 Tohgo Kanoh
Tohgo Kanoh Takamasa Mizoguchi1
Takamasa Mizoguchi1 Motoyuki Itoh
Motoyuki Itoh
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Aging Neurosci., 03 May 2024
Sec. Cellular and Molecular Mechanisms of Brain-aging
Volume 16 - 2024 | https://doi.org/10.3389/fnagi.2024.1399098
Many age-related neurological diseases still lack effective treatments, making their understanding a critical and urgent issue in the globally aging society. To overcome this challenge, an animal model that accurately mimics these diseases is essential. To date, many mouse models have been developed to induce age-related neurological diseases through genetic manipulation or drug administration. These models help in understanding disease mechanisms and finding potential therapeutic targets. However, some age-related neurological diseases cannot be fully replicated in human pathology due to the different aspects between humans and mice. Although zebrafish has recently come into focus as a promising model for studying aging, there are few genetic zebrafish models of the age-related neurological disease. This review compares the aging phenotypes of humans, mice, and zebrafish, and provides an overview of age-related neurological diseases that can be mimicked in mouse models and those that cannot. We presented the possibility that reproducing human cerebral small vessel diseases during aging might be difficult in mice, and zebrafish has potential to be another animal model of such diseases due to their similarity of aging phenotype to humans.
The quest to understand the intricacies of human age-related neurological diseases has led scientists to explore various animal models, each offering unique insights into the pathophysiology of diseases. Among these, the mouse (Mus musculus) models have been used as powerful tools in studying neurological disorder.
The mouse model has been a cornerstone in biomedical research for decades. Mice share about 85% of their DNA with humans and have similar nervous systems, making them excellent models for studying the age-related neurological disease (Waterston et al., 2002; Xu et al., 2022). The availability of sophisticated genetic manipulation techniques in mice further enhances their utility in disease modeling. Mouse models of age-related neurological diseases have provided deep insight into these diseases in humans. However, no single model can perfectly recapitulate all aspects of human age-related neurological disease, and it is necessary to combine insights from several models to understand the pathophysiology of these diseases.
Zebrafish (Danio rerio), a small tropical freshwater fish, has gained prominence in the scientific community due to its genetic and physiological similarities to humans. Approximately 70% of human genes have at least one obvious zebrafish orthologue, making it a valuable model for studying human diseases (Choi et al., 2021). Moreover, their prolific breeding capabilities allow for the generation of large sample sizes. The transparency of zebrafish larvae or adult zebrafish of mutants lacking melanocytes and iridophores permits live imaging of cellular and molecular processes in vivo, providing a dynamic view of disease progression and therapeutic effects that is not easily achievable in other model organisms (White et al., 2008). In addition to these merits, recent studies revealed the similarities of neurological disease-related aging phenotypes between zebrafish and humans, suggesting that zebrafish has a potential as a age-related neurological disease model (Arslan-Ergul et al., 2013).
In this review, we compared the neurological disease-related aging phenotypes that are common or different among humans, mice, and zebrafish. Subsequently, we discuss the advantages and limitations of mouse models for age-related neurological diseases and explore the potential of zebrafish to overcome these limitations.
When considering the creation of model organisms for neurodegenerative diseases accelerated by aging, it is critical to assess whether humans and the model organisms follow similar aging processes, as this can significantly impact the applicability of the research findings to humans. Therefore, it is useful to summarize the similarities and differences in aging phenotypes. Here, we compared the normal aging phenotypes across humans, mice, and zebrafish (Table 1). In mice and zebrafish, aging impairs various physiological functions similar to those in humans, such as basal metabolism (Kitazoe et al., 2019; Yang et al., 2019; Li et al., 2022), locomotor activity (Hunter et al., 2016; Yanai and Endo, 2021; Rutkove et al., 2023), cognitive function (Aartsen et al., 2002; Ruhl et al., 2015; Yang et al., 2018; Yanai and Endo, 2021), bone metabolism (Szulc et al., 2001; Monma et al., 2019; Wan et al., 2021), regenerative capacity (Tsai et al., 2007; Loforese et al., 2017; Rando and Jones, 2021; Tower et al., 2022), and reproductive capacity (Franks and Payne, 1970; Little, 1997; Tsai et al., 2007; Aitken, 2023), indicating a degree of conserved aging mechanisms across these species (Table 1). In leveraging the advantages of each model organism to mimic or overcome human age-related neurological diseases, it is essential to understand differences that may lead to divergent phenotypes with brain aging. The differences in normal age-related phenotypes between mice and zebrafish include a lifespan and clearance systems in the brain.
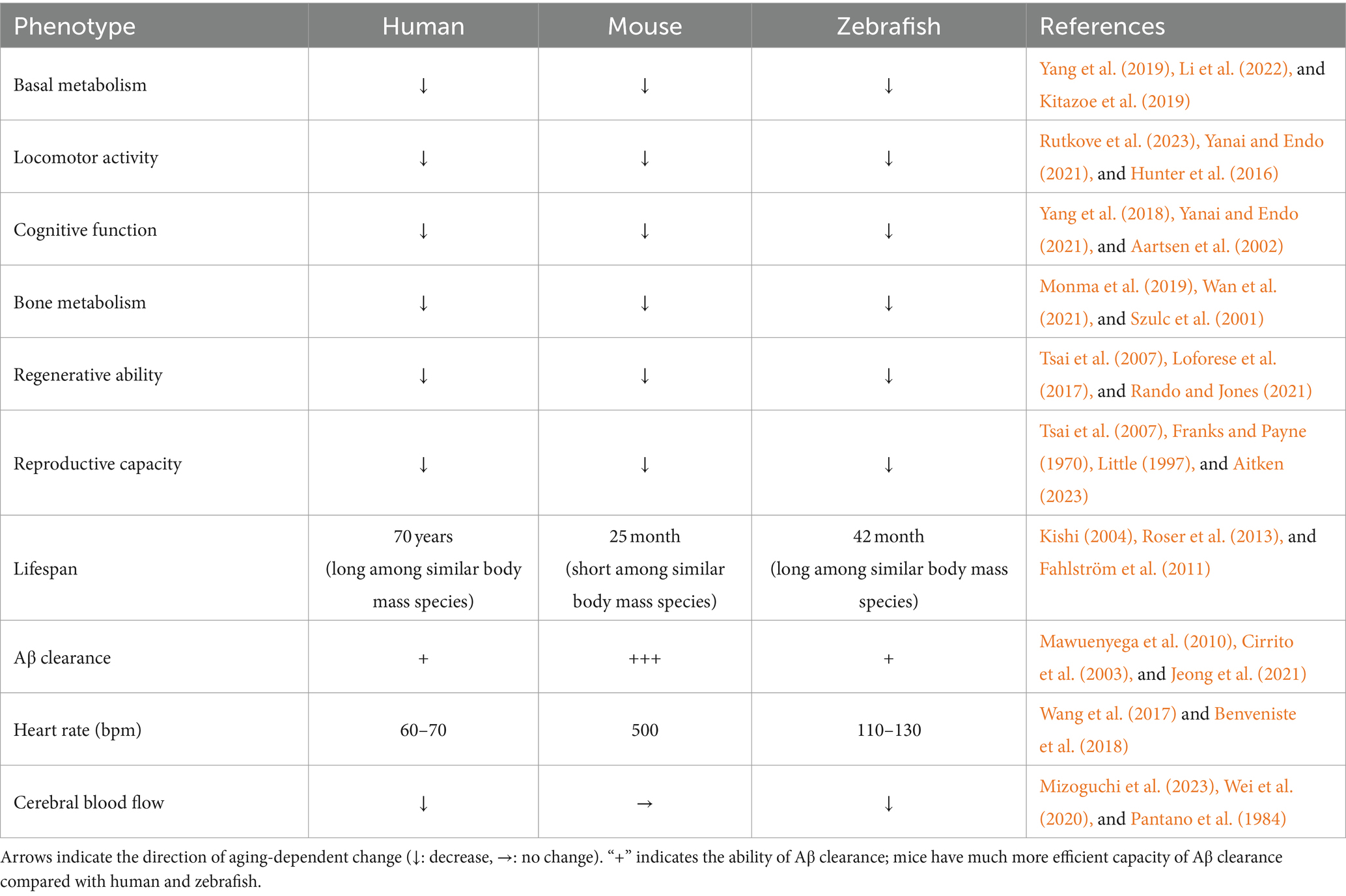
Table 1. The characteristics associated with aging among humans, mice, and zebrafish.
