 Ella Dunn
Ella Dunn Biqin Zhang
Biqin Zhang Virender K. Sahota
Virender K. Sahota Hrvoje Augustin
Hrvoje Augustin- Department of Biological Sciences, Centre for Biomedical Sciences, Royal Holloway University of London, Egham, United Kingdom
Neurodegenerative diseases are a large class of neurological disorders characterized by progressive dysfunction and death of neurones. Examples include Alzheimer’s disease, Parkinson’s disease, frontotemporal dementia, and amyotrophic lateral sclerosis. Aging is the primary risk factor for neurodegeneration; individuals over 65 are more likely to suffer from a neurodegenerative disease, with prevalence increasing with age. As the population ages, the social and economic burden caused by these diseases will increase. Therefore, new therapies that address both aging and neurodegeneration are imperative. Ketogenic diets (KDs) are low carbohydrate, high-fat diets developed initially as an alternative treatment for epilepsy. The classic ketogenic diet provides energy via long-chain fatty acids (LCFAs); naturally occurring medium chain fatty acids (MCFAs), on the other hand, are the main components of the medium-chain triglyceride (MCT) ketogenic diet. MCT-based diets are more efficient at generating the ketone bodies that are used as a secondary energy source for neurones and astrocytes. However, ketone levels alone do not closely correlate with improved clinical symptoms. Recent findings suggest an alternative mode of action for the MCFAs, e.g., via improving mitochondrial biogenesis and glutamate receptor inhibition. MCFAs have been linked to the treatment of both aging and neurodegenerative disease via their effects on metabolism. Through action on multiple disease-related pathways, MCFAs are emerging as compounds with notable potential to promote healthy aging and ameliorate neurodegeneration. MCFAs have been shown to stimulate autophagy and restore mitochondrial function, which are found to be disrupted in aging and neurodegeneration. This review aims to provide insight into the metabolic benefits of MCFAs in neurodegenerative disease and healthy aging. We will discuss the use of MCFAs to combat dysregulation of autophagy and mitochondrial function in the context of “normal” aging, Parkinson’s disease, amyotrophic lateral sclerosis and Alzheimer’s disease.
1. Introduction
Neurodegeneration is defined as the progressive dysfunction, structural impairment, and eventual death of neuronal cells. Neurodegenerative diseases (NDs) are predominantly adult-onset pathologies that can have familial (genetic) or sporadic causes and form a large class of neurological disorders that are the main cause of disability and the second leading cause of death worldwide (Erkkinen et al., 2018; Bannick et al., 2019). Globally, the most common NDs are Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS).
The ketogenic diet is a low-carbohydrate, high-fat diet. The classic KD utilizes long chain triglycerides (LCTs)–consisting of long chain fatty acids (LCFAs)–to provide between 60 and 80% of dietary energy. The alternative type of ketogenic diet relies on medium chain triglycerides (MCTs) that are hydrolyzed to medium chain fatty acids (MCFAs) as a primary energy source (Neal, 2017). LCFAs and MCFAs differ by the number of carbon atoms: LCFAs have between 16 and 20 carbon atoms, whereas MCFAs have 6–12 (Neal, 2017; Table 1). After hydrolysis of triglycerides to fatty acids in the gut, ketone bodies (known as ketones) are generated in the liver via β-oxidation. Compared to LCFAs, MCFAs are more rapidly metabolized and generate ketones more efficiently, allowing for a more palatable diet due to the higher carbohydrate content (Huttenlocher et al., 1971).
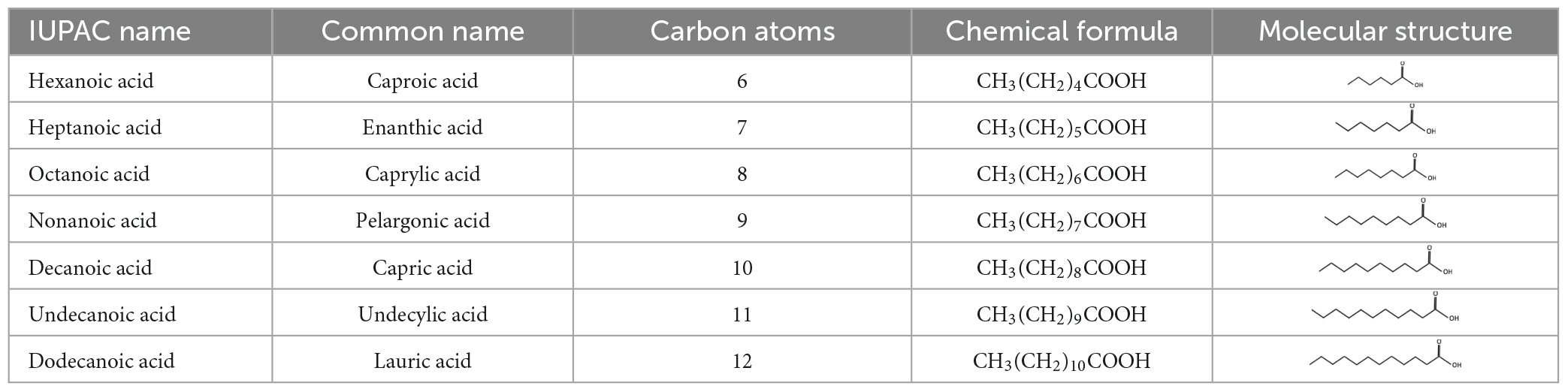
Table 1. Medium chain fatty acids.
For almost a century, the ketogenic diet has been used in the treatment of drug-resistant epilepsy (Martin et al., 2016). Recent in vitro and in vivo studies suggest a wider, neuroprotective role of the KD in the context of cancer, neurodegenerative diseases, and normal, healthy aging (Moreno and Mobbs, 2017). The mechanisms of action of the KD are not fully understood but are likely related to improved mitochondrial function and energy metabolism, and enhanced autophagy.
In this review, we discuss how impaired autophagy and mitochondrial dynamics affect aging and pathogenesis of NDs, focusing on AD, PD, and ALS, and provide an overview of experimental evidence suggesting novel therapeutic avenues aimed at exploiting the beneficial effects of MCFAs on these processes (Table 2).
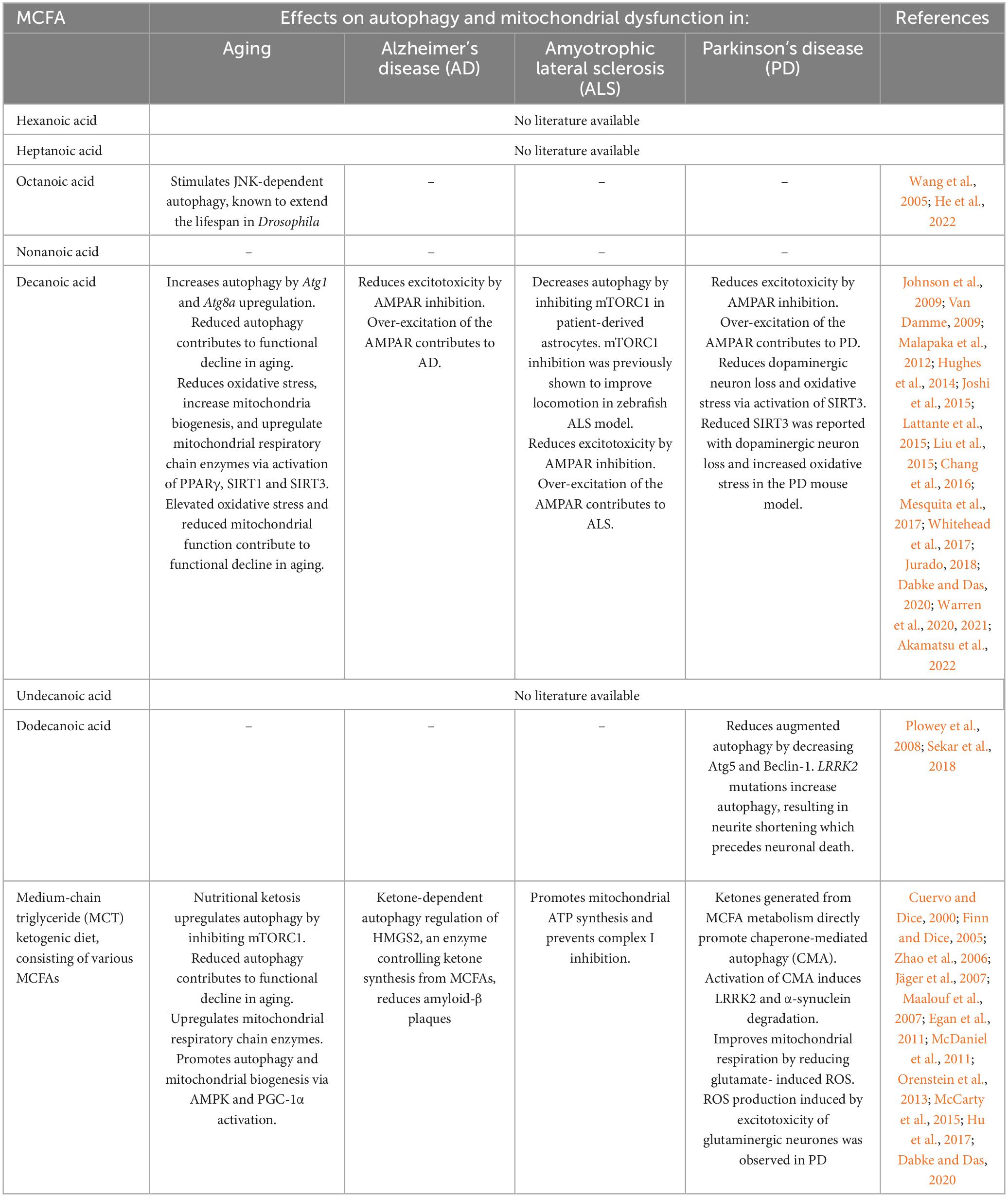
Table 2. The potential effects of medium chain fatty acids (MCFAs) on autophagy and mitochondrial function in aging and neurodegenerative diseases.
2. MCT-based diets, neurodegenerative diseases, and aging
2.1. Neurodegenerative diseases and “healthy” aging
Multiple disease-causing factors have been identified for AD, PD, and ALS, ranging from environmental to genetic. A pathological feature shared by all three NDs is the accumulation of protein inclusions that are believed to play a critical role in the onset of neurodegeneration (Longhena et al., 2017). AD is the most common ND, causing progressively severe and irreversible cognitive and physical decline. The major hallmarks of AD pathology are accumulation of β-amyloid plaques in the brain, and the hyperphosphorylation and aggregation of tau protein to form neurofibrillary tangles, both of which trigger neuronal cell death (Luque and Jaffe, 2009). The loss of dopaminergic neurones and the accumulation of Lewy bodies (LB), cytoplasmic inclusions consisting of α -synuclein in the brain, leads to PD, the fastest growing neurological disorder in the world (Volpicelli-Daley et al., 2014; Poewe et al., 2017; Raza et al., 2019). ALS, the most common form of motor neurone disease (MND), is characterized by a progressive, paralysis-causing loss of motor neurones in the brain and spinal cord (Hulisz, 2018) and the presence of inclusions containing TDP-43, a protein that becomes pathological when hyper-phosphorylated or hyper-ubiquinated (Lillo and Hodges, 2009). At the molecular level, human NDs are characterized by proteotoxic stress, oxidative stress, neuroinflammation, mitochondrial dysfunction, excitotoxicity and autophagy dysregulation (Dong et al., 2009; Dugger and Dickson, 2017). The convergence of biological processes implicated in different NDs indicates common mechanisms driving neurodegeneration. Currently, there are no treatments able to sufficiently alter any ND, signifying a need for further research and funding.
The greatest risk factor for all major NDs is aging. Biological aging is described as the gradual functional decline of cells, tissues, and organs, leading to physical, physiological, metabolic and psychological changes over time and increased vulnerability to death. The molecular and cellular processes underlying aging including genomic instability, telomere loss, epigenetic changes, compromised protein homeostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion and altered intercellular communication (López-Otín et al., 2013). A large body of experimental evidence suggests that amelioration of these hallmarks of aging may extend lifespan and improve the health status in various animal models of “healthy” aging and human diseases, prompting the search for genetic, pharmacological and dietary modifiers of these processes. Due to their ability to modulate many of the cellular mechanisms underlying aging and pathobiology of NDs, MCFAs emerged as a promising class of molecules for intervention into these processes.
2.2. MCT-based diet
The MCT diet relies on three main ketones to elicit metabolic effects: acetone, acetoacetate and β-hydroxybutyrate (BHB) (Schonfeld and Wojtczak, 2016). Ketone-based diets are known to modulate levels of GABA and glutamate, inhibit voltage dependent calcium channel regulation and alter mitochondrial function and ATP availability (Bough and Rho, 2007; Lutas and Yellen, 2013; Kadowaki et al., 2017). Under normal dietary conditions, glucose is utilized as a main energy source. In conditions where glucose is unavailable, such as in fasting conditions, ketones become the main energy source. Whereas ketone bodies can be neuroprotective (Kim et al., 2007; Yang et al., 2019), there is a lack of clear correlation between plasma ketone levels in mouse seizure models (Tan et al., 2017), and ex vivo studies indicate that fatty acids, rather than ketones, provide anti-seizure activity in rat models of epilepsy (Chang et al., 2016). These experimental data suggest the existence of additional modes of action through which ketogenic diets can exert their beneficial effects on the nervous system.
Octanoic acid and decanoic acid are medium chain fatty acids (MCFAs; Table 1) and components of the medium chain triglyceride (MCT) ketogenic diet. These MCFAs can provide an alternate energy source to glucose for neurones and astrocytes (Cunnane et al., 2020). NDs are associated with metabolic dysfunction, specifically with altered glucose and lipid metabolism in the brain, indicating MCFAs as a potential treatment option (Wlaz et al., 2012; Cunnane et al., 2020; Estes et al., 2021). Importantly, recent findings demonstrate a pharmacological mode of action for MCFAs through its inhibitory action on α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)-type glutamate receptors (Chang et al., 2016) and activation of the peroxisomal proliferator-activated receptor gamma (PPARγ) (Hughes et al., 2014).
Medium chain fatty acids and the MCT diet improve cognitive performance of patients suffering from AD and mild cognitive impairment (Krikorian et al., 2012; Taylor et al., 2018). In addition, MCFAs have been shown to improve mitochondrial function and promote autophagy, both of which are known to be dysregulated in aging and NDs (Rangaraju et al., 2019; Stavoe and Holzbaur, 2019).
Importantly, MCFAs can cross the blood brain barrier (BBB) (Wlaz et al., 2012, 2015), raising the possibility that MCFAs may also be used to treat conditions affecting the central nervous system, including AD, ALS, PD, and even some types of brain tumors (Castellano et al., 2015; Croteau et al., 2017; Tefera et al., 2017; Manzo et al., 2018; Zeng et al., 2019). The ability of MCFAs to traverse the BBB provides an advantage from a pharmacological perspective, especially as drug delivery to the nervous system can be problematic. MCFAs can be administered via multiple routes. Patients can adhere to the MCT diet by restricting carbohydrates and eating foods that supply them with the specific MCFAs (Grammatikopoulou et al., 2020). Ready-to-consume drink supplements are available, as well as pre-prepared ketogenic formulas and meals (Schoeler et al., 2021); patients may also choose to supplement their diet with capsules and oils containing the MCFAs they require (Grammatikopoulou et al., 2020).
