 Mayue Yu
Mayue Yu Manqing Zhang3†
Manqing Zhang3† Moxin Wu
Moxin Wu Xiaoping Yin
Xiaoping Yin Zhiying Chen
Zhiying Chen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Aging Neurosci., 03 August 2023
Sec. Cellular and Molecular Mechanisms of Brain-aging
Volume 15 - 2023 | https://doi.org/10.3389/fnagi.2023.1224633
Chronic cerebral ischemia (CCI), a condition that can result in headaches, dizziness, cognitive decline, and stroke, is caused by a sustained decrease in cerebral blood flow. Statistics show that 70% of patients with CCI are aged > 80 years and approximately 30% are 45–50 years. The incidence of CCI tends to be lower, and treatment for CCI is urgent. Studies have confirmed that CCI can activate the corresponding mechanisms that lead to mitochondrial dysfunction, which, in turn, can induce mitophagy to maintain mitochondrial homeostasis. Simultaneously, mitochondrial dysfunction can aggravate the insufficient energy supply to cells and various diseases caused by CCI. Regulation of mitophagy has become a promising therapeutic target for the treatment of CCI. This article reviews the latest progress in the important role of mitophagy in CCI and discusses the induction pathways of mitophagy in CCI, including ATP synthesis disorder, oxidative stress injury, induction of reactive oxygen species, and Ca2+ homeostasis disorder, as well as the role of drugs in CCI by regulating mitophagy.
Chronic cerebral ischemia (CCI) is considered low-efficiency functional congestion caused by long-term vascular disease or circulatory disorders. It plays a crucial role in cerebrovascular and neurodegenerative diseases and can lead to diseases such as vascular dementia (VD) and Alzheimer’s disease (AD) (Gao, 2018; Ciacciarelli et al., 2020; Li et al., 2022). Studies have shown that symptoms such as headache and dizziness caused by CCI are reversible when cerebral blood supply insufficiency is relieved (Calabrese et al., 2016). Active secondary prevention can reduce ischemic stroke recurrence by approximately 80% (Hankey, 2014). In contrast, the risk of acute stroke, vascular cognitive impairment, and dementia increases if the ongoing decline in cerebral blood flow is not corrected in a timely manner (Liao et al., 2016; Rajeev et al., 2022). According to the Global Burden of Disease (2020) statistical report, the incidence of ischemic stroke worldwide accounted for 64.8%, with a prevalence of 76.5%.
In addition to serving, as a source of bioenergy, mitochondria directly regulate programmed cell death (Kislin et al., 2017). Mitochondrial damage has been reported as a pathological mechanism leading to ischemic neuronal death (Anzell et al., 2018). Autophagy, an intracellular lysosomal degradation pathway, can be classified into canonical and non-canonical pathways. Autophagy processes have been shown to include autophagosome induction and formation and autophagic flux (Xie and Klionsky, 2007). Autophagic flux consists of autophagosome trafficking and fusion with lysosomes to form autophagolysosomes, in which autophagic contents are broken down (Xie and Klionsky, 2007). Mitophagy is the process of targeting damaged or dysfunctional mitochondria and delivering them to lysosomes for degradation, complete self-renewal, and maintaining homeostasis (Pickles et al., 2018). Several CCI-induced neurodegenerative diseases, including VD and AD, are significantly influenced by mitophagy (Arun et al., 2016). Mitochondrial autophagy has a dual function. Its negative effect is the induction of neuronal death (cytodestructive autophagy), while its protective function is to prevent the accumulation of damaged mitochondria (cytoprotective autophagy) (Zhang et al., 2021). If its protective properties can be used effectively, the regulation of mitochondrial autophagy may be a valuable therapeutic target. However, compared to research on the mechanism of mitophagy in acute cerebral ischemia, insufficient research has been conducted on this mechanism in CCI nationally and internationally (Nguyen et al., 2018; Wang et al., 2021; Li et al., 2023). In light of these circumstances, this study aimed to explore the mechanism of mitophagy and its function in CCI and to offer new suggestions for clinical management.
The brain uses more oxygen than any other organ and is highly metabolically active. Although it makes up only 2% of the human body by weight, brain tissue delivers 25% of the glucose and approximately 20% of the oxygen required by the body (Siwicka-Gieroba et al., 2022). Brief periods of ischemia and hypoxia can seriously harm the brain. Mitochondria play a crucial role in cellular energy stations such as ATP production, reactive oxygen species production, Ca2+ homeostasis, and apoptosis (Tang et al., 2016). A detailed diagram of this mechanism is shown in Figure 1. As a result, the normal physiological activities of brain cells are closely related to the normal function of mitochondria.
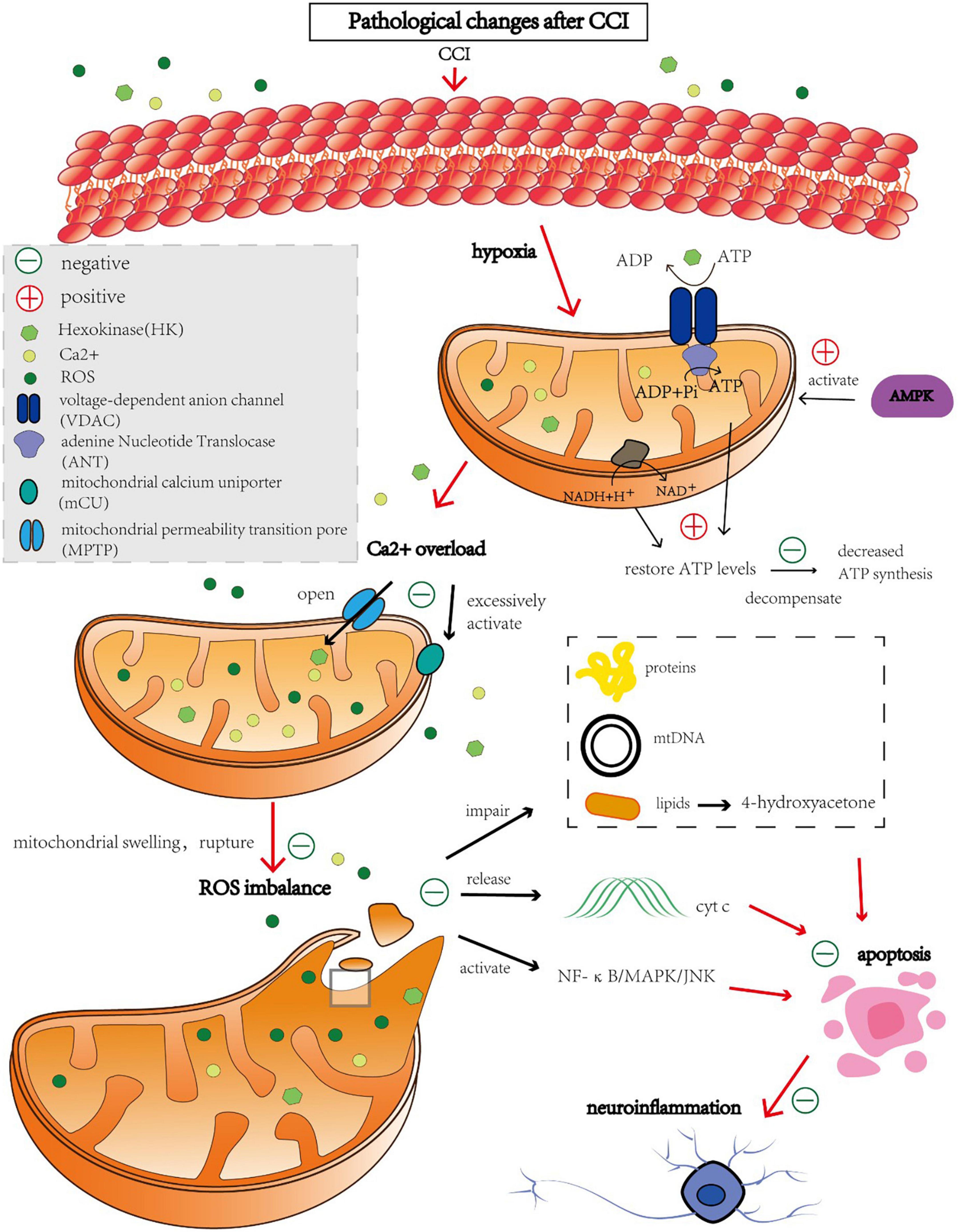
Figure 1. Molecules causing mitochondrial autophagy after CCI and regulatory protective targets.