Zebrafish is known to have an average lifespan of 42 months (Gerhard et al., 2002; Kishi, 2004), while the lifespan of mice typically ranges from 25 to 26 months (Fahlström et al., 2011) (Table 1). Interestingly, even genetically modified mice designed for longevity live as long as the wild-type zebrafish (Bartke et al., 2001). It is known that there is a positive correlation between body weight and a lifespan in vertebrates (Kuparinen et al., 2023), suggesting that zebrafish, despite their relatively small size, is inherently long-lived species compared to other animals including mice. In addition, humans tend to live longer among mammals of equivalent body weight, while mice demonstrate a relatively shorter lifespan (Roser et al., 2013; Kowalczyk et al., 2020) (Table 1). These observations suggest that zebrafish offers a unique opportunity to replicate aging-related phenotypes that are not observable in shorter-lived species. This aspect of zebrafish biology underscores the potential of using them to explore complex aging processes and their implications for human health, offering insights into longevity and mechanisms underlying age-related diseases.
In the context of age-related neurological diseases, the efficiency of clearance systems in the brain plays a pivotal role in maintaining brain homeostasis. As organisms age, the efficiency of autophagy and the ubiquitin-proteasome system declines, leading to a disruption in proteostasis and subsequent accumulation of abnormal proteins in the brain (Kaushik and Cuervo, 2015). To clear these substances, the brain has several clearance mechanisms including the glymphatic system, which can drain these substances along cerebral small vessels (Nedergaard, 2013). Benveniste et al. (2018) suggest significant differences in the clearance efficiency of the glymphatic system between humans and mice (Table 1). This assertion is supported by the several studies investigating the half-life of amyloid-beta (Aβ), mainly cleared by the glymphatic system (Wang et al., 2023). In humans with Alzheimer’s disease, the half-life of Aβ is around 13 h, whereas in Alzheimer’s disease model mice, even aged individuals show a half-life of merely 2–4 h, indicating that the clearance of Aβ from the brain in mice is at least three times faster than that in humans (Cirrito et al., 2003; Barten et al., 2005; Mawuenyega et al., 2010; Qosa et al., 2014). Research utilizing zebrafish larvae, in which Aβ was injected into the brain, and the amount of Aβ was measured between 5 h-post-injection and 24 h-post-injection, showed only a 40% reduction in Aβ levels, indicating the half-life of Aβ is over 19 h in zebrafish larvae (Jeong et al., 2021). These studies suggest that zebrafish possesses a capability for brain clearance that is similar to humans, while mice exhibit a significantly higher capacity compared to the other two species (Table 1).
One of the possible causes of this difference is variations in heart rate, which directly impact the efficiency of the glymphatic system. It has been demonstrated that the glymphatic system is stimulated upon artificially elevating the heart rate in mice through the administration of dobutamine (Iliff et al., 2013). The resting heart rate of mice is around 500 bpm, while it is around 60–70 bpm in humans (Wang et al., 2017; Benveniste et al., 2018) (Table 1). Therefore, the clearance capacity in the brain in humans might be much weaker than in mice. Notably, the resting heart rate of adult zebrafish is around 120 bpm (Table 1), suggesting that zebrafish may possess brain clearance mechanisms closer to humans than to mice (Mousavi and Patil, 2020). Moreover, it is implied that dysregulation of cerebral blood flow affects glymphatic system (Sepehrinezhad et al., 2023). The cerebral blood flow is decreased with aging in humans and zebrafish, whereas this does not change in mice (Pantano et al., 1984; Wei et al., 2020; Mizoguchi et al., 2023) (Table 1). This suggests that the efficiency of glymphatic system is decreased with aging in humans and zebrafish, but less in mice.
These could imply that neurovascular aging related to neurodegenerative diseases may occur earlier in humans and zebrafish compared to mice. Thus, considering the difference in brain clearance capacity points toward the potential of zebrafish as a more representative model for studying human cerebrovascular aging and clearance mechanisms in the context of aging-related neurodegenerative diseases.
For some age-accelerated disorders, such as Alzheimer’s disease and cerebral small vessel diseases, mouse models do not fully mimic the human disease. These diseases are known to be related to cerebrovascular pathology, and aging might be a large risk factor of onset and progression of these diseases (described in detail in section 4).
Alzheimer’s disease (AD) is the most common type of dementia. Two main pathological hallmarks of AD are Aβ plaques and neurofibrillary tangles (NFT) (Blennow et al., 2006). The Aβ plaques are formed by deposition of Aβ protein, and the NFT is formed by intracellular tau protein hyperphosphorylation, which is induced by Aβ (Blennow et al., 2006). There are several AD risk genes such as APP, PSEN1, and PSEN2. The single missense mutation of these genes causes the AD pathology in human (Bagyinszky et al., 2016).
Transgenic mouse models have significantly contributed to our understanding of Alzheimer’s disease (AD), with multiple types of transgenic mice of AD overexpressing mutant forms of AD risk genes (Sanchez-Varo et al., 2022; Yokoyama et al., 2022). These models exhibit key features of AD pathology, including Aβ plaques and cognitive decline. However, they have limitations, including artificial temporal or spatial expression patterns in transgenic overexpression systems, leading to complex outcomes that may not accurately represent the human condition. For example, as reported by Jankowsky et al. (2005), analyzing cognitive behavior was difficult due to their severe hyperactivity which is not a human AD symptom, using transgenic mice expressing chimeric mouse/human APP Swedish/Indiana (carrying KM570, 571NL, and V617F mutation). The authors described that hyperactivity, which is not observed in human, might be caused by neuronal alterations due to transgene expression during early postnatal development (Jankowsky et al., 2005). Moreover, most transgenic mice, such as single transgenic mice (carrying mutant APP) or double transgenic mice (carrying both mutant APP and mutant PSEN1), did not show the formation of NFT, despite exhibiting cognitive decline (Metaxas and Kempf, 2016; Drummond and Wisniewski, 2017; Sasaguri et al., 2017). These limitations highlight the need for alternative AD models, which substitute for transgenic models.
To overcome the issues associated with transgenic models, researchers have attempted to create models using knock-in techniques. These techniques aim to mimic wild-type expression levels and patterns more closely. However, the expected Aβ plaque deposition was not detected in APP Swedish (carrying KM670, 671NL mutation) or London (carrying V717I mutation), and memory impairment was not observed in the knock-in model that carries a single APP mutation (carrying V642I) (Kawasumi et al., 2004; Köhler et al., 2005). Subsequent efforts led to the development of APP knock-in mice models incorporating multiple familial Alzheimer’s mutations. These models include the Swedish (NL), Beyreuther/Iberian (F), and Arctic (G) mutations. The APP NL-G-F mice, which harbor all three mutations, began to develop Aβ plaques at two months and showed memory impairment from six months (Saito et al., 2014). However, further research revealed that in some cases, APP NL-G-F mice did not exhibit the expected decline in memory abilities [refer to the discussion in Sakakibara et al. (2019)]. These suggest that introducing mutated forms of APP through knock-in techniques may not fully replicate the symptoms of AD. In terms of PSEN1/2, there are a few studies on familial mutant PSEN1 knock-in mouse models. These mouse models carry a single familial mutation such as L435F, I213T or R278I, and have shown Aβ plaques but exhibit no or mild memory impairment (Lalonde and Strazielle, 2005; Saito et al., 2011; Xia et al., 2015). Therefore, it is difficult to conclude the PSEN1 knock-in AD mouse models fully replicate the AD pathology.
Cerebral Small Vessel Diseases (CSVDs) are the collective term for diseases that affect the cerebral small vessels. Damage to small vessels lead to lesions in subcortical structures like lacunar infarcts, white matter lesions, large hemorrhages, and microbleeds, leading to dementia (Pantoni, 2010). The progression of CSVDs is highly age-associated (Chung et al., 2023). Characteristic pathologies of CSVDs include enlarged perivascular spaces and formation of abluminal protein deposits (Pantoni, 2010). Recent studies have suggested that the glymphatic system plays a pivotal role in the pathophysiology of CSVDs (Mestre et al., 2017; Benveniste and Nedergaard, 2022). The mouse models of monogenic CSVDs such as CADASIL, CARASIL, Fabry disease, and RVCL, are discussed in the following section.
Cerebral autosomal dominant arteriopathy with subcortical infarct and leukoencephalopathy (CADASIL) is a prototypical CSVD caused by mutations in the NOTCH3 gene (Chabriat et al., 2009; André, 2010). CADASIL is characterized by the accumulation of granular osmiophilic material (GOM) and the extracellular domain of NOTCH3 in the vascular walls, leading to the loss of perivascular cell (vascular smooth muscle cell and pericyte), vascular dysfunction, recurrent lacunar infarcts, cognitive impairments, depressive symptoms, and motor deficits (Kalaria et al., 2004; Chabriat et al., 2009; Dziewulska and Lewandowska, 2012). Despite its clinical importance, the precise pathogenesis of CADASIL remains elusive, and there is currently no effective treatment, emphasizing the need for animal models to better understand and address this condition.
