 Martha R. C. Bhattacharya
Martha R. C. Bhattacharya
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Aging Neurosci., 08 August 2023
Sec. Neuroinflammation and Neuropathy
Volume 15 - 2023 | https://doi.org/10.3389/fnagi.2023.1166146
This article is part of the Research TopicModel Organisms in Neuroinflammation and Neuropathy: Drosophila melanogasterView all 5 articles
The degeneration of axons and their terminals occurs following traumatic, toxic, or genetically-induced insults. Common molecular mechanisms unite these disparate triggers to execute a conserved nerve degeneration cascade. In this review, we will discuss how models of peripheral nerve injury and neuropathy in Drosophila have led the way in advancing molecular understanding of axon degeneration and nerve injury pathways. Both neuron-intrinsic as well as glial responses to injury will be highlighted. Finally, we will offer perspective on what additional questions should be answered to advance these discoveries toward clinical interventions for patients with neuropathy.
Peripheral nerve damage and subsequent degeneration of axons occurs in a diverse group of syndromes and, taken together, makes peripheral neuropathy one of the most frequent human neurodegenerative conditions (Singer et al., 2012). Peripheral neuropathy and peripheral nerve injury occur due to localized injuries like sciatica and carpal tunnel syndrome and via neurotoxicity due to high glucose levels or exposure to microtubule-disrupting chemotherapeutic agents (Theiss and Meller, 2000; LaPointe et al., 2013; Menorca et al., 2013; Albers and Pop-Busui, 2014; Fukuda et al., 2017). In addition, spontaneous or hereditary forms of peripheral neuropathy (including both sensory and motor impairment) can disconnect or disrupt function of circuits (Züchner et al., 2004; Timmerman et al., 2013). Finally, neuroinflammatory diseases like multiple sclerosis cause demyelination and degeneration of axons (Singh et al., 2017). Because of the relative ease of access to peripheral structures compared to those in the central nervous system, peripheral nerves are an attractive setting for studies of degenerative pathways that also affect the brain.
Pathological axon degeneration is also called Wallerian degeneration due to its initial description in the frog hypoglossal nerve by Waller (1851). Even Waller (1851) recorded macroscopic findings that match what we see today: a delay period following injury, a rapid axon fragmentation process, and the subsequent clearance of the fragmented nerve. We now have more sophisticated tools to watch this process both in vitro and in vivo in many organisms (Tao and Rolls, 2011; Shin et al., 2012; Babetto et al., 2013; Vargas et al., 2015; Tian et al., 2016; Li et al., 2018). The molecular study of axon degeneration was launched by a serendipitous discovery of the Wallerian Degeneration Slow (Wlds) mouse phenotype and, later, the responsible genomic alteration [triplication of the NAD-producing enzyme, nicotinamide mononucleotide adenylyltransferase (NMNAT1)] (Lunn et al., 1989; Coleman et al., 1998; Mack et al., 2001). These discoveries showed that axon degeneration, rather than a passive process, was actively controlled: the initiation, pace, and extent are determined by the activation state of cellular pathways. This led to a flurry of research that has identified the key components of the Wallerian degeneration pathway (summarized below) (Miller et al., 2009; Bhattacharya et al., 2012, 2016; Osterloh et al., 2012; Shin et al., 2012; Xiong et al., 2012; Neukomm et al., 2017) and led to emerging strategies for clinical intervention in peripheral neuropathy (DiAntonio, 2019; Feldman et al., 2022; Shi et al., 2022).
Drosophila melanogaster has led the way in identifying the molecular pathways downstream of axon injury that result in axonal fragmentation and disassembly. Using fly models of genetic, traumatic, or toxic nerve injury, mutations in multiple fly genes including Drosophila sterile alpha and TIR motif containing protein (dSarm), highwire (hiw), and wallenda (wnd) genes were first identified as axo-protective (Miller et al., 2009; Osterloh et al., 2012; Xiong et al., 2012). Following initial descriptions of pathways in fly, later work in mouse has universally confirmed the key Drosophila discoveries (Table 1). Given this excellent foundation, fly models of peripheral neuropathy and axon degeneration continue to be a key source of information about the mechanisms activated by nerve injury and strategies to mitigate its effects.
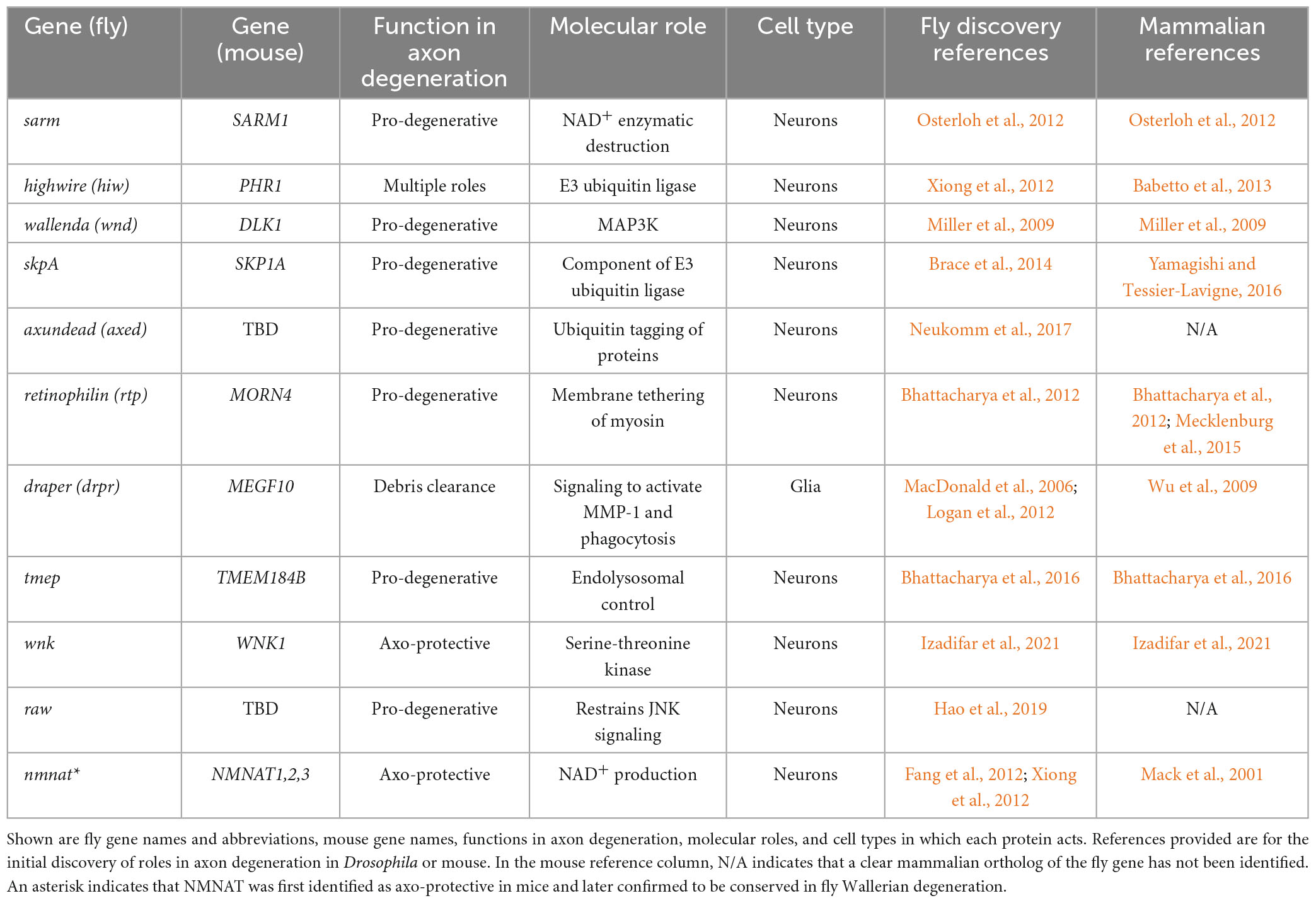
Table 1. Genes discovered in Drosophila for roles in axon degeneration.
