 Mandana Amelimojarad
Mandana Amelimojarad Melika Amelimojarad
Melika Amelimojarad Xiaonan Cui
Xiaonan Cui
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Aging Neurosci. , 03 July 2024
Sec. Alzheimer's Disease and Related Dementias
Volume 16 - 2024 | https://doi.org/10.3389/fnagi.2024.1391517
This article is part of the Research Topic A comprehensive look at biomarkers in neurodegenerative diseases: from early diagnosis to treatment response assessment View all 23 articles
As the most common cause of dementia, Alzheimer’s disease (AD) is characterized by neurodegeneration and synaptic loss with an increasing prevalence in the elderly. Increased inflammatory responses triggers brain cells to produce pro-inflammatory cytokines and accelerates the Aβ accumulation, tau protein hyper-phosphorylation leading to neurodegeneration. Therefore, in this paper, we discuss the current understanding of how inflammation affects brain activity to induce AD pathology, the inflammatory biomarkers and possible therapies that combat inflammation for AD.
Dementia is a noxious neurodegenerative disorder, and Alzheimer’s disease (AD), is the most common cause of dementia with increasing prevalence among the elderly (Aarsland, 2020). According to the World Health Organization (WHO), by 2050 the number of people with dementia will reach 132 million in the world (Porsteinsson et al., 2021; Ren et al., 2022).
AD is characterized by progressive neuropathological processes including cognitive function impairment and memory loss principally caused by increased accumulation of amyloid-β (Aβ) plaques, and hyperphosphorylated tau protein (Nakamura et al., 2018; Silva et al., 2019).
Different risk factors have been detected for AD development such as age, cardiovascular changes, metabolic disorders, increased metal ions accumulation, and brain injury (Baltes et al., 2011; Silva et al., 2019). Despite the vicious role of amyloid plaques and neurofibrillary tangles in the brain, the significant role of abnormal inflammation in inducing the inflammatory mediators release from brain cells, neurodegeneration, and loss of neuronal synapses is considered as the new hallmarks of AD pathology (Gouras et al., 2015; Newcombe et al., 2018; Muralidar et al., 2020).
Currently, nonsteroidal anti-inflammatory drugs (NSAIDs) and cholinesterase inhibitors are approved drugs to delay AD but none of them could cure the disease (Moride et al., 2003; Long and Holtzman, 2019). Therefore, identification of core pathologies mechanism responsible for AD, different proteins and genes associating with neuroinflammation and potential therapeutic targets is essential (Weggen et al., 2003; Benito-León et al., 2019).
In this review, we focused on an in-depth evaluation of the Blood–Brain Barrier (BBB), the brain cells especially the microglia modification in inducing the inflammatory responses as a new interest target of AD pathogenesis research. In addition, we highlighted all the inflammatory biomarkers with the potential to be used for targeted therapy.
Microglia and astrocytes are the two main neuroglial cells, playing critical functions in Homeostasis, neuron development, differentiation, survival, synaptic plasticity, and neuronal metabolism (Zhang et al., 2021; McKee et al., 2023). The activation process of microglia and astrocytes is followed by a series of morphological and biological functions leading to the release of pro-inflammatory mediators and phagocytic activity (Weggen et al., 2003; Zhou and Hu, 2013; Greten and Grivennikov, 2019).
Microglia are one of macrophagic immune cells that reside in the central nervous system (CNS) and play important roles in surveillance and phagocytosis (Sheng et al., 2019). by recruiting other innate immune cells like neutrophils, dendritic cells, monocytes, invasive macrophages, and natural killer (NK) cells, Microglia are considered as important modulators of the innate immune response in the brain.
In response to infection, the inflammatory response activates resting microglia and encourages the release of free radicals (NO), reactive oxygen species (ROS), and pro-inflammatory cytokines (e.g., IL-1β, IL6, TNF). There are two types of activated microglia states: pro-inflammatory (M1-like; neurotoxic) and anti-inflammatory (M2-like; neuroprotective). Therefore, M1 and M2 polarization switches play the most significant role in the proper activation of microglia and release of pro-inflammatory mediators (Ransohoff, 2016; Greten and Grivennikov, 2019; Ho et al., 2020).
Although, Activation of microglia, seems to help in the clearance of Aβ during the chronic phase of neuroinflammation and early development of AD through phagocytosis (Villeda et al., 2011; Long and Holtzman, 2019). According to Newcombe et al. (2018), the pathogenesis of AD may be advanced by the microglia’s continuous brain stimulation in response to the accumulation of Aβ plaque, tau protein phosphorylation, and inflammatory responses which impairs their ability to phagocytose, produces pro-inflammatory mediators, and exacerbates tau and Aβ pathology (Leng and Edison, 2020).
Mutations in microglia-related genes have a substantial impact on the ability of microglia, causing them to become permanently activated, reducing their capacity for phagocytosis, and ultimately resulting in neuroinflammation and neurodegeneration (Zhang et al., 2021). Therefore, understanding the molecular mechanism of microglia is highly important to detect their dual roles in either Aβ plaques accumulation or degradation (Baltes et al., 2011).
Recently certain molecular regulators of microglial proliferation have been directly demonstrated to exist including triggering receptor expressed on myeloid cells 2 (TREM2) and apolipoprotein E (APOE) which are both among AD risk factors for Late Onset AD (LOAD) (Wolfe et al., 2018).
In the central nervous system, APOE plays multiple roles, such as maintaining lipid homeostasis, healing damaged neurons, eliminating toxins like Aβ, and immune responses modulator (Bertram et al., 2008). Among all APOE isoforms, APOE4 has been shown to exacerbate tau-mediated neurodegeneration, while the absence of APOE is protective in Patients with AD (Liu et al., 2013). Patients who carry at least one APOEε4 allele shows faster disease progression, and increased brain atrophy compared to non-APOEε4 carriers (LaDu et al., 1995; Shi et al., 2017). As previously mentioned, it inhibits the gene that produces SirT1, a molecule that has been associated with longer lifespans and has anti-Alzheimer’s properties and instead It’s linked to nuclear factor kappa B (NF-κB) activation, which encourages inflammation (Shi et al., 2017). This explains why ApoE4 is linked to an elevated inflammatory response: it suppresses multiple genes that inhibit inflammation while accelerating the NF-κB that stimulates it (Teng et al., 2017).
Aβ binding to APOE and other apolipoproteins was tested in different in vitro (Shi et al., 2017; Zhang et al., 2021). Even though the binding was consistently verified, none of those investigations suggested that variations in APOE-Aβ binding were linked to an increased risk of AD (Keren-Shaul et al., 2017). According to Yuan et al., TREM2 deficiency increased the amount of diffuse amyloid plaques that covered a greater surface area due to longer and more branched amyloid fibrils (Yuan et al., 2016a). Through TREM2 binding APOE evaluates the phagocytosis and APOE-Aβ uptake, while the TREM2 R47H variant has less affinity to bind with APOE (Tao et al., 2018; Sheng et al., 2019). Due to its dysregulation of neuroinflammation and elevation of AD risk, the missense mutation R47H of TREM2 is linked to AD risk (Ruganzu et al., 2021). A dose-dependent reduction in TREM2 inhibits the accumulation of myeloid cells surrounding Aβ plaques. In addition, plaque number and size are decreased in TREM2 deficiency (Wang et al., 2016; Yeh et al., 2016).
Microglia in plaque-loaded brain areas of AD transgenic mice expressed more TREM2, suggesting a significant role for TREM2 against AD (Yuan et al., 2016a). Growing data indicates that TREM2 deficiency support microglial phagocytosis and maintain microglial responses to Aβ deposition through inhibit the transition of microglia from a homeostatic to a disease-oriented state (Wang et al., 2016; Yuan et al., 2016b). TREM2 in blood and CSF can act as biomarker for the diagnosis of early AD since, the TREM2 levels in CSF increase in the early stages of AD, while it decreases in late stages (Wang et al., 2016).
Beside the two last popular AD hallmark genes, recent data suggests that the fractalkine ligand and its microglial receptor (CX3CL1/CX3CR1) can influence pathologies related to tau by controlling microglial migration and attracting monocytes to the brain (Lyons et al., 2009; Joaquín Merino et al., 2016).
Microglia most likely proliferate more quickly and assemble around fibrillar amyloid plaques because of dysregulated fractalkine/CX3CR1 signaling, brought on by CX3CR1 receptor deletion, indicating that CX3CR1 has been found to maintain microglia in an inactive, non-neurotoxic condition (Lyons et al., 2009; Bhaskar et al., 2010; Lee et al., 2010).
Mice deficient in CX3CR1 showed a alters the inflammatory milieu, decreased neuronal loss, and increase of the amount of Aβ phagocytosis mediated by microglia however an aggravated tau phosphorylation was also detected (Yin et al., 2017).
Similarly, colony-stimulating factor 1 receptor (CSF1R), inhibition has attenuated the neurodegeneration process caused by tau proteins (Chadarevian et al., 2023). Mutation of IFNγ receptors increases Aβ synthesis and microglial activation (Orihuela et al., 2016; Huang et al., 2018).
The CSF-1-CSF-1R pathway, which is mainly active in reactive microgliosis conditions has also been connected to microglia survival in the context of TREM2 expression (Öst et al., 2006; Spangenberg et al., 2019). This pathway affects Aβ clearance. A similar mechanism may also be involved in microglial survival, as it has been shown that TREM2 promote macrophage survival via CSF-1R pathway (Chadarevian et al., 2023). The role of CSF-1R signaling in microglia survival is detected by a study indicating that TREM2-deficient microglia to exhibit reduced survival at low CSF-1 concentrations (Mancuso et al., 2019).
The genetic deletion of the inflammatory NLR family pyrin domain containing 3 (NLRP3) facilitates the synthesis of IL-1β and improves Aβ clearance by microglia as well as cognitive function in AD mice (Wang Z. et al., 2020; Bai and Zhang, 2021). NLRP3 activation increase the AD pathogenesis by damaging the microglia mitochondrial aggregation and impairs the structural and functional integrity of mitochondria by increasing the release of proinflammatory cytokines (Liang et al., 2022). All the genes related to microglia activity are listed in Table 1.