Metabolic disorders are believed to be the first causal factor of CCI. Following cerebral ischemia, partial pressure of oxygen in the brain decreases. Most aerobic oxidation pathways switch to anaerobic glycolysis, and adenosine monophosphate-activated protein kinase (AMPK) is activated. Active AMPK phosphorylates multiple downstream substrate proteins, inhibits the biosynthetic pathway of ATP consumption, and negatively regulates ATP regeneration to restore cellular energy levels as much as possible (Andjelkovic et al., 2019). However, as the duration of ischemia increases, this negative feedback cannot compensate for the loss of mitochondrial energy and downregulation of the expression of proteases involved in oxidative phosphorylation complexes, such as nicotinamide adenine dinucleotide dehydrogenase and cytochrome oxidase, which reduce ATP synthesis (He et al., 2012).
Mitochondrial permeability transition pore (MPTP) is a non-specific voltage-dependent special protein complex that crosses the mitochondrial outer membrane and controls mitochondrial permeability (Halestrap, 2009). In the physiological state, MPTP is switched off. However, the MPTP is open during ischemia, which is triggered by Ca2+ overload and elevated oxidative stress in the mitochondrial matrix (Kushnareva and Sokolove, 2000; Zhao et al., 2019). The opening of the MPTP leads to an increase in mitochondrial permeability, which allows solutes such as water, macromolecules, and ions to freely enter the mitochondrial matrix, resulting in mitochondrial swelling, outer membrane rupture, and the release of large amounts of reactive oxygen species (ROS) (Krasnikov et al., 2005). In addition, increased mitochondrial permeability also leads to the loss of membrane potential, which in turn lowers cellular mitochondrial ATP levels, enhances intracellular Ca2+ concentration, and activates the endogenous apoptotic pathway, thereby inducing neuronal damage caused by ischemia and hypoxia (Zorov et al., 2000; Broughton et al., 2009).
Nuclear respiratory factor 2 (Nrf2) is a key transcription factor of antioxidants (Hannan et al., 2020). When cells undergo oxidative stress, Nrf2 is activated, enters the nucleus, binds to promoters of antioxidant response genes and promotes their transcription and expression (Yen et al., 2016). These genes include superoxide dismutase, glutathione peroxidase, and glutathione S-transferase, which scavenge free radicals and other oxidative substances, reducing damage from oxidative stress in cells (Yang et al., 2019). URB597 alleviates ischemic cerebrovascular disease by activating the Nrf2 pathway, reducing mitochondrial oxidative stress and inflammation (Wang et al., 2022).
When entering equilibrium with the antioxidant system, ROS cause minimal damage under typical circumstances (Takizawa et al., 1998). However, after CCI, the activity of the respiratory chain enzyme complex is inhibited, mitochondrial respiratory dysfunction occurs, and excessive ROS (Yu et al., 2014). Excessive ROS damage to proteins, mtDNA, and lipids leads to apoptosis, neuroinflammation, and destruction of the blood-brain barrier in the ischemic brain (Shirley et al., 2014). Excess ROS levels induce apoptosis through lipid peroxidation. In rats with cerebral ischemia, 4-hydroxyacetone, a by-product of lipid peroxidation, increases and induces axonal damage and oligodendrocyte apoptosis (Mccracken et al., 2000; Matsuda et al., 2009). ROS can also lead to cell apoptosis by releasing cytochrome c (Cyt c), improving mitochondrial permeability and activating the NF-κB/MAPK/JNK pathway (Kim et al., 2006, 2010). In particular, Cyt c is a soluble protein anchored to the inner mitochondrial membrane, and upon release from mitochondria, it triggers a cascade of apoptotic signaling, which typically peaks after ischemia in Cyt c release (Hüttemann et al., 2011; Tajiri et al., 2016). Mammalian target of rapamycin (mTOR) is an important cell signal transduction pathway, which is involved in the regulation of cell growth, metabolism and autophagy (Saxton and Sabatini, 2017). Activation of mTOR signaling pathway can inhibit ROS production. Ethidium bromide, for example, induces mitochondrial clearance through the autophagy pathway (Luo et al., 2013). However, inhibition of mTOR with rapamycin preserved mitochondrial membrane potential and reduced the production of ROS (Nacarelli et al., 2014). In addition, sertraline is a selective serotonin reuptake inhibitor (SSRI) that regulates AMPK-mTOR signal-mediated autophagy by targeting the mitochondrial voltage-dependent anion channels 1 protein (VDAC1) (Hwang et al., 2021). To sum up, there is a complex interaction between mTOR, ROS and mitophagy. Most importantly, Autophagy is regulated in the nervous system by activating ROS and mediating the Akt-mTOR signaling pathway (Gao et al., 2019; Liu et al., 2019a,b).
After CCI, cells are unable to maintain a negative membrane potential due to the lack of ATP, and neuronal depolarization results in an influx of calcium ions into the cell (Gouriou et al., 2011). An excessive increase in calcium ion concentration activates the mitochondrial calcium uniporter (mCU) in cells, which changes mitochondrial permeability, impairs its ability to generate ATP, and leads to the release of proapoptotic factors (Gouriou et al., 2011). Preclinical research is currently being conducted on medications that block mCU, such as Ru360 (García-Rivas et al., 2006). Even partial inhibition of calcium uptake prevents mitochondrial depolarization, the opening of large mitochondrial channels, and cytochrome c release.
Hexokinase is a six-carbon sugar phosphorylase involved in glycolysis, from which ATP is produced (Tan and Miyamoto, 2015). Furthermore, after CCI, the levels of VDAC, especially VDAC1, have been found to decrease and the interaction between VDAC1 and hexokinase has been reduced. These changes may result in a reduction in ATP/ADP exchange and affect the transport of small-molecule metabolites required for oxidative phosphorylation to mitochondria, thus inhibiting respiration and affecting mitochondrial energy supply and mitochondrial-mediated apoptosis (He et al., 2012).
The regulation of mitophagy has a wide range of potential applications for the treatment of CCI and the defense of injured brain tissue, as mitophagy mediates a number of signaling pathways that play an important role in the disease. Reviewing prior regulation of mitophagy signaling pathways and regulatory variables has provided information on the study and development of new medications. The mitochondrial autophagy pathway in CCI is shown in Figure 2.

Figure 2. Mitochondrial autophagy pathway in CCI.