2.3. Current uses of MCT-based diets, preclinical studies, and caveats
The MCT diet was originally developed as a treatment for epilepsy as starvation, a condition in which ketones are utilized as the main energy source, had long been observed to reduce the frequency of seizures (Martin et al., 2016). For patients that have developed resistance to the standard, pharmacological epilepsy treatments, the MCT diet is especially important as it provides an alternative treatment option (Loscher et al., 2020). Furthermore, an MCT diet containing only decanoic and octanoic acids has been shown to improve attention and structural connectivity in the brains of patients with mild cognitive impairment (Roy et al., 2022).
In pre-clinical trials, the classic ketogenic diet has been proven beneficial in the treatment of multiple diseases and disorders. For example, ketone-producing diet can prolong life and reduce seizures in mouse models of Dravet syndrome, an early onset childhood developmental disorder with severe epileptic encephalopathy (Jancovski et al., 2021). A reduction in anxiety is seen in rats on the MCT diet, along with an increase in social competitiveness (Hollis et al., 2018). The ketogenic diet is under assessment for treating multiple sclerosis, as a 3-day fasting cycle in which ketones are used as a primary source of energy appears to alleviate symptoms associated with MS-related autoimmunity (Choi et al., 2016). In mice, a ketogenic diet suppresses insulin resistance and inflammation in a high fat diet-induced model of obesity (Geng et al., 2016). The ketogenic diet is also beneficial in Duchenne muscular dystrophy, where muscle function was restored in a rat model (Fujikura et al., 2021). An oil-based MCT diet has also been used to treat hypertrophic cardiomyopathy in a patient with a deficiency in the gene coding for acyl-coA dehydrogenase (Pervaiz et al., 2011).
Although both OA and decanoic acid (DA) constitute the main fatty acids in the MCT-diet, they appear to have distinct roles. The effect of DA on mitochondrial function appears to be stronger when compared to OA (discussed later). In the brain, β-oxidation of OA is favored, whereas DA preferentially stimulates lactate production through glycolysis which is then utilized as an energy source by brain cells. β-oxidation of both OA and DA occurs through the action of carnitine palmitoyltransferase, (CPT) but it is thought that OA also undergoes oxidation that is independent of CPT (Khabbush et al., 2017). CPT levels in neurones is low, which may explain why DA is spared from oxidation and appears to be present at higher levels than OA.
Despite their advantages and clinical benefits, ketogenic diets should be administered with caution. As they mimic starvation conditions, patients must be very carefully monitored to avoid possible side effects such as acidosis and hypoglycemia (Sankar and Sotero de Menezes, 1999). Ketogenic dietary regimens are known to result in weight loss and can be very effective to help patients with obesity reach a healthy weight. However, if prescribed to an individual with a healthy weight, extra weight loss could be detrimental to the patients’ health (Bueno et al., 2013). The ketogenic diet is not recommended for patients with liver failure, porphyria, pyruvate kinase deficiency, carnitine translocase deficiency, primary carnitine deficiency, pancreatitis, and carnitine palmitoyl transferase deficiency (Maswood et al., 2020). The ketogenic diet also comes with a range of short-term side effects, commonly referred to as the “keto flu,” including fatigue, dizziness, nausea, vomiting and headaches (Maswood et al., 2020). Short-term symptoms usually subside when the body has adjusted to the diet, however, long-term side effects such as the build-up of kidney stones are not uncommon; in addition, most patients will require supplementation to prevent vitamin deficiencies (Wilong, 2007; Maswood et al., 2020). Recent clinical findings demonstrate a correlation between a long-term ketogenic diet and reduced bone density (Simm et al., 2017) and experiments in mice suggest a significant negative effect on bone health by octanoic acid, a major component of the MCT diet (Jain et al., 2021). Overall, following a ketogenic or MCT-based diet has the potential to compromise the nutritional status of some patients necessitating regular monitoring and nutrient supplementation.
Another important consideration is that some ketogenic diets are very hard to follow. Due to their restrictiveness, patients are limited on what they can eat, and compliance issues are very prominent. The retention rate can be partially improved by enhancing the taste of pre-prepared foods and increasing patient support (Tong et al., 2022).
3. Mitochondrial dysfunction in aging and neurodegenerative diseases
Mitochondria are responsible for generating most of the adenosine triphosphate (ATP) supply in the cell through oxidative respiration (Mookerjee et al., 2010). ATP production in mitochondria is achieved by transferring electrons from NADH and FADH2 generated by glycolysis and the Krebs cycle through Complexes I to IV of the Electron Transport Chain (ETC) (Martínez-Reyes and Chandel, 2020). Complex I, III, and IV use high energy electrons to pump protons to the intermembrane space creating a proton gradient across the inner mitochondria membrane. The transfer of protons back to the mitochondria matrix drives the production of ATP via Complex V (Zhao et al., 2019). Interestingly, muscle and brain tissues of aged mice fed a ketogenic diet exhibited increased levels of the Krebs cycle protein citrate synthase and Complex I and IV proteins (Zhou et al., 2021; Figure 1).
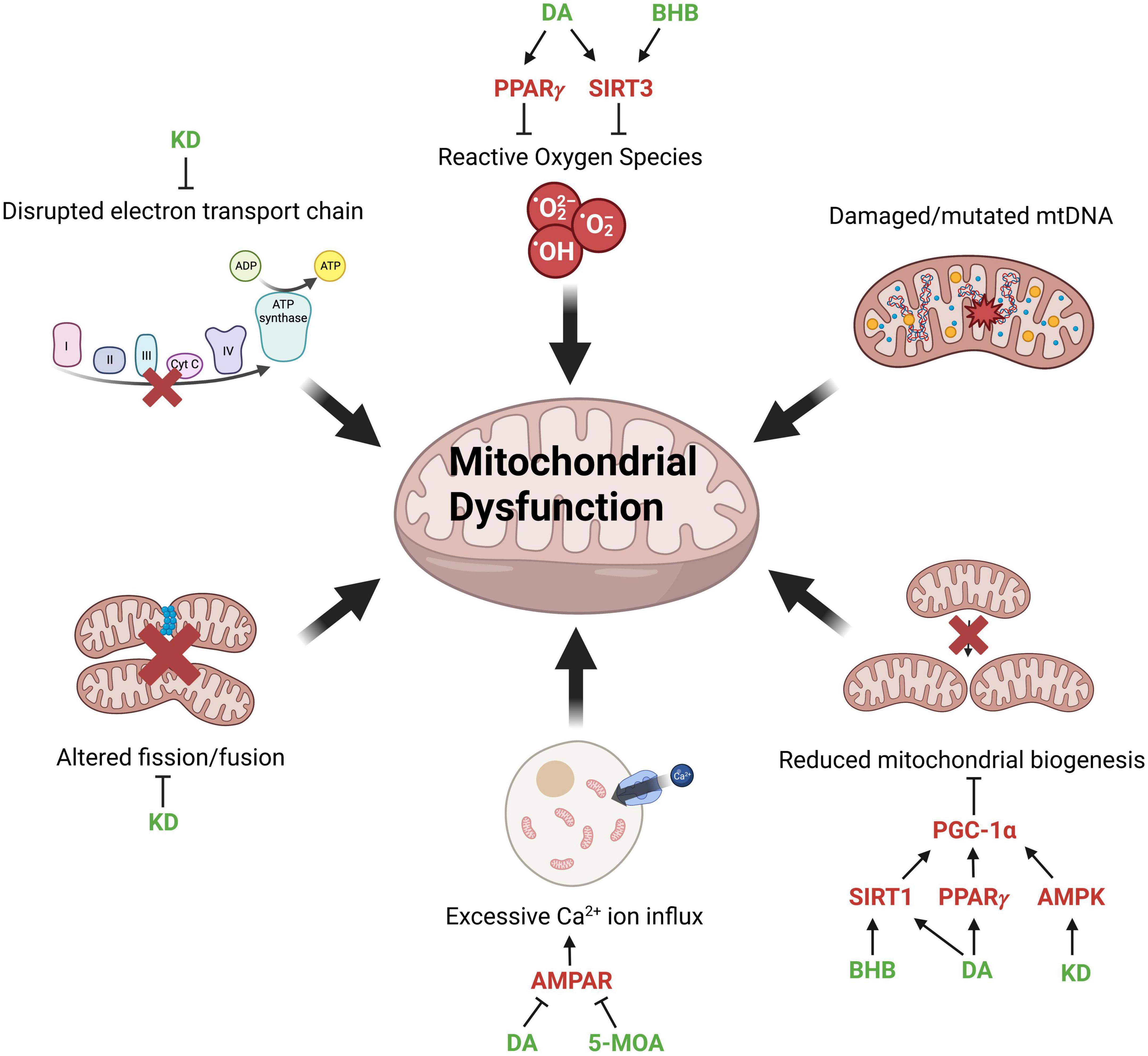
Figure 1. Factors contributing to mitochondrial dysfunction and potential points of intervention. Mitochondrial dysfunction associated with aging and neurodegenerative diseases and molecular modulators of mitochondrial function (red) through which MCFAs (medium-chain fatty acids), MCT (medium-chain triglyceride) diet or KD (ketogenic diet) (green) can potentially reverse the pathological phenotype. The production of ROSs (reactive oxygen species) is mitigated by DA (decanoic acid) and BHB (β-hydroxybutyrate) activating PPARγ (peroxisome proliferator-activated receptors gamma) and SIRT3 (sirtuin 3). Reduced mitochondrial biogenesis is restored by BHB, DA, and KD through the activation of SIRT1 (sirtuin 1), PPARγ, and AMPK (AMP-activated protein kinase), which subsequently activate PGC-1α (PPARγ coactivator 1-alpha) to promote mitochondrial biogenesis. Excessive calcium influx into cells is reduced by DA and 5-MOA (5-methyl octanoic acid), which inhibit AMPAR (AMPA-type glutamate receptors). KD reverses the disrupted mitochondrial dynamics and ETC (electron transport chain) function via inhibition of Drp1 (dynamin-related protein 1) and enhances the expression of ETC complexes, respectively. Created with BioRender.com.
Mitochondrial dysfunction is a hallmark of both NDs and normal, “healthy” aging (Figure 1). As an organism ages, mitochondrial ATP production declines due to increased electron leakage and consequent uncoupling of the ETC (Mookerjee et al., 2010). The brain requires a large amount of energy and neuronal cells have a limited glycolytic capability, making them extremely reliant on the energy produced by mitochondria and vulnerable to mitochondrial dysfunction (Moreira et al., 2009). Treatments that can improve mitochondrial output may therefore be beneficial in rescuing symptoms of aging and NDs. Mitochondria are dynamic and constantly undergo fission and fusion (Zhu et al., 2013). However, in AD, ALS, and PD, mitochondrial dynamics is disrupted, resulting in mitochondrial fragmentation and reduced energy output (Wang et al., 2009; Lim et al., 2012; Jiang et al., 2015). Mitochondrial fission is regulated by the dynamin-related protein 1 (Drp1). Interestingly, KD has been shown to promote mitochondrial integrity by suppressing the mitochondrial translocation of Drp1 and reducing ER stress (Guo et al., 2018; Figure 1). On the other hand, although damaged or mutated mtDNA contributes to mitochondrial dysfunction, there is minimal evidence suggesting that MCFAs are directly beneficial in restoring the structural integrity of mtDNA.
3.1. MCFAs may promote healthy aging by upregulating mitochondrial biogenesis and reducing oxidative stress
The 10-carbon DA can activate pathways or receptors related to mitochondrial function, such as peroxisome proliferator-activated receptors (PPARs) (Michalik et al., 2006; Malapaka et al., 2012). PPARs are involved in various physiological processes, including energy metabolism and inflammation (Varga et al., 2011). Mice fed with PPARγ agonists exhibited extended lifespan, improved cognitive ability, reduced inflammation, and enhanced mitochondrial function (Xu et al., 2020). Decanoic acid is identified as a modulator that directly binds and activates PPARγ (Malapaka et al., 2012). Moreover, DA stimulates complex I activity and increases mitochondrial number in cultured neurones through the regulation of PPARγ (Hughes et al., 2014; Figure 1).
Sirtuins are a family of proteins involved in stress response, epigenetic modification, cellular metabolism and longevity (Houtkooper et al., 2012). Three of the seven known sirtuins, SIRT3, SIRT 4, and SIRT5 are predominantly expressed in the mitochondria and believed to function as a link between metabolism and aging (Ji et al., 2022). SIRT1 and SIRT3 display a reduced expression with age (Donmez and Guarente, 2010; Jing et al., 2013), resulting in increased oxidative stress and reduced mitochondrial biogenesis due to reduced activation of PGC-1α (PPARγ coactivator 1-alpha), a key regulator of mitochondrial biogenesis (Onyango et al., 2002; Nemoto et al., 2005). Interestingly, cultured hippocampal neurones treated with BHB or DA show activation of SIRT1 and SIRT3, as well as upregulation of mitochondrial respiratory chain enzymes (Dabke and Das, 2020; Figure 1 and Table 2).
The AMP-activated protein kinase (AMPK), a critical sensor of intracellular ATP levels and regulator of energy homeostasis, is another protein known to decline with age (Reznick et al., 2007; Hardie et al., 2012; Herzig and Shaw, 2018). Age-related decline of AMPK results in reduced activation of PGC-1α, with a negative effect on mitochondrial biogenesis (Jäger et al., 2007; Figure 1) and autophagy (Egan et al., 2011). Little is known regarding the effect of MCFAs on AMPK activity, however, rats with ad libitum access to KD showed an elevated level of AMPK signaling in the liver (McDaniel et al., 2011), warranting further evaluation of the role of MCT-based diets in modulating this pathway.
3.2. MCFAs may improve symptoms of Alzheimer’s disease by promoting mitochondrial biogenesis
Multiple studies have linked mitochondrial dysfunction to Alzheimer’s disease (AD). PGC-1α, a crucial regulator of mitochondrial biogenesis, is suppressed in both AD models and patients (Qin et al., 2009; Sheng et al., 2012); SIRT1 expression and activity are also found to be reduced in the brains of AD patients (Julien et al., 2009; Lalla and Donmez, 2013). Reduced PGC-1 activation and downregulation of SIRT1 also promote amyloid-beta (Aβ) production, impaired Aβ clearance and tau hyperphosphorylation, resulting in the formation of Aβ plaques and neurofibrillary tangles (Qin et al., 2006; Min et al., 2010). The accumulation of Aβ has been shown to affect mitochondrial bioenergetics, dynamics, distribution and clearance in human cell cultures, yeast, fruit fly and mice (Iijima-Ando et al., 2009; Sinclair et al., 2021; Lee et al., 2022; Epremyan et al., 2023; Zyśk et al., 2023). Altered glucose metabolism is one of the hallmarks of AD (Hoyer, 1991; Schubert, 2005; Calsolaro and Edison, 2016). Due to its positive effect on glucose levels, KD has been proposed as a potential treatment for AD (Broom et al., 2019). Furthermore, KD-induced elevated ketones have been reported to have a neuroprotective effect by reducing oxidative stress in AD and PD models (Kashiwaya et al., 2000; Kim et al., 2007; Yang et al., 2019). Indeed, MCT diet improved cognitive function in AD patients with mild to moderate symptoms of AD (Ota et al., 2019). Another study also showed that patients who followed the MCT diet had an improved brain energy metabolism (Croteau et al., 2018). Although the underlying mechanisms remain unclear, it was proposed that the beneficial effect of the MCT diet on AD stems from the ability of the ketones to provide an alternative fuel source to the brain cells that are damaged or impaired due to metabolic dysfunction (Takeishi et al., 2021). Ketones in the brain are also shown to reduce inflammation and oxidative stress, two known contributors to the development of AD (Pinto et al., 2018). In addition, the brains of AD patients exhibit altered levels of mitochondrial respiratory complexes, along with impaired axonal transport and synaptic morphology, and increased ROS production and oxidative stress (Manczak et al., 2004; Rice et al., 2014; Pickett et al., 2018; Butterfield and Halliwell, 2019).