Larval and adult stage Drosophila have many accessible peripheral nerves from which to choose for neuropathy studies (Figure 1). In larvae, the body is organized in repeating hemi-segments, and nerves innervate each hemi-segment in a stereotyped pattern. The larval nerves contain both sensory and motor axons, as in mice. Because these nerves leave the VNC in close proximity to each other, a researcher can injure 6–8 nerves simultaneously in one animal (or leave them as uninjured controls) (Brace and DiAntonio, 2020). In adults, nerves entering appendages such as antennae, legs, and wings are ideal models for neuropathy studies due to their accessibility (Phelps et al., 2021). Two other advantages of adult axon degeneration models (versus larval studies) are (1) they allow for a longer analysis of axon injury effects (up to 60 days), and (2) the function of these axons can be read out behaviorally using olfactory, locomotor, or grooming assays (Wang et al., 2011; Neukomm et al., 2017; François-Moutal et al., 2019; Paglione et al., 2020).
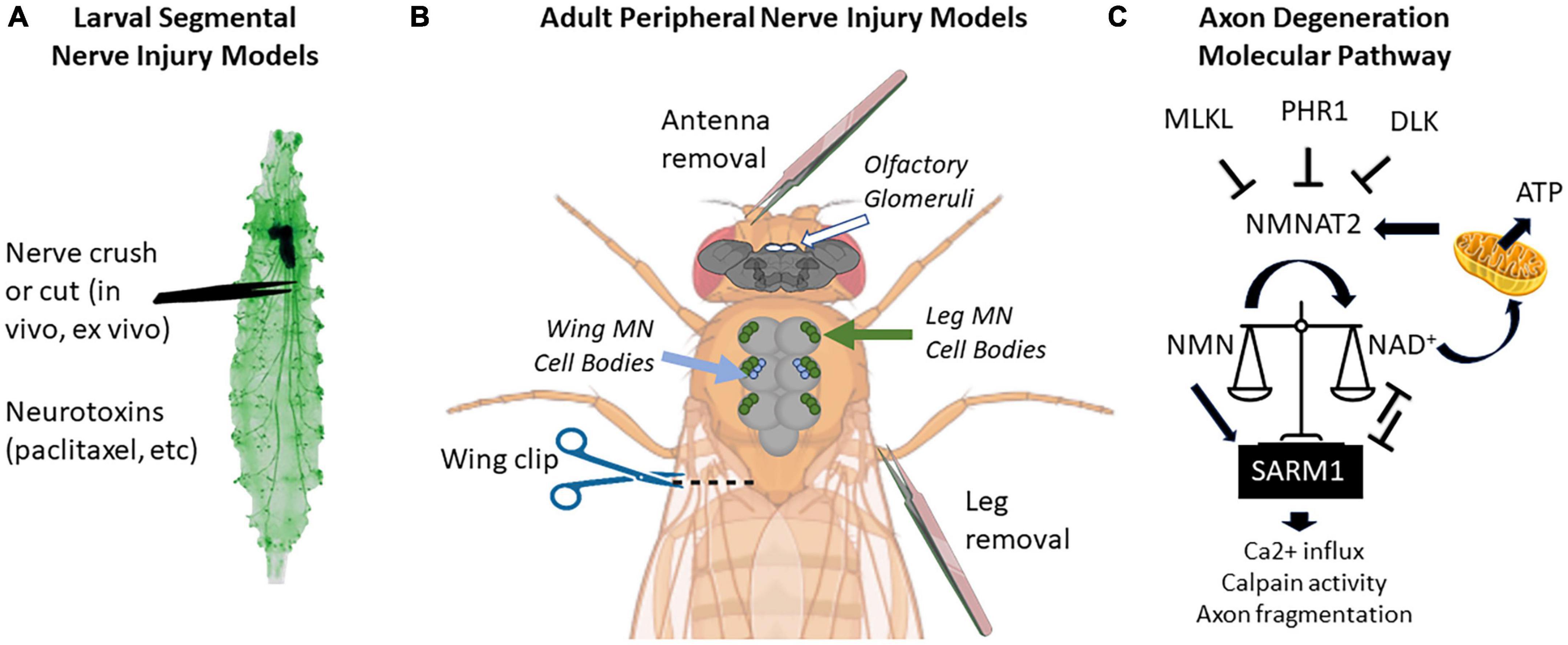
Figure 1. Locations of peripheral nerve injuries commonly used in Drosophila melanogaster and molecular pathway of axon degeneration. (A) Larval segmental nerves (green) can be crushed with forceps or cut with scissors. Larvae can also be fed drugs that cause nervous system toxicity, such as paclitaxel. (B) Peripherally projecting axons of central nervous system structures (brain and thoracic ganglion) can be severed by removing appendages such as antennas or legs or by cutting through the wing margin. On the brain, the location of olfactory glomeruli is shown with white circles. On the thoracic ganglion, the location of cell bodies of motor neurons (MN) from the legs is shown in green, while the cell bodies from wing neurons are shown in blue. (C) Current proposed pathway of axon degeneration centered around regulation of NMNAT2 and SARM. Gene names use mammalian nomenclature. A high NMN/NAD+ ratio triggers Sarm1 activation, which triggers a positive feedback loop to further reduce NAD+ via enzymatic cleavage. NMNAT2 is the predominant axonal form in mammals, but axonally localized NMNAT1 can provide a similar function. Not pictured: Axed, Wnk1, or intermediate MAP kinase pathway members [MKK4/MKK7 and c-Jun n-terminal kinase (JNK)]. Larval image modified from Balapagos and used under Creative Commons ShareAlike Genetic license (CC BY-SA 2.0). Image creation was assisted by Biorender.com.
In this review, I will highlight new findings from Drosophila that have potential to offer both better mechanistic understanding of different types of neuropathy and relevant directions for future patient care. The main (though not exclusive) focus will be on peripheral nerve injuries and disease models in Drosophila, though axon degeneration certainly occurs in central nervous system disorders such as Alzheimer’s Disease and tauopathies (Law et al., 2022). I will also offer some caveats of the Drosophila models that should be considered when extrapolating findings to mice and man.
Physical crush or transection of fly nerves is perhaps the most utilized neuropathy-inducing insult because it permits synchronization of the injury time course in multiple axons and also because the injury is localized to a single site for each nerve. Using olfactory antennal axotomy (by removal of one of the two antennae, which retains viability), early candidate approaches and unbiased screens identified Wallerian degeneration factors such as dSarm1 and Wallenda (Miller et al., 2009; Osterloh et al., 2012). In a parallel screening paradigm using transection of wing axons, the axonal localization of mitochondria was found to be critical for axonal maintenance following injury (Fang et al., 2012). Together, these three screens set the stage for a large expansion of this field and, importantly, established Drosophila as a strong gene discovery platform for evolutionarily conserved nerve injury factors (Miller et al., 2009; Sasaki et al., 2009; Bhattacharya et al., 2012, 2016; Xiong et al., 2012; Babetto et al., 2013; Neukomm et al., 2014).
Sarm1 (the mammalian ortholog of dSarm) is widely appreciated to be the “central executioner” for axon degeneration (Osterloh et al., 2012; DiAntonio, 2019). Initially characterized as an adaptor for Toll-like receptors (Couillault et al., 2004; Liberati et al., 2004; Carty et al., 2006), Sarm1 was unexpectedly found to have enzymatic activity that cleaves NAD+ molecules, reducing their availability in neurons and causing metabolic catastrophe (Essuman et al., 2017). It is this NAD-destroying function that precipitates the rapid degeneration of axons in vitro and in vivo (Geisler et al., 2019; Bosanac et al., 2021). To probe the domains responsible for this action, multiple groups have built transgenic Drosophila lines that express structurally altered variants of dSarm or Sarm1 (Neukomm et al., 2017; Brace et al., 2022; Herrmann et al., 2022). These lines enabled the discovery of separate signaling functions of Sarm1 in developmental versus degenerative signaling, highlighting multiple ways that this NAD-cleaving enzyme contributes to nervous system integrity (Brace et al., 2022; Herrmann et al., 2022).
