double-blind, placebo-controlled clinical studies. Arthritis Rheum. 60, 1232–1241. doi: 10.1002/art.24485
Davatzikos, C., Bhatt, P., Shaw, L. M., Batmanghelich, K. N., and Trojanowski, J. Q. (2011). Prediction of MCI to AD conversion, via MRI, CSF biomarkers, and pattern classification. Neurobiol. Aging 32, 2322.e19–2322.e27. doi: 10.1016/j.neurobiolaging.2010.05.023
de la Monte, S. M., and Tong, M. (2014). Brain metabolic dysfunction at the core of Alzheimer's disease. Biochem. Pharmacol. 88, 548–559. doi: 10.1016/j.bcp.2013.12.012
Denny, W. A. (2022). Inhibitors and activators of the p38 mitogen-activated MAP kinase (MAPK) family as drugs to treat Cancer and inflammation. Curr. Cancer Drug Targets 22, 209–220. doi: 10.2174/1568009622666220215142837
Doody, R. S., Raman, R., Farlow, M., Iwatsubo, T., Vellas, B., Joffe, S., et al. (2013). A phase 3 trial of semagacestat for treatment of Alzheimer's disease. N. Engl. J. Med. 369, 341–350. doi: 10.1056/NEJMoa1210951
Egan, M. F., Kost, J., Tariot, P. N., Aisen, P. S., Cummings, J. L., Vellas, B., et al. (2018). Randomized trial of Verubecestat for mild-to-moderate Alzheimer's disease. N. Engl. J. Med. 378, 1691–1703. doi: 10.1056/NEJMoa1706441
Fischer, R., and Maier, A. (2015). Interrelation of oxidative stress and inflammation in neurodegenerative disease: role of TNF. Oxidative Med. Cell. Longev. 2015, 1–18. doi: 10.1155/2015/610813
Friedle, S. A., Brautigam, V. M., Nikodemova, M., Wright, M. L., and Watters, J. J. (2011). The P2X7-Egr pathway regulates nucleotide-dependent inflammatory gene expression in microglia. Glia 59, 1–13. doi: 10.1002/glia.21071
Gee, M. S., Son, S. H., Jeon, S. H., Do, J., Kim, N., Ju, Y. J., et al. (2020). A selective p38alpha/beta MAPK inhibitor alleviates neuropathology and cognitive impairment, and modulates microglia function in 5XFAD mouse. Alzheimers Res. Ther. 12:45. doi: 10.1186/s13195-020-00617-2
Gold, M., Alderton, C., Zvartau-Hind, M., Egginton, S., Saunders, A. M., Irizarry, M., et al. (2010). Rosiglitazone monotherapy in mild-to-moderate Alzheimer’s disease: results from a randomized, double-blind, placebo-controlled phase III study. Dement. Geriatr. Cogn. Disord. 30, 131–146. doi: 10.1159/000318845
Goldman, J. W., Rosen, L. S., Tolcher, A. W., Papadopoulos, K., Beeram, M., Shi, P., et al. (2018). Phase 1 and pharmacokinetic study of LY3007113, a p38 MAPK inhibitor, in patients with advanced cancer. Investig. New Drugs 36, 629–637. doi: 10.1007/s10637-017-0532-2
Grande, G., Vanacore, N., Vetrano, D. L., Cova, I., Rizzuto, D., Mayer, F., et al. (2018). Free and cued selective reminding test predicts progression to Alzheimer's disease in people with mild cognitive impairment. Neurol. Sci. 39, 1867–1875. doi: 10.1007/s10072-018-3507-y
Granic, I., Dolga, A. M., Nijholt, I. M., van Dijk, G., and Eisel, U. L. (2009). Inflammation and NF-kappaB in Alzheimer's disease and diabetes. J. Alzheimers Dis. 16, 809–821. doi: 10.3233/JAD-2009-0976