Several transgenic mouse models have been developed that can partially mimic CADASIL pathology. Transgenic mice with the rat Notch3 R169C mutation exhibit GOM lesions, Notch3 accumulation, pericyte loss, and memory impairment (Joutel et al., 2010; Ghosh et al., 2015; Ehret et al., 2021). Mice with the human NOTCH3 R90C mutation also showed GOM lesions, vascular dysfunction, and memory deficits (Ruchoux et al., 2003; Lacombe et al., 2005; Liu et al., 2015). However, Ruchoux et al. (2003) showed that these mice did not exhibit significant brain parenchyma damage. These suggest that the observed memory deficits might arise from mechanisms different from those in human CADASIL. Another transgenic model expressing the human NOTCH3 R182C mutation has been established, which develops GOM lesions, but does not show the white matter lesions, changes in cerebral blood flow, or memory impairment seen in human patients (Rutten et al., 2015; Gravesteijn et al., 2020). To more accurately mimic the pathological conditions of human CADASIL, knock-in models are increasingly developed. There are two types of knock-in CADASIL mouse models; one is Notch3 R170C (corresponding to human R169C) knock-in mice, and another is Notch3 R142C (corresponding to human R141C) knock-in mice (Lundkvist et al., 2005; Wallays et al., 2011). However, it is important to note that, to our knowledge, none of these CADASIL knock-in mouse models have yet exhibited memory impairments.
Cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL) is a hereditary disease caused by loss-of-function mutations in the Htra1 gene, characterized by baldness, strokes, white matter lesions, and early-onset dementia (Tikka et al., 2014). The abnormal accumulation of extracellular matrix proteins and TGF-β1, which are degraded by Htra1 was observed around small cerebral arteries in CARASIL patients (Hara et al., 2009). While aged Htra1 knockout mice showed the abnormal protein accumulation in cerebral arteries, they have not successfully replicated white matter lesions, strokes, or smooth muscle cell loss seen in the human condition (Beaufort et al., 2014; Kato et al., 2021).
Fabry disease results from mutations in the gene encoding α-galactosidase A (α-GalA), a lysosomal hydrolase enzyme (Germain, 2010). This leads to decreased enzyme activity and the accumulation of its substrate, globotriaosylceramide (GL-3), within the lysosomes of various organs, including blood vessels, kidneys, heart, and dorsal root ganglia (Choi, 2015). The primary symptoms of Fabry disease in humans are burning pain, autonomic dysfunctions, posterior circulation stroke, cognitive impairment, and depression (Bolsover et al., 2014; Choi, 2015). α-GalA knockout mice have been developed to study Fabry disease. While these mice exhibit the accumulation of GL-3 (Ohshima et al., 1997; Bangari et al., 2015), they did not show depressive-like behavior or learning and memory deficits (Hofmann et al., 2017).
Retinal vasculopathy with cerebral leukodystrophy (RVCL) is caused by mutations in a 3′-5′ DNA exonuclease TREX1 (Richards et al., 2007). The primary symptoms in human RVCL patients include activation of immune system, leukoencephalopathy, lacunar infarcts, retinopathy, nephropathy, and migraines (Schuh et al., 2015; Søndergaard et al., 2017). While the pathomechanism remains unknown, vascular basement membranes were found to be thicker and multi-layered (Søndergaard et al., 2017). This suggests that the clearance system in the brain might be impaired in RVCL patients. Frame-shift mutant TREX1 knock-in mice have been developed as RVCL models. Although these mice replicated activation of immune system, they did not exhibit key manifestations such as retinopathy and neurological symptoms (Sakai et al., 2017).
There are several diseases in which most human symptoms can be mimicked in mouse models including behavioral or cognitive dysfunctions. These diseases are highly associated with neuropathological changes, rather than vascular ones. In addition, aging might be a risk factor of these diseases, but the onset age is relatively younger compared to the diseases introduced in section 2 (described in detail in section 4).
Parkinson’s disease (PD) is a neurodegenerative disorder characterized by the accumulation of α-Synuclein in the neurons of the substantia nigra and striatum, and damage to dopaminergic neurons (Kalia and Lang, 2015; Balestrino and Schapira, 2020). PD primarily manifests as motor dysfunction, and approximately 40% of PD patients suffer from dementia (Cummings, 1988). The exact cause of dopaminergic neuron impairment in PD remains unclear, highlighting the importance of animal model research in elucidating these mechanisms. Genetic mouse models replicating PD often utilize genes considered to be causative, such as LRRK2, PRKN, and PINK1. Transgenic mouse models of these genes consistently exhibit motor dysfunction, and most of these models show age-related cognitive impairments (Magen and Chesselet, 2011; Magen et al., 2012; Pischedda et al., 2021; Dovonou et al., 2023). Several knock-in mouse models harboring mutations of pathogenic LRRK2 variants have also been developed. These models typically replicate the characteristic neuronal damage and motor dysfunctions observed in PD (Chang et al., 2022; Dovonou et al., 2023). It is reported that LRRK2 G2019S knock-in mice successfully mimic the neuronal pathology in striatum and cognitive impairments, further contributing to our understanding of the broader impact of PD on cognitive functions (Hussein et al., 2022).
Huntington’s disease (HD) is a disorder resulting from abnormal amplification of CAG repeats in the Htt gene, leading to the formation of insoluble aggregates and subsequent neuronal loss (Walker, 2007; Ross and Tabrizi, 2011). HD is characterized by involuntary, dance-like movements of the limbs, known as chorea, cognitive impairments, and psychiatric symptoms (Walker, 2007). Studies using transgenic mice that overexpress Htt with amplified CAG repeats have demonstrated the manifestation of motor and cognitive impairments (Lione et al., 1999; Lüesse et al., 2001; Giralt et al., 2011; Kaye et al., 2021). These models have been instrumental in mirroring the symptomatology of HD, providing valuable insights into the disease mechanisms and progression. Similar to transgenic models, knock-in mice carrying HD-like mutations in the Htt gene consistently exhibit stable motor and cognitive impairments (Simmons et al., 2009; Giralt et al., 2012; Menalled et al., 2012; Yhnell et al., 2016). This suggests that, similar to PD, HD is a disorder where phenotypic traits are relatively easier to replicate in mouse models.
As mentioned above, some diseases can be accurately replicated in mouse models, while others cannot.
A common trait among diseases less effectively modeled in mice is vascular impairment. CADASIL, CARASIL, Fabry disease and RVCL are known as CSVDs, and 80% of AD patients also present with cerebral amyloid angiopathy, a type of CSVD characterized by the accumulation of Aβ deposits in brain arteries (Boyle et al., 2015; Mestre et al., 2017; Greenberg et al., 2020). The pathology of CSVDs is closely related to the glymphatic system, and mice have a more efficient glymphatic system compared to humans, which may contribute to their reduced capacity to accurately phenocopy CSVDs. For diseases like PD and HD, studies showed that the accumulation of abnormal protein such as α-Synuclein and Huntingtin may be involved in cerebrovascular pathology (Drouin-Ouellet et al., 2015; Paul and Elabi, 2022). However, the primary pathology of these diseases is neuronal, a fact supported by the predominant expression of α-Synuclein and Huntingtin in neurons (Young, 2003; Gil and Rego, 2008; Stefanis, 2012; Wong and Krainc, 2017). These suggest that the abnormal aggregated proteins might have a more significant impact on neurons in PD and HD than in CSVDs and AD.
Another commonality is the variability in disease onset. Some previous studies suggested the typical onset age of familial AD ranges from 30s to over 70 years (Percy et al., 1991; Duara et al., 1993; Lopera et al., 1997; Quiroz et al., 2010). Furthermore, while CADASIL has a relatively young onset age, the range is quite broad, with migraines manifesting between 5 and 61 years and lacunar infarcts occurring between 26 and 81 years, indicating that onset at an older age is not uncommon (Tan and Markus, 2016). The wide range of the onset age extending into later years implies that there are individuals who may not exhibit symptoms until they are into old age. On the other hand, the onset age for HD is correlated with the number of CAG repeat amplifications; with over 50 repeats, the onset age is around 20 years (Brinkman et al., 1997; Wexler, 2004). Genetic models of HD in mice possess at least 50 CAG repeats, with some models exhibiting upwards of 150 CAG repeats (Kaye et al., 2021). This suggests that the HD mouse models might exhibit the age-related symptoms in human HD at a younger age. In the case of PD, the typical onset age of familial PD ranges from 20s to 50s, suggesting that familial PD predominantly manifests at a relatively younger age (Spira et al., 2001; Shojaee et al., 2009; Lin et al., 2019). Considering these variations and the relatively short lifespan of mice, mice might not be ideal for accurately replicating human age-related symptoms, potentially limiting their effectiveness in disease modeling.