Synaptic dysfunction is one of the early signs of AD and other neurodegenerative diseases (Taoufik et al., 2018). AMPA-type glutamate receptors (AMPARs) are Ca2+ permeable channels that mediate excitatory synaptic transmission in the brain and contribute to glutamate receptor-mediated neurodegeneration (Greig et al., 2000; Palmer et al., 2005; Joshi et al., 2011). Over-excitation of the AMPAR leads to excitotoxicity, which is implicated in AD, ALS and PD (Johnson et al., 2009; Van Damme, 2009; Joshi et al., 2015; Whitehead et al., 2017; Jurado, 2018; Akamatsu et al., 2022), leading to the idea that inhibition of AMPARs could potentially improve the symptoms and progression of AD and other NDs. A recent study identified DA as a selective inhibitor of excitatory synapses in hippocampal slices and a non-competitive antagonist of AMPA receptors, explaining its anti-convulsant effect in the in vitro seizure model (Chang et al., 2016). OA, another key MCT diet component (Masino, 2022), does not inhibit AMPARs (Chang et al., 2016). However, its modified version, 5-methyl octanoic acid (5-MOA), inhibits AMPARs both in vitro and in vivo, suggesting potential novel therapeutic interventions for AD (Chang et al., 2015; Figure 1).
3.3. MCFAs could ameliorate symptoms of amyotrophic lateral sclerosis via improved mitochondrial biogenesis
The exact molecular mechanisms behind motor neurone degeneration in ALS are currently unclear, however, there is increasing evidence implicating mitochondrial dysfunction in the pathogenesis of ALS (Zhao et al., 2022). Aggregation of swollen mitochondria in the neurones of ALS patients was one of the first pathological changes seen in the disease, providing clear evidence of dysfunction (Atsumi, 1981; Taylor et al., 2016). Morphologically abnormal mitochondria have been observed in multiple cellular and animal models of ALS; mitochondria appear fragmented, vacuolated and more spherical, and are seen in atypical clusters along the axon (Higgins et al., 2003; Magrané et al., 2014).
Defective mitochondrial respiration, ATP production and oxidative phosphorylation have also been widely reported in ALS patients. Indeed, post-mortem analysis of sporadic ALS patients demonstrated reduced complex I-IV activity in the spinal cord, and impairment of I and IV activity in skeletal muscle, leading to a reduction in ATP production (Wiedemann et al., 1998, 2002). Elevated ROS production and ROS-associated damage are common pathological features of ALS. Both post-mortem and biofluid analysis of ALS patients has demonstrated markers of ROS damage (Shaw et al., 1995; Smith et al., 1998). It is thought that oxidative damage via an increase in ROS production results in aggregation of TDP-43, one of the main pathological hallmarks in ALS (Cohen et al., 2012).
Amyotrophic lateral sclerosis mouse models subjected to the ketogenic diet have larger numbers of motor neurones in the spinal cord compared to mice on a normal diet (Zhao et al., 2006). In addition, motor performance of the “KD mice” was significantly enhanced due to preservation of motor neurones in the spinal cord. The same study detected a higher concentration of BHB (a ketone generated via MCFA metabolism) in the blood of KD-fed mice (Zhao et al., 2006). BHB was able to promote mitochondrial ATP synthesis and prevent the inhibition of complex I in vitro, likely contributing to the increased motor performance observed in the KD-treated ALS model mice (Zhao et al., 2006; Table 2). Dietary supplementation of OA, a key component of the MCT diet, had no effect on the survival rate of ALS mice (Zhao et al., 2012), but significantly improved motor performance, by protecting against spinal cord motor neurone loss (Zhao et al., 2012). This study concluded that OA treatment significantly promoted oxygen consumption rate, restoring energy metabolism (Zhao et al., 2012).
3.4. MCFAs may alleviate symptoms of Parkinson’s disease by elevating ketone levels
Mitochondrial dysfunction has long been associated with PD (Mullin and Schapira, 2013; Park et al., 2018). Mutations in the Leucine Rich Repeat Kinase 2 (LRRK2) gene are a common genetic cause of late-onset familial and sporadic PD (Kumari and Tan, 2009). The G2019S mutation increases LRRK2 kinase activity (Chen and Wu, 2018). Overexpression of either wild-type LRRK2 or LRRK2G2019S leads to increased ROS production and reduced mitochondrial function in mice, fruit flies, iPSC cell cultures and PD patients (Mortiboys et al., 2010; Cooper et al., 2012; Ng et al., 2012; Yue et al., 2015). SIRT3, capable of reducing oxidative stress (Nemoto et al., 2005), is also decreased in the mouse model of AD (Yang et al., 2015). The formation of Lewy bodies, composed of aggregated α-Synuclein (α-Syn) protein, is another PD hallmark. Mutations in the SNCA gene, encoding α-Syn, induce ROS production and mitochondrial fragmentation (Ryan et al., 2015). α-Syn is located on mitochondria-associated membranes (MAM), which regulate Ca2+ signaling and apoptosis (Guardia-Laguarta et al., 2014). Excessive α-Syn disrupts mitochondria-ER interactions, impairing ATP output and Ca2+ exchange (Paillusson et al., 2017). α-Syn also suppresses mitochondrial biogenesis through PGC-1α inhibition (Ryan et al., 2013). Interestingly, PD patients exhibit under-expression of PGC-1α, resulting in the loss of dopaminergic neurones (Zheng et al., 2010).
Decanoic acid has been shown to reduce the degeneration of dopaminergic neurons in the C. elegans PD model by modulating the insulin signaling pathway—a critical regulator of longevity in both invertebrates and vertebrates—resulting in the enhanced transcription of genes encoding antioxidant and heat-shock proteins (Sanguanphun et al., 2022). Interestingly, the polyphenolic compound resveratrol has been shown to have an antioxidant effect by acting as a free radical scavenger, providing a neuroprotective effect via binding and activating SIRT1 (Iuga et al., 2012; Cao et al., 2015; Salehi et al., 2018). Resveratrol can also protect neuronal cells against oxidative stress and toxicity specifically induced by α-synuclein via SIRT1 activation (Albani et al., 2009). In line with these studies, SIRT3 is required to reduce dopaminergic neuron loss in a mouse model of PD, enhances mitochondrial antioxidant capacity and reduces oxidative stress (Liu et al., 2015). Although few studies directly investigated the effect of MCFAs on PD, multiple reports provide indirect evidence supporting MCFAs’ beneficial effect on PD via elevating the level of ketone bodies. Similar to AD, the KD has been shown to alleviate symptoms of PD (Kashiwaya et al., 2000). In one study, rat neuronal cells were treated with ketone bodies, specifically beta-hydroxybutyrate (BHB) and acetoacetate, resulting in decreased production of glutamate-induced ROS. This reduction was due to an increase in the NAD+/NADH ratio, which in turn led to improved mitochondrial respiration (Maalouf et al., 2007; Table 2). Previous study also showed enhanced mitochondrial respiration and a reduction of PD symptoms in PD model mice treated with D-BHB through subcutaneous injections (Tieu et al., 2003; Table 2). These findings suggest that the use of MCFAs may have potential therapeutic benefits in the treatment of PD.
Overall, MCFAs have shown potential in treating NDs and functional decline caused by aging via restoring mitochondrial function through the activation of the PPARγ and the inhibition of the AMPAR.
4. Autophagy in aging and neurodegenerative disease
Dysfunctional protein homeostasis is a key feature of both aging and NDs, resulting in the accumulation of misfolded, mislocalized and aggregated proteins within neurones (Aman et al., 2021). There are two major pathways responsible for degradation of cellular proteins and maintenance of homeostasis: the ubiquitin proteasome system (UPS) and the autophagy-lysosome pathway (Lilienbaum, 2013). Normally, around 90% of cellular proteins throughout the body are degraded by the UPS, however, when the UPS is defective, autophagy can be upregulated to help clear larger protein aggregates (Rock et al., 1994; Kageyama et al., 2014). Autophagy is thought to be the primary method of protein clearance in ND patient neurones, as the UPS is unable to degrade the large pathological aggregates of proteins (Chandran and Rochet, 2022). Autophagy was first described in the 1960s as a lysosome-dependent pathway for degradation of damaged or unnecessary cellular components (De Duve and Wattiaux, 1966). There are three main types of autophagy: macroautophagy, chaperone mediated autophagy (CMA) and microautophagy (Figure 2). In microautophagy, cellular components requiring degradation are taken up directly by the lysosome through invagination of the membrane, whereas in CMA, proteins targeted for degradation are translocated across the lysosomal membrane with the help of a chaperone protein (Figure 2; Glick et al., 2010). Macroautophagy (hereafter referred to as autophagy) involves the delivery of cytoplasmic components to lysosomes for degradation via an autophagosome—an intermediary vesicle that binds to and fuses with the lysosome to form an autolysosome (Figure 2; Glick et al., 2010).
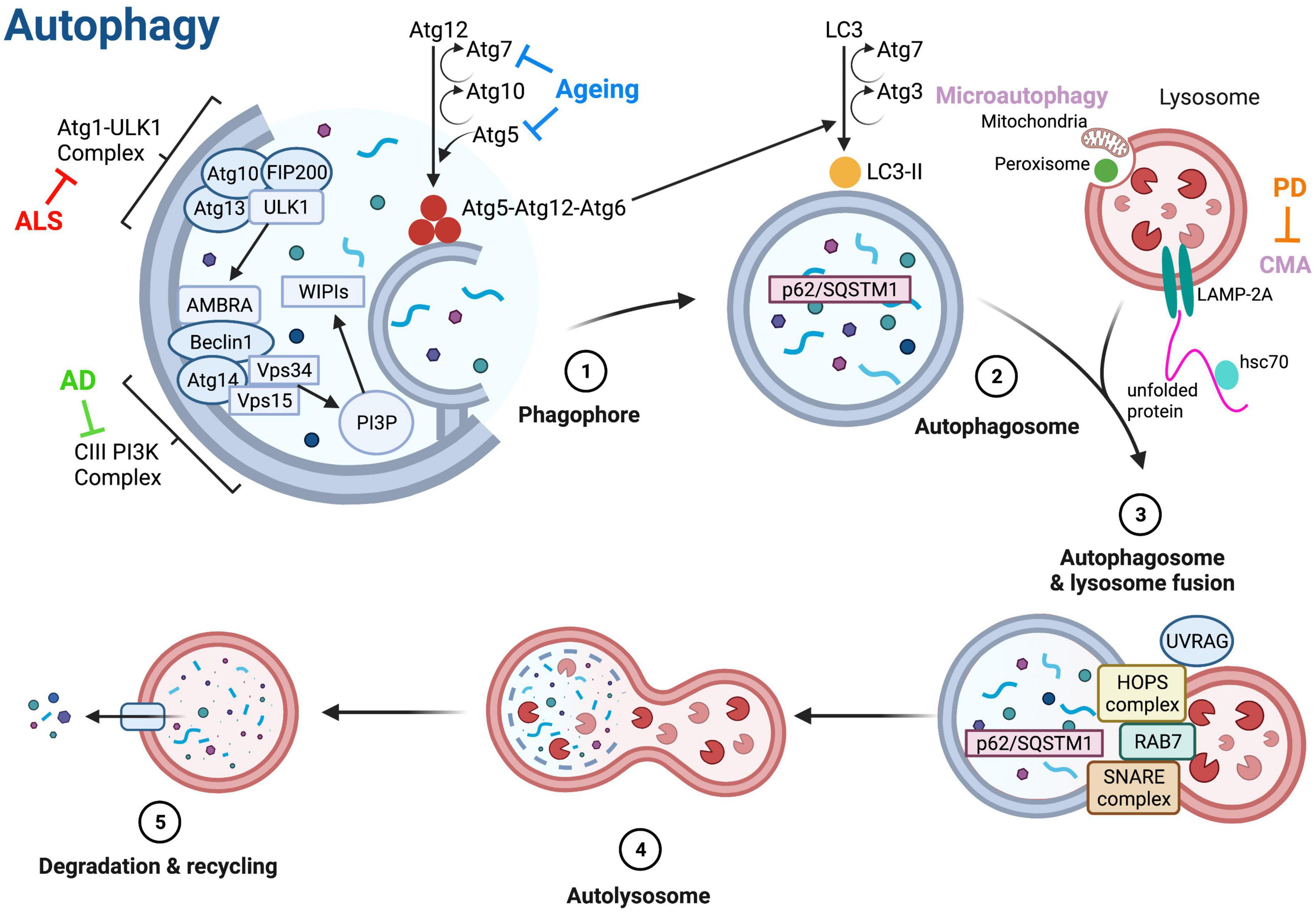
Figure 2. Molecular regulation of autophagy. The three main types of autophagy are macroautophagy (referred to as autophagy), microautophagy and chaperone-mediated autophagy (CMA). Autophagy is initiated via the autophagy-related (Atg) 1-Unc51-like kinase (ULK) complex that includes ULK1, Atg13, FIP200 (focal adhesion kinase family interacting protein of 200 kD) and Atg101. Phosphorylation of ULK1 stimulates the translocation of the class III phosphoinositide 3-kinase (PI3K-III) complex containing Beclin-1 from the cytoskeleton to a pre-autophagosomal structure, initiating the formation of the autophagosome (Suzuki et al., 2007; Mizushima, 2009; Di Bartolomeo et al., 2010). The activity of the PI3K-III complex generates phosphatidylinositol-3-phosphate (PI3P), which binds to the WD repeat domain phosphoinositide-interacting 1 (WIPI1) and WIPI2 complexes, catalyzing two ubiquitin-like reactions that allow elongation of the autophagosome membrane (Ohsumi and Mizushima, 2004). Atg5 and Atg12 are conjugated together in the first reaction, in the presence of Atg7 and Atg10 (Ohsumi et al., 2000). During the second reaction, a complex containing Atg5, Atg12, and Atg16 attaches to the membrane and induces the conjugation of phosphatidylethanolamine to LC3 (microtubule-associated protein 1A/1B-light chain 3), forming LC3-II and facilitating membrane closure (Fujita et al., 2008). LC3-II binds to autophagy receptors, such as Sequestosome 1 (p62/SQSTM1), that are bound to components targeted for degradation (Pankiv et al., 2007). Once autophagosomes are fully formed, the UV radiation resistance gene (UVRAG) protein is phosphorylated and combines with the homotypic fusion and protein sorting (HOPS) complex to assist with membrane trafficking and fusion, thus allowing fusion of the autophagosome with a lysosome (Liu and Sabatini, 2020). Ras-associated binding (Rab) proteins and soluble N-ethylmaleimide-sensitive-factor attachment protein receptor (SNARE) proteins also assist in fusion of autophagosomes and lysosomes (Aman et al., 2021). After fusion, the lysosomal content is degraded by lysosomal hydrolases, and degradation products are released into the cell for re-use (Aman et al., 2021). Microautophagy involves degradation of peroxisomes and mitochondria via invagination of the lysosome membrane (Kunz et al., 2004). In CMA, protein substrates are unfolded by heat shock cognate 71 kDa protein (hsc70) chaperones and translocated across the lysosome membrane via interaction with lysosome-associated membrane protein 2A (LAMP-2A) (Majeski and Fred Dice, 2004). Blunt arrows indicate main proteins/processes downregulated in ageing (blue), amyotrophic lateral sclerosis (ALS, red) and Alzheimer’s disease (AD, green), and Parkinson’s disease (PD, orange). Created with BioRender.com.