Griffin, W. S. T., and Mrak, R. (2002). Interleukin-1 in the genesis and progression of and risk for development of neuronal degeneration in Alzheimer’s disease. J. Leukoc. Biol. 72, 233–238. doi: 10.1189/jlb.72.2.233
Grimm, A., Friedland, K., and Eckert, A. (2016). Mitochondrial dysfunction: the missing link between aging and sporadic Alzheimer's disease. Biogerontology 17, 281–296. doi: 10.1007/s10522-015-9618-4
Hardingham, G. E., and Bading, H. (2010). Synaptic versus extra-synaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat. Rev. Neurosci. 11, 682–696. doi: 10.1038/nrn2911
Henley, D. B., Sundell, K. L., Sethuraman, G., Dowsett, S. A., and May, P. C. (2014). Safety profile of semagacestat, a gamma-secretase inhibitor: IDENTITY trial findings. Curr. Med. Res. Opin. 30, 2021–2032. doi: 10.1185/03007995.2014.939167
Hensley, K., Floyd, R. A., Zheng, N. Y., Nael, R., Robinson, K. A., Nguyen, X., et al. (1999). p38 kinase is activated in the Alzheimer's disease brain. J. Neurochem. 72, 2053–2058. doi: 10.1046/j.1471-4159.1999.0722053.x
Hippius, H., and Neundorfer, G. (2003). The discovery of Alzheimer's disease. Dialogues Clin. Neurosci. 5, 101–108. doi: 10.31887/DCNS.2003.5.1/hhippius
Holbrook, J. A., Jarosz-Griffiths, H. H., Caseley, E., Lara-Reyna, S., Poulter, J. A., Williams-Gray, C. H., et al. (2021). Neurodegenerative disease and the NLRP3 Inflammasome. Front. Pharmacol. 12:643254. doi: 10.3389/fphar.2021.643254
Hyman, B. T., Phelps, C. H., Beach, T. G., Bigio, E. H., Cairns, N. J., Carrillo, M. C., et al. (2012). National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement. 8, 1–13. doi: 10.1016/j.jalz.2011.10.007
Insel, P. S., Hansson, O., Mackin, R. S., Weiner, M., and Mattsson, N. (2018). Amyloid pathology in the progression to mild cognitive impairment. Neurobiol. Aging 64, 76–84. doi: 10.1016/j.neurobiolaging.2017.12.018
Jagust, W. (2018). Imaging the evolution and pathophysiology of Alzheimer disease. Nat. Rev. Neurosci. 19, 687–700. doi: 10.1038/s41583-018-0067-3
Kelly, A., Vereker, E., Nolan, Y., Brady, M., Barry, C., Loscher, C. E., et al. (2003). Activation of p38 plays a pivotal role in the inhibitory effect of lipopolysaccharide and interleukin-1 beta on long term potentiation in rat dentate gyrus. J. Biol. Chem. 278, 19453–19462. doi: 10.1074/jbc.M301938200
Kempuraj, D., Thangavel, R., Natteru, P. A., Selvakumar, G. P., Saeed, D., Zahoor, H., et al. (2016). Neuroinflammation induces neurodegeneration. J. Neurol. Neurosurg. Spine 1:1003. doi: 10.1016/j.drudis.2016.08.001.S1359-6446(16)30288-4
Kheiri, G., Dolatshahi, M., Rahmani, F., and Rezaei, N. (2018). Role of p38/MAPKs in Alzheimer's disease: implications for amyloid beta toxicity targeted therapy. Rev. Neurosci. 30, 9–30. doi: 10.1515/revneuro-2018-0008
Lacalle-Aurioles, M., and Iturria-Medina, Y. (2023). Fornix degeneration in risk factors of Alzheimer's disease, possible trigger of cognitive decline. Cereb. Circ. Cogn. Behav. 4:100158. doi: 10.1016/j.cccb.2023.100158