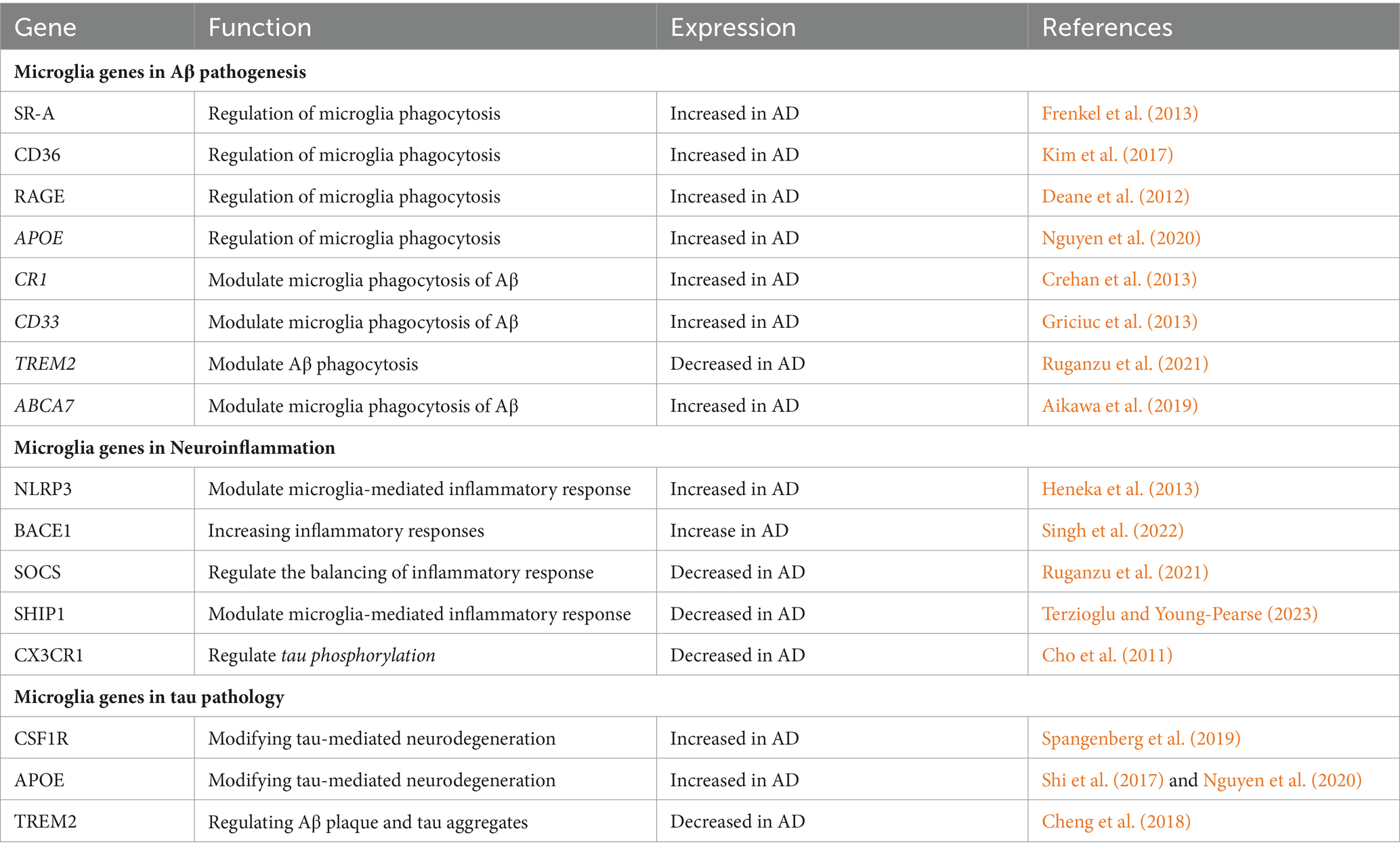
Table 1. Summarizes all the genes related to microglial activity and their functions in AD.
The vascular blood–brain barrier (BBB), which serves as the brain’s primary interface with the outside world, is vital to maintaining brain homeostasis, it regulates the entry and exit of biological substances and is essential for shielding the brain parenchyma from blood-borne pathogens or exogenous substances into the central nervous system (Takechi et al., 2017). The BBB is composed of both molecular (the glycocalyx and basement membrane, junction complex) and cellular components (endothelial cells, pericytes, and astrocytes), The brain microvascular endothelial cells have developed a junction complex such as tight junction which sounds to be a very early feature of BBB development, separating blood from CNS by brain endothelial cells and provide the best conditions for synaptic and neural activity by certain ion channels and variety of efflux transporters (Halliday et al., 2000; Du et al., 2018; Khan et al., 2023). Under normal conditions, the BBB is relatively impermeable, the disruption of BBB and vascular dysfunction by the release of Many vasoactive substances, cytokines, and chemical mediators including glutamate, aspartate, taurine, ATP, endothelin-1, NO, TNF-α, and macrophage-inflammatory protein 2 (MIP2). Bradykinin, 5HT, histamine, thrombin, UTP, UMP, substance P, quinolinic acid, platelet-activating factor, and free radicals under pathologic circumstances such as AD have been associated with multiple molecular changes result in increased BBB permeability (Kadry et al., 2020).
Growing body of research indicates that BBB disruption is an early indicator of neurodegeneration, including AD.
Considering the major role of BBB to clear around 85% of AD-related forms of Aβ from the brain, BBB breakdown can dysregulate efflux and influx of Aβ transporters result in Aβ accumulation and decrease tight junction protein expression, which causes a greater influx of peripheral immune cells into the brain and capillary degeneration (Winkler et al., 2015).
Clinical researches have demonstrated that decrease in pericyte quantity and coverage in the cortex and hippocampus of AD patients and mouse might be a reason for breaking BBB integrity through reducing brain microcirculation (Sweeney et al., 2018). Therefore, in AD patients numerous circulating soluble inflammatory mediators may impact on BBB malfunction specially during systemic inflammation and/or infection. Which is demonstrated by the fact that serum from mice treated with lipopolysaccharide (LPS) weakened the integrity of an in vitro BBB model more than serum from mice treated with a vehicle. Also aging can cause alterations in BBB as well as the immune system’s reactions. Aging cells usually take on a senescence-associated secretory phenotype which is associated with a transcriptional program that promotes Immune cells activation, migration, and infiltration by producing growth factors, cytokines, chemokines, and extracellular matrix proteases affect the BBB integrity (Lasry and Ben-Neriah, 2015; Figure 1).
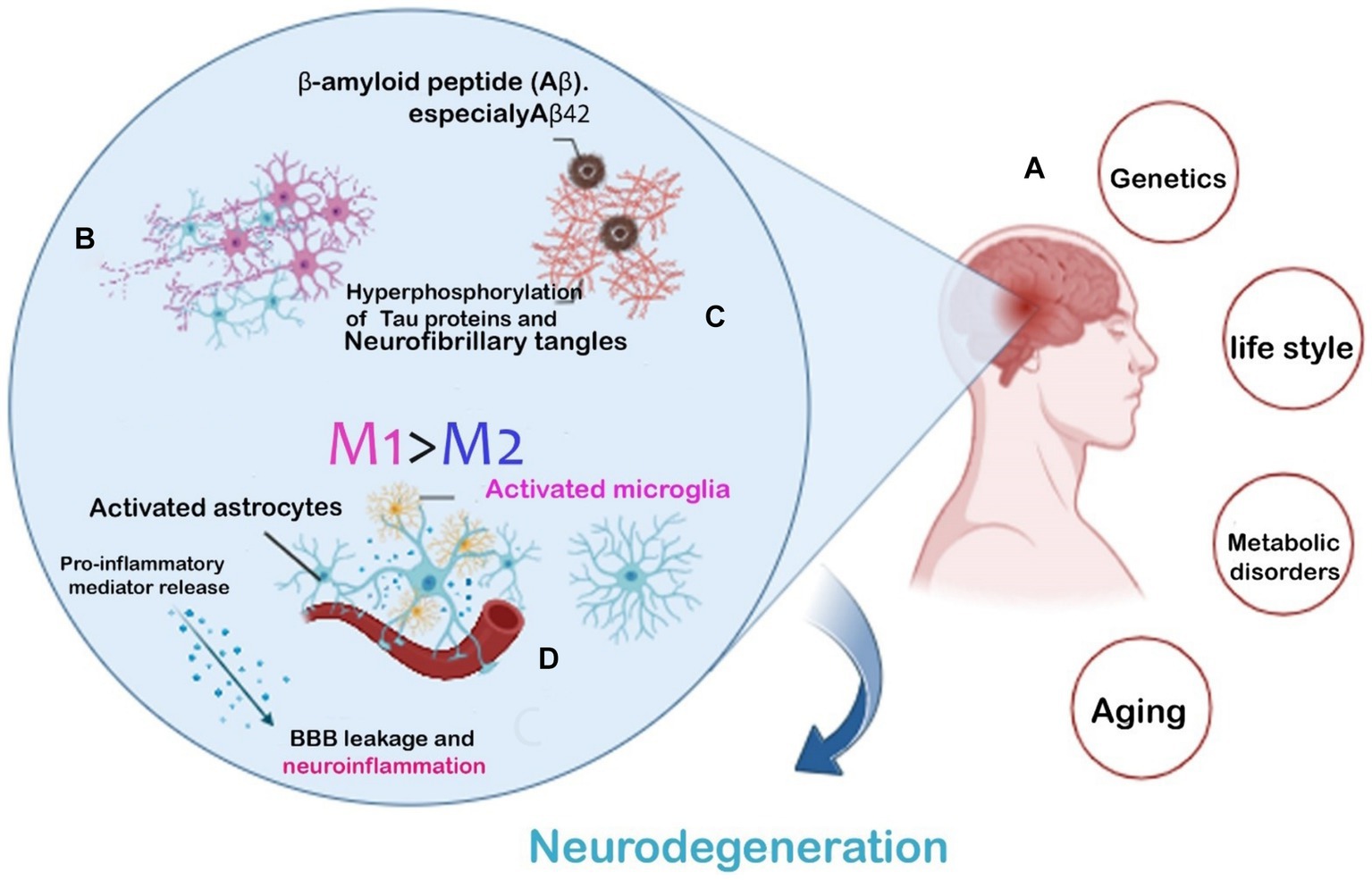
Figure 1. AD hallmarks and risk factors leading to neurodegeneration. Common risk factors leading to Two AD (A). The main pathogenic hallmarks of AD are the extra-accumulation of amyloid-β plaques and Tau phosphorylation (B). Microglial phenotype modification accelerating the neuroinflammatory response (C). Inflammatory cytokines released from activated microglia causing BBB leakage and neurodegeneration (D).
Different clinical studies indicate the role of inflammation in cognitive decline especially in AD pathogenesis. Currently, inflammation is considered the third main hallmark of AD besides the hyper-phosphorylated tau protein and amyloid-beta (Aβ) protein accumulation (Bhaskar et al., 2010; Das and Ganesh, 2023). The molecules responsible for inflammation can be generally divided into cytokines and transcription factors (Šimić et al., 2016). Although the inflammatory response can be beneficial via accelerating the Aβ clearance, at the same time they can increase the Aβ and tau production, and promote neurodegeneration and synapse loss (Šimić et al., 2016).