The pathway for activation of Sarm1 is still under investigation, but clues have recently emerged. In one study by Izadifar et al. (2021), With-No-Lysine (K) (Wnk) kinase was discovered to be an inhibitor of Sarm1 activity in both Drosophila and mice. Loss of dWnk causes axon degeneration in multiple model systems, and its over-expression prevents some Sarm1-mediated disruption of neural circuits (Izadifar et al., 2021). Recently it was also found that nicotinamide mononucleotide (NMN), a synthetic precursor of NAD+, binds to an allosteric site on the armadillo (ARM) repeat domain and promotes Sarm1 activation. As the NMN/NAD+ ratio increases, Sarm1 activity also increases (Figley et al., 2021). It will be of future interest to examine how Wnk kinase expression affects the metabolic state of the neuron, which could link these two observations.
Intersecting with the dSarm/Sarm1 pathway, the proteins Highwire [PAM-Highwire-Rpm-1 (Phr1) in mammals] and Wallenda [dual leucine zipper kinase 1 (Dlk1) in mammals] also promote injury-induced degeneration and were first recognized for this role using injuries in the fly larval segmental nerves and the olfactory nerves, respectively (Miller et al., 2009; Xiong et al., 2012). Work in both fly and mouse systems demonstrated that Hiw/Phr1, an E3 ubiquitin ligase, targets an axonally-transported NMNAT variant, NMNAT2, for degradation (Gilley and Coleman, 2010; Xiong et al., 2012; Babetto et al., 2013). Thus, when Hiw/Phr1 is absent, NAD+ is produced in higher levels in the axon, providing axo-protection after injury. The E3 ubiquitin ligase complex containing Highwire also downregulates a pro-degenerative pathway controlled by the MAP triple kinase (MAP3K) Wallenda/DLK (Collins et al., 2006). Downstream of Wallenda/DLK and other MAP3Ks, multiple factors ensure the fragmentation of injured or NAD+ -depleted axons (Shin et al., 2012; Huntwork-Rodriguez et al., 2013; Larhammar et al., 2017).
Axon degeneration in Drosophila can also be initiated by treatment with drugs that induce nerve damage. For example, in humans, microtubule-disrupting components of conventional chemotherapy cocktails disrupt axon transport in long axons, leading to their demise. Using the drug paclitaxel (taxol) to trigger peripheral axon degeneration in Drosophila larvae, additional neuronal axon degeneration factors were identified including membrane occupation and recognition nexus 4 (MORN4) and transmembrane protein 184b (TMEM184B) (Bhattacharya et al., 2012, 2016). How these factors fit into known signaling pathways, specifically those controlled by Sarm and by MAP kinase cascades, remains to be determined.
Recent studies have expanded the focus from neuron-intrinsic factors to reveal information on the role of glia in injury responses. Flies have glia that cover the range of functions expected of mammalian glial types, including those for nerve insulation (wrapping glia), barrier function (subperineurial and perineurial glia), synaptic regulation (astrocyte-like glia) and neuronal cell body contacts (cortex glia) (Logan and Freeman, 2007; Yildirim et al., 2019). Drosophila subperineurial glia also have engulfment functions in both development and disease (Sonnenfeld and Jacobs, 1995). In the absence of injury, peripheral axons in Drosophila must be maintained via appropriate wrapping and metabolic signaling; these processes are controlled by the discoidin domain receptor (Ddr) and TGFβ signaling, respectively (Corty et al., 2022; Lassetter et al., 2023).
Following axonal injury, the Drosophila glial surface receptor Draper initiates the phagocytic pathway involved in severed axon clearance (MacDonald et al., 2006). Using appendage axotomy to robustly activate glial injury responses, the evolutionarily conserved signaling pathway downstream of Draper was recently identified (Lu et al., 2017; Purice et al., 2017). Draper signaling initiates transcriptional changes in glia following nerve injury (Lu et al., 2017; Purice et al., 2017). This cascade proceeds via transcriptional activation by STAT92E and AP-1. Through this transcriptional regulation, matrix metalloproteinase MMP-1 gene expression is upregulated and is necessary for remodeling of tissues and glial membrane expansion following injury (Purice et al., 2017). Other differentially activated glial genes have yet to be fully characterized in this system, but overall the analysis points to both Draper and Toll receptor signaling as key determinants of phagocytic and immune responses to injury.
Drosophila models of Charcot-Marie-Tooth (CMT) syndrome have yielded important insights into the pathological mechanisms causing sensory and motor neuropathy. CMT is linked to mutations in over 100 genes (WU Neuromuscular, 2023) and can be classified into myelin-affecting (CMT1, CMT4) and axon-affecting (CMT2, CMTX) phenotypes (Bolino and D’Antonio, 2023). Regardless of the subtype, all CMTs ultimately result in axon degeneration of peripheral nerves. Drosophila models of CMT2-associated mutations in Mitofusin2 (MFN2), have revealed that both loss- and gain-of-function human variants of MFN2 cause CMT-associated neurodegeneration and have implicated excessive mitochondrial fusion and disrupted mitochondria-ER contacts in disease pathogenesis (El Fissi et al., 2018; Shen et al., 2021). In another CMT subtype (CMT4J), autosomal recessive mutations in the PIP2 phosphatase FIG4 in humans cause a syndrome showing degeneration both in central and peripheral neuron populations (Chow et al., 2007). In mice, FIG4 mutation reduces the number of large diameter myelinated axons and impairs nerve conduction and action potential generation (Chow et al., 2007). By altering Drosophila Fig4 to introduce mutations corresponding to human patient variants, we now know that impaired phosphatase activity is not the reason for the patient syndrome and that its role in the nervous system involves the maintenance of lysosomal membrane integrity (Bharadwaj et al., 2016). By taking advantage of the “rough eye” phenotype caused by dFig4 mutations in flies, a modifier screen recently resulted in the identification of long non-coding RNAs that whose knockdown can suppress some phenotypes of dFig4 (Muraoka et al., 2018; Shimada et al., 2020). Whether this phenotype is due to a direct effect on Fig4 expression levels or on other related pathways has not yet been determined, but it may offer clues for how to molecularly target the Fig4 pathway in CMT 4J patients.
Spinal muscular atrophy (SMA), which causes motor neuron loss, can be modeled in Drosophila by depleting the survival motor neuron (SMN) protein; these models also exhibit motor neuron death. By taking advantage of the extremely well-described trajectory of motor neuron cell fate decisions during Drosophila embryonic development (Mark et al., 2021; Meng and Heckscher, 2021), Grice and Liu (2011, 2022) were able to show that early neurogenesis disruption, in addition to later neuromuscular junction (NMJ) dysfunction, contribute to locomotor phenotypes in SMA.
Some limitations to extrapolation of Drosophila findings to mammalian systems should be considered. First, Drosophila do not myelinate their nerves. In some peripheral neuropathies such as CMT Type 1A, Schwann cell disruption by mutations in peripheral myelin protein 22 (PMP22) or myelin protein zero (MPZ) cause secondary axon degeneration (Bolino and D’Antonio, 2023). The lack of myelination in Drosophila suggests this subtype is better modeled in vertebrates. Second, Drosophila lack an adaptive immune system. Therefore, non-neuronal responses to nervous system injury are likely simplified when compared to their mammalian counterparts. In the mouse, for example, sequential post-injury infiltration of neutrophils, macrophages and lymphocytes into injured nerves contribute to sensitization and pain (Marchand et al., 2005).