Lebovitz, H. E. (2002). Differentiating members of the thiazolidinedione class: a focus on safety. Diabetes Metab. Res. Rev. 18, S23–S29. doi: 10.1002/dmrr.252
Lee, J. K., and Kim, N. J. (2017). Recent advances in the inhibition of p38 MAPK as a potential strategy for the treatment of Alzheimer's disease. Molecules 22:1287. doi: 10.3390/molecules22081287
Li, X., Feng, X., Sun, X., Hou, N., Han, F., and Liu, Y. (2022). Global, regional, and national burden of Alzheimer's disease and other dementias, 1990-2019. Front. Aging Neurosci. 14:937486. doi: 10.3389/fnagi.2022.937486
Linial, M., Stern, A., and Weinstock, M. (2020). Effect of ladostigil treatment of aging rats on gene expression in four brain areas associated with regulation of memory. Neuropharmacology 177:108229. doi: 10.1016/j.neuropharm.2020.108229
Mahase, E. (2021). Aducanumab: European agency rejects Alzheimer’s drug over efficacy and safety concerns. BMJ 375:n3127. doi: 10.1136/bmj.n3127
Mahase, E. (2022). Lecanemab trial finds slight slowing of cognitive decline, but clinical benefits are uncertain. BMJ 379:o2912. doi: 10.1136/bmj.o2912
Mahley, R. W. (2016). Apolipoprotein E: from cardiovascular disease to neurodegenerative disorders. J. Mol. Med. (Berl) 94, 739–746. doi: 10.1007/s00109-016-1427-y
Malynn, B. A., and Ma, A. (2019). A20: a multifunctional tool for regulating immunity and preventing disease. Cell. Immunol. 340:103914. doi: 10.1016/j.cellimm.2019.04.002
Marber, M. S., Rose, B., and Wang, Y. (2011). The p38 mitogen-activated protein kinase pathway--a potential target for intervention in infarction, hypertrophy, and heart failure. J. Mol. Cell. Cardiol. 51, 485–490. doi: 10.1016/j.yjmcc.2010.10.021
Maruyama, W., Weinstock, M., Youdim, M. B., Nagai, M., and Naoi, M. (2003). Anti-apoptotic action of anti-Alzheimer drug, TV3326 [(N-propargyl)-(3R)-aminoindan-5-yl]-ethyl methyl carbamate, a novel cholinesterase-monoamine oxidase inhibitor. Neurosci. Lett. 341, 233–236. doi: 10.1016/s0304-3940(03)00211-8
Matsunaga, S., Kishi, T., and Iwata, N. (2015). Memantine monotherapy for Alzheimer’s disease: a systematic review and meta-analysis. PLoS One 10:e0123289. doi: 10.1371/journal.pone.0123289
Mayerson, A. B., Hundal, R. S., Dufour, S., Lebon, V., Befroy, D., Cline, G. A., et al. (2002). The effects of rosiglitazone on insulin sensitivity, lipolysis, and hepatic and skeletal muscle triglyceride content in patients with type 2 diabetes. Diabetes 51, 797–802. doi: 10.2337/diabetes.51.3.797
McShane, R., Westby, M. J., Roberts, E., Minakaran, N., Schneider, L., Farrimond, L. E., et al. (2019). Memantine for dementia. Cochrane Database Syst. Rev. 3:CD003154. doi: 10.1002/14651858.CD003154.pub6
Mohanty, P., Aljada, A., Ghanim, H., Hofmeyer, D., Tripathy, D., Syed, T., et al. (2004). Evidence for a potent anti-inflammatory effect of rosiglitazone. J. Clin. Endocrinol. Metab. 89, 2728–2735. doi: 10.1210/jc.2003-032103