The balance between initiation and termination of immune response ensures the prompt removal of invasive pathogens and the cessation of excessive response within the central nervous system. This is crucial for the prevention of many diseases including the (Zheng et al., 2016). The inappropriate activation of inflammatory cytokines may lead to long-lasting alteration of regulatory neural gene expression. For instance, cytokines by interacting with different immune molecule groups such as the major histocompatibility complex class I (MHC I) can adversely affect the synaptic plasticity necessary for synapse formation and activity-dependent synaptic pruning (Ljunggren and Anderson, 1998). It is believed that these changes in synaptogenesis are fundamental to the causes of dementia. Additionally, cytokines can strongly stimulate the hypothalamic-pituitary-adrenal (HPA) axis, and increase the hormones release (Brosseron et al., 2014).Pro-inflammatory cytokines that cause chronic inflammation, like TNF-α, IL-6, and IL-1β, can influence and penetrate the blood–brain barrier (BBB), causing it to release proinflammatory mediators and increasing cell permeability, which permits leukocytes to enter the brain (Szczepanik et al., 2001; Swardfager et al., 2010). While anti-inflammatory cytokines are also produced. These include IL-1 receptor antagonist, IL-4, IL-10, and IL-11. These cytokines may be a part of a complex mechanism that prevents excessive neuroinflammation (Pousset et al., 2001; Guillot-Sestier et al., 2015). Activating the NF-κB pathway in microglia, can subsequently increase the amount of tau seeding and spreading and most AD patients are detected with considerably higher levels of NF-κB (Kaltschmidt et al., 1997). The silencing of microglial NF-κB cognitive abnormalities and homeostatic were restored. Hence, inhibiting the NF-κB pathway may offer a therapeutic approach to lessen AD pathogenesis (Sun et al., 2022). Finally, the other factor that can directly or indirectly increase inflammation and neuroinflammatory mediators is the overproduction of neutrophil extracellular traps (NETs) that induce macrophage activation and tissue damage (Brosseron et al., 2014; Swanson et al., 2018). Therefore, as shown in Figure 2, the permanent activation of astrocytes and Microglia can cause chronic inflammation. Chronic inflammation can be also caused by specific environmental factors, bacterial and viral infections, and Aging (Zhao et al., 2021). In chronic inflammation there is a major change in inflammatory pathway activation, leading to different immune responses and excessive production of inflammatory cytokines which lead to neuroinflammation (Figure 2; Neurath and Finotto, 2011).
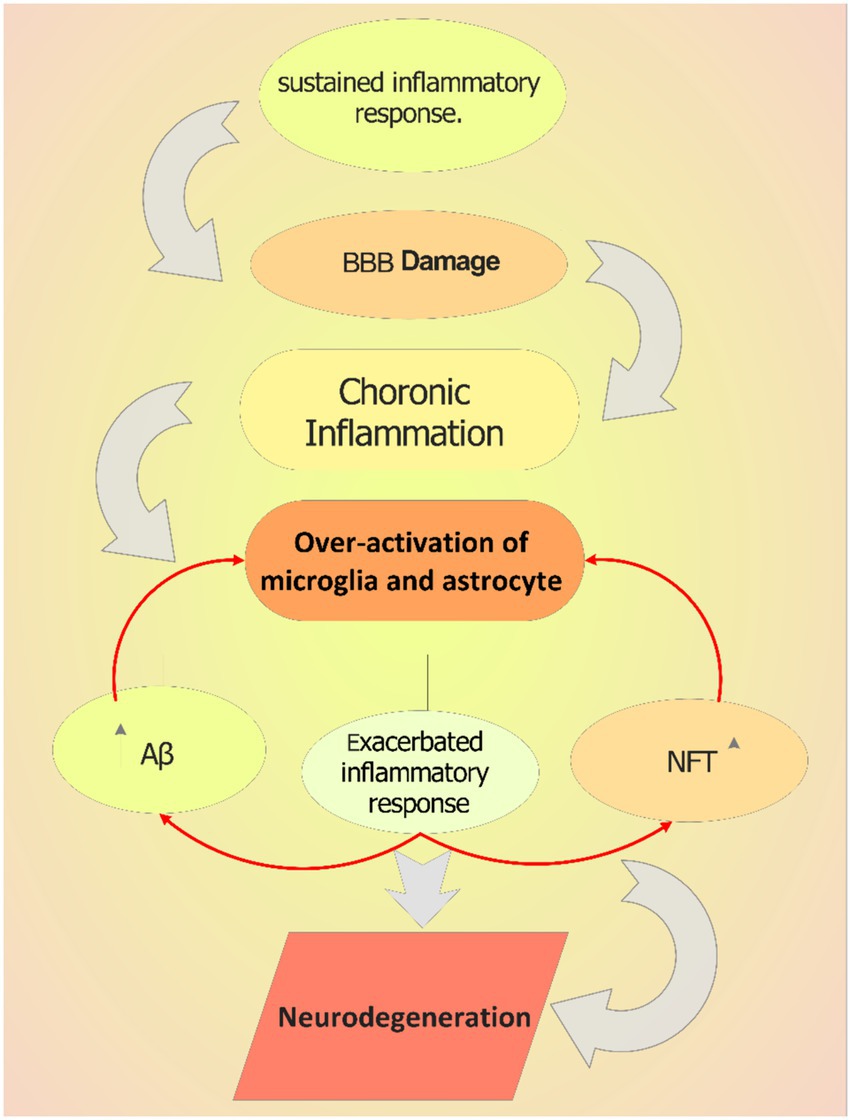
Figure 2. The role of chronic inflammation in AD pathology. Sustained inflammatory response can cause blood–brain barrier (BBB) damage, which increases the entrance and activity of other immune cells in brain. This over-activates microglia in the brain and triggers them to produce more inflammatory mediators including cytokines, which increase the extracellular plaques accumulation triggering neuroinflammation.
Currently, Aβ42and phosphorylated tau proteins are the main fluid-based biomarkers of Cerebrospinal fluid (CSF) in clinical practice (Bălaşa et al., 2020). However, there are still limitations in their specific detection based on their low concentration in blood (Noble et al., 2014; Galizzi and Di Carlo, 2023). As mentioned, inflammation plays a major role in AD development and among all different neuroinflammatory biomarkers which can be considered as therapeutic targets for drug design, cytokines, chemokines and transcription factors for their precise roles in the various stages of AD, possible medical applications, and easy isolation from blood or CSF have attracted a lot of attention (Zheng et al., 2016; AmeliMojarad et al., 2022; Park et al., 2022).
Different research groups indicated the cytokine levels alternation in AD patients. For example, IL-1β, TNF-α, NF-κB and chemokines like CCL2 has found to be increasing in AD patients which can also be used as inflammatory markers (Bălaşa et al., 2020).
Fast-progressing AD is linked to IFN-γ polymorphism implies that this cytokine may actively contribute to accelerating the progression of AD specially the LOAD (O’Bryant et al., 2017).
Dysregulation of the cytokines and chemokines can cause neuroinflammatory modulation, altering the microglia phenotype, and reducing microgliosis which accelerate the AD progression (Swanson et al., 2018; Zhang et al., 2021). Nonetheless, the most recent meta-analysis revealed substantial heterogeneity in certain comparisons but no significant differences in cytokines, such as IL-1β, IL-6, IL-8, IL-10, or TNF-α, were discovered between AD patients and healthy controls (Blennow and Hampel, 2003; Newcombe et al., 2018).
Other inflammatory biomarkers in Alzheimer’s disease may include IL-33 and the soluble form of its receptor ST2 (sST2). In animal models of Alzheimer’s disease, IL-33 stimulates microglia and protects against Aβ plaques, despite its association with inflammation (Fu et al., 2016).
A 1-year follow-up study indicated that MCI and AD patients with positive IL-33 expression in serum performed better on cognitive tests, adding to the evidence for IL-33’s benefit. The explanation for the increase in IL-33 in AD and MCI patients’ plasma is surprising, given higher levels of this cytokine have been related to improved cognitive function. Recent research suggests that higher levels of sST2 in AD patients buffer the physiological effects of IL-33 and may play a role in the cognitive function impairment associated with AD (Fu et al., 2016; Liang et al., 2020). Moreover, based on the damaged blood–brain barrier (BBB), different proteins can pass through BBB therefore, the blood of AD patients can reflect the AD progression-related targets. More importantly, the large surface area of the blood–brain barrier can be considered as potential for therapeutic intervention (Sweeney et al., 2018; Niculescu et al., 2020).
Therefore, detecting the well-established inflammatory biomarkers and methods for early diagnosis and monitoring of AD patients can be considered as alternative method of AD identification. However, cytokines may not be sufficient to demonstrate that an imbalance in cytokine levels is the sole cause of AD based on their overlapping with other neurodegenerative disease and aging. Therefore, it makes more sense to combine the use of several proteins given the unpredictable results of using a single cytokine level.
But few sets of biomarkers have demonstrated consistent performance and good reproducibility since the first AD prediction model comprising 18 plasma biomarkers with multiple cytokines was proposed. Using hypersensitive methods, such as immunoprecipitation-mass spectrometry (IP-MS), and single-molecular mass analysis (SIMOA) can detect the minor changes in the Aβ plasma level in patients with AD (Wu et al., 2021; Nijakowski et al., 2024). A more sensible strategy is to use multiple proteins in combination (Ray et al., 2007; Zheng et al., 2016). However, only a small number of biomarker sets have demonstrated consistent performance and good reproducibility since the first AD prediction model comprising 18 plasma biomarkers with multiple cytokines was proposed (Ray et al., 2007). Furthermore, a combination of soluble IL-6 receptor (sIL-6R), tissue inhibitor of metalloproteinases-1 (TIMP-1), and soluble TNF-α receptor I (sTNFR-I) in CSF was found to provide the best prediction to AD among other molecules after screening 120 inflammatory molecules in CSF and serum of AD, MCI, and healthy controls using protein-array analysis (Richens et al., 2014; Delaby et al., 2015). Future research on AD should look at pathogens other than Aβ and examine how cytokines interact with other players. New genes and proteins can only be discovered through the creation of brain banks, while genome-wide association studies and online database analysis will continually update polymorphism information linked to AD (Delaby et al., 2015; Khan and Alkon, 2015). Table 2 summarized the recent neuroinflammatory biomarkers related with AD.