Autophagy is an intricate, multi-stage process; the main stages and critical proteins mediating autophagy are illustrated in Figure 2. Autophagic clearance is initiated via the autophagy-related 1 (Atg1)-Unc51-like kinase (ULK) complex; ULK1 phosphorylation via the activation of AMP-activated protein kinase (AMPK), or the inhibition of mechanistic target of rapamycin complex 1 (mTORC1) initiates phagophore formation (Figure 2; Shang and Wang, 2011; Liu and Sabatini, 2020). The UPS can control the magnitude of the autophagy response via ULK1 and mTOR kinases (Liu C. et al., 2016; Nazio et al., 2016). Once autophagy is initiated, the phagophore membrane elongates and matures to become an autophagosome, which then fuses with a lysosome to form an autolysosome that degrades and recycles cargo (Figure 2).
Neurones are long-living, post-mitotic cells critically dependent on autophagy for maintaining their homeostasis and functionality (Stavoe and Holzbaur, 2019). Unsurprisingly, autophagy dysfunction is widely reported in ALS, AD, PD, and in aging. Neuronal cells are especially vulnerable to impaired autophagy—autophagosomes containing cargo to be degraded must be transported along the axon to the cell body to fuse with lysosomes, as lysosomes are rarely found in distal axons (Maday et al., 2012; Cheng et al., 2015). This vulnerability results in opportunity for proteins to aggregate and form pathological inclusions—a hallmark pathology of many NDs. A large body of evidence indicates MCFAs as a suitable intervention for combating symptoms of NDs and aging due in part to their autophagy-promoting effects. MCFAs are also known to inhibit autophagy when it is pathologically augmented, indicating that MCFAs may have a modulatory effect on autophagy in NDs, dependent on the context. One of the difficulties in reviewing the use of MCFAs in treating NDs, is the variety of different cell types and models used to study autophagy. We attempt to reconcile the dysregulation of autophagy in various model systems, and how the involvement of MCFAs affect autophagy.
4.1. MCFAs may promote healthy aging via upregulation of autophagy
Reduced autophagic activity is thought to be responsible for accumulating damaged or dysfunctional cellular components during aging, significantly contributing to the organismal functional decline. A study using S. cerevisiae to determine autophagy-related genes in aging identified several short-lived mutants as having defective autophagy (Matecic et al., 2010). Deletion or suppression of Atg genes can reduce the lifespan of various model organisms, and essential autophagy genes such as Atg5 and Atg7 are downregulated in the human brain during normal aging (Figure 2; Komatsu et al., 2005; Simonsen et al., 2008; Tóth et al., 2008; Lipinski et al., 2010).
Nutritional ketosis, brought upon by the MCT diet, is known to upregulate autophagy throughout the brain, via inhibition of mTORC1 (Table 2; McCarty et al., 2015). DA has been demonstrated to stimulate autophagy in Dictyostelium, a widely used model to study autophagic clearance, seen via the increase in autophagosome number and enhanced autophagic flux, likely due to increased expression of Atg1 and Atg8a (Table 2; Mesquita et al., 2017; Warren et al., 2021). The term “autophagic flux” describes the dynamic process of autophagy from phagophore initiation to lysosome recycling and is often used as a measurement of degradation activity. Interestingly, these results were not seen with OA, suggesting that the effect on autophagy may be specific to DA (Warren et al., 2021). However, OA can stimulate c-Jun N-terminal kinase (JNK)-dependent autophagy in rats (Table 2; He et al., 2022). JNK activation is known to extend the lifespan in Drosophila, indicating that OA can promote healthy aging, albeit through a different mechanism to DA (Table 2; Wang et al., 2005). Furthermore, activation of JNK-dependent autophagy increases Beclin-1 expression, a key protein in autophagosome membrane elongation (Figure 2; Park et al., 2009) suggesting an mTOR-independent modulation of autophagy by OA.
In addition to its role in maintaining cellular homeostasis, autophagy has also been shown to have anti-inflammatory and antioxidant effects (Giordano et al., 2014; Qian et al., 2017). This suggests that a decline in autophagic activity may contribute to the chronic inflammation and oxidative stress associated with aging. Low grade, chronic inflammation is known to promote aging; referred to as “inflammageing,” this condition is characterized by increased levels of inflammatory markers throughout the body (Franceschi et al., 2000). The possible beneficial impact of MCFAs on aging may therefore stem from their autophagy-promoting effects, mediated by decreased inflammation and oxidative stress.
4.2. MCFAs could correct dysregulated autophagic flux in Alzheimer’s disease
Autophagy dysregulation is a common occurrence in AD. Early studies detected large amounts of aggregated tau protein and subcellular vesicles within swollen neurites (a common AD pathology) in AD patient brains; these vesicles were later identified to be immature autophagosomes (Suzuki and Terry, 1967; Nixon et al., 2005). Similar results were seen in AD mice, in which immature autophagosomes were shown to accumulate in neurites before β-amyloid (Aβ) plaques began to form, suggesting that autophagy dysfunction precedes, and possibly causes, pathological inclusion formation (Yu et al., 2005).
Medium-chain triglyceride diets have a positive effect on Aβ plaques, via the regulation of autophagy. The activity of HMGCS2, an enzyme that controls the synthesis of ketones from MCFAs, is mediated by the mTOR pathway (Hu et al., 2017). HMGCS2 induces the autophagic clearance of Aβ precursor protein (APP), thus reducing the occurrence of Aβ plaques (Table 2; Hu et al., 2017). Autophagy regulation by HMGCS2 is dependent on ketones, therefore, an MCT diet, or indeed MCFA supplementation, will increase the activity of HMGCS2 and clearance of APP (Table 2; Hu et al., 2017). Aβ accumulation is largely caused by the imbalance between its production and clearance (Selkoe and Hardy, 2016). It is plausible to assume that, by preventing the production of Aβ plaques, MCFA treatment may delay AD onset and progression.
Normal autophagic flux is essential for neuronal homeostasis (Zhang et al., 2013), and its impairment is known to correlate with AD progression (Chung et al., 2019). Proper formation and degradation of autophagosomes is essential for normal autophagic flux and the PI3K-CIII complex plays a vital role in autophagosome formation. The expression of Beclin-1, a core subunit of the PI3K-CIII complex, is significantly reduced in AD; consequently, the generation of PI3P, the product of PI3K-CIII complex activity, is also downregulated in the patients’ brains (Figure 2; Pickford et al., 2008; Lucin et al., 2013; Morel et al., 2013). A recent study showed that a diet high in MCFAs—achieved by replacing animal fat with coconut oil—can restore impaired autophagic flux in mouse hepatocytes (Wang et al., 2017). Interestingly, this functional restoration is independent of AMPK and mTOR signaling, suggesting that MCFAs, unlike short-chain fatty acids (Iannucci et al., 2016), may not act directly on the classic autophagy-regulating pathways (Wang et al., 2017). As MCFAs can restore normal autophagic flux, it is possible that treatment will slow down or even prevent disease progression in AD.
4.3. MCFAs may improve motor symptoms in amyotrophic lateral sclerosis by upregulating autophagy
The ketogenic diet has proven to be an effective therapy in mouse models of ALS by promoting mitochondrial energy production and membrane stabilization (Zhao et al., 2006). However, whether MCFAs influence autophagy in ALS remains unclear. Several ALS-associated genes are functionally implicated in autophagy, including SQSTM1, C9orf72, Ubiquilin-2, and VAPB (Mao et al., 2019; Şentürk et al., 2019; Wu et al., 2020; Cozzi and Ferrari, 2022). The C9orf72 protein binds GTPases Rab7, and Rab11 to aid endosome maturation and recycling, interacts with Atg1-ULK complex to aid autophagosome formation, and regulates autophagic flux via Rab8a and Rab39b (Sellier et al., 2016; Sullivan et al., 2016; Farg et al., 2017). Mutations in C9orf72 impair autophagic flux, resulting in protein aggregation leading to motor neurone dysfunction and death (Sellier et al., 2016). Toxic dipeptide repeats aberrantly translated from the hexanucleotide repeat expansion region within intron 1 of the C9orf72 gene disrupt the VAPB-PTPIP51 interaction at the mitochondrial-endoplasmic reticulum interface (Gomez-Suaga et al., 2022), thereby linking mitochondrial function to autophagy, through two ALS disease-causing genes, C9orf72 and VAPB. When functioning normally, the autophagy receptor p62/SQSTM1 interacts with proteins targeted for degradation and brings them into the autophagosome where the receptor and its cargo are degraded upon fusion with a lysosome (Figure 2; Klionsky et al., 2016). The presence of p62/SQSTM1-positive inclusions in ALS patient motor neurones indicates that autophagic flux has been disrupted, as degradation of cargo has not been achieved (Al-Sarraj et al., 2011). As MCFAs are proven to restore autophagic flux in hepatocytes, it is possible that the same effect will be seen in motor neurones (Wang et al., 2017). Furthermore, MCFAs may also combat the decrease in autophagy seen in ALS. Rapamycin, a known inhibitor of mTORC1 and a potent autophagy inducer in many cell types including neurones (Noda and Ohsumi, 1998; Rangaraju et al., 2010), can rescue impaired locomotion in a zebrafish model of ALS (Table 2; Lattante et al., 2015). Preclinical data show that decanoic acid can inhibit mTORC1 in patient-derived astrocytes, much in the same way as rapamycin, resulting in an overall increase in autophagy (Table 2; Warren et al., 2020). These results suggest that MCFAs have the possibility to alleviate motor symptoms of ALS by increasing autophagy levels.
Due to their positive effects on both autophagy and mitochondrial function, MCFAs hold promise as a novel, multi-target therapeutic strategy for ALS. However, this may not be true for all types of ALS. Mouse models of ALS caused by mutations in the copper-zinc superoxide dismutase (SOD1) enzyme demonstrate pathological induction of autophagy in the spinal cord in the early, pre-symptomatic stages of the disease (Li et al., 2008; Zhang et al., 2011). When SOD1 ALS mice were treated with rapamycin, motor neurone degeneration was accelerated and the lifespan was significantly shortened (Zhang et al., 2011), providing evidence that stimulation of autophagy is not always beneficial in ALS. Clearly, the relationship between autophagy and neurodegeneration is complex, and a delicate balance of autophagy is likely required for health benefits in NDs.
4.4. MCFAs may modulate autophagy in Parkinson’s disease, and increase the clearance of pathological inclusions by promoting chaperone-mediated autophagy
Mutations in α-synuclein (SNCA) and leucine-rich repeat kinase 2 (LRRK2, PARK8) are associated with genetic forms of PD, and have been widely studied for their involvement in autophagy dysfunction. The mechanisms by which these genes affect autophagic clearance and contribute to PD pathology are still being uncovered, but the upregulation of autophagy is a promising therapeutic intervention for PD.
Mutations in LRRK2 are the most common genetic cause of PD. LRRK2 is a kinase that regulates many autophagy-related processes within a cell, including vesicular trafficking and endosomal transport (Dodson et al., 2012; Steger et al., 2016; Connor-Robson et al., 2019). Disease-causing LRRK2 mutations increase the kinase activity of LRRK2, contributing to PD pathology (Jeong and Lee, 2020). LRRK2 is normally degraded in lysosomes via CMA; the G2019S mutation inhibits this process (Figure 2; Orenstein et al., 2013). BHB, one of the main ketones generated via MCFA metabolism, directly promotes CMA activation by increasing protein oxidation, resulting in increased clearance of proteins damaged by oxidation (Table 2; Finn and Dice, 2005). An activation of CMA by MCFAs may therefore increase the degradation of mutated LRRK2, preventing increased kinase activity (Table 2).
Interestingly, the G2019S mutation in LRRK2 also increases autophagy in cultured human neurons, possibly to compensate for the reduction in CMA, resulting in neurite shortening—a pathological hallmark that precedes neuronal death in many NDs (Table 2; Plowey et al., 2008). It is possible that MCFAs are a suitable treatment to simultaneously promote CMA and decrease autophagy in PD patients. Fatty acids are known to inhibit autophagy regulated by AMPK activation in mice (Liu T. et al., 2016) and mTOR suppression in rat and human hepatocytes (Vinciguerra et al., 2008), it remains unclear, however, whether this is mediated by MCFAs. Treatment with lauric acid (LA), an MCFA with 12 carbon atoms, can decrease pathologically augmented autophagy in human chondrocytes (cells responsible for cartilage formation) as shown by the reduced expression of Atg5 and Beclin-1, proteins involved in the elongation and maturation of the autophagosome (Figure 2; Sekar et al., 2018). LCFAs such as palmitic acid, myristic acid and stearic acid further increased autophagy in chondrocytes, indicating that LA, and perhaps other MCFAs, may be better at modulating autophagy (Sekar et al., 2018). LCFAs also induced the expression of LC3, a key player in autophagosome formation, whereas LA treatment returned LC3 expression to the control levels, suggesting stabilization of autophagosome production (Sekar et al., 2018). On the other hand, a recent study demonstrated that LA could induce autophagy in wild-type pig intestinal cells, resulting in dysregulated autophagic flux and cell death (Yang et al., 2020). These studies show that LA can promote or reduce autophagy depending on the context, indicating LA may be an ideal treatment to normalize autophagy homeostasis in ND patients (Table 2).
Accumulation of α-synuclein is a hallmark pathology in both sporadic and familial PD. Although the normal function of α-synuclein has not been fully elucidated, it is thought that this protein plays a role in the formation of synaptic vesicles (Burré et al., 2010). The A53T mutation in SNCA has been linked to autophagy; cells expressing A53T mutant α-synuclein display accumulation of autophagic vesicles, suggesting an impairment of autophagic flux (Stefanis et al., 2001). Like LRRK2, α-synuclein is also degraded via CMA; A53T mutations prevent this CMA-associated degradation and increase autophagy, likely as a compensatory response (Table 2; Cuervo and Dice, 2000). Considering the autophagy-stimulating activity of MCFAs, it is possible that this class of lipids may promote the degradation of α-synuclein via an increase in CMA (Finn and Dice, 2005). LA treatment may also be successful in ameliorating the compensatory increase in autophagy, restoring autophagic flux (Sekar et al., 2018; Yang et al., 2020). Overall, it seems that MCFA treatment may be advantageous in both sporadic and familial PD.
5. Discussion
Medium chain fatty acids have a direct role in regulating autophagy and mitochondrial function, processes that are dysfunctional in aging and NDs. This review highlights evidence that MCFAs have the potential to be sufficient treatments for some NDs, and promote healthy aging, due to their effects on autophagic processes and the function of mitochondria. It is possible that MCFAs affect dysregulated autophagy and mitochondrial function together as well as in parallel, providing more evidence that they are an advantageous treatment option. It is worth noting that there is an overlap of disease-causing genes involved various neurodegenerative diseases. Thus, MCFA treatments that modulate the severity of AD and PD, may also be effective in treating ALS and possibly other neurodegenerative diseases. However, this is an under-researched area and there are currently very few studies reporting a link between MCFAs and autophagy and mitochondrial function in neurones. Adverse side effects associated with MCFAs and the MCT diet have also been reported, warranting further studies into the mode of action and safety aspects of these compounds.
5.1. Avenues for future research and limitations of research into MCFAs
A systematic analysis of each MCFA using animal models will be required to gain a clearer picture of the effects that these compounds have on different neurodegenerative diseases. Drosophila would be particularly amenable to this type of analysis as the disease models are well established, and tissue-specific effects as well as whole organism effects can be easily determined. In the case of ALS, Drosophila could be used to assess differences between motor neurone and brain degeneration as neural cell type-specific toolsets are readily available to the research community (Jenett et al., 2012; Aso et al., 2014).
In addition, modifications to individual MCFAs may provide compounds with more specific biological effects. For example, 4-MOA and 5-MOA are both methyl-modified derivatives of octanoic acid, and it would be interesting to compare the different effects that these molecules have on neuronal biology at the molecular level.