Mullard, A. (2019). Anti-amyloid failures stack up as Alzheimer antibody flops. Nat. Rev. Drug Discov. 327. doi: 10.1038/d41573-019-00064-1
Nathan, D. M. (2007). Rosiglitazone and cardiotoxicity--weighing the evidence. N. Engl. J. Med. 357, 64–66. doi: 10.1056/NEJMe078117
Novak, G., Streffer, J. R., Timmers, M., Henley, D., Brashear, H. R., Bogert, J., et al. (2020). Long-term safety and tolerability of atabecestat (JNJ-54861911), an oral BACE1 inhibitor, in early Alzheimer's disease spectrum patients: a randomized, double-blind, placebo-controlled study and a two-period extension study. Alzheimers Res. Ther. 12:58. doi: 10.1186/s13195-020-00614-5
Panarsky, R., Luques, L., and Weinstock, M. (2012). Anti-inflammatory effects of ladostigil and its metabolites in aged rat brain and in microglial cells. J. Neuroimmune Pharmacol. 7, 488–498. doi: 10.1007/s11481-012-9358-z
Papaconstantinou, J. (2019). The role of signaling pathways of inflammation and oxidative stress in development of senescence and aging phenotypes in cardiovascular disease. Cells 8:1383. doi: 10.3390/cells8111383
Pathan, A. R., Gaikwad, A. B., Viswanad, B., and Ramarao, P. (2008). Rosiglitazone attenuates the cognitive deficits induced by high fat diet feeding in rats. Eur. J. Pharmacol. 589, 176–179. doi: 10.1016/j.ejphar.2008.06.016
Penninkilampi, R., Brothers, H. M., and Eslick, G. D. (2016). Pharmacological agents targeting gamma-secretase increase risk of Cancer and cognitive decline in Alzheimer's disease patients: a systematic review and Meta-analysis. J. Alzheimers Dis. 53, 1395–1404. doi: 10.3233/JAD-160275
Petersen, R. C. (2004). Mild cognitive impairment as a diagnostic entity. J. Intern. Med. 256, 183–194. doi: 10.1111/j.1365-2796.2004.01388.x
Prins, N. D., Harrison, J. E., Chu, H. M., Blackburn, K., Alam, J. J., Scheltens, P., et al. (2021). A phase 2 double-blind placebo-controlled 24-week treatment clinical study of the p38 alpha kinase inhibitor neflamapimod in mild Alzheimer's disease. Alzheimers Res. Ther. 13:106. doi: 10.1186/s13195-021-00843-2
Reichert, F., Zohar, K., Lezmi, E., Eliyahu, T., Rotshenker, S., Linial, M., et al. (2024). Ladostigil reduces the Adenoside triphosphate/lipopolysaccharide-induced secretion of pro-inflammatory cytokines from microglia and modulate-immune regulators, TNFAIP3, and EGR1. Biomol. Ther. 14:112. doi: 10.3390/biom14010112
Risner, M. E., Saunders, A. M., Altman, J. F., Ormandy, G. C., Craft, S., Foley, I. M., et al. (2006). Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer’s disease. Pharmacogenomics J. 6, 246–254. doi: 10.1038/sj.tpj.6500369
Rivera-Cervantes, M. C., Castaneda-Arellano, R., Castro-Torres, R. D., Gudino-Cabrera, G., y Velasco, A. I. F., Camins, A., et al. (2015). P38 MAPK inhibition protects against glutamate neurotoxicity and modifies NMDA and AMPA receptor subunit expression. J. Mol. Neurosci. 55, 596–608. doi: 10.1007/s12031-014-0398-0