As zebrafish possesses the less effective clearance capacity in the brain and a long lifespan, diseases that are not fully replicable in mouse models might be more successfully modeled in zebrafish. Although there are currently only a few examples of genetic models used to analyze adult disease states in zebrafish, some studies suggest they offer advantages over mice. In zebrafish with a knockout of PSEN1, a risk gene for AD, adult fish exhibit anxiety-like behaviors, a contrast to mice with PSEN1 knockout, which do not show changes in memory capabilities or anxiety-like behaviors (Saura et al., 2004; Sundvik et al., 2013; Soto-Faguás et al., 2021). Another study established that a zebrafish model expressing human APP carrying the Swedish mutation under the control of zebrafish appb promotor (Pu et al., 2017). This model showed the Aβ deposition and neuron loss in the telencephalon which controls zebrafish memory, subsequently learning ability was impaired (Pu et al., 2017). In contrast, the mouse model carrying APP Swedish mutant showed Aβ deposits but did not exhibit neuronal loss and profound impairment in learning ability, even in their old age (King and Arendash, 2002; Walker et al., 2002).
A recent study established a zebrafish model for Fabry disease by knocking out the gla gene encoding α-GalA (Elsaid et al., 2022a). This study found that this zebrafish model could replicate the nephropathy phenotype seen in adult stage, a typical pathology of Fabry disease. Another study showed that the changes in gene expression in the gla knockout zebrafish is consistent to that in the gla knockout human cell line (Consolato et al., 2022; Elsaid et al., 2022b). However, this zebrafish model has not been analyzed for the neuronal pathology.
In contrast, the genetically modified zebrafish models for CADASIL, CARASIL and RVCL have not been established.
The zebrafish models for PD and HD are well established (Kumar et al., 2021; Doyle and Croll, 2022). These models can replicate human pathology like as mouse models. By employing zebrafish, it is possible to conduct analyses that are not feasible with mice such as drug screening and live imaging (Zhan et al., 2024). Therefore, it is meaningful to develop zebrafish models for diseases for which mouse models already have been well established.
Collectively, as discussed above, zebrafish have potential for modeling age-related neurological diseases particularly accompanied by the vascular pathology, and some zebrafish models of AD show the symptoms that cannot be replicated in mouse models. Further studies are needed to establish the genetic model in zebrafish that closely mirrors human patients. It is crucial that we integrate the insights from various models to unravel the pathomechanism of human age-related neurological diseases.
Exploring the therapeutic target for the age-related neurological disease is one of the most urgent challenges in today’s global aging society. To address this challenge, researchers should integrate insights obtained from various animal models because each has advantages and disadvantages. We discussed the normal age-related phenotypes of zebrafish which shows similarities to humans, but not mice in aspects such as a lifespan and clearance systems in the brain. In addition, we provided an overview of age-related neurological diseases that can be mimicked in mouse models and those that cannot, using specific examples. Based on the discussion above, it is suggested that diseases that cannot be effectively replicated in mouse models often involve brain vascular pathology. This might be due to the more efficient clearance system in the mouse brain compared to humans and zebrafish. Another reason why zebrafish mimics human age-related neurological disease is their longer lifespan compared to mice. The longevity of zebrafish enables replication of symptoms and pathologies that worsen with age. In conclusion, zebrafish has a great potential for mimicking human age-related neurological disease, due to their similar clearance system in the brain and lifespan to humans.
TK: Conceptualization, Resources, Writing – original draft, Writing – review and editing. TM: Writing – review and editing. AT: Writing – review and editing. MI: Conceptualization, Resources, Supervision, Writing – review and editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by JST SPRING Grant Number JPMJSP2109 and Innovative Medicine CHIBA Doctoral WISE Program to TK; JSPS KAKENHI Grant Numbers JP19K06454 to TM, JP22H02715 and JP22H05485 to AT, JP18H02568 and JP21H02621 to MI; AMED under Grant Number JP23gm6710006h0002 for AT.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aartsen, M. J., Smits, C. H. M., Van Tilburg, T., Knipscheer, K. C. P. M., and Deeg, D. J. H. (2002). Activity in older adults: cause or consequence of cognitive functioning? A longitudinal study on everyday activities and cognitive performance in older adults. J. Gerontol. Ser. B Psychol. Sci. Soc. Sci. 57, P153–P162. doi: 10.1093/geronb/57.2.p153
Aitken, R. J. (2023). Male reproductive ageing: a radical road to ruin. Hum. Reprod. 38, 1861–1871. doi: 10.1093/humrep/dead157
André, C. (2010). CADASIL: pathogenesis, clinical and radiological findings and treatment. Arq. Neuropsiquiatr. 68, 287–299. doi: 10.1590/s0004-282x2010000200026
Arslan-Ergul, A., Ozdemir, A. T., and Adams, M. M. (2013). Aging, neurogenesis, and caloric restriction in different model organisms. Aging Dis. 4, 221–232.
Bagyinszky, E., Youn, Y. C., An, S. S. A., and Kim, S. (2016). Mutations, associated with early-onset Alzheimer’s disease, discovered in Asian countries. Clin. Interv. Aging 11, 1467–1488. doi: 10.2147/cia.s116218
Balestrino, R., and Schapira, A. H. V. (2020). Parkinson disease. Eur. J. Neurol. 27, 27–42. doi: 10.1111/ene.14108
Bangari, D. S., Ashe, K. M., Desnick, R. J., Maloney, C., Lydon, J., Piepenhagen, P., et al. (2015). α-Galactosidase a knockout mice. Am. J. Pathol. 185, 651–665. doi: 10.1016/j.ajpath.2014.11.004
Barten, D. M., Guss, V. L., Corsa, J. A., Loo, A., Hansel, S. B., Zheng, M., et al. (2005). Dynamics of β-amyloid reductions in brain, cerebrospinal fluid, and plasma of β-amyloid precursor protein transgenic mice treated with a γ-secretase inhibitor. J. Pharmacol. Exp. Ther. 312, 635–643. doi: 10.1124/jpet.104.075408
Bartke, A., Coschigano, K., Kopchick, J., Chandrashekar, V., Mattison, J., Kinney, B., et al. (2001). Genes that prolong life: relationships of growth hormone and growth to aging and life span. J. Gerontol. A Biol. Sci. Med. Sci. 56, B340–B349. doi: 10.1093/gerona/56.8.B340
Beaufort, N., Scharrer, E., Kremmer, E., Lux, V., Ehrmann, M., Huber, R., et al. (2014). Cerebral small vessel disease-related protease HtrA1 processes latent TGF-β binding protein 1 and facilitates TGF-β signaling. Proc. Natl. Acad. Sci. 111, 16496–16501. doi: 10.1073/pnas.1418087111
Benveniste, H., Liu, X., Koundal, S., Sanggaard, S., Lee, H., and Wardlaw, J. (2018). The Glymphatic system and waste clearance with brain aging: a review. Gerontology 65, 106–119. doi: 10.1159/000490349
Benveniste, H., and Nedergaard, M. (2022). Cerebral small vessel disease: a glymphopathy? Curr. Opin. Neurobiol. 72, 15–21. doi: 10.1016/j.conb.2021.07.006
Blennow, K., de Leon, M. J., and Zetterberg, H. (2006). Alzheimer's disease. Lancet 368, 387–403. doi: 10.1016/S0140-6736(06)69113-7