One of the limitations of the research into the biological effects of MCFAs is the lack of a standardized model that can be used to compare the effects of different compounds on neural function. As a result, many of the observations are specific to a given cell, or a given organism. Given that Drosophila has both motor neurones and a complex brain, this model appears ideally positioned for studying the effects of MCFAs on tissue specific and organ-specific alterations. In addition, the complete neural circuitry of the fly brain has recently been mapped (Winding et al., 2023), aiding the efforts to understand how MCFAs modulate neurodegeneration at the level of neurons and connecting synapses.
Author contributions
ED and BZ wrote the manuscript, summarized literature, and prepared figures. VKS wrote and revised the manuscript. HA conceptualized and designed the article and wrote and revised the manuscript. All authors contributed to the article and approved the submitted version.
Acknowledgments
We would like to thank Prof. Robin Williams (Royal Holloway, University of London) for providing useful comments and feedback on the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Akamatsu, M., Yamashita, T., Teramoto, S., Huang, Z., Lynch, J., Toda, T., et al. (2022). Testing of the therapeutic efficacy and safety of AMPA receptor RNA aptamers in an ALS mouse model. Life Sci Alli. 5:e202101193. doi: 10.26508/lsa.202101193
Albani, D., Polito, L., Batelli, S., Mauro, S. D., Fracasso, C., Martelli, G., et al. (2009). The SIRT1 activator resveratrol protects SK-N-BE cells from oxidative stress and against toxicity caused by alpha-synuclein or amyloid-beta (1-42) peptide. J. Neurochem. 110, 1445–1456. doi: 10.1111/j.1471-4159.06228.x
Al-Sarraj, S., King, A., Troakes, C., Smith, B., Maekawa, S., Bodi, I., et al. (2011). p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol. 122, 691–702. doi: 10.1007/s00401-011-0911-2
Aman, Y., Schmauck-Medina, T., Hansen, M., Morimoto, R. I., Simon, A. K., Bjedov, I., et al. (2021). Autophagy in healthy aging and disease. Nat. Aging 1, 634–650. doi: 10.1038/s43587-021-00098-4
Aso, Y., Hattori, D., Yu, Y., Johnston, R. M., Iver, N. A., Ngo, T. T., et al. (2014). The neuronal architecture of the mushroom body provides a logic for associative learning. eLife 3:e04577. doi: 10.7554/eLife.04577
Atsumi, T. (1981). The ultrastructure of intramuscular nerves in amyotrophic lateral sclerosis. Acta neuropathologica 55, 193–198. doi: 10.1007/BF00691318
Bannick, M. S., Beghi, E., Blake, N., Ellenbogen, R. G., Afarideh, M., Ahmadi, A., et al. (2019). Global, regional, and national burden of neurological disorders, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 18, 459–480. doi: 10.1016/S1474-4422(18)30499-X
Bough, K. J., and Rho, J. M. (2007). Anticonvulsant mechanisms of the ketogenic diet. Epilepsia 48, 43–58.
Broom, G. M., Shaw, I. C., and Rucklidge, J. J. (2019). The ketogenic diet as a potential treatment and prevention strategy for Alzheimer’s disease. Nutrition 60, 118–121. doi: 10.1016/j.nut.2018.10.003
Bueno, N. B., de Melo, I. S. V., de Oliveira, S. L., and da Rocha Ataide, T. (2013). Very-low-carbohydrate ketogenic diet v. low-fat diet for long-term weight loss: A meta-analysis of randomised controlled trials. Br. J. Nutr. 110, 1178–1187. doi: 10.1017/S0007114513000548
Burré, J., Sharma, M., Tsetsenis, T., Buchman, V., Etherton, M. R., and Südhof, T. C. (2010). α-Synuclein promotes SNARE-complex assembly in Vivo and in Vitro. Science 329, 1663–1667. doi: 10.1126/science.1195227
Butterfield, D. A., and Halliwell, B. (2019). Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 20, 148–160. doi: 10.1038/s41583-019-0132-6
Calsolaro, V., and Edison, P. (2016). Alterations in glucose metabolism in Alzheimer’s disease. Recent Patents Endocr. Metab. Immune Drug Discov. 10, 31–39. doi: 10.2174/1872214810666160615102809
Cao, D., Wang, M., Qiu, X., Liu, D., Jiang, H., Yang, N., et al. (2015). Structural basis for allosteric, substrate-dependent stimulation of SIRT1 activity by resveratrol. Genes Dev. 29, 1316–1325. doi: 10.1101/gad.265462.115
Castellano, C. A., Nugent, S., Paquet, N., Tremblay, S., Bocti, C., Lacombe, G., et al. (2015). Lower brain 18F-fluorodeoxyglucose uptake but normal 11C-acetoacetate metabolism in mild Alzheimer’s disease dementia. J. Alzheimers Dis. 43, 1343–1353.
Chandran, A., and Rochet, J. (2022). Shining a light on autophagy in neurodegenerative diseases. J. Biol. Chem. 298:101437. doi: 10.1016/j.jbc.2021.101437
Chang, P., Augustin, K., Boddum, K., Williams, S., Sun, M., Terschak, J. A., et al. (2016). Seizure control by decanoic acid through direct AMPA receptor inhibition. Brain 139, 431–443. doi: 10.1093/brain/awv325
Chang, P., Zuckermann, A. M. E., Williams, S., Close, A. J., Cano-Jaimez, M., McEvoy, J. P., et al. (2015). Seizure control by derivatives of medium chain fatty acids associated with the ketogenic diet show novel branching-point structure for enhanced potency. J. Pharmacol. Exp. Therap. 352, 43–52. doi: 10.1124/jpet.114.218768
Chen, M., and Wu, R. (2018). LRRK 2 gene mutations in the pathophysiology of the ROCO domain and therapeutic targets for Parkinson’s disease: A review. J. Biomed. Sci. 25:52. doi: 10.1186/s12929-018-0454-0
Cheng, X., Zhou, B., Lin, M., Cai, Q., and Sheng, Z. (2015). Axonal autophagosomes recruit dynein for retrograde transport through fusion with late endosomes. J. Cell Biol. 209, 377–386. doi: 10.1083/jcb.201412046
Choi, I. Y., Piccolo, L., Childress, P., Bollman, B., Ghosh, A., Brandhorst, S., et al. (2016). A diet mimicking fasting promotes regeneration and reduces autoimmunity and multiple sclerosis symptoms. Cell Rep. 15, 2136–2146.
Chung, K. M., Hernández, N., Sproul, A. A., and Yu, W. H. (2019). Alzheimer’s disease and the autophagic-lysosomal system. Neurosci. Lett. 697, 49–58. doi: 10.1016/j.neulet.2018.05.017
Cohen, T. J., Hwang, A. W., Unger, T., Trojanowski, J. Q., and Lee, V. M. Y. (2012). Redox signalling directly regulates TDP-43 via cysteine oxidation and disulphide cross-linking. EMBO J. 31, 1241–1252. doi: 10.1038/emboj.2011.471
Connor-Robson, N., Booth, H., Martin, J. G., Gao, B., Li, K., Doig, N., et al. (2019). An integrated transcriptomics and proteomics analysis reveals functional endocytic dysregulation caused by mutations in LRRK2. Neurobiol. Dis. 127, 512–526. doi: 10.1016/j.nbd.2019.04.005
Cooper, O., Seo, H., Andrabi, S., Guardia-Laguarta, C., Graziotto, J., Sundberg, M., et al. (2012). Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Sci. Transl. Med. 4:3003985. doi: 10.1126/scitranslmed.3003985
Cozzi, M., and Ferrari, V. (2022). Autophagy dysfunction in ALS: From transport to protein degradation. J. Molec. Neurosci. 72, 1456–1481. doi: 10.1007/s12031-022-02029-3
Croteau, E., Castellano, C., Richard, M. A., Fortier, M., Nugent, S., Lepage, M., et al. (2018). Ketogenic medium chain triglycerides increase brain energy metabolism in Alzheimer’s disease. J. Alzheimers Dis. 64, 551–561. doi: 10.3233/JAD-180202
Croteau, E., Castellano, C. A., Fortier, M., Bocti, C., Fulop, T., Paquet, N., et al. (2017). A cross-sectional comparison of brain glucose and ketone metabolism in cognitively healthy older adults, mild cognitive impairment and early Alzheimer’s disease. Exp. Gerontol. 107, 18–26. doi: 10.1016/j.exger.2017.07.004
Cuervo, A. M., and Dice, J. F. (2000). Age-related decline in chaperone-mediated autophagy. J. Biol. Chem. 275, 31505–31513. doi: 10.1074/jbc.M002102200
Cunnane, S. C., Trushina, E., Morland, C., Prigione, A., Casadesus, G., Andrews, Z. B., et al. (2020). Brain energy rescue: An emerging therapeutic concept for neurodegenerative disorders of ageing. Nat. Rev. 19, 609–633. doi: 10.1038/s41573-020-0072-x
Dabke, P., and Das, A. M. (2020). Mechanism of action of ketogenic diet treatment: Impact of decanoic acid and beta-hydroxybutyrate on sirtuins and energy metabolism in hippocampal murine neurones. Nutrients 12:379. doi: 10.3390/nu12082379
De Duve, C., and Wattiaux, R. (1966). Functions of lysosomes. Annu. Rev. Physiol. 28, 435–492. doi: 10.1146/annurev.ph.28.030166.002251
Di Bartolomeo, S., Corazzari, M., Nazio, F., Oliverio, S., Lisi, G., Antonioli, M., et al. (2010). Dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J. Cell Biol. 191, 155–168. doi: 10.1083/jcb.201002100
Dodson, M. W., Zhang, T., Jiang, C., Chen, S., and Guo, M. (2012). Roles of the Drosophila LRRK2 homolog in Rab7-dependent lysosomal positioning. Hum. Mol. Genet. 21, 1350–1363. doi: 10.1093/hmg/ddr573
Dong, X. X., Wang, Y., and Qin, Z. H. (2009). Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 30, 379–387. doi: 10.1038/aps.2009.24
Donmez, G., and Guarente, L. (2010). Aging and disease: Connections to sirtuins. Aging Cell 9, 285–290. doi: 10.1111/j.1474-9726.2010.00548.x
Dugger, B. N., and Dickson, D. W. (2017). Pathology of neurodegenerative diseases. Cold Spring Harbor Perspect. Biol. 9:a028035. doi: 10.1101/cshperspect.a028035
Egan, D. F., Shackelford, D. B., Mihaylova, M. M., Gelino, S., Kohnz, R. A., Mair, W., et al. (2011). Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331, 456–461. doi: 10.1126/science.1196371
Epremyan, K. K., Rogov, A. G., Goleva, T. N., Lavrushkina, S. V., Zinovkin, R. A., and Zvyagilskaya, R. A. (2023). Altered mitochondrial morphology and bioenergetics in a new yeast model expressing Aβ42. Int. J. Mol. Sci. 24:24020900. doi: 10.3390/ijms24020900
Erkkinen, M. G., Kim, M., and Geschwind, M. D. (2018). Clinical neurology and epidemiology of the major neurodegenerative diseases. Cold Spring Harbor Persp. Biol. 10:a033118. doi: 10.1101/cshperspect.a033118
Estes, R. E., Lin, B., Khera, A., and Davis, M. Y. (2021). Lipid metabolism influence on neurodegenerative disease progression: Is the vehicle as important as the cargo? Front. Mol. Neurosci. 14:788695. doi: 10.3389/fnmol.2021.788695
Farg, M. A., Sundaramoorthy, V., Sultana, J. M., Yang, S., Atkinson, R. A. K., Levina, V., et al. (2017). C9ORF72, implicated in amyotrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum. Mol. Genet. 26, 4093–4094. doi: 10.1093/hmg/ddx309
Finn, P. F., and Dice, J. F. (2005). Ketone bodies stimulate chaperone-mediated autophagy. J. Biol. Chem. 280, 25864–25870. doi: 10.1074/jbc.M502456200
Franceschi, C., Bonafe, M., Valensin, S., Olivieri, F. I, De Luca, M., Ottaviani, E., et al. (2000). Inflamm-aging: An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 908, 244–254. doi: 10.1111/j.1749-6632.2000.tb06651.x
Fujikura, Y., Sugihara, H., Hatakeyama, M., Oishi, K., and Yamanouchi, K. (2021). Ketogenic diet with medium-chain triglycerides restores skeletal muscle function and pathology in a rat model of Duchenne muscular dystrophy. FASEB 35:e21861.
Fujita, N., Hayashi-Nishino, M., Fukumoto, H., Omori, H., Yamamoto, A., Noda, T., et al. (2008). An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol. Biol. Cell 19, 4651–4659. doi: 10.1091/mbc.E08-03-0312
Geng, S., Zhu, W., Xie, C., Li, X., Wu, J., Liang, Z., et al. (2016). Medium-chain triglyceride ameliorates insulin resistance in high fat diet-induced obese mice. Eur. J. Nutr. 55, 931–940.
Giordano, S., Darley-Usmar, V., and Zhang, J. (2014). Autophagy as an essential cellular antioxidant pathway in neurodegenerative disease. Redox Biol. 2, 82–90. doi: 10.1016/j.redox.2013.12.013
Glick, D., Barth, S., and Macleod, K. F. (2010). Autophagy: Cellular and molecular mechanisms. J. Pathol. 221, 3–12. doi: 10.1002/path.2697
Gomez-Suaga, P., Morotz, G. M., Markovinovic, A., Martin-Guerrero, S. M., Preza, E., Arias, N., et al. (2022). Disruption of ER-mitochondria tethering and signalling in C9orf72-associated amyotrophic lateral sclerosis and frontotemporal dementia. Aging Cell 21:e13549. doi: 10.1111/acel.13549
Grammatikopoulou, M. G., Goulis, D. G., Gkiouras, K., Theodoridis, X., Gkouskou, K. K., Evangeliou, A., et al. (2020). To Keto or Not to Keto? A Systematic Review of Randomized Controlled Trials Assessing the Effects of Ketogenic Therapy on Alzheimer Disease. Adv. Nutr. 11, 1583–1602. doi: 10.1093/advances/nmaa073
Greig, A., Donevan, S. D., Mujtaba, T. J., Parks, T. N., and Rao, M. S. (2000). Characterization of the AMPA-activated receptors present on motoneurones. J. Neurochem. 74, 179–191. doi: 10.1046/j.1471-4159.2000.0740179.x
Guardia-Laguarta, C., Area-Gomez, E., Rüb, C., Liu, Y., Magrané, J., Becker, D., et al. (2014). α-Synuclein is localized to mitochondria-associated ER membranes. J. Neurosci. 34, 249–259. doi: 10.1523/JNEUROSCI.2507-13.2014
Guo, M., Wang, X., Zhao, Y., Yang, Q., Ding, H., Dong, Q., et al. (2018). Ketogenic diet improves brain ischemic tolerance and inhibits NLRP3 inflammasome activation by preventing drp1-mediated mitochondrial fission and endoplasmic reticulum stress. Front. Mol. Neurosci. 11:86. doi: 10.3389/fnmol.2018.00086
Hardie, D. G., Ross, F. A., and Hawley, S. A. (2012). AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 13, 251–262. doi: 10.1038/nrm3311
He, K., Cao, C., Xu, X., Ye, Z., Ma, X., Chen, W., et al. (2022). Octanoic acid–rich enteral nutrition prevented lipopolysaccharide-induced acute liver injury through c-Jun N-terminal kinase–dependent autophagy. J. Parent. Enter. Nutr. 46, 1353–1360. doi: 10.1002/jpen.2297
Herzig, S., and Shaw, R. J. (2018). AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 19, 121–135. doi: 10.1038/nrm.2017.95
Higgins, C., Jung, C., and Xu, Z. (2003). ALS-associated mutant SOD1 super (G93A) causes mitochondrial vacuolation by expansion of the intermembrane space and by involvement of SOD1 aggregation and peroxisomes. BMC Neurosci. 4:16. doi: 10.1186/1471-2202-4-16
Hollis, F., Mitchell, E. S., Canto, C., Wang, D., and Sandi, C. (2018). Medium chain triglyceride diet reduces anxiety-like behaviors and enhance social competitiveness in rats. Neuropharmacology 138, 245–256.