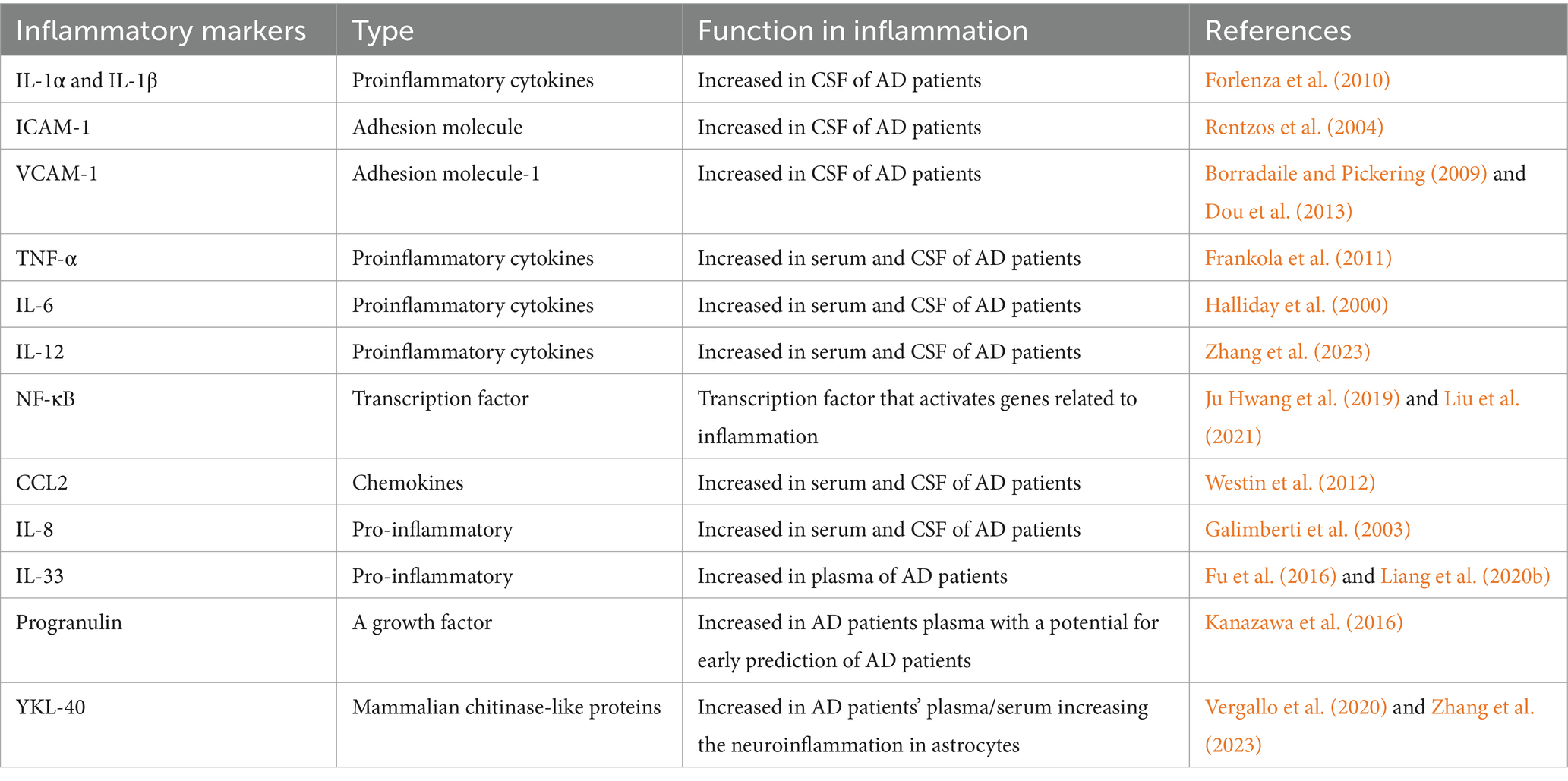
Table 2. List of neuroinflammatory biomarkers for AD.
Novel therapeutics are being offered by the recently made connections between inflammation and neurodegeneration (Wu et al., 2021). Currently, a major treatment strategy for AD is the reduction of toxic Aβ plaque accumulation and generation and reducing the inflammatory responses (Muralidar et al., 2020; Wang Z. et al., 2020). Even though there is still no known treatment for AD, NSAIDs are commonly used drugs for AD with the ability to decrease of Aβ plaque load, microglial activation, and proinflammatory cytokine levels. Currently the most promising drugs in reducing inflammation are COX-2 inhibitors Celecoxib and roficoxib which attenuate the neuroinflammation in AD (Moride et al., 2003; Miguel-Álvarez et al., 2015).
COX-2 inhibitors work by inhibiting the cyclooxygenase (COX-1 and COX-2 enzyme), arachidonic acid cannot be converted into prostaglandins, or prostacyclin without cyclooxygenase which have degenerative effect. And can raise Aβ levels (Moride et al., 2003; Benito-León et al., 2019).
It’s interesting to note that degenerative brain cells express high levels of COX-2; therefore, blocking COX may lessen AD. Aβ-induced microglial activation may occur directly or indirectly, leading to an increase in COX-2 which can be found during inflammation (Moride et al., 2003). Compared to control brains, AD brains exhibit higher levels of COX-1 and COX-2 (Moussa and Dayoub, 2023).
Research using animal models of AD has demonstrated the potential benefit of NSAIDs against AD. For instance, oral administration of ibuprofen, a nonspecific COX inhibitor, at the outset of amyloid plaque formation in transgenic mice overexpressing APP reduced glial activation and plaque density (Moussa and Dayoub, 2023). In a different experiment, treated rats with indomethacin, reduced microglial activation, improved the hippocampus over time, and avoided working memory problems. Furthermore, and elevated COX-2 levels were generated in mice given an intracerebroventricular injection of Aβ (Karkhah et al., 2021). In addition, pretreatment with the specific COX-2 inhibitor NS398 reduced COX-2 levels and cognitive impairment (Minter et al., 2003). Further studies have demonstrated that therapy with ibuprofen and naproxen in transgenic mice models of AD Other studies of NSAIDs in human cell cultures have raised hopes for its usage in AD treatment (Wilkinson et al., 2012; Linda and Hershey, 2019; Steven Karceski, 2019). For instance, the overexpress APP695NL, in human neuroglioma cells identified different NSAIDs that can selectively reduce Aβ42 such as sulindac, ibuprofen, and diclofenac (Weggen et al., 2003).
Activating PPARγ, a transcriptional factor that suppresses the expression of proinflammatory genes by blocking the activity of other transcription factors like NFκB, AP-1, and STAT1, is another potential neuroprotective mechanism of NSAIDs. Additionally, proinflammatory genes can be suppressed by PPARγ in the vasculature and myeloid lineage cells like macrophages and microglia (Daynes and Jones, 2002; Heneka et al., 2011).
Consequently, pioglitazone, a PPARγ agonist, has been used in clinical AD research suppressing the expression of genes that promote inflammation to regulate transcription (Geldmacher et al., 2011).
However, NSAID usage is only beneficial in the early stages of AD, because, with the start of the Aβ deposition process, NSAIDs are ineffective and even dangerous because they decrease microglial inflammation, which mediates the clearance of A despite its negative effects (Ho et al., 2006). Targeting NLRP3 inflammasome of microglia is another strategy against AD and AD-related inflammatory responses, a small molecule NLRP3 inhibitor such as JC-124, and MCC950 has been discovered which vigorously pro-inflammatory cytokines, chemokines, and ROS in AD however, along with more comprehensive evaluations of the outcomes, could produce delightfully unexpected results (Yin et al., 2018; Kelley et al., 2019; Zhang et al., 2021; Sharma et al., 2023). Minocycline is a tetracycline with anti-inflammatory qualities that can cross the blood–brain barrier (BBB) (Garcez et al., 2019). An in vivo study suggests that minocycline reduces Aβ accumulation and attenuates microglial activation because it inhibits the NLRP3 inflammasome (Li et al., 2016; Garcez et al., 2019).
Nicodipine (P2X7R antagonists), a dihydropyridine calcium channel antagonist, has also been shown to confer neuroprotective effects by reducing the levels of activated NF-κB and inhibiting the release of mature IL-1β in Aβ-stimulated microglia (whose potential target is P2X7R), which plays a permissive role in NLRP3 inflammasome activation and cytokines release (Ryu and McLarnon, 2008; Di Virgilio et al., 2017; Huang et al., 2023). The list of recent agents for treatment strategy of AD is provided in Table 3.
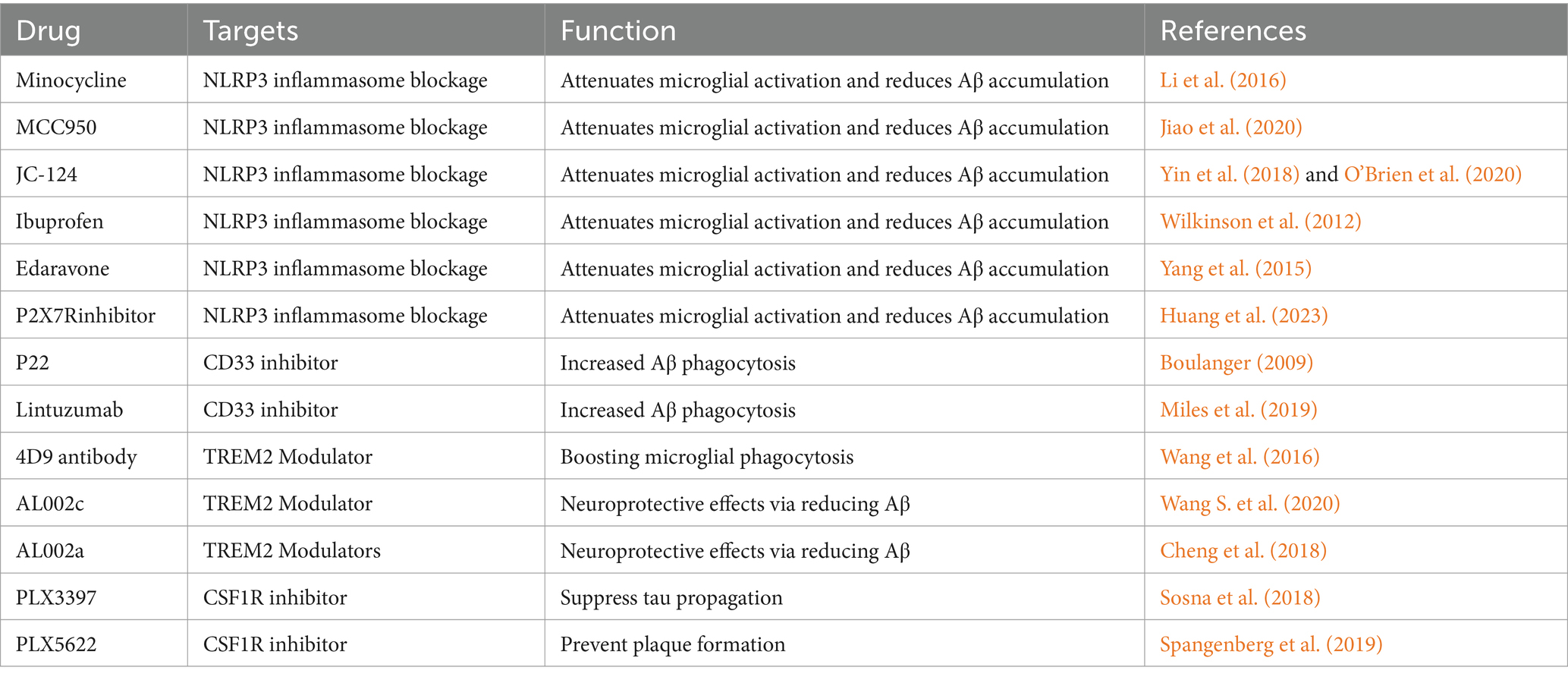
Table 3. Current agents for therapeutic strategies in AD.