Bolsover, F. E., Murphy, E., Cipolotti, L., Werring, D. J., and Lachmann, R. H. (2014). Cognitive dysfunction and depression in Fabry disease: a systematic review. J. Inherit. Metab. Dis. 37, 177–187. doi: 10.1007/s10545-013-9643-x
Boyle, P. A., Yu, L., Nag, S., Leurgans, S., Wilson, R. S., Bennett, D. A., et al. (2015). Cerebral amyloid angiopathy and cognitive outcomes in community-based older persons. Neurology 85, 1930–1936. doi: 10.1212/wnl.0000000000002175
Brinkman, R. R., Mezei, M. M., Theilmann, J., Almqvist, E., and Hayden, M. R. (1997). The likelihood of being affected with Huntington disease by a particular age, for a specific CAG size. Am. J. Hum. Genet. 60, 1202–1210
Chabriat, H., Joutel, A., Dichgans, M., Tournier-Lasserve, E., and Bousser, M.-G. (2009). CADASIL. Lancet Neurol. 8, 643–653. doi: 10.1016/s1474-4422(09)70127-9
Chang, E. E. S., Ho, P. W.-L., Liu, H.-F., Pang, S. Y.-Y., Leung, C.-T., Malki, Y., et al. (2022). LRRK2 mutant knock-in mouse models: therapeutic relevance in Parkinson's disease. Translational. Neurodegeneration 11:10. doi: 10.1186/s40035-022-00285-2
Choi, J. C. (2015). Genetics of cerebral small vessel disease. J. Stroke 17, 7–16. doi: 10.5853/jos.2015.17.1.7
Choi, T.-Y., Choi, T.-I., Lee, Y.-R., Choe, S.-K., and Kim, C.-H. (2021). Zebrafish as an animal model for biomedical research. Exp. Mol. Med. 53, 310–317. doi: 10.1038/s12276-021-00571-5
Chung, C.-P., Ihara, M., Hilal, S., and Chen, L.-K. (2023). Targeting cerebral small vessel disease to promote healthy aging: preserving physical and cognitive functions in the elderly. Arch. Gerontol. Geriatr. 110:104982. doi: 10.1016/j.archger.2023.104982
Cirrito, J. R., May, P. C., O'Dell, M. A., Taylor, J. W., Parsadanian, M., Cramer, J. W., et al. (2003). In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changes in amyloid-beta metabolism and half-life. J. Neurosci. 23, 8844–8853. doi: 10.1523/jneurosci.23-26-08844.2003
Consolato, F., De Fusco, M., Schaeffer, C., Pieruzzi, F., Scolari, F., Gallieni, M., et al. (2022). α-Gal a missense variants associated with Fabry disease can lead to ER stress and induction of the unfolded protein response. Mol. Genet. Metab. Rep. 33:100926. doi: 10.1016/j.ymgmr.2022.100926
Cummings, J. L. (1988). Intellectual impairment in Parkinson's disease: clinical, pathologic, and biochemical correlates. J. Geriatr. Psychiatry Neurol. 1, 24–36. doi: 10.1177/089198878800100106
Dovonou, A., Bolduc, C., Soto Linan, V., Gora, C., Peralta Iii, M. R., and Lévesque, M. (2023). Animal models of Parkinson’s disease: bridging the gap between disease hallmarks and research questions. Translational. Neurodegeneration 12:36. doi: 10.1186/s40035-023-00368-8
Doyle, J. M., and Croll, R. P. (2022). A critical review of zebrafish models of Parkinson’s disease. Front. Pharmacol. 13:835827. doi: 10.3389/fphar.2022.835827
Drouin-Ouellet, J., Sawiak, S. J., Cisbani, G., Lagacé, M., Kuan, W.-L., Saint-Pierre, M., et al. (2015). Cerebrovascular and blood–brain barrier impairments in Huntington's disease: potential implications for its pathophysiology. Ann. Neurol. 78, 160–177. doi: 10.1002/ana.24406
Drummond, E., and Wisniewski, T. (2017). Alzheimer’s disease: experimental models and reality. Acta Neuropathol. 133, 155–175. doi: 10.1007/s00401-016-1662-x
Duara, R., Lopez-Alberola, R. F., Barker, W. W., Loewenstein, D. A., Zatinsky, M., Eisdorfer, C. E., et al. (1993). A comparison of familial and sporadic Alzheimer's disease. Neurology 43:1377. doi: 10.1212/WNL.43.7.1377
Dziewulska, D., and Lewandowska, E. (2012). Pericytes as a new target for pathological processes in CADASIL. Neuropathology 32, 515–521. doi: 10.1111/j.1440-1789.2011.01290.x
Ehret, F., Moreno Traspas, R., Neumuth, M.-T., Hamann, B., Lasse, D., and Kempermann, G. (2021). Notch3-dependent effects on adult neurogenesis and Hippocampus-dependent learning in a modified transgenic model of CADASIL. Front. Aging Neurosci. 13:617733. doi: 10.3389/fnagi.2021.617733
Elsaid, H. O. A., Furriol, J., Blomqvist, M., Diswall, M., Leh, S., Gharbi, N., et al. (2022a). Reduced α-galactosidase a activity in zebrafish (Danio rerio) mirrors distinct features of Fabry nephropathy phenotype. Mol. Genet. Metab. Rep. 31:100851. doi: 10.1016/j.ymgmr.2022.100851
Elsaid, H. O. A., Tjeldnes, H., Rivedal, M., Serre, C., Eikrem, Ø., Svarstad, E., et al. (2022b). Gene expression analysis in gla-mutant zebrafish reveals enhanced Ca2+ signaling similar to Fabry disease. Int. J. Mol. Sci. 24:358. doi: 10.3390/ijms24010358
Fahlström, A., Yu, Q., and Ulfhake, B. (2011). Behavioral changes in aging female C57BL/6 mice. Neurobiol. Aging 32, 1868–1880. doi: 10.1016/j.neurobiolaging.2009.11.003
Franks, L. M., and Payne, J. (1970). The influence of age on reproductive capacity in C57BL mice. Reproduction 21, 563–565. doi: 10.1530/jrf.0.0210563
Gerhard, G. S., Kauffman, E. J., Wang, X., Stewart, R., Moore, J. L., Kasales, C. J., et al. (2002). Life spans and senescent phenotypes in two strains of zebrafish (Danio rerio). Exp. Gerontol. 37, 1055–1068. doi: 10.1016/S0531-5565(02)00088-8
Ghosh, M., Balbi, M., Hellal, F., Dichgans, M., Lindauer, U., and Plesnila, N. (2015). Pericytes are involved in the pathogenesis of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Ann. Neurol. 78, 887–900. doi: 10.1002/ana.24512
Gil, J. M., and Rego, A. C. (2008). Mechanisms of neurodegeneration in Huntington’s disease. Eur. J. Neurosci. 27, 2803–2820. doi: 10.1111/j.1460-9568.2008.06310.x
Giralt, A., Puigdellivol, M., Carreton, O., Paoletti, P., Valero, J., Parra-Damas, A., et al. (2012). Long-term memory deficits in Huntington's disease are associated with reduced CBP histone acetylase activity. Hum. Mol. Genet. 21, 1203–1216. doi: 10.1093/hmg/ddr552
Giralt, A., Saavedra, A., Carretón, O., Xifró, X., Alberch, J., and Pérez-Navarro, E. (2011). Increased PKA signaling disrupts recognition memory and spatial memory: role in Huntington's disease. Hum. Mol. Genet. 20, 4232–4247. doi: 10.1093/hmg/ddr351
Gravesteijn, G., Munting, L. P., Overzier, M., Mulder, A. A., Hegeman, I., Derieppe, M., et al. (2020). Progression and classification of granular Osmiophilic material (GOM) deposits in functionally characterized human NOTCH3 transgenic mice. Transl. Stroke Res. 11, 517–527. doi: 10.1007/s12975-019-00742-7
Greenberg, S. M., Bacskai, B. J., Hernandez-Guillamon, M., Pruzin, J., Sperling, R., and Van Veluw, S. J. (2020). Cerebral amyloid angiopathy and Alzheimer disease — one peptide, two pathways. Nat. Rev. Neurol. 16, 30–42. doi: 10.1038/s41582-019-0281-2
Hara, K., Shiga, A., Fukutake, T., Nozaki, H., Miyashita, A., Yokoseki, A., et al. (2009). Association of HTRA1 mutations and familial ischemic cerebral small-vessel disease. N. Engl. J. Med. 360, 1729–1739. doi: 10.1056/nejmoa0801560
Hofmann, L., Karl, F., Sommer, C., and Üçeyler, N. (2017). Affective and cognitive behavior in the alpha-galactosidase a deficient mouse model of Fabry disease. PLoS One 12:e0180601. doi: 10.1371/journal.pone.0180601
Hunter, S. K., Pereira, H. M., and Keenan, K. G. (2016). The aging neuromuscular system and motor performance. J. Appl. Physiol. 121, 982–995. doi: 10.1152/japplphysiol.00475.2016