Patients with poorly controlled diabetes often experience peripheral neuropathy, which presents as hyperalgesia (extra sensitivity to normally innocuous stimuli), which can later progress to numbness. Inter-epidermal nerve fibers in the skin that subserve these sensations can show dystrophic terminals and withdrawal from the epidermis (Cheung et al., 2015). Patients with painful diabetic neuropathy also show decreases in intraepidermal nerve fiber density, indicating that nerve terminals are disrupted (Cheung et al., 2015). Drosophila fed a high sugar diet show thermal nociceptive hypersensitivity, similar to painful neuropathy in humans (Dabbara et al., 2021). This phenomenon is driven by sensory neuron function: when insulin receptor expression is impaired specifically in sensory neurons, overall dendrite length of multidendritic Class IV sensory neurons (which tile the cuticle wall) is reduced and animals show persistent hyperalgesia (Im et al., 2018). Thus the Drosophila system is well suited to examine not only pathological axon damage but also dendrite dysfunction.
Recent work has highlighted the similarities and differences between axon and dendrite degeneration by taking advantage of the class IV sensory neurons described above. In this system, Sarm over-expression or NMNAT knockout cause dendrite degeneration, suggesting that dendrites also degenerate via NAD+ depletion (Ji et al., 2022). The process of fragmentation was found to be separable from that of phagocytic clearance, done by endothelial cells in the larval cuticle. The authors describe a dendrite degeneration model which proposes three NAD+ “checkpoints:” one which activates Sarm, one in which additional Sarm-induced NAD+ exposes phosphatidylserine (PS) on the surface of injured dendrites to recruit phagocytic cells, and finally an ultra-low NAD+ level that causes dendrite breakdown independent of phagocytosis (Ji et al., 2022). These intriguing results suggest that there are more levels of control of neurite breakdown than previously appreciated. Further work will be required to confirm the alterations in NAD+ levels directly and to examine whether this model extends to mechanisms in axons or in other locations where glia rather than epidermal cells are responsible for phagocytosis.
The Drosophila larval neuromuscular junction has long been appreciated as a phenomenal location for studies of synaptic function; indeed, significant findings on the genes controlling the synaptic vesicle cycle were initially identified using this platform (Zhang, 2003; Frank et al., 2020). Using flies to model motor neuropathies such as amyotrophic lateral sclerosis (ALS) thus has been very well received. One caveat to the point-by-point comparison to mammalian systems is that, in mammals, the neuromuscular junction is cholinergic rather than glutamatergic. However, basic principles of neural toxicity of ALS-linked proteins are easily modeled in Drosophila larval and adult peripheral nerves (Yang et al., 2015). Some studies also use photoreceptor neurons or the central brain to investigate neuron death associated with ALS-linked gene expression (Ihara et al., 2013; Cunningham et al., 2020; Dubey et al., 2022). Here we will focus on axonal and synaptic effects of ALS genes in Drosophila.
The simplicity of Drosophila transgenic creation has permitted many models of ALS in which human or Drosophila versions of disease-associated protein variants can be expressed in spatially and temporally specific ways using binary expression systems like GAL4-UAS (Brand and Perrimon, 1993). For example, TDP-43, a protein whose aggregation is a hallmark of ALS and other neurodegenerative diseases, can be expressed in Drosophila larval motor neurons or in accessible adult structures (eyes, legs, or wings) to evaluate the pathways leading to motor neuron dysfunction and loss (Li et al., 2010; Estes et al., 2011; Sreedharan et al., 2015). Using pupal lethality as a readout of TDP-43 toxicity, an innovative drug screen for ALS suppressors identified pioglitazone, a PPARγ agonist, as a compound that mitigates TDP-43-dependent locomotor dysfunction (Joardar et al., 2015). However, this compound does not protect against all genetic causes of ALS and its mechanism is still unclear. Metabolomic profiling of larvae with TDP-43 over-expression has also revealed a role for the Tricarboxylic acid (TCA) cycle in the pathology of ALS (Loganathan et al., 2022).
In 2011, two groups studying ALS cohorts identified a hexanucleotide expansion in chromosome 9, open reading frame 72 (C9orf72) as a cause of ALS, and this alteration has since been appreciated to be the most common genetic alteration in sporadic ALS cases (DeJesus-Hernandez et al., 2011; Renton et al., 2011). While no C9orf72 homolog exists in flies, Drosophila models have been instrumental in understanding how this hexanucleotide expansion, which causes expression of multiple dipeptide repeat proteins, cause neurodegeneration. One of the biggest strengths of fly models is the ability to perform in vivo forward genetic screens, which have identified mediators and modifiers of dipeptide repeat toxicity (Xu et al., 2013; Solomon et al., 2018; Goodman et al., 2019). Drosophila models are also used to validate in vitro screening results in cultured cells (Donnelly et al., 2013). For example, stress granule accumulation occurs in ALS-affected neurons and disrupts critical cellular functions like nucleocytoplasmic transport and RNA metabolism (Li et al., 2013; Zhang et al., 2015, 2018). Markmiller et al. (2018) performed a screen in the Drosophila eye for suppressors of the ALS-associated “rough eye” phenotype and identified a number of novel candidates. Candidates were also studied directly in peripheral nerves of the Drosophila wing, and 5 novel stress granule proteins were identified as modifiers of the ALS phenotype (Markmiller et al., 2018).
Whole genome sequencing of rare disease patient samples, and genome-wide association studies, are continuously generating predictions of the importance of individual genetic loci to disease risk or severity. In order to demonstrate a true effect of these genetic variants, it is often required to modify the gene’s expression (up or down), either alone or in the background of a disease model. The costs and time associated with doing these rigorous experiments in mice in vivo is substantial. In contrast, Drosophila is a simple and powerful system in which genetic manipulations are simple and time-efficient, and appropriate disease models (such as those described above) are easily identified. Because it is predicted that roughly 65% of human disease genes have orthologs in Drosophila (Ugur et al., 2016), the fly has become a preferred model in which to first test the consequences of altering genes predicted by human genomic analyses (Sarkar and Feany, 2021; Wang et al., 2021).
The use of the fly to test disease-associated variants can be done in multiple ways such as transgenic expression of disease variants, knockdown of fly ortholog(s) of the human genes, or directly modifying the analogous amino acids in the fly ortholog via CRISPR. To give a few examples: Sorbitol dehydrogenase (SORD) homozygous mutations were recently identified in a cohort of patients clinically diagnosed with CMT. By creating a Drosophila model in which the orthologs (Sord1 and Sord2) were disrupted, the authors showed not only that the role of SORD in axon maintenance is conserved but that a pharmacological reversal of the phenotype is possible (Cortese et al., 2020). In another example, Cytochrome c oxidase assembly factor 7 (COA7) was identified from patient genomic analysis as a new causative gene for peripheral neuropathy (Higuchi et al., 2018). To model this disorder in vivo, the authors created a Drosophila model of COA7 impairment and showed defects in synaptic branches of the peripheral terminals of motor neurons as well as impaired locomotion (Higuchi et al., 2018). These results supported the causality of the variant-phenotype relationship in vivo.
Considering the advantages in accessibility, cost, and evolutionary conservation described above, Drosophila has become appreciated as one of the best models for understanding both neuron-intrinsic and extrinsic contributions to peripheral nerve maintenance and pathological forms of peripheral nerve loss. The number of human genomics studies who complete their first in vivo validations in Drosophila is growing. Additionally, a network of laboratories has united to use model organisms as rare disease profiling platforms, with Drosophila as one of the leaders in this movement (Gahl et al., 2015; Huang et al., 2022; Morimoto et al., 2023; Srivastava et al., 2023). For more established models of diseases such as ALS, flies will continue to provide a platform for unbiased, in vivo screening for enhancers and suppressors of nerve degeneration and cell loss.
A goal for future work on nerve injury in Drosophila is to unite our understanding of how glial cells contribute to axonal damage responses. This field is still in its infancy. However, because of the conservation of functions of glia in Drosophila and mammals, glial-neuronal communication can be addressed well in flies using advanced genetic tools and forward screening approaches (Yildirim et al., 2019). Another challenge that lays ahead is to integrate the pathways identified through human disease cohorts into known signaling pathways mediating axon degeneration (for example, are processes disrupted by new neuropathy genes like SORD or COA7 upstream or downstream of Sarm1?). An exciting era is before us as we begin to answer these questions.