Rodrigue, K. M., Kennedy, K. M., Devous, M. D. Sr., Rieck, J. R., Hebrank, A. C., Diaz-Arrastia, R., et al. (2012). beta-amyloid burden in healthy aging: regional distribution and cognitive consequences. Neurology 78, 387–395. doi: 10.1212/WNL.0b013e318245d295
Romero-Sevilla, R., Lopez-Espuela, F., Fuentes, J. M., de San Juan, B. D., Portilla-Cuenca, J. C., Hijon, C. C., et al. (2022). Role of inflammatory cytokines in the conversion of mild cognitive impairment to dementia: a prospective study. Curr. Alzheimer Res. 19, 68–75. doi: 10.2174/1567205019666220127102640
Schneider, L. S., Geffen, Y., Rabinowitz, J., Thomas, R. G., Schmidt, R., Ropele, S., et al. (2019). Low-dose ladostigil for mild cognitive impairment: a phase 2 placebo-controlled clinical trial. Neurology 93, e1474–e1484. doi: 10.1212/WNL.0000000000008239
Shankar, G. M., Li, S., Mehta, T. H., Garcia-Munoz, A., Shepardson, N. E., Smith, I., et al. (2008). Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842. doi: 10.1038/nm1782
Shoham, S., Linial, M., and Weinstock, M. (2019). Age-induced spatial memory deficits in rats are correlated with specific brain region alterations in microglial morphology and gene expression. J. Neuroimmune Pharmacol. 14, 251–262. doi: 10.1007/s11481-018-9817-2
Shoji, M., Golde, T. E., Ghiso, J., Cheung, T. T., Estus, S., Shaffer, L. M., et al. (1992). Production of the Alzheimer amyloid beta protein by normal proteolytic processing. Science 258, 126–129. doi: 10.1126/science.1439760
Sims, J. R., Zimmer, J. A., Evans, C. D., Lu, M., Ardayfio, P., Sparks, J., et al. (2023). Donanemab in early symptomatic Alzheimer disease: the TRAILBLAZER-ALZ 2 randomized clinical trial. JAMA 330, 512–527. doi: 10.1001/jama.2023.13239
Smith, J. A., Das, A., Ray, S. K., and Bani, N. L. (2012). Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res. Bull. 87, 10–20. doi: 10.1016/j.brainresbull.2011.10.004
Spencer, C. M., and Noble, S. (1998). Rivastigmine. Review of its use in Alzheimer’s disease. Drugs Aging 13, 391–411. doi: 10.2165/00002512-199813050-00005
Sperling, R. A., Donohue, M. C., Raman, R., Rafii, M. S., Johnson, K., Masters, C. L., et al. (2023). Trial of Solanezumab in preclinical Alzheimer's disease. N. Engl. J. Med. 389, 1096–1107. doi: 10.1056/NEJMoa2305032
Sterling, J., Herzig, Y., Goren, T., Finkelstein, N., Lerner, D., Goldenberg, W., et al. (2002). Novel dual inhibitors of AChE and MAO derived from hydroxy aminoindan and phenethylamine as potential treatment for Alzheimer's disease. J. Med. Chem. 45, 5260–5279. doi: 10.1021/jm020120c
Strittmatter, W. J., Saunders, A. M., Schmechel, D., Pericak-Vance, M., Enghild, J., Salvesen, G. S., et al. (1993). Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. USA 90, 1977–1981. doi: 10.1073/pnas.90.5.1977
Tobinick, E., Gross, H., Weinberger, A., and Cohen, H. (2006). TNF-alpha modulation for treatment of Alzheimer's disease: a 6-month pilot study. Med. Gen. Med. 8:25.