Chronic inflammation is the third core pathology in the progression of Alzheimer’s disease, alongside the well-known activities of Aβ and tau. Microglia play a crucial part in this process, activated microglia are thought to be the primary source of pro-inflammatory mediators released, such as cytokines, which drive inflammatory cascades in the CNS, resulting in neuroinflammatory modulation. Activated microglia can also enhance blood–brain barrier (BBB) permeability, synaptic loss, and neurodegeneration in the brain, accelerating the AD pathogenesis. Since there are still no effective therapies in terms of disease attenuation or prevention, further research is needed to unrevealing the potential reliable biomarkers for monitoring AD in early stages (Leng and Edison, 2020).
Inflammatory markers alternation in patients with AD can be considered as a new means to track AD progression. Novel biomarkers related to neuroinflammation such as proinflammatory cytokines and chemokines are mainly altered in in patients with AD. However, there are still limitation for considering proinflammatory markers as AD specific biomarkers, since many neurodegenerative diseases have similar clinical presentations, it is possible that their changes be explained by aging or other systemic disease. However, based on their easy extraction and interpretation, they can still be considered the best first-step biomarkers in the multi-step AD process. As a result, we can improve the accuracy of AD diagnosis and treatment plans in the near future by using the multiplex model, which combines various blood markers and proteins of AD patients.
MaA: Conceptualization, Data curation, Writing – review & editing. MeA: Visualization, Writing – original draft, Writing – review & editing. XC: Funding acquisition, Supervision, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by XC, the host of the Capacity Construction Project of Major Clinical (specialized) Departments of Traditional Chinese Medicine of Liaoning Province (No. LNZYXZK201909), and the host of the Distinguished Professor Program of Liaoning Province.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
AD, Alzheimer’s disease; APOE, Apolipoprotein E; TREM2, Triggering receptor expressed on myeloid cells 2; Aβ, β-amyloid peptide; APP, Amyloid precursor protein; BBB, Blood–brain barrier; TNF, Tumor necrosis factor; NSAIDs, Nonsteroidal anti-inflammatory drugs; TGF-β, Transforming growth factor; PGE2, Prostaglandin E2; IL, Interleukin; CNS, Central nervous system; NF-kB, Nuclear factor kappa activated light chain B.
Aarsland, D. (2020). Epidemiology and pathophysiology of dementia-related psychosis. J. Clin. Psychiatry 81:AD19038BR1C. doi: 10.4088/JCP.AD19038BR1C
Aikawa, T., Ren, Y., Yamazaki, Y., Tachibana, M., Johnson, M. R., Anderson, C. T., et al. (2019). ABCA7 haplodeficiency disturbs microglial immune responses in the mouse brain. Proc. Natl. Acad. Sci. USA 116, 23790–23796. doi: 10.1073/pnas.1908529116
AmeliMojarad, M., AmeliMojarad, M., and Nourbakhsh, M. (2022). Circulating circular RNA ADAM9 a potential biomarker for human colorectal cancer. Gene Rep. 26:101516. doi: 10.1016/j.genrep.2022.101516
Bai, H., and Zhang, Q. (2021). Activation of NLRP3 Inflammasome and onset of Alzheimer’s disease. Front. Immunol. 12:701282. doi: 10.3389/fimmu.2021.701282
Bălaşa, A. F., Chircov, C., and Grumezescu, A. M. (2020). Body fluid biomarkers for Alzheimer’s disease—an up-to-date overview. Biomedicines 8, 1–21. doi: 10.3390/BIOMEDICINES8100421
Baltes, C., Princz-Kranz, F., Rudin, M., and Mueggler, T. (2011). Detecting amyloid-β plaques in Alzheimer’s disease. Methods Mol. Biol. 711, 511–533. doi: 10.1007/978-1-61737-992-5_26
Benito-León, J., Contador, I., Vega, S., Villarejo-Galende, A., and Bermejo-Pareja, F. (2019). Non-steroidal anti-inflammatory drugs use in older adults decreases risk of Alzheimer’s disease mortality. PLoS One 14:e0222505. doi: 10.1371/journal.pone.0222505
Bertram, L., Lange, C., Mullin, K., Parkinson, M., Hsiao, M., Hogan, M. F., et al. (2008). Genome-wide association analysis reveals putative Alzheimer’s disease susceptibility loci in addition to APOE. Am. J. Hum. Genet. 83:623. doi: 10.1016/j.ajhg.2008.10.008
Bhaskar, K., Konerth, M., Kokiko-Cochran, O. N., Cardona, A., Ransohoff, R. M., and Lamb, B. T. (2010). Regulation of tau pathology by the microglial Fractalkine receptor. Neuron 68:19. doi: 10.1016/j.neuron.2010.08.023
Blennow, K., and Hampel, H. (2003). CSF markers for incipient Alzheimer’s disease. Lancet Neurol. 2, 605–613. doi: 10.1016/S1474-4422(03)00530-1
Borradaile, N. M., and Pickering, G. J. (2009). Nicotinamide phosphoribosyltransferase imparts human endothelial cells with extended replicative lifespan and enhanced angiogenic capacity in a high glucose environment. Aging Cell 8, 100–112. doi: 10.1111/j.1474-9726.2009.00453.x
Boulanger, L. M. (2009). Immune proteins in brain development and synaptic plasticity. Neuron 64, 93–109. doi: 10.1016/j.neuron.2009.09.001
Brosseron, F., Krauthausen, M., Kummer, M., and Heneka, M. T. (2014). Body fluid cytokine levels in mild cognitive impairment and Alzheimer’s disease: a comparative overview. Mol. Neurobiol. 50, 534–544. doi: 10.1007/s12035-014-8657-1
Chadarevian, J. P., Lombroso, S. I., Peet, G. C., Hasselmann, J., Tu, C., Marzan, D. E., et al. (2023). Engineering an inhibitor-resistant human CSF1R variant for microglia replacement. J. Exp. Med. 220:e20220857. doi: 10.1084/jem.20220857
Cheng, Q., Danao, J., Talreja, S., Wen, P., Yin, J., Sun, N., et al. (2018). TREM2-activating antibodies abrogate the negative pleiotropic effects of the Alzheimer’s disease variant Trem2R47H on murine myeloid cell function. J. Biol. Chem. 293:12620. doi: 10.1074/jbc.RA118.001848
Cho, S. H., Sun, B., Zhou, Y., Kauppinen, T. M., Halabisky, B., Wes, P., et al. (2011). CX3CR1 protein signaling modulates microglial activation and protects against plaque-independent cognitive deficits in a mouse model of Alzheimer disease. J. Biol. Chem. 286:32713. doi: 10.1074/jbc.M111.254268
Crehan, H., Hardy, J., and Pocock, J. (2013). Blockage of CR1 prevents activation of rodent microglia. Neurobiol. Dis. 54, 139–149. doi: 10.1016/j.nbd.2013.02.003
Das, T. K., and Ganesh, B. P. (2023). Interlink between the gut microbiota and inflammation in the context of oxidative stress in Alzheimer’s disease progression. Gut Microbes 15:2206504. doi: 10.1080/19490976.2023.2206504
Daynes, R. A., and Jones, D. C. (2002). Emerging roles of PPARs in inflammation and immunity. Nat. Rev. Immunol. 2, 748–759. doi: 10.1038/nri912
Deane, R., Singh, I., Sagare, A. P., Bell, R. D., Ross, N. T., LaRue, B., et al. (2012). A multimodal RAGE-specific inhibitor reduces amyloid β–mediated brain disorder in a mouse model of Alzheimer disease. J. Clin. Invest. 122:1377. doi: 10.1172/JCI58642
Delaby, C., Gabelle, A., Blum, D., Schraen-Maschke, S., Moulinier, A., Boulanghien, J., et al. (2015). Central nervous system and peripheral inflammatory processes in Alzheimer’s disease: biomarker profiling approach. Front. Neurol. 6:181. doi: 10.3389/fneur.2015.00181
Di Virgilio, F., Dal Ben, D., Sarti, A. C., Giuliani, A. L., and Falzoni, S. (2017). The P2X7 receptor in infection and inflammation. Immunity 47, 15–31. doi: 10.1016/j.immuni.2017.06.020
Dou, W., Zhang, J., Sun, A., Zhang, E., Ding, L., Mukherjee, S., et al. (2013). Protective effect of naringenin against experimental colitis via suppression of toll-like receptor 4/NF-κB signalling. Br. J. Nutr. 110, 599–608. doi: 10.1017/S0007114512005594
Du, X., Wang, X., and Geng, M. (2018). Alzheimer’s disease hypothesis and related therapies. Transl Neurodegener. 7:2. doi: 10.1186/s40035-018-0107-y
Forlenza, O. V., Diniz, B. S., Talib, L. L., Mendonça, V. A., Ojopi, E. B., Gattaz, W. F., et al. (2010). Increased serum IL-1β level in Alzheimer’s disease and mild cognitive impairment. Dement. Geriatr. Cogn. Disord. 28, 507–512. doi: 10.1159/000255051
Frankola, K. A., Greig, N. H., Luo, W., and Tweedie, D. (2011). Targeting TNF-alpha to elucidate and ameliorate neuroinflammation in neurodegenerative diseases. CNS Neurol. Disord. Drug Targets 10:391. doi: 10.2174/187152711794653751
Frenkel, D., Wilkinson, K., Zhao, L., Hickman, S. E., Means, T. K., Puckett, L., et al. (2013). Scara1 deficiency impairs clearance of soluble amyloid-β by mononuclear phagocytes and accelerates Alzheimer’s-like disease progression. Nat. Commun. 4:2030. doi: 10.1038/NCOMMS3030
Fu, A. K. Y., Hung, K. W., Yuen, M. Y. F., Zhou, X., Mak, D. S. Y., Chan, I. C. W., et al. (2016). IL-33 ameliorates Alzheimer’s disease-like pathology and cognitive decline. Proc. Natl. Acad. Sci. USA 113, E2705–E2713. doi: 10.1073/PNAS.1604032113
Galimberti, D., Schoonenboom, N., Scarpini, E., and Scheltens, P. (2003). Chemokines in serum and cerebrospinal fluid of Alzheimer’s disease patients. Ann. Neurol. 53, 547–548. doi: 10.1002/ana.10531
Galizzi, G., and Di Carlo, M. (2023). Mitochondrial DNA and inflammation in Alzheimer’s disease. Curr. Issues Mol. Biol. 45:8586. doi: 10.3390/cimb45110540
Garcez, M. L., Mina, F., Bellettini-Santos, T., da Luz, A. P., Schiavo, G. L., Macieski, J. M. C., et al. (2019). The involvement of NLRP3 on the effects of minocycline in an AD-like pathology induced by β-amyloid oligomers administered to mice. Mol. Neurobiol. 56, 2606–2617. doi: 10.1007/s12035-018-1211-9
Geldmacher, D. S., Fritsch, T., McClendon, M. J., and Landreth, G. (2011). A randomized pilot clinical trial of the safety of pioglitazone in treatment of patients with Alzheimer disease. Arch. Neurol. 68, 45–50. doi: 10.1001/archneurol.2010.229