Mitophagy is initiated during neuronal apoptosis following CCI through the BNIP3-Cyt c-related pathway and parkin-mediated signaling (Su et al., 2018). After CCI induction, Parkin and BNIP3 expression increased, and Cyt c was released from the mitochondria into the cytoplasm; however, the first two phenomena were significantly attenuated after treatment with the autophagy inhibitor 3-MA. Similarly, URB597 (an orally biocompatible inhibitor of fatty acid amide hydrolase) treatment significantly reversed the increase in Beclin-1, parkin, and BNIP3 protein expression and the decrease in autophagy-related proteins after CCI (Su et al., 2018). Autophagy consists of three major sequential steps: sequestration, transport, and degradation (Mizushima, 2007). During degradation, autophagosomes and their cargo are degraded by lysosomal hydrolases, and lysosomal dysfunction can lead to accumulation of autophagosomes (Mizushima et al., 2008). Some researchers have argued that this accumulation should be treated as an abnormally excessive form of autophagy (Su et al., 2018). Further, the beneficial effects of URB597 on chronic ischemic brain injury occur by inhibiting impaired autophagic degradation and disruption of the Beclin-1/Bcl-2 complex, followed by severing BNIP3-Cyt c and parkin-mediated mitophagy; this ultimately prevents abnormal excessive autophagy and mitophagy (Su et al., 2018).
The biological function of PPAR depends on the coactivation of PPAR-γ coactivator 1α (PGC-1α) (Haemmerle et al., 2011). PGC-1α is a master transcription factor in the regulation of antioxidant enzymes, clearance systems, and mitochondrial biogenesis (Kaarniranta et al., 2020). Once activated by phosphorylation or deacetylation, PGC-1α activates the transcription of NRF 1 and 2, which regulates mitochondrial transcription factor A (TFAM) (Li et al., 2017). TFAM then translocates to the mitochondrial matrix and stimulates mtDNA replication and mitochondrial gene expression (Tang, 2016). The upstream transcription factor Sirt1 regulates PGC-1α by increasing its expression and decreasing its acetylation (Iwabu et al., 2010). Nicotinamide adenine dinucleotide (NAD), a substrate of Sirt1 that regulates Sirt1 expression, improves cognitive function and reduces neuroinflammation in in vivo and in vitro CCI models (Mouchiroud et al., 2013). Furthermore, these therapeutic effects were associated with mitochondrial protection and inhibition of ROS by activating the Sirt1/PGC-1α pathway (Zhao et al., 2021).
Akt activation enhanced GSK-3β phosphorylation, leading to mTOR activation, and the autophagic protein Beclin-1 expression was significantly downregulated, inhibiting cell cytodestructive autophagy (Wang R. C. et al., 2012; Liu et al., 2015). Akt phosphorylation prevents Bax translocation to mitochondria and inhibits Cyt c release as well as destructive autophagy, attenuating CCI-induced neuronal injury (Sadidi et al., 2009; Castillo et al., 2011). Meanwhile, ERK activation upregulates Bcl-2 expression, which negatively regulates destructive autophagy through a combination of Beclin-1 and Bax (Subramanian and Shaha, 2007). Activation of the γ-aminobutyric acid B receptor (GABAB) can attenuate CCI-induced increases in atg5 and atg7 expression and inhibit cytodestructive autophagy and neuronal apoptosis (Lindqvist et al., 2014). Baclofen-induced ERK1/2 phosphorylation can accelerate cytoprotective autophagy by moderately increasing the expression of Beclin-1. Activation of GABAB receptors improves the surface expression of the GABAA receptor α1 subunit, leading to the downregulation of astrocytes and neurons surface and mitochondrial expression, which in turn enhances cytoprotective autophagy (Liu et al., 2015).
The opening of mitoKATP channels is related to potassium uptake from the mitochondrial matrix and maintains the volume of the mitochondrial matrix by reducing the Ca2+ load. Reduced mitochondrial Ca2+ load can inhibit MPTP opening, prevent ROS production in the mitochondria, and inhibit excitatory oxidative stress and cell death (Fornazari et al., 2008). mitoKATP consists of two subunits, Kir6.1, 6.2, and SUR1 or SUR2 (Zhou et al., 2010). Chronic intermittent hypobaric hypoxia (CIHH) can upregulate the protein expression of Kir6.2 and SUR1 in the mitochondria of the hippocampal CA1 region induced by ischemia, thus improving learning and memory dysfunction induced by ischemia in the hippocampal CA1 region. Additionally, CIHH alleviates delay neuronal death (DND) by maintaining mitoKATP activity, thus inhibiting Cyt c-induced apoptosis (Zhang et al., 2016).
Bilateral common carotid artery occlusion (2VO) has been used to create a CCI animal model in most trials to investigate the underlying mechanism (Du et al., 2017). The pathogenic role of cerebral hypoperfusion in neurodegenerative diseases can be understood from data collected using a rat 2VO model (Farkas et al., 2007). The 2VO model has shown that neuronal function is directly affected by mitochondrial bioenergetic abnormalities, which may trigger the onset of VD (Du et al., 2013). CCI is difficult to diagnose because it rarely occurs by itself and frequently cooccurs with other brain lesions (Zhao and Gong, 2015). A summary of drug treatment mechanisms is presented in Table 1.
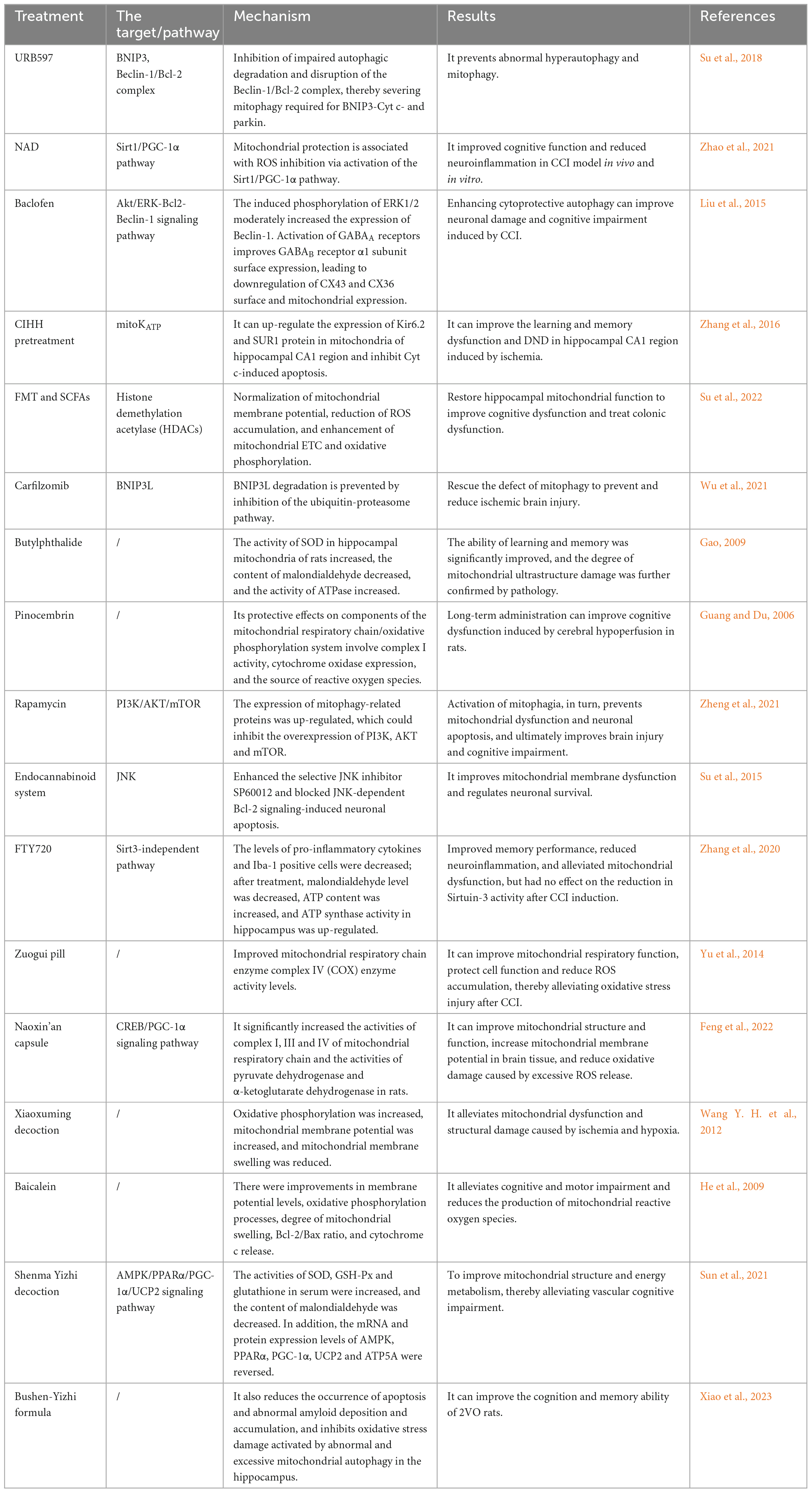
Table 1. Specific performance of CCI therapeutic drugs.