Houtkooper, R. H., Pirinen, E., and Auwerx, J. (2012). Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 13, 225–238. doi: 10.1038/nrm3293
Hoyer, S. (1991). Abnormalities of glucose metabolism in Alzheimer’s disease. Ann. N. Y. Acad. Sci. 640, 53–58. doi: 10.1111/j.1749-6632.1991.tb00190.x
Hu, L., Zhu, B., Lai, Y., Long, Y., Zha, J., Hu, X., et al. (2017). HMGCS2 promotes autophagic degradation of the amyloid-β precursor protein through ketone body-mediated mechanisms. Biochem. Biophys. Res. Commun. 486, 492–498. doi: 10.1016/j.bbrc.2017.03.069
Hughes, S. D., Kanabus, M., Anderson, G., Hargreaves, I. P., Rutherford, T., O’Donnell, M., et al. (2014). The ketogenic diet component decanoic acid increases mitochondrial citrate synthase and complex I activity in neuronal cells. J. Neurochem. 129, 426–433. doi: 10.1111/jnc.12646
Hulisz, D. (2018). Amyotrophic lateral sclerosis: Disease state overview. Am. J. Manag. Care 15:S320.
Huttenlocher, P., Wilbourn, A., and Signore, J. (1971). Medium-chain triglycerides as a therapy for intractable childhood epilepsy. Neurology 21, 1097–1103.
Iannucci, L. F., Sun, J., Singh, B., Zhou, J., Kaddai, V., Lanni, A., et al. (2016). Short chain fatty acids induce UCP2-mediated autophagy in hepatic cells. Biochem. Biophys. Res. Commun. 480, 461–467. doi: 10.1016/j.bbrc.2016.10.072
Iijima-Ando, K., Hearn, S. A., Shenton, C., Gatt, A., Zhao, L., and Iijima, K. (2009). Mitochondrial mislocalization underlies Abeta42-induced neuronal dysfunction in a Drosophila model of Alzheimer’s disease. PLoS One 4:8310. doi: 10.1371/journal.pone.0008310
Iuga, C., Alvarez-Idaboy, J. R., and Russo, N. (2012). ‘Antioxidant activity of trans-resveratrol toward hydroxyl and hydroperoxyl radicals: A quantum chemical and computational kinetics study’. J. Organ. Chem. 77, 3868–3877. doi: 10.1021/jo3002134
Jäger, S., Handschin, C., St-Pierre, J., and Spiegelman, B. M. (2007). AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. U.S.A. 104, 12017–12022. doi: 10.1073/pnas.0705070104
Jain, S., Rai, R., Singh, D., and Vohora, D. (2021). Octanoic acid, a major component of widely consumed medium-chain triglyceride ketogenic diet is detrimental to bone. Sci. Rep. 11:7003. doi: 10.1038/s41598-021-86468-9
Jancovski, N., Baldwin, T., Orford, M., Li, M., Jones, G. D., Burbano, L. E., et al. (2021). Protective effects of medium chain triglyceride diet in a mouse model of Dravet syndrome. Epilepsia 62, 3131–3142.
Jenett, A., Rubin, G. M., Ngo, T. T., Shepherd, D., Murphy, C., Dionne, H., et al. (2012). A Gal4-driver line resource for Drosophila neurobiology. Cell Rep. 2, 991–1001.
Jeong, G. R., and Lee, B. D. (2020). Pathological Functions of LRRK2 in Parkinson’s Disease. Cells 9:2565. doi: 10.3390/cells9122565
Ji, Z., Liu, G. H., and Qu, J. (2022). Mitochondrial sirtuins, metabolism, and aging. J. Genet. Genom. 49, 287–298.
Jiang, Z., Wang, W., Perry, G., Zhu, X., and Wang, X. (2015). Mitochondrial dynamic abnormalities in amyotrophic lateral sclerosis. Transl. Neurodegener. 4:14. doi: 10.1186/s40035-015-0037-x
Jing, E., O’Neill, B. T., Rardin, M. J., Kleinridders, A., Ilkeyeva, O. R., Ussar, S., et al. (2013). Sirt3 regulates metabolic flexibility of skeletal muscle through reversible enzymatic deacetylation. Diabetes 62, 3404–3417. doi: 10.2337/db12-1650
Johnson, K. A., Conn, P. J., and Niswender, C. M. (2009). Glutamate receptors as therapeutic targets for Parkinson’s disease. CNS Neurol. Disord. Drug Targets 8, 475–491. doi: 10.2174/187152709789824606
Joshi, D. C., Singh, M., Krishnamurthy, K., Joshi, P. G., and Joshi, N. B. (2011). AMPA induced Ca2+ influx in motor neurones occurs through voltage gated Ca2+ channel and Ca2+ permeable AMPA receptor. Neurochem. Int. 59, 913–921. doi: 10.1016/j.neuint.2011.06.023
Joshi, D. C., Tewari, B. P., Singh, M., Joshi, P. G., and Joshi, N. B. (2015). AMPA receptor activation causes preferential mitochondrial Ca2+ load and oxidative stress in motor neurones. Brain Res. 1616, 1–9. doi: 10.1016/j.brainres.2015.04.042
Julien, C., Tremblay, C., Emond, V., Lebbadi, M., Salem, N. J., Bennett, D. A., et al. (2009). Sirtuin 1 reduction parallels the accumulation of tau in Alzheimer disease. J. Neuropathol. Exp. Neurol. 68, 48–58. doi: 10.1097/NEN.0b013e3181922348
Jurado, S. (2018). AMPA receptor trafficking in natural and pathological aging. Front. Mol. Neurosci. 10:446. doi: 10.3389/fnmol.2017.00446
Kadowaki, A., Sada, N., Juge, N., Wakasa, A., Moriyama, Y., and Inoue, T. (2017). Neuronal inhibition and seizure suppression by acetoacetate and its analog, 2-phenylbutyrate. Epilepsia 58, 845–857.
Kageyama, S., Sou, Y., Uemura, T., Kametaka, S., Saito, T., Ishimura, R., et al. (2014). Proteasome dysfunction activates autophagy and the keap1-Nrf2 pathway. J. Biol. Chem. 289, 24944–24955. doi: 10.1074/jbc.M114.580357
Kashiwaya, Y., Takeshima, T., Mori, N., Nakashima, K., Clarke, K., and Veech, R. L. (2000). d-β-Hydroxybutyrate protects neurones in models of Alzheimer’s and Parkinson’s disease. Proc. Natl. Acad. Sci. U.S.A. 97, 5440–5444. doi: 10.1073/pnas.97.10.5440
Khabbush, A., Orford, M., Tsai, Y., Rutherford, T., O’Donnell, M., Eaton, S., et al. (2017). Neuronal decanoic acid oxidation is markedly lower than that of octanoic acid: A mechanistic insight into the medium-chain triglyceride ketogenic diet. Epilepsia 58, 1423–1429.
Kim, D. Y., Davis, L. M., Sullivan, P. G., Maalouf, M., Simeone, T. A., Brederode, J v, et al. (2007). Ketone bodies are protective against oxidative stress in neocortical neurons. J. Neurochem. 101, 1316–1326. doi: 10.1111/j.1471-4159.2007.04483.x
Klionsky, D. J., Askanas, V., Aurelian, L., Bae, J., Baek, S., Balaji, K. N., et al. (2016). Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). AUTOPHAGY 12, 1–222. doi: 10.1080/15548627.2015.1100356
Komatsu, M., Waguri, S., Ueno, T., Iwata, J., Murata, S., Tanida, I., et al. (2005). Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 169, 425–434. doi: 10.1083/jcb.200412022
Krikorian, R., Shidler, M. D., Dangelo, K., Couch, S. C., Benoit, S. C., and Clegg, D. J. (2012). Dietary ketosis enhances memory in mild cognitive impairment. Neurobiol. Aging 33, .e19–.e425. doi: 10.1016/j.neurobiolaging.2010.10.006
Kumari, U., and Tan, E. K. (2009). LRRK2 in Parkinson’s disease: Genetic and clinical studies from patients. FEBS J. 276, 6455–6463. doi: 10.1111/j.1742-4658.2009.07344.x
Kunz, J. B., Schwarz, H., and Mayer, A. (2004). Determination of four sequential stages during microautophagy in vitro. J. Biol. Chem. 279, 9987–9996. doi: 10.1074/jbc.M307905200
Lalla, R., and Donmez, G. (2013). The role of sirtuins in Alzheimer’s disease. Front. Aging Neurosci. 5:16. doi: 10.3389/fnagi.2013.00016
Lattante, S., de Calbiac, H., Le Ber, I., Brice, A., Ciura, S., and Kabashi, E. (2015). Sqstm1 knock-down causes a locomotor phenotype ameliorated by rapamycin in a zebrafish model of ALS/FTLD. Hum. Mol. Genet. 24, 1682–1690. doi: 10.1093/hmg/ddu580
Lee, A., Kondapalli, C., Virga, D. M., Lewis, T. L., Koo, S. Y., Ashok, A., et al. (2022). Aβ42 oligomers trigger synaptic loss through CAMKK2-AMPK-dependent effectors coordinating mitochondrial fission and mitophagy. Nat. Commun. 13:4444. doi: 10.1038/s41467-022-32130-5
Li, L., Zhang, X., and Le, W. (2008). Altered macroautophagy in the spinal cord of SOD1 mutant mice. Autophagy 4, 290–293. doi: 10.4161/auto.5524
Lilienbaum, A. (2013). Relationship between the proteasomal system and autophagy. Int. J. Biochem. Mol. Biol. 4, 1–26.
Lillo, P., and Hodges, J. R. (2009). Frontotemporal dementia and motor neurone disease: Overlapping clinic-pathological disorders. J. Clin. Neurosci. 16, 1131–1135. doi: 10.1016/j.jocn.2009.03.005
Lim, K., Ng, X., Grace, L. G., and Yao, T. (2012). Mitochondrial dynamics and Parkinson’s disease: Focus on parkin. Antioxid. Redox Signal. 16, 935–949. doi: 10.1089/ars.2011.4105
Lipinski, M. M., Zheng, B., Lu, T., Yan, Z., Py, B. F., Ng, A., et al. (2010). Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 107, 14164–14169. doi: 10.1073/pnas.1009485107
Liu, C., Lin, Y., Chen, Y., Chen, C., Pang, L., Chen, H., et al. (2016). Cul3-KLHL20 ubiquitin ligase governs the turnover of ULK1 and VPS34 complexes to control autophagy termination. Mol. Cell 61, 84–97. doi: 10.1016/j.molcel.2015.11.001
Liu, G. Y., and Sabatini, D. M. (2020). mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 21, 183–203. doi: 10.1038/s41580-019-0199-y
Liu, L., Peritore, C., Ginsberg, J., Kayhan, M., and Donmez, G. (2015). SIRT3 attenuates MPTP-induced nigrostriatal degeneration via enhancing mitochondrial antioxidant capacity. Neurochem. Res. 40, 600–608. doi: 10.1007/s11064-014-1507-8
Liu, T., Xiong, X., Ren, X., Zhao, M., Shi, C., Wang, J., et al. (2016). FNDC5 alleviates hepatosteatosis by restoring AMPK/mTOR-mediated autophagy, fatty acid oxidation, and lipogenesis in mice. Diabetes 65, 3262–3275. doi: 10.2337/db16-0356
Longhena, F., Spano, P., and Bellucci, A. (2017). “Targeting of Disordered Proteins by Small Molecules in Neurodegenerative Diseases,” in Targeting Trafficking in Drug Development. Handbook of Experimental Pharmacology, eds A. Ulloa-Aguirre and Y. Tao (Cham: Springer), doi: 10.1007/164_2017_60
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M., and Kroemer, G. (2013). The hallmarks of aging. Cell 153, 1194–1217. doi: 10.1016/j.cell.2013.05.039
Loscher, W., Potschka, H., Sisodiya, S. M., and Vezzani, A. (2020). Drug resistance in epilepsy: Clinical impact, potential mechanisms, and new innovative treatment options. Pharmacol. Rev. 71, 606–638.
Lucin, K. M., O’Brien, C. E., Bieri, G., Czirr, E., Mosher, K. I., Abbey, R. J., et al. (2013). Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer’s disease. Neuron 79, 873–886.
Luque, F. A., and Jaffe, S. L. (2009). The molecular and cellular pathogenesis of dementia of the Alzheimer’s type an overview. Int. Rev. Neurobiol. 84:151.
Lutas, A., and Yellen, G. (2013). The ketogenic diet: Metabolic influences on brain excitability and epilepsy. Trends Neurosci. 36, 32–40.
Maalouf, M., Sullivan, P. G., Davis, L., Kim, D. Y., and Rho, J. M. (2007). Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience 145, 256–264. doi: 10.1016/j.neuroscience.2006.11.065
Maday, S., Wallace, K. E., and Holzbaur, E. L. F. (2012). Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurones. J. Cell Biol. 196, 407–417. doi: 10.1083/jcb.201106120
Magrané, J., Cortez, C., Gan, W., and Manfredi, G. (2014). Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum. Mol. Genet. 23, 1413–1424. doi: 10.1093/hmg/ddt528
Majeski, A. E., and Fred Dice, J. (2004). Mechanisms of chaperone-mediated autophagy. Amsterdam: Elsevier Ltd.
Malapaka, R. R. V., Khoo, S., Zhang, J., Choi, J. H., Zhou, X. E., Xu, Y., et al. (2012). Identification and mechanism of 10-carbon fatty acid as modulating ligand of peroxisome proliferator-activated receptors. J. Biol. Chem. 287, 183–195. doi: 10.1074/jbc.M111.294785
Manczak, M., Park, B. S., Jung, Y., and Reddy, P. H. (2004). Differential expression of oxidative phosphorylation genes in patients with Alzheimer’s disease: Implications for early mitochondrial dysfunction and oxidative damage. Neuromolec. Med. 5:147.
Manzo, E., O’Conner, A. G., Barrows, J. M., Shreiner, D. D., Birchak, G. J., and Zarnescu, D. C. (2018). Medium-chain fatty acids, beta-hydroxybutyric acid and genetic modulation of the carnitine shuttle are protective in a Drosophila model of ALS based on TDP-43. Front. Mol. Neurosci. 11:182. doi: 10.3389/fnmol.2018.00182
Mao, D., Lin, G., Tepe, B., Zuo, Z., Tan, K. L., Senturk, M., et al. (2019). VAMP associated proteins are required for autophagic and lysosomal degradation by promoting PtdIns4P-mediated endosomal pathway. Autophagy 15, 1214–1233.
Martin, K., Jackson, C. F., Levy, R. G., and Cooper, P. N. (2016). Ketogenic diet and other dietary treatments for epilepsy. Cochrane Database Syst. Rev. 2, CD001903. doi: 10.1002/14651858.CD001903.pub3
Martínez-Reyes, I., and Chandel, N. S. (2020). Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 11:102. doi: 10.1038/s41467-019-13668-3
Masino, S. A. (2022). Ketogenic diet and metabolic therapies: Expanded roles in health and disease. Oxford: Oxford University Press.
Maswood, W., Annamaraju, P., and Uppaluri, K. R. (2020). Ketogenic Diet. Treasure Island, FL: StatPearls Publishing.
Matecic, M., Smith, D. L., Pan, X., Maqani, N., Bekiranov, S., Boeke, J. D., et al. (2010). A microarray-based genetic screen for yeast chronological aging factors. PLoS Genet. 6:1000921. doi: 10.1371/journal.pgen.1000921
McCarty, M. F., DiNicolantonio, J. J., and O’Keefe, J. H. (2015). Ketosis may promote brain macroautophagy by activating Sirt1 and hypoxia-inducible factor-1. Med. Hypoth. 85, 631–639. doi: 10.1016/j.mehy.2015.08.002
McDaniel, S. S., Rensing, N. R., Thio, L. L., Yamada, K. A., and Wong, M. (2011). The ketogenic diet inhibits the mammalian target of rapamycin (mTOR) pathway. Epilepsia 52, 7. doi: 10.1111/j.1528-1167.2011.02981.x
Mesquita, A., Cardenal-Muñoz, E., Dominguez, E., Muñoz-Braceras, S., Nuñez-Corcuera, B., Phillips, B. A., et al. (2017). Autophagy in Dictyostelium: Mechanisms, regulation and disease in a simple biomedical model. Autophagy 13, 24–40. doi: 10.1080/15548627.2016.1226737
Michalik, L., Auwerx, J., Berger, J. P., Chatterjee, V. K., Glass, C. K., Gonzalez, F. J., et al. (2006). International Union of Pharmacology. LXI. Peroxisome Proliferator-Activated Receptors. Pharmacol. Rev. 58:726. doi: 10.1124/pr.58.4.5
Min, S., Cho, S., Zhou, Y., Schroeder, S., Haroutunian, V., Seeley, W. W., et al. (2010). Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 67, 953–966. doi: 10.1016/j.neuron.2010.08.044
Mizushima, N. (2009). The role of the Atg1/ULK1 complex in autophagy regulation. Curr. Opin. Cell Biol. 22, 132–139. doi: 10.1016/j.ceb.2009.12.004
Mookerjee, S. A., Divakaruni, A. S., Jastroch, M., and Brand, M. D. (2010). Mitochondrial uncoupling and lifespan. Mech. Ageing Dev. 131, 463–472. doi: 10.1016/j.mad.2010.03.010
Moreira, P. I., Duarte, A. I., Santos, M. S., Rego, A. C., and Oliveira, C. R. (2009). An Integrative View of the Role of Oxidative Stress, Mitochondria and Insulin in Alzheimer’s Disease. J. Alzheimers Dis. 16, 741–761. doi: 10.3233/JAD-2009-0972
Morel, E., Chamoun, Z., Lasiecka, Z. M., Chan, R. B., Williamson, R. L., Vetanovetz, C., et al. (2013). Phosphatidylinositol-3-phosphate regulates sorting and processing of amyloid precursor protein through the endosomal system. Nat. Commun. 4:2250. doi: 10.1038/ncomms3250
Moreno, C. L., and Mobbs, C. V. (2017). Epigenetic mechanisms underlying lifespan and age-related effects of dietary restriction and the ketogenic diet. Mol. Cell Endocrinol. 455, 33–40.
Mortiboys, H., Johansen, K. K., Aasly, J. O., and Bandmann, O. (2010). Mitochondrial impairment in patients with Parkinson disease with the G2019S mutation in LRRK2. Neurology 75, 2017–2020. doi: 10.1212/WNL.0b013e3181ff9685
Mullin, S., and Schapira, A. (2013). α-Synuclein and mitochondrial dysfunction in Parkinson’s disease. Mol. Neurobiol. 47, 587–597. doi: 10.1007/s12035-013-8394-x
Nazio, F., Carinci, M., Valacca, C., Bielli, P., Strappazzon, F., Antonioli, M., et al. (2016). Fine-tuning of ULK1 mRNA and protein levels is required for autophagy oscillation. J. Cell Biol. 215, 841–856. doi: 10.1083/jcb.201605089
Neal, E. (2017). “Alternative ketogenic diets,” in Ketogenic diet and metabolic therapies, ed. S. Masino (New York, NY: Oxford University Press).
Nemoto, S., Fergusson, M. M., and Finkel, T. (2005). SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha}. J. Biol. Chem. 280, 16456–16460. doi: 10.1074/jbc.M501485200
Ng, C., Guan, M. S. H., Koh, C., Ouyang, X., Yu, F., Tan, E., et al. (2012). AMP Kinase Activation Mitigates Dopaminergic Dysfunction and Mitochondrial Abnormalities in Drosophila Models of Parkinson’s Disease. J. Neurosci. 32:14311. doi: 10.1523/JNEUROSCI.0499-12.2012
Nixon, R., Wegiel, J., Kumar, A., Yu, W., Peterhoff, C., Cataldo, A., et al. (2005). Extensive Involvement of Autophagy in Alzheimer Disease: An Immuno-Electron Microscopy Study. J. Neuropathol. Exp. Neurol. 64, 113–122. doi: 10.1093/jnen/64.2.113
Noda, T., and Ohsumi, Y. (1998). ‘Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast’. J. Biol. Chem. 273, 3963–3966. doi: 10.1074/jbc.273.7.3963
Ohsumi, Y., and Mizushima, N. (2004). Two ubiquitin-like conjugation systems essential for autophagy. Semin. Cell Dev. Biol. 15, 231–236. doi: 10.1016/j.semcdb.2003.12.004
Ohsumi, Y., Ichimura, Y., Kirisako, T., Takao, T., Satomi, Y., Shimonishi, Y., et al. (2000). A ubiquitin-like system mediates protein lipidation. Nature 408, 488–492. doi: 10.1038/35044114
Onyango, P., Celic, I., McCaffery, J. M., Boeke, J. D., and Feinberg, A. P. (2002). SIRT3, a human SIR2 homologue, is an NAD-dependent deacetylase localized to mitochondria. Proc. Natl. Acad. Sci. U.S.A. 99, 13653–13658. doi: 10.1073/pnas.222538099
Orenstein, S. J., Kuo, S., Tasset, I., Arias, E., Koga, H., Fernandez-Carasa, I., et al. (2013). Interplay of LRRK2 with chaperone-mediated autophagy. Nat. Neurosci. 16, 394–406. doi: 10.1038/nn.3350
Ota, M., Matsuo, J., Ishida, I., Takano, H., Yokoi, Y., Hori, H., et al. (2019). Effects of a medium-chain triglyceride-based ketogenic formula on cognitive function in patients with mild-to-moderate Alzheimer’s disease. Neurosci. Lett. 690, 232–236. doi: 10.1016/j.neulet.2018.10.048
Paillusson, S., Gomez-Suaga, P., Stoica, R., Little, D., Gissen, P., Devine, M. J., et al. (2017). α-Synuclein binds to the ER-mitochondria tethering protein VAPB to disrupt Ca (2+) homeostasis and mitochondrial ATP production. Acta Neuropathol. 134, 129–149. doi: 10.1007/s00401-017-1704-z
Palmer, C. L., Cotton, L., and Henley, J. M. (2005). The molecular pharmacology and cell biology of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors. Pharmacol. Rev. 57, 253–277. doi: 10.1124/pr.57.2.7
Pankiv, S., Clausen, T. H., Lamark, T., Brech, A., Bruun, J., Outzen, H., et al. (2007). p62/SQSTM1 Binds Directly to Atg8/LC3 to Facilitate Degradation of Ubiquitinated Protein Aggregates by Autophagy. J. Biol. Chem. 282, 24131–24145. doi: 10.1074/jbc.M702824200
Park, J., Davis, R. L., and Sue, C. M. (2018). Mitochondrial Dysfunction in Parkinson’s Disease: New Mechanistic Insights and Therapeutic Perspectives. Curr. Neurol. Neurosci. Rep. 18, 18–13. doi: 10.1007/s11910-018-0829-3
Park, K., Lee, S., Lee, C., Jang, J., Chung, J., Kwon, M., et al. (2009). Upregulation of Beclin-1 expression and phosphorylation of Bcl-2 and p53 are involved in the JNK-mediated autophagic cell death. Biochem. Biophys. Res. Commun. 382, 726–729. doi: 10.1016/j.bbrc.2009.03.095
Pervaiz, M. A., Kendal, F., Hegde, M., and Singh, R. H. (2011). MCT oil-based diet reverses hypertrophic cardiomyopathy in a patient with very long chain acyl-coA dehydrogenase deficiency. Indian J. Hum. Genet. 17, 29–32.
Pickett, E. K., Rose, J., McCrory, C., McKenzie, C., King, D., Smith, C., et al. (2018). Region-specific depletion of synaptic mitochondria in the brains of patients with Alzheimer’s disease. Acta Neuropathol. 136, 747–757. doi: 10.1007/s00401-018-1903-2
Pickford, F., Masliah, E., Britschgi, M., Lucin, K., Narasimhan, R., Jaeger, P. A., et al. (2008). The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J. Clin. Invest. 118, 2190–2199.
Pinto, A., Bonucci, A., Maggi, E., Corsi, M., and Businaro, R. (2018). Anti-Oxidant and Anti-Inflammatory Activity of Ketogenic Diet: New Perspectives for Neuroprotection in Alzheimer’s Disease. Antioxidants 7:7050063. doi: 10.3390/antiox7050063
Plowey, E. D., Cherra, S. J. I., Liu, Y., and Chu, C. T. (2008). Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J. Neurochem. 105, 1048–1056. doi: 10.1111/j.1471-4159.2008.05217.x
Poewe, W., Seppi, K., Tanner, C. M., Halliday, G. M., Brundin, P., Volkmann, J., et al. (2017). Parkinson disease. Nat. Rev. Dis. Prim. 3:17014. doi: 10.1038/nrdp.2017.14
Qian, M., Fang, X., and Wang, X. (2017). Autophagy and inflammation. Clin. Transl. Med. 6, 17–15. doi: 10.1186/s40169-017-0154-5
Qin, W., Haroutunian, V., Katsel, P., Cardozo, C. P., Ho, L., Buxbaum, J. D., et al. (2009). PGC-1alpha expression decreases in the Alzheimer disease brain as a function of dementia. Arch. Neurol. 66, 352–361. doi: 10.1001/archneurol.2008.588
Qin, W., Yang, T., Ho, L., Zhao, Z., Wang, J., Chen, L., et al. (2006). Neuronal SIRT1 activation as a novel mechanism underlying the prevention of Alzheimer disease amyloid neuropathology by calorie restriction. J. Biol. Chem. 281, 21745–21754. doi: 10.1074/jbc.M602909200
Rangaraju, S., Verrier, J. D., Madorsky, I., Nicks, J., Dunn, J., William, A., et al. (2010). Rapamycin Activates Autophagy and Improves Myelination in Explant Cultures from Neuropathic Mice. J. Neurosci. 30, 11388–11397. doi: 10.1523/JNEUROSCI.1356-10.2010
Rangaraju, V., Lewis, T. L. J., Hirabayashi, Y., Bergami, M., Motori, E., Cartoni, R., et al. (2019). Pleiotropic Mitochondria: The Influence of Mitochondria on Neuronal Development and Disease. J. Neurosci. 39, 8200–8208. doi: 10.1523/JNEUROSCI.1157-19.2019
Raza, C., Anjum, R., and Shakeel, N. U. A. (2019). Parkinson’s disease: Mechanisms, translational models and management strategies. Life Sci. 226, 77–90. doi: 10.1016/j.lfs.2019.03.057
Reznick, R. M., Zong, H., Li, J., Morino, K., Moore, I. K., Yu, H. J., et al. (2007). Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell Metab. 5, 151–156. doi: 10.1016/j.cmet.2007.01.008
Rice, A. C., Keeney, P. M., Algarzae, N. K., Ladd, A. C., Thomas, R. R., and Bennett, J. P. J. (2014). Mitochondrial DNA copy numbers in pyramidal neurones are decreased and mitochondrial biogenesis transcriptome signalling is disrupted in Alzheimer’s disease hippocampi. J. Alzheimers Dis. 40, 319–330. doi: 10.3233/JAD-131715
Rock, K. L., Gramm, C., Rothstein, L., Clark, K., Stein, R., Dick, L., et al. (1994). Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell 78, 761–771. doi: 10.1016/S0092-8674(94)90462-6
Roy, M., Edde, M., Fortier, M., Croteau, E., Castellano, C., St-Pierre, V., et al. (2022). A ketogenic intervention improves dorsal attention network functional and structural connectivity in mild cognitive impairment. Neurobiol. Aging 115, 77–87.
Ryan, B. J., Hoek, S., Fon, E. A., and Wade-Martins, R. (2015). Mitochondrial dysfunction and mitophagy in Parkinson’s: From familial to sporadic disease. Trends Biochem. Sci. 40, 200–210. doi: 10.1016/j.tibs.2015.02.003
Ryan, S. D., Dolatabadi, N., Chan, S. F., Zhang, X., Akhtar, M. W., Parker, J., et al. (2013). Isogenic human iPSC Parkinson’s model shows nitrosative stress-induced dysfunction in MEF2-PGC1α transcription. Cell 155, 1351–1364. doi: 10.1016/j.cell.2013.11.009
Salehi, B., Mishra, A. P., Nigam, M., Sener, B., Kilic, M., Sharifi-Rad, M., et al. (2018). Resveratrol: A Double-Edged Sword in Health Benefits. Biomedicines 6:91.
Sanguanphun, T., Sornkaew, N., Malaiwong, N., Chalorak, P., Jattujan, P., Niamnont, N., et al. (2022). Neuroprotective effects of a medium chain fatty acid, decanoic acid, isolated from H. leucospilota against Parkinsonism in C. elegans PD model. Front. Pharmacol. 13:1004568. doi: 10.3389/fphar.2022.1004568
Sankar, R., and Sotero de Menezes, M. (1999). Metabolic and endocrine aspects of the ketogenic diet. Epilepsy Res. 37, 191–201. doi: 10.1016/S0920-1211(99)00071-6
Schoeler, N. E., Orford, M., Vivekananda, U., and K.Vita Study Group. (2021). K.Vita: A feasibility study of a blend of medium chain triglycerides to manage drug-resistant epilepsy. Brain Commun. 3, fcab160. doi: 10.1093/braincomms/fcab160
Schonfeld, P., and Wojtczak, L. (2016). Short- and medium-chain fatty acids in energy metabolism: The cellular perspective. J. Lipid Res. 57, 943–954.
Schubert, D. (2005). Glucose metabolism and Alzheimer’s disease. Ageing Res. Rev. 4, 240–257. doi: 10.1016/j.arr.2005.02.003
Sekar, S., Wu, X., Friis, T., Crawford, R., Prasadam, I., and Xiao, Y. (2018). Saturated fatty acids promote chondrocyte matrix remodelling through reprogramming of autophagy pathways. Nutrition 54, 144–152. doi: 10.1016/j.nut.2018.02.018
Selkoe, D. J., and Hardy, J. (2016). The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 8, 595–608. doi: 10.15252/emmm.201606210
Sellier, C., Campanari, M., Julie Corbier, C., Gaucherot, A., Kolb-Cheynel, I., Oulad-Abdelghani, M., et al. (2016). Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neurone dysfunction and cell death. EMBO J. 35, 1276–1297. doi: 10.15252/embj.201593350
Şentürk, M., Mao, B., and Bellen, H. J. (2019). Loss of proteins associated with amyotrophic lateral sclerosis affects lysosomal acidification via different routes. Autophagy 15, 1467–1469.