Gouras, G. K., Olsson, T. T., and Hansson, O. (2015). β-Amyloid peptides and amyloid plaques in Alzheimer’s disease. Neurotherapeutics 12, 3–11. doi: 10.1007/s13311-014-0313-y
Greten, F. R., and Grivennikov, S. I. (2019). Inflammation and Cancer: triggers, mechanisms, and consequences. Immunity 51, 27–41. doi: 10.1016/j.immuni.2019.06.025
Griciuc, A., Serrano-Pozo, A., Parrado, A. R., Lesinski, A. N., Asselin, C. N., Mullin, K., et al. (2013). Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid Beta. Neuron 78:631. doi: 10.1016/j.neuron.2013.04.014
Guillot-Sestier, M. V., Doty, K. R., Gate, D., Rodriguez, J., Leung, B. P., Rezai-Zadeh, K., et al. (2015). Il10 deficiency rebalances innate immunity to mitigate Alzheimer-like pathology. Neuron 85, 534–548. doi: 10.1016/j.neuron.2014.12.068
Halliday, G., Robinson, S. R., Shepherd, C., and Kril, J. (2000). Alzheimer’s disease and inflammation: a review of cellular and therapeutic mechanisms. Clin. Exp. Pharmacol. Physiol. 27, 1–8. doi: 10.1046/j.1440-1681.2000.03200.x
Heneka, M. T., Kummer, M. P., Stutz, A., Delekate, A., Schwartz, S., Vieira-Saecker, A., et al. (2013). NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493:674. doi: 10.1038/nature11729
Heneka, M. T., Reyes-Irisarri, E., Hüll, M., and Kummer, M. P. (2011). Impact and therapeutic potential of PPARs in Alzheimer’s disease. Curr. Neuropharmacol. 9:643. doi: 10.2174/157015911798376325
Ho, T. T. B., Nasti, A., Seki, A., Komura, T., Inui, H., Kozaka, T., et al. (2020). Combination of gemcitabine and anti-PD-1 antibody enhances the anticancer effect of M1 macrophages and the Th1 response in a murine model of pancreatic cancer liver metastasis. J. Immunother. Cancer 8:e001367. doi: 10.1136/jitc-2020-001367
Ho, L., Qin, W., Stetka, B. S., and Pasinetti, G. M. (2006). Is there a future for cyclo-oxygenase inhibitors in Alzheimer’s disease? CNS Drugs 20, 85–98. doi: 10.2165/00023210-200620020-00001
Huang, J. L., Xu, Z. H., Yang, S. M., Yu, C., Zhang, F., Qin, M. C., et al. (2018). Identification of differentially expressed profiles of Alzheimer’s disease associated circular RNAs in a Panax Notoginseng Saponins-treated Alzheimer’s disease mouse model. Comput. Struct. Biotechnol. J. 16:523. doi: 10.1016/j.csbj.2018.10.010
Huang, Q., Ying, J., Yu, W., Dong, Y., Xiong, H., Zhang, Y., et al. (2023). P2X7 receptor: an emerging target in Alzheimer’s disease. Mol. Neurobiol. 2023, 1–15. doi: 10.1007/S12035-023-03699-9
Jiao, J., Zhao, G., Wang, Y., Ren, P., and Wu, M. (2020). MCC950, a selective inhibitor of NLRP3 Inflammasome, reduces the inflammatory response and improves neurological outcomes in mice model of spinal cord injury. Front. Mol. Biosci. 7:37. doi: 10.3389/fmolb.2020.00037
Joaquín Merino, J., Muñetón-Gómez, V., Alvárez, M.-I., and Toledano-Díaz, A. (2016). Effects of CX3CR1 and Fractalkine chemokines in amyloid Beta clearance and p-tau accumulation in Alzheimer’s disease (AD) rodent models: is Fractalkine a systemic biomarker for AD? Curr. Alzheimer Res. 13, 403–412. doi: 10.2174/1567205013666151116125714
Ju Hwang, C., Choi, D.-Y., Park, M. H., and Hong, J. T. (2019). NF-κB as a key mediator of brain inflammation in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 18, 3–10. doi: 10.2174/1871527316666170807130011
Kadry, H., Noorani, B., and Cucullo, L. (2020). A blood–brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers 17, 1–24. doi: 10.1186/S12987-020-00230-3
Kaltschmidt, B., Uherek, M., Volk, B., Baeuerle, P. A., and Kaltschmidt, C. (1997). Transcription factor NF-kappaB is activated in primary neurons by amyloid beta peptides and in neurons surrounding early plaques from patients with Alzheimer disease. Proc. Natl. Acad. Sci. USA 94, 2642–2647. doi: 10.1073/pnas.94.6.2642
Kanazawa, M., Kawamura, K., Takahashi, T., Nishizawa, M., and Shimohata, T. (2016). Progranulin. Nihon Rinsho 74, 579–582. doi: 10.1212/WNL.0000000000004840
Karkhah, A., Saadi, M., Pourabdolhossein, F., Saleki, K., and Nouri, H. R. (2021). Indomethacin attenuates neuroinflammation and memory impairment in an STZ-induced model of Alzheimer’s like disease. Immunupharmacol. Immubol. 43, 758–766. doi: 10.1080/08923973.2021.1981374
Kelley, N., Jeltema, D., Duan, Y., and He, Y. (2019). The NLRP3 Inflammasome: an overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 20:3328. doi: 10.3390/ijms20133328
Keren-Shaul, H., Spinrad, A., Weiner, A., Matcovitch-Natan, O., Dvir-Szternfeld, R., Ulland, T. K., et al. (2017). A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169, 1276–1290.e17. doi: 10.1016/j.cell.2017.05.018
Khan, T. K., and Alkon, D. L. (2015). Peripheral biomarkers of Alzheimer’s disease. J. Alzheimers Dis. 44, 729–744. doi: 10.3233/JAD-142262
Khan, K. M., Balasubramanian, N., Gaudencio, G., Wang, R., Pushpavathi Selvakumar, G., Kolling, L., et al. (2023). Human tau-overexpressing mice recapitulate brainstem involvement and neuropsychiatric features of early Alzheimer’s disease. Acta Neuropathol Commun. 11:57. doi: 10.1186/s40478-023-01546-5
Kim, S. M., Mun, B. R., Lee, S. J., Joh, Y., Lee, H. Y., Ji, K. Y., et al. (2017). TREM2 promotes Aβ phagocytosis by upregulating C/EBPα-dependent CD36 expression in microglia. Sci. Rep. 7:11118. doi: 10.1038/s41598-017-11634-x
LaDu, M. J., Pederson, T. M., Frail, D. E., Reardon, C. A., Getz, G. S., and Falduto, M. T. (1995). Purification of apolipoprotein E attenuates isoform-specific binding to beta-amyloid. J. Biol. Chem. 270, 9039–9042. doi: 10.1074/jbc.270.16.9039
Lasry, A., and Ben-Neriah, Y. (2015). Senescence-associated inflammatory responses: aging and cancer perspectives. Trends Immunol. 36, 217–228. doi: 10.1016/j.it.2015.02.009
Lee, S., Varvel, N. H., Konerth, M. E., Xu, G., Cardona, A. E., Ransohoff, R. M., et al. (2010). CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer’s disease mouse models. Am. J. Pathol. 177, 2549–2562. doi: 10.2353/ajpath.2010.100265
Leng, F., and Edison, P. (2020). Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat. Rev. Neurol. 17, 157–172. doi: 10.1038/s41582-020-00435-y
Li, J., Chen, J., Mo, H., Chen, J., Qian, C., Yan, F., et al. (2016). Minocycline protects against NLRP3 Inflammasome-induced inflammation and P53-associated apoptosis in early brain injury after subarachnoid hemorrhage. Mol. Neurobiol. 53, 2668–2678. doi: 10.1007/s12035-015-9318-8
Liang, C. S., Su, K. P., Tsai, C. L., Lee, J. T., Chu, C. S., Yeh, T. C., et al. (2020). The role of interleukin-33 in patients with mild cognitive impairment and Alzheimer’s disease. Alzheimers Res. Ther. 12, 157–172. doi: 10.1186/S13195-020-00652-Z
Liang, T., Zhang, Y., Wu, S., Chen, Q., and Wang, L. (2022). The role of NLRP3 Inflammasome in Alzheimer’s disease and potential therapeutic targets. Front. Pharmacol. 13:845185. doi: 10.3389/fphar.2022.845185
Linda, A., and Hershey, R. B. L. (2019). Naproxen for presymptomatic Alzheimer disease: Is this the end, or shall we try again? Neurology 92, 829–830. doi: 10.1212/WNL.0000000000007233
Liu, C. C., Kanekiyo, T., Xu, H., and Bu, G. (2013). Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat. Rev. Neurol. 9, 106–118. doi: 10.1038/nrneurol.2012.263
Liu, X., Wang, K., Wei, X., Xie, T., Lv, B., Zhou, Q., et al. (2021). Interaction of NF-κB and Wnt/β-catenin signaling pathways in Alzheimer’s disease and potential active drug treatments. Neurochem. Res. 46, 711–731. doi: 10.1007/s11064-021-03227-y
Ljunggren, G., and Anderson, D. J. (1998). Cytokine induced modulation of MHC class I and class II molecules on human cervical epithelial cells. J. Reprod. Immunol. 38, 123–138. doi: 10.1016/S0165-0378(98)00009-6
Long, J. M., and Holtzman, D. M. (2019). Alzheimer disease: an update on pathobiology and treatment strategies. Cell 179, 312–339. doi: 10.1016/j.cell.2019.09.001
Lyons, A., Lynch, A. M., Downer, E. J., Hanley, R., O’Sullivan, J. B., Smith, A., et al. (2009). Fractalkine-induced activation of the phosphatidylinositol-3 kinase pathway attentuates microglial activation in vivo and in vitro. J. Neurochem. 110, 1547–1556. doi: 10.1111/j.1471-4159.2009.06253.x
Mancuso, R., Fryatt, G., Cleal, M., Obst, J., Pipi, E., Monzón-Sandoval, J., et al. (2019). CSF1R inhibitor JNJ-40346527 attenuates microglial proliferation and neurodegeneration in P301S mice. Brain 142:3243. doi: 10.1093/brain/awz241
McKee, C. G., Hoffos, M., Vecchiarelli, H. A., and Tremblay, M. È. (2023). Microglia: a pharmacological target for the treatment of age-related cognitive decline and Alzheimer’s disease. Front. Pharmacol. 14:1125982. doi: 10.3389/FPHAR.2023.1125982/BIBTEX
Miguel-Álvarez, M., Santos-Lozano, A., Sanchis-Gomar, F., Fiuza-Luces, C., Pareja-Galeano, H., Garatachea, N., et al. (2015). Non-steroidal anti-inflammatory drugs as a treatment for Alzheimer’s disease: a systematic review and meta-analysis of treatment effect. Drugs Aging 32, 139–147. doi: 10.1007/s40266-015-0239-z
Miles, L. A., Hermans, S. J., Crespi, G. A. N., Gooi, J. H., Doughty, L., Nero, T. L., et al. (2019). Small molecule binding to Alzheimer risk factor CD33 promotes Aβ phagocytosis. iScience 19:110. doi: 10.1016/J.ISCI.2019.07.023
Minter, H. A., Eveson, J. W., Huntley, S., Elder, D. J. E., and Hague, A. (2003). The cyclooxygenase 2-selective inhibitor NS398 inhibits proliferation of oral carcinoma cell lines by mechanisms dependent and independent of reduced prostaglandin E2 synthesis. Clin. Cancer Res. 9, 1885–1897.