Short-chain fatty acids (SCFAs) produced by bacteria include acetate, propionate, and butyrate. These SCFAs can penetrate the blood-brain barrier and have a considerable impact on the brain due to their effects on numerous neuronal functions and gut-brain signaling pathways. FMT and SCFAs significantly altered Ndufb2 and Atp5mc1 levels, indicating that electron transport chain (ETC) complexes I and V are the main sites for the regulation of oxidative phosphorylation. FMT and SCFAs alleviate mitochondrial dysfunction by increasing acetate, acetyl-CoA, and ATP contents, as well as the activities of complexes I and V of mitochondrial ETC (Su et al., 2022).
Carfilzomib is a proteasome inhibitor that is used to treat multiple myeloma. It forms a covalent irreversible bond with the LMP2 and LMP7 catalytic subunits of the 20S proteasome, which are two intracellular receptors (Sin et al., 1999). Carfilzomib prevents defects in BCL/adenovirus E1B interacting protein 3-like (BNIP3L) degradation and mitophagy deficiency (Wu et al., 2021). Defective mitophagy caused by BNIP3L deletion has significant implications for ischemic neuronal injury. This is because restored BNIP3L has been observed to reduce cerebellar infarct volume, alleviating ischemic brain injury (Wu et al., 2021).
Butylphthalide is a chemical component of celery oil. Superoxide dismutase (SOD) activity increased, malondialdehyde levels decreased, and ATPase activity increased in the hippocampal mitochondria of CCI rats after therapy, significantly improving learning and memory. Pathological results provided additional evidence that injection reduced mitochondrial ultrastructural destruction. Butylphthalide injection has a protective effect on the structure and function of mitochondria in brain tissue, which may be related to its influence on mitochondrial oxidative damage and energy metabolism dysfunction (Gao, 2009).
Pinocembrin is a flavonoid found in propolis that can potentially strengthen the central nervous system. A decrease in transmembrane potential during hypoxia greatly affects mitochondrial function, producing excessive ROS (Nohl et al., 2005). In animal experiments, the expression of Cyt c oxidase in the hippocampus of rats in the 2VO group decreased significantly; meanwhile, mitochondrial membrane potential levels decreased. Pinocembrin significantly reversed these phenomena (Guang and Du, 2006). Cyt c oxidase is a metabolic indicator of neuronal oxidative activity; therefore, this raises the possibility that pinocembrin shields the rat’s brain mitochondria. In addition, pinocembrin can greatly reduce the degree of mitochondrial swelling, increase the mitochondrial membrane potential, and protect the mitochondrial structure and ROS production, which may explain why pinocembrin protects mitochondria from oxidative stress (Guang and Du, 2006).
Rapamycin is a popular allosteric mTOR inhibitor that binds directly to the mTOR complex and promotes autophagy in several eukaryotes. PINK1, Parkin, and LC3B expression levels have been reported to increase after rapamycin treatment in animal studies, stimulating mitophagy and preventing mitochondrial dysfunction and neuronal apoptosis. Together with experimental treatment control of MHY1485 (an mTOR activator) and the initial notion that the mTOR pathway increases autophagy, it also affects the expression of PI3K, AKT, and mTOR (Bartolomé et al., 2017). These findings suggest that rapamycin exerts its neuroprotective effects by suppressing the PI3K/AKT/mTOR signaling pathway, which increases autophagy (Zheng et al., 2021).
The cannabinoid receptor agonist WIN55212-2 (WIN) and the fatty acid amide hydrolase inhibitor URB597 were administered to counteract the effects of CCI on JNK phosphorylation, lowering the Bcl-2/Bax ratio and caspase-3 activation, all of which are involved in controlling neuronal survival. Moreover, WIN and URB597 inhibit neuronal death induced by JNK-dependent Bcl-2 signaling and improve mitochondrial membrane dysfunction by increasing the selective JNK inhibitor SP600125 (Su et al., 2015).
In 2010, the US Food and Drug Administration approved FTY720, a sphingosine-1-phosphate receptor agonist with potent anti-inflammatory properties, as the first oral medication for the treatment of multiple sclerosis (Wang et al., 2020). Moreover, recent studies have shown that it effectively reduces mitochondrial dysfunction and spatial memory impairment (Wang et al., 2020). FTY720 protects the brain from damage by lowering oxidative stress and neuroinflammation and enhancing synaptic function. According to a study in 2VO animals, FTY720 can improve hippocampal mitochondrial function and enhance ATP synthase activity. ATP levels and ATP synthase activity in the hippocampus are increased, suggesting that FTY720 could reduce CCI-induced mitochondrial dysfunction (Zhang et al., 2020). However, p62 expression, which is crucial for the transfer of ubiquitylated substrates to autophagosomes, and SIRT3, the primary regulator of mitochondrial activity, did not show an effect after the intervention (Zhang et al., 2020).
Traditional Chinese medicine has been reported to improve the activity of the ETC complex, decrease calcium overload following excitability toxicity, and restore the self-regulation function of mitochondria by focusing on mitochondrial dysfunction. This preserves the integrity of mitochondrial structure and function, promotes the reconstruction of energy metabolism, and ultimately improves brain injury and cognitive impairment (Wang et al., 2023). For example, the Zuogui pill and Naoxin capsule improve mitochondrial structure and function and reduce ROS accumulation by improving mitochondrial respiratory chain enzyme complexes (Yu et al., 2014; Feng et al., 2022). Xiaoxuming decoction and baicalein have significantly improved oxidative phosphorylation and mitochondrial membrane potential (He et al., 2009; Wang Y. H. et al., 2012). The Shenma Yizhi decoction and Bushen-Yizhi formula can improve mitochondrial dysfunction by regulating the expression levels of various proteins (Sun et al., 2021; Xiao et al., 2023).
The pathogenesis of persistent cerebral ischemia is complex. One of the main reasons for brain injury and aberrant alterations in brain function caused by prolonged cerebral ischemia is the impairment of brain energy metabolism. Increased free radical production, oxidative stress damage, and altered mitochondrial structure and function contribute significantly to the pathophysiology of CCI (Zhou et al., 2021). Therefore, the significance of mitochondrial dysfunction in CCI has received considerable attention, and it is crucial to investigate changes in mitochondrial structure and function to better understand the effect of medications on chronic cerebral ischemia.
Few clinical studies on pharmacological therapy for CCI are currently available, with the majority focusing on the development of new medications to treat cerebral ischemia-reperfusion injury. Most medications play a limited clinical role in the management of persistent cerebral ischemia (Lana et al., 2014; Kim et al., 2016; Yan et al., 2022). According to recent studies, URB597 blocks the Parkin route to restrict mitophagy, NAD stimulates the PPAR pathway to prevent ROS release, and Baclofen-induced ERK1/2 phosphorylation can accelerate cytoprotective autophagy. Whether there are any further pathways for the treatment of CCI remains unknown. Therefore, it is important to understand the mechanisms, identify newer and more potent therapeutic targets, introduce pharmaceuticals into trials in humans for clinical evaluation, and improve the efficacy and safety of medications.