As mentioned above, caveats do exist for this model involving lack of myelination or an adaptive immune system. However, the myriad advantages of using Drosophila to identify and characterize factors contributing to axon, dendrite, and synapse disruption will continue to keep flies at the leading edge of discoveries that, in turn, will improve our ability to mitigate the suffering of patients suffering from peripheral neuropathy.
MB performed the research, wrote the manuscript, and approved the submitted version.
This work was provided by a grant from the National Institutes of Health (R01 NS105680) to MB.
We would like to acknowledge the members of her lab for helpful discussions in the development of this manuscript.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Albers, J. W., and Pop-Busui, R. (2014). Diabetic neuropathy: Mechanisms, emerging treatments, and subtypes. Curr. Neurol. Neurosci. Rep. 14:473.
Babetto, E., Beirowski, B., Russler, E. V., Milbrandt, J., and DiAntonio, A. (2013). The Phr1 ubiquitin ligase promotes injury-induced axon self-destruction. Cell Rep. 3, 1422–1429. doi: 10.1016/j.celrep.2013.04.013
Bharadwaj, R., Cunningham, K. M., Zhang, K., and Lloyd, T. E. (2016). FIG4 regulates lysosome membrane homeostasis independent of phosphatase function. Hum. Mol. Genet. 25, 681–692.
Bhattacharya, M. R. C., Geisler, S., Pittman, S. K., Doan, R. A., Weihl, C. C., Milbrandt, J., et al. (2016). TMEM184b promotes axon degeneration and neuromuscular junction maintenance. J. Neurosci. 36, 4681–4689. doi: 10.1523/JNEUROSCI.2893-15.2016
Bhattacharya, M. R. C., Gerdts, J., Naylor, S. A., Royse, E. X., Ebstein, S. Y., Sasaki, Y., et al. (2012). A model of toxic neuropathy in Drosophila reveals a role for MORN4 in promoting axonal degeneration. J. Neurosci. 32, 5054–5061. doi: 10.1523/JNEUROSCI.4951-11.2012
Bolino, A., and D’Antonio, M. (2023). Recent advances in the treatment of Charcot-Marie-Tooth neuropathies. J. Peripher. Nerv. Syst. 28, 134–149.
Bosanac, T., Hughes, R., Engber, T., Devraj, R., Brearley, A., Danker, K., et al. (2021). Pharmacological SARM1 inhibition protects axon structure and function in paclitaxel-induced peripheral neuropathy. Brain 144, 3226–3238. doi: 10.1093/brain/awab184
Brace, E. J., and DiAntonio, A. (2020). Models of axon degeneration in Drosophila larvae. Methods Mol. Biol. 2143, 311–320. doi: 10.1007/978-1-0716-0585-1_23
Brace, E. J., Essuman, K., Mao, X., Palucki, J., Sasaki, Y., Milbrandt, J., et al. (2022). Distinct developmental and degenerative functions of SARM1 require NAD+ hydrolase activity. PLoS Genet. 18:e1010246. doi: 10.1371/journal.pgen.1010246
Brace, E. J., Wu, C., Valakh, V., and DiAntonio, A. (2014). SkpA restrains synaptic terminal growth during development and promotes axonal degeneration following injury. J. Neurosci. 34, 8398–8410.
Brand, A. H., and Perrimon, N. (1993). Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118, 401–405. doi: 10.1242/dev.118.2.401
Carty, M., Goodbody, R., Schröder, M., Stack, J., Moynagh, P., and Bowie, A. (2006). The human adaptor SARM negatively regulates adaptor protein TRIF-dependent Toll-like receptor signaling. Nat. Immunol. 7, 1074–1081.
Cheung, A., Podgorny, P., Martinez, J. A., Chan, C., and Toth, C. (2015). Epidermal axonal swellings in painful and painless diabetic peripheral neuropathy. Muscle Nerve 51, 505–513.
Chow, C. Y., Zhang, Y., Dowling, J., Jin, N., Adamska, M., Shiga, K., et al. (2007). Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature 448, 68–72.
Coleman, M. P., Conforti, L., Buckmaster, E., Tarlton, A., Ewing, R., Brown, M., et al. (1998). An 85-kb tandem triplication in the slow Wallerian degeneration (Wlds) mouse. Proc. Natl. Acad. Sci. U.S.A. 95, 9985–9990.
Collins, C. A., Wairkar, Y. P., Johnson, S. L., and DiAntonio, A. (2006). Highwire restrains synaptic growth by attenuating a MAP kinase signal. Neuron 51, 57–69.
Cortese, A., Zhu, Y., Rebelo, A., Negri, S., Courel, S., Abreu, L., et al. (2020). Biallelic mutations in SORD cause a common and potentially treatable hereditary neuropathy with implications for diabetes. Nat. Genet. 52, 473–481.
Corty, M. M., Hulegaard, A., Hill, J., Sheehan, A., Aicher, S., and Freeman, M. (2022). Discoidin domain receptor regulates ensheathment, survival and caliber of peripheral axons. Development 149:dev200636. doi: 10.1242/dev.200636
Couillault, C., Pujol, N., Reboul, J., Sabatier, L., Guichou, J., Kohara, Y., et al. (2004). TLR-independent control of innate immunity in Caenorhabditis elegans by the TIR domain adaptor protein TIR-1, an ortholog of human SARM. Nat. Immunol. 5, 488–494. doi: 10.1038/ni1060
Cunningham, K. M., Maulding, K., Ruan, K., Senturk, M., Grima, J., Sung, H., et al. (2020). TFEB/Mitf links impaired nuclear import to autophagolysosomal dysfunction in C9-ALS. Elife 9:e59419. doi: 10.7554/eLife.59419
Dabbara, H., Schultz, A., and Im, S. H. (2021). Drosophila insulin receptor regulates diabetes-induced mechanical nociceptive hypersensitivity. MicroPubl. Biol. 2021:10.17912/micropub.biology.000456. doi: 10.17912/micropub.biology.000456
DeJesus-Hernandez, M., Mackenzie, I., Boeve, B., Boxer, A., Baker, M., Rutherford, N., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
DiAntonio, A. (2019). Axon degeneration: Mechanistic insights lead to therapeutic opportunities for the prevention and treatment of peripheral neuropathy. Pain 160, S17–S22. doi: 10.1097/j.pain.0000000000001528
Donnelly, C. J., Zhang, P., Pham, J., Haeusler, A., Mistry, N., Vidensky, S., et al. (2013). RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80, 415–428. doi: 10.1016/j.neuron.2013.10.015
Dubey, S. K., Maulding, K., Sung, H., and Lloyd, T. E. (2022). Nucleoporins are degraded via upregulation of ESCRT-III/Vps4 complex in Drosophila models of C9-ALS/FTD. Cell Rep. 40:111379. doi: 10.1016/j.celrep.2022.111379
El Fissi, N., Rojo, M., Aouane, A., Karatas, E., Poliacikova, G., David, C., et al. (2018). Mitofusin gain and loss of function drive pathogenesis in Drosophila models of CMT2A neuropathy. EMBO Rep. 19:e45241.
