
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
METHODS article
Front. Aging Neurosci. , 20 December 2023
Sec. Alzheimer's Disease and Related Dementias
Volume 15 - 2023 | https://doi.org/10.3389/fnagi.2023.1296919
This article is part of the Research Topic Current Chemical Approaches in Combating Neuroinflammation in Alzheimer's Disease (AD) View all 5 articles
 Manisha Prajapat1†
Manisha Prajapat1† Gurjeet Kaur1†Gajendra Choudhary1†Paras Pahwa1
Gurjeet Kaur1†Gajendra Choudhary1†Paras Pahwa1 Seema Bansal2Rupa Joshi1Gitika Batra3
Seema Bansal2Rupa Joshi1Gitika Batra3 Abhishek Mishra4Rubal Singla1Harminder Kaur1Praisy K. Prabha1Ajay Prakash Patel1
Abhishek Mishra4Rubal Singla1Harminder Kaur1Praisy K. Prabha1Ajay Prakash Patel1 Bikash Medhi1*‡
Bikash Medhi1*‡Alzheimer’s disease (AD) is the most common progressive neurodegenerative disease and is associated with dementia. Presently, various chemical and environmental agents are used to induce in-vitro models of Alzheimer disease to investigate the efficacy of different therapeutic drugs. We screened literature from databases such as PubMed, ScienceDirect, and Google scholar, emphasizing the diverse targeting mechanisms of neuro degeneration explored in in-vitro models. The results revealed studies in which different types of chemicals and environmental agents were used for in-vitro development of Alzheimer-targeting mechanisms of neurodegeneration. Studies using chemically induced in-vitro AD models included in this systematic review will contribute to a deeper understanding of AD. However, none of these models can reproduce all the characteristics of disease progression seen in the majority of Alzheimer’s disease subtypes. Additional modifications would be required to replicate the complex conditions of human AD in an exact manner. In-vitro models of Alzheimer’s disease developed using chemicals and environmental agents are instrumental in providing insights into the disease’s pathophysiology; therefore, chemical-induced in-vitro AD models will continue to play vital role in future AD research. This systematic screening revealed the pivotal role of chemical-induced in-vitro AD models in advancing our understanding of AD pathophysiology and is therefore important to understand the potential of these chemicals in AD pathogenesis.
Alzheimer’s disease (AD) is a neurodegenerative disease that is considered the most common cause of dementia. The key pathological hallmarks of AD are plaque and tangle formation arising due to amyloid beta deposition and hyper phosphorylation of tau in brain tissues, respectively (Basaly et al., 2021; Grant et al., 2021). The main clinical symptoms of Alzheimer’s disease include cognitive dysfunction and progressive loss of memory. There are multiple factors responsible for AD, such as genetic mutations, epigenetic factors, aging, and external environmental factors, leading to abnormal neuronal function (Tiwari et al., 2019; Basaly et al., 2021). The German neurologist Dr. Alois Alzheimer discovered this neurodegenerative disease in 1906 while treating a 51-year-old patient who was suffering from memory loss, language problems, hallucinations, and disorientation (Tiwari et al., 2019; Reitz et al., 2020). AD is a major contributor to the overall global epidemic, with approximately 48.6 million people suffering from AD or dementia worldwide. Approximately 10% of people 65 years of age or above are affected by AD (Wang et al., 2021). It is expected that the cases of AD will rise at a highly accelerated rate by 2050. To date, there is no proper cure or prevention for Alzheimer’s disease. Furthermore, the exact mechanism of AD pathogenesis is still elusive. According to previous studies, the pathological characteristics of AD are defined by the extracellular accumulation of insoluble amyloid beta protein, intracellular aggregation of hyperphosphorylated tau protein, mitochondrial dysfunction, increased inflammation, and oxidative stress–induced apoptotic activity (McCarthy et al., 2004; Gan et al., 2014; Rai et al., 2020b). Many environmental factors and chemicals have been identified in the past for inducing AD models in different in vitro and in vivo studies (Wang B. R. et al., 2018). Furthermore, the increasing prevalences of various metabolic diseases such as diabetes have also been linked to neurodegeneration as well the pathogenesis of AD (Martins, 2018).
To date, there is no systematic review study that has thoroughly investigated the chemical agents that can be used to induce in vitro AD models. Thus, this is the first study to compile all the AD inducible agents in in-vitro models. The first objective of this study is to determine the number of chemical and environmental agents that have been utilized to induce Alzheimer’s disease (AD) in in vitro models up to the present time. The second objective is to identify those agents that have demonstrated the potential to induce pathological features of AD, such as amyloid beta accumulation and tau hyperphosphorylation, in cell culture models. Thirdly, we seek to uncover previously unknown in-vitro agents that can be employed to establish a reliable and replicable AD in-vitro model.
To achieve these three objectives, we conducted a systematic review of all original research studies focused on Alzheimer’s disease induced in in vitro models. Therefore, in this study, we systematically reviewed all in-vitro studies involving AD models to enlist different types of AD inducing agents. Through this study, we compared various AD-inducing agents and also explored the underlying pathological pathways associated with the disease.
The search strategy was based on the PICO (Population, Intervention, Control, and Outcomes) format, with the approach as follows: P—In vitro inducible AD model; I—None; C—None; and O—Presence of amyloid beta accumulation/tau hyperphosphorylation/neuronal toxicity. Four authors independently searched the selected databases for inclusion of relevant studies in the systematic review.
We screened PubMed, Science Direct, and Google Scholar for the keywords “chemically induced Alzheimer disease model in vitro study,” “Alzheimer disease” AND “cell culture,” “inducing agent” AND “Alzheimer disease,” and “Alzheimer disease” AND “in vitro,” from the year 2001 to 24 March 2023. The search result files were extracted and initially screened based on the title and abstract using Rayyan QCRI. Further screening of full text articles was carried out to identify studies for inclusion in the systematic review. Research articles involving the use of an induced in vitro AD model were included for further reviewing. The studies were screened as per the criteria described in the Prisma chart (Figure 1). The basis of inclusion was primarily the identification of different chemicals, metals, and environmental agents that were being used for in vitro studies.
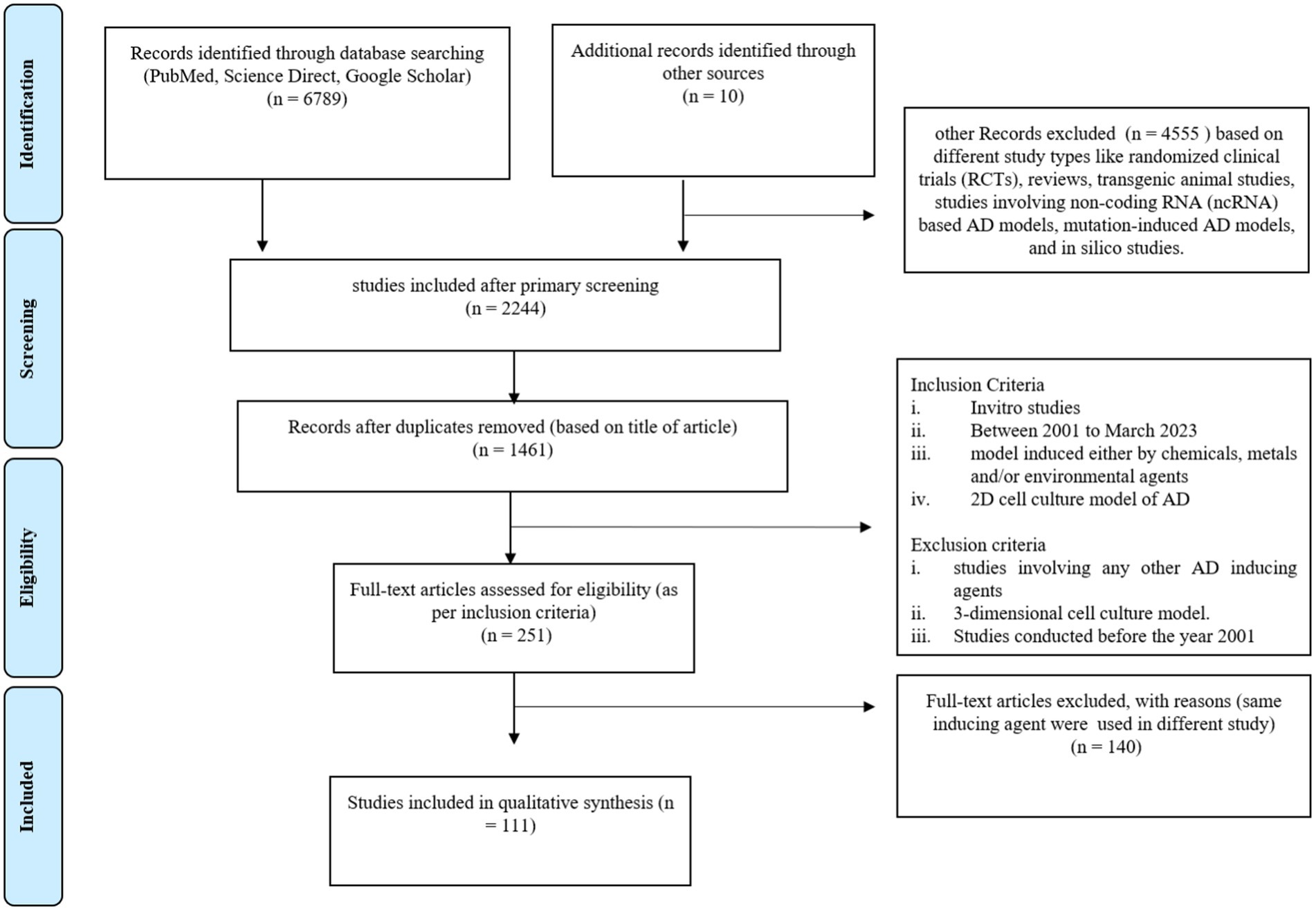
Figure 1. Prisma chart of this systematic review study.
A total of 6,799 articles were extracted after preliminary screening of the databases, out of which, 98 duplicate studies were removed. In the initial screening, 4,555 articles were excluded on the basis of a different study type, such as randomized clinical trials (RCTs), reviews, transgenic animal studies, studies involving non-coding RNA (ncRNA)–based AD models, mutation-induced AD models, and in silico studies. The primary screening results revealed 2,244 studies. Further, based on the title of the article, 1,461 studies were included and a total of 484 articles were excluded. Further, 1,210 articles were excluded based on different study types, such as randomized clinical trials (RCTs), reviews, transgenic animal studies, studies involving non-coding RNA (ncRNA)–based AD models, mutation-induced AD models, and in silico studies. The remaining 251 articles were screened for inclusion of in vitro and in vivo studies with only chemical- and environmental-based agents to induce the AD model. Based on the use of different inducing agents in in-vitro studies, we selected 111 articles for this systematic review. These articles were only related to original research including in-vitro models of AD research. The details of the included studies, such as the inducing agents used and the molecular markers studied for expression are summarized in Table 1.
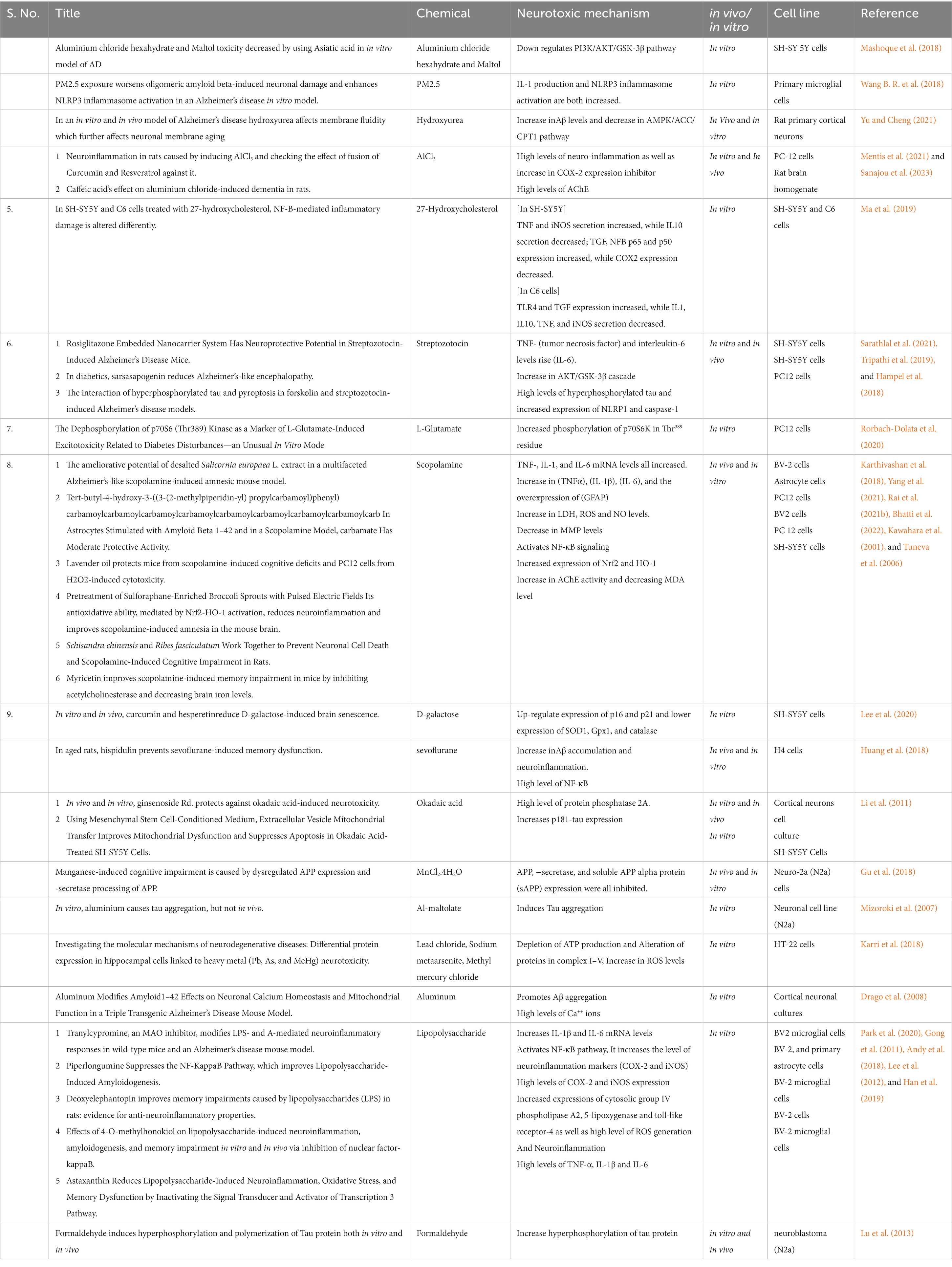
Table 1. Details of different studies on inducing agent and their molecular marker expression.
There are several pathological pathways that are directly or indirectly involved in the neurodegeneration process of Alzheimer’ disease.
The most widely accepted hypothesis for AD pathogenesis is based on the cholinergic pathway, amyloid beta cascade, and tau hyperphosphorylation. These three hypotheses are linked by various interconnected pathway mechanisms.
According to the cholinergic hypothesis, the dysfunction of acetylcholine neurotransmission in neurons is majorly due to a reduction in acetylcholine-transferase enzyme, a reduction of acetylcholine receptors, or reduction of acetylcholine itself. Moreover, cholinergic loss is based on the degeneration of cholinergic neurons and their projecting axons to the cerebral cortex. The main components for acetylcholine synthesis are choline, acetyl coenzyme, and choline acetyltransferase (ChAT). Normally, acetylcholine is released from presynaptic neurons into the synapse, from where it further binds to postsynaptic receptors (M1) that subsequently transmit the signal to connecting neurons. Another enzyme, acetyl cholinesterase, is also responsible for the breakdown of acetylcholine into choline and acetate in the synaptic cleft, which are taken by cells for the process of recycling (Srivastava et al., 2019). Therefore, the deregulation of the enzymatic activity of acetylcholinesterase may also lead to AD (Hampel et al., 2018; Gothwal et al., 2019; Tripathi et al., 2019). In the case of the amyloid beta cascade hypothesis, Aβ is formed when the APP protein is digested by alpha, beta, and gamma secretase. Previous studies have shown that in the case of the non-amyloidogenic pathway, alpha and gamma-secretase enzymes produce a C-terminal fragment, soluble ectodomain, and cleave the APP transmembrane protein. Another pathway is the pathological amyloidogenic pathway, in which the APP protein is cleaved by beta and gamma-secretase enzymes, producing amyloid beta peptide fragments that are more prone to aggregation, thus leading to the development of insoluble oligomers and amyloid plaques. The accumulation of the insoluble oligomers induce synaptic dysfunction, inflammation, mitochondrial damage, hyperphosphorylation, and dysregulation of calcium homeostasis, ultimately leading to neuronal toxicity and degeneration (Esmieu et al., 2019; Fan et al., 2020; Rai et al., 2020a). Tau is a protein expressed by the MAPT (microtubule associated tau) gene and is found in the neuronal axons of the brain. The tau protein has a main roles in the maintenance of microtubule structure, transport function, and synapse structure. The phosphorylation and dephosphorylation status of the tau protein is dependent on the activity of protein kinase and protein phosphatase. In disease conditions, hyperphosphorylation of the tau protein results in structural alteration, leading to its detachment from microtubules, further resulting in a loss of microtubule stability and polymerization capacity (Ismail et al., 2017). This results in elevated tau protein levels in the cytoplasm, leading to tau aggregation and the formation of insoluble paired helical filaments (PHFs). The deposition of PHFs in the form of fibrillar neurofibrillary tangles (NFTs) causes neurotoxicity, cell dysfunction, and loss of synapses (Fan et al., 2020; Rai et al., 2020b).
Other routes underlying the etiology of AD, such as excitatory glutamatergic neurotransmission through N-methyl-d-aspartate receptor (NMDAR), result in triggered neuro excitotoxicity (Wang and Reddy, 2017). Dysregulation of Wnt signaling (wingless/integrated), the AKT pathway, MAPK/ERK pathway, JNK pathway (Jun N-terminal kinase), RAGE (receptor for advanced glycation end products) pathway, and AMPK (AMP-activated protein kinase) pathway have also been reported in previous studies and have been linked to the pathogenesis of Alzheimer’s disease (Atzori et al., 2001; Greco et al., 2009; Exil et al., 2014; Loera-Valencia et al., 2021; Ramakrishna et al., 2023).
Other pathways, such as activation of glial cells (like microglia and astrocytes), can induce oxidative and inflammatory reactions, which lead to neuronal dysfunction and apoptosis, and eventually Alzheimer’s disease. The microglial cells have a role in the clearance of amyloid plaque; however, aberrant gene expression of ABCA7 (ATP-binding cassette sub-family A member 7), CD3 (cluster of differentiation-3), CR1 (complement receptor type 1), SPI1, and TREM2c (triggering receptors expressed on myeloid cells 2c) can expedite the neuronal degeneration process. The disrupted phagocytic activity of microglia cells enhances the accumulation of amyloid beta deposition, subsequently leading to neuronal degeneration (McCarthy et al., 2004; Wang et al., 2021). Several studies have also reported the role of the transcription factor AP1 (activator protein 1), NFkB (nuclear factor k beta), SP 1, NRF2 (nuclear factor-erythroid factor 2), and TFEB (transcription factor EB) in initiating the process of neuro degeneration that might lead to the development of AD (Rai et al., 2021b).
The emerging world faces a genetic risk of Alzheimer’s disease (AD) due to mutations in PSEN1 (located on chromosome 14), PSEN2 (located on chromosome 1), and APP (located on chromosome 21), which are known to cause familial Alzheimer’s disease (FAD). Some non-genetic factors have also been identified to be involved in AD pathogenesis, such as smoking, hypercholesterolemia, obesity, vitamin deficiency, diabetes, hypertension, head trauma, stroke, and depression. The severity of metabolic diseases and traumatic injuries was shown to increase as a result of exposure to pesticides, an electromagnetic field, aluminium, lead, iron, copper, zinc, mercury, particulate matter, and solvents, which, directly or indirectly, might pose a risk for developing AD (Rahman et al., 2020; Rai et al., 2021a). Hence, all these environmental and chemical factors were able to induce plaque and tangle formation, increase oxidative stress, and decrease neprilysin (amyloid beta degrading enzyme), thereby increasing neuronal death. Thus, pre- and post-natal exposure to these environmental factors may promote the development of this neurodegenerative disease in later life (Chin-Chan et al., 2015; Esmieu et al., 2019). Nonetheless, numerous protective factors, including social contact, mental activity, nonsteroidal anti-inflammatory (NSAIDs) usage, coffee intake, moderate alcohol use, physical activity, and past vaccinations may lessen the chance of developing AD (Mentis et al., 2021). The pathophysiology of AD is represented in Figure 2.
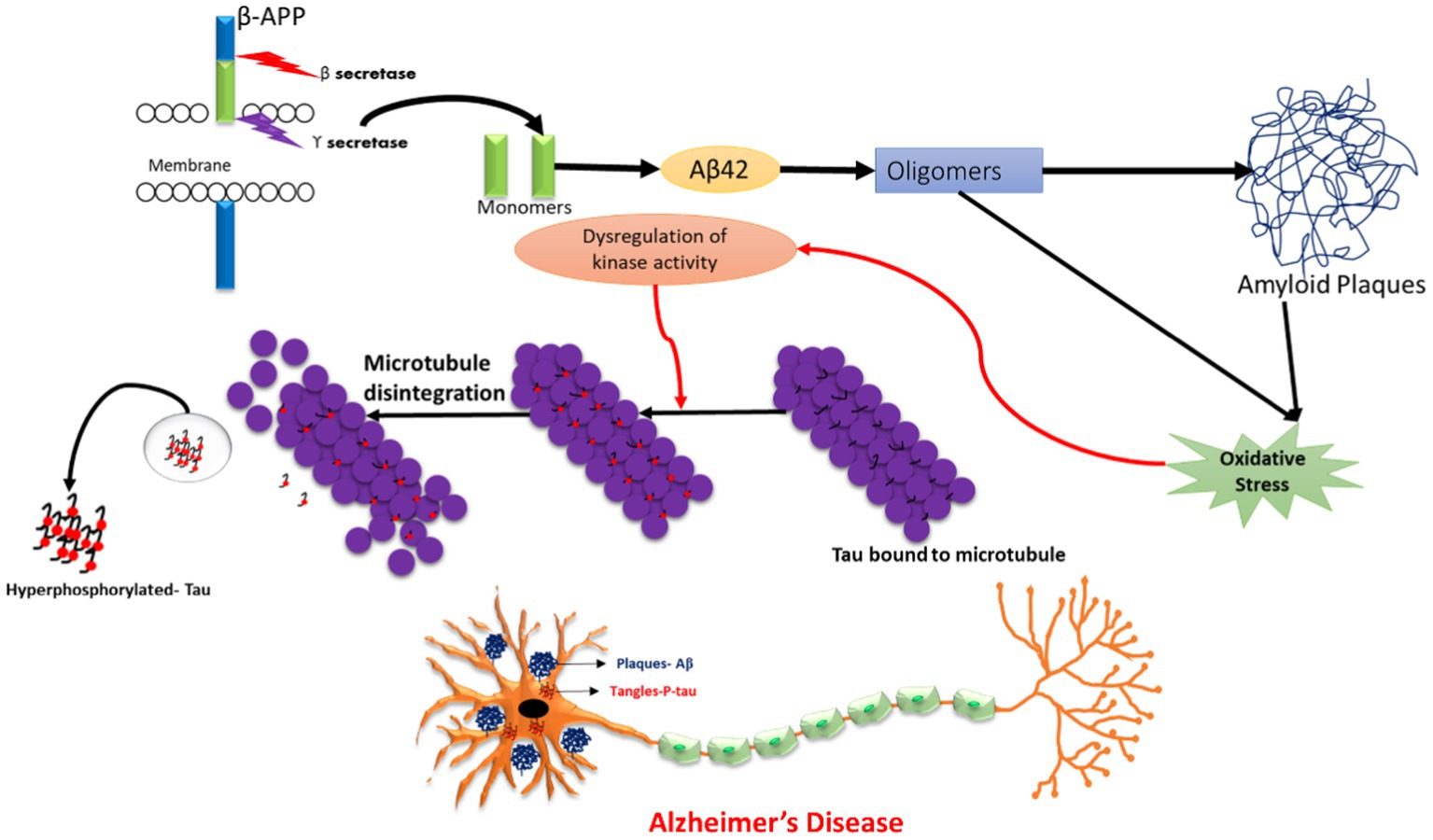
Figure 2. Schematic representation of Alzheimer’s Disease (AD) pathology.
The toxic effects of many agents, such as environmental, chemical, and xenobiotic agents, have been identified in previous in vitro and in vivo studies (Lu et al., 2013; Martins, 2018). Longer exposure to these toxic agents can be hazardous as they induce neuroinflammation and neuropathology, opening the door for AD development (Wang B. R. et al., 2018).
AD inducing agents can be divided into two categories: (1) environmental inducing agents and (2) chemically inducing agents (Figure 3).
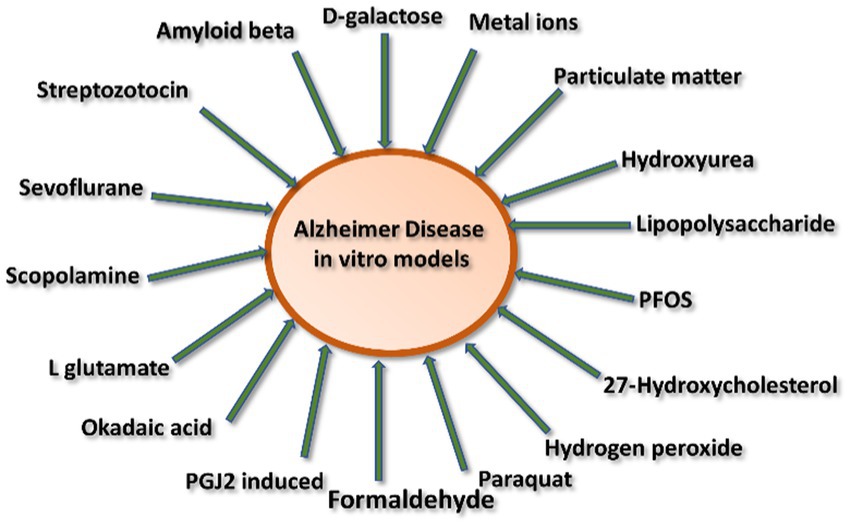
Figure 3. Schematic representation of various inducing agents involved in Alzheimer’s disease.
Aluminum chloride hexahydrate (AlCl3H12O6), maltol, PM2.5 (particulate matter 2.5), hydroxyurea, AlCl3, 27-hydroxycholesterol, H2O2, MnCl2.4H2O, Al-maltolate, lead, arsenic, and mercury.
Excessive exposure to heavy metals has been found to be involved in neurotoxicity and neuronal damage.
Aluminum is abundantly present as an environmental toxicant and is associated with neuropathological disease. Aluminum chloride enters the body via soil, food, pharmaceutical agents, and water, from where it may further accumulate in the brain and affect neurotransmitter synthesis, axonal transport, disturb the balance of protein phosphorylation and dephosphorylation, protein degradation, and gene expression, thereby inducing inflammation, apoptotic activity, and amyloid beta deposition. Al can also cause mitochondrial and endoplasmic reticulum dysfunction, leading to an increase in oxidative stress and apoptosis (Kawahara et al., 1994; Mustafa Rizvi et al., 2014). Therefore, aluminum is a possible inducing agent to cause neurotoxicity as well as neurodegenerative diseases such as Alzheimer’s disease (Tiwari et al., 2019).
In the mechanistic pathway, Al is able to decrease the nucleus translocation of NRF2, either directly or by indirectly activating GSK3 beta. Increased NRF2 levels in the cytoplasm phosphorylate the BAX apoptotic protein, which is further involved in the release of cytochrome-c from the mitochondria, leading to apoptosis activity. On the other hand, Al also activates GSK3 beta by inhibiting AKT and Wnt signaling pathways, which are further responsible for hyperphosphorylation of the tau protein, leading to synaptoxicity (Kawahara et al., 1994; Hong and An, 2018; Sanajou et al., 2023).
Previous studies have shown that aluminum reacts with maltolate, which is extensively present in normal human food items (baked cereals, bread, browned food, cakes, chocolate milk, and coffee), leading to the formation of aluminum maltolate Al (mal)3 in the intestine, which further increases the Al level in the brain and subsequently induces oxidative stress and mitochondrial dysfunction (Mashoque et al., 2018; Reitz et al., 2020; Bhatti et al., 2022). Al (mal)3 is further involved in transcriptional disruption rather than translational modification (Mustafa Rizvi et al., 2014). Through many in-vitro studies, it was found that the chronic exposure of cells to 50–100 mM AlCl3 caused a disruption in synapse development and promoted tau protein build-up. According to a previous study, Al (mal)3 and Al (acac)3 (aluminium acetylacetonate) can easily penetrate cells, thereby increasing the cellular toxicity. Axonal transport in cultured rat cortical neurons was found to be affected after 1-h pulse exposure to 250 mM Al (mal)3 (Kawahara et al., 2001). Another in vitro study found that Al (≤ 50 μM) enhanced the survival of cerebellar granule neuronal cells; however, higher levels of Al (≥ 100 μM) induced cell death (Tuneva et al., 2006; Figure 4).
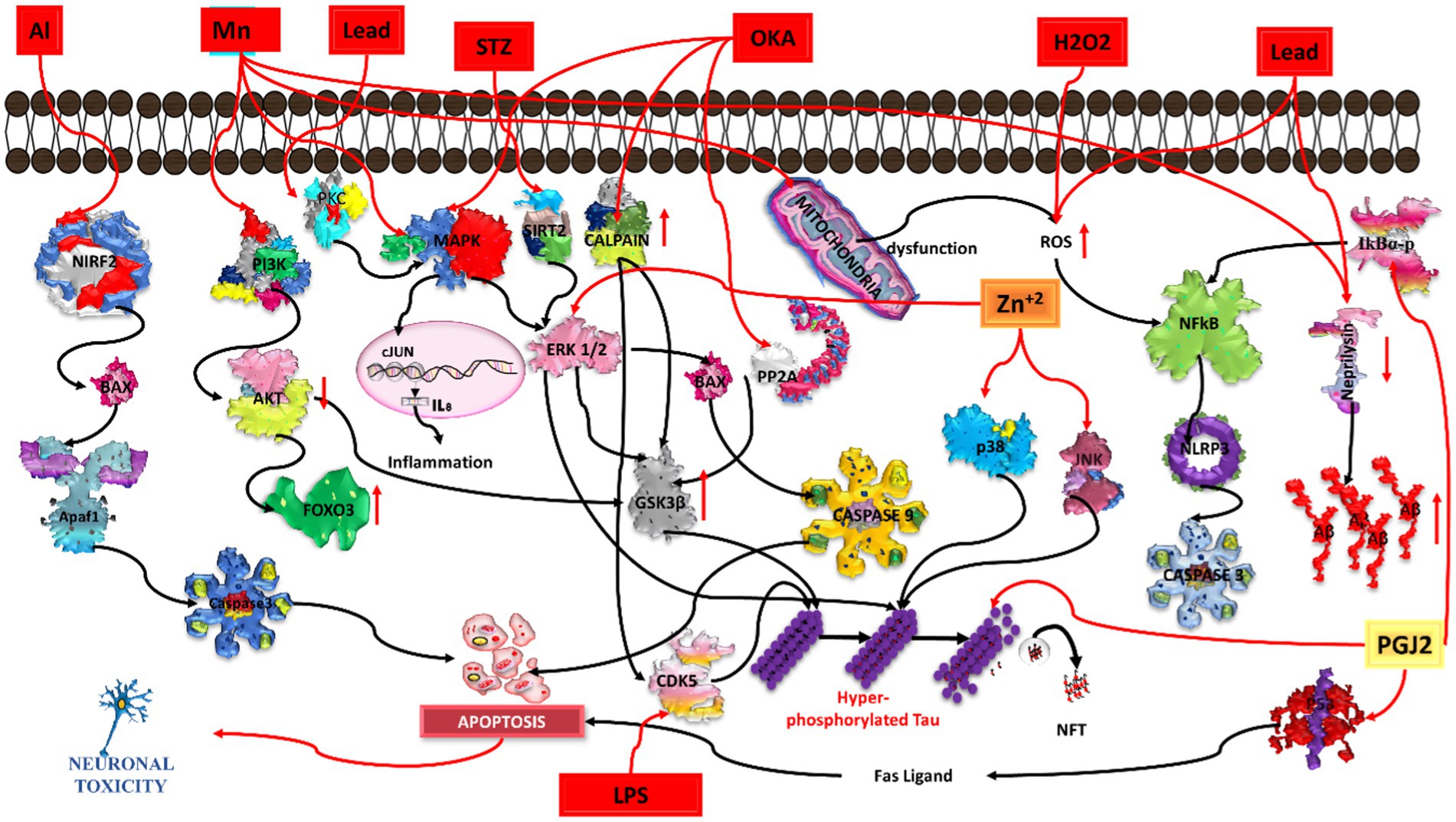
Figure 4. Illustration of various chemicals inducing Alzheimer’s pathology in in-vitro conditions. Aluminium (Al) triggers oxidative stress and neuroinflammation, contributing to beta-amyloid plaques and tau tangles. Manganese (Mn) induces mitochondrial dysfunction and neuronal damage. Lead (Pb) disrupts calcium homeostasis, promoting beta-amyloid aggregation. Streptozocin (STZ) impairs glucose metabolism, exacerbating tau pathology. Okadaic acid (OKA) activates protein phosphatases, causing hyperphosphorylation of tau. Hydrogen peroxide (H2O2) generates oxidative stress, leading to beta-amyloid accumulation. Lipopolysaccharide (LPS) induces neuroinflammation, contributing to beta-amyloid deposition and synaptic dysfunction. Common pathways include oxidative stress, neuroinflammation, and protein aggregation, while each chemical initiates specific pathways leading to neurodegeneration.
Increased exposure to Mn can cause cognitive impairment, which leads to Alzheimer disease. Mn enters the brain via diet, soil, and water, inducing apoptosis and inflammation and leading to neuronal degeneration. A higher concentration of Mn can activate MAPK/ERK/GSK3 beta signaling for hyperphosphorylation of the tau protein (Cai et al., 2011). Mn can also produce reactive oxygen species (ROS) through mitochondrial dysfunction, which further follow the activation of the NfKb/NLRP3/caspase1 signaling pathway and results in neuronal apoptosis. FOXO3 is a transcript factor expressed in the brain. It has a role in maintaining protein homeostasis; however, it is also implicated in neurodegenerative disease. A previous study suggested that Mn-exposed cells markedly increased the phosphorylation of MAPK/ERK, which further phosphorylated FOXO3; it is not able to enter the nucleus and could not transcribe the genes involved in antioxidant activity, DNA repair, and anti-apoptotic activities (Exil et al., 2014). A previous in vitro study found that the viability of N2a cells gradually decreases when exposed to Mn at concentrations of 50, 100, 200, 400, 800, 1,000, and 1,200 μM for 24 h. Mn also inhibited the processing of APP (amyloid precursor protein) and produced amyloid beta aggregations; however, it did not show any effect on BACE1 (beta-secretase1)–based APP processing or amyloid beta trafficking. Mn was found to decrease the expression of the synaptic protein and decrease neprilysin expression (Tong et al., 2014; Yang et al., 2021; Figure 4).
Lead may induce amyloid beta accumulation in differentiated SHSY5Y cells. According to a previous study, when differentiated SHSY5Y cells were treated with different concentrations (ranging between 0–5,000 μM) of lead for 24 to 72 h, no significant toxicity was observed for the lower concentrations (1, 5, 10, 20, or 50 μM) at 48 and 72 h. However, higher concentrations of lead (above 1,000 μM) reduced the cell viability at 72 h. Treatment with Pb also induces morphological alterations in neurons, thereby leading to cell damage. Higher exposure of Pb may increase AβPP (amyloid β-protein precursor) gene expression, producing more AβPP protein and further increasing the amyloid beta levels. Moreover, Pb decreases NEP (neutral endopeptidase) gene expression, reducing the production of NEP protein. The NEP protein acts as a degradation enzyme for amyloid beta. Hence, higher exposure to lead can induce increased amyloid beta levels, thereby playing a vital role in the onset of AD (Huang et al., 2011; Figure 4).
Arsenic is a metalloid and an environmental risk factor for AD. Various studies have highlighted the role of arsenic and its derivatives in the induction and progression of AD pathogenesis. In previous studies, sodium arsenite exposure to differentiated SHSY5Y cells at a low concentration range (i.e., 1 to 10 μM) was able to decrease tau1 expression via dephosphorylation at 189–207 residues and phosphorylation at 202 residues. Furthermore, the study observed that arsenic increases GSK3 beta kinase activity and also increases the phosphorylation of ERK1/2 in neurons, thereby inducing hyperphosphorylation of the tau protein and subsequently leading to neurotoxicity as well as apoptosis (Wisessaowapak et al., 2021). Another study reported that arsenite induces neurotoxicity in the transfected Hela-p35 cell line by increasing the calcium level and activating the calpain protein (calcium dependent proteases). Calcium-induced activated calpain causes cleavage of p35 to p25, resulting in the formation of a hyper-activated complex of CDK5.p25. This complex was found to be responsible for hyperphosphorylation of tau and NFT formation (Vahidnia et al., 2008). Thus, higher exposure to arsenic upregulates the hyperphosphorylated tau protein, a hallmark feature of AD pathogenesis. A study by Rehman et al. observed that exposure of PC12 cells to arsenic (10 μM) causes neurotoxicity by increasing ROS production, leading to mitochondrial dysfunction, thereby increasing cytochrome C release and activating caspase3. Therefore, activated caspase3 causes apoptosis of neuronal cells. Additionally, arsenic also altered the protein expression of apoptosis and autophagy markers such as the downregulation of Nrf2, Bcl2/Bclx, mTOR, and Akt and the upregulation of P38 MAPK, JNK, LC3, and Bax (Rahaman et al., 2020). Hence, all these factors contribute to neurotoxicity, neuroinflammation, and neuronal loss and, ultimately, the progression of AD pathogenesis (Figure 5).
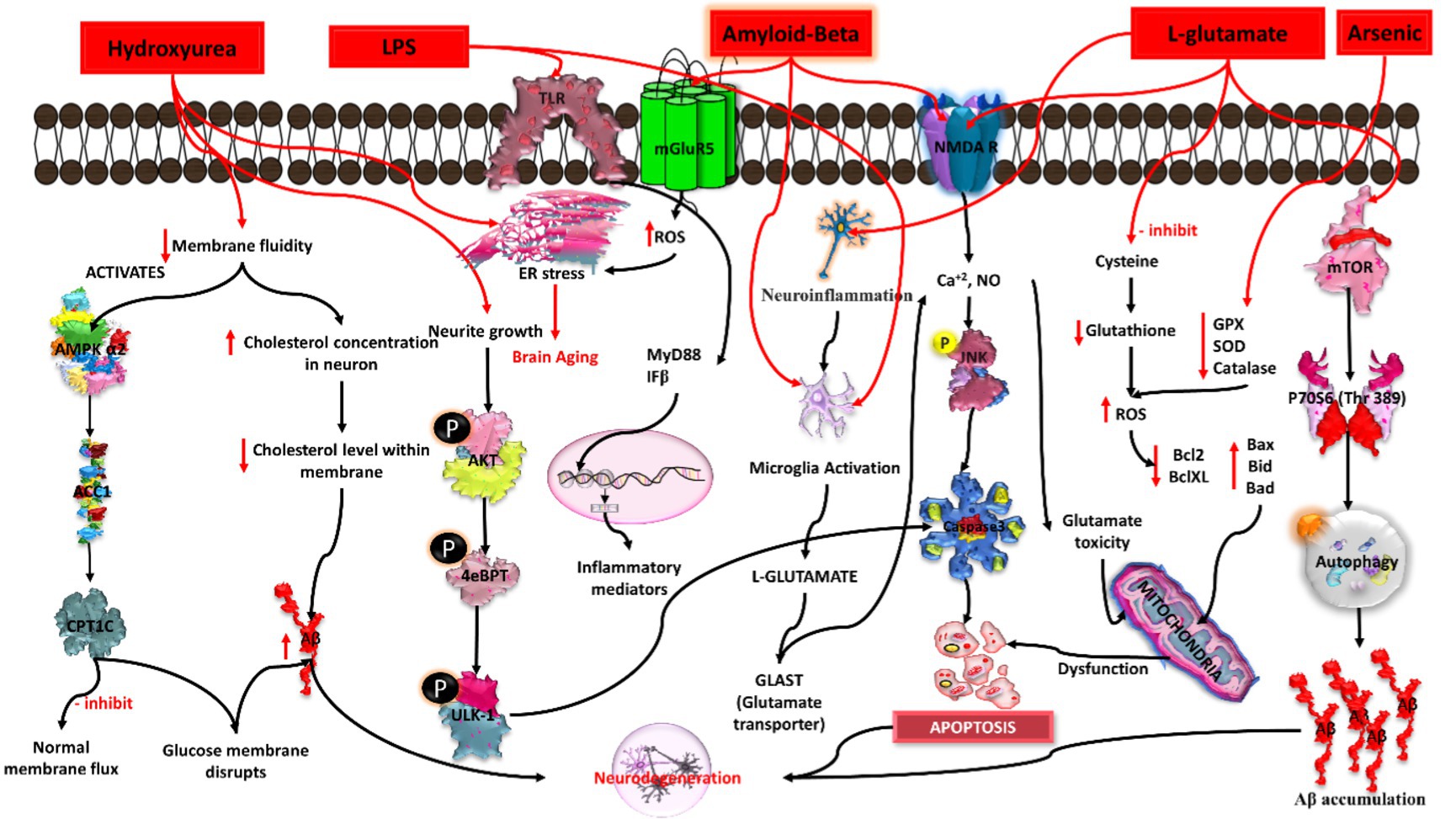
Figure 5. Mechanistic pathways of Alzheimer’s induction by chemical agents in in-vitro conditions. Hydroxyurea triggers DNA damage, lipopolysaccharide (LPS) induces neuroinflammation, amyloid-beta (Aβ) promotes plaque formation, L-glutamate contributes to excitotoxicity, and arsenic disrupts cellular homeostasis. Each agent elicits specific pathways leading to Alzheimer’s pathology, collectively portraying a comprehensive understanding of the diverse molecular mechanisms involved in disease onset.
Mercury is able to induce glutamate-mediated excitotoxicity, which further leads to neuronal degeneration. Many previous studies have revealed that mercury affects glutamatergic signaling by reducing the re-uptake of glutamate, enhancing spontaneous glutamate release from neurons, thereby inhibiting glutamine synthetase, which subsequently enhances the glutamate level in the synaptic cleft. Moreover, the postsynaptic NMDA (N-methyl-D-aspartic acid or N-methyl-D-aspartate) receptor also has also been found to be affected by MeHg (methylmercury). When cortical neuronal cultures were exposed to HgCl2 at different concentrations, i.e., 25 nM,100 nM, and 25 μM for 3 days, neurite growth is inhibited, leading to neuronal degeneration (Xu et al., 2012). Evidence suggests that HgCl2 (mercury chloride) can induce oxidative stress and increase amyloid beta production, leading to hyperphosphorylation of the tau protein via that induces cellular toxicity, with reduced cell viability (Olivieri et al., 2000; Song and Choi, 2013). Mercury is also used for the development of a BBB (blood brain barrier) in vitro model (Toimela et al., 2004). Additionally, mercury chloride has the ability to increase β-secretase levels, which further elevate the expression of beta secretase-1 (BACE 1) and neprilysin (NEP) proteins, leading to high levels of APP (Figure 6).
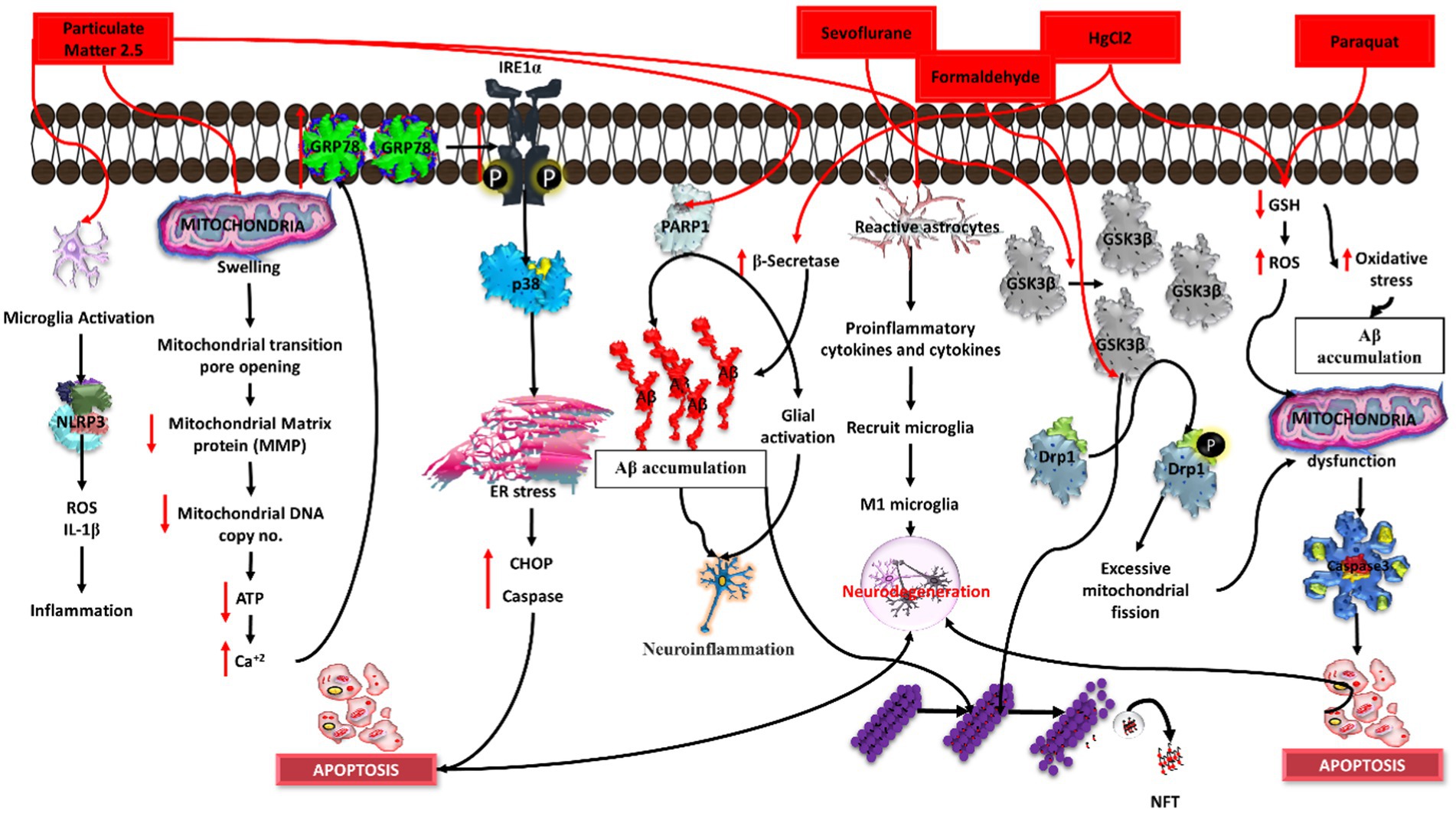
Figure 6. Induction of Alzheimer’s disease (AD) by distinct chemical agents in an in-vitro context. Particulate matter 2.5 initiates neuroinflammation, sevoflurane disrupts synaptic function, formaldehyde promotes tau hyperphosphorylation, magnesium chloride (MgCl2) induces oxidative stress, and paraquat triggers mitochondrial dysfunction. Each agent instigates specific pathways, collectively illuminating the intricate mechanisms underlying AD pathology in response to environmental and chemical exposures.
Zinc may regulate various pathways such as MAPK (mitogen-activated protein kinase), PI3K (phosphoinositide 3-kinase), and rapamycin-dependent pathways. When the SHSY5Y neuroblastoma cell line was exposed to 100 μM zinc sulphate, it influenced the ribosomal protein kinase and phosphorylation status of GSK3 beta. The long-term exposure of SHSY5Y cells to zinc can induce phosphorylation of various proteins such as p-PKB, p-ERK1/2, p-JNK, and p-p38, which can further alter the tau protein (An et al., 2005). Another study showed that SN56 neuroblastoma cholinergic cells uptake more zinc ions, which leads to decreased acetyl-CoA levels, ATP, and acetylcholine synthesis (Zyśk et al., 2018). Moreover, nanoparticles of zinc oxide are known to be involved in the induction of oxidative stress and inflammation in different cell types. Zinc oxide can induce apoptosis through the PI3K-AKT-caspase3/7 pathway and necrosis via LOX-mediated ROS level elevation (Kim et al., 2015; Figure 4).
Currently, research is focused on the association between AD and air pollutants, as well as the development of in vitro and in vivo study models using air pollutants. It was found that long-term exposure to certain air pollutants can induce inflammation and neuropathology, which pave the way for AD development (Wang B. R. et al., 2018). Various air pollutant particulate matter have severe health threats. Particulate matter (PM) is usually categorized into three types: ultra-fine PM (less than 0.1 μM), fine PM (less than 2.5 μM), and coarse PM (PM10 or less than 10 μM). Among these three types, PM2.5 is composed of different inorganic and organic matter, such as carbon, ammonium, hydrogen ions, metals, water, sulphates, and lipopolysaccharides (Wang B. R. et al., 2018; Figure 6).
Exposure to PM2.5 causes amyloid beta–induced neuronal injury, microglial inflammation, and an increase in the production of IL-1β and ROS, triggering cytotoxicity (Wang B. R. et al., 2018). In a previous study, PM2.5 led to apoptosis in the neuroblastoma cell line by inducing endoplasmic reticulum (ER) stress due to the overexpression of the ER molecule chaperone GRP78. Exposure to PM2.5 leads to a significant increase in IRE1α, activating p38MAPK, which promotes apoptosis by increasing the expression of the ER stress-related protein CHOP, caspase, and GRP78, further accelerating the mechanism of the neurological disease (Wang et al., 2019; Zhang M. et al., 2022). It was validated through cell culture experiment that PM2.5 was able to cross the BBB (Kang et al., 2021). PM2.5 exposure to human bronchial epithelial (HBE) cells was able to decrease SIRT1 (sirtuin1) expression. SIRT1 has a role in deacetylase activity, which further decreases tau accumulation (Lai et al., 2019). The treatment of PM2.5 in a human neuroblastoma cell line activates poly (ADP-ribose) polymerase (PARP-1), which causes Aβ accumulation and glial activation, resulting in neuroinflammation (Jang et al., 2018). The PMs evaluated in J774A.1 proliferative cells were shown to exert a cytotoxic effect after 72 h at different concentrations. Furthermore, PMs were also able to induce IL-6 and TNF-alpha secretion in J774A.1 cells (Osornio-Vargas et al., 2003). Another in vitro investigation found that when neuroblastoma cells (N2a-APP/Swe) were exposed to PM, their APP processing increased. Short-term PM exposure to neuronal N2a-APP/Swe cells promotes pro-amyloidogenic APP processing, as measured by the increased sAPP/ratio and Aβ42 production (Cacciottolo et al., 2017).
Exposure of human SHSY5Y cells to different concentrations of particulate matter (25, 100, and 250 μg/mL) for 24 h (Zhang M. et al., 2022) influenced the morphology of the cells, increased the mitochondrial permeability, reduced the ATP production, decreased the mtDNA replication, and increased the expression of mitochondrial genes such as Drp1 (dynamin-related protein 1), OPA1 (optic atrophy-1), CypD (cyclophilin D), COX IV (cyclooxygenase IV), and SIRT3, thereby increasing the calcium levels. This cascade of events eventually leads to apoptosis by upregulating the apoptotic pathway (CHOP/caspase-12/caspase-8) in response to an increase in oxidative stress, which causes endoplasmic reticulum stress, neurotoxicity, and neuroinflammation (Lee et al., 2021; You et al., 2022).
Therefore, PM2.5 acts as an environmental inducer of Alzheimer’s disease by penetrating the BBB and disrupting its integrity, resulting in an increase in pro-inflammatory mediators (IL-1, IL-6, and TNF-alpha), ROS, amyloid-beta production, astrogliosis, microglial activation, mitochondrial dysfunction, and ER stress. All these factors contribute to neuroinflammation and neuronal injury (synaptic impairment, tau accumulation, and neuronal death), which lead to neurological diseases such as Alzheimer’s (Kawahara et al., 2001; Huang et al., 2011; Rai et al., 2020b; Figure 6).
Recent in vitro and in vivo studies show that hydroxyurea-induced “membrane aging” may be exploited to create an Alzheimer’s disease model. Hydroxyurea is primarily used to treat sickle cell disease and functions as an antineoplastic agent because it decreases deoxyribonucleotides and inhibits the ribonucleotide reductase enzyme. Hydroxyurea can induce breakage near the replication sites of dsDNA and can also break the mt-DNA, leading to mitochondrial dysfunction and stress conditions and subsequently leading to senescence-like changes in cells lines (Yu and Cheng, 2021). Previous studies revealed that hydroxyurea induced Alzheimer’s disease by decreasing the membrane fluidity via the AMP-activated protein kinase (AMPK) pathway. It activates AMPK 2 and reduces the expression of its downregulating genes, acetyl CoA carboxylase (ACC1), and carnitine palmitoyl transferase 1 (CPT1C), which inhibit normal membrane flux and disturb glucose metabolism. Normal membrane flux inhibition disrupts cytoskeleton anchoring, cellular transport, intracellular communication, and neuronal transmission. Because of the disturbance in glucose metabolism, there is an increase in amyloid-beta, which leads to neuronal degeneration. Because membrane fluidity is involved in the maintenance of cholesterol levels in neurons, studies have shown that a decrease in membrane fluidity causes an increase in neuronal cholesterol levels, which decrease cholesterol levels within membrane fractions and thus cause an increase in amyloid-beta (Vahidnia et al., 2008). Induced pluripotent stem cells (iPSCs), when treated with hydroxyurea at a concentration of 1 mM, 8 mM, and 16 mM, show ER stress, a decrease in neurite outgrowth, a decrease in the expression of p-AKT, p-4EBP1, and p-ULK-1, and an increase in the expression of caspase-3, leading to apoptosis and finally causing death of neurons (Rahaman et al., 2020; Figure 5).
27-hydroxycholesterol (27-OHC) has a role in decreasing glucose metabolism in neurons. A previous study explained that administration of 1 μM 27-OHC to rat primary neurons for 24 h leads to dampening of glucose uptake. It affects the insulin signaling pathway by decreasing GLUT-4 and increasing IRAP (interleukin-1 receptor antagonist protein) mRNA expression in cortical and hippocampal primary neurons. Because of the decrease in GLUT-4, there is a decrease in glucose uptake by neurons, which causes an imbalance in the glucose-insulin signaling pathway and leads to spatial memory deficits, an early clinical sign of AD (Ismail et al., 2017). Higher concentrations of 27-OHC were found to affect the RAGE receptor by mediating the nuclear receptor RXRγ. It influences RAGE by increasing the levels of S100A8, a calcium binding protein that acts as a ligand for the RAGE receptor. The increased level of RAGE in neurons, astrocytes, microglia, and endothelial cells leads to an inflammatory response that causes synaptic dysfunction and neurodegeneration. 27-OHC has the ability to cause oxidative stress, neuronal malfunction, and short-term memory impairment (Loera-Valencia et al., 2021). 27-OHC causes damage to astrocytic neuronal cells by activating the TGFβ/NFkB pathway, resulting in the subsequent release of inflammatory cytokines (Wang et al., 2020). Another study has shown that SHSY5Y cells treated with 27-OHC at a concentration of 5 μM, 10 μM, and 20 μM have increased levels of NFkBp65, NFkBp50, and TGF-beta and decreased expression of COX-2 (Ma et al., 2019) as well as elevated amyloid beta levels (Wang et al., 2020). Furthermore, it was found that STAT3 phosphorylated at the Tyr705 position after treatment with 27-OHC induces nerve cell senescence (Liu et al., 2021; Figure 7).
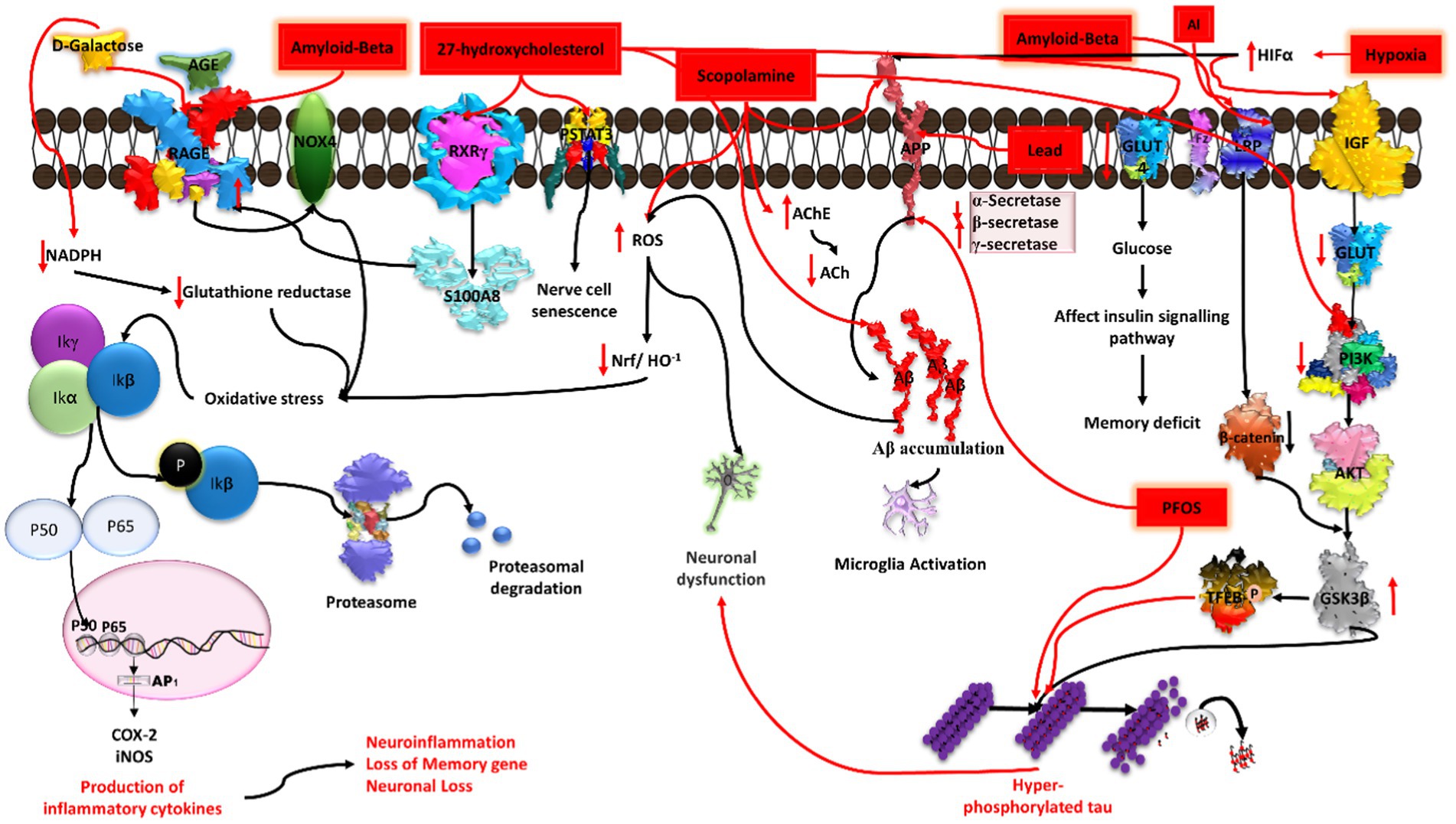
Figure 7. Illustration of the induction of Alzheimer’s disease (AD) by diverse chemical agents in an in-vitro setting. Amyloid-beta contributes to plaque formation, 27-hydroxycholesterol influences cholesterol metabolism, scopolamine disrupts cholinergic signaling, aluminium (Al) and lead (Pb) trigger neurotoxicity and oxidative stress, while hypoxia induces cellular oxygen deprivation. Each chemical agent initiates specific pathways, collectively providing insights into the multifaceted mechanisms underlying AD pathology.
At higher concentrations, H2O2, a significant source of reactive oxygen species (ROS), can lead to oxidative stress in various cell lines. This oxidative stress can trigger apoptosis and produce alterations in signaling pathways. Previous studies have reported that H2O2 can influence amyloid precursor protein (APP) expression and gamma secretase via JNK signaling, leading to an increase in amyloid-beta (Aβ) production by reducing the protein levels of both APP and C99. Therefore, under pathological conditions, JNK may serve as a key mediator, playing a central role in facilitating the stimulatory effect of various stress signals on γ-secretase activity (Shen et al., 2008). SK-N-SH cells treated with H2O2 for 24 h showed a dose-dependent cytotoxic effect, an increase in oxidative stress, activated caspase 3 activity, and acetylcholinesterase (AChE) inhibition activity (Sattayasai et al., 2013). In another study, the administration of 100 μm of H2O2 to PC12 cells for 2 h suppressed the proliferation of cells and promoted shrinkage in the cell body, the release of LDH, increases in intra-cellular ROS, extra cellular NO, loss of MMP, which further induce apoptosis and damage dendritic networking, thereby inducing cell death (Xu et al., 2016; Zhou et al., 2018; Figure 4).
The role of chronic hypoxia/deprivation of vascular oxygen in the induction of AD pathogenesis is quite well explored, whereas low or intermittent hypoxia has a neuroprotective role. Previous studies have documented that hypoxia resulting from vascular insufficiency can increase the expression of APP. A study by Sun et al. showed the effect of hypoxia (2% O2 and 5% CO2) on SH-SY5Y cells, N2A, and HEK293 cells. The authors observed that a hypoxia-like condition activates HIF-1α, which further modulates the amyloidogenic processing of APP via increasing BACE1/α-secretase and simultaneously decreasing the β-secretase activity, ultimately leading to the formation of Aβ oligomers/aggregates. Aβ oligomers/aggregates have been found to be associated with microglia activation and thus cause neuronal cell death and inflammation (Sun et al., 2006; Lall et al., 2019).
HIF-1α also increases the ROS levels, which further leads to activation of NFκBp65, resulting in an increase in pro-inflammatory mediators. From previous studies, it has been reported that a chronic hypoxic condition also alters GLUTs and decreases ATP, which alter the insulin signaling pathway as well as PI3K/Akt/GSK3β AXIS. Activated GSK3β increases the hyperphosphorylation of tau, ultimately resulting in subsequent neuron damage/apoptosis (Lall et al., 2019). Hypoxia-induced activated HIF-1α also decreases neprilysin (a metalloendopeptidase protein) expression, leading to Aβ accumulation. Additionally, it also causes calcium dysfunction via increasing intracellular calcium concentration, subsequently leading to neuronal excitability and loss of synaptic plasticity (Zhang et al., 2007; Figure 7).
Amyloid beta, streptozotocin, L-glutamate, scopolamine, D-galactose, sevoflurane, okadaic acid, forskolin (FSK), lipopolysaccharides (LPS), PGJ2 (prostaglandin J2), and formaldehyde.
Amyloid beta deposition occurs in the extra cellular region of neurons, further leading to AD (Stancu et al., 2014; Ramakrishna et al., 2023). Many previous studies have validated that direct injection of amyloid beta may induce alterations in primary hippocampal cortical neurons. Amyloid beta peptides of different sizes (Aβ1-40, 1.43, and 25–35) at varying concentrations are used to develop in vitro models of AD. Various experimental studies have highlighted the use of either synthetic amyloid beta or naturally extracted amyloid-beta peptide fragments directly from the brain of patients with AD (Jin et al., 2011). In the in-vitro model, amyloid beta alters tau conformation and increases tau phosphorylation as well as GSK3 beta activation (Cacciottolo et al., 2017). SHSY5Y cells exposed to an amyloid beta oligomer at concentrations of 0.01 nM to 1 nM showed decreased neurite density, morphological alteration, increased hyperphosphorylation of tau, activation of apoptotic pathway, and loss of cell viability (de Medeiros et al., 2019). Another study unveiled that amniotic membrane (AM) neuronal cells treated with amyloid beta 42 fragments (2.5 μM) for 24 h induce an AD model. The authors found a detrimental impact on the Wnt signaling pathway, impeding the neurogenesis process and triggering the activation of Gsk3 beta. This activation is involved in the hyperphosphorylation of the tau protein (Batista et al., 2019; Prajapat et al., 2020; Patil et al., 2023). Amyloid beta 1–40 and amyloid beta 1–42 at varying concentrations of 0.5 to 20 uM were able to induce apoptotic activity in PC12 cells for 24 h (Kumaran et al., 2018; Wang H. et al., 2018). Islet amyloid polypeptide (IAPP), another agent produced by pancreatic beta cells, has the ability to cross the BBB and interact with tau fibrils, thereby promoting amyloid beta production (Zhang G. et al., 2022; Figure 7).
Streptozotocin (STZ) is a glucosamine-nitrosourea compound used as an antibiotic. An in-vitro model of Alzheimer’s disease has been developed using streptozotocin in various studies. Streptozotocin can modulate the activity of GSK3 alpha/beta (Bagaméry et al., 2021) and secretase, as observed in both in vitro and in vivo studies. Wang et al. found that insulin is able to inhibit betaA4 production by modulating amyloid precursor protein processing via an increase in the activity of alpha secretase and a simultaneous decrease in beta secretase activity in the APP overexpressing mutant SHSY5Y cell line. STZ was shown to exert a toxic effect in a dose dependent manner. SHSY5Y neuroblastoma cells exposed to STZ (concentration: 0.3, 1, 3, 5, and 10 mM) caused neuronal cell damage, increased oxidative stress, depolarized the mitochondrial membrane and apoptosis, and caused tau hyperphosphorylation, thereby influencing the insulin receptor expression (Bagaméry et al., 2020). A concentration of 1 mM of STZ was able to reduce the ATP concentration without cell death at 24 and 48 h (Plaschke and Kopitz, 2015). Moreover, STZ has shown toxicity in immature neuronal cells (Isaev et al., 2018). Streptozotocin inhibits the action of insulin, thereby increasing beta secretase and decreasing alpha secretase, which ultimately leads to increased amyloid beta fragments (Plaschke and Kopitz, 2015). STZ is also used to induce beta cell toxicity in the pancreas. The STZ drug is validated to generate early pathophysiological changes in cell cultures and animal AD models. STZ injected into an animal through the intraperitoneal and intracerebroventricular routes was able to reduce the cognitive behavior and increase the aggregation and deposition of amyloid beta oligomers, as well as increase the total tau protein and change the phosphorylation/total tau level ratio. Other alterations, such as a neuroinflammation, oxidative stress, and biochemical changes, have also been observed in the brain (Kamat, 2015; Sun et al., 2018; Bagaméry et al., 2020; Bansal et al., 2021; Figure 4).
Okadaic acid (OKA) is a polyether toxin produced by algae (Kamat et al., 2013). The main function of OKA is to inhibit PP2A (protein phosphatase 2A) and PP1A (protein phosphatase 1A) activity, and further activation of GSK3 beta, CDK5, and calpain leads to induction of abnormal hyperphosphorylation of the tau protein at positions ser202 and ser396 (Boban et al., 2019), causing tau tangle formation. Inhibition of this phosphatase activity and activation of calpain have been implicated in neurodegenerative diseases such as Alzheimer’s disease. OKA is able to activate MAPK signaling, which further activates apoptotic signaling of ERK/BAD/CASPASE9 for neuronal damage. OKA regulates various other neurotoxicity mechanisms, as observed in both in vitro and in vivo studies (Xu et al., 2004; Kamat et al., 2013). Previous in vitro and in vivo studies have shown that OKA is an oxidative stress inducer (Sachdeva and Chopra, 2015). A previous study found that exposure of primary cortical culture neurons to 50 μM of OKA and 50 μM of glutamate induces cell death (Yi et al., 2009). Further, okadaic acid was able to induce tau hyperphosphorylation, causing tangle formation, which ultimately resulted in amyloid beta deposition; therefore, it can be used to induce an in vitro Alzheimer’s disease model. Prior studies have shown that PC12 cells treated with OA for 24 h showed decreased cell viability, increased tau hyperphosphorylation, increased amyloid beta 1–42, and beta secretase, as well as affecting the expression of P-GSK3 beta, P-AKT, and MEF2D (Huang et al., 2020; Amonruttanapun et al., 2022; Figure 4).
Prostaglandins (PG) such as PGJ2 play a key role in the inactivation of GSK3 beta by phosphorylation at the ser9 position. Numerous studies have shown that PGJ2 was able to induce hyperphosphorylation of tau in mouse cortical primary neurons. PGJ2 inactivates NFKB activity via stabilization of IKBα. PGJ2 phosphorylates IKBα for stabilization rather than degradation. Inactivated NFkB was not able to activate antiapoptotic genes or neuronal survival genes, therefore leading to neuronal cell death (Li et al., 2004). As reported in a previous study, PGJ2-treated SHSY5Y cells were responsible for the activation of the P53-dependent Fas/FasL pathway (Kondo et al., 2002).
When cells were treated with PGJ2 at concentrations of 10 μM and 20 μM, tau proteins with hyperphosphorylation at ser396, ser404, and thr205 were found to be upregulated (Mpousis et al., 2016; Thysiadis et al., 2018). PGJ2 acts as an inducing model for AD. On the other hand, AcPHF6 (acetyl PHF6) derivatives are able to induce fibrillation of cross beta sheet structures and tau aggregation in cell cultures. Therefore, it can be a suitable agent for the induction of an in vitro model for Alzheimer’s disease (Lunven et al., 2016; Figure 4).
Scopolamine is a muscarinic acetylcholine receptor antagonist that induces cognitive impairment by increasing oxidative stress, inflammation, and the inhibition of cholinergic neurotransmission. Exposure of human neuroblastoma SHSY5Y cells to 1 to 3 mM concentration of scopolamine for 24 h was able to decrease the cell viability. In another study, it upregulated the AChE activity in rat PC12 cells (Puangmalai et al., 2017). Exposure to scopolamine decreases PI3K/Akt, which activates GSK3β, ultimately resulting in subsequent neuron damage/apoptosis via increasing the hyperphosphorylation of tau. Scopolamine exposure to IMR32 neuronal and C6 glioma cells at a dose of 3 mM for 2 h induces oxidative stress that leads to an alteration in downstream signaling pathways such as NFκB-mediated CREB/BDNF and GFAP activity, leading to neuroinflammation, apoptosis, synaptic dysfunction, and loss of memory genes (Konar et al., 2011; Figure 7).
The excitatory neurotransmitter in CNS is glutamate, which can cause neurodegeneration in animal models, primary cultures of brain cells, and cell lines such as PC12. In a study involving the PC12 cell line, a high concentration of glutamate was able to inhibit the uptake of cystine and reduced glutathione intracellularly, which lead to the induction of ROS, ultimately causing cell death (Yu et al., 2005). Studies have found that L-glutamate leads to excitotoxicity in human neuroblastoma SH-SY5Y cells when treated with different concentrations (0.1–10 mM) for 12 h. The overactivation of NMDA receptors induces glutamate excitotoxicity due to the large influx of Ca+2 and NO. This causes activation of the JNK signaling pathway, which further activates caspase-3 and pro apoptotic genes, ultimately leading to apoptosis and neuronal cell death (Bagaméry et al., 2021). Previous research has also linked glutamate excitotoxicity to a variety of neurodegenerative diseases, retinal diseases, hyperglycemia, diabetic retinopathy, and type 3 and type 4 diabetes (Wang et al., 2019). A high concentration of L-glutamate not only causes excitotoxicity but also affects the tau protein in rat primary culture neurons (Chen et al., 1995). It has been suggested that there is involvement of MAPK, PI3K/Akt, and mTOR signaling pathways in L-glutamate-induced excitotoxicity (Plaschke and Kopitz, 2015). Microglial cells were also found to release a high concentration of glutamate, which decreases the level of glutamate transporters (GLAST1) and results in glutamate-induced excitotoxicity (Takaki et al., 2012; Figure 5).
Mild Alzheimer’s and dementia can also be induced by D-galactose, which is a reducing sugar naturally occurring in our body and present in various food products. However, an excess amount of D-galactose can induce oxidative stress, apoptosis, and inflammation in neuronal cells. Previously, it was reported that D-galactose can induce senescence and further neuronal damage in both in vivo and in vitro AD models. One such study highlighted that human SHSY5Y cells exposed to D-galactose at a concentration of 300 mM for 48 h exhibited enhanced inflammation, apoptosis, oxidative stress, and mitochondrial dysfunction since D-galactose is used to mimic the natural aging process.
Other studies by Wang et al. and Rahimi et al. suggested that exposure of 200 mM to 400 mM D-galactose to retinoic acid induced differentiated SHSY5Y cells that can cause senescence and oxidative stress by decreasing NADPH glutathione reductase (Lee et al., 2020). A high amount of D-galactose reacts with peptides and forms advanced glycation end products (AGEs) in the body, which interact with receptor of advanced glycation end products (RAGE). This RAGE is present in the membranes of various cells, such as dendritic cells, neuronal cells, macrophages lymphocytes, glial cells, smooth muscles cells, and endothelial cells. RAGE was found to cause oxidative stress and inflammation by acting on Nox4 (Rahimi et al., 2018). Astrocytic CRT cells and rat primary astrocytes treated with D-gal were found to have increased NFκBp65 and therefore increased pro-inflammatory mediators. Furthermore, cells showed decreased cell viability and pronounced occurrence of cellular senescence due to an increase in caspase3 (Hou et al., 2019; Figure 7).
According to previous in vivo studies, sevoflurane can induce cognitive impairment and tau hyperphosphorylation. In vitro studies have shown that when a neuronal culture was exposed to sevoflurane, it increased the amount of tau and also induced phosphorylation of tau at 202 and 205 residues (Dong et al., 2021). Other studies reported that sevoflurane was able to induce caspase-3 activity, causing cytochrome C release and apoptosis. It can alter the processing of the amyloid precursor protein (APP) and can also increase the levels of amyloid beta peptide fragments, as well as elevate the intracellular BACE (beta-secretase 1) levels in the brain tissue of mice, leading to cellular damage (Wei et al., 2014). SK-N-SH cells treated with an approximate concentration of 10 mM of sevoflurane showed upregulation of GSK3 beta protein, accelerating the phosphorylation of Drp1 at ser616 residues, which further leads to damaged mitochondria and ultimately leads to apoptosis via caspase 3 activation (Boost et al., 2009; Liu et al., 2022). Sevoflurane-induced neuronal toxicity has been one of the pieces of evidence for AD pathogenesis in cell culture and animal studies (Huang et al., 2018; Figure 6).
Formaldehyde is a common environmental agent found in various products such as paint, cloth, exhaust gas, medicinal items, and industrial goods. It is a natural by-product of metabolism in living organisms. DNA/protein crosslinks are produced when exposed to formaldehyde, suggesting a key mechanism of DNA damage. Surprisingly, these crosslinks have been utilized to calculate medicine dose. Formaldehyde also works as a crosslinking agent, interacting with thiol and amino groups to cause protein polymerization.
Furthermore, formaldehyde may interact with Aβ, resulting in the development of neurotoxic amyloid-like complexes that are permanently cross-linked. The effects of formaldehyde-induced tau aggregation on human neuroblastoma cells (SHSY-5Y cell line) and rat hippocampus cells were studied. The researchers discovered that very modest quantities of formaldehyde (0.01–0.1%) are enough to cause the production of amyloid-like tau clumps. Consequently, both SH-SY5Y and hippocampus cells undergo apoptosis as a result of these aggregates (Nie et al., 2007). According to a study by Lu et al., a higher concentration of formaldehyde can damage the central nervous system. It can cross the BBB. In this study, the authors exposed N2A cells to 0.5 mM formaldehyde for 24 h. Formaldehyde can trigger hyper phosphorylation of the tau (p396) protein through the activation of GSK3 beta, which further leads to neuronal cell death (Lu et al., 2013; Figure 6).
However, apart from environmental and chemical inducing agents, xenobiotics and xenometals can also be considered as important inducing agents for the pathogenesis of Alzheimer’s disease. Previous in vivo studies have demonstrated the significance of such agents in a rodent model of Alzheimer’s disease (Mahdi et al., 2019). In the context of xenobiotics and xenometals, they were shown to repress anti-aging genes such as sirtuin 1 (Martins, 2016, 2017). Sirtuin 1 plays a very important role in mitophagy, amyloid beta aggregation, and programmed cell death, and is a critically important gene for AD (Martins, 2018).
Paraquat is a herbicide that was found to be involved in neurodegeneration. According to a previous study, when differentiated SHSY5Y cells were treated with paraquat, it induces downregulation of GSH levels, which in turn leads to high levels of ROS in the mitochondria. Further, paraquat also has the ability to elevate caspase 3 levels and proteasome, which leads to apoptosis and further leads to neuronal cell death (McCarthy et al., 2004; Figure 6).
PFOS is an organic pollutant able to cause neuronal toxicity and is a potential risk factor for AD in late life. Exposure of SHSY5Y cells to PFOS induced tau pathology via the induction of specific phosphorylation of the protein, resulting in a high expression of GSK3 beta, thereby increasing the expression of APP and inducing high APoE levels (Basaly et al., 2021; Figure 7).
Lipopolysaccharide (LPS) is located in the outer membrane of Gram-negative bacteria, with the toll like receptor (TLR) as its main receptor, although it exerts its effects via other receptors. LPS interacts with TLR to further recruit downstream adaptors, such as MyD88, TIR-domain-containing adaptor-inducing interferon-β, and/or triggering receptors expressed on myeloid cells (TRAM). All these adaptors can activate transcription factors responsible for the activation of pro-inflammatory genes. LPS can also induce microglial astrocytic cells as both of them express the TLR4 receptor. Indeed, activation of this receptor leads to the production of different inflammatory mediators. LPS-induced inflammation exacerbates tau pathology through cdk5 kinase activity (Sun et al., 2006), resulting in amyloid beta deposition, as observed in both in vitro and in vivo studies (Bansal et al., 2021). Astrocytes are major players in the development of diverse neurodegenerative diseases. Many of the neuroinflammatory models developed in vitro (cultured astrocytes) and in vivo (mouse) were accomplished using LPS in numerous studies (Liu et al., 2019; Creative Biolabs, n.d.; Figure 5).
The lack of ability to perform a systematic analysis on in vitro studies makes the extrapolation of data to a patient population much more challenging. The present study aims to identify the different compounds that were used to generate in-vitro models of Alzheimer’s disease due to their toxic nature toward humans—generating a model that can more closely imitate the pathological features of AD subtypes. However, various in vivo Alzheimer’s models have been employed to explore the molecular pathways, and testing the efficacy of drugs has also been successfully carried out. As neuronal toxicity models exhibit physiological relevance in terms of amyloid beta plaque formation and tau hyperphosphorylation, their clinical prognostic utility will be helpful in evaluating single drug effects as well as drug–drug combinations. A method to develop an in vitro Alzheimer’s disease model using multiple chemicals has been established to untangle AD pathogenesis, identify molecular processes, and test numerous prospective therapeutic drugs against AD. This applies especially to metal ions, particulate matter, and chemical inducers that have potential to cause Aβ plaques and neurofibril tangles. Nonetheless, none of the existing models accurately reproduce the pathology of human Alzheimer’s disease, even though each in-vitro model is recognized to have its own advantages.
Heavy metals, PGJ2, okadaic acid, scopolamine, amyloid beta, and LPS are among the chemicals with proven neurotoxicity and have been used to upregulate Alzheimer-specific genes in differentiated neuronal cells. However, to trigger cellular AD pathology, each chemical may work through different approaches and different signaling pathways. The aim of the present study is to identify a common molecular mechanism and the advantages of each model need to be considered while screening drugs for AD research. Each type of chemical that is being used for creating in vitro AD models replicates certain stages of AD pathophysiological alterations and their associated molecular mechanisms, which should be carefully considered when choosing an in vitro AD model.
Cerebral organoids that allow the production of ordered structures akin to the human brain have recently become the newest route for in vitro AD research (Kumaran et al., 2018). These latest 3D in vitro models might have the potential to offer new prospects toward the exploration of personalized therapeutic regimes and new strategies.
The development of methods for obtaining a functionally meaningful AD model will aid in increasing the predictive capacity of new therapeutic drug screening agents. However, for these models to be accepted by industry, higher-throughput screening procedures that can be assessed by an automated system would be required.
The inducible in vitro AD models reviewed in this comprehensive review have helped us to identify different compounds that are suitable to be used for the development of in-vitro models of Alzheimer’s disease capable of more closely imitating the pathological features of AD subtypes. Regardless, none of these models can effectively imitate all aspects of disease development in most AD subtypes. As a result, current models will need to be modified to adequately replicate the complex scenarios occurring during the actual manifestation of Alzheimer’s disease. Nevertheless, chemically induced AD models will continue to play an essential part in future Alzheimer’s disease research.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
MP: Conceptualization, Methodology, Writing – original draft. GK: Conceptualization, Data curation, Methodology, Writing – original draft. GC: Data curation, Investigation, Software, Writing – original draft. PP: Methodology, Writing – original draft. SB: Investigation, Software, Writing – review & editing. RJ: Software, Visualization, Writing – review & editing. GB: Visualization, Writing – original draft. AM: Investigation, Visualization, Writing – review & editing. RS: Writing – original draft. HK: Writing – review & editing. PKP: Writing – review & editing. AP: Supervision, Validation, Writing – review & editing. BM: Supervision, Validation, Writing – original draft.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Amonruttanapun, P., Chongthammakun, S., and Chamniansawat, S. (2022). The effects of okadaic acid-treated SH-SY5Y cells on microglia activation and phagocytosis. Cell Biol. Int. 46, 234–242. doi: 10.1002/cbin.11722
An, W. L., Bjorkdahl, C., Liu, R., Cowburn, R. F., Winblad, B., and Pei, J. J. (2005). Mechanism of zinc-induced phosphorylation of p70 S6 kinase and glycogen synthase kinase 3β in SH-SY5Y neuroblastoma cells. J. Neurochem. 92, 1104–1115. doi: 10.1111/j.1471-4159.2004.02948.x
Andy, S. N., Pandy, V., Alias, Z., and Kadir, H. A. (2018). Deoxyelephantopin ameliorates lipopolysaccharides (LPS)-induced memory impairments in rats: evidence for its anti-neuroinflammatory properties. Life Sci. 206, 45–60. doi: 10.1016/j.lfs.2018.05.035
Atzori, C., Ghetti, B., Piva, R., Srinivasan, A. N., Zolo, P., Delisle, M. B., et al. (2001). Activation of the JNK/p 38 pathway occurs in diseases characterized by tau protein pathology and is related to tau phosphorylation but not to apoptosis. J. Neuropathol. Exp. Neurol. 60, 1190–1197. doi: 10.1093/jnen/60.12.1190
Bagaméry, F., Varga, K., Kecsmár, K., Vincze, I., Szökő, É., and Tábi, T. (2020). Lack of insulin resistance in response to streptozotocin treatment in neuronal SH-SY5Y cell line. J. Neural Transm. 127, 71–80. doi: 10.1007/s00702-019-02118-5
Bagaméry, F., Varga, K., Kecsmár, K., Vincze, I., Szökő, É., and Tábi, T. (2021). The impact of differentiation on cytotoxicity and insulin sensitivity in Streptozotocin treated SH-SY5Y cells. Neurochem. Res. 46, 1350–1358. doi: 10.1007/s11064-021-03269-2
Bansal, S., Agrawal, M., Mahendiratta, S., Kumar, S., Arora, S., Joshi, R., et al. (2021). Everolimus: a potential therapeutic agent targeting PI3K/Akt pathway in brain insulin system dysfunction and associated neurobehavioral deficits. Fundam. Clin. Pharmacol. 35, 1018–1031. doi: 10.1111/fcp.12677
Basaly, V., Hill, J., Bihaqi, S. W., Marques, E., Slitt, A. L., and Zawia, N. H. (2021). Developmental Perfluorooctanesulfonic acid (PFOS) exposure as a potential risk factor for late-onset Alzheimer’s disease in CD-1 mice and SH-SY5Y cells. Neurotoxicology 86, 26–36. doi: 10.1016/j.neuro.2021.06.008
Batista, C. R. A., Gomes, G. F., Candelario-Jalil, E., Fiebich, B. L., and de Oliveira, A. C. P. (2019). Lipopolysaccharide-induced Neuroinflammation as a bridge to understand neurodegeneration. Int. J. Mol. Sci. 20:2293. doi: 10.3390/ijms20092293
Bhatti, G. K., Gupta, A., Pahwa, P., Khullar, N., Singh, S., Navik, U., et al. (2022). Targeting mitochondrial bioenergetics as a promising therapeutic strategy in metabolic and neurodegenerative diseases. Biom. J. 45, 733–748. doi: 10.1016/j.bj.2022.05.002
Boban, M., Babić Leko, M., Miškić, T., Hof, P. R., and Šimić, G. (2019). Human neuroblastoma SH-SY5Y cells treated with okadaic acid express phosphorylated high molecular weight tau-immunoreactive protein species. J. Neurosci. Methods 319, 60–68. doi: 10.1016/j.jneumeth.2018.09.030
Boost, K. A., Leipold, T., Scheiermann, P., Hoegl, S., Sadik, C. D., Hofstetter, C., et al. (2009). Sevoflurane and isoflurane decrease TNF-alpha-induced gene expression in human monocytic THP-1 cells: potential role of intracellular IkappaBalpha regulation. Int. J. Mol. Med. 23, 665–671. doi: 10.3892/ijmm_00000178
Cacciottolo, M., Wang, X., Driscoll, I., Woodward, N., Saffari, A., Reyes, J., et al. (2017). Particulate air pollutants, APOE alleles and their contributions to cognitive impairment in older women and to amyloidogenesis in experimental models. Transl. Psychiatry 7:e1022. doi: 10.1038/tp.2016.280
Cai, T., Che, H., Yao, T., Chen, Y., Huang, C., Zhang, W., et al. (2011). Manganese induces tau hyperphosphorylation through the activation of ERK MAPK pathway in PC12 cells. Toxicol. Sci. 119, 169–177. doi: 10.1093/toxsci/kfq308
Chen, J., Wang, H., Ying, Y., Juarez, L., Binder, L., Ghanbari, H., et al. (1995). “The effects of sodium nitroprusside and L-glutamate on the regulation of the cytoskeletal associated protein tau in primary cultured neurons from rat” in Biochemical, pharmacological, and clinical aspects of nitric oxide. eds. B. A. Weissman, N. Allon, and S. Shapira (Boston, MA: Springer US), 205–212.
Chin-Chan, M., Navarro-Yepes, J., and Quintanilla-Vega, B. (2015). Environmental pollutants as risk factors for neurodegenerative disorders: Alzheimer and Parkinson diseases. Front. Cell Neurosci. 9:124. doi: 10.3389/fncel.2015.00124
Creative Biolabs. (n.d.) LPS-Induced Neuroinflammation Model – Creative biolabs. Available at: https://www.creative-biolabs.com/drug-discovery/therapeutics/lps-induced-neuroinflammation-model.htm
de Medeiros, L. M., De Bastiani, M. A., Rico, E. P., Schonhofen, P., Pfaffenseller, B., Wollenhaupt-Aguiar, B., et al. (2019). Cholinergic differentiation of human neuroblastoma SH-SY5Y cell line and its potential use as an in vitro model for Alzheimer’s disease studies. Mol. Neurobiol. 56, 7355–7367. doi: 10.1007/s12035-019-1605-3
Dong, Y., Liang, F., Huang, L., Fang, F., Yang, G., Tanzi, R. E., et al. (2021). The anesthetic sevoflurane induces tau trafficking from neurons to microglia. Commun. Biol. 4, 1–13. doi: 10.1038/s42003-021-02047-8
Drago, D., Cavaliere, A., Mascetra, N., Ciavardelli, D., di Ilio, C., Zatta, P., et al. (2008). Aluminum modulates effects of beta amyloid (1-42) on neuronal calcium homeostasis and mitochondria functioning and is altered in a triple transgenic mouse model of Alzheimer’s disease. Rejuvenation Res. 11, 861–871. doi: 10.1089/rej.2008.0761
Esmieu, C., Guettas, D., Conte-Daban, A., Sabater, L., Faller, P., and Hureau, C. (2019). Copper-targeting approaches in Alzheimer’s disease: how to improve the fallouts obtained from in vitro studies. Inorg. Chem. 58, 13509–13527. doi: 10.1021/acs.inorgchem.9b00995
Exil, V., Ping, L., Yu, Y., Chakraborty, S., Caito, S. W., Wells, K. S., et al. (2014). Activation of MAPK and fox O by manganese (Mn) in rat neonatal primary astrocyte cultures. PLoS One 9:e94753. doi: 10.1371/journal.pone.0094753
Fan, L., Mao, C., Hu, X., Zhang, S., Yang, Z., Hu, Z., et al. (2020). New insights into the pathogenesis of Alzheimer’s disease. Front. Neurol. 10:1312. doi: 10.3389/fneur.2019.01312
Gan, X., Huang, S., Wu, L., Wang, Y., Hu, G., Li, G., et al. (2014). Inhibition of ERK-DLP1 signaling and mitochondrial division alleviates mitochondrial dysfunction in Alzheimer’s disease cybrid cell. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 1842, 220–231. doi: 10.1016/j.bbadis.2013.11.009
Gong, Q. H., Wang, Q., Pan, L. L., Liu, X. H., Xin, H., and Zhu, Y. Z. (2011). S-propargyl-cysteine, a novel hydrogen sulfide-modulated agent, attenuates lipopolysaccharide-induced spatial learning and memory impairment: involvement of TNF signaling and NF-κB pathway in rats. Brain Behav. Immun. 25, 110–119. doi: 10.1016/j.bbi.2010.09.001
Gothwal, A., Kumar, H., Nakhate, K. T., Ajazuddin, D. A., Dutta, A., Borah, A., et al. (2019). Lactoferrin coupled lower generation PAMAM dendrimers for brain targeted delivery of Memantine in aluminum-chloride-induced Alzheimer’s disease in mice. Bioconjug. Chem. 30, 2573–2583. doi: 10.1021/acs.bioconjchem.9b00505
Grant, P., Kumar, J., Kar, S., and Overduin, M. (2021). Effects of specific inhibitors for CaMK1D on a primary neuron model for Alzheimer’s disease. Molecules 26:7669. doi: 10.3390/molecules26247669
Greco, S. J., Sarkar, S., Johnston, J. M., and Tezapsidis, N. (2009). Leptin regulates tau phosphorylation and amyloid through AMPK in neuronal cells. Biochem. Biophys. Res. Commun. 380, 98–104. doi: 10.1016/j.bbrc.2009.01.041
Gu, S. M., Lee, H. P., Ham, Y. W., Son, D. J., Kim, H. Y., Oh, K. W., et al. (2018). Piperlongumine improves lipopolysaccharide-induced Amyloidogenesis by suppressing NF-kappa B pathway. NeuroMolecular Med. 20, 312–327. doi: 10.1007/s12017-018-8495-9
Hampel, H., Mesulam, M. M., Cuello, A. C., Farlow, M. R., Giacobini, E., Grossberg, G. T., et al. (2018). The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 141, 1917–1933. doi: 10.1093/brain/awy132
Han, J. H., Lee, Y. S., Im, J. H., Ham, Y. W., Lee, H. P., Han, S. B., et al. (2019). Astaxanthin ameliorates lipopolysaccharide-induced Neuroinflammation, oxidative stress and memory dysfunction through inactivation of the signal transducer and activator of transcription 3 pathway. Mar. Drugs 17:E123. doi: 10.3390/md17020123
Hong, Y., and An, Z. (2018). Hesperidin attenuates learning and memory deficits in APP/PS1 mice through activation of Akt/Nrf2 signaling and inhibition of RAGE/NF-κB signaling. Arch. Pharm. Res. 41, 655–663. doi: 10.1007/s12272-015-0662-z
Hou, J., Yun, Y., Xue, J., Sun, M., and Kim, S. (2019). D-galactose induces astrocytic aging and contributes to astrocytoma progression and chemoresistance via cellular senescence. Mol. Med. Rep. 20, 4111–4118. doi: 10.3892/mmr.2019.10677
Huang, H., Bihaqi, S. W., Cui, L., and Zawia, N. H. (2011). In vitro Pb exposure disturbs the balance between Aβ production and elimination: the role of AβPP and Neprilysin. Neurotoxicology 32, 300–306. doi: 10.1016/j.neuro.2011.02.001
Huang, L., Huang, K., and Ning, H. (2018). Hispidulin prevents sevoflurane- induced memory dysfunction in aged rats. Biomed. Pharmacother. Biomed. Pharmacother. 97, 412–422. doi: 10.1016/j.biopha.2017.10.142
Huang, L., Zhong, X., Qin, S., and Deng, M. (2020). Protocatechuic acid attenuates β-secretase activity and okadaic acid-induced autophagy via the Akt/GSK-3β/MEF2D pathway in PC12 cells. Mol. Med. Rep. 21, 1328–1335. doi: 10.3892/mmr.2019.10905
Isaev, N. K., Genrikhs, E. E., Voronkov, D. N., Kapkaeva, M. R., and Stelmashook, E. V. (2018). Streptozotocin toxicity in vitro depends on maturity of neurons. Toxicol. Appl. Pharmacol. 348, 99–104. doi: 10.1016/j.taap.2018.04.024
Ismail, M. A. M., Mateos, L., Maioli, S., Merino-Serrais, P., Ali, Z., Lodeiro, M., et al. (2017). 27-hydroxycholesterol impairs neuronal glucose uptake through an IRAP/GLUT4 system dysregulation. J. Exp. Med. 214, 699–717. doi: 10.1084/jem.20160534
Jang, S., Kim, E. W., Zhang, Y., Lee, J., Cho, S. Y., Ha, J., et al. (2018). Particulate matter increases beta-amyloid and activated glial cells in hippocampal tissues of transgenic Alzheimer’s mouse: involvement of PARP-1. Biochem. Biophys. Res. Commun. 500, 333–338. doi: 10.1016/j.bbrc.2018.04.068
Jin, M., Shepardson, N., Yang, T., Chen, G., Walsh, D., and Selkoe, D. J. (2011). Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce tau hyperphosphorylation and neuritic degeneration. Proc. Natl. Acad. Sci. U. S. A. 108, 5819–5824. doi: 10.1073/pnas.1017033108
Kamat, P. K. (2015). Streptozotocin induced Alzheimer’s disease like changes and the underlying neural degeneration and regeneration mechanism. Neural Regen. Res. 10, 1050–1052. doi: 10.4103/1673-5374.160076
Kamat, P. K., Rai, S., and Nath, C. (2013). Okadaic acid induced neurotoxicity: an emerging tool to study Alzheimer’s disease pathology. Neurotoxicology 37, 163–172. doi: 10.1016/j.neuro.2013.05.002
Kang, Y. J., Tan, H. Y., Lee, C. Y., and Cho, H. (2021). An air particulate pollutant induces Neuroinflammation and neurodegeneration in human brain models. Adv. Sci. 8:2101251. doi: 10.1002/advs.202101251
Karri, V., Ramos, D., Martinez, J. B., Odena, A., Oliveira, E., Coort, S. L., et al. (2018). Differential protein expression of hippocampal cells associated with heavy metals (Pb, as, and MeHg) neurotoxicity: deepening into the molecular mechanism of neurodegenerative diseases. J. Proteome 187, 106–125. doi: 10.1016/j.jprot.2018.06.020
Karthivashan, G., Park, S. Y., Kweon, M. H., Kim, J., Haque, M. E., Cho, D. Y., et al. (2018). Ameliorative potential of desalted Salicornia europaea L. extract in multifaceted Alzheimer’s-like scopolamine-induced amnesic mice model. Sci. Rep. 8:7174. doi: 10.1038/s41598-018-25381-0
Kawahara, M., Kato, M., and Kuroda, Y. (2001). Effects of aluminum on the neurotoxicity of primary cultured neurons and on the aggregation of beta-amyloid protein. Brain Res. Bull. 55, 211–217. doi: 10.1016/S0361-9230(01)00475-0
Kawahara, M., Muramoto, K., Kobayashi, K., Mori, H., and Kuroda, Y. (1994). Aluminum promotes the aggregation of Alzheimer′s amyloid β-protein in vitro. Biochem. Biophys. Res. Commun. 198, 531–535. doi: 10.1006/bbrc.1994.1078
Kim, J. H., Jeong, M. S., Kim, D. Y., Her, S., and Wie, M. B. (2015). Zinc oxide nanoparticles induce lipoxygenase-mediated apoptosis and necrosis in human neuroblastoma SH-SY5Y cells. Neurochem. Int. 90, 204–214. doi: 10.1016/j.neuint.2015.09.002
Konar, A., Shah, N., Singh, R., Saxena, N., Kaul, S. C., Wadhwa, R., et al. (2011). Protective role of Ashwagandha leaf extract and its component withanone on scopolamine-induced changes in the brain and brain-derived cells. PLoS One 6:e27265. doi: 10.1371/journal.pone.0027265
Kondo, M., Shibata, T., Kumagai, T., Osawa, T., Shibata, N., Kobayashi, M., et al. (2002). 15-deoxy-Δ12,14-prostaglandin J2: the endogenous electrophile that induces neuronal apoptosis. Proc. Natl. Acad. Sci. U. S. A. 99, 7367–7372. doi: 10.1073/pnas.112212599
Kumaran, A., Ho, C. C., and Hwang, L. S. (2018). Protective effect of Nelumbo nucifera extracts on beta amyloid protein induced apoptosis in PC12 cells, in vitro model of Alzheimer’s disease. J. Food Drug Anal. 26, 172–181. doi: 10.1016/j.jfda.2017.01.007
Lai, T., Wen, X., Wu, D., Su, G., Gao, Y., Chen, C., et al. (2019). SIRT1 protects against urban particulate matter-induced airway inflammation. Int. J. Chron. Obstruct. Pulmon. Dis. 14, 1741–1752. doi: 10.2147/COPD.S202904
Lall, R., Mohammed, R., and Ojha, U. (2019). What are the links between hypoxia and Alzheimer’s disease? Neuropsychiatr. Dis. Treat. 15, 1343–1354. doi: 10.2147/NDT.S203103
Lee, S. H., Chen, Y. H., Chien, C. C., Yan, Y. H., Chen, H. C., Chuang, H. C., et al. (2021). Three month inhalation exposure to low-level PM2.5 induced brain toxicity in an Alzheimer’s disease mouse model. PLoS One 16:e0254587. doi: 10.1371/journal.pone.0254587
Lee, Y. J., Choi, D. Y., Choi, I. S., Kim, K. H., Kim, Y. H., Kim, H. M., et al. (2012). Inhibitory effect of 4-O-methylhonokiol on lipopolysaccharide-induced neuroinflammation, amyloidogenesis and memory impairment via inhibition of nuclear factor-kappa B in vitro and in vivo models. J. Neuroinflammation 9:35. doi: 10.1186/1742-2094-9-35
Lee, J., Kim, Y. S., Kim, E., Kim, Y., and Kim, Y. (2020). Curcumin and hesperetin attenuate D-galactose-induced brain senescence in vitro and in vivo. Nutr. Res. Pract. 14, 438–452. doi: 10.4162/nrp.2020.14.5.438
Li, Z., Jansen, M., Ogburn, K., Salvatierra, L., Hunter, L., Mathew, S., et al. (2004). Neurotoxic prostaglandin J2 enhances cyclooxygenase-2 expression in neuronal cells through the p38MAPK pathway: a death wish? J. Neurosci. Res. 78, 824–836. doi: 10.1002/jnr.20346
Li, L., Liu, J., Yan, X., Qin, K., Shi, M., Lin, T., et al. (2011). Protective effects of ginsenoside Rd against okadaic acid-induced neurotoxicity in vivo and in vitro. J. Ethnopharmacol. 138, 135–141. doi: 10.1016/j.jep.2011.08.068
Liu, J., Jiao, K., Zhou, Q., Yang, J., Yang, K., Hu, C., et al. (2021). Resveratrol alleviates 27-hydroxycholesterol-induced senescence in nerve cells and affects zebrafish locomotor behavior via activation of SIRT1-mediated STAT3 signaling. Oxidative Med. Cell. Longev. 2021:e6673343, 1–17. doi: 10.1155/2021/6673343
Liu, J., Li, L., Xie, P., Zhao, X., Shi, D., Zhang, Y., et al. (2022). Sevoflurane induced neurotoxicity in neonatal mice links to a GSK3β/Drp1-dependent mitochondrial fission and apoptosis. Free Radic. Biol. Med. 181, 72–81. doi: 10.1016/j.freeradbiomed.2022.01.031
Liu, L., Qu, Y., Liu, Y., Zhao, H., Ma, H.-C., and Noor, A. F. (2019). Atsttrin reduces lipopolysaccharide-induced neuroinflammation by inhibiting the nuclear factor kappa B signaling pathway. Neural. Regen. Res. 14, 1994–2002. doi: 10.4103/1673-5374.259623
Loera-Valencia, R., Ismail, M. A. M., Goikolea, J., Lodeiro, M., Mateos, L., Björkhem, I., et al. (2021). Hypercholesterolemia and 27-hydroxycholesterol increase S100A8 and RAGE expression in the brain: a link between cholesterol, Alarmins, and neurodegeneration. Mol. Neurobiol. 58, 6063–6076. doi: 10.1007/s12035-021-02521-8
Lu, J., Miao, J., Su, T., Liu, Y., and He, R. (2013). Formaldehyde induces hyperphosphorylation and polymerization of Tau protein both in vitro and in vivo. Biochim. Biophys. Acta 1830, 4102–4116. doi: 10.1016/j.bbagen.2013.04.028
Lunven, L., Bonnet, H., Yahiaoui, S., Yi, W., Da Costa, L., Peuchmaur, M., et al. (2016). Disruption of fibers from the tau model AcPHF6 by naturally occurring Aurones and synthetic analogues. ACS Chem. Neurosci. 7, 995–1003. doi: 10.1021/acschemneuro.6b00102
Ma, W. W., Li, C. Q., Zhao, L., Wang, Y. S., and Xiao, R. (2019). NF-κB-mediated inflammatory damage is differentially affected in SH-SY5Y and C6 cells treated with 27-hydroxycholesterol. Food Sci. Nutr. 7, 1685–1694. doi: 10.1002/fsn3.1005
Mahdi, O., Baharuldin, M. T., Nor, N. H., Chiroma, S. M., Jagadeesan, S., and Moklas, M. A. (2019). Chemicals used for the induction of Alzheimer’s disease-like cognitive dysfunctions in rodents. Biomed. Res. Ther. 6, 3460–3484.
Martins, I. J. (2016). Anti-aging genes improve appetite regulation and reverse cell senescence and apoptosis in global populations. Adv Aging Res. 5, 9–26. doi: 10.4236/aar.2016.51002
Martins, I. J. (2017). Single gene inactivation with implications to diabetes and multiple organ dysfunction syndrome. J. Clin. Epigenetics 3
Martins, I. (2018). “Increased risk for obesity and diabetes with neurodegeneration in developing countries” in 10 contributions on Genetics, 2–35.
Mashoque, A. R., Justin Thenmozhi, A., Manivasagam, T., Nataraj, J., Essa, M. M., and Chidambaram, S. B. (2018). Asiatic acid nullified aluminium toxicity in in vitro model of Alzheimer’s disease. Front. Biosci. 10, 287–299. doi: 10.2741/e823
McCarthy, S., Somayajulu, M., Sikorska, M., Borowy-Borowski, H., and Pandey, S. (2004). Paraquat induces oxidative stress and neuronal cell death; neuroprotection by water-soluble coenzyme Q10. Toxicol. Appl. Pharmacol. 201, 21–31. doi: 10.1016/j.taap.2004.04.019
Mentis, A. F. A., Dardiotis, E., Efthymiou, V., and Chrousos, G. P. (2021). Non-genetic risk and protective factors and biomarkers for neurological disorders: a meta-umbrella systematic review of umbrella reviews. BMC Med. 19:6. doi: 10.1186/s12916-020-01873-7
Mizoroki, T., Meshitsuka, S., Maeda, S., Murayama, M., Sahara, N., and Takashima, A. (2007). Aluminum induces tau aggregation in vitro but not in vivo. J. Alzheimers Dis. 11, 419–427. doi: 10.3233/JAD-2007-11401
Mpousis, S., Thysiadis, S., Avramidis, N., Katsamakas, S., Efthimiopoulos, S., and Sarli, V. (2016). Synthesis and evaluation of gallocyanine dyes as potential agents for the treatment of Alzheimer’s disease and related neurodegenerative tauopathies. Eur. J. Med. Chem. 108, 28–38. doi: 10.1016/j.ejmech.2015.11.024
Mustafa Rizvi, S. H., Parveen, A., Verma, A. K., Ahmad, I., Arshad, M., and Mahdi, A. A. (2014). Aluminium induced endoplasmic reticulum stress mediated cell death in SH-SY5Y neuroblastoma cell line is independent of p 53. PLoS One 9:e98409. doi: 10.1371/journal.pone.0098409
Nie, C. L., Wei, Y., Chen, X., Liu, Y. Y., Dui, W., Liu, Y., et al. (2007). Formaldehyde at low concentration induces protein tau into globular amyloid-like aggregates in vitro and in vivo. PLoS One 2:e629. doi: 10.1371/journal.pone.0000629
Olivieri, G., Brack, C., Müller-Spahn, F., Stähelin, H. B., Herrmann, M., Renard, P., et al. (2000). Mercury induces cell cytotoxicity and oxidative stress and increases β-amyloid secretion and tau phosphorylation in SHSY5Y neuroblastoma cells. J. Neurochem. 74, 231–236. doi: 10.1046/j.1471-4159.2000.0740231.x
Osornio-Vargas, A. R., Bonner, J. C., Alfaro-Moreno, E., Martínez, L., García-Cuellar, C., Ponce-de-León Rosales, S., et al. (2003). Proinflammatory and cytotoxic effects of Mexico City air pollution particulate matter in vitro are dependent on particle size and composition. Environ. Health Perspect. 111, 1289–1293. doi: 10.1289/ehp.5913
Park, H., Han, K. M., Jeon, H., Lee, J. S., Lee, H., Jeon, S. G., et al. (2020). The MAO inhibitor tranylcypromine alters LPS- and Aβ-mediated Neuroinflammatory responses in wild-type mice and a mouse model of AD. Cells 9:E1982. doi: 10.3390/cells9091982
Patil, M., Gamit, N., Dharmarajan, A., Sethi, G., and Warrier, S. (2023). Identification of a novel Wnt antagonist based therapeutic and diagnostic target for Alzheimer’s disease using a stem cell-derived model. Bioengineering 10:192. doi: 10.3390/bioengineering10020192
Plaschke, K., and Kopitz, J. (2015). In vitro streptozotocin model for modeling Alzheimer-like changes: effect on amyloid precursor protein secretases and glycogen synthase kinase-3. J. Neural. Transm. Vienna Austria 1996 122, 551–557. doi: 10.1007/s00702-014-1319-7
Prajapat, M., Sarma, P., Shekhar, N., Kaur, H., Singh, S., Kumar, S., et al. (2020). In silico docking and comparative ADMET profile of different glycogen synthase kinase 3 beta inhibitors as the potential leads for the development of anti-Alzheimer drug therapy. J. Adv. Pharm. Technol. Res. 11, 194–201. doi: 10.4103/japtr.JAPTR_178_19
Puangmalai, N., Thangnipon, W., Soi-Ampornkul, R., Suwanna, N., Tuchinda, P., and Nobsathian, S. (2017). Neuroprotection of N-benzylcinnamide on scopolamine-induced cholinergic dysfunction in human SH-SY5Y neuroblastoma cells. Neural Regen. Res. 12, 1492–1498. doi: 10.4103/1673-5374.215262
Rahaman, M. S., Banik, S., Akter, M., Rahman, M. M., Sikder, M. T., Hosokawa, T., et al. (2020). Curcumin alleviates arsenic-induced toxicity in PC12 cells via modulating autophagy/apoptosis. Ecotoxicol. Environ. Saf. 200:110756. doi: 10.1016/j.ecoenv.2020.110756
Rahimi, V. B., Askari, V. R., and Mousavi, S. H. (2018). Ellagic acid reveals promising anti-aging effects against d-galactose-induced aging on human neuroblastoma cell line, SH-SY5Y: a mechanistic study. Biomed. Pharmacother. 108, 1712–1724. doi: 10.1016/j.biopha.2018.10.024
Rahman, M. A., Rahman, M. S., Uddin, M. J., Mamum-Or-Rashid, A. N. M., Pang, M. G., and Rhim, H. (2020). Emerging risk of environmental factors: insight mechanisms of Alzheimer’s diseases. Environ. Sci. Pollut. Res. 27, 44659–44672. doi: 10.1007/s11356-020-08243-z
Rai, S. N., Chaturvedi, V. K., Singh, B. K., and Singh, M. P. (2020a). Commentary: Trem 2 deletion reduces late-stage amyloid plaque accumulation, elevates the Aβ42: Aβ40 ratio, and exacerbates axonal dystrophy and dendritic spine loss in the PS2APP Alzheimer’s mouse model. Front. Aging Neurosci. 12:219. doi: 10.3389/fnagi.2020.00219
Rai, S. N., Singh, C., Singh, A., Singh, M. P., and Singh, B. K. (2020b). Mitochondrial dysfunction: a potential therapeutic target to treat Alzheimer’s disease. Mol. Neurobiol. 57, 3075–3088. doi: 10.1007/s12035-020-01945-y
Rai, S. N., Singh, P., Steinbusch, H. W. M., Vamanu, E., Ashraf, G., and Singh, M. P. (2021a). The role of vitamins in neurodegenerative disease: An update. Biomedicine 9:1284. doi: 10.3390/biomedicines9101284
Rai, S. N., Tiwari, N., Singh, P., Mishra, D., Singh, A. K., Hooshmandi, E., et al. (2021b). Therapeutic potential of vital transcription factors in Alzheimer’s and Parkinson’s disease with particular emphasis on transcription factor EB mediated autophagy. Front. Neurosci. 15:777347. doi: 10.3389/fnins.2021.777347
Ramakrishna, K., Nalla, L. V., Naresh, D., Venkateswarlu, K., Viswanadh, M. K., Nalluri, B. N., et al. (2023). WNT-β catenin signaling as a potential therapeutic target for neurodegenerative diseases: current status and future perspective. Diseases 11:89. doi: 10.3390/diseases11030089
Reitz, C., Rogaeva, E., and Beecham, G. W. (2020). Late-onset vs nonmendelian early-onset Alzheimer disease. Neurol. Genet. 6:e512. doi: 10.1212/NXG.0000000000000512
Rorbach-Dolata, A., Kicinska, A., and Piwowar, A. (2020). The Dephosphorylation of p70S6 (Thr389) kinase as a marker of L-glutamate-induced excitotoxicity related to diabetes disturbances-an unconventional in vitro model. Neurotox. Res. 37, 628–639. doi: 10.1007/s12640-019-00155-2
Sachdeva, A. K., and Chopra, K. (2015). Naringin mitigate okadaic acid-induced cognitive impairment in an experimental paradigm of Alzheimer’s disease. J. Funct. Foods 19, 110–125. doi: 10.1016/j.jff.2015.08.024
Sanajou, S., Yirün, A., Demirel, G., Çakir, D. A., Şahin, G., Erkekoğlu, Ü. P., et al. (2023). Antioxidant dihydrolipolic acid protects against in vitro aluminum-induced toxicity. J. Appl. Toxicol. 43, 1793–1805. doi: 10.1002/jat.4513
Sarathlal, K. C., Kakoty, V., Marathe, S., Chitkara, D., and Taliyan, R. (2021). Exploring the neuroprotective potential of rosiglitazone embedded Nanocarrier system on Streptozotocin induced mice model of Alzheimer’s disease. Neurotox. Res. 39, 240–255. doi: 10.1007/s12640-020-00258-1
Sattayasai, J., Chaonapan, P., Arkaravichie, T., Soi-ampornkul, R., Junnu, S., Charoensilp, P., et al. (2013). Protective effects of Mangosteen extract on H2O2-induced cytotoxicity in SK-N-SH cells and scopolamine-induced memory impairment in mice. PLoS One 8:e85053. doi: 10.1371/journal.pone.0085053
Shen, C., Chen, Y., Liu, H., Zhang, K., Zhang, T., Lin, A., et al. (2008). Hydrogen peroxide promotes Aβ production through JNK-dependent activation of γ-secretase*. J. Biol. Chem. 283, 17721–17730. doi: 10.1074/jbc.M800013200
Song, J. W., and Choi, B. S. (2013). Mercury induced the accumulation of amyloid Beta (Aβ) in PC12 cells: the role of production and degradation of Aβ. Toxicol. Res. 29, 235–240. doi: 10.5487/TR.2013.29.4.235
Srivastava, P., Tripathi, P. N., Sharma, P., Rai, S. N., Singh, S. P., Srivastava, R. K., et al. (2019). Design and development of some phenyl benzoxazole derivatives as a potent acetylcholinesterase inhibitor with antioxidant property to enhance learning and memory. Eur. J. Med. Chem. 163, 116–135. doi: 10.1016/j.ejmech.2018.11.049
Stancu, I. C., Vasconcelos, B., Terwel, D., and Dewachter, I. (2014). Models of β-amyloid induced tau-pathology: the long and “folded” road to understand the mechanism. Mol. Neurodegener. 9:51. doi: 10.1186/1750-1326-9-51
Sun, X., He, G., Qing, H., Zhou, W., Dobie, F., Cai, F., et al. (2006). Hypoxia facilitates Alzheimer’s disease pathogenesis by up-regulating BACE1 gene expression. Proc. Natl. Acad. Sci. 103, 18727–18732. doi: 10.1073/pnas.0606298103
Sun, P., Ortega, G., Tan, Y., Hua, Q., Riederer, P. F., Deckert, J., et al. (2018). Streptozotocin impairs proliferation and differentiation of adult hippocampal neural stem cells in vitro-correlation with alterations in the expression of proteins associated with the insulin system. Front. Aging Neurosci. 10:145. doi: 10.3389/fnagi.2018.00145
Takaki, J., Fujimori, K., Miura, M., Suzuki, T., Sekino, Y., and Sato, K. (2012). L-glutamate released from activated microglia downregulates astrocytic L-glutamate transporter expression in neuroinflammation: the ‘collusion’ hypothesis for increased extracellular L-glutamate concentration in neuroinflammation. J. Neuroinflammation 9:275. doi: 10.1186/1742-2094-9-275
Thysiadis, S., Katsamakas, S., Mpousis, S., Avramidis, N., Efthimiopoulos, S., and Sarli, V. (2018). Design and synthesis of gallocyanine inhibitors of DKK1/LRP6 interactions for treatment of Alzheimer’s disease. Bioorg. Chem. 80, 230–244. doi: 10.1016/j.bioorg.2018.06.018
Tiwari, S., Atluri, V., Kaushik, A., Yndart, A., and Nair, M. (2019). Alzheimer’s disease: pathogenesis, diagnostics, and therapeutics. Int. J. Nanomedicine 14, 5541–5554. doi: 10.2147/IJN.S200490
Toimela, T., Mäenpää, H., Mannerström, M., and Tähti, H. (2004). Development of an in vitro blood–brain barrier model—cytotoxicity of mercury and aluminum. Toxicol. Appl. Pharmacol. 195, 73–82. doi: 10.1016/j.taap.2003.11.002
Tong, Y., Yang, H., Tian, X., Wang, H., Zhou, T., Zhang, S., et al. (2014). High manganese, a risk for Alzheimer’s disease: high manganese induces amyloid-β related cognitive impairment. J. Alzheimers Dis. 42, 865–878. doi: 10.3233/JAD-140534
Tripathi, P. N., Srivastava, P., Sharma, P., Tripathi, M. K., Seth, A., Tripathi, A., et al. (2019). Biphenyl-3-oxo-1, 2, 4-triazine linked piperazine derivatives as potential cholinesterase inhibitors with anti-oxidant property to improve the learning and memory. Bioorg. Chem. 85, 82–96. doi: 10.1016/j.bioorg.2018.12.017
Tuneva, J., Chittur, S., Boldyrev, A. A., Birman, I., and Carpenter, D. O. (2006). Cerebellar granule cell death induced by aluminum. Neurotox. Res. 9, 297–304. doi: 10.1007/BF03033320
Vahidnia, A., van der Straaten, R. J. H. M., Romijn, F., van Pelt, J., van der Voet, G. B., and de Wolff, F. A. (2008). Mechanism of arsenic-induced neurotoxicity may be explained through cleavage of p 35 to p 25 by calpain. Toxicol. In Vitro 22, 682–687. doi: 10.1016/j.tiv.2007.12.010
Wang, Y., An, Y., Ma, W., Yu, H., Lu, Y., Zhang, X., et al. (2020). 27-hydroxycholesterol contributes to cognitive deficits in APP/PS1 transgenic mice through microbiota dysbiosis and intestinal barrier dysfunction. J. Neuroinflammation 17:199. doi: 10.1186/s12974-020-01873-7
Wang, H., Jiang, T., Li, W., Gao, N., and Zhang, T. (2018). Resveratrol attenuates oxidative damage through activating mitophagy in an in vitro model of Alzheimer’s disease. Toxicol. Lett. 282, 100–108. doi: 10.1016/j.toxlet.2017.10.021
Wang, R., and Reddy, P. H. (2017). Role of glutamate and NMDA receptors in Alzheimer’s disease. J. Alzheimers Dis. 57, 1041–1048. doi: 10.3233/JAD-160763
Wang, B. R., Shi, J. Q., Ge, N. N., Ou, Z., Tian, Y. Y., Jiang, T., et al. (2018). PM2.5 exposure aggravates oligomeric amyloid beta-induced neuronal injury and promotes NLRP3 inflammasome activation in an in vitro model of Alzheimer’s disease. J. Neuroinflammation 15:132. doi: 10.1186/s12974-018-1178-5
Wang, H., Yang, F., Zhang, S., Xin, R., and Sun, Y. (2021). Genetic and environmental factors in Alzheimer’s and Parkinson’s diseases and promising therapeutic intervention via fecal microbiota transplantation. NPJ Parkinsons Dis. 7, 70–10. doi: 10.1038/s41531-021-00213-7
Wang, Y., Zhang, M., Li, Z., Yue, J., Xu, M., Zhang, Y., et al. (2019). Fine particulate matter induces mitochondrial dysfunction and oxidative stress in human SH-SY5Y cells. Chemosphere 218, 577–588. doi: 10.1016/j.chemosphere.2018.11.149
Wang, Y., Zhang, X., Wang, T., Liu, W., Wang, L., Hao, L., et al. (2020). 27-hydroxycholesterol promotes the transfer of astrocyte-derived cholesterol to neurons in co-cultured SH-SY5Y cells and C6 cells. Front. Cell Dev. Biol. 8:580599. doi: 10.3389/fcell.2020.580599
Wei, G. H., Zhang, J., Liao, D. Q., Li, Z., Yang, J., Luo, N. F., et al. (2014). The common anesthetic, sevoflurane, induces apoptosis in A549 lung alveolar epithelial cells. Mol. Med. Rep. 9, 197–203. doi: 10.3892/mmr.2013.1806
Wisessaowapak, C., Visitnonthachai, D., Watcharasit, P., and Satayavivad, J. (2021). Prolonged arsenic exposure increases tau phosphorylation in differentiated SH-SY5Y cells: the contribution of GSK3 and ERK1/2. Environ. Toxicol. Pharmacol. 84:103626. doi: 10.1016/j.etap.2021.103626
Xu, F., Farkas, S., Kortbeek, S., Zhang, F. X., Chen, L., Zamponi, G. W., et al. (2012). Mercury-induced toxicity of rat cortical neurons is mediated through N-methyl-D-aspartate receptors. Mol. Brain 5:30. doi: 10.1186/1756-6606-5-30
Xu, Y. F., Jie Zhang, Y., Hong Zhang, A., Zhang, Q., Wu, T., and Wang, J. Z. (2004). Attenuation of okadaic acid–induced hyperphosphorylation of cytoskeletal proteins by heat preconditioning and its possible underlying mechanisms. Cell Stress Chaperones 9, 304–312. doi: 10.1379/CSC-23R1.1
Xu, P., Wang, K., Lu, C., Dong, L., Gao, L., Yan, M., et al. (2016). Protective effect of lavender oil on scopolamine induced cognitive deficits in mice and H2O2 induced cytotoxicity in PC12 cells. J. Ethnopharmacol. 193, 408–415. doi: 10.1016/j.jep.2016.08.030
Yang, Y., Zhang, J., Yang, X., Li, Z., Wang, J., Lu, C., et al. (2021). Dysregulated APP expression and α-secretase processing of APP is involved in manganese-induced cognitive impairment. Ecotoxicol. Environ. Saf. 220:112365. doi: 10.1016/j.ecoenv.2021.112365
Yi, K. D., Covey, D. F., and Simpkins, J. W. (2009). Mechanism of Okadaic acid induced neuronal death and the effect of estrogens. J. Neurochem. 108, 732–740. doi: 10.1111/j.1471-4159.2008.05805.x
You, R., Ho, Y. S., and Chang, R. C. C. (2022). The pathogenic effects of particulate matter on neurodegeneration: a review. J. Biomed. Sci. 29:15. doi: 10.1186/s12929-022-00799-x
Yu, Q., and Cheng, X. (2021). Hydroxyurea-induced membrane fluidity decreasing as a characterization of neuronal membrane aging in Alzheimer’s disease. Aging 13, 12817–12832. doi: 10.18632/aging.202949
Yu, D., Duan, Y., Bao, Y., Wei, C., and An, L. (2005). Isoflavonoids from Astragalus mongholicus protect PC12 cells from toxicity induced by l-glutamate. J. Ethnopharmacol. 98, 89–94. doi: 10.1016/j.jep.2004.12.027
Zhang, G., Meng, L., Wang, Z., Peng, Q., Chen, G., Xiong, J., et al. (2022). Islet amyloid polypeptide cross-seeds tau and drives the neurofibrillary pathology in Alzheimer’s disease. Mol. Neurodegener. 17:12. doi: 10.1186/s13024-022-00518-y
Zhang, M., Wang, Y., Wong, R. M. S., Yung, K. K. L., and Li, R. (2022). Fine particulate matter induces endoplasmic reticulum stress-mediated apoptosis in human SH-SY5Y cells. Neurotoxicology 88, 187–195. doi: 10.1016/j.neuro.2021.11.012
Zhang, X., Zhou, K., Wang, R., Cui, J., Lipton, S. A., Liao, F. F., et al. (2007). Hypoxia-inducible factor 1α (HIF-1α)-mediated hypoxia increases BACE1 expression and β-amyloid generation*. J. Biol. Chem. 282, 10873–10880. doi: 10.1074/jbc.M608856200
Zhou, H., Qu, Z., Zhang, J., Jiang, B., Liu, C., and Gao, W. (2018). Shunaoxin dropping pill, a Chinese herb compound preparation, attenuates memory impairment in d -galactose-induced aging mice. RSC Adv. 8, 10163–10171. doi: 10.1039/C7RA13726E
Keywords: Alzheimer’s disease, in-vitro models, neurodegeneration, tau protein, amyloid beta
Citation: Prajapat M, Kaur G, Choudhary G, Pahwa P, Bansal S, Joshi R, Batra G, Mishra A, Singla R, Kaur H, Prabha PK, Patel AP and Medhi B (2023) A systematic review for the development of Alzheimer’s disease in in vitro models: a focus on different inducing agents. Front. Aging Neurosci. 15:1296919. doi: 10.3389/fnagi.2023.1296919
Edited by:
Prasenjit Mondal, Harvard Medical School, United StatesReviewed by:
Vina Tikiyani, National Institute of Diabetes and Digestive and Kidney Diseases (NIH), United StatesCopyright © 2023 Prajapat, Kaur, Choudhary, Pahwa, Bansal, Joshi, Batra, Mishra, Singla, Kaur, Prabha, Patel and Medhi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bikash Medhi, ZHJiaWthc2h1c0B5YWhvby5jb20=
†These authors have contributed equally to this work
‡ORCID: Bikash Medhi orcid.org/0000-0002-4017-641X
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.