ZC and MZ: conception and design. ZC and XY: administrative support. MY: provision of study materials, collection, and assembly of data. MY, PF, and MW: data analysis and interpretation. ZC: revised the final version. All authors contributed in manuscript writing and approved the final version of manuscript.
This study was supported partially by the National Natural Science Foundation of China (81960221 and 82260249 to XY), the National Science & Technology Fundamental Resource Investigation Program of China (2018FY100903 to XY), the Jiangxi Provincial Health Commission Science and Technology Plan project (202311506 to ZC), the Jiangxi Provincial Administration of Traditional Chinese Medicine Science and Technology Plan project (2022A322 to ZC), the Key Projects of Jiangxi Provincial Department of Education (GJJ2201902 to ZC), and the Youth Foundation of Natural Science Foundation of Jiangxi Province (20224BAB216045 to ZC).
We sincerely thank the staff of the Jiujiang Precision Clinical Medicine Research Center and its students Ketao Tu, Jinming Ma, Qinghua Huang, and Weixin Zhou.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
ERK, extracellular regulated protein kinases; CCI, chronic cerebral ischemia; VD, vascular dementia; AD, Alzheimer’s disease; AMPK, AMP-activated protein kinase; MPTP, mitochondrial permeability transition pore; ROS, reactive oxygen species; Cyt c, cytochrome c; mTOR, mammalian target of rapamycin; NF-κB, nuclear factor-kappaB; JNK, jun-terminal kinase; mCU, mitochondrial calcium uniporter; VDAC, voltage-dependent anion channel; PPAR, peroxisome proliferator-activated receptor; PGC-1α, PPAR-γ coactivator 1α; NRF, nuclear respiratory factor; TFAM, mitochondrial transcription factor A; NAD, nicotinamide adenine dinucleotide; GABAB, γ-aminobutyric acid B receptor; mitoKATP, mitochondrial membrane ATP-sensitive potassium channel; CIHH, chronic intermittent hypobaric hypoxia; DND, delay neuronal death; 2VO, bilateral common carotid arteries occlusion; FMT, fecal microbiota transplantation; SCFAs, short-chain fatty acids; ETC, electron transport chain; BNIP3L, BCL/adenovirus E1B interacting protein 3-like; SOD, superoxide dismutase; ECS, endocannabinoid system; WIN, WIN55212-2.
Andjelkovic, A. V., Xiang, J., Stamatovic, S. M., Hua, Y., Xi, G., Wang, M. M., et al. (2019). Endothelial targets in stroke: Translating animal models to human. Arterioscler. Thromb. Vasc. Biol. 39, 2240–2247. doi: 10.1161/ATVBAHA.119.312816
Anzell, A. R., Maizy, R., Przyklenk, K., and Sanderson, T. H. (2018). Mitochondrial quality control and disease: Insights into ischemia-reperfusion injury. Mol. Neurobiol. 55, 2547–2564. doi: 10.1007/s12035-017-0503-9
Arun, S., Liu, L., and Donmez, G. (2016). Mitochondrial biology and neurological diseases. Curr. Neuropharmacol. 14, 143–154. doi: 10.2174/1570159x13666150703154541
Bartolomé, A., García-Aguilar, A., Asahara, S. I., Kido, Y., Guillén, C., Pajvani, U. B., et al. (2017). MTORC1 regulates both general autophagy and mitophagy induction after oxidative phosphorylation uncoupling. Mol. Cell. Biol. 37, e00441–17. doi: 10.1128/MCB.00441-17
Broughton, B. R., Reutens, D. C., and Sobey, C. G. (2009). Apoptotic mechanisms after cerebral ischemia. Stroke 40, e331–e339. doi: 10.1161/STROKEAHA.108.531632
Calabrese, V., Giordano, J., Signorile, A., Laura Ontario, M., Castorina, S., De Pasquale, C., et al. (2016). Major pathogenic mechanisms in vascular dementia: Roles of cellular stress response and hormesis in neuroprotection. J. Neurosci. Res. 94, 1588–1603. doi: 10.1002/jnr.23925
Castillo, K., Rojas-Rivera, D., Lisbona, F., Caballero, B., Nassif, M., Court, F. A., et al. (2011). BAX inhibitor-1 regulates autophagy by controlling the IRE1α branch of the unfolded protein response. EMBO J. 30, 4465–4478. doi: 10.1038/emboj.2011.318
Ciacciarelli, A., Sette, G., Giubilei, F., and Orzi, F. (2020). Chronic cerebral hypoperfusion: An undefined, relevant entity. J. Clin. Neurosci. 73, 8–12. doi: 10.1016/j.jocn.2020.01.026
Du, J., Ma, M., Zhao, Q., Fang, L., Chang, J., Wang, Y., et al. (2013). Mitochondrial bioenergetic deficits in the hippocampi of rats with chronic ischemia-induced vascular dementia. Neuroscience 231, 345–352. doi: 10.1016/j.neuroscience.2012.11.062
Du, S. Q., Wang, X. R., Xiao, L. Y., Tu, J. F., Zhu, W., He, T., et al. (2017). Molecular mechanisms of vascular dementia: What can be learned from animal models of chronic cerebral hypoperfusion? Mol. Neurobiol. 54, 3670–3682. doi: 10.1007/s12035-016-9915-1
Farkas, E., Luiten, P. G., and Bari, F. (2007). Permanent, bilateral common carotid artery occlusion in the rat: A model for chronic cerebral hypoperfusion-related neurodegenerative diseases. Brain Res. Rev. 54, 162–180. doi: 10.1016/j.brainresrev.2007.01.003
Feng, Y. J., Wang, L., Han, G. H., Yu, H. N., Li, D. Y., Zhen, W. Z., et al. (2022). Naoxin’an capsule alleviates mitochondrial and oxidative damage in chronic cerebral ischemia-induced VCI in rats via activating CREB/PGC-1α pathway. Chin. J. Exp. Trad. Med. Formulae 28, 19–29.
Fornazari, M., De Paula, J. G., Castilho, R. F., and Kowaltowski, A. J. (2008). Redox properties of the adenoside triphosphate-sensitive K+ channel in brain mitochondria. J. Neurosci. Res. 86, 1548–1556. doi: 10.1002/jnr.21614
Gao, L. (2018). Expert consensus on the diagnosis and treatment of chronic cerebral ischemia in integrated traditional Chinese and Western medicine. Chin. J. Integr. Med. 38, 1161–1167.
Gao, X., Yang, J., Li, Y., Yu, M., Liu, S., Han, Y., et al. (2019). Lanthanum chloride induces autophagy in rat hippocampus through ROS-mediated JNK and AKT/mTOR signaling pathways. Metallomics 11, 439–453. doi: 10.1039/c8mt00295a
Gao, Z. S. (2009). Mitochondrial structure and function changes with chronic cerebral hypoperfusion in rat brain and the effects of butylphthalide injection. Master’s thesis. Hebei: Hebei Medical University.
García-Rivas, J., Carvajal, K., Correa, F., and Zazueta, C. (2006). Ru360, a specific mitochondrial calcium uptake inhibitor, improves cardiac post-ischaemic functional recovery in rats in vivo. Br. J. Pharmacol. 149, 829–837. doi: 10.1038/sj.bjp.0706932
Global Burden of Disease (2020). Global burden of disease study, Institute for Health Metrics and Evaluation. Seattle, WA: University of Washington. Available online at: https://ghdx.healthdata.org/ (accessed August 1, 2021).