Essuman, K., Summers, D., Sasaki, Y., Mao, X., DiAntonio, A., and Milbrandt, J. (2017). The SARM1 toll/interleukin-1 receptor domain possesses intrinsic NAD+ cleavage activity that promotes pathological axonal degeneration. Neuron 93, 1334–1343.e5. doi: 10.1016/j.neuron.2017.02.022
Estes, P. S., Boehringer, A., Zwick, R., Tang, J., Grigsby, B., and Zarnescu, D. (2011). Wild-type and A315T mutant TDP-43 exert differential neurotoxicity in a Drosophila model of ALS. Hum. Mol. Genet. 20, 2308–2321. doi: 10.1093/hmg/ddr124
Fang, Y., Soares, L., Teng, X., Geary, M., and Bonini, N. M. (2012). A novel Drosophila model of nerve injury reveals an essential role of Nmnat in maintaining axonal integrity. Curr. Biol. 22, 590–595. doi: 10.1016/j.cub.2012.01.065
Feldman, H. C., Merlini, E., Guijas, C., DeMeester, K., Njomen, E., Kozina, E., et al. (2022). Selective inhibitors of SARM1 targeting an allosteric cysteine in the autoregulatory ARM domain. Proc. Natl. Acad. Sci. U.S.A. 119:e2208457119. doi: 10.1073/pnas.2208457119
Figley, M. D., Gu, W., Nanson, J., Shi, Y., Sasaki, Y., Cunnea, K., et al. (2021). SARM1 is a metabolic sensor activated by an increased NMN/NAD+ ratio to trigger axon degeneration. Neuron 109, 1118–1136.e11. doi: 10.1016/j.neuron.2021.02.009
François-Moutal, L., Felemban, R., Scott, D., Sayegh, M., Miranda, V., Perez-Miller, S., et al. (2019). Small molecule targeting TDP-43’s RNA recognition motifs reduces locomotor defects in a Drosophila model of amyotrophic lateral sclerosis (ALS). ACS Chem. Biol. 14, 2006–2013. doi: 10.1021/acschembio.9b00481
Frank, C. A., James, T. D., and Müller, M. (2020). Homeostatic control of Drosophila neuromuscular junction function. Synapse 74:e22133. doi: 10.1002/syn.22133
Fukuda, Y., Li, Y., and Segal, R. A. (2017). A mechanistic understanding of axon degeneration in chemotherapy-induced peripheral neuropathy. Front. Neurosci. 11:481. doi: 10.3389/fnins.2017.00481
Gahl, W. A., Wise, A. L., and Ashley, E. A. (2015). The undiagnosed diseases network of the National Institutes of Health: A national extension. JAMA 314, 1797–1798. doi: 10.1001/jama.2015.12249
Geisler, S., Huang, S., Strickland, A., Doan, R., Summers, D., Mao, X., et al. (2019). Gene therapy targeting SARM1 blocks pathological axon degeneration in mice. J. Exp. Med. 216, 294–303.
Gilley, J., and Coleman, M. P. (2010). Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS Biol. 8:e1000300. doi: 10.1371/journal.pbio.1000300
Goodman, L. D., Prudencio, M., Kramer, N., Martinez-Ramirez, L., Srinivasan, A., Lan, M., et al. (2019). Toxic expanded GGGGCC repeat transcription is mediated by the PAF1 complex in C9orf72-associated FTD. Nat. Neurosci. 22, 863–874.
Grice, S. J., and Liu, J.-L. (2011). Survival motor neuron protein regulates stem cell division, proliferation, and differentiation in Drosophila. PLoS Genet. 7:e1002030. doi: 10.1371/journal.pgen.1002030
Grice, S. J., and Liu, J.-L. (2022). Motor defects in a Drosophila model for spinal muscular atrophy result from SMN depletion during early neurogenesis. PLoS Genet. 18:e1010325. doi: 10.1371/journal.pgen.1010325
Hao, Y., Waller, T. J., Nye, D. M., Li, J., Zhang, Y., Hume, R. I., et al. (2019). Degeneration of injured axons and dendrites requires restraint of a protective JNK signaling pathway by the transmembrane protein raw. J. Neurosci. 39, 8457–8470.
Herrmann, K. A., Liu, Y., Llobet-Rosell, A., McLaughlin, C., Neukomm, L., Coutinho-Budd, J., et al. (2022). Divergent signaling requirements of dSARM in injury-induced degeneration and developmental glial phagocytosis. PLoS Genet. 18:e1010257. doi: 10.1371/journal.pgen.1010257
Higuchi, Y., Okunushi, R., Hara, T., Hashiguchi, A., Yuan, J., Yoshimura, A., et al. (2018). Mutations in COA7 cause spinocerebellar ataxia with axonal neuropathy. Brain 141, 1622–1636.
Huang, Y., Lemire, G., Briere, L., Liu, F., Wessels, M., Wang, X., et al. (2022). The recurrent de novo c.2011C>T missense variant in MTSS2 causes syndromic intellectual disability. Am. J. Hum. Genet. 109, 1923–1931.
Huntwork-Rodriguez, S., Wang, B., Watkins, T., Ghosh, A., Pozniak, C., Bustos, D., et al. (2013). JNK-mediated phosphorylation of DLK suppresses its ubiquitination to promote neuronal apoptosis. J. Cell Biol. 202, 747–763. doi: 10.1083/jcb.201303066
Ihara, R., Matsukawa, K., Nagata, Y., Kunugi, H., Tsuji, S., Chihara, T., et al. (2013). RNA binding mediates neurotoxicity in the transgenic Drosophila model of TDP-43 proteinopathy. Hum. Mol. Genet. 22, 4474–4484. doi: 10.1093/hmg/ddt296
Im, S. H., Patel, A. A., Cox, D. N., and Galko, M. J. (2018). Drosophila insulin receptor regulates the persistence of injury-induced nociceptive sensitization. Dis. Mod. Mech. 11:dmm034231. doi: 10.1242/dmm.034231
Izadifar, A., Courchet, J., Virga, D., Verreet, T., Hamilton, S., Ayaz, D., et al. (2021). Axon morphogenesis and maintenance require an evolutionary conserved safeguard function of Wnk kinases antagonizing Sarm and Axed. Neuron 109, 2864–2883.e8. doi: 10.1016/j.neuron.2021.07.006
Ji, H., Sapar, M. L., Sarkar, A., Wang, B., and Han, C. (2022). Phagocytosis and self-destruction break down dendrites of Drosophila sensory neurons at distinct steps of Wallerian degeneration. Proc. Natl. Acad. Sci. U.S.A 119:e2111818119. doi: 10.1073/pnas.2111818119
Joardar, A., Menzl, J., Podolsky, T., Manzo, E., Estes, P., Ashford, S., et al. (2015). PPAR gamma activation is neuroprotective in a Drosophila model of ALS based on TDP-43. Hum. Mol. Genet. 24, 1741–1754. doi: 10.1093/hmg/ddu587
LaPointe, N. E., Morfini, G., Brady, S., Feinstein, S., Wilson, L., and Jordan, M. (2013). Effects of eribulin, vincristine, paclitaxel and ixabepilone on fast axonal transport and kinesin-1 driven microtubule gliding: Implications for chemotherapy-induced peripheral neuropathy. Neurotoxicology 37, 231–239. doi: 10.1016/j.neuro.2013.05.008
Larhammar, M., Huntwork-Rodriguez, S., Rudhard, Y., Sengupta-Ghosh, A., and Lewcock, J. W. (2017). The Ste20 family kinases MAP4K4, MINK1, and TNIK converge to regulate stress-induced JNK signaling in neurons. J. Neurosci. 37, 11074–11084. doi: 10.1523/JNEUROSCI.0905-17.2017
Lassetter, A. P., Corty, M., Barria, R., Sheehan, A., Hill, J., Aicher, S., et al. (2023). Glial TGFβ activity promotes neuron survival in peripheral nerves. J. Cell Biol. 222:e202111053.
Law, A. D., Cassar, M., Long, D., Chow, E., Giebultowicz, J., Venkataramanan, A., et al. (2022). FTD-associated mutations in tau result in a combination of dominant and recessive phenotypes. Neurobiol. Dis. 170:105770. doi: 10.1016/j.nbd.2022.105770
Li, D., Li, F., Guttipatti, P., and Song, Y. (2018). A Drosophila in vivo injury model for studying neuroregeneration in the peripheral and central nervous system. J. Vis. Exp. 135:57557. doi: 10.3791/57557
Li, Y. R., King, O. D., Shorter, J., and Gitler, A. D. (2013). Stress granules as crucibles of ALS pathogenesis. J. Cell Biol. 201, 361–372.