Hussein, A., Tielemans, A., Baxter, M. G., Benson, D. L., and Huntley, G. W. (2022). Cognitive deficits and altered cholinergic innervation in young adult male mice carrying a Parkinson's disease Lrrk2(G2019S) knockin mutation. Exp. Neurol. 355:114145. doi: 10.1016/j.expneurol.2022.114145
Iliff, J. J., Wang, M., Zeppenfeld, D. M., Venkataraman, A., Plog, B. A., Liao, Y., et al. (2013). Cerebral arterial pulsation drives Paravascular CSF–interstitial fluid exchange in the murine brain. J. Neurosci. 33, 18190–18199. doi: 10.1523/jneurosci.1592-13.2013
Jankowsky, J. L., Slunt, H. H., Gonzales, V., Savonenko, A. V., Wen, J. C., Jenkins, N. A., et al. (2005). Persistent amyloidosis following suppression of Aβ production in a transgenic model of Alzheimer disease. PLoS Med. 2:e355. doi: 10.1371/journal.pmed.0020355
Jeong, Y.-M., Lee, J.-G., Cho, H.-J., Lee, W. S., Jeong, J., and Lee, J.-S. (2021). Differential clearance of Aβ species from the brain by brain lymphatic endothelial cells in zebrafish. Int. J. Mol. Sci. 22:11883. doi: 10.3390/ijms222111883
Joutel, A., Monet-Leprêtre, M., Gosele, C., Baron-Menguy, C., Hammes, A., Schmidt, S., et al. (2010). Cerebrovascular dysfunction and microcirculation rarefaction precede white matter lesions in a mouse genetic model of cerebral ischemic small vessel disease. J. Clin. Invest. 120, 433–445. doi: 10.1172/jci39733
Kalaria, R. N., Viitanen, M., Kalimo, H., Dichgans, M., and Tabira, T. (2004). The pathogenesis of CADASIL: an update. J. Neurol. Sci. 226, 35–39. doi: 10.1016/j.jns.2004.09.008
Kalia, L. V., and Lang, A. E. (2015). Parkinson's disease. Lancet 386, 896–912. doi: 10.1016/s0140-6736(14)61393-3
Kato, T., Manabe, R.-I., Igarashi, H., Kametani, F., Hirokawa, S., Sekine, Y., et al. (2021). Candesartan prevents arteriopathy progression in cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy model. J. Clin. Invest. 131:e140555. doi: 10.1172/jci140555
Kaushik, S., and Cuervo, A. M. (2015). Proteostasis and aging. Nat. Med. 21, 1406–1415. doi: 10.1038/nm.4001
Kawasumi, M., Chiba, T., Yamada, M., Miyamae-Kaneko, M., Matsuoka, M., Nakahara, J., et al. (2004). Targeted introduction of V642I mutation in amyloid precursor protein gene causes functional abnormality resembling early stage of Alzheimer's disease in aged mice. Eur. J. Neurosci. 19, 2826–2838. doi: 10.1111/j.0953-816x.2004.03397.x
Kaye, J., Reisine, T., and Finkbeiner, S. (2021). Huntington’s disease mouse models: unraveling the pathology caused by CAG repeat expansion. Faculty Rev. 10:77. doi: 10.12703/r/10-77
King, D. L., and Arendash, G. W. (2002). Behavioral characterization of the Tg2576 transgenic model of Alzheimer's disease through 19 months. Physiol. Behav. 75, 627–642. doi: 10.1016/S0031-9384(02)00639-X
Kishi, S. (2004). Functional aging and gradual senescence in zebrafish. Ann. N. Y. Acad. Sci. 1019, 521–526. doi: 10.1196/annals.1297.097
Kitazoe, Y., Kishino, H., Tanisawa, K., Udaka, K., and Tanaka, M. (2019). Renormalized basal metabolic rate describes the human aging process and longevity. Aging Cell 18:e12968. doi: 10.1111/acel.12968
Köhler, C., Ebert, U., Baumann, K., and Schröder, H. (2005). Alzheimer's disease-like neuropathology of gene-targeted APP-SLxPS1mut mice expressing the amyloid precursor protein at endogenous levels. Neurobiol. Dis. 20, 528–540. doi: 10.1016/j.nbd.2005.04.009
Kowalczyk, A., Partha, R., Clark, N. L., and Chikina, M. (2020). Pan-mammalian analysis of molecular constraints underlying extended lifespan. eLife 9:e51089. doi: 10.7554/eLife.51089
Kumar, V., Singh, C., and Singh, A. (2021). Zebrafish an experimental model of Huntington’s disease: molecular aspects, therapeutic targets and current challenges. Mol. Biol. Rep. 48, 8181–8194. doi: 10.1007/s11033-021-06787-y
Kuparinen, A., Yeung, E., and Hutchings, J. A. (2023). Correlation between body size and longevity: new analysis and data covering six taxonomic classes of vertebrates. Acta Oecol. 119:103917. doi: 10.1016/j.actao.2023.103917
Lacombe, P., Oligo, C., Domenga, V. R., Tournier-Lasserve, E., and Joutel, A. (2005). Impaired cerebral Vasoreactivity in a transgenic mouse model of cerebral autosomal dominant Arteriopathy with subcortical infarcts and leukoencephalopathy Arteriopathy. Stroke 36, 1053–1058. doi: 10.1161/01.str.0000163080.82766.eb
Lalonde, R., and Strazielle, C. (2005). PS1 knockin mice with the Japanese I213T mutation: effects on exploratory activity, motor coordination, and spatial learning. Behav. Brain Res. 162, 182–190. doi: 10.1016/j.bbr.2005.02.037
Li, X., Wang, X., Zhang, C., Wang, J., Wang, S., and Hu, L. (2022). Dysfunction of metabolic activity of bone marrow mesenchymal stem cells in aged mice. Cell Prolif. 55:e13191. doi: 10.1111/cpr.13191
Lin, C.-H., Chen, P.-L., Tai, C.-H., Lin, H.-I., Chen, C.-S., Chen, M.-L., et al. (2019). A clinical and genetic study of early-onset and familial parkinsonism in Taiwan: An integrated approach combining gene dosage analysis and next-generation sequencing. Mov. Disord. 34, 506–515. doi: 10.1002/mds.27633
Lione, L. A., Carter, R. J., Hunt, M. J., Bates, G. P., Morton, A. J., and Dunnett, S. B. (1999). Selective discrimination learning impairments in mice expressing the human Huntington's disease mutation. J. Neurosci. 19, 10428–10437. doi: 10.1523/jneurosci.19-23-10428.1999
Little, T. D. (1997). Mean and covariance structures (MACS) analyses of cross-cultural data: practical and theoretical issues. Multivar. Behav. Res. 32, 53–76. doi: 10.1207/s15327906mbr3201_3
Liu, X.-Y., Gonzalez-Toledo, M. E., Fagan, A., Duan, W.-M., Liu, Y., Zhang, S., et al. (2015). Stem cell factor and granulocyte colony-stimulating factor exhibit therapeutic effects in a mouse model of CADASIL. Neurobiol. Dis. 73, 189–203. doi: 10.1016/j.nbd.2014.09.006
Loforese, G., Malinka, T., Keogh, A., Baier, F., Simillion, C., Montani, M., et al. (2017). Impaired liver regeneration in aged mice can be rescued by silencing hippo core kinases MST1 and MST2. EMBO Mol. Med. 9, 46–60. doi: 10.15252/emmm.201506089
Lopera, F., Ardilla, A., Martinez, A., Madrigal, L., Arango-Viana, J. C., Lemere, C. A., et al. (1997). Clinical features of early-onset Alzheimer disease in a large kindred with an E280A presenilis-1 mutation. Am. J. Ophthalmol. 124, 137–138. doi: 10.1016/S0002-9394(14)71677-0
Lüesse, H.-G., Schiefer, J., Spruenken, A., Puls, C., Block, F., and Kosinski, C. M. (2001). Evaluation of R6/2 HD transgenic mice for therapeutic studies in Huntington's disease: behavioral testing and impact of diabetes mellitus. Behav. Brain Res. 126, 185–195. doi: 10.1016/S0166-4328(01)00261-3
Lundkvist, J., Zhu, S., Hansson, E. M., Schweinhardt, P., Miao, Q., Beatus, P., et al. (2005). Mice carrying a R142C notch 3 knock-in mutation do not develop a CADASIL-like phenotype. Genesis 41, 13–22. doi: 10.1002/gene.20091
Magen, I., and Chesselet, M.-F. (2011). Mouse models of cognitive deficits due to alpha-Synuclein pathology. J. Parkinsons Dis. 1, 217–227. doi: 10.3233/jpd-2011-11043
Magen, I., Fleming, S. M., Zhu, C., Garcia, E. C., Cardiff, K. M., Dinh, D., et al. (2012). Cognitive deficits in a mouse model of pre-manifest Parkinson’s disease. Eur. J. Neurosci. 35, 870–882. doi: 10.1111/j.1460-9568.2012.08012.x
Mawuenyega, K. G., Sigurdson, W., Ovod, V., Munsell, L., Kasten, T., Morris, J. C., et al. (2010). Decreased clearance of CNS β-amyloid in Alzheimer’s disease. Science 330:1774. doi: 10.1126/science.1197623