Moride, Y., Ducruet, T., Rochon, S., and Lavoie, F. (2003). Persistency of use of COX-2-specific inhibitors and non-specific non-steroidal anti-inflammatory drugs (NSAIDs) in Quebec. Rheumatology 42, iii17–iii22. doi: 10.1093/rheumatology/keg494
Moussa, N., and Dayoub, N. (2023). Exploring the role of COX-2 in Alzheimer’s disease: potential therapeutic implications of COX-2 inhibitors. Saudi Pharm. J. 31:101729. doi: 10.1016/j.jsps.2023.101729
Muralidar, S., Ambi, S. V., Sekaran, S., Thirumalai, D., and Palaniappan, B. (2020). Role of tau protein in Alzheimer’s disease: the prime pathological player. Int. J. Biol. Macromol. 163, 1599–1617. doi: 10.1016/j.ijbiomac.2020.07.327
Nakamura, A., Kaneko, N., Villemagne, V. L., Kato, T., Doecke, J., Doré, V., et al. (2018). High performance plasma amyloid-β biomarkers for Alzheimer’s disease. Nature 554, 249–254. doi: 10.1038/nature25456
Neurath, M. F., and Finotto, S. (2011). IL-6 signaling in autoimmunity, chronic inflammation and inflammation-associated cancer. Cytokine Growth Factor Rev. 22, 83–89. doi: 10.1016/j.cytogfr.2011.02.003
Newcombe, E. A., Camats-Perna, J., Silva, M. L., Valmas, N., Huat, T. J., and Medeiros, R. (2018). Inflammation: the link between comorbidities, genetics, and Alzheimer’s disease. J. Neuroinflammation 15:276. doi: 10.1186/S12974-018-1313-3
Nguyen, A. T., Wang, K., Hu, G., Wang, X., Miao, Z., Azevedo, J. A., et al. (2020). APOE and TREM2 regulate amyloid responsive microglia in Alzheimer’s disease. Acta Neuropathol. 140:477. doi: 10.1007/s00401-020-02200-3
Niculescu, A. B., Le-Niculescu, H., Roseberry, K., Wang, S., Hart, J., Kaur, A., et al. (2020). Blood biomarkers for memory: toward early detection of risk for Alzheimer disease, pharmacogenomics, and repurposed drugs. Mol. Psychiatry 25:1651. doi: 10.1038/s41380-019-0602-2
Nijakowski, K., Owecki, W., Jankowski, J., and Surdacka, A. (2024). Salivary biomarkers for Alzheimer’s disease: a systematic review with Meta-analysis. Int. J. Mol. Sci. 25:1168. doi: 10.3390/ijms25021168
Noble, J. M., Scarmeas, N., Celenti, R. S., Elkind, M. S. V., Wright, C. B., Schupf, N., et al. (2014). Serum IgG antibody levels to periodontal microbiota are associated with incident Alzheimer disease. PLoS One 9:e114959. doi: 10.1371/journal.pone.0114959
O’Brien, W. T., Pham, L., Symons, G. F., Monif, M., Shultz, S. R., and McDonald, S. J. (2020). The NLRP3 inflammasome in traumatic brain injury: potential as a biomarker and therapeutic target. J. Neuroinflammation 17, 1–12. doi: 10.1186/S12974-020-01778-5
O’Bryant, S. E., Mielke, M. M., Rissman, R. A., Lista, S., Vanderstichele, H., Zetterberg, H., et al. (2017). Blood-based biomarkers in Alzheimer disease: current state of the science and a novel collaborative paradigm for advancing from discovery to clinic. Alzheimers Dement. 13, 45–58. doi: 10.1016/j.jalz.2016.09.014
Orihuela, R., McPherson, C. A., and Harry, G. J. (2016). Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 173, 649–665. doi: 10.1111/bph.13139
Öst, M., Nylén, K., Csajbok, L., Öhrfelt, A. O., Tullberg, M., Wikkelsö, C., et al. (2006). Initial CSF total tau correlates with 1-year outcome in patients with traumatic brain injury. Neurology 67, 1600–1604. doi: 10.1212/01.wnl.0000242732.06714.0f
Park, J. E., Gunasekaran, T. I., Cho, Y. H., Choi, S. M., Song, M. K., Cho, S. H., et al. (2022). Diagnostic blood biomarkers in Alzheimer’s disease. Biomedicines 10:169. doi: 10.3390/biomedicines10010169
Porsteinsson, A. P., Isaacson, R. S., Knox, S., Sabbagh, M. N., and Rubino, I. (2021). Diagnosis of early Alzheimer’s disease: clinical practice in 2021. J. Prev Alzheimers Dis. 8, 371–386. doi: 10.14283/JPAD.2021.23
Pousset, F., Cremona, S., Dantzer, R., Kelley, K. W., and Parnet, P. (2001). IL-10 and IL-4 regulate type-I and type-II IL-1 receptors expression on IL-1β-activated mouse primary astrocytes. J. Neurochem. 79, 726–736. doi: 10.1046/j.1471-4159.2001.00569.x
Ransohoff, R. M. (2016). A polarizing question: do M1 and M2 microglia exist? Nat. Neurosci. 19, 987–991. doi: 10.1038/nn.4338
Ray, S., Britschgi, M., Herbert, C., Takeda-Uchimura, Y., Boxer, A., Blennow, K., et al. (2007). Classification and prediction of clinical Alzheimer’s diagnosis based on plasma signaling proteins. Nat. Med. 13, 1359–1362. doi: 10.1038/nm1653
Ren, R., Qi, J., Lin, S., Liu, X., Yin, P., Wang, Z., et al. (2022). The China Alzheimer report 2022. Gen. Psychiatry 35:e100751. doi: 10.1136/gpsych-2022-100751
Rentzos, M., Michalopoulou, M., Nikolaou, C., Cambouri, C., Rombos, A., Dimitrakopoulos, A., et al. (2004). Serum levels of soluble intercellular adhesion Molecule-1 and soluble endothelial leukocyte adhesion Molecule-1 in Alzheimer’s disease. J. Geriatr. Psychiatry Neurol. 17, 225–231. doi: 10.1177/0891988704269822
Richens, J. L., Vere, K. A., Light, R. A., Soria, D., Garibaldi, J., Smith, A. D., et al. (2014). Practical detection of a definitive biomarker panel for Alzheimer’s disease; comparisons between matched plasma and cerebrospinal fluid. Int. J. Mol. Epidemiol. Genet. 5:53
Ruganzu, J. B., Zheng, Q., Wu, X., He, Y., Peng, X., Jin, H., et al. (2021). TREM2 overexpression rescues cognitive deficits in APP/PS1 transgenic mice by reducing neuroinflammation via the JAK/STAT/SOCS signaling pathway. Exp. Neurol. 336:113506. doi: 10.1016/j.expneurol.2020.113506
Ryu, J. K., and McLarnon, J. G. (2008). Block of purinergic P2X(7) receptor is neuroprotective in an animal model of Alzheimer’s disease. Neuroreport 19, 1715–1719. doi: 10.1097/WNR.0b013e3283179333
Sharma, B., Satija, G., Madan, A., Garg, M., Alam, M. M., Shaquiquzzaman, M., et al. (2023). Role of NLRP3 Inflammasome and its inhibitors as emerging therapeutic drug candidate for Alzheimer’s disease: a review of mechanism of activation, regulation, and inhibition. Inflammation 46, 56–87. doi: 10.1007/s10753-022-01730-0
Sheng, L., Chen, M., Cai, K., Song, Y., Yu, D., Zhang, H., et al. (2019). Microglial Trem2 induces synaptic impairment at early stage and prevents amyloidosis at late stage in APP/PS1 mice. FASEB J. 33, 10425–10442. doi: 10.1096/fj.201900527R
Shi, Y., Yamada, K., Liddelow, S. A., Smith, S. T., Zhao, L., Luo, W., et al. (2017). ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549, 523–527. doi: 10.1038/nature24016
Silva, M. V. F., Loures, C. D. M. G., Alves, L. C. V., De Souza, L. C., Borges, K. B. G., and Carvalho, M. D. G. (2019). Alzheimer’s disease: risk factors and potentially protective measures. J. Biomed. Sci. 26:33. doi: 10.1186/s12929-019-0524-y
Šimić, G., Babić Leko, M., Wray, S., Harrington, C., Delalle, I., Jovanov-Milošević, N., et al. (2016). Tau protein hyperphosphorylation and aggregation in Alzheimer’s disease and other Tauopathies, and possible neuroprotective strategies. Biomol. Ther. 6, 2–28. doi: 10.3390/BIOM6010006
Singh, N., Das, B., Zhou, J., Hu, X., and Yan, R. (2022). Targeted BACE-1 inhibition in microglia enhances amyloid clearance and improved cognitive performance. Sci. Adv. 8:eabo3610. doi: 10.1126/sciadv.abo3610
Sosna, J., Philipp, S., Albay, R. I., Reyes-Ruiz, J. M., Baglietto-Vargas, D., LaFerla, F. M., et al. (2018). Early long-term administration of the CSF1R inhibitor PLX3397 ablates microglia and reduces accumulation of intraneuronal amyloid, neuritic plaque deposition and pre-fibrillar oligomers in 5XFAD mouse model of Alzheimer’s disease. Mol. Neurodegener. 13:11. doi: 10.1186/s13024-018-0244-x
Spangenberg, E., Severson, P. L., Hohsfield, L. A., Crapser, J., Zhang, J., Burton, E. A., et al. (2019). Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat. Commun. 10:3758. doi: 10.1038/S41467-019-11674-Z