Li, Y., Ray, P., Rao, E., Shi, C., Guo, W., Chen, X., et al. (2010). A Drosophila model for TDP-43 proteinopathy. Proc. Natl. Acad. Sci. U.S.A. 107, 3169–3174. doi: 10.1073/pnas.0913602107
Liberati, N. T., Fitzgerald, K., Kim, D., Feinbaum, R., Golenbock, D., and Ausubel, F. (2004). Requirement for a conserved toll/interleukin-1 resistance domain protein in the Caenorhabditis elegans immune response. Proc. Natl. Acad. Sci. U.S.A. 101, 6593–6598. doi: 10.1073/pnas.0308625101
Logan, M. A., and Freeman, M. R. (2007). The scoop on the fly brain. Neuron Glia Biol. 3, 63–74. doi: 10.1017/S1740925X07000646
Logan, M. A., Hackett, R., Doherty, J., Sheehan, A., Speese, S. D., and Freeman, M. R. (2012). Negative regulation of glial engulfment activity by Draper terminates glial responses to axon injury. Nat. Neurosci. 15, 722–730.
Loganathan, S., Wilson, B., Carey, S., Manzo, E., Joardar, A., Ugur, B., et al. (2022). TDP-43 proteinopathy causes broad metabolic alterations including TCA cycle intermediates and dopamine levels in Drosophila models of ALS. Metabolites 12:101. doi: 10.3390/metabo12020101
Lu, T.-Y., MacDonald, J., Neukomm, L., Sheehan, A., Bradshaw, R., Logan, M., et al. (2017). Axon degeneration induces glial responses through Draper-TRAF4-JNK signalling. Nat. Commun. 8:14355. doi: 10.1038/ncomms14355
Lunn, E. R., Perry, V. H., Brown, M. C., Rosen, H., and Gordon, S. (1989). Absence of Wallerian degeneration does not hinder regeneration in peripheral nerve. Eur. J. Neurosci. 1, 27–33.
MacDonald, J. M., Beach, M., Porpiglia, E., Sheehan, A., Watts, R., and Freeman, M. (2006). The Drosophila cell corpse engulfment receptor Draper mediates glial clearance of severed axons. Neuron 50, 869–881. doi: 10.1016/j.neuron.2006.04.028
Mack, T. G., Reiner, M., Beirowski, B., Mi, W., Emanuelli, M., Wagner, D., et al. (2001). Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat. Neurosci. 4, 1199–1206. doi: 10.1038/nn770
Marchand, F., Perretti, M., and McMahon, S. B. (2005). Role of the Immune system in chronic pain. Nat. Rev. Neurosci. 6, 521–532.
Mark, B., Lai, S., Zarin, A., Manning, L., Pollington, H., Litwin-Kumar, A., et al. (2021). A developmental framework linking neurogenesis and circuit formation in the Drosophila CNS. Elife 10:e67510. doi: 10.7554/eLife.67510
Markmiller, S., Soltanieh, S., Server, K., Mak, R., Jin, W., Fang, M., et al. (2018). Context-dependent and disease-specific diversity in protein interactions within stress granules. Cell 172, 590–604.e13. doi: 10.1016/j.cell.2017.12.032
Mecklenburg, K. L., Freed, S. A., Raval, M., Quintero, O. A., Yengo, C. M., and O’Tousa, J. E. (2015). Invertebrate and vertebrate class III myosins interact with MORN repeat-containing adaptor proteins. PLoS One 10:e0122502.
Meng, J. L., and Heckscher, E. S. (2021). Development of motor circuits: From neuronal stem cells and neuronal diversity to motor circuit assembly. Curr. Top. Dev. Biol. 142, 409–442.
Menorca, R. M., Fussell, T. S., and Elfar, J. C. (2013). Nerve physiology: Mechanisms of injury and recovery. Hand. Clin. 29, 317–330.
Miller, B. R., Press, C., Daniels, R., Sasaki, Y., Milbrandt, J., and DiAntonio, A. (2009). A dual leucine kinase-dependent axon self-destruction program promotes Wallerian degeneration. Nat. Neurosci. 12, 387–389. doi: 10.1038/nn.2290
Morimoto, M., Bhambhani, V., Gazzaz, N., Davids, M., Sathiyaseelan, P., Macnamara, E., et al. (2023). Bi-allelic ATG4D variants are associated with a neurodevelopmental disorder characterized by speech and motor impairment. NPJ Genom. Med. 8:4. doi: 10.1038/s41525-022-00343-8
Muraoka, Y., Nakamura, A., Tanaka, R., Suda, K., Azuma, Y., Kushimura, Y., et al. (2018). Genetic screening of the genes interacting with Drosophila FIG4 identified a novel link between CMT-causing gene and long noncoding RNAs. Exp. Neurol. 310, 1–13. doi: 10.1016/j.expneurol.2018.08.009
Neukomm, L. J., Burdett, T. C., Gonzalez, M. A., Züchner, S., and Freeman, M. R. (2014). Rapid in vivo forward genetic approach for identifying axon death genes in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 111, 9965–9970. doi: 10.1073/pnas.1406230111
Neukomm, L. J., Burdett, T., Seeds, A., Hampel, S., Coutinho-Budd, J., Farley, J., et al. (2017). Axon death pathways converge on axundead to promote functional and structural axon disassembly. Neuron 95, 78–91.e5. doi: 10.1016/j.neuron.2017.06.031
Osterloh, J. M., Yang, J., Rooney, T., Fox, A., Adalbert, R., Powell, E., et al. (2012). dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science 337, 481–484. doi: 10.1126/science.1223899
Paglione, M., Rosell, A. L., Chatton, J.-Y., and Neukomm, L. J. (2020). Morphological and functional evaluation of axons and their synapses during axon death in Drosophila melanogaster. J. Vis. Exp. 157. doi: 10.3791/60865
Phelps, J. S., Hildebrand, D., Graham, B., Kuan, A., Thomas, L., Nguyen, T., et al. (2021). Reconstruction of motor control circuits in adult Drosophila using automated transmission electron microscopy. Cell 184, 759–774.e18. doi: 10.1016/j.cell.2020.12.013
Purice, M. D., Ray, A., Münzel, E., Pope, B., Park, D., Speese, S., et al. (2017). A novel Drosophila injury model reveals severed axons are cleared through a draper/MMP-1 signaling cascade. Elife 6:e23611. doi: 10.7554/eLife.23611
Renton, A. E., Majounie, E., Waite, A., Simón-Sánchez, J., Rollinson, S., Gibbs, J., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268.