Menalled, L. B., Kudwa, A. E., Miller, S., Fitzpatrick, J., Watson-Johnson, J., Keating, N., et al. (2012). Comprehensive behavioral and molecular characterization of a new Knock-in mouse model of Huntington’s disease: zQ175. PLoS One 7:e49838. doi: 10.1371/journal.pone.0049838
Mestre, H., Kostrikov, S., Mehta, I., and Nedergaard, M. (2017). Perivascular spaces, glymphatic dysfunction, and small vessel disease. Clin. Sci. 131, 2257–2274. doi: 10.1042/cs20160381
Metaxas, A., and Kempf, S. J. (2016). Neurofibrillary tangles in Alzheimer's disease: elucidation of the molecular mechanism by immunohistochemistry and tau protein phospho-proteomics. Neural Regen. Res. 11, 1579–1581. doi: 10.4103/1673-5374.193234
Mizoguchi, T., Okita, M., Minami, Y., Fukunaga, M., Maki, A., and Itoh, M. (2023). Age-dependent dysfunction of the cerebrovascular system in the zebrafish telencephalon. Exp. Gerontol. 178:112206. doi: 10.1016/j.exger.2023.112206
Monma, Y., Shimada, Y., Nakayama, H., Zang, L., Nishimura, N., and Tanaka, T. (2019). Aging-associated microstructural deterioration of vertebra in zebrafish. Bone Rep. 11:100215. doi: 10.1016/j.bonr.2019.100215
Mousavi, S. E., and Patil, J. G. (2020). Light-cardiogram, a simple technique for heart rate determination in adult zebrafish, Danio rerio. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 246:110705. doi: 10.1016/j.cbpa.2020.110705
Nedergaard, M. (2013). Garbage truck of the brain. Science 340, 1529–1530. doi: 10.1126/science.1240514
Ohshima, T., Murray, G. J., Swaim, W. D., Longenecker, G., Quirk, J. M., Cardarelli, C. O., et al. (1997). α-Galactosidase a deficient mice: a model of Fabry disease. Proc. Natl. Acad. Sci. 94, 2540–2544. doi: 10.1073/pnas.94.6.2540
Pantano, P., Baron, J. C., Lebrun-Grandié, P., Duquesnoy, N., Bousser, M. G., and Comar, D. (1984). Regional cerebral blood flow and oxygen consumption in human aging. Stroke 15, 635–641. doi: 10.1161/01.str.15.4.635
Pantoni, L. (2010). Cerebral small vessel disease: from pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol. 9, 689–701. doi: 10.1016/S1474-4422(10)70104-6
Paul, G., and Elabi, O. F. (2022). Microvascular changes in Parkinson’s disease- focus on the neurovascular unit. Front. Aging Neurosci. 14:853372. doi: 10.3389/fnagi.2022.853372
Percy, M. E., Markovic, V. D., McLachlan, D. R. C., Berg, J. M., Hummel, J. T., Laing, M. E., et al. (1991). Family with 22-derived marker chromosome and late-onset dementia of the Alzheimer type: I. Application of a new model for estimation of the risk of disease associated with the marker. Am. J. Med. Genet. 39, 307–313. doi: 10.1002/ajmg.1320390312
Pischedda, F., Cirnaru, M. D., Ponzoni, L., Sandre, M., Biosa, A., Carrion, M. P., et al. (2021). LRRK2 G2019S kinase activity triggers neurotoxic NSF aggregation. Brain 144, 1509–1525. doi: 10.1093/brain/awab073
Pu, Y. Z., Liang, L., Fu, A. L., Liu, Y., Sun, L., Li, Q., et al. (2017). Generation of Alzheimer's disease transgenic zebrafish expressing human APP mutation under control of zebrafish appb promotor. Curr. Alzheimer Res. 14, 668–679. doi: 10.2174/1567205013666161201202000
Qosa, H., Abuasal, B. S., Romero, I. A., Weksler, B., Couraud, P.-O., Keller, J. N., et al. (2014). Differences in amyloid-β clearance across mouse and human blood–brain barrier models: kinetic analysis and mechanistic modeling. Neuropharmacology 79, 668–678. doi: 10.1016/j.neuropharm.2014.01.023
Quiroz, Y. T., Budson, A. E., Celone, K., Ruiz, A., Newmark, R., Castrillón, G., et al. (2010). Hippocampal hyperactivation in presymptomatic familial Alzheimer's disease. Ann. Neurol. 68, 865–875. doi: 10.1002/ana.22105
Rando, T. A., and Jones, D. L. (2021). Regeneration, rejuvenation, and replacement: turning Back the clock on tissue aging. Cold Spring Harb. Perspect. Biol. 13:a040907. doi: 10.1101/cshperspect.a040907
Richards, A., Van Den Maagdenberg, A. M. J. M., Jen, J. C., Kavanagh, D., Bertram, P., Spitzer, D., et al. (2007). C-terminal truncations in human 3′-5′ DNA exonuclease TREX1 cause autosomal dominant retinal vasculopathy with cerebral leukodystrophy. Nat. Genet. 39, 1068–1070. doi: 10.1038/ng2082
Ross, C. A., and Tabrizi, S. J. (2011). Huntington's disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 10, 83–98. doi: 10.1016/s1474-4422(10)70245-3
Ruchoux, M. M., Domenga, V., Brulin, P., Maciazek, J., Limol, S., Tournier-Lasserve, E., et al. (2003). Transgenic mice expressing mutant Notch3 develop vascular alterations characteristic of cerebral autosomal dominant Arteriopathy with subcortical infarcts and leukoencephalopathy. Am. J. Pathol. 162, 329–342. doi: 10.1016/s0002-9440(10)63824-2
Ruhl, T., Jonas, A., Seidel, N. I., Prinz, N., Albayram, O., Bilkei-Gorzo, A., et al. (2015). Oxidation and cognitive impairment in the aging zebrafish. Gerontology 62, 47–57. doi: 10.1159/000433534
Rutkove, S. B., Callegari, S., Concepcion, H., Mourey, T., Widrick, J., Nagy, J. A., et al. (2023). Electrical impedance myography detects age-related skeletal muscle atrophy in adult zebrafish. Sci. Rep. 13:7191. doi: 10.1038/s41598-023-34119-6
Rutten, J. W., Klever, R. R., Hegeman, I. M., Poole, D. S., Dauwerse, H. G., Broos, L. A. M., et al. (2015). The NOTCH3 score: a pre-clinical CADASIL biomarker in a novel human genomic NOTCH3 transgenic mouse model with early progressive vascular NOTCH3 accumulation. Acta Neuropathol. Commun. 3:89. doi: 10.1186/s40478-015-0268-1
Saito, T., Matsuba, Y., Mihira, N., Takano, J., Nilsson, P., Itohara, S., et al. (2014). Single app knock-in mouse models of Alzheimer's disease. Nat. Neurosci. 17, 661–663. doi: 10.1038/nn.3697
Saito, T., Suemoto, T., Brouwers, N., Sleegers, K., Funamoto, S., Mihira, N., et al. (2011). Potent amyloidogenicity and pathogenicity of Aβ43. Nat. Neurosci. 14, 1023–1032. doi: 10.1038/nn.2858
Sakai, T., Miyazaki, T., Shin, D.-M., Kim, Y.-S., Qi, C.-F., Fariss, R., et al. (2017). DNase-active TREX1 frame-shift mutants induce serologic autoimmunity in mice. J. Autoimmun. 81, 13–23. doi: 10.1016/j.jaut.2017.03.001
Sakakibara, Y., Sekiya, M., Saito, T., Saido, T. C., and Iijima, K. M. (2019). Amyloid-β plaque formation and reactive gliosis are required for induction of cognitive deficits in app knock-in mouse models of Alzheimer's disease. BMC Neurosci. 20:13. doi: 10.1186/s12868-019-0496-6
Sanchez-Varo, R., Mejias-Ortega, M., Fernandez-Valenzuela, J. J., Nuñez-Diaz, C., Caceres-Palomo, L., Vegas-Gomez, L., et al. (2022). Transgenic mouse models of Alzheimer’s disease: An integrative analysis. Int. J. Mol. Sci. 23:5404. doi: 10.3390/ijms23105404
Sasaguri, H., Nilsson, P., Hashimoto, S., Nagata, K., Saito, T., De Strooper, B., et al. (2017). APP mouse models for Alzheimer's disease preclinical studies. EMBO J. 36, 2473–2487. doi: 10.15252/embj.201797397
Saura, C. A., Choi, S.-Y., Beglopoulos, V., Malkani, S., Zhang, D., Rao, B. S. S., et al. (2004). Loss of Presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron 42, 23–36. doi: 10.1016/s0896-6273(04)00182-5
Schuh, E., Ertl-Wagner, B., Lohse, P., Wolf, W., Mann, J. F., Lee-Kirsch, M. A., et al. (2015). Multiple sclerosis–like lesions and type I interferon signature in a patient with RVCL. Neurol. Neuroimmunol. Neuroinflamm. 2:e55. doi: 10.1212/NXI.0000000000000055
Sepehrinezhad, A., Stolze Larsen, F., Ashayeri Ahmadabad, R., Shahbazi, A., and Sahab Negah, S. (2023). The Glymphatic system May play a vital role in the pathogenesis of hepatic encephalopathy: a narrative review. Cells 12:979. doi: 10.3390/cells12070979