Torres-Acosta, N., O’Keefeb, J. H., O’Keefec, E. L., Isaacsond, R., and Smalle, G. (2020). Therapeutic potential of TNF-α inhibition for Alzheimer’s disease prevention. J. Alzheimers Dis. 78, 619–626. doi: 10.3233/JAD-200711
van Dyck, C. H., Swanson, C. J., Aisen, P., Bateman, R. J., Chen, C., Gee, M., et al. (2023). Lecanemab in early Alzheimer's disease. N. Engl. J. Med. 388, 9–21. doi: 10.1056/NEJMoa2212948
Varatharaj, A., and Galea, I. (2017). The blood-brain barrier in systemic inflammation. Brain Behav. Immun. 60, 1–12. doi: 10.1016/j.bbi.2016.03.010
Verdile, G., Keane, K. N., Cruzat, V. F., Medic, S., Sabale, M., Rowles, J., et al. (2015). Inflammation and oxidative stress: the molecular connectivity between insulin resistance, obesity, and Alzheimer’s disease. Mediators Inflamm 2015:105828. doi: 10.1155/2015/105828
Voet, S., Mc Guire, C., Hagemeyer, N., Martens, A., Schroeder, A., Wieghofer, P., et al. (2018). A20 critically controls microglia activation and inhibits inflammasome-dependent neuroinflammation. Nat. Commun. 9:2036. doi: 10.1038/s41467-018-04376-5
Watkins, P. B., Zimmerman, H. J., Knapp, M. J., Gracon, S. I., and Lewis, K. W. (1994). Hepatotoxic effects of tacrine administration in patients with Alzheimer’s disease. JAMA 271, 992–998. doi: 10.1001/jama.1994.03510370044030
Watz, H., Barnacle, H., Hartley, B. F., and Chan, R. (2014). Efficacy and safety of the p38 MAPK inhibitor losmapimod for patients with chronic obstructive pulmonary disease: a randomised, double-blind, placebo-controlled trial. Lancet Respir. Med. 2, 63–72. doi: 10.1016/S2213-2600(13)70200-5
Weinstock, M., Bejar, C., Schorer-Apelbaum, D., Panarsky, R., Luques, L., and Shoham, S. (2013). Dose-dependent effects of ladostigil on microglial activation and cognition in aged rats. J. Neuroimmune Pharmacol. 8, 345–355. doi: 10.1007/s11481-013-9433-0
Weinstock, M., Goren, T., and Youdim, M. B. H. (2000). Development of a novel neuroprotective drug (TV3326) for the treatment of Alzheimer’s disease, with cholinesterase and monoamine oxidase inhibitory activities. Drug Dev. Res. 50, 216–222. doi: 10.1002/1098-2299(200007/08)50:3/4<216::AID-DDR4>3.0.CO;2-Z
Weinstock, M., and Groner, E. (2008). Rational design of a drug for Alzheimer's disease with cholinesterase inhibitory and neuroprotective activity. Chem. Biol. Interact. 175, 216–221. doi: 10.1016/j.cbi.2008.03.014
Wessels, A. M., Lines, C., Stern, R. A., Kost, J., Voss, T., Mozley, L. H., et al. (2020). Cognitive outcomes in trials of two BACE inhibitors in Alzheimer's disease. Alzheimers Dement. 16, 1483–1492. doi: 10.1002/alz.12164
Whitehouse, P. J., Price, D. L., Struble, R. G., Clark, A. W., Coyle, J. T., and Delon, M. R. (1982). Alzheimer’s disease and senile dementia: loss of neurons in the basal forebrain. Science 215, 1237–1239. doi: 10.1126/science.7058341
Yi, J. H., Park, S. W., Brooks, N., Lang, B. T., and Vemuganti, R. (2008). PPARgamma agonist rosiglitazone is neuroprotective after traumatic brain injury via anti-inflammatory and anti-oxidative mechanisms. Brain Res. 1244, 164–172. doi: 10.1016/j.brainres.2008.09.074
Zhou, J. P., Yang, X. N., Song, Y., Zhou, F., Liu, J. J., Hu, Y. Q., et al. (2021). Rosiglitazone alleviates lipopolysaccharide-induced inflammation in RAW264.7 cells via inhibition of NF-κB and in a PPARγ-dependent manner. Exp. Ther. Med. 22:743. doi: 10.3892/etm.2021.10175
Keywords: antibodies against beta amyloid, β and γ secretase inhibitors, cholinesterase inhibitors, ladostigil, rosiglitazone, TNFα antagonists
Citation: Weinstock M (2024) Therapeutic agents for Alzheimer’s disease: a critical appraisal. Front. Aging Neurosci. 16:1484615. doi: 10.3389/fnagi.2024.1484615
Edited by:
Antonella Tramutola, Sapienza University of Rome, ItalyReviewed by:
Martina Gabrielli, National Research Council (CNR), ItalyRavinder Gulia, University of California, Irvine, United States
Copyright © 2024 Weinstock. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marta Weinstock, bWFydGFyQGVrbWQuaHVqaS5hYy5pbA==