Sarkar, S., and Feany, M. B. (2021). Precision medicine on the fly: Using Drosophila to decipher gene-environment interactions in Parkinson’s disease. Toxicol. Sci. 182, 159–167. doi: 10.1093/toxsci/kfab060
Sasaki, Y., Vohra, B. P. S., Baloh, R. H., and Milbrandt, J. (2009). Transgenic mice expressing the Nmnat1 protein manifest robust delay in axonal degeneration in vivo. J. Neurosci. 29, 6526–6534. doi: 10.1523/JNEUROSCI.1429-09.2009
Shen, J. L., Fortier, T., Zhao, Y., Wang, R., Burmeister, M., and Baehrecke, E. (2021). Vmp1, Vps13D, and Marf/Mfn2 function in a conserved pathway to regulate mitochondria and ER contact in development and disease. Curr. Biol. 31, 3028–3039.e7. doi: 10.1016/j.cub.2021.04.062
Shi, Y., Kerry, P., Nanson, J., Bosanac, T., Sasaki, Y., Krauss, R., et al. (2022). Structural basis of SARM1 activation, substrate recognition, and inhibition by small molecules. Mol. Cell 82, 1643–1659.e10. doi: 10.1016/j.molcel.2022.03.007
Shimada, S., Muraoka, Y., Ibaraki, K., Takano-Shimizu-Kouno, T., Yoshida, H., and Yamaguchi, M. (2020). Identification of CR43467 encoding a long non-coding RNA as a novel genetic interactant with dFIG4, a CMT-causing gene. Exp. Cell Res. 386:111711. doi: 10.1016/j.yexcr.2019.111711
Shin, J. E., Miller, B., Babetto, E., Cho, Y., Sasaki, Y., Qayum, S., et al. (2012). SCG10 is a JNK target in the axonal degeneration pathway. Proc. Natl. Acad. Sci. U.S.A. 109, E3696–E3705. doi: 10.1073/pnas.1216204109
Singer, M. A., Vernino, S. A., and Wolfe, G. I. (2012). Idiopathic neuropathy: New paradigms, new promise. J. Peripher. Nerv. Syst. 17, 43–49. doi: 10.1111/j.1529-8027.2012.00395.x
Singh, S., Dallenga, T., Winkler, A., Roemer, S., Maruschak, B., Siebert, H., et al. (2017). Relationship of acute axonal damage, Wallerian degeneration, and clinical disability in multiple sclerosis. J. Neuroinflammation 14:57. doi: 10.1186/s12974-017-0831-8
Solomon, D. A., Stepto, A., Au, W. H., Adachi, Y., Diaper, D. C., Hall, R., et al. (2018). A feedback loop between dipeptide-repeat protein, TDP-43 and karyopherin-α mediates C9orf72-related neurodegeneration. Brain 141, 2908–2924. doi: 10.1093/brain/awy241
Sonnenfeld, M. J., and Jacobs, J. R. (1995). Macrophages and glia participate in the removal of apoptotic neurons from the Drosophila embryonic nervous system. J. Comp. Neurol. 359, 644–652. doi: 10.1002/cne.903590410
Sreedharan, J., Neukomm, L. J., Brown, R. H., and Freeman, M. R. (2015). Age-dependent TDP-43-mediated motor neuron degeneration requires GSK3, hat-trick, and xmas-2. Curr. Biol. 25, 2130–2136. doi: 10.1016/j.cub.2015.06.045
Srivastava, S., Shaked, H. M., Gable, K., Gupta, S. D., Pan, X., Somashekarappa, N., et al. (2023). SPTSSA variants alter sphingolipid synthesis and cause a complex hereditary spastic paraplegia. Brain 146, 1420–1435. doi: 10.1093/brain/awac460
Tao, J., and Rolls, M. M. (2011). Dendrites have a rapid program of injury-induced degeneration that is molecularly distinct from developmental pruning. J. Neurosci. 31, 5398–5405. doi: 10.1523/JNEUROSCI.3826-10.2011
Theiss, C., and Meller, K. (2000). Taxol impairs anterograde axonal transport of microinjected horseradish peroxidase in dorsal root ganglia neurons in vitro. Cell Tissue Res. 299, 213–224. doi: 10.1007/s004419900120
Tian, F., Yang, W., Mordes, D. A., Wang, J. Y., Salameh, J. S., Mok, J., et al. (2016). Monitoring peripheral nerve degeneration in ALS by label-free stimulated Raman scattering imaging. Nat. Commun. 7:13283. doi: 10.1038/ncomms13283
Timmerman, V., Clowes, V. E., and Reid, E. (2013). Overlapping molecular pathological themes link Charcot–Marie–Tooth neuropathies and hereditary spastic paraplegias. Exp. Neurol. 246, 14–25. doi: 10.1016/j.expneurol.2012.01.010
Ugur, B., Chen, K., and Bellen, H. J. (2016). Drosophila tools and assays for the study of human diseases. Dis. Model Mech. 9, 235–244. doi: 10.1242/dmm.023762
Vargas, M. E., Yamagishi, Y., Tessier-Lavigne, M., and Sagasti, A. (2015). Live imaging of calcium dynamics during axon degeneration reveals two functionally distinct phases of calcium influx. J. Neurosci. 35, 15026–15038. doi: 10.1523/JNEUROSCI.2484-15.2015
Waller, A. (1851). Experiments on the section of the glosso-pharyngeal and hypoglossal nerves of the frog, and observations of the alterations produced thereby in the structure of their primitive fibres. Edinb. Med. Surg. J. 76, 369–376.
Wang, J.-W., Brent, J. R., Tomlinson, A., Shneider, N. A., and McCabe, B. D. (2011). The ALS-associated proteins FUS and TDP-43 function together to affect Drosophila locomotion and life span. J. Clin. Invest. 121, 4118–4126. doi: 10.1172/JCI57883
Wang, M., Li, A., Sekiya, M., Beckmann, N. D., Quan, X., Schrode, N., et al. (2021). Transformative network modeling of multi-omics data reveals detailed circuits, key regulators, and potential therapeutics for Alzheimer’s disease. Neuron 109, 257–272.e14. doi: 10.1016/j.neuron.2020.11.002
Wu, H.-H., Bellmunt, E., Scheib, J. L., Venegas, V., Burkert, C., Reichardt, L. F., et al. (2009). Glial precursors clear sensory neuron corpses during development via Jedi-1, an engulfment receptor. Nat. Neurosci. 12, 1534–1541.
WU Neuromuscular (2023). Hereditary motor sensory neuropathies: Charcot-Marie-Tooth. Available online at: https://neuromuscular.wustl.edu/time/hmsn.html#cmtmol (accessed April 23, 2023).
Xiong, X., Hao, Y., Sun, K., Li, J., Li, X., Mishra, B., et al. (2012). The highwire ubiquitin ligase promotes axonal degeneration by tuning levels of Nmnat protein. PLoS Biol. 10:e1001440. doi: 10.1371/journal.pbio.1001440
Xu, Z., Poidevin, M., Li, X., Li, Y., Shu, L., Nelson, D., et al. (2013). Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc. Natl. Acad. Sci. U.S.A. 110, 7778–7783.
Yamagishi, Y., and Tessier-Lavigne, M. (2016). An atypical SCF-like ubiquitin ligase complex promotes Wallerian degeneration through regulation of axonal Nmnat2. Cell Rep. 17, 774–782.
Yang, D., Abdallah, A., Li, Z., Lu, Y., Almeida, S., and Gao, F. (2015). FTD/ALS-associated poly(GR) protein impairs the Notch pathway and is recruited by poly(GA) into cytoplasmic inclusions. Acta Neuropathol. 130, 525–535. doi: 10.1007/s00401-015-1448-6
Yildirim, K., Petri, J., Kottmeier, R., and Klämbt, C. (2019). Drosophila glia: Few cell types and many conserved functions. Glia 67, 5–26. doi: 10.1002/glia.23459
Zhang, B. (2003). Genetic and molecular analysis of synaptic vesicle recycling in Drosophila. J. Neurocytol. 32, 567–589. doi: 10.1023/B:NEUR.0000020611.44254.86
Zhang, K., Daigle, J., Cunningham, K., Coyne, A., Ruan, K., Grima, J., et al. (2018). Stress granule assembly disrupts nucleocytoplasmic transport. Cell 173, 958–971.e17.
Zhang, K., Donnelly, C., Haeusler, A., Grima, J., Machamer, J., Steinwald, P., et al. (2015). The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525, 56–61.
Keywords: Drosophila, peripheral neuropathy, axon degeneration, ALS, Charcot-Marie Tooth disease
Citation: Bhattacharya MRC (2023) A nerve-wracking buzz: lessons from Drosophila models of peripheral neuropathy and axon degeneration. Front. Aging Neurosci. 15:1166146. doi: 10.3389/fnagi.2023.1166146
Received: 14 February 2023; Accepted: 24 July 2023;
Published: 08 August 2023.
Edited by:
Yan Kong, Southeast University, ChinaReviewed by:
Teresa Niccoli, University College London, United KingdomCopyright © 2023 Bhattacharya. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Martha R. C. Bhattacharya, bWFydGhhYjFAYXJpem9uYS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.