Shojaee, S., Sina, F., Farboodi, N., Fazlali, Z., Ghazavi, F., Ghorashi, S. A., et al. (2009). A clinic-based screening of mutations in exons 31, 34, 35, 41, and 48 of LRRK2 in Iranian Parkinson's disease patients. Mov. Disord. 24, 1023–1027. doi: 10.1002/mds.22503
Simmons, D. A., Rex, C. S., Palmer, L., Pandyarajan, V., Fedulov, V., Gall, C. M., et al. (2009). Up-regulating BDNF with an ampakine rescues synaptic plasticity and memory in Huntington's disease knockin mice. Proc. Natl. Acad. Sci. 106, 4906–4911. doi: 10.1073/pnas.0811228106
Søndergaard, C. B., Nielsen, J. E., Hansen, C. K., and Christensen, H. (2017). Hereditary cerebral small vessel disease and stroke. Clin. Neurol. Neurosurg. 155, 45–57. doi: 10.1016/j.clineuro.2017.02.015
Soto-Faguás, C. M., Sanchez-Molina, P., and Saura, C. A. (2021). Loss of presenilin function enhances tau phosphorylation and aggregation in mice. Acta Neuropathol. Commun. 9:162. doi: 10.1186/s40478-021-01259-7
Spira, P. J., Sharpe, D. M., Halliday, G., Cavanagh, J., and Nicholson, G. A. (2001). Clinical and pathological features of a parkinsonian syndrome in a family with an Ala53Thr α-synuclein mutation. Ann. Neurol. 49, 313–319. doi: 10.1002/ana.67
Stefanis, L. (2012). α-Synuclein in Parkinson's disease. Cold Spring Harb. Perspect. Med. 2:a009399. doi: 10.1101/cshperspect.a009399
Sundvik, M., Chen, Y. C., and Panula, P. (2013). Presenilin1 regulates histamine neuron development and behavior in zebrafish, danio rerio. J. Neurosci. 33, 1589–1597. doi: 10.1523/jneurosci.1802-12.2013
Szulc, P., Garnero, P., Munoz, F., Marchand, F., and Delmas, P. D. (2001). Cross-sectional evaluation of bone metabolism in men. J. Bone Miner. Res. 16, 1642–1650. doi: 10.1359/jbmr.2001.16.9.1642
Tan, R. Y. Y., and Markus, H. S. (2016). CADASIL: migraine, encephalopathy, stroke and their inter-relationships. PLoS One 11:e0157613. doi: 10.1371/journal.pone.0157613
Tikka, S., Baumann, M., Siitonen, M., Pasanen, P., Pöyhönen, M., Myllykangas, L., et al. (2014). CADASIL and CARASIL. Brain Pathol. 24, 525–544. doi: 10.1111/bpa.12181
Tower, R. J., Busse, E., Jaramillo, J., Lacey, M., Hoffseth, K., Guntur, A. R., et al. (2022). Spatial transcriptomics reveals metabolic changes underly age-dependent declines in digit regeneration. eLife 11:e71542. doi: 10.7554/eLife.71542
Tsai, S. B., Tucci, V., Uchiyama, J., Fabian, N. J., Lin, M. C., Bayliss, P. E., et al. (2007). Differential effects of genotoxic stress on both concurrent body growth and gradual senescence in the adult zebrafish. Aging Cell 6, 209–224. doi: 10.1111/j.1474-9726.2007.00278.x
Walker, L. C., Bian, F., Callahan, M. J., Lipinski, W. J., Durham, R. A., and Levine, H. (2002). Modeling Alzheimer's disease and other proteopathies in vivo: is seeding the key? Amino Acids 23, 87–93. doi: 10.1007/s00726-001-0113-7
Wallays, G., Nuyens, D., Silasi-Mansat, R., Souffreau, J., Callaerts-Vegh, Z., Van Nuffelen, A., et al. (2011). Notch3 Arg170Cys Knock-in mice display pathologic and clinical features of the neurovascular disorder cerebral autosomal dominant Arteriopathy with subcortical infarcts and leukoencephalopathy. Arterioscler. Thromb. Vasc. Biol. 31, 2881–2888. doi: 10.1161/atvbaha.111.237859
Wan, D., Ai, S., Ouyang, H., and Cheng, L. (2021). Activation of 4-1BB signaling in bone marrow stromal cells triggers bone loss via the p-38 MAPK-DKK1 axis in aged mice. Exp. Mol. Med. 53, 654–666. doi: 10.1038/s12276-021-00605-y
Wang, L. W., Huttner, I. G., Santiago, C. F., Kesteven, S. H., Yu, Z.-Y., Feneley, M. P., et al. (2017). Standardized echocardiographic assessment of cardiac function in normal adult zebrafish and heart disease models. Dis. Model. Mech. 10, 63–76. doi: 10.1242/dmm.026989
Wang, Y.-J., Sun, Y.-R., Pei, Y.-H., Ma, H.-W., Mu, Y.-K., Qin, L.-H., et al. (2023). The lymphatic drainage systems in the brain: a novel target for ischemic stroke? Neural Regen. Res. 18:485. doi: 10.4103/1673-5374.346484
Waterston, R. H., Lindblad-Toh, K., Birney, E., Rogers, J., Abril, J. F., Agarwal, P., et al. (2002). Initial sequencing and comparative analysis of the mouse genome. Nature 420, 520–562. doi: 10.1038/nature01262
Wei, Z., Chen, L., Hou, X., Van Zijl, P. C. M., Xu, J., and Lu, H. (2020). Age-related alterations in brain perfusion, venous oxygenation, and oxygen metabolic rate of mice: a 17-month longitudinal MRI study. Front. Neurol. 11:559. doi: 10.3389/fneur.2020.00559
Wexler, N. S. (2004). Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington's disease age of onset. Proc. Natl. Acad. Sci. 101, 3498–3503. doi: 10.1073/pnas.0308679101
White, R. M., Sessa, A., Burke, C., Bowman, T., Leblanc, J., Ceol, C., et al. (2008). Transparent adult zebrafish as a tool for in vivo transplantation analysis. Cell Stem Cell 2, 183–189. doi: 10.1016/j.stem.2007.11.002
Wong, Y. C., and Krainc, D. (2017). α-Synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat. Med. 23, 1–13. doi: 10.1038/nm.4269
Xia, D., Watanabe, H., Wu, B., Lee, H., Li, Y., Tsvetkov, E., et al. (2015). Presenilin-1 Knockin mice reveal loss-of-function mechanism for familial Alzheimer’s disease. Neuron 85, 967–981. doi: 10.1016/j.neuron.2015.02.010
Xu, N., Lagrow, T. J., Anumba, N., Lee, A., Zhang, X., Yousefi, B., et al. (2022). Functional connectivity of the brain across rodents and humans. Front. Neurosci. 16:816331. doi: 10.3389/fnins.2022.816331
Yanai, S., and Endo, S. (2021). Functional aging in male C57BL/6J mice across the life-span: a systematic behavioral analysis of motor, emotional, and memory function to define an aging phenotype. Front. Aging Neurosci. 13:697621. doi: 10.3389/fnagi.2021.697621
Yang, P., Kajiwara, R., Tonoki, A., and Itoh, M. (2018). Successive and discrete spaced conditioning in active avoidance learning in young and aged zebrafish. Neurosci. Res. 130, 1–7. doi: 10.1016/j.neures.2017.10.005
Yang, P., Yamaki, M., Kuwabara, S., Kajiwara, R., and Itoh, M. (2019). A newly developed feeder and oxygen measurement system reveals the effects of aging and obesity on the metabolic rate of zebrafish. Exp. Gerontol. 127:110720. doi: 10.1016/j.exger.2019.110720
Yhnell, E., Dunnett, S. B., and Brooks, S. P. (2016). A longitudinal operant assessment of cognitive and Behavioural changes in the HdhQ111 mouse model of Huntington’s disease. PLoS One 11:e0164072. doi: 10.1371/journal.pone.0164072
Yokoyama, M., Kobayashi, H., Tatsumi, L., and Tomita, T. (2022). Mouse models of Alzheimer’s disease. Front. Mol. Neurosci. 15, 1171–1183. doi: 10.3389/fnmol.2022.912995
Young, A. B. (2003). Huntingtin in health and disease. J. Clin. Invest. 111, 299–302. doi: 10.1172/jci17742
Keywords: memory, aging, mouse, zebrafish, lifespan, cerebral blood vessel, Alzheimer’s disease
Citation: Kanoh T, Mizoguchi T, Tonoki A and Itoh M (2024) Modeling of age-related neurological disease: utility of zebrafish. Front. Aging Neurosci. 16:1399098. doi: 10.3389/fnagi.2024.1399098
Edited by:
Liz Girardi Müller, Regional Community University of Chapecó, BrazilReviewed by:
Dong Yang, The Scripps Research Institute, United StatesCopyright © 2024 Kanoh, Mizoguchi, Tonoki and Itoh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Motoyuki Itoh, bWl0b0BjaGliYS11Lmpw
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.