Shang, L., and Wang, X. (2011). AMPK and mTOR coordinate the regulation of Ulk1 and mammalian autophagy initiation. Autophagy 7, 924–926. doi: 10.4161/auto.7.8.15860
Shaw, P. J., Ince, P. G., Falkous, G., and Mantle, D. (1995). Oxidative damage to protein in sporadic motor neurone disease spinal cord. Ann. Neurol. 38, 691–695. doi: 10.1002/ana.410380424
Sheng, B., Wang, X., Su, B., Lee, H., Casadesus, G., Perry, G., et al. (2012). Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer’s disease. J. Neurochem. 120, 419–429. doi: 10.1111/j.1471-4159.2011.07581.x
Simm, P. J., Bicknell-Royle, J., Lawrie, J., Nation, J., Draffin, K., Stewart, K. G., et al. (2017). The effect of the ketogenic diet on the developing skeleton. Epilepsy Res. 136, 62–66.
Simonsen, A., Cumming, R. C., Brech, A., Isakson, P., Schubert, D. R., and Finley, K. D. (2008). Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy 4, 176–184. doi: 10.4161/auto.5269
Sinclair, P., Baranova, A., and Kabbani, N. (2021). Mitochondrial Disruption by Amyloid Beta 42 Identified by Proteomics and Pathway Mapping. Cells 10:10092380. doi: 10.3390/cells10092380
Smith, R. G., Henry, Y. K., Mattson, M. P., and Appel, S. H. (1998). Presence of 4-hydroxynonenal in cerebrospinal fluid of patients with sporadic amyotrophic lateral sclerosis. Ann. Neurol. 44, 696–699. doi: 10.1002/ana.410440419
Stavoe, A. K. H., and Holzbaur, E. L. F. (2019). Autophagy in Neurons. Annu. Rev. Cell Dev. Biol, 35, 477–500. doi: 10.1146/annurev-cellbio-100818-125242
Stefanis, L., Larsen, K. E., Rideout, H. J., Sulzer, D., and Greene, L. A. (2001). Expression of A53T Mutant But Not Wild-Type alpha -Synuclein in PC12 Cells Induces Alterations of the Ubiquitin-Dependent Degradation System, Loss of Dopamine Release, and Autophagic Cell Death. J. Neurosci. 21, 9549–9560. doi: 10.1523/JNEUROSCI.21-24-09549.2001
Steger, M., Tonelli, F., Ito, G., Davies, P., Trost, M., Vetter, M., et al. (2016). Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. eLife 5:e1281. doi: 10.7554/eLife.12813
Sullivan, P. M., Zhou, X., Robins, A. M., Paushter, D. H., Kim, D., Smolka, M. B., et al. (2016). The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta Neuropathol. Commun. 4:51. doi: 10.1186/s40478-016-0324-5
Suzuki, K., and Terry, R. D. (1967). Fine structural localization of acid phosphatase in senile plaques in Alzheimer’s presenile dementia. Acta Neuropathol. 8, 276–284. doi: 10.1007/BF00688828
Suzuki, K., Kubota, Y., Sekito, T., and Ohsumi, Y. (2007). Hierarchy of Atg proteins in pre-autophagosomal structure organization. Genes Cells 12, 209–218. doi: 10.1111/j.1365-2443.2007.01050.x
Takeishi, J., Tatewaki, Y., Nakase, T., Takano, Y., Tomita, N., Yamamoto, S., et al. (2021). Alzheimer’s disease and type 2 diabetes mellitus: The use of MCT oil and the ketogenic diet. Int. J. Mol. Sci. 22:12310. doi: 10.3390/ijms222212310
Tan, K., Carrasco-Pozo, C., McDonald, T., Puchowicz, M., and Borges, K. (2017). Tridecanoin is anticonvulsant, antioxidant, and improves mitochondrial function. J. Cereb. Blood Flow Metab. 37, 2035–2048. doi: 10.1177/0271678X16659498
Taoufik, E., Kouroupi, G., Zygogianni, O., and Matsas, R. (2018). Synaptic dysfunction in neurodegenerative and neurodevelopmental diseases: An overview of induced pluripotent stem-cell-based disease models. Open Biol. 8, 180138. doi: 10.1098/rsob.180138
Taylor, J. P., Brown, J., Robert, H., and Cleveland, D. W. (2016). Decoding ALS: From genes to mechanism. Nature 539, 197–206. doi: 10.1038/nature20413
Taylor, M. K., Sullivan, D. K., Mahnken, J. D., Burns, J. M., and Swerdlow, R. H. (2018). Feasibility and efficacy data from a ketogenic diet intervention in Alzheimer’s disease. Alzheimers Dement. 4, 28–36. doi: 10.1016/j.trci.2017.11.002
Tefera, T. W., Tan, K. N., McDonald, T. S., and Borges, K. (2017). Alternative fuels in Epilepsy and Amyotrophic Lateral Sclerosis. Neurochem. Res. 42, 1610–1620.
Tieu, K., Perier, C., Caspersen, C., Teismann, P., Wu, D., Yan, S., et al. (2003). D-beta-hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J. Clin. Investig. 112, 892–901. doi: 10.1172/JCI18797
Tong, X., Deng, Y., Liu, L., Tang, X., Yu, T., Gan, J., et al. (2022). Clinical implementation of ketogenic diet in children with drug-resistant epilepsy: Advantages, disadvantages, and difficulties. Seizure 99, 75–81. doi: 10.1016/j.seizure.2022.04.015
Tóth, M. L., Sigmond, T., Borsos, E., Barna, J., Erdélyi, P., Takács-Vellai, K., et al. (2008). Longevity pathways converge on autophagy genes to regulate life span in Caenorhabditis elegans. Autophagy 4, 330–338. doi: 10.4161/auto.5618
Van Damme, P. (2009). The role of AMPA receptors and VEGF in ALS. Verh. K Acad. Geneeskd. Belg. 71, 241–250.
Varga, T., Czimmerer, Z., and Nagy, L. (2011). PPARs are a unique set of fatty acid regulated transcription factors controlling both lipid metabolism and inflammation. Biochim. Biophys. Acta 1812, 1007–1022. doi: 10.1016/j.bbadis.2011.02.014
Vinciguerra, M., Veyrat-Durebex, C., Moukil, M. A., Rubbia-Brandt, L., Rohner-Jeanrenaud, F., and Foti, M. (2008). PTEN down-regulation by unsaturated fatty acids triggers hepatic steatosis via an NF-kappaBp65/mTOR-dependent mechanism. Gastroenterology 134, 268–280. doi: 10.1053/j.gastro.2007.10.010
Volpicelli-Daley, L. A., Luk, K. C., and Lee, V. M. (2014). Addition of exogenous α-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous α-synuclein to Lewy body and Lewy neurite–like aggregates. Nat. Protoc. 9, 2135–2146. doi: 10.1038/nprot.2014.143
Wang, M. C., Bohmann, D., and Jasper, H. (2005). JNK Extends Life Span and Limits Growth by Antagonizing Cellular and Organism-Wide Responses to Insulin Signalling. Cell 121, 115–125. doi: 10.1016/j.cell.2005.02.030
Wang, M., Singh, B. K., Hsu, M., Huang, C., Yen, P. M., Wu, L., et al. (2017). Increasing Dietary Medium-Chain Fatty Acid Ratio Mitigates High-fat Diet-Induced Non-Alcoholic Steatohepatitis by Regulating Autophagy. Sci. Rep. 7, 13999–13913. doi: 10.1038/s41598-017-14376-y
Wang, X., Su, B., Lee, H., Li, X., Perry, G., Smith, M. A., et al. (2009). Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 29, 9090–9103. doi: 10.1523/JNEUROSCI.1357-09.2009
Warren, E. C., Dooves, S., Lugarà, E., Damstra-Oddy, J., Schaf, J., Heine, V. M., et al. (2020). Decanoic acid inhibits mTORC1 activity independent of glucose and insulin signalling. Proc. Natl. Acad. Sci. U.S.A. 117:23617. doi: 10.1073/pnas.2008980117
Warren, E. C., Kramár, P., Lloyd-Jones, K., and Williams, R. S. B. (2021). Decanoic Acid Stimulates Autophagy in D. discoideum. Cells 10:2946. doi: 10.3390/cells10112946
Whitehead, G., Regan, P., Whitcomb, D. J., and Cho, K. (2017). Ca (2+)-permeable AMPA receptor: A new perspective on amyloid-beta mediated pathophysiology of Alzheimer’s disease. Neuropharmacology 112, 221–227. doi: 10.1016/j.neuropharm.2016.08.022
Wiedemann, F. R., Manfredi, G., Mawrin, C., Beal, M. F., and Schon, E. A. (2002). Mitochondrial DNA and respiratory chain function in spinal cords of ALS patients. J. Neurochem. 80, 616–625. doi: 10.1046/j.0022-3042.2001.00731.x
Wiedemann, F. R., Winkler, K., Kuznetsov, A. V., Bartels, C., Vielhaber, S., Feistner, H., et al. (1998). Impairment of mitochondrial function in skeletal muscle of patients with amyotrophic lateral sclerosis. J. Neurol. Sci. 156, 65–72.
Wilong, A. A. (2007). Complications and Consequences of Epilepsy Surgery, Ketogenic Diet, and Vagus Nerve Stimulation. Semin. Pediatr. Neurol. 14, 201–203. doi: 10.1016/j.spen.2007.08.007
Winding, M., Pedigo, B. D., Barnes, C. L., Patsolic, H. G., Park, Y., Kazimiers, T., et al. (2023). The connectome of an insect brain. Science 379, eadd9330. doi: 10.1126/science.add9330
Wlaz, P., Socala, K., Nieoczym, D., Luszczki, J. J., Zarnowska, I., Zarnowski, T., et al. (2012). Anticonvulsant profile of caprylic acid, a main constituent of the medium-chain triglyceride (MCT) ketogenic diet, in mice. Neuropharmacology 62, 1882–1889.
Wlaz, P., Socala, K., Nieoczym, D., Zarnowski, T., Zarnowska, I., Czuczwar, S. J., et al. (2015). Acute anticonvulsant effects of capric acid in seizure tests in mice. Prog. Neuropsychopharmacol. Biol. Psychiatry 57, 110–116.
Wu, J. J., Cai, A., Greenslade, J. E., Higgins, N. R., Fan, C., Le, N. T. T., et al. (2020). ALS/FTD mutations in UBQLN2 impede autophagy by reducing autophagosome acidification through loss of function. Proc. Natl. Acad. Sci. U.S.A. 117, 15230–15241.
Xu, L., Ma, X., Verma, N., Perie, L., Pendse, J., Shamloo, S., et al. (2020). PPARγ agonists delay age-associated metabolic disease and extend longevity. Aging Cell 19:e13267.
Yang, H., Shan, W., Zhu, F., Wu, J., and Wang, Q. (2019). Ketone bodies in neurological diseases: Focus on neuroprotection and underlying mechanisms. Front. Neurol. 10:585. doi: 10.3389/fneur.2019.00585
Yang, W., Zou, Y., Zhang, M., Zhao, N., Tian, Q., Gu, M., et al. (2015). Mitochondrial Sirt3 Expression is Decreased in APP/PS1 Double Transgenic Mouse Model of Alzheimer’s Disease. Neurochem. Res. 40, 1576–1582. doi: 10.1007/s11064-015-1630-1
Yang, Y., Huang, J., Li, J., Yang, H., and Yin, Y. (2020). The Effects of Lauric Acid on IPEC-J2 Cell Differentiation, Proliferation, and Death. Curr. Mol. Med. 20, 572–581. doi: 10.2174/1566524020666200128155115
Yu, W. H., Cuervo, A. M., Kumar, A., Peterhoff, C. M., Schmidt, S. D., Lee, J., et al. (2005). Macroautophagy: A Novel β-Amyloid Peptide-Generating Pathway Activated in Alzheimer’s Disease. J. Cell Biol. 171, 87–98. doi: 10.1083/jcb.200505082
Yue, M., Hinkle, K. M., Davies, P., Trushina, E., Fiesel, F. C., Christenson, T. A., et al. (2015). Progressive dopaminergic alterations and mitochondrial abnormalities in LRRK2 G2019S knock-in mice. Neurobiol. Dis. 78, 172–195. doi: 10.1016/j.nbd.2015.02.031
Zeng, Q., Stylianou, T., Preston, J., Glover, S., O’Neill, K., Woolf, E. C., et al. (2019). A ketogenic diet alters the epigenetic landscape of GBM to potentiate the effects of chemotherapy and radiotherapy. Neuro Oncol. 21, iv8. doi: 10.1093/neuonc/noz167.033
Zhang, X., Li, L., Chen, S., Yang, D., Wang, Y., Zhang, X., et al. (2011). Rapamycin treatment augments motor neuron degeneration in SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Autophagy 7, 412–425. doi: 10.4161/auto.7.4.14541
Zhang, X. J., Chen, S., Huang, K. X., and Le, W. D. (2013). Why should autophagic flux be assessed? Acta Pharmacol. Sin. 34, 595–599.
Zhao, J., Wang, X., Huo, Z., Chen, Y., Liu, J., Zhao, Z., et al. (2022). The impact of mitochondrial dysfunction in amyotrophic lateral sclerosis. Cells 11:2049.
Zhao, R., Jiang, S., Zhang, L., and Yu, Z. (2019). Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 44, 3–15. doi: 10.3892/ijmm.2019.4188
Zhao, W., Varghese, M., Vempati, P., Dzhun, A., Cheng, A., Wang, J., et al. (2012). Caprylic triglyceride as a novel therapeutic approach to effectively improve the performance and attenuate the symptoms due to the motor neurone loss in ALS disease. PLoS One 7:e49191. doi: 10.1371/journal.pone.0049191
Zhao, Z., Lange, D. J., Voustianiouk, A., MacGrogan, D., Ho, L., Suh, J., et al. (2006). A ketogenic diet as a potential novel therapeutic intervention in amyotrophic lateral sclerosis. BMC Neurosci. 7:29. doi: 10.1186/1471-2202-7-29
Zheng, B., Liao, Z., Locascio, J. J., Lesniak, K. A., Roderick, S. S., Watt, M. L., et al. (2010). PGC-1α, A Potential Therapeutic Target for Early Intervention in Parkinson’s Disease. Sci. Transl. Med. 2:52ra73. doi: 10.1126/scitranslmed.3001059
Zhou, Z., Hagopian, K., López-Domínguez, J. A., Kim, K., Jasoliya, M., Roberts, M. N., et al. (2021). A ketogenic diet impacts markers of mitochondrial mass in a tissue specific manner in aged mice. Aging 13, 7914–7930. doi: 10.18632/aging.202834
Zhu, X., Perry, G., Smith, M. A., and Wang, X. (2013). Abnormal mitochondrial dynamics in the pathogenesis of Alzheimer’s disease. J. Alzheimers Dis. 33, 253–262. doi: 10.3233/JAD-2012-129005
Keywords: ageing, amyotrophic lateral sclerosis, Parkinson’ s disease, Alzheimer’s disease, medium chain fatty acid (MCFA), autophagy, mitochondria, ketogenic diet (KD)
Citation: Dunn E, Zhang B, Sahota VK and Augustin H (2023) Potential benefits of medium chain fatty acids in aging and neurodegenerative disease. Front. Aging Neurosci. 15:1230467. doi: 10.3389/fnagi.2023.1230467
Received: 29 May 2023; Accepted: 07 August 2023;
Published: 23 August 2023.
Edited by:
Alexey Moskalev, Komi Scientific Center (RAS), RussiaReviewed by:
Heather M. Wilkins, University of Kansas Medical Center Research Institute, United StatesCristina Pintado, University of Castilla-La Mancha, Spain
Copyright © 2023 Dunn, Zhang, Sahota and Augustin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hrvoje Augustin, aHJ2b2plLmF1Z3VzdGluQHJodWwuYWMudWs=
†These authors have contributed equally to this work and share first authorship