Gouriou, Y., Demaurex, N., Bijlenga, P., and De Marchi, U. (2011). Mitochondrial calcium handling during ischemia-induced cell death in neurons. Biochimie 93, 2060–2067. doi: 10.1016/j.biochi.2011.08.001
Guang, H. M., and Du, G. H. (2006). Protections of pinocembrine on brain mitochondria contribute to cognitive improvement in chronic cerebral hypoperfused rats. Eur. J. Pharmacol. 542, 77–83. doi: 10.1016/j.ejphar.2006.04.054
Haemmerle, G., Moustafa, T., Woelkart, G., Büttner, S., Schmidt, A., van de Weijer, T., et al. (2011). ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-α and PGC-1. Nat. Med. 17, 1076–1085. doi: 10.1038/nm.2439
Halestrap, A. P. (2009). What is the mitochondrial permeability transition pore? J. Mol. Cell. Cardiol. 46, 821–831. doi: 10.1016/j.yjmcc.2009.02.021
Hankey, G. J. (2014). Secondary stroke prevention. Lancet Neurol. 13, 178–194. doi: 10.1016/S1474-4422(13)70255-2
Hannan, M. A., Dash, R., Sohag, A. A. M., Haque, M. N., and Moon, I. S. (2020). Neuroprotection against oxidative stress: Phytochemicals targeting TrkB signaling and the Nrf2-ARE antioxidant system. Front. Mol. Neurosci. 13:116. doi: 10.3389/fnmol.2020.00116
He, X. L., Bi, M. G., and Du, G. H. (2012). Correlation between mitochondrial proteome and energy metabolism in rat brain tissue of rats with chronic cerebral ischemia. Chin. Pharmacol. Bull. 28, 1200–1205.
He, X. L., Wang, Y. H., Gao, M., Li, X. X., Zhang, T. T., and Du, G. H. (2009). Baicalein protects rat brain mitochondria against chronic cerebral hypoperfusion-induced oxidative damage. Brain Res. 1249, 212–221. doi: 10.1016/j.brainres.2008.10.005
Hüttemann, M., Pecina, P., Rainbolt, M., Sanderson, T. H., Kagan, V. E., Samavati, L., et al. (2011). The multiple functions of cytochrome c and their regulation in life and death decisions of the mammalian cell: From respiration to apoptosis. Mitochondrion 11, 369–381. doi: 10.1016/j.mito.2011.01.010
Hwang, H. Y., Shim, J. S., Kim, D., and Kwon, H. J. (2021). Antidepressant drug sertraline modulates AMPK-MTOR signaling-mediated autophagy via targeting mitochondrial VDAC1 protein. Autophagy 17, 2783–2799. doi: 10.1080/15548627.2020.1841953
Iwabu, M., Yamauchi, T., Okada-Iwabu, M., Sato, K., Nakagawa, T., Funata, M., et al. (2010). Adiponectin and AdipoR1 regulate PGC-1alpha and mitochondria by Ca(2+) and AMPK/SIRT1. Nature 464, 1313–1319. doi: 10.1038/nature08991
Kaarniranta, K., Uusitalo, H., Blasiak, J., Felszeghy, S., Kannan, R., Kauppinen, A., et al. (2020). Mechanisms of mitochondrial dysfunction and their impact on age-related macular degeneration. Prog. Retin. Eye Res. 79:100858. doi: 10.1016/j.preteyeres.2020.100858
Kim, E. J., Raval, A. P., Hirsch, N., and Perez-Pinzon, M. A. (2010). Ischemic preconditioning mediates cyclooxygenase-2 expression via nuclear factor-kappa B activation in mixed cortical neuronal cultures. Transl. Stroke Res. 1, 40–47. doi: 10.1007/s12975-009-0006-8
Kim, J. S., Jin, Y., and Lemasters, J. J. (2006). Reactive oxygen species, but not Ca2+ overloading, trigger pH- and mitochondrial permeability transition-dependent death of adult rat myocytes after ischemia-reperfusion. Am. J. Physiol. Heart Circ. Physiol. 290, H2024–H2034. doi: 10.1152/ajpheart.00683.2005
Kim, M. S., Bang, J. H., Lee, J., Han, J. S., Baik, T. G., and Jeon, W. K. (2016). Ginkgo biloba L. extract protects against chronic cerebral hypoperfusion by modulating neuroinflammation and the cholinergic system. Phytomedicine 23, 1356–1364. doi: 10.1016/j.phymed.2016.07.013
Kislin, M., Sword, J., Fomitcheva, I. V., Croom, D., Pryazhnikov, E., Lihavainen, E., et al. (2017). Reversible disruption of neuronal mitochondria by ischemic and traumatic injury revealed by quantitative two-photon imaging in the neocortex of anesthetized mice. J. Neurosci. 37, 333–348. doi: 10.1523/JNEUROSCI.1510-16.2016
Krasnikov, B. F., Zorov, D. B., Antonenko, Y. N., Zaspa, A. A., Kulikov, I. V., Kristal, B. S., et al. (2005). Comparative kinetic analysis reveals that inducer-specific ion release precedes the mitochondrial permeability transition. Biochim. Biophys. Acta. 1708, 375–392. doi: 10.1016/j.bbabio.2005.05.009
Kushnareva, Y. E., and Sokolove, P. M. (2000). Prooxidants open both the mitochondrial permeability transition pore and a low-conductance channel in the inner mitochondrial membrane. Arch. Biochem. Biophys. 376, 377–388. doi: 10.1006/abbi.2000.1730
Lana, D., Melani, A., Pugliese, A. M., Cipriani, S., Nosi, D., Pedata, F., et al. (2014). The neuron-astrocyte-microglia triad in a rat model of chronic cerebral hypoperfusion: Protective effect of dipyridamole. Front. Aging Neurosci. 6:322. doi: 10.3389/fnagi.2014.00322
Li, J. Z., Zhang, J. W., and Liu, H. F. (2022). Expert consensus on the clinical diagnosis and treatment of chronic cerebral ischemia. Chin. J. Pract. Nerv. Dis. 25, 661–667.