Steven Karceski, S. K. (2019). Can naproxen slow the progression of Alzheimer disease? Neurology 92, e2181–e2184. doi: 10.1212/WNL.0000000000007418
Sun, E., Motolani, A., Campos, L., and Lu, T. (2022). The pivotal role of NF-kB in the pathogenesis and therapeutics of Alzheimer’s disease. Int. J. Mol. Sci. 23:8972. doi: 10.3390/ijms23168972
Swanson, A., Wolf, T., Sitzmann, A., and Willette, A. A. (2018). Neuroinflammation in Alzheimer’s disease: pleiotropic roles for cytokines and neuronal pentraxins. Behav. Brain Res. 347, 49–56. doi: 10.1016/j.bbr.2018.02.015
Swardfager, W., Lanctt, K., Rothenburg, L., Wong, A., Cappell, J., and Herrmann, N. (2010). A meta-analysis of cytokines in Alzheimer’s disease. Biol. Psychiatry 68, 930–941. doi: 10.1016/j.biopsych.2010.06.012
Sweeney, M. D., Sagare, A. P., and Zlokovic, B. V. (2018). Blood–brain barrier breakdown in Alzheimer’s disease and other neurodegenerative disorders. Nat. Rev. Neurol. 14:133. doi: 10.1038/nrneurol.2017.188
Szczepanik, A. M., Funes, S., Petko, W., and Ringheim, G. E. (2001). IL-4, IL-10 and IL-13 modulate Aβ(1–42)-induced cytokine and chemokine production in primary murine microglia and a human monocyte cell line. J. Neuroimmunol. 113, 49–62. doi: 10.1016/S0165-5728(00)00404-5
Takechi, R., Lam, V., Brook, E., Giles, C., Fimognari, N., Mooranian, A., et al. (2017). Blood-brain barrier dysfunction precedes cognitive decline and neurodegeneration in diabetic insulin resistant mouse model: an implication for causal link. Front. Aging Neurosci. 9:399. doi: 10.3389/fnagi.2017.00399
Tao, Q., Ang, T. F. A., DeCarli, C., Auerbach, S. H., Devine, S., Stein, T. D., et al. (2018). Association of Chronic low-grade Inflammation with Risk of Alzheimer disease in ApoE4 carriers. JAMA Netw. Open 1:e183597. doi: 10.1001/jamanetworkopen.2018.3597
Teng, Z., Guo, Z., Zhong, J., Cheng, C., Huang, Z., Wu, Y., et al. (2017). ApoE influences the blood-brain barrier through the NF-κB/MMP-9 pathway after traumatic brain injury. Sci. Rep. 7:6649. doi: 10.1038/s41598-017-06932-3
Terzioglu, G., and Young-Pearse, T. L. (2023). Microglial function, INPP5D/SHIP1 signaling, and NLRP3 inflammasome activation: implications for Alzheimer’s disease. Mol. Neurodegener. 18, 1–18. doi: 10.1186/S13024-023-00674-9
Vergallo, A., Lista, S., Lemercier, P., Chiesa, P. A., Zetterberg, H., Blennow, K., et al. (2020). Association of plasma YKL-40 with brain amyloid-β levels, memory performance, and sex in subjective memory complainers. Neurobiol. Aging 96, 22–32. doi: 10.1016/j.neurobiolaging.2020.07.009
Villeda, S. A., Luo, J, Mosher, K. I., Zou, B., Britschgi, B, Bieri, G., et al. (2011). The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature. 477, 90–94. doi: 10.1038/nature10357
Wang, S., Mustafa, M., Yuede, C. M., Salazar, S. V., Kong, P., Long, H., et al. (2020). Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer’s disease model. J. Exp. Med. 217:e20200785. doi: 10.1084/JEM.20200785
Wang, Y., Ulland, T. K., Ulrich, J. D., Song, W., Tzaferis, J. A., Hole, J. T., et al. (2016). TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J. Exp. Med. 213, 667–675. doi: 10.1084/JEM.20151948
Wang, Z., Zhang, S., Xiao, Y., Zhang, W., Wu, S., Qin, T., et al. (2020). NLRP3 Inflammasome and inflammatory diseases. Oxidative Med. Cell. Longev. 2020:4063562. doi: 10.1155/2020/4063562
Weggen, S., Eriksen, J. L., Sagi, S. A., Pietrzik, C. U., Ozols, V., Fauq, A., et al. (2003). Evidence that nonsteroidal anti-inflammatory drugs decrease amyloid beta 42 production by direct modulation of gamma-secretase activity. J. Biol. Chem. 278, 31831–31837. doi: 10.1074/JBC.M303592200
Westin, K., Buchhave, P., Nielsen, H., Minthon, L., Janciauskiene, S., and Hansson, O. (2012). CCL2 is associated with a faster rate of cognitive decline during early stages of Alzheimer’s disease. PLoS One 7:e30525. doi: 10.1371/journal.pone.0030525
Wilkinson, B. L., Cramer, P. E., Varvel, N. H., Reed-Geaghan, E., Jiang, Q., Szabo, A., et al. (2012). Ibuprofen attenuates oxidative damage through NOX2 inhibition in Alzheimer’s disease. Neurobiol. Aging 33, 197.e21–197.e32. doi: 10.1016/j.neurobiolaging.2010.06.014
Winkler, E. A., Nishida, Y., Sagare, A. P., Rege, S. V., Bell, R. D., Perlmutter, D., et al. (2015). GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat. Neurosci. 18, 521–530. doi: 10.1038/nn.3966
Wolfe, C. M., Fitz, N. F., Nam, K. N., Lefterov, I., and Koldamova, R. (2018). The role of APOE and TREM2 in Alzheimer′s disease—current understanding and perspectives. Int. J. Mol. Sci. 20:81. doi: 10.3390/IJMS20010081
Wu, X., Xiao, Z., Yi, J., Ding, S., Gu, H., Wu, W., et al. (2021). Development of a plasma biomarker diagnostic model incorporating ultrasensitive digital immunoassay as a screening strategy for Alzheimer disease in a Chinese population. Clin. Chem. 67, 1628–1639. doi: 10.1093/CLINCHEM/HVAB192
Yang, R., Wang, Q., Li, F., Li, J., and Liu, X. (2015). Edaravone injection ameliorates cognitive deficits in rat model of Alzheimer’s disease. Neurol. Sci. 36, 2067–2072. doi: 10.1007/s10072-015-2314-y
Yeh, F. L., Wang, Y., Tom, I., Gonzalez, L. C., and Sheng, M. (2016). TREM2 binds to Apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-Beta by microglia. Neuron 91, 328–340. doi: 10.1016/j.neuron.2016.06.015
Yin, Z., Raj, D., Saiepour, N., Van Dam, D., Brouwer, N., Holtman, I. R., et al. (2017). Immune hyperreactivity of Aβ plaque-associated microglia in Alzheimer’s disease. Neurobiol. Aging 55, 115–122. doi: 10.1016/j.neurobiolaging.2017.03.021
Yin, J., Zhao, F., Chojnacki, J. E., Fulp, J., Klein, W. L., Zhang, S., et al. (2018). NLRP3 Inflammasome inhibitor ameliorates amyloid pathology in a mouse model of Alzheimer’s disease. Mol. Neurobiol. 55, 1977–1987. doi: 10.1007/s12035-017-0467-9
Yuan, P., Condello, C., Keene, C. D., Wang, Y., Bird, T. D., Paul, S. M., et al. (2016a). TREM2 Haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron 92, 252–264. doi: 10.1016/J.NEURON.2016.09.016
Yuan, P., Condello, C., Keene, C. D., Wang, Y., Bird, T. D., Paul, S. M., et al. (2016b). TREM2 Haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron 90, 724–739. doi: 10.1016/J.NEURON.2016.05.003
Zhang, Y., Tian, J., Ni, J., Wei, M., Li, T., and Shi, J. (2023). Peripheral blood and cerebrospinal fluid levels of YKL-40 in Alzheimer’s disease: a systematic review and Meta-analysis. Brain Sci. 13:1364. doi: 10.3390/brainsci13101364
Zhang, G., Wang, Z., Hu, H., Zhao, M., and Sun, L. (2021). Microglia in Alzheimer’s disease: a target for therapeutic intervention. Front. Cell. Neurosci. 15:749587. doi: 10.3389/fncel.2021.749587
Zhao, H., Wu, L., Yan, G., Chen, Y., Zhou, M., Wu, Y., et al. (2021). Inflammation and tumor progression: signaling pathways and targeted intervention. Signal Transduct. Target. Ther. 6, 1–46. doi: 10.1038/s41392-021-00658-5
Zheng, C., Zhou, X. W., and Wang, J. Z. (2016). The dual roles of cytokines in Alzheimer’s disease: update on interleukins, TNF-α, TGF-β and IFN-γ. Transl. Neurodegener. 5, 1–15. doi: 10.1186/S40035-016-0054-4
Keywords: neuroinflammation, Alzheimer’s disease, microglia, NSAIDs, inflammation
Citation: Amelimojarad M, Amelimojarad M and Cui X (2024) The emerging role of brain neuroinflammatory responses in Alzheimer’s disease. Front. Aging Neurosci. 16:1391517. doi: 10.3389/fnagi.2024.1391517
Edited by:
Yi Min Wan, National University Health System, SingaporeReviewed by:
Aashutosh Shetti, Northwestern University, United StatesCopyright © 2024 Amelimojarad, Amelimojarad and Cui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaonan Cui, Q1hOMjNAc2luYS5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.