 Jeongyeon Nam
Jeongyeon Nam Youngdae Gwon
Youngdae Gwon- Department of Molecular Cell Biology, Sungkyunkwan University School of Medicine, Suwon, Republic of Korea
Biomolecular condensates are subcellular organizations where functionally related proteins and nucleic acids are assembled through liquid–liquid phase separation, allowing them to develop on a larger scale without a membrane. However, biomolecular condensates are highly vulnerable to disruptions from genetic risks and various factors inside and outside the cell and are strongly implicated in the pathogenesis of many neurodegenerative diseases. In addition to the classical view of the nucleation-polymerization process that triggers the protein aggregation from the misfolded seed, the pathologic transition of biomolecular condensates can also promote the aggregation of proteins found in the deposits of neurodegenerative diseases. Furthermore, it has been suggested that several protein or protein-RNA complexes located in the synapse and along the neuronal process are neuron-specific condensates displaying liquid-like properties. As their compositional and functional modifications play a crucial role in the context of neurodegeneration, further research is needed to fully understand the role of neuronal biomolecular condensates. In this article, we will discuss recent findings that explore the pivotal role of biomolecular condensates in the development of neuronal defects and neurodegeneration.
1. Introduction
Compartmentalization is a critical feature that benefits eukaryotic cells, allowing them to concentrate functionally relevant biomolecules in specific subcellular organelles. Over time, eukaryotic cells have evolved to create distinct environments that promote the function of organelle-specific biomolecules, such as lysosomes or mitochondria. While membrane-bound organelles separated from the other cellular parts with lipid bilayers mainly composed of amphiphilic phospholipids have long been investigated, membrane-less assemblies of proteins and nucleic acids, defined as biomolecular condensates, have emerged as another paradigm to give rise to cellular organizations as a result of numerous studies over the last decade (Banani et al., 2017). De-mixed with the surrounding phase, biomolecular condensates embrace a subset of molecules of related functions, promoting many biological processes in themselves (Lyon et al., 2021). Now, the formation of biomolecular condensates is understood by the principle of liquid–liquid phase separation (LLPS).
Although biomolecular condensates are found in many eukaryotic cells, neurons, which differ structurally from other cells, exhibit a number of unique biomolecular condensates (Gopal et al., 2017; Milovanovic et al., 2018; Zeng et al., 2018; Wu et al., 2019; Mcdonald et al., 2020). Neurons are polarized cells with an axon that extends from the soma to transmit electrochemical information and multiple dendrites that receive inputs from adjacent neurons. In the interneuronal synapse, there are several microorganizations, including synaptic vesicle (SV) clusters, presynaptic active zones (AZ), and postsynaptic densities (PSD) (Feng et al., 2019; Emperador-Melero and Kaeser, 2020; Reshetniak and Rizzoli, 2021). Also, since axon and dendrites make up more than 90% of neuronal volumes, neurons face challenges in maintaining the structural and compositional integrity of synapses (Kasthuri et al., 2015). To overcome this, mRNAs encoding synaptic proteins are transported through neurite projections in a form of ribonucleoprotein (RNP) granule, or RNA transport granule, for localized translation (Das et al., 2021). Of note, LLPS plays a vital role in the assembly and functional regulation of these structures.
In this review, we will summarize recent updates about biomolecular condensates found exclusively in neurons. We will describe how multivalent interactions among components, facilitated by the principle of LLPS, generate biomolecular condensates. We will also discuss how the solidification of these condensates, from liquid to solid-like phase, is frequently found in the pathogenesis of neurodegenerative diseases. Additionally, we will describe the specialized roles of neuronal biomolecular condensates and their modification under disease-relevant contexts. This review will provide a comprehensive understanding of neuronal biomolecular condensates and their role in the mechanism of neurodegeneration.
2. Multivalency-driven LLPS As the molecular momentum to assemble biomolecular condensates
A de-mixing event work against the spontaneous increment in entropy to minimize the free energy in the system. Therefore, the contribution of enthalpy which offsets the loss of entropy is crucial for this de-mixing process as suggested by Flory-Huggins lattice models for polymer-solvent mixtures (Brangwynne et al., 2015; Nott et al., 2015; Workman and Pettitt, 2021). Enthalpic momentums are determined by the net change in intra-and intermolecular interactions during the de-mixing processes. Typically, for a given set of molecules to separate into a different phase, the sum of the magnitude of interactions among them exceeds the magnitude of interactions with other molecules. These molecules co-exist in two distinct phases following the de-mixing process of such solutions, one phase having a dense concentration and the other having a diluted concentration. LLPS is defined as a switch from 1-phase regime to 2-phase regime that results in a local enrichment of macromolecules above the saturation concentration in droplet-like structures (Hyman et al., 2014). In biological systems, these droplet-shaped foci composed mostly of proteins and nucleic acids that exhibit different phase properties from their surroundings are termed as biomolecular condensates (Banani et al., 2017; Lyon et al., 2021). The proteins in biomolecular condensates exhibit weak and transient interactions with multiple partners, which occur in both folded and intrinsically disordered regions (IDRs) (Banani et al., 2017; Shin and Brangwynne, 2017).
2.1. Folded domain
LLPS of sequentially positioned domains of one protein and short linear motifs (SLiM) of another protein is an example of how folded protein domains contribute to the valency-dependent assembly of biomolecular condensates (Hastings and Boeynaems, 2021). In the actin regulatory signaling system, SH3 domains of cytoplasmic protein NCK interact with proline-rich motifs, a prominent type of SLiM, of neuronal Wiskkott-Aldrich syndrome protein (N-WASP) (Li et al., 2012). Additionally, NCK binds to phosphorylated tyrosine residues of either Nephrin in kidney podocytes or SLP-76 in T cells, respectively through its SH2 domains (Li et al., 2012; Su et al., 2016). As the saturation threshold for phase separation in these signaling complexes, which are frequently represented as mesh networks, can be altered by varying the valency of connections, multivalency is crucial for encouraging protein condensation (Li et al., 2012; Banani et al., 2016; Su et al., 2016). Other examples of LLPS which require folded domain-mediated interactions include the LSm domain of Edc3 with leucine-rich motifs of Dcp2 and/or Pdc1 in p-bodies, and BTB and BACK domains of SPOP in nuclear speckles (Fromm et al., 2014; Bouchard et al., 2018).
2.2. Intrinsically disordered regions
Intrinsically disordered regions plays a crucial role for LLPS. IDRs do not have static tertiary structures and can be varied within a broad range of conformations whose energy states are similar to one another (Wright and Dyson, 2015). The conformational variance of IDR is often derived from the low complexity (LC) of its amino acid sequences (Uversky, 2002). Certain kinds of amino acids are enriched in IDRs depending on their chemical characteristics. Polar amino acids (e.g., serine and glutamine) and charged amino acids (e.g., glutamate, lysine, and arginine) are enriched in IDRs and contribute to LLPS driven by electrostatic interaction with other IDR-containing proteins or nucleic acids (Alberti et al., 2009; Pak et al., 2016). For example, the condensation of DDX4 in germ granules relies on the charged blocks in its IDR, and neutralizing the charges in these clusters inhibits the phase separation of DDX4 (Nott et al., 2015). Additionally, tyrosine and phenylalanine which have aromatic structures are frequently positioned in the IDR and enable IDR to have cation-π or π-π interactions required for LLPS, though tyrosine showed the better affinity with lysine or arginine compared to phenylalanine (Wang et al., 2018; Schuster et al., 2020). Otherwise, IDR could interact with SLiM as demonstrated by the interaction between the oligomerization domain of nucleophosmin (NPM1) and arginine-rich motifs of a group of nucleolar proteins in the granular component of nucleoli (Mitrea et al., 2016). Collectively, the sum of short-lived and weak interactions among biomolecules that happen simultaneously act as a driving momentum for LLPS.
3. Facilitation of protein fibrilization in the phase-deformed biomolecular condensates
Neurons become postmitotic during early development and typically remained differentiated for their entire lifespan, except in cases of cell cycle re-entry found in the brains of neurodegenerative diseases (Lee et al., 2009; Anda et al., 2016). Along with the closed nature of the brain accomplished by the limited molecular exchange across the blood–brain barrier, long-lived neurons often accumulate toxic protein fibrils and aggregates through the aging process. Therefore, it is important to study how soluble and functional proteins evolve into insoluble and dysfunctional aggregates to better understand the pathogenesis of neurodegenerative diseases (Soto and Pritzkow, 2018).
Nucleation-dependent fibrilization is a well-defined model to describe the formation of many biological polymers such as actin and tubulin cytoskeletons but also has long been thought to generate pathologic protein fibrils often found in the neurodegenerative diseases (Roychaudhuri et al., 2009). Misfolded proteins escaping from the proteostasis system often become the seed of this process (Hipp et al., 2019). Protein amyloids and filamentous protein aggregates with cross-beta structures are accumulated as insoluble deposits inside or outside of the cells. In particular, favorable environments for protein fibrilization are fostered in the nervous system with postmitotic neurons where seeds for amyloid formation can be enriched as aging proceeds (Hipp et al., 2019). Amyloid plaque and Lewy body found in the brains of Alzheimer’s disease (AD) and Parkinson’s diseases (PD), respectively, are well-known examples of protein amyloids.
Furthermore, recent studies have demonstrated that a liquid-to-solid phase transition is another route for building up the pathologic protein aggregates (Nedelsky and Taylor, 2019; Mathieu et al., 2020). Liquid-like biomolecular condensates become more dense and gel-like, and eventually transitioning into a solid-like phase during the pathogenic circumstances of human diseases (Alberti and Hyman, 2021). Especially, much evidence reflecting these pathologic transitions are readily identified in the process of neurodegenerative diseases including amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), huntington’s disease (HD), and tauopathy (Molliex et al., 2015; Patel et al., 2015; Lee et al., 2016; Ambadipudi et al., 2017; Jain and Vale, 2017; Peskett et al., 2018; Wegmann et al., 2018; Nedelsky and Taylor, 2019; Alberti and Hyman, 2021).
3.1. Amyotrophic lateral sclerosis–Frontotemporal dementia
Amyotrophic lateral sclerosis–frontotemporal dementia, collectively now accounted as a spectrum disorder of ALS and FTD, is a representative research field where the phase transition from liquid to solid states of biomolecular condensates initiates the pathogenesis (Ling et al., 2013). Multiple genetic causes of ALS–FTD have been found in the genes encoding RNA binding proteins (RBPs) such as transactive response DNA-binding protein 43 kDa (TDP43), Fused in Sarcoma (FUS), HNRNPA1, and TIA1 which can undergo LLPS in vitro (Molliex et al., 2015; Patel et al., 2015; Mackenzie et al., 2017). Given that most RBP mutations causing ALS–FTD are located in IDR, it is not surprising that ALS–FTD-associated mutant FUS, HNRNPA1 and TIA1 exhibit a greater propensity to undergo LLPS at lower saturation concentration. Mutant proteins are also more likely to form denser, solid-like phases, and even insoluble fibrils (Figure 1A) (Molliex et al., 2015; Patel et al., 2015; Mackenzie et al., 2017).
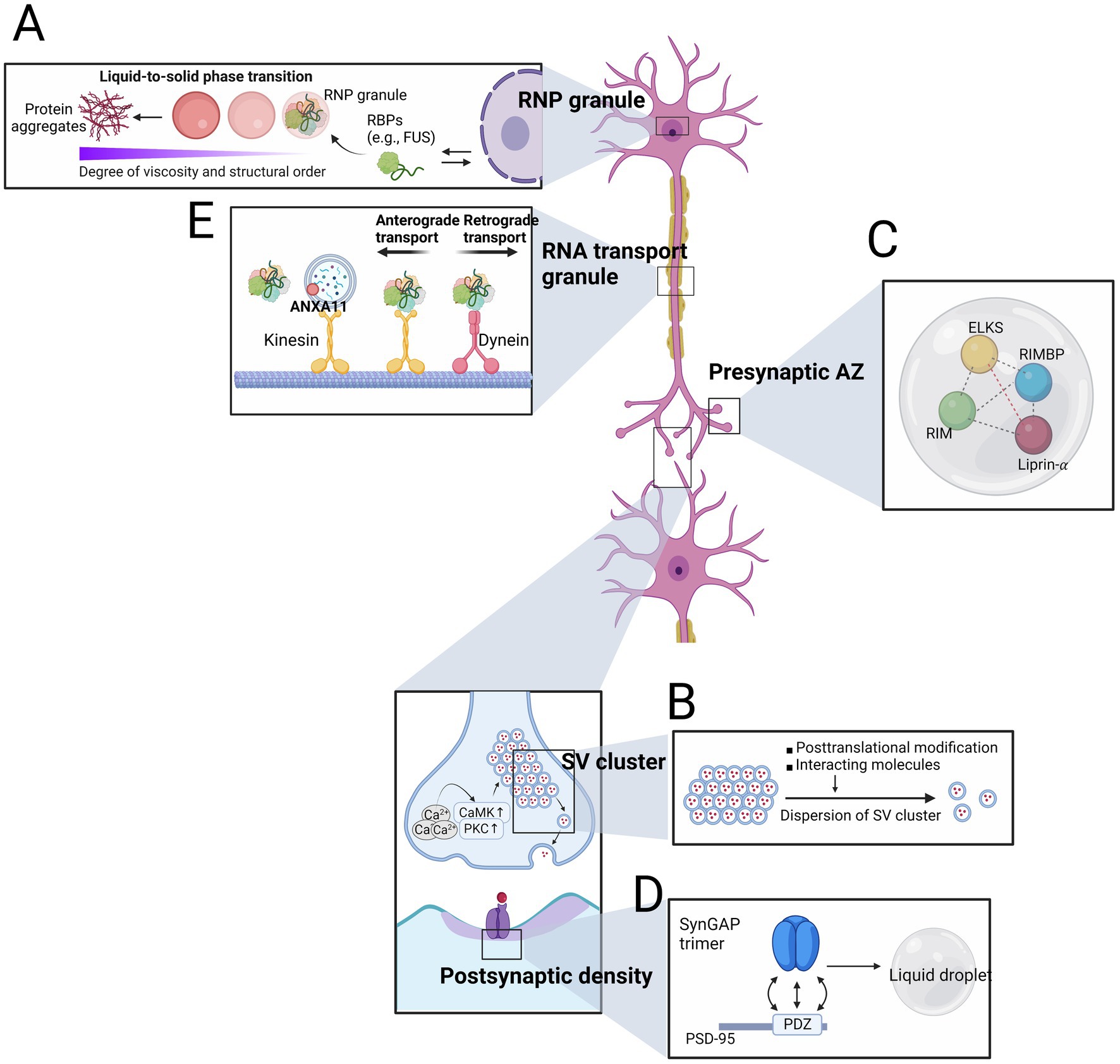
Figure 1. Neuronal condensates at the physiologic and pathologic contexts. (A) RNP granules, which are composed of mutant proteins encoded by amyotrophic lateral sclerosis (ALS)–frontotemporal dementia (FTD) causing genes, undergo a phase transition to solid states, resulting in the formation of protein aggregates. (B) Both posttranslational modification such as phosphorylation of synapsin by CaMKII dispersion and changes in interacting molecules (e.g., α-synuclein) lead to the dispersion of synaptic vesicle (SV) cluster. (C) Coexisting phase of scaffolding proteins serve as a hub to accommodate other presynaptic active zones (AZ) proteins. (D) The interaction between SynGAP trimer and PSD-95 plays a crucial role in assembling postsynaptic density. (E) RNA transport granules, either alone or with lysosomes, travel bi-directionally along the microtubule tract.
Liquid-to-solid phase transition is also observed in the RNP granules, prominent types of biomolecular condensates. RNP granules are composed of RNA and RBPs and play a vital role in RNA regulation processes such as storage, splicing, decapping, and degradation. Wild-type TDP43, FUS, HNRNPA1, and TIA1 are present in RNP granules such as stress granules, p-bodies, and nucleoli and serve a general function. However, RNP granules harboring mutated RBPs had lower dynamicity, in accordance with in vitro experiments. Given that pathologic aggregates could be incubated in solidified RNP granules that offer an adequate niche for protein fibrilization, pathologic phase transition is necessary for the progression of ALS–FTD (Molliex et al., 2015; Patel et al., 2015; Zhang et al., 2019).
The expansion of GGGGCC hexanucleotide repeat in the first intron of C9ORF72 is the most prevalent genetic cause of ALS–FTD (Dejesus-Hernandez et al., 2011; Renton et al., 2011). Though C9ORF72 protein levels are decreased in the patients with hexanucleotide expansion, the most striking feature is the accumulation of protein aggregates containing five dipeptide repeats (DPR), (GA)n, (GP)n, (GR)n, (PA)n, and (PR)n, which are synthesized by the repeat-associated non-AUG translation of GGGGCC repeat (Mori et al., 2013a,b; Saberi et al., 2018). Each DPR has a different level of toxicity. While (GP)n and (PA)n with uncharged and coiled structures are relatively non-toxic, (GR)n, and (PR)n with the positively charged coiled structures are highly toxic with unique interactome (GA)n is moderately toxic with compacted structure similar to the amyloid protein with cross-beta structure (Freibaum and Taylor, 2017). Arginine-containing (GR)n, and (PR)n can separate with RBPs and RNA into droplets in vitro, and thus are distributed in RNP granules like nucleoli and stress granules (Kwon et al., 2014; Lee et al., 2016; Lin et al., 2016; Boeynaems et al., 2017; White et al., 2019). Furthermore, recruitment of (GR)n, and (PR)n triggers the arrested dynamicity of RNP granules, leading to cellular toxicity (Lee et al., 2016; Boeynaems et al., 2017; White et al., 2019). Molecular findings related to the neuropathogenic role of arginine-containing DPRs have been studied in animal models, but more research is needed. (Choi et al., 2019; Cook et al., 2020).
Apart from the expedited protein fibrilization, phase transition in the disease-relevant context also affects the physiologic function of RNP granules. A lot of evidence supported that the function of biomolecular condensates is closely related to their material properties (Elbaum-Garfinkle et al., 2015; Zhu et al., 2019; Dorone et al., 2021; Lasker et al., 2022). ALS–FTD causing mutations also disrupts the function of RNP granules (Murakami et al., 2015). For example, ribosome biogenesis and concomitant translation rates are impaired by (GR)n and (PR)n DPR species that affect the integrating role of nucleophosmin in the LLPS of a nucleolar network (White et al., 2019).
Cells have regulatory mechanisms to eliminate aberrant RNP granules with altered material properties because they are associated with cellular dysfunction and toxicity. Protein quality control (PQC) systems such as chaperone, unfoldase, proteasome, and autophagy systems are involved in disassembling or clearing aberrant RNP granules (Buchan et al., 2013; Ganassi et al., 2016; Turakhiya et al., 2018; Wang et al., 2019; Gwon et al., 2021). It is worth noting that another functional category of ALS–FTD-causing genes, other than RBPs, is PQC, including VCP, SQSTM1, OPTN, and UBQLN2. Among them, de-mixing of SQSTM1 and UBQLN2 in both cell-free and cellular systems were identified and mutations linked to ALS–FTD lead to reduced fluidity (SQSTOM1) and even liquid-to-solid phase transition (UBQLN2) (Dao et al., 2019; Faruk et al., 2021). Mutated VCP gene is associated with persistent stress granules lacking reversibility and is implicated in the disease-relevant phenotypes (Buchan et al., 2013).
3.2. Huntington’s disease
Huntington’s disease is caused by the accumulation of mutant huntingtin proteins with a polyglutamine (poly Q) tract. The expansion of the CAG trinucleotide in the first exon of HTT gene is responsible for this abnormal protein expression. Protein products of the first exon of HTT separate into liquid droplets when the number of glutamine repeats is less than those in HD patients (Peskett et al., 2018). As the poly Q length increases, HTT exon 1 protein assemblies are converted to solid-like structures, indicating that phase transition is the underlying process of how the pathogenic HTT protein species exert cellular toxicity (Peskett et al., 2018). Molecular factors such as protein methylation and chaperone function affect the phase behavior of HTT protein (Aktar et al., 2019; Ratovitski et al., 2022). It will also be intriguing to investigate the crosstalk between HTT condensates and other biomolecular condensates in the development of HD (Sønmez et al., 2021).
3.3. Tauopathy
Neurofibrillary tangles, or intra-neuronal tau deposits, are present in various neurodegenerative diseases such as AD, progressive supranuclear palsy, FTD, and parkinsonism linked to chromosome 17 (FTDP-17) (Gøtz et al., 2019). The progression of tauopathies is associated with the instability of microtubules, which occurs in tandem with the toxicity of pathologic conformations of tau, from oligomers to fibrils (Zhang et al., 2022). Recent studies have illustrated several structural and pathogenic factors governing tau LLPS (Boyko and Surewicz, 2022). First, an amphoteric characteristic of tau possessing a negatively charged N-terminal part and positively charged middle and C-terminal part allows for electrostatic interactions with other tau molecules (homotypic interaction), other proteins, and RNA (Zhang et al., 2017; Boyko et al., 2019; Ukmar-Godec et al., 2019; Siegert et al., 2021). Acetylation on lysine residues neutralizes the charge of tau, perturbing tau-RNA interactions and reducing LLPS propensity (Ukmar-Godec et al., 2019). Second, modification associated with tauopathies affects the LLPS behavior of tau. Both P310L mutation found in FTDP-17 and pathogenic phosphorylation promote phase separation and generate tau droplets with less dynamicity (Ambadipudi et al., 2017; Wegmann et al., 2018; Kanaan et al., 2020). Third, alternative splicing on exon 10 of tau producing fewer microtubule-binding repeats inhibits tau LLPS (Ambadipudi et al., 2017). Fourth, tau and microtubule/microtubule-associated proteins reciprocally regulate in the separated phase, linking to functional aspects of tau (Hernãndez-Vega et al., 2017; Siahaan et al., 2019; Tan et al., 2019; Zhang et al., 2020; Savastano et al., 2021). Overall, the pathologic phenotypes of tauopathies are influenced by differential tau LLPS depending on normal or disease-relevant context.
Apart from the homogenous tau condensates, the interaction of tau with RBPs from the proteomic analysis and recruitment of tau to stress granules were revealed (Vanderweyde et al., 2016). Among these RBPs, TIA1 potentiates tau LLPS, and loss of TIA1 ameliorates the neurodegeneration in P301S mutant tau-expressing mice (Apicco et al., 2018; Ash et al., 2021). Tau condensation through TIA1 is understood as the molecular basis for facilitating tau oligomerization and causing neurodegeneration (Jiang et al., 2019; Ash et al., 2021). Additionally, tau aggregates ensemble with small nuclear RNAs and small nucleolar RNAs are found in serine arginine repetitive matrix protein 2 (SRRM2)-positive nuclear splicing speckle and required for alterations in splicing process (Lester et al., 2021). Therefore, reciprocal regulation between tau and biomolecular condensates underlies much of tauopathies.
3.4. Synucleinopathy
Aberrant accumulation of α-synuclein is a hallmark of several neurodegenerative diseases including Parkinson’s disease, dementia with Lewy bodies, and multiple system atrophy, collectively characterized by motor symptoms including tremor, rigidity, bradykinesia, and posture instability and cognitive impairments (Magalhåes and Lashuel, 2022). Recent research has shown that α-synuclein undergoes phase separation, which is connected to its aggregation (Ray et al., 2020). α-synuclein liquid droplets lose their dynamicity after prolonged incubation or when familial mutations of PD are introduced. Also, cellular α-synuclein condensates are converted to a solid-like phase and further evolve into the aggresome upon the addition of metal ion, which promote the aggregation of α-synuclein. α-synuclein LLPS occurs through homotypic interactions among non-amyloid-β component (NAC) domains, but intramolecular interactions between the N-terminal region and C-terminal tails masks the NAC domain under physiologic conditions, making the protein soluble (Sawner et al., 2021). Certain changes in the milieu, including pH and ionic composition, can convert the conformational state of α-synuclein and trigger phase separation, further leading to aggregation.
4. Synaptic vesicle cluster, presynaptic active zone, and postsynaptic density: Biomolecular condensates in synaptic connection
Synapses are the connection between presynaptic and postsynaptic neurons, and are composed of a presynaptic terminus, synaptic cleft, and postsynaptic dendritic spine, each enriched with distinct profiles of proteins according to their functions. The presynaptic axon terminus has SV clusters as an inventory of SV and triggers the release of neurotransmitters at the presynaptic active zone (AZ). Proper assembly of SV cluster and presynaptic AZ is governed by LLPS, which implies that they are biomolecular condensates (Milovanovic et al., 2018; Wu et al., 2019; Mcdonald et al., 2020). Understanding synaptic LLPS would give a unique viewpoint on neuropathies because cognitive dysfunctions are easily identified in the brains of neurodegenerative disease patients at the synapse level.
4.1. SV cluster
The SV cluster is a subsynaptic structure where dozens to thousands of SVs are closely placed near the presynaptic AZ without a boundary (Reshetniak and Rizzoli, 2021). In response to releasing stimuli, such as Ca2+ influx that can be translated into the activation of calcium/calmodulin-dependent protein kinase family or protein kinase C (PKC), SV exocytosis results in the release of neurotransmitters to the synaptic cleft (Cesca et al., 2010). The spatial confinement of SVs within SV cluster remained elusive until LLPS of synapsin, the major constituent of SV, was discovered (Milovanovic et al., 2018). The IDR of synapsin is essential for this process by recruiting SH3 domain-containing proteins to synapsin condensates. Furthermore, phosphorylation of synapsin by CaMKII disperses the synapsin condensates, recapitulating the dissolution event of SVs from the cluster (Figure 1B). Abrogation of synapsin LLPS by adding an IDR-targeting antibody disrupts SV clusters in the lamprey, supporting the crucial role of synapsin LLPS in organizing SV clusters (Pechstein et al., 2020). While synapsin condensates coexist with liposome in vitro, cation-π interaction with SV membrane protein synaptophysin potentiates the SV organizing ability based on the observation of SV-like clusters even in the fibroblast cells (Kim et al., 2021; Park et al., 2021). Another partner of synapsin condensates is α-synuclein, and excessive α-synuclein to synapsin reduces the synapsin condensate formation (Hoffmann et al., 2021). Because α-synuclein and synapsin are functionally linked in terms of SV regulation, further investigation of their relationship in PD contexts, in which α-synuclein is accumulated, will provide a novel insight of PD pathogenesis (Kramer and Schulz-Schaeffer, 2007; Atias et al., 2019; Bridi et al., 2021).
4.2. Presynaptic AZ
The Presynaptic AZ is located beneath the presynaptic membrane and is where SV exocytosis occurs (Sþdhof, 2012). Presynaptic AZ is seen as dense marks under electron microscopy and has a concentrated proteome, making them distinct from the surroundings. Scaffold proteins such as Glutamine/leucine/lysine/serine-rich protein (ELKS), Liprin-α, Rab3-interacting molecule (RIM), and RIM-binding protein (RIMBP) interact with each other during SV fusion events to recruit other AZ proteins including voltage-gated Ca2+ channel (VGCC) and cell adhesion molecules (Sþdhof, 2012). Recent reports have demonstrated that these scaffold molecules undergo LLPS dominated by both structured domain-and IDR-affiliated multivalent interactions (Wu et al., 2019; Mcdonald et al., 2020; Liang et al., 2021). In vitro reconstitution systems have revealed co-phase separation of RIM and RIMBP, driven by the multivalent interactions between the proline-rich motif of RIM and SH3 domain of RIMBP. This event is further coupled to the incorporation of VGCCs into their separated phase (Figure 1C).
Liprin-α plays a pivotal role in regulating AZ condensates profiles. Oligomeric Liprin-α assembled upon a coiled-coiled region can be de-mixed with ELKS (Liang et al., 2021). ELKS and Liprin-α condensates were identified in the developing synapses of Caenorhabditis elegans (Mcdonald et al., 2020). Interestingly, Liprin-α determines whether RIM, RIMBP, and VGCC are distributed with ELKS condensates (Liang et al., 2021). ELKS-Liprin-α coexisting droplets accommodate RIM and RIMBP, while liquid droplets composed of ELKS alone cannot recruit both. However, RIM and RIMBP in the ELKS-resident droplets failed to incorporate VGCC. The heterogeneity between the condensates provides the molecular basis for distinct protein–protein interaction networks found in the synapse (Lautenschlæger, 2022).
4.3. Postsynaptic densities
Similar to the presynaptic AZ, PSD is an electron-dense area at the postsynaptic membrane proximal to AZ (Sþdhof, 2012). PSD contains a significant number of proteins that respond to the released neurotransmitters, including glutamate receptors, downstream signaling molecules, and scaffold proteins (Feng and Zhang, 2009). LLPS also plays an essential role in the assembly of PSD. The multivalent interaction between the trimeric complex of Ras/Rap GTPase-activating protein SynGAP and PDZ domain of postsynaptic density protein 95 (PSD-95) forms liquid-like droplets (Figure 1D). These two highly abundant proteins act on the normal structure and function of the postsynaptic neuron (Vazquez et al., 2004; Chen et al., 2015; Zeng et al., 2016). Replacing endogenous SynGAP with a trimerization-defective mutant SynGAP or endogenous PSD-95 with mutant PSD-95 that fails to interact with SynGAP reduces spine volume and postsynaptic condensates, underscoring the functional role of SynGAP/PSD-95 condensates in regulating postsynaptic activity.
Phase separation also contributes to the differential assembly of excitatory PSD (ePSD) and inhibitory PSD (iPSD) (Zeng et al., 2018). In the supported membrane bilayer system, co-phase separation of four enriched scaffold proteins of ePSD, PSD-95, guanylate kinase-associated protein (GKAP), SH3 and multiple ankyrin repeat domains protein (Shank), and Homer scaffold protein (Homer), was achieved. These condensates also recruit SynGAP and N-methyl D-aspartate receptor subtype 2B (NR2B) but repel gephyrin, a key scaffold of iPSD. Therefore, ePSD formation by LLPS enables an augmented glutamate receptor response in a condensed area. On the contrary, iPSD is formed by the LLPS of gephyrin and glycine or GABA receptors (Bai et al., 2021).
5. RNA transport granule relays RNA and RBP to neurites for localized translation
Ribonucleic acid (RNA) transport granules are specialized ribonucleoprotein granules found in neurons that transport mRNA to axons and dendrites for localized translation (Fernandopulle et al., 2021). In order to travel to the proximal parts of neuron, they rely on microtubule-dependent transport machinery which bi-directionally dispatch biomolecules along with the track assembled with microtubules by aid of motor proteins such as dynein and kinesin (Abouward and Schiavo, 2021). Recent findings has suggested that functional failures in RNA transport granules are caused by mutations in RBPs that are genetically associated with neurodegenerative diseases (Figure 1E) (Liao et al., 2019; Fernandopulle et al., 2021).
One such RBP is Annexin A11 (ANXA11), which is a genetic risk for both familial and sporadic cases of ALS–FTD (Smith et al., 2017). Mutations in ANXA11 result in impaired Ca2+ homeostasis and protein translation, as well as hinder proper elimination of stress granules (Nahm et al., 2020). In the neuronal context, ANXA11 is found in the proteomes labeled in the vicinity of both lysosome and RNA transport granules and links them during the cargo transport (Liao et al., 2019). Since ANXA11 proteins mutated in annexin repeats region less interacts with the lysosome, the travel distances of RNA cargo in ANXA11 mutation harboring neurons are reduced as well.
Another cause of the functional impairment of RNA transport granules is mutations in TAR DNA-binding domain (TARDBP) that result in ALS–FTD and code for TDP-43. In addition to the pathogenic aggregation of TDP-43, its physiologic functions that regulate gene expression in both transcriptional and translational levels are also affected. Cellular dysfunctions such aberrant splicing and abnormal RNP granules are brought on by the loss of TDP-43 functions and increased cytoplasmic localizations that are related to changes in the interplay between the proteome and transcriptome (Tollervey et al., 2011; Ling et al., 2015). In addition, TDP-43 directs RNA trafficking to synaptic processes in a microtubule-dependent manner (Alami et al., 2014). RNA transport granules with TDP-43 demonstrate anterograde transport, whereas those without TDP-43 undergo retrograde transport. Moreover, a kymograph from fruit fly motor neurons expressing TDP-43 A315T or M337V mutants showed RNA transport granules travel retrogradely, causing the cytoplasmic accumulation of TDP-43 which are reminiscent of ALS histology (Neumann et al., 2006; Alami et al., 2014). Moreover, the loss of TDP-43 results in poor local translation, which compromises synaptic transmission. (Diaper et al., 2013; Wong et al., 2021).
6. Conclusion and future perspectives
Upon the dramatic shift in the distribution of biomolecules between mixed and de-mixed states accomplished by weak and multivalent interactions, biomolecular condensates are understood as functional units of biological phenomena. Multiple pathogenic factors including genetic risks of neurodegenerative disease or cellular stresses alter the material properties and dynamicity of biomolecular condensates. In the neuronal context, fibrilization of pathogenic proteins are promoted at aberrant biomolecular condensates where phase transition occurs. In addition, the phase transition of biomolecular condensates is often involved in their loss of function. There is still much to be understood because the research on neuronal condensates is still in its early stages. This work will unveil how abnormal alteration of neuronal condensates triggers neuronal dysfunction such as impaired axonal transport, loss of synaptic plasticity, and excitotoxicity. Many genetic loci for synaptic proteins that are identified as neurodegenerative disease-causing variants can be attractive topics in that a considerable portion of disease-associated genes encode condensate-forming proteins (Stein et al., 2010; Liu et al., 2021; Prokopenko et al., 2021; Banani et al., 2022). Furthermore, it will be crucial to reflect the actual pathophysiology of neurodegenerative disease by establishing a higher order system than in vitro and cellular levels using multicellular or animal models to analyze the material properties and dynamics of biomolecular condensates. Those approaches will broaden our knowledge and be utilized as a bridge to develop therapeutic interventions from molecular findings.
Author contributions
JN and YG wrote the manuscript. YG conceived and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. 2022R1C1C1012534).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abouward, R., and Schiavo, G. (2021). Walking the line: mechanisms underlying directional mRNA transport and localisation in neurons and beyond. Cell. Mol. Life Sci. 78, 2665–2681. doi: 10.1007/s00018-020-03724-3
Aktar, F., Burudpakdee, C., Polanco, M., Pei, S., Swayne, T. C., Lipke, P. N., et al. (2019). The huntingtin inclusion is a dynamic phase-separated compartment. Life Sci. Alliance 2:e201900489. doi: 10.26508/lsa.201900489
Alami, N. H., Smith, R. B., Carrasco, M. A., Williams, L. A., Winborn, C. S., Han, S. S. W., et al. (2014). Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 81, 536–543. doi: 10.1016/j.neuron.2013.12.018
Alberti, S., Halfmann, R., King, O., Kapila, A., and Lindquist, S. (2009). A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cells 137, 146–158. doi: 10.1016/j.cell.2009.02.044
Alberti, S., and Hyman, A. A. (2021). Biomolecular condensates at the nexus of cellular stress, protein aggregation disease and ageing. Nat. Rev. Mol. Cell Biol. 22, 196–213. doi: 10.1038/s41580-020-00326-6
Ambadipudi, S., Biernat, J., Riedel, D., Mandelkow, E., and Zweckstetter, M. (2017). Liquid-liquid phase separation of the microtubule-binding repeats of the Alzheimer-related protein tau. Nat. Commun. 8:275. doi: 10.1038/s41467-017-00480-0
Anda, F. C., Madabhushi, R., Rei, D., Meng, J., Græff, J., Durak, O., et al. (2016). Cortical neurons gradually attain a post-mitotic state. Cell Res. 26, 1033–1047. doi: 10.1038/cr.2016.76
Apicco, D. J., Ash, P. E. A., Maziuk, B., Leblang, C., Medalla, M., Abdullatif, A. L., et al. (2018). Reducing the RNA binding protein TIA1 protects against tau-mediated neurodegeneration in vivo. Nat. Neurosci. 21, 72–80. doi: 10.1038/s41593-017-0022-z
Ash, P. E. A., Lei, S., Shattuck, J., Boudeau, S., Carlomagno, Y., Medalla, M., et al. (2021). TIA1 potentiates tau phase separation and promotes generation of toxic oligomeric tau. Proc. Natl. Acad. Sci. U. S. A. 118:e2014188118. doi: 10.1073/pnas.2014188118
Atias, M., Tevet, Y., Sun, J., Stavsky, A., Tal, S., Kahn, J., et al. (2019). Synapsins regulate α-synuclein functions. Proc. Natl. Acad. Sci. U. S. A. 116, 11116–11118. doi: 10.1073/pnas.1903054116
Bai, G., Wang, Y., and Zhang, M. (2021). Gephyrin-mediated formation of inhibitory postsynaptic density sheet via phase separation. Cell Res. 31, 312–325. doi: 10.1038/s41422-020-00433-1
Banani, S. F., Afeyan, L. K., Hawken, S. W., Henninger, J. E., Dall'agnese, A., Clark, V. E., et al. (2022). Genetic variation associated with condensate dysregulation in disease. Dev. Cell 57, 1776–1788.e8. doi: 10.1016/j.devcel.2022.06.010
Banani, S. F., Lee, H. O., Hyman, A. A., and Rosen, M. K. (2017). Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 18, 285–298. doi: 10.1038/nrm.2017.7
Banani, S. F., Rice, A. M., Peeples, W. B., Lin, Y., Jain, S., Parker, R., et al. (2016). Compositional control of phase-separated cellular bodies. Cells 166, 651–663.
Boeynaems, S., Bogaert, E., Kovacs, D., Konijnenberg, A., Timmerman, E., Volkov, A., et al. (2017). Phase separation of C9orf72 dipeptide repeats perturbs stress granule dynamics. Mol. Cell 65, 1044–1055.e5. doi: 10.1016/j.molcel.2017.02.013
Bouchard, J. J., Otero, J. H., Scott, D. C., Szulc, E., Martin, E. W., Sabri, N., et al. (2018). Cancer mutations of the tumor suppressor SPOP disrupt the formation of active, phase-separated compartments. Mol. Cell 72, 19–36.e8. doi: 10.1016/j.molcel.2018.08.027
Boyko, S., Qi, X., Chen, T. H., Surewicz, K., and Surewicz, W. K. (2019). Liquid-liquid phase separation of tau protein: the crucial role of electrostatic interactions. J. Biol. Chem. 294, 11054–11059. doi: 10.1074/jbc.AC119.009198
Boyko, S., and Surewicz, W. K. (2022). Tau liquid-liquid phase separation in neurodegenerative diseases. Trends Cell Biol. 32, 611–623. doi: 10.1016/j.tcb.2022.01.011
Brangwynne, C. P., Tompa, P., and Pappu, R. V. (2015). Polymer physics of intracellular phase transitions. Nat. Phys. 11, 899–904. doi: 10.1038/nphys3532
Bridi, J. C., Bereczki, E., Smith, S. K., Poéas, G. M., Kottler, B., Domingos, P. M., et al. (2021). Presynaptic accumulation of α-synuclein causes synaptopathy and progressive neurodegeneration in drosophila. Brain Commun. 3:fcab049. doi: 10.1093/braincomms/fcab049
Buchan, J. R., Kolaitis, R. M., Taylor, J. P., and Parker, R. (2013). Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cells 153, 1461–1474. doi: 10.1016/j.cell.2013.05.037
Cesca, F., Baldelli, P., Valtorta, F., and Benfenati, F. (2010). The synapsins: key actors of synapse function and plasticity. Prog. Neurobiol. 91(4), 313–348. doi: 10.1016/j.pneurobio.2010.04.006
Chen, X., Levy, J. M., Hou, A., Winters, C., Azzam, R., Sousa, A. A., et al. (2015). PSD-95 family MAGUKs are essential for anchoring AMPA and NMDA receptor complexes at the postsynaptic density. Proc. Natl. Acad. Sci. U. S. A. 112, E6983–E6992. doi: 10.1073/pnas.1517045112
Choi, S. Y., Lopez-Gonzalez, R., Krishnan, G., Phillips, H. L., Li, A. N., Seeley, W. W., et al. (2019). C9ORF72-ALS/FTD-associated poly(GR) binds Atp5a1 and compromises mitochondrial function in vivo. Nat. Neurosci. 22, 851–862. doi: 10.1038/s41593-019-0397-0
Cook, C. N., Wu, Y., Odeh, H. M., Gendron, T. F., Jansen-West, K., Del Rosso, G., et al. (2020). C9orf72 poly(GR) aggregation induces TDP-43 proteinopathy. Sci. Transl. Med. 12. doi: 10.1126/scitranslmed.abb3774
Dao, T. P., Martyniak, B., Canning, A. J., Lei, Y., Colicino, E. G., Cosgrove, M. S., et al. (2019). ALS-linked mutations affect UBQLN2 oligomerization and phase separation in a position-and amino acid-dependent manner. Structure 27, 937–951.e5. doi: 10.1016/j.str.2019.03.012
Das, S., Vera, M., Gandin, V., Singer, R. H., and Tutucci, E. (2021). Intracellular mRNA transport and localized translation. Nat. Rev. Mol. Cell Biol. 22, 483–504. doi: 10.1038/s41580-021-00356-8
Dejesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
Diaper, D. C., Adachi, Y., Sutcliffe, B., Humphrey, D. M., Elliott, C. J., Stepto, A., et al. (2013). Loss and gain of drosophila TDP-43 impair synaptic efficacy and motor control leading to age-related neurodegeneration by loss-of-function phenotypes. Hum. Mol. Genet. 22, 1539–1557. doi: 10.1093/hmg/ddt005
Dorone, Y., Boeynaems, S., Flores, E., Jin, B., Hateley, S., Bossi, F., et al. (2021). A prion-like protein regulator of seed germination undergoes hydration-dependent phase separation. Cells 184, 4284–4298.e27. doi: 10.1016/j.cell.2021.06.009
Elbaum-Garfinkle, S., Kim, Y., Szczepaniak, K., Chen, C. C., Eckmann, C. R., Myong, S., et al. (2015). The disordered P granule protein LAF-1 drives phase separation into droplets with tunable viscosity and dynamics. Proc. Natl. Acad. Sci. U. S. A. 112, 7189–7194. doi: 10.1073/pnas.1504822112
Emperador-Melero, J., and Kaeser, P. S. (2020). Assembly of the presynaptic active zone. Curr. Opin. Neurobiol. 63, 95–103. doi: 10.1016/j.conb.2020.03.008
Faruk, M. O., Ichimura, Y., Kageyama, S., Komatsu-Hirota, S., El-Gowily, A. H., Sou, Y. S., et al. (2021). Phase-separated protein droplets of amyotrophic lateral sclerosis-associated p62/SQSTM1 mutants show reduced inner fluidity. J. Biol. Chem. 297:101405. doi: 10.1016/j.jbc.2021.101405
Feng, Z., Chen, X., Zeng, M., and Zhang, M. (2019). Phase separation as a mechanism for assembling dynamic postsynaptic density signalling complexes. Curr. Opin. Neurobiol. 57, 1–8. doi: 10.1016/j.conb.2018.12.001
Feng, W., and Zhang, M. (2009). Organization and dynamics of PDZ-domain-related supramodules in the postsynaptic density. Nat. Rev. Neurosci. 10, 87–99. doi: 10.1038/nrn2540
Fernandopulle, M. S., Lippincott-Schwartz, J., and Ward, M. E. (2021). RNA transport and local translation in neurodevelopmental and neurodegenerative disease. Nat. Neurosci. 24, 622–632. doi: 10.1038/s41593-020-00785-2
Freibaum, B. D., and Taylor, J. P. (2017). The role of dipeptide repeats in C9ORF72-related ALS-FTD. Front. Mol. Neurosci. 10:35. