
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Aging Neurosci. , 20 December 2023
Sec. Parkinson’s Disease and Aging-related Movement Disorders
Volume 15 - 2023 | https://doi.org/10.3389/fnagi.2023.1282174
 Zied Landoulsi1†
Zied Landoulsi1† Sinthuja Pachchek1†
Sinthuja Pachchek1† Dheeraj Reddy Bobbili1Lukas Pavelka2,3
Dheeraj Reddy Bobbili1Lukas Pavelka2,3 Patrick May1*Rejko Krüger1,2,3* and on behalf of the NCER-PD Consortium
Patrick May1*Rejko Krüger1,2,3* and on behalf of the NCER-PD ConsortiumObjectives: To explore the genetic architecture of PD in the Luxembourg Parkinson’s Study including cohorts of healthy people and patients with Parkinson’s disease (PD) and atypical parkinsonism (AP).
Methods: 809 healthy controls, 680 PD and 103 AP were genotyped using the Neurochip array. We screened and validated rare single nucleotide variants (SNVs) and copy number variants (CNVs) within seven PD-causing genes (LRRK2, SNCA, VPS35, PRKN, PARK7, PINK1 and ATP13A2). Polygenic risk scores (PRSs) were generated using the latest genome-wide association study for PD. We then estimated the role of common variants in PD risk by applying gene-set-specific PRSs.
Results: We identified 60 rare SNVs in seven PD-causing genes, nine of which were pathogenic in LRRK2, PINK1 and PRKN. Eleven rare CNVs were detected in PRKN including seven duplications and four deletions. The majority of PRKN SNVs and CNVs carriers were heterozygous and not differentially distributed between cases and controls. The PRSs were significantly associated with PD and identified specific molecular pathways related to protein metabolism and signal transduction as drivers of PD risk.
Conclusion: We performed a comprehensive genetic characterization of the deep-phenotyped individuals of the Luxembourgish Parkinson’s Study. Heterozygous SNVs and CNVs in PRKN were not associated with higher PD risk. In particular, we reported novel digenic variants in PD related genes and rare LRRK2 SNVs in AP patients. Our findings will help future studies to unravel the genetic complexity of PD.
Parkinson’s disease (PD) is the fastest growing neurodegenerative disorder, affecting more than 8.5 million people (Dorsey and Bloem, 2018). The main pathological hallmarks of PD include loss of dopaminergic neurons in the substantia nigra and the presence of intraneural Lewy bodies, with motor and non-motor symptoms (Bloem et al., 2021). The etiology of sporadic PD is complex and influenced by both environmental and genetic factors. Familial monogenic forms defined by rare and pathogenic variants in autosomal dominant (e.g., SNCA, LRRK2, VPS35) or recessive (PRKN, PINK1, PARK7) PD-related genes, account for less than 10% of Mendelian cases (Lesage and Brice, 2009; Karimi-Moghadam et al., 2018). The contribution of genetics in the remaining patients with sporadic forms of PD is not yet well defined. Common variants have also been described as a risk factor for PD (Nalls et al., 2019). The presence of heterozygous variants in the GBA1 gene has emerged as a common risk factor for PD, estimated to occur in about 4–12% of PD patients (Pachchek et al., 2023). The major haplotype (H1) of the microtubule-associated protein Tau (MAPT) gene has been also associated with increased risk of PD (Skipper et al., 2004; Zabetian et al., 2007). Additionally, a specific MAPT H1 sub-haplotype (H1c) has been strongly linked with progressive supranuclear palsy (PSP; Myers et al., 2005). Disease susceptibility may be influenced by a combined effect of more than 90 common low-risk genetic loci defined by large genome-wide association studies (Nalls et al., 2019; Blauwendraat et al., 2020), including those in the SNCA and LRRK2 genes. Although less explored than common single-nucleotide variants (SNVs), copy number variants (CNVs) have been reported, especially in PD-associated genes where pathogenic deletions and duplications have been identified using either a gene candidate approach (PRKN, SNCA, PINK1, PARK7 and ATP13A2) (Toft and Ross, 2010; Pankratz et al., 2011; La Cognata et al., 2017) or genome-wide burden analysis (Liu et al., 2013; Sarihan et al., 2021).
Despite ongoing global scientific efforts in genetic analysis, improvements are still needed in terms of early diagnosis and prognosis, causative treatments, and new therapeutic approaches. As the population ages, the number of PD patients will increase dramatically. It is therefore important to generate reliable evidence on the epidemiology and genetic etiology of PD to enable precision medicine and prevention for neurodegeneration in PD. In particular, three genetic discoveries that have led to new therapeutic approaches (targeting alpha-synuclein, glucocerebrosidase and LRRK2 pathway) are now in clinical development (Sardi et al., 2018).
We had previously performed a comprehensive screening of GBA1 gene variants (Pachchek et al., 2023). Here, we sought to genetically characterize patients with PD or atypical parkinsonism (AP) in the Luxembourg Parkinson’s Study screening for rare SNVs, CNVs, and estimated the effect of common SNVs using polygenic risk scores (PRS).
A total of 1,592 individuals (783 cases and 809 neurologically healthy controls) were recruited from March 2015 to December 2022 as part of the Luxembourg Parkinson’s Study, a large longitudinal monocentric study within the framework of the NCER-PD [National Centre for Excellence in Research in PD (Hipp et al., 2018; Pavelka et al., 2022)]. The most up-to-date diagnostic status of the participants was used at the time of export (July 2023). Assignment of diagnosis was based on the following diagnostic criteria: for PD UKPDSBB (Litvan et al., 2003); for progressive supranuclear palsy (PSP) Institute of Neurological Disorders and Stroke/Society criteria (Litvan et al., 1996); for frontotemporal dementia with parkinsonism (FTD-P) (Neary et al., 1998); for multiple system atrophy (MSA) (Gilman et al., 2008); for dementia with Lewy bodies (DLB) (McKeith et al., 2005). Of these individuals, 680 fulfilled the criteria for PD and 103 for AP (52 for PSP, 26 for LBD, 14 for MSA, 10 for corticobasal syndrome (CBS), and one for FTD-P). All subjects gave written informed consent. The study was approved by the National Research Ethics Committee (CNER Ref: 201407/13).
Samples were genotyped with the customized NeuroChip array, which was designed to contain tagging rare and common variants associated with neurodegenerative diseases [v.1.0 and v1.1; Illumina, San Diego, CA (Blauwendraat et al., 2017)]. These disease-targeted variants include loci from the largest completed meta-analysis of PD cases and controls, which identified many of the known PD mutations and additional rare high-risk variants. Using PLINK v1.9 (Chang et al., 2015), we performed two rounds of quality control (QC). The first round included the following steps: samples with call rates <95%, missingness rates >5%, Hardy–Weinberg equilibrium p-value <1e-6 and whose genetically determined sex deviated from the sex reported in clinical data were excluded from the analysis. We also removed samples exhibiting excess heterozygosity (F statistic >0.2). After these steps, the remaining samples were used for rare variant screening and validation process. Next, we performed a second round of QC steps where the filtered variants were checked for relatedness [using KING (Manichaikul et al., 2010)] and samples with first-degree relatedness were excluded. To determine the genetic ancestry, we calculated the first 10 principal components (PCs) using PLINK and merged our data with the 1,000 genomes dataset. We selected only samples of European ancestrys, excluding those with a value >3SD based on the first and the second PCs. Samples passing QC were then imputed using the Haplotype Reference Consortium r1.1 2016 on a local instance of the Michigan Imputation Server (Das et al., 2016) and filtered for imputation quality (R2 > 0.3). Imputed genetic variants passing QC numbered 19,490,906 SNPs.
Using the imputed NeuroChip genotyping data, we selected rs1800547 to differentiate the MAPT H1 and H2 haplotypes and six tagging variants (rs1467967, rs242557, rs3785883, rs2471738, rs8070723, and rs7521) to define the H1 specific sub-haplotypes (Pittman, 2005). H1c sub-haplotype is defined by the following alleles (rs1467967(A), rs242557(A), rs3785883(G), rs2471738(T), rs8070723(A) and rs7521(G)). The association between MAPT haplotypes genotypic and allelic frequency in cases and controls was tested using the Chi-squared and Fisher’s exact tests, respectively. P values were considered significant at p < 0.05.
We annotated the variants with ANNOVAR [v 2020-06-08 (Wang et al., 2010)]. We searched for variants within a list of “PD genes” including the major causal genes according to MDS gene classification1 (1) Dominant forms of classical parkinsonism, LRRK2, SNCA, VPS35, (2) Recessive forms of early-onset parkinsonism: PRKN, PARK7, and PINK1; (3) Atypical parkinsonism: ATP13A2. We then selected rare nonsynonymous and splicing (+/−2 bp) rare variants based on the minor alleles frequency (MAF) <1% in the Genome Aggregation Database [gnomAD r2.1 (Karczewski et al., 2020)] exomes and genomes for the non-Finnish European (NFE) population. We performed Sanger sequencing to confirm all rare variants within these PD genes. The pathogenicity of the validated rare variants was assigned based on ClinVar (Landrum et al., 2014), the MDSgenes pathogenicity score (see text footnote 1), the Combined Annotation Dependent Depletion (CADD) (Rentzsch et al., 2019) and the Rare Exome Variant Ensemble Learner REVEL scores (Ioannidis et al., 2016). CADD provides ranking scores that predict the deleteriousness of variants, considering conservation and functional information, and variants with scores equal to or greater than a CADD score of 20 are in the 1% most deleterious. REVEL is an ensemble method for predicting the pathogenicity of missense variants by integrating multiple scores. Scores range from 0 to 1 and variants with higher scores are more likely to be pathogenic. Scores greater than 0.5 are predicted to be ‘likely disease causing’, as 75.4% of known disease mutations but only 10.9% of neutral variants have a score greater than 0.5 (Ioannidis et al., 2016).
We generated a custom population B-allele frequency (BAF) and GC wave-adjusted log R ratio (LRR) intensity file using GenomeStudio (v2.0.5 Illumina) for all the samples that passed genotyping QC steps and used PennCNV (v1.0.5, Wang et al., 2007) to detect CNVs. Only autosomal CNV were targeted for CNV calling, as calls from sex chromosomes are often of poor quality. Adjacent CNV calls were merged into one single call if the number of overlapping markers between them was less than 20% of the total number when the two segments were combined. We conducted an intensity-based QC to exclude samples with low-quality data. After intensity-based QC, all samples had an LRR standard deviation <0.25, an absolute value of the waviness factor < 0.05 and a BAF drift <0.01. Spurious CNV calls in known problematic genomic regions (provided by PennCNV) were also removed prior the analysis. We excluded additional samples with a total number of CNVs calls greater than 80 (this threshold corresponds to the median + 3 SDs of the total number of CNVs per sample). Called CNVs were removed from the dataset if they spanned <20 SNPs, were < 20 kilobases (kb) in length and had a SNP density < 0.0001 (number of markers/length of CNVs). Additionally, SNP density was not considered for CNVs spanning ≥20 SNPs and ≥ 1 Mb in length. CNVs were then annotated for gene content using refGene including gene name and the corresponding exonic coordinates in the hg19 assembly using ANNOVAR (v 2020-06-08). We then searched for CNVs in the same list of “PD genes” used to screen for rare SNVs. We assessed the frequency of CNVs based on complete overlap with CNVs of the same copy number reported in gnomAD-SV (Collins et al., 2020) and in the Database of Genomic Variants (DGV) (MacDonald et al., 2014). We evaluated the clinical impact of the detected CNVs using the CNV-ClinViewer (Macnee et al., 2023), which integrates clinical interpretation of CNVs according to the ACMG guideline and the ClassifyCNV scores. Selected CNVs were validated using the multiplex ligation-dependent probe amplification (MLPA) assay.
PRSs were calculated for healthy controls and PD cases using the R package PRSice2 (Choi and O’Reilly, 2019) with default parameters. PRSs for each sample were generated using the imputed genotyping data from the Luxembourg Parkinson’s Study and the summary statistics of 90 genome-wide significant SNVs that were previously reported to be associated with PD risk in the largest PD genome-wide association study (GWAS) statistics to date (Nalls et al., 2019). PRSice2 implements the clumping and thresholding method. The criteria for linkage disequilibrium (LD) clumping of SNPs were pairwise LD r2 < 0.1 within a 250 kb window. PRSs were computed at different GWAS p-value thresholds (from 5e-08 to 5e-01). PRSice2 identified the best p-value threshold for selecting variants that explained the maximum variance in the target sample. The predictive accuracy of the PRS model was determined by two methods: by the observed phenotypic variance (PRS model fit, R2) calculated by PRSice2 and by the area under the receiver operating curve (AUC, pROC R package). The phenotypic variance R2 was adjusted for a PD prevalence of 0.005 (Bandres-Ciga et al., 2020). The PRS distributions between healthy controls and PD cases were compared using the Wilcoxon rank-sum test. PRSs for curated gene-sets were generated using the msigdb function implemented in PRSice2, based on a collection of 3,090 canonical pathways from the molecular signature database (MSigDB2, “c2.cp.v2023.1.Hs.symbols.gmt”) with an MAF threshold of 0.01. The summary statistics of PD GWAS from Nalls et al. (2019) (excluding 23&me data) were used as the base dataset. The mapping file “Homo_sapiens.GRCh37.87.gtf” was used as the universal background for gene-set analysis. Resulting gene-sets with a value of p less than 0.05, corrected for Bonferroni multiple testing, were considered significant. In order to understand which biological processes were associated with PD after excluding known risk factors, we performed the same analysis after removing the 90 PD GWAS hits (Nalls et al., 2019) and additional SNVs that were located 1 Mb upstream and downstream. We used a logistic regression model to calculate the odds ratio (OR) to assess whether PRS could predict PD risk. Age, sex and the first five PCs were included as covariates.
After the quality control procedure, the final dataset for the Luxembourg Parkinson’s study comprised 1,490 individuals (667 PD cases, 99 atypical PD cases and 724 healthy controls). Detailed demographic data are summarized in Table 1. The control group had a mean age at assessment of 65.8 ± 11.6 years. The PD patients had a mean age of onset (AAO) of 62.3 ± 11.8 years. To illustrate the ethnic composition of our cohort, we performed PCA using 1,000 Genome populations as a reference (The 1000 Genomes Project Consortium, 2012) and showed that all our samples clustered strongly with the European ancestry (Supplementary Figure 1).
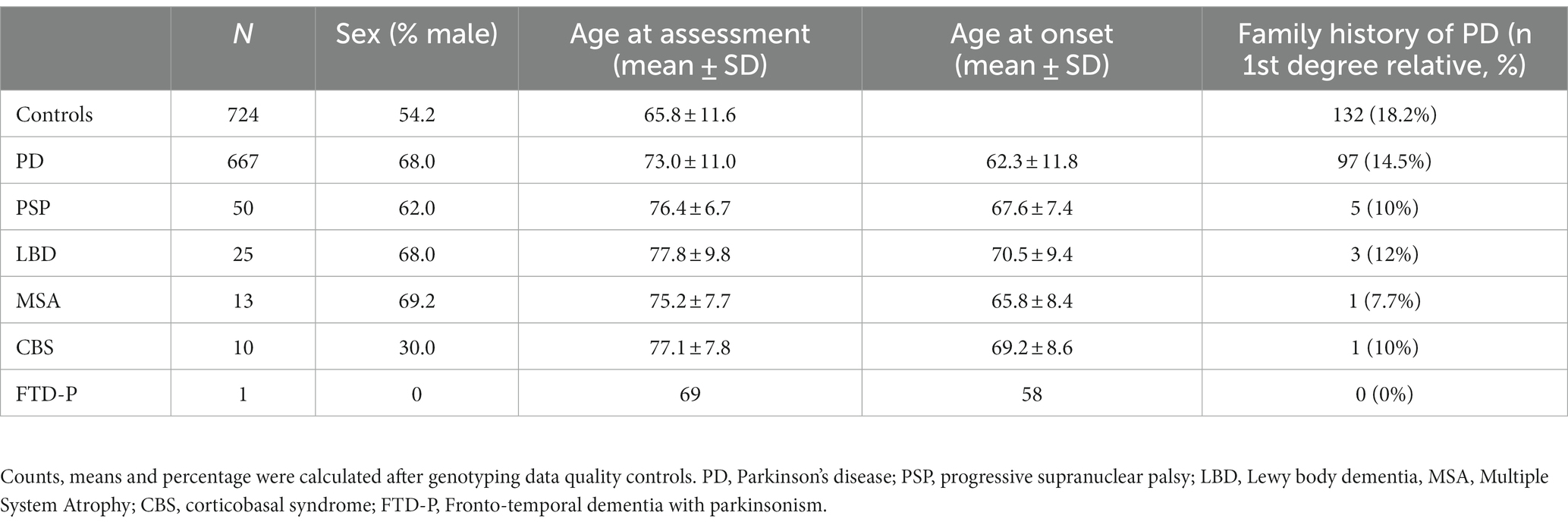
Table 1. Demographic data of cases and healthy controls from the Luxembourg Parkinson’s study.
We screened for rare (gnomAD NFE MAF < 1%) exonic and splice region variants in seven PD causal genes and validated these findings by Sanger sequencing. We identified 60 rare variants (59 missense and one frameshift) in all PD-related genes except for SNCA (Table 2), in 119 individuals including 52 controls, 57 PD and 10 AP patients, representing 7.9% of the total cohort (Table 3). All carriers were heterozygous, except two PD patients that were homozygous for LRRK2 p.I723V and PINK1 p.L369P, respectively.
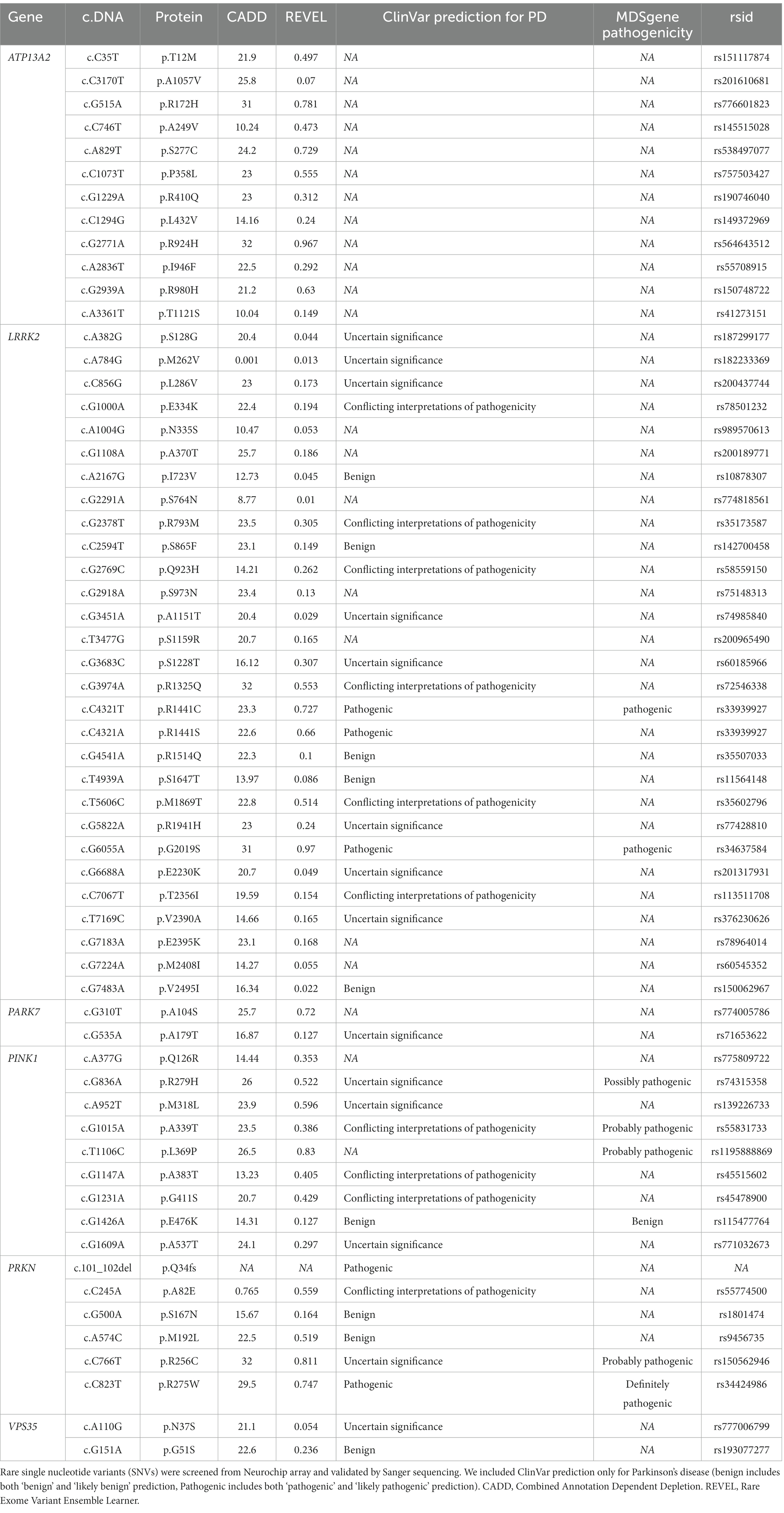
Table 2. Rare single nucleotide variants in PD related genes in the Luxembourg Parkinson’s study.
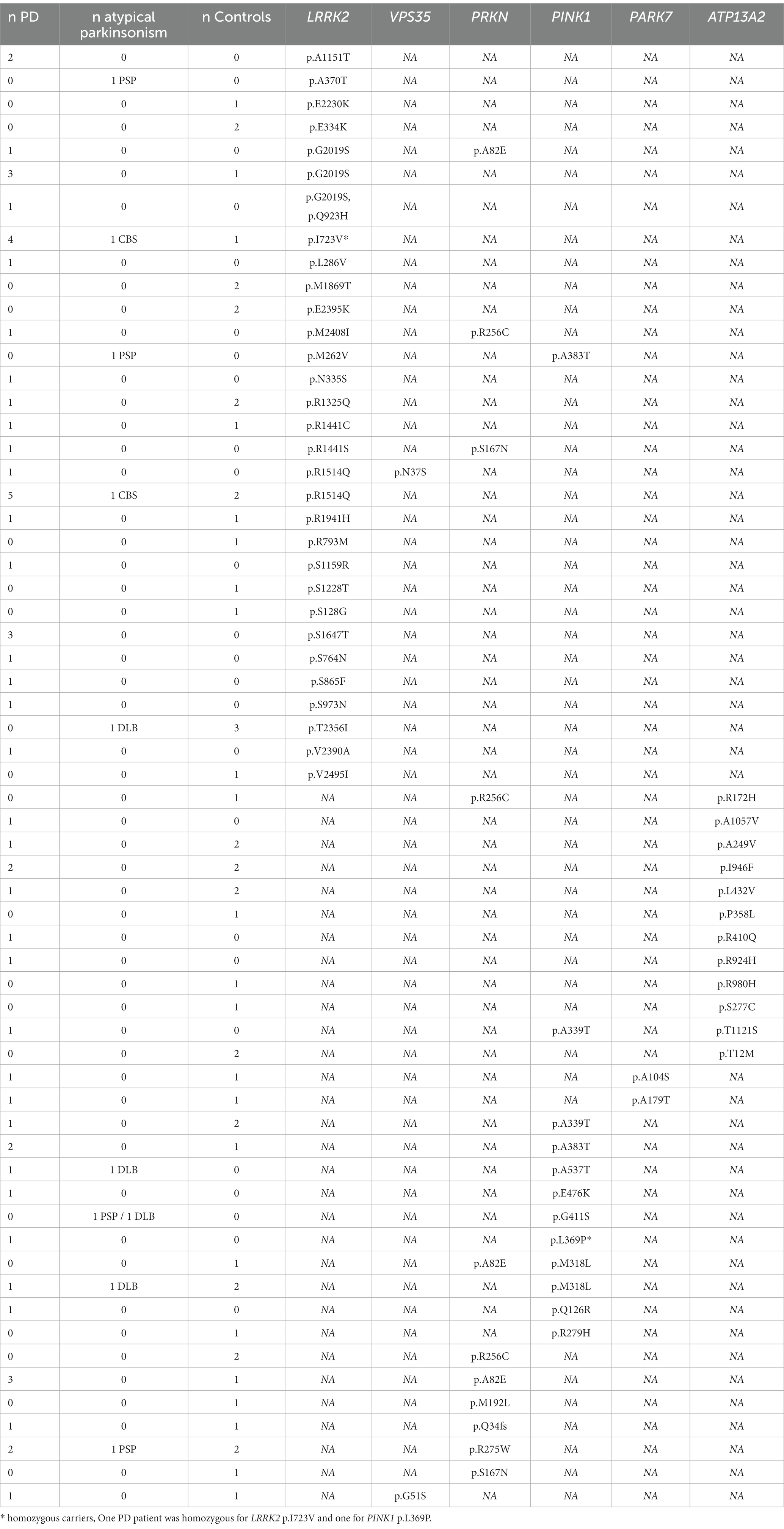
Table 3. Number and phenotypes of rare variant carriers in PD related genes.
Among the 29 rare variants identified in LRRK2, five variants have a CADD score > 20 and REVEL score > 0.5 (p.R1325Q, p.R1441S, p.R1441C, p.M1869T, and p.G2019S) showing high evidence for pathogenicity. Three of these variants were classified as pathogenic for PD in ClinVar (Table 2) and were present in nine individuals representing 0.60% of the total cohort (Table 3). Among these variants, five PD patients carried the extensively studied pathogenic variant p.G2019S while two PD patients carried the pathogenic p.R1441C and p.R1441S variant.
Two control individuals with family history of PD (Table 3) had rare LRRK2 variants. One control individual carried the variant p.G2019S (38 years old) and has a high probability of developing PD (Healy et al., 2008). Another control individual (77 years old) carried the p.R1441C, although this variant is described as highly penetrant [more than 90% of carriers had PD by the age of 75 (Haugarvoll and Wszolek, 2009)].
In the autosomal recessive PD-causing genes (PRKN, PARK7, PINK1, and ATP13A2), we identified 28 heterozygous rare variant carriers and only one homozygous rare variant carrier (PINK1 p.L369P, Table 2). The distribution of these variants was similar between cases and controls (27 PD, six AP and 29 controls, value of p = 0.39, Table 3). Four controls and 10 patients had a first-degree family-history of PD. The age of the control individuals carrying these heterozygous variants ranged from 52 to 85 years (mean = 67.8 years). The AAO of the PD patients carrying these heterozygous variants ranged from 39 to 87 (mean = 65.5). One PD patients was younger than 40 years (carrying PINK1 p.A383T), all the others were older than 50 years.
According to ClinVar, two pathogenic PRKN variants (p.R275W and p.Q34fs) were found in three PD patients, one PSP patient and three controls (all heterozygous, representing 0.46% of the total cohort, Table 3). Three PRKN variants (p.M192L, p.R256C and p.R275W) were predicted to be likely pathogenic with CADD and REVEL scores above the selected threshold. However, we noted the occurence of heterozygous p.R256C in three controls (age 80, 81 and 85 years) and one PD patient (AAO = 52 years), which is classified as ‘probably pathogenic’ according to the MDSgenes pathogenicity score. For PINK1 we found no pathogenic variant classified in ClinVar. However, p.R279H, p.A339T and p.L369P are ‘probably pathogenic’ according to the MDSgene pathogenicity scores, but only when homozygous. Two of these variants (p.R279H, p.L369P) and p.M318L were classified as ‘likely pathogenic’ based on CADD and REVEL scores. In addition, the PARK7 p.A104S and ATP13A2 p.R172H, p.S277C, p.P358L, p.R924H and p.R980H heterozygous variants had higher CADD and REVEL scores but were not reported to be pathogenic for PD in ClinVar or MDSgene.
Overall, we described nine pathogenic variants from databases of clinical interest in LRRK2, PRKN and PINK1 in a total of 26 samples (13 PD, 1 PSP and 12 controls, all heterozygous) representing 1.7% of the total cohort (Table 3). Given the zygosity of the variants, only variants in LRRK2 can be responsible for the disease. AP patients were heterozygous carriers of probably benign variants in LRRK2 and PINK1 and a pathogenic variant in PRKN.
An extensive screening of GBA1 variants was previously performed by our team (Pachchek et al., 2023) using GBA1-targeted PacBio sequencing in individuals from the Luxembourg Parkinson’s study (660 PD patients, 100 patients with other forms of parkinsonism and 808 controls). We identified 21 rare GBA1 variants (20 missense and one splice site) in 37 PD patients and 16 controls (representing 5.6% of PD patients and 1.9% controls), which were validated by Sanger sequencing. Eleven rare variants were classified as pathogenic while the others were classified as variants of unknown significance (VUS). For the samples that were both genotyped and screened by targeted GBA1-sequencing, we found that none of carriers of rare variants in the studied PD-causing genes, identified within the NeuroChip, harbored an additional pathogenic GBA1 variant.
We initially detected 25,299 CNVs, including 13,862 duplications and 11,437 deletions in 728 controls and 757 PD cases. After all QC and filtering steps, the final number of CNVs was 1,079 CNVs, including 737 duplications and 342 deletions in 373 controls and 366 cases. CNV analysis showed that almost half of the samples (49.7%) carried at least one QC-passed CNV. The length of the CNVs in the entire cohort ranged from 20 kb to 3.0 megabases (Mb) with a median size of 160 kb. The characteristics of our CNV analysis are shown in Table 4.
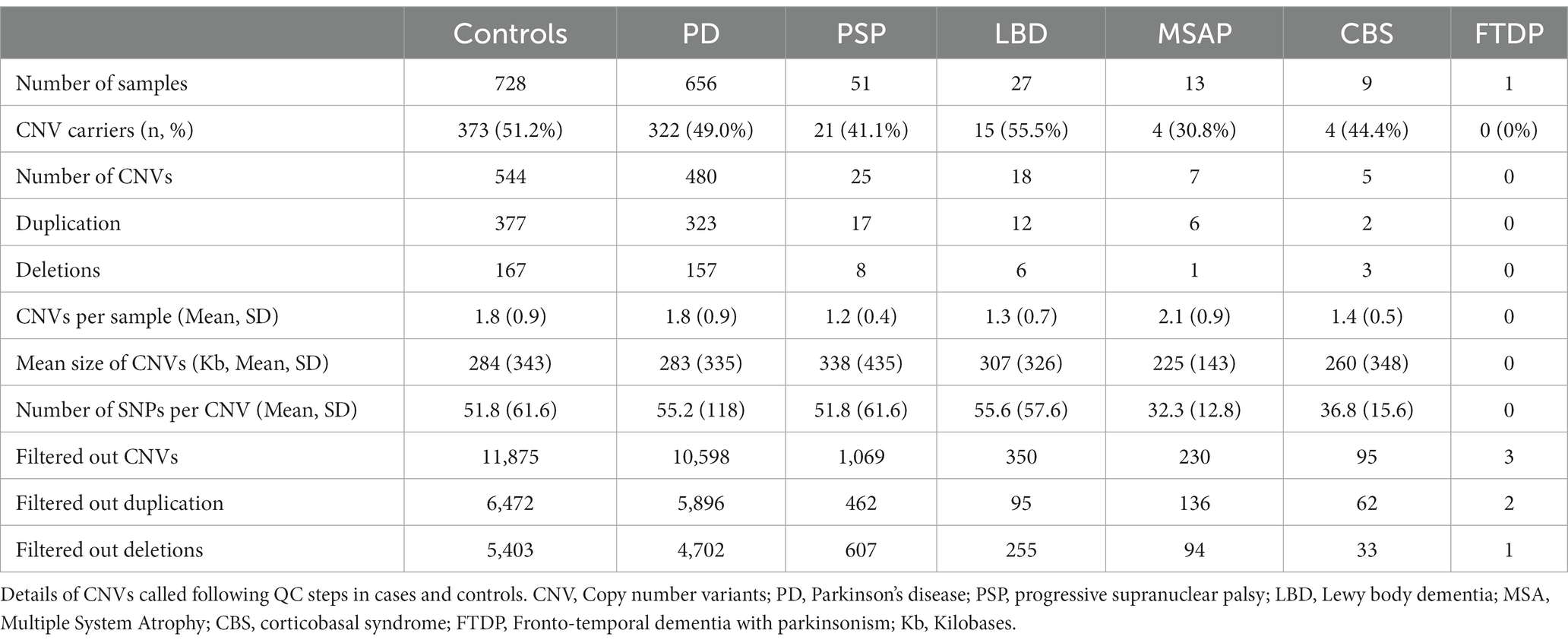
Table 4. Summary of CNV calls from the Luxembourg Parkinson’s study.
We then explored CNVs overlapping known PD genes and identified 15 CNVs in 18 samples (six controls and 12 PD cases) that were exclusively in the PRKN gene (Table 5). None of the PRKN CNV carriers had a rare variant in the same gene. We tested the presence of five CNVs by MLPA. As MLPA only covers exonic regions of PRKN, three MLPA results were consistent with PennCNV results (Table 5). One duplication was located in exon 2 rather than in a nearby intronic region and one duplication was found to be homozygous covering exon1 rather than heterozygous covering exon 2 (Table 5). After MLPA validation, of the 15 PRKN CNVs, eight were single copy deletions, six were single copy duplications and only one was a probably pathogenic homozygous duplication in a late-onset PD patient (AAO = 69 years). Among the PD cases, three PRKN heterozygous CNV carriers had an AAO ≤ 50 years (including one patient diagnosed with a juvenile form of PD at the age of 18). One CNV was detected in four samples, while the others were detected in only a single sample (Table 5). Eleven CNVs were considered as rare, since they were not reported in DGV and were spanning structural variants reported in European descent gnomAD_SV dataset with a frequency of less than 1% (Table 5). No clear clinical impact was observed for all the PRKN CNVs (uncertain significance in CNV-ClinViewer).
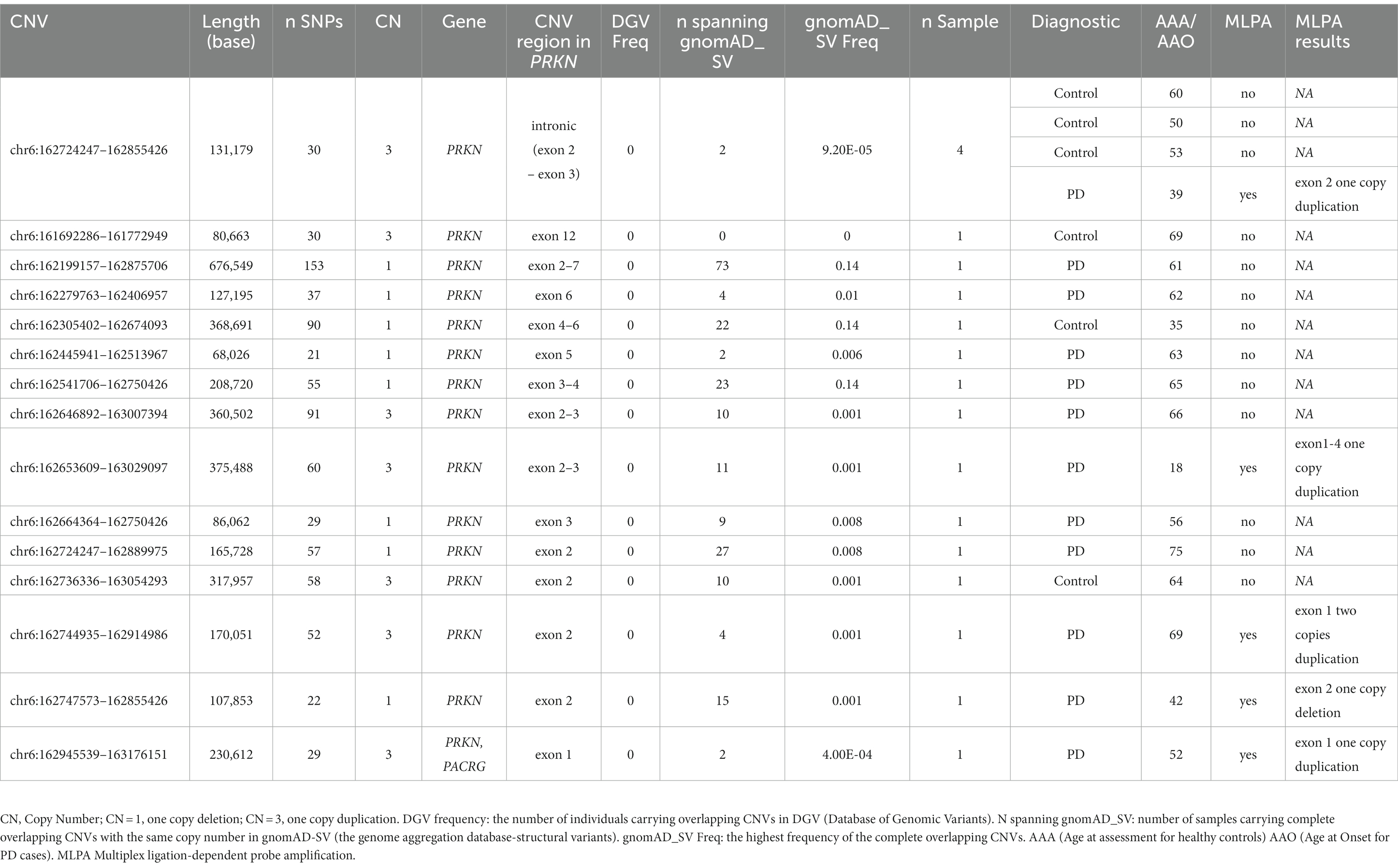
Table 5. CNVs detected in any of the PD genes in the Luxembourg Parkinson’s study.
Eight individuals (five PD cases, one with PSP and two controls) carried two variants in two different PD-related genes (Table 3). The AAO of the patients ranged from 52 to 71 (mean = 64.3). In particular, in autosomal recessive PD genes, pathogenic PRKN p.R256C and PINK1 p.A339T (in heterozygous state) were detected in the same individual with another probably benign variant. One PD patient (AAO = 62 years) carried the heterozygous PRKN deletion (chr6:162,279,763–162,406,957) and also the benign LRRK2 variant p.R1514Q. Moreover, two controls were carriers of two variants in PRKN-ATP13A2 (81 years old) and in PRKN-PINK1 (70 years old) respectively (Table 3).
The number of heterozygous rare pathogenic PRKN SNVs (p.Q34fs, p.R256C and p.R275W) was not significantly different between controls (n = 6, 0.82%) and PD cases (n = 4, 0.5%, value of p = 0.6). If we consider all the rare heterozygous CNV deletions as pathogenic loss-of-function variations together with the homozygous duplication, we counted seven PD cases each carrying one rare pathogenic deletion. Overall, the number of heterozygous pathogenic SNVs and CNVs was slightly higher in PD (n = 11, 1.64%) than in controls (n = 6, 0.82%), but the difference is still not significant (value of p = 0.16). The sample size is too small to examine a significant burden of these rare variants on PD risk.
We found a statistically significant over-representation of the MAPT H1 haplotype in PD (value of p = 0.018) and PSP (value of p = 0.008) cases, present in 80.5% of PD, 88.0% of PSP cases compared to 76.8% of controls (Supplementary Table 1). No significant association was found between sub-haplotype H1c and any of the investigated diseases (Supplementary Table 1).
Using significant common SNVs from the largest PD GWAS summary statistics (Nalls et al., 2019), we calculated the PRS in the Luxembourg Parkinson’s study for 724 controls and 667 PD patients. The PRS model was calculated based on 75 clumped SNPs that showed the best prediction at the GWAS value of p threshold of 5e-08 and an observed phenotypic variance R2 of 5.3% (1.9% after adjustment for PD prevalence of 5e-03, empirical value of p = 9.9e-05) with an AUC of 62.8%. We found a significant association of PRS with higher PD risk (OR = 1.70[1.50–1.93], p = 5.9e-17). The distribution of PRS scores in PD cases and healthy controls was significantly different (Wilcoxon test value of p <2.2e-16, Figure 1A). Individuals with the 5 and 10% of highest PRS values had a 9.5-fold [3.9–26.3] (p = 1.4e-08) and 5.6-fold [3.3–9.7] (p = 1.8e-12) increased risk, respectively, compared to individuals with the lowest 5 and 10% PRS values (Figure 1A). Out of the 3,090 canonical pathways gene-sets representing the most important biological processes and diseases, 17 gene-sets were significantly associated with PD risk (Bonferroni adjusted value of p <0.05, Figure 1B; Supplementary Table 2). Among the enriched pathways, the majority were associated with PD (showing the highest R2 values, Figure 1B) and PD pathogenesis (Alpha synuclein, Parkin and ubiquitination related pathways), Alzheimer disease (AD), signal transduction and metabolism of proteins. No gene-set remained significant after excluding the 90 PD GWAS hits region (1 Mb upstream and downstream each locus), indicating the absence of other risk loci acting independently of the known ones.
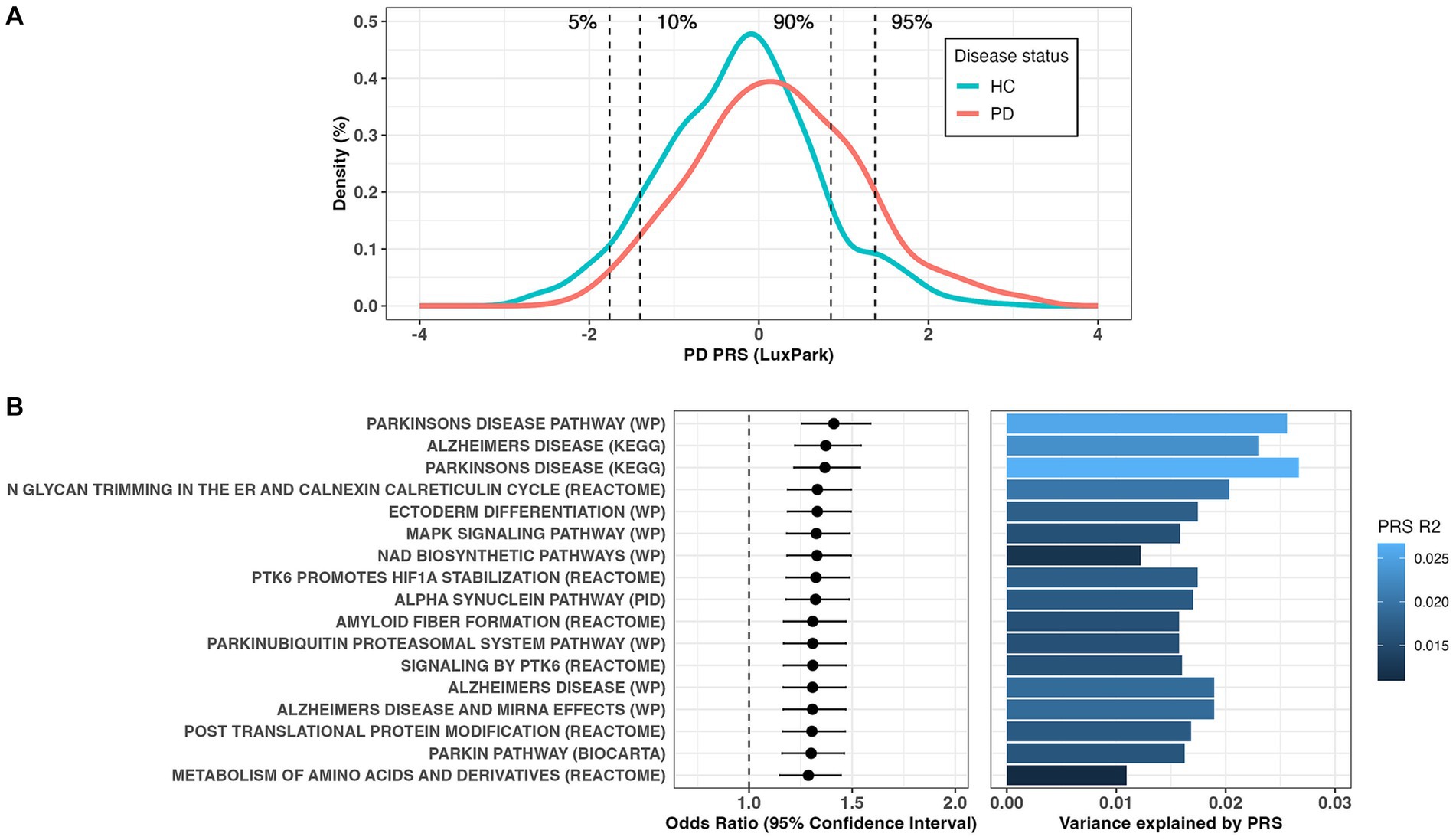
Figure 1. (A) Distribution of the polygenic risk score (PRS) between Parkinson’s disease (PD) patients and controls. (B) Forest plots showing PRS odds ratio (OR) and 95% confidence interval for the significant canonical pathways associated with PD risk (left panel) and the estimation of variance explained by PRS (right panel).
The current report is a comprehensive genetic description of participants recruited within the monocentric case–control Luxembourg Parkinson’s study, including patients with PD and atypical parkinsonism, with previously described recruitment design and clinical characteristics (Hipp et al., 2018; Pavelka et al., 2022). Previous long-read sequencing of GBA1 gene in our cohort revealed that 12.1% of PD patients carried GBA1 variants (Pachchek et al., 2023). Analyzing now the complete Neurochip genotyping data, we investigated the potential effect of rare variants, common low-risk variants and CNVs on the PD pathogenesis. Our findings are consistent with those previously reported in European ancestry datasets.
In the LRRK2 gene, two well-established pathogenic SNVs were found in five PD patients and two controls with a frequency similar to previous European ancestry datasets (Correia Guedes et al., 2010; Shu et al., 2019). Pathogenic and probably pathogenic variants in the ATP13A2, PARK7, PRKN and PINK1 genes associated with autosomal recessive PD were found in the heterozygous state, except in one PD patient. The latter carried a homozygous pathogenic PINK1 variant (p.L369P) and had an AAO of 32 years (Arena and Valente, 2017) reported a similar finding, where homozygous variants in PINK1 associated with early-onset PD (EOPD) were present in the patient before the age of 45 years. In our study, the number of heterozygous SNVs in the recessive PARK7, ATP13A2, PRKN and PINK1 genes was not significantly different between PD cases and controls. Controls carrying these variants were over 50 years of age. The majority had no family history of PD and most of PD patients were not of young onset (AAO > 50 years). Patients with AP carried probably benign heterozygous variants, mainly in LRRK2 and PINK1. Pathogenic LRRK2 variants have been described in patients with primary tauopathies, although at a low frequency (Wen et al., 2021). In particular, LRRK2 has recently emerged as a genetic risk factor associated with PSP progression (Jabbari et al., 2021).
We called CNVs from the genotyping data of individuals in the Luxembourg Parkinson’s study and after a stringent quality control and filtering steps, we screened for CNVs overlapping PD causal genes. We identified 12 PD patients who carried CNVs exclusively in the PRKN gene, of which three CNVs were validated by MLPA and were reported in patients having a disease AAO ≤ 50 years. Especially, we described a heterozygous exon1-4 duplication in a patient with EOPD (AAO of 18 years) who did not present any rare variant in the PD-related genes studied here. Moreover, we validated by MLPA a homozygous duplication of PRKN exon1 in another PD patient with a late disease-onset (69 years). Duplications of PRKN exons were previously reported as ‘likely pathogenic’ (Schüle et al., 2015). Indeed, both homozygous and compound heterozygous PRKN deletions and duplications have previously been associated with early-onset and familial forms of PD (Elfferich et al., 2011; Kim et al., 2012; Huttenlocher et al., 2015; Ahmad et al., 2023). This was recently reproduced in a large CNV study of 4,800 clinical exome sequencing reports (Pennings et al., 2023). In a Latin American PD cohort, CNVs in PRKN were significantly associated with disease progression, with a prevalence of 5.6% in EOPD cases (Sarihan et al., 2021).
We found that six PD cases and one PSP case carried digenic variants in two different PD-related genes (LRRK2-PRKN, LRRK2-PINK1, PINK1-ATP13A2) with AAO greater than 50 years. Hitherto, only a few studies have identified digenic variants of PD-related genes [LRRK2-PRKN (Dächsel et al., 2006), PINK1-PARK7 (Tang et al., 2006) or PRKN-PINK1 (Hayashida et al., 2021)]. Previous studies reported that carriers of digenic variants in PRKN and PINK1 develop the disease at a younger age and exhibit distinctive symptoms such as schizophrenia, facial dyskinesia, grimacing and severe dysarthria (Funayama et al., 2008) and also epilepsy and essential tremor (Hayashida et al., 2021). However, the digenic variants reported in this study differ from those previously described, and carriers of these variants have an older age at onset. Nonetheless, the ambiguity surrounding digenic variants persists due to the limited number of reported cases. A detailed familial and clinical study could be carried for every individual, to confirm that the combination of these heterozygous variants, in the context of a digenic inheritance, may point out the phenotype observed in PD and PSP cases.
In our study, we did not find a significant overrepresentation of rare heterozygous SNVs and CNVs in PRKN. In particular, heterozygous pathogenic PRKN variants were not significantly more frequent in controls than in PD cases. Homozygous or compound heterozygous variants in this gene were the most common cause of EOPD (Kilarski et al., 2012), while heterozygous loss of PRKN function may be a potential risk factor for developing PD (Klein et al., 2007; Huttenlocher et al., 2015; Castelo Rueda et al., 2021; Lubbe et al., 2021) and therefore identifying individuals at increased risk might be useful in the prodromal phase. However, this role of heterozygous PRKN is still under debate, as previous reports suggested a lack of association with PD (Kay et al., 2010). Recently, in a larger association study, Yu and colleagues fully sequenced PRKN in a PD cases/controls cohort from European ancestry, including 1965 late-onset and 553 early-onset, and concluded that heterozygous SNVs or CNVs in PRKN are not associated with EOPD (Yu et al., 2021). They reported that 1.52% of PD and 1.8% of controls were carriers. Here, using a SNP array based on CNVs and SNPs screening, we showed similar percentages (1.64% of PD and 0.82% of controls) with non-significant differences between controls and mainly late-onset PD cases.
Potential neuroprotective PD therapies and clinical trials are now targeting specific PD subtypes based on genetic markers causing or increasing the disease risk, such as therapies targeting LRRK2, GBA1 and alpha-synuclein (Sardi et al., 2018). Parkin-proved disease is characterized by a slow motor progression, preserved cognition and a limited increase in dopaminergic medication over time (Menon et al., 2023). Moreover, severe loss of dopaminergic neurons was observed in homozygous PRKN carriers without Lewy bodies formation, which is one of the major markers of idiopathic PD (Mata, 2004). Confirming the potential role of heterozygous PRKN variants in the pathogenesis of PD will be crucial, despite the lack of data describing PD conversion of individuals carrying these genetic risk factors.
Beyond the effects of rare variants, we have demonstrated that individuals carrying the MAPT H1 haplotype are at higher risk to develop PD and PSP. These findings are consistent with previous studies that have assessed the H1 haplotype as a PD (Zabetian et al., 2007) and PSP (Baker et al., 1999) risk factor. We did not detect the association of PD or any forms of atypical Parkinsonism with H1c sub-haplotype which was strongly associated with risk for PSP and CBD (Myers et al., 2005; Kouri et al., 2015) but not PD (Zabetian et al., 2007). Next, we estimated the total cumulative contribution of common low-risk SNVs by calculating the PRS. Our PRS model of disease risk showed an expected trend similar to previous reports showing that PRS discriminates PD cases from unaffected individuals (Dehestani et al., 2021). Several polygenic analyses have become standard tools for assessing the risk for complex disorders and an accurate method for predicting disease status and identifying high-risk individuals (Lewis and Vassos, 2020). Next, we looked up at how thousands of biological pathways might contribute to the risk of developing PD. In addition to pathways already associated with PD and AD, molecular processes underlying proteins metabolism, signal transduction and post-translational protein modification were among the most important contributors to PD risk. The metabolic dysfunction, energy failure and redox imbalance observed in PD were considered obvious features to qualify PD as a complex metabolic disorder (Anandhan et al., 2017). In addition, disruption of any stage in the protein life cycle could engender PD pathology (Langston and Cookson, 2020). Comparing our results with a previous large-scale gene set-specific PRS studies that reported the involvement of multiple processes in the etiology of PD (Bandres-Ciga et al., 2020), similar molecular processes were found here. However, other processes such as immune response, synaptic transmission and endosomal-lysosomal dysfunction were not highlighted which may be due to the smaller sample size in our dataset. Pathway PRSs are expected to provide important insights into the complex heterogeneity of PD and how patients respond to treatment, by generating biologically traceable therapeutic targets from polygenic signals (Choi et al., 2023). We are aware that our study has several limitations: (1) the sample size was not large enough to have sufficient statistical power to perform further analysis, such as GWAS for PD risk or AAO, genome-wide CNV burden or human leukocyte antigen (HLA) association; (2) not all known variants associated with PD can be accurately assessed by the NeuroChip and we might have missed some mutated alleles, even though we confirmed all the identified variants by Sanger sequencing; (3) we used best practices to call CNVs from genotyping data (Sarihan et al., 2021), and thus we will always miss small CNVs that are systematically filtered out. Moreover, we could validate only few of the called CNVs with MLPA and (4) our analysis revealed a higher incidence of first-degree family history among controls. Therefore, caution must be exercised when searching for recessive disease forms. Although proxy cases have proven their effectiveness in large scale study investigating common variants (Nalls et al., 2019) and have also highlighted their usefulness in detecting rare variants (Makarious et al., 2023).
In conclusion, our study has successfully performed a comprehensive genetic baseline characterization of the Luxembourgish PD case–control cohort, investigating rare variants, CNVs and PRSs. Our findings do not support an association between PD risk and rare heterozygous PRKN variants. We also described a possible role of LRRK2 in AP and new possible digenic inheritance patterns in PD. Together with other studies in different European populations, our findings will advance the understanding of PD pathogenesis and genetics and could redefine the development of future therapeutic targets and therapies.
Genetic and clinical data for this manuscript are not publicly available as they are linked to the internal regulations of the Luxembourg Parkinson’s Study. Requests for accessing the datasets can be directed to request bmNlci1wZEB1bmkubHU=. The code behind the analyses is available under Apache-2.0 license. More information on how to request access to the data and the code is available at https://doi.org/10.17881/ea65-t027.
The studies involving humans were approved by the National Research Ethics Committee (CNER Ref: 201407/13). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Geeta Acharya, Gloria Aguayo, Myriam Alexandre, Wim Ammerlann, Katy Beaumont, Camille Bellora, Jessica Calmes, Lorieza Castillo, Gessica Contesotto, Daniela Esteves, Guy Fagherazzi, Jean-Yves Ferrand, Manon Gantenbein, Marijus Giraitis, Jérôme Graas, Gaël Hammot, Anne-Marie Hanff, Estelle Henry, Michael Heymann, Alexander Hundt, Sonja Jónsdóttir, Jochen Klucken, Rejko Krüger, Pauline Lambert, Victoria Lorentz, Paula Cristina Lupu, Guilherme Marques, Deborah Mcintyre, Chouaib Mediouni, Myriam Menster, Maura Minelli, Ulf Nehrbass, Fozia Noor, Magali Perquin, Rosalina Ramos Lima, Eduardo Rosales, Estelle Sandt, Margaux Schmitt, Amir Sharify, Kate Sokolowska, Hermann Thien, Johanna Trouet, Olena Tsurkalenko, Michel Vaillant, Mesele Valenti, Luxembourg Institute of Health, Strassen, Luxembourg; Muhammad Ali, Giuseppe Arena, Rudi Balling, Michele Bassis, Regina Becker, Ibrahim Boussaad, Kathrin Brockmann, Piotr Gawron, Soumyabrata Ghosh, Enrico Glaab, Elisa Gómez De Lope, Valentin Groues, Anne Grünewald, Wei Gu, Linda Hansen, Michael Heneka, Sascha Herzinger, Jacek Jaroslaw Lebioda, Yohan Jaroz, Quentin Klopfenstein, Jochen Klucken, Rejko Krüger, Zied Landoulsi, Tainá M. Marques, Patricia Martins Conde, Patrick May, Francoise Meisch, Michel Mittelbronn, Michel Mittelbronn, Sarah Nickels, Marek Ostaszewski, Clarissa P. C. Gomes, Sinthuja Pachchek, Claire Pauly, Laure Pauly, Lukas Pavelka, Armin Rauschenberger, Rajesh Rawal, Dheeraj Reddy Bobbili, Kirsten Roomp, Isabel Rosety, Stefano Sapienza, Venkata Satagopam, Sabine Schmitz, Reinhard Schneider, Jens Schwamborn, Ekaterina Soboleva, Rebecca Ting Jiin Loo, Christophe Trefois, Carlos Vega, Maharshi Vyas, Paul Wilmes, Evi Wollscheid-Lengeling, Luxembourg Centre for Systems Biomedicine, University of Luxembourg, Esch-sur-Alzette, Luxembourg; Guy Berchem, Nico Diederich, Marijus Giraitis, Linda Hansen, Jochen Klucken, Rejko Krüger, Laura Longhino, Romain Nati, Beatrice Nicolai, Claire Pauly, Lukas Pavelka, Elodie Thiry, Liliana Vilas Boas, Gelani Zelimkhanov, Centre Hospitalier de Luxembourg, Strassen, Luxembourg; Michel Mittelbronn, Friedrich Mühlschlegel, Laboratoire National de Santé, Dudelange, Luxembourg; Alexandre Bisdorff, Rene Dondelinger, Centre Hospitalier Emile Mayrisch, Esch-sur-Alzette, Luxembourg; Sylvia Herbrink, Centre Hospitalier du Nord, Ettelbrück, Luxembourg; Roseline Lentz, Parkinson Luxembourg Association, Leudelange, Luxembourg; Michele Hu, Oxford Parkinson’s Disease Centre, Nuffield Department of Clinical Neurosciences, University of Oxford, Oxford, United Kingdom; Richard Wade-Martins, Oxford Parkinson’s Disease Centre, Department of Physiology, Anatomy and Genetics, University of Oxford, Oxford, United Kingdom; Clare Mackay, Oxford Centre for Human Brain Activity, Wellcome Centre for Integrative Neuroimaging, Department of Psychiatry, University of Oxford, Oxford, United Kingdom; Daniela Berg, Kathrin Brockmann, Thomas Gasser, Inga Liepelt, Center of Neurology and Hertie Institute for Clinical Brain Research, Department of Neurodegenerative Diseases, University Hospital Tübingen, Tübingen, Germany; Brit Mollenhauer, Paracelsus-Elena-Klinik, Kassel, Germany; Katrin Marcus, Ruhr-University of Bochum, Bochum, Germany; Robert Liszka, Westpfalz-Klinikum GmbH, Kaiserslautern, Germany; Walter Maetzler, Department of Neurology, University Medical Center Schleswig-Holstein, Kiel, Germany; Mariella Graziano, Association of Physiotherapists in Parkinson’s Disease Europe, Esch-sur-Alzette, Luxembourg; Nadine Jacoby, Private Practice, Ettelbruck, Luxembourg; Jean-Paul Nicolay, Private Practice, Luxembourg, Luxembourg.
ZL: Conceptualization, Data curation, Investigation, Methodology, Writing – original draft. SP: Conceptualization, Data curation, Investigation, Methodology, Writing – review & editing. DB: Data curation, Writing – review & editing. LP: Data curation, Writing – review & editing. PM: Conceptualization, Supervision, Writing – review & editing, Investigation, Methodology. RK: Conceptualization, Supervision, Writing – review & editing, Investigation, Methodology.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The National Centre of Excellence in Research on Parkinson’s Disease (NCER-PD) is funded by the Luxembourg National Research Fund (FNR/NCER13/BM/11264123), the PEARL program (FNR/P13/6682797 to RK), MotaSYN (12719684 to RK), MAMaSyn (to RK), MiRisk-PD (C17/BM/11676395 to RK and PM), the FNR/DFG Core INTER (ProtectMove, FNR11250962 to PM and ZL), and the PARK-QC DTU (PRIDE17/12244779/PARK-QC to RK and SP).
We would like to thank all participants of the Luxembourg Parkinson’s Study for their important support of our research. Furthermore, we acknowledge the joint effort of the National Centre of Excellence in Research on Parkinson’s Disease (NCER-PD).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnagi.2023.1282174/full#supplementary-material
Ahmad, A., Nkosi, D., and Iqbal, M. A. (2023). PARK2 microdeletion or duplications have been implicated in different neurological disorders including early onset Parkinson disease. Genes 14:600. doi: 10.3390/genes14030600
Anandhan, A., Jacome, M. S., Lei, S., Hernandez-Franco, P., Pappa, A., Panayiotidis, M. I., et al. (2017). Metabolic dysfunction in Parkinson’s disease: bioenergetics, redox homeostasis and central carbon metabolism. Brain Res. Bull. 133, 12–30. doi: 10.1016/j.brainresbull.2017.03.009
Arena, G., and Valente, E. M. (2017). PINK1 in the limelight: multiple functions of an eclectic protein in human health and disease: functions of PINK1 in human pathology. J. Pathol. 241, 251–263. doi: 10.1002/path.4815
Baker, M., Litvan, I., Houlden, H., Adamson, J., Dickson, D., Perez-Tur, J., et al. (1999). Association of an extended haplotype in the tau gene with progressive supranuclear palsy. Hum. Mol. Genet. 8, 711–715. doi: 10.1093/hmg/8.4.711
Bandres-Ciga, S., Saez-Atienzar, S., Kim, J. J., Makarious, M. B., Faghri, F., Diez-Fairen, M., et al. (2020). Large-scale pathway specific polygenic risk and transcriptomic community network analysis identifies novel functional pathways in Parkinson disease. Acta Neuropathol. 140, 341–358. doi: 10.1007/s00401-020-02181-3
Blauwendraat, C., Faghri, F., Pihlstrom, L., Geiger, J. T., Elbaz, A., Lesage, S., et al. (2017). NeuroChip, an updated version of the NeuroX genotyping platform to rapidly screen for variants associated with neurological diseases. Neurobiol. Aging 57, 247.e9–247.e13. doi: 10.1016/j.neurobiolaging.2017.05.009
Blauwendraat, C., Nalls, M. A., and Singleton, A. B. (2020). The genetic architecture of Parkinson’s disease. Lancet Neurol. 19, 170–178. doi: 10.1016/S1474-4422(19)30287-X
Bloem, B. R., Okun, M. S., and Klein, C. (2021). Parkinson’s disease. Lancet 397, 2284–2303. doi: 10.1016/S0140-6736(21)00218-X
Castelo Rueda, M. P., Raftopoulou, A., Gögele, M., Borsche, M., Emmert, D., Fuchsberger, C., et al. (2021). Frequency of heterozygous parkin (PRKN) variants and penetrance of Parkinson’s disease risk markers in the population-based CHRIS cohort. Front. Neurol. 12:706145. doi: 10.3389/fneur.2021.706145
Chang, C. C., Chow, C. C., Tellier, L. C., Vattikuti, S., Purcell, S. M., and Lee, J. J. (2015). Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaSci 4:7. doi: 10.1186/s13742-015-0047-8
Choi, S. W., García-González, J., Ruan, Y., Wu, H. M., Porras, C., Johnson, J., et al. (2023). PRSet: pathway-based polygenic risk score analyses and software. PLoS Genet. 19:e1010624. doi: 10.1371/journal.pgen.1010624
Choi, S. W., and O’Reilly, P. F. (2019). PRSice-2: polygenic risk score software for biobank-scale data. GigaScience 8:giz082. doi: 10.1093/gigascience/giz082
Collins, R. L., Brand, H., Karczewski, K. J., Zhao, X., Alföldi, J., Francioli, L. C., et al. (2020). A structural variation reference for medical and population genetics. Nature 581, 444–451. doi: 10.1038/s41586-020-2287-8
Correia Guedes, L., Ferreira, J. J., Rosa, M. M., Coelho, M., Bonifati, V., and Sampaio, C. (2010). Worldwide frequency of G2019S LRRK2 mutation in Parkinson’s disease: a systematic review. Parkinsonism Relat. Disord. 16, 237–242. doi: 10.1016/j.parkreldis.2009.11.004
Dächsel, J. C., Mata, I. F., Ross, O. A., Taylor, J. P., Lincoln, S. J., Hinkle, K. M., et al. (2006). Digenic parkinsonism: investigation of the synergistic effects of PRKN and LRRK2. Neurosci. Lett. 410, 80–84. doi: 10.1016/j.neulet.2006.06.068
Das, S., Forer, L., Schönherr, S., Sidore, C., Locke, A. E., Kwong, A., et al. (2016). Next-generation genotype imputation service and methods. Nat. Genet. 48, 1284–1287. doi: 10.1038/ng.3656
Dehestani, M., Liu, H., and Gasser, T. (2021). Polygenic risk scores contribute to personalized medicine of Parkinson’s disease. JPM 11:1030. doi: 10.3390/jpm11101030
Dorsey, E. R., and Bloem, B. R. (2018). The Parkinson pandemic—a call to action. JAMA Neurol. 75:9. doi: 10.1001/jamaneurol.2017.3299
Elfferich, P., Verleun-Mooijman, M. C., Maat-Kievit, J. A., van de Warrenburg, B. P. C., Abdo, W. F., Eshuis, S. A., et al. (2011). Breakpoint mapping of 13 large parkin deletions/duplications reveals an exon 4 deletion and an exon 7 duplication as founder mutations. Neurogenetics 12, 263–271. doi: 10.1007/s10048-011-0302-9
Funayama, M., Li, Y., Tsoi, T., Lam, C., Ohi, T., Yazawa, S., et al. (2008). Familial parkinsonism with digenic parkin and PINK1 mutations. Mov. Disord. 23, 1461–1465. doi: 10.1002/mds.22143
Gilman, S., Wenning, G. K., Low, P. A., Brooks, D. J., Mathias, C. J., Trojanowski, J. Q., et al. (2008). Second consensus statement on the diagnosis of multiple system atrophy. Neurology 71, 670–676. doi: 10.1212/01.wnl.0000324625.00404.15
Haugarvoll, K., and Wszolek, Z. K. (2009). Clinical features of LRRK2 parkinsonism. Parkinsonism Relat. Disord. 15, S205–S208. doi: 10.1016/S1353-8020(09)70815-6
Hayashida, A., Li, Y., Yoshino, H., Daida, K., Ikeda, A., Ogaki, K., et al. (2021). The identified clinical features of Parkinson’s disease in homo-, heterozygous and digenic variants of PINK1. Neurobiol. Aging 97, 146.e1–146.e13. doi: 10.1016/j.neurobiolaging.2020.06.017
Healy, D. G., Falchi, M., O’Sullivan, S. S., Bonifati, V., Durr, A., Bressman, S., et al. (2008). Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol. 7, 583–590. doi: 10.1016/S1474-4422(08)70117-0
Hipp, G., Vaillant, M., Diederich, N. J., Roomp, K., Satagopam, V. P., Banda, P., et al. (2018). The Luxembourg Parkinson’s study: a comprehensive approach for stratification and early diagnosis. Front. Aging Neurosci. 10:326. doi: 10.3389/fnagi.2018.00326
Huttenlocher, J., Stefansson, H., Steinberg, S., Helgadottir, H. T., Sveinbjörnsdóttir, S., Riess, O., et al. (2015). Heterozygote carriers for CNVs in PARK2 are at increased risk of Parkinson’s disease. Hum. Mol. Genet. 24, 5637–5643. doi: 10.1093/hmg/ddv277
Ioannidis, N. M., Rothstein, J. H., Pejaver, V., Middha, S., McDonnell, S. K., Baheti, S., et al. (2016). REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 99, 877–885. doi: 10.1016/j.ajhg.2016.08.016
Jabbari, E., Koga, S., Valentino, R. R., Reynolds, R. H., Ferrari, R., Tan, M. M. X., et al. (2021). Genetic determinants of survival in progressive supranuclear palsy: a genome-wide association study. Lancet Neurol. 20, 107–116. doi: 10.1016/S1474-4422(20)30394-X
Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alföldi, J., Wang, Q., et al. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443. doi: 10.1038/s41586-020-2308-7
Karimi-Moghadam, A., Charsouei, S., Bell, B., and Jabalameli, M. R. (2018). Parkinson disease from mendelian forms to genetic susceptibility: new molecular insights into the neurodegeneration process. Cell. Mol. Neurobiol. 38, 1153–1178. doi: 10.1007/s10571-018-0587-4
Kay, D. M., Stevens, C. F., Hamza, T. H., Montimurro, J. S., Zabetian, C. P., Factor, S. A., et al. (2010). A comprehensive analysis of deletions, multiplications, and copy number variations in PARK2. Neurology 75, 1189–1194. doi: 10.1212/WNL.0b013e3181f4d832
Kilarski, L. L., Pearson, J. P., Newsway, V., Majounie, E., Knipe, M. D. W., Misbahuddin, A., et al. (2012). Systematic review and UK-based study of PARK2 (parkin), PINK1, PARK7 (DJ-1) and LRRK2 in early-onset Parkinson’s disease: PARK2, PINK1, PARK7, LRRK2 in EOPD. Mov. Disord. 27, 1522–1529. doi: 10.1002/mds.25132
Kim, S. Y., Seong, M. W., Jeon, B. S., Kim, S. Y., Ko, H. S., Kim, J. Y., et al. (2012). Phase analysis identifies compound heterozygous deletions of the PARK2 gene in patients with early-onset Parkinson disease. Clin. Genet. 82, 77–82. doi: 10.1111/j.1399-0004.2011.01693.x
Klein, C., Lohmann-Hedrich, K., Rogaeva, E., Schlossmacher, M. G., and Lang, A. E. (2007). Deciphering the role of heterozygous mutations in genes associated with parkinsonism. Lancet Neurol. 6, 652–662. doi: 10.1016/S1474-4422(07)70174-6
Kouri, N., Ross, O. A., Dombroski, B., Younkin, C. S., Serie, D. J., Soto-Ortolaza, A., et al. (2015). Genome-wide association study of corticobasal degeneration identifies risk variants shared with progressive supranuclear palsy. Nat. Commun. 6:7247. doi: 10.1038/ncomms8247
La Cognata, V., Morello, G., D’Agata, V., and Cavallaro, S. (2017). Copy number variability in Parkinson’s disease: assembling the puzzle through a systems biology approach. Hum. Genet. 136, 13–37. doi: 10.1007/s00439-016-1749-4
Landrum, M. J., Lee, J. M., Riley, G. R., Jang, W., Rubinstein, W. S., Church, D. M., et al. (2014). ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 42, D980–D985. doi: 10.1093/nar/gkt1113
Langston, R. G., and Cookson, M. R. (2020). Pathways of protein synthesis and degradation in PD pathogenesis. Prog. Brain Res. 252, 217–270. doi: 10.1016/bs.pbr.2020.01.002
Lesage, S., and Brice, A. (2009). Parkinson’s disease: from monogenic forms to genetic susceptibility factors. Hum. Mol. Genet. 18, R48–R59. doi: 10.1093/hmg/ddp012
Lewis, C. M., and Vassos, E. (2020). Polygenic risk scores: from research tools to clinical instruments. Genome Med. 12:44. doi: 10.1186/s13073-020-00742-5
Litvan, I., Agid, Y., Calne, D., Campbell, G., Dubois, B., Duvoisin, R. C., et al. (1996). Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop*. Neurology 47, 1–9. doi: 10.1212/WNL.47.1.1
Litvan, I., Bhatia, K. P., Burn, D. J., Goetz, C. G., Lang, A. E., McKeith, I., et al. (2003). SIC task force appraisal of clinical diagnostic criteria for parkinsonian disorders. Mov. Disord. 18, 467–486. doi: 10.1002/mds.10459
Liu, X., Cheng, R., Ye, X., Verbitsky, M., Kisselev, S., Mejia-Santana, H., et al. (2013). Increased rate of sporadic and recurrent rare genic copy number variants in Parkinson’s disease among Ashkenazi Jews. Mol. Genet. Genomic Med. 1, 142–154. doi: 10.1002/mgg3.18
Lubbe, S. J., Bustos, B. I., Hu, J., Krainc, D., Joseph, T., Hehir, J., et al. (2021). Assessing the relationship between monoallelic PRKN mutations and Parkinson’s risk. Hum. Mol. Genet. 30, 78–86. doi: 10.1093/hmg/ddaa273
MacDonald, J. R., Ziman, R., Yuen, R. K. C., Feuk, L., and Scherer, S. W. (2014). The database of genomic variants: a curated collection of structural variation in the human genome. Nucl. Acids Res. 42, D986–D992. doi: 10.1093/nar/gkt958
Macnee, M., Pérez-Palma, E., Brünger, T., Klöckner, C., Platzer, K., Stefanski, A., et al. (2023). CNV-ClinViewer: enhancing the clinical interpretation of large copy-number variants online. Bioinformatics 39:btad290. doi: 10.1093/bioinformatics/btad290
Makarious, M. B., Lake, J., Pitz, V., Ye Fu, A., Guidubaldi, J. L., Solsberg, C. W., et al. (2023). Large-scale rare variant burden testing in Parkinson’s disease. Brain 146, 4622–4632. doi: 10.1093/brain/awad214
Manichaikul, A., Mychaleckyj, J. C., Rich, S. S., Daly, K., Sale, M., and Chen, W.-M. (2010). Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873. doi: 10.1093/bioinformatics/btq559
Mata, I. F. (2004). Parkin genetics: one model for Parkinson’s disease. Hum. Mol. Genet. 13, 127R–133R. doi: 10.1093/hmg/ddh089
McKeith, I. G., Dickson, D. W., Lowe, J., Emre, M., O’Brien, J. T., Feldman, H., et al. (2005). Diagnosis and management of dementia with Lewy bodies: third report of the DLB consortium. Neurology 65, 1863–1872. doi: 10.1212/01.wnl.0000187889.17253.b1
Menon, P. J., Sambin, S., Criniere-Boizet, B., Courtin, T., Casse, F., Tesson, C., et al. (2023). Genotype-Phenotype correlation in Parkin-Parkinson’s disease (P3-11.012). Presented at the 2023 Annual Meeting Abstracts, Lippincott Williams & Wilkins, p. 2970.
Myers, A. J., Kaleem, M., Marlowe, L., Pittman, A. M., Lees, A. J., Fung, H. C., et al. (2005). The H1c haplotype at the MAPT locus is associated with Alzheimer’s disease. Hum. Mol. Genet. 14, 2399–2404. doi: 10.1093/hmg/ddi241
Nalls, M. A., Blauwendraat, C., Vallerga, C. L., Heilbron, K., Bandres-Ciga, S., Chang, D., et al. (2019). Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 18, 1091–1102. doi: 10.1016/S1474-4422(19)30320-5
Neary, D., Snowden, J. S., Gustafson, L., Passant, U., Stuss, D., Black, S., et al. (1998). Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 51, 1546–1554. doi: 10.1212/WNL.51.6.1546
Pachchek, S., Landoulsi, Z., Pavelka, L., Schulte, C., Buena-Atienza, E., Gross, C., et al. (2023). Accurate long-read sequencing identified GBA1 as major risk factor in the Luxembourgish Parkinson’s study. NPJ Parkinsons Dis 9:156. doi: 10.1038/s41531-023-00595-w
Pankratz, N., Dumitriu, A., Hetrick, K. N., Sun, M., Latourelle, J. C., Wilk, J. B., et al. (2011). Copy number variation in familial Parkinson disease. PLoS One 6:e20988. doi: 10.1371/journal.pone.0020988
Pavelka, L., Rauschenberger, A., Landoulsi, Z., Pachchek, S., May, P., Glaab, E., et al. (2022). Publisher correction: age at onset as stratifier in idiopathic Parkinson’s disease – effect of ageing and polygenic risk score on clinical phenotypes. NPJ Parkinsons Dis. 8:112. doi: 10.1038/s41531-022-00378-9
Pennings, M., Meijer, R. P. P., Gerrits, M., Janssen, J., Pfundt, R., De Leeuw, N., et al. (2023). Copy number variants from 4800 exomes contribute to ~7% of genetic diagnoses in movement disorders, muscle disorders and neuropathies. Eur. J. Hum. Genet. 31, 654–662. doi: 10.1038/s41431-023-01312-0
Pittman, A. M. (2005). Linkage disequilibrium fine mapping and haplotype association analysis of the tau gene in progressive supranuclear palsy and corticobasal degeneration. J. Med. Genet. 42, 837–846. doi: 10.1136/jmg.2005.031377
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J., and Kircher, M. (2019). CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47, D886–D894. doi: 10.1093/nar/gky1016
Sardi, S. P., Cedarbaum, J. M., and Brundin, P. (2018). Targeted therapies for Parkinson’s disease: from genetics to the clinic: targeted therapies for PD. Mov. Disord. 33, 684–696. doi: 10.1002/mds.27414
Sarihan, E. I., Pérez-Palma, E., Niestroj, L., Loesch, D., Inca-Martinez, M., Horimoto, A. R. V. R., et al. (2021). Genome-wide analysis of copy number variation in Latin American Parkinson’s disease patients. Mov. Disord. 36, 434–441. doi: 10.1002/mds.28353
Schüle, B., Hatchwell, E., Eis, P. S., and Langston, J. W. (2015). Comparative genomic hybridization solves a 14-year-old PARKIN mystery. Ann. Neurol. 78, 663–664. doi: 10.1002/ana.24478
Shu, L., Zhang, Y., Sun, Q., Pan, H., and Tang, B. (2019). A comprehensive analysis of population differences in LRRK2 variant distribution in Parkinson’s disease. Front. Aging Neurosci. 11:13. doi: 10.3389/fnagi.2019.00013
Skipper, L., Wilkes, K., Toft, M., Baker, M., Lincoln, S., Hulihan, M., et al. (2004). Linkage disequilibrium and association of MAPT H1 in Parkinson disease. Am. J. Hum. Genet. 75, 669–677. doi: 10.1086/424492
Tang, B., Xiong, H., Sun, P., Zhang, Y., Wang, D., Hu, Z., et al. (2006). Association of PINK1 and DJ-1 confers digenic inheritance of early-onset Parkinson’s disease. Hum. Mol. Genet. 15, 1816–1825. doi: 10.1093/hmg/ddl104
The 1000 Genomes Project Consortium (2012). An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65. doi: 10.1038/nature11632
Toft, M., and Ross, O. A. (2010). Copy number variation in Parkinson’s disease. Genome Med. 2:62. doi: 10.1186/gm183
Wang, K., Li, M., Hadley, D., Liu, R., Glessner, J., Grant, S. F. A., et al. (2007). PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 17, 1665–1674. doi: 10.1101/gr.6861907
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38:e164. doi: 10.1093/nar/gkq603
Wen, Y., Zhou, Y., Jiao, B., and Shen, L. (2021). Genetics of progressive supranuclear palsy: a review. JPD 11, 93–105. doi: 10.3233/JPD-202302
Yu, E., Rudakou, U., Krohn, L., Mufti, K., Ruskey, J. A., Asayesh, F., et al. (2021). Analysis of heterozygous PRKN variants and copy-number variations in Parkinson’s disease. Mov. Disord. 36, 178–187. doi: 10.1002/mds.28299
Keywords: Parkinson’s disease, genetics, Luxembourg, polygenic risk score, copy number variants
Citation: Landoulsi Z, Pachchek S, Bobbili DR, Pavelka L, May P, Krüger R and the NCER-PD Consortium (2023) Genetic landscape of Parkinson’s disease and related diseases in Luxembourg. Front. Aging Neurosci. 15:1282174. doi: 10.3389/fnagi.2023.1282174
Edited by:
Bogdan O. Popescu, Carol Davila University of Medicine and Pharmacy, RomaniaCopyright © 2023 Landoulsi, Pachchek, Bobbili, Pavelka, May, Krüger and the NCER-PD Consortium. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Patrick May, cGF0cmljay5tYXlAdW5pLmx1; Rejko Krüger, cmVqa28ua3J1ZWdlckBsaWgubHU=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.