Li, P. A., Hou, X., and Hao, S. (2017). Mitochondrial biogenesis in neurodegeneration. J. Neurosci. Res. 95, 2025–2029. doi: 10.1002/jnr.24042
Li, Y., Zheng, W. Q., Pan, L., Liu, M. R., Zhou, X. Y., Chen, Z., et al. (2023). The role of autophagy and related mechanisms in ischemic brain injury: An update. Sci. China 53, 19–29. doi: 10.1360/SSV-2021-0194
Liao, Y., Yang, R., Lin, J., Wu, S. Y., Wu, C. H., Shang, S.-L., et al. (2016). Analysis of cerebrovascular function in patients with chronic cerebral circulation insufficiency. Int. J. Psychiatry Neurol. 05, 9–13. doi: 10.12677/IJPN.2016.51002
Lindqvist, L. M., Heinlein, M., Huang, D. C., and Vaux, D. L. (2014). Prosurvival Bcl-2 family members affect autophagy only indirectly, by inhibiting Bax and Bak. Proc. Natl. Acad. Sci. U.S.A. 111, 8512–8517. doi: 10.1073/pnas.1406425111
Liu, L., Li, C. J., Lu, Y., Zong, X. G., Luo, C., Sun, J., et al. (2015). Baclofen mediates neuroprotection on hippocampal CA1 pyramidal cells through the regulation of autophagy under chronic cerebral hypoperfusion. Sci. Rep. 5:14474. doi: 10.1038/srep14474
Liu, X., Zhao, P., Wang, X., Wang, L., Zhu, Y., and Gao, W. (2019a). Triptolide induces glioma cell autophagy and apoptosis via upregulating the ROS/JNK and downregulating the Akt/mTOR signaling pathways. Front. Oncol. 9:387. doi: 10.3389/fonc.2019.00387
Liu, X., Zhao, P., Wang, X., Wang, L., Zhu, Y., Song, Y., et al. (2019b). Correction to: Celastrol mediates autophagy and apoptosis via the ROS/JNK and Akt/mTOR signaling pathways in glioma cells. J. Exp. Clin. Cancer Res. 38:284. doi: 10.1186/s13046-019-1285-x
Luo, Y., Hu, Y. D., Zhang, M. H., Xiao, Y., Song, Z. C., and Xu, Y. (2013). EtBr-induced selective degradation of mitochondria occurs via autophagy. Oncol. Rep. 30, 1201–1208. doi: 10.3892/or.2013.2590
Matsuda, S., Umeda, M., Uchida, H., Kato, H., and Araki, T. (2009). Alterations of oxidative stress markers and apoptosis markers in the striatum after transient focal cerebral ischemia in rats. J. Neural Transm. 116, 395–404. doi: 10.1007/s00702-009-0194-0
Mccracken, E., Valeriani, V., Simpson, C., Jover, T., McCulloch, J., and Dewar, D. (2000). The lipid peroxidation by-product 4-hydroxynonenal is toxic to axons and oligodendrocytes. J. Cereb. Blood Flow Metab. 20, 1529–1536. doi: 10.1097/00004647-200011000-00002
Mizushima, N. (2007). Autophagy: Process and function. Genes Dev. 21, 2861–2873. doi: 10.1101/gad.1599207
Mizushima, N., Levine, B., Cuervo, A. M., and Klionsky, D. J. (2008). Autophagy fights disease through cellular self-digestion. Nature 451, 1069–1075. doi: 10.1038/nature06639
Mouchiroud, L., Houtkooper, R. H., Moullan, N., Katsyuba, E., Ryu, D., Cantó, C., et al. (2013). The NAD(+)/sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell 154, 430–441. doi: 10.1016/j.cell.2013.06.016
Nacarelli, T., Azar, A., and Sell, C. (2014). Inhibition of mTOR prevents ROS production initiated by ethidium bromide-induced mitochondrial DNA depletion. Front. Endocrinol. 5:122. doi: 10.3389/fendo.2014.00122
Nguyen, H., Zarriello, S., Rajani, M., Tuazon, J., Napoli, E., and Borlongan, C. V. (2018). Understanding the role of dysfunctional and healthy mitochondria in stroke pathology and its treatment. Int. J. Mol. Sci. 19:2127. doi: 10.3390/ijms19072127
Nohl, H., Gille, L., and Staniek, K. (2005). Intracellular generation of reactive oxygen species by mitochondria. Biochem. Pharmacol. 69, 719–723. doi: 10.1016/j.bcp.2004.12.002
Pickles, S., Vigié, P., and Youle, R. J. (2018). Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol. 28, R170–R185. doi: 10.1016/j.cub.2018.01.004
Rajeev, V., Fann, D. Y., Dinh, Q. N., Kim, H. A., De Silva, T. M., Lai, M. K. P., et al. (2022). Pathophysiology of blood brain barrier dysfunction during chronic cerebral hypoperfusion in vascular cognitive impairment. Theranostics 12, 1639–1658. doi: 10.7150/thno.68304
Sadidi, M., Lentz, S. I., and Feldman, E. L. (2009). Hydrogen peroxide-induced Akt phosphorylation regulates Bax activation. Biochimie 91, 577–585. doi: 10.1016/j.biochi.2009.01.010
Saxton, R. A., and Sabatini, D. M. (2017). mTOR signaling in growth, metabolism, and disease. Cell 168, 960–976. doi: 10.1016/j.cell.2017.02.004
Shirley, R., Ord, E. N., and Work, L. M. (2014). Oxidative stress and the use of antioxidants in stroke. Antioxidants 3, 472–501. doi: 10.3390/antiox3030472
Sin, N., Kim, K. B., Elofsson, M., Meng, L., Auth, H., Kwok, B. H., et al. (1999). Total synthesis of the potent proteasome inhibitor epoxomicin: A useful tool for understanding proteasome biology. Bioorg. Med. Chem. Lett. 9, 2283–2288. doi: 10.1016/s0960-894x(99)00376-5
Siwicka-Gieroba, D., Robba, C., Gołacki, J., Badenes, R., and Dabrowski, W. (2022). Cerebral oxygen delivery and consumption in brain-injured patients. J. Pers. Med. 12:1763. doi: 10.3390/jpm12111763
Su, S. H., Wu, Y. F., Lin, Q., Yu, F., and Hai, J. (2015). Cannabinoid receptor agonist WIN55,212-2 and fatty acid amide hydrolase inhibitor URB597 suppress chronic cerebral hypoperfusion-induced neuronal apoptosis by inhibiting c-Jun N-terminal kinase signaling. Neuroscience 301, 563–575. doi: 10.1016/j.neuroscience.2015.03.021
Su, S. H., Wu, Y. F., Lin, Q., Zhang, L., Wang, D. P., and Hai, J. (2022). Fecal microbiota transplantation and replenishment of short-chain fatty acids protect against chronic cerebral hypoperfusion-induced colonic dysfunction by regulating gut microbiota, differentiation of Th17 cells, and mitochondrial energy metabolism. J. Neuroinflammation 19:313. doi: 10.1186/s12974-022-02675-9
Su, S. H., Wu, Y. F., Wang, D. P., and Hai, J. (2018). Inhibition of excessive autophagy and mitophagy mediates neuroprotective effects of URB597 against chronic cerebral hypoperfusion. Cell Death Dis. 9:733. doi: 10.1038/s41419-018-0755-y
Subramanian, M., and Shaha, C. (2007). Up-regulation of Bcl-2 through ERK phosphorylation is associated with human macrophage survival in an estrogen microenvironment. J. Immunol. 179, 2330–2338.
Sun, C., Liu, M., Liu, J., Zhang, T., Zhang, L., Li, H., et al. (2021). ShenmaYizhi decoction improves the mitochondrial structure in the brain and ameliorates cognitive impairment in VCI rats via the AMPK/UCP2 signaling pathway. Neuropsychiatr. Dis. Treat. 17, 1937–1951. doi: 10.2147/NDT.S302355
Tajiri, N., Borlongan, C. V., and Kaneko, Y. (2016). Cyclosporine A treatment abrogates ischemia-induced neuronal cell death by preserving mitochondrial integrity through upregulation of the Parkinson’s disease-associated protein DJ-1. CNS Neurosci. Ther. 22, 602–610. doi: 10.1111/cns.12546
Takizawa, S., Hirabayashi, H., Matsushima, K., Tokuoka, K., and Shinohara, Y. (1998). Induction of heme oxygenase protein protects neurons in cortex and striatum, but not in hippocampus, against transient forebrain ischemia. J. Cereb. Blood Flow Metab. 18, 559–569. doi: 10.1097/00004647-199805000-00011
Tan, V. P., and Miyamoto, S. (2015). HK2/hexokinase-II integrates glycolysis and autophagy to confer cellular protection. Autophagy 11, 963–964. doi: 10.1080/15548627.2015.1042195
Tang, B. L. (2016). Sirt1 and the mitochondria. Mol. Cells 39, 87–95. doi: 10.14348/molcells.2016.2318
Tang, Y. C., Tian, H. X., Yi, T., and Chen, H. B. (2016). The critical roles of mitophagy in cerebral ischemia. Protein Cell 7, 699–713. doi: 10.1007/s13238-016-0307-0
Wang, D. P., Kang, K., Sun, J., Lin, Q., Lv, Q. L., and Hai, J. (2022). URB597 and andrographolide improve brain microvascular endothelial cell permeability and apoptosis by reducing oxidative stress and inflammation associated with activation of Nrf2 signaling in oxygen-glucose deprivation. Oxid. Med. Cell. Longev. 2022:4139330. doi: 10.1155/2022/4139330
Wang, R. C., Wei, Y., An, Z., Zou, Z., Xiao, G., Bhagat, G., et al. (2012). Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science 338, 956–959. doi: 10.1126/science.1225967
Wang, X., Fang, Y., Huang, Q., Xu, P., Lenahan, C., Lu, J., et al. (2021). An updated review of autophagy in ischemic stroke: From mechanisms to therapies. Exp. Neurol. 340:113684. doi: 10.1016/j.expneurol.2021.113684
Wang, Y., Wang, Y., Li, S., Jin, H., Duan, J., Lu, X., et al. (2023). Insights of Chinese herbal medicine for mitochondrial dysfunction in chronic cerebral hypoperfusion induced cognitive impairment: Existed evidences and potential directions. Front. Pharmacol. 14:1138566. doi: 10.3389/fphar.2023.1138566
Wang, Y. H., He, X. L., Li, X. X., Qin, H. L., and Du, G. H. (2012). Effects of the effective component group of Chinese herbal medicine Xiaoxuming Decoction on brain mitochondria in rats with chronic cerebral ischemia. Zhong Xi Yi Jie He Xue Bao 10, 569–576. doi: 10.3736/jcim20120513
Wang, Z., Kawabori, M., and Houkin, K. (2020). FTY720 (fingolimod) ameliorates brain injury through multiple mechanisms and is a strong candidate for stroke treatment. Curr. Med. Chem. 27, 2979–2993. doi: 10.2174/0929867326666190308133732
Wu, X., Zheng, Y., Liu, M., Li, Y., Ma, S., Tang, W., et al. (2021). BNIP3L/NIX degradation leads to mitophagy deficiency in ischemic brains. Autophagy 17, 1934–1946. doi: 10.1080/15548627.2020.1802089
Xiao, Q., Liu, H., Yang, C., Chen, Y., Huang, Y., Xiao, X., et al. (2023). Bushen-Yizhi formula exerts neuroprotective effect via inhibiting excessive mitophagy in rats with chronic cerebral hypoperfusion. J. Ethnopharmacol. 310:116326. doi: 10.1016/j.jep.2023.116326
Xie, Z. P., and Klionsky, D. J. (2007). Autophagosome formation: Core machinery and adaptations. Nat. Cell Biol. 9, 1102–1109. doi: 10.1038/ncb1007-1102
Yan, F., Tian, Y., Huang, Y. Y., Wang, Q., Liu, P., Wang, N. Q., et al. (2022). Xi-Xian-Tong-Shuan capsule alleviates vascular cognitive impairment in chronic cerebral hypoperfusion rats by promoting white matter repair, reducing neuronal loss, and inhibiting the expression of pro-inflammatory factors. Biomed. Pharmacother. 145:112453. doi: 10.1016/j.biopha.2021.112453
Yang, M. Y., Yu, Q. L., Huang, Y. S., and Yang, G. (2019). Neuroprotective effects of andrographolide derivative CX-10 in transient focal ischemia in rat: Involvement of Nrf2/AE and TLR/NF-κB signaling. Pharmacol. Res. 144, 227–234. doi: 10.1016/j.phrs.2019.04.023
Yen, T. L., Chen, R. J., Jayakumar, T., Lu, W. J., Hsieh, C. Y., Hsu, M. J., et al. (2016). Andrographolide stimulates p38 mitogen-activated protein kinase-nuclear factor erythroid-2-related factor 2-heme oxygenase 1 signaling in primary cerebral endothelial cells for definite protection against ischemic stroke in rats. Transl. Res. 170, 57–72. doi: 10.1016/j.trsl.2015.12.002
Yu, M., Wang, Q. Y., Gu, Q., Wang, X., Deng, B., Jiang, D. D., et al. (2014). Effects of Zuogui Pill on the activities of mitochondrial respiratory chain complexes in rats with chronic cerebral ischemia. J. Neurol. Neurorehab. 11, 52–55.
Zhang, L., Dai, L., and Li, D. (2021). Mitophagy in neurological disorders. J. Neuroinflammation 18:297. doi: 10.1186/s12974-021-02334-5
Zhang, M., Hu, Y., Zhang, J., and Zhang, J. (2020). FTY720 prevents spatial memory impairment in a rat model of chronic cerebral hypoperfusion via a SIRT3-independent pathway. Front. Aging Neurosci. 12:593364. doi: 10.3389/fnagi.2020.593364
Zhang, S. X., Guo, Z., Yang, S. J., Ma, H. J., Fu, C. R., Wang, S., et al. (2016). Chronic intermittent hybobaric hypoxia protects against cerebral ischemia via modulation of mitoKATP. Neurosci. Lett. 635, 8–16. doi: 10.1016/j.neulet.2016.10.025
Zhao, X. Y., Lu, M. H., Yuan, D. J., Xu, D. E., Yao, P. P., Ji, W. L., et al. (2019). Mitochondrial dysfunction in neural injury. Front. Neurosci. 13:30. doi: 10.3389/fnins.2019.00030
Zhao, Y., and Gong, C. X. (2015). From chronic cerebral hypoperfusion to Alzheimer-like brain pathology and neurodegeneration. Cell. Mol. Neurobiol. 35, 101–110. doi: 10.1007/s10571-014-0127-9
Zhao, Y., Zhang, J., Zheng, Y., Zhang, Y., Zhang, X. J., Wang, H., et al. (2021). NAD+ improves cognitive function and reduces neuroinflammation by ameliorating mitochondrial damage and decreasing ROS production in chronic cerebral hypoperfusion models through Sirt1/PGC-1α pathway. J. Neuroinflammation 18:207. doi: 10.1186/s12974-021-02250-8
Zheng, G. M., Wang, L., Li, X. Q., Niu, X. L., Xu, G. D., and Lv, P. Y. (2021). Rapamycin alleviates cognitive impairment in murine vascular dementia: The enhancement of mitophagy by PI3K/AKT/mTOR axis. Tissue Cell 69:101481. doi: 10.1016/j.tice.2020.101481
Zhou, M., He, H. J., Hirano, M., Sekiguchi, M., Tanaka, O., Kawahara, K., et al. (2010). Localization of ATP-sensitive K+ channel subunits in rat submandibular gland. J. Histochem. Cytochem. 58, 499–507. doi: 10.1369/jhc.2009.955047
Zhou, Y., Zhang, S. S., and Fan, X. (2021). Role of Polyphenols as Antioxidant Supplementation in Ischemic Stroke. Oxid. Med. Cell. Longevity 2021:5471347. doi: 10.1155/2021/5471347
Keywords: chronic cerebral ischemia, stroke, mitochondrial autophagy, oxidative stress, treatment
Citation: Yu M, Zhang M, Fu P, Wu M, Yin X and Chen Z (2023) Research progress of mitophagy in chronic cerebral ischemia. Front. Aging Neurosci. 15:1224633. doi: 10.3389/fnagi.2023.1224633
Received: 18 May 2023; Accepted: 20 July 2023;
Published: 03 August 2023.
Edited by:
Sandeep Singh, Salk Institute for Biological Studies, United StatesReviewed by:
Prince Kumar Singh, Hebrew University of Jerusalem, IsraelCopyright © 2023 Yu, Zhang, Fu, Wu, Yin and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhiying Chen, Y2hlbnpoaXlpbmdAY2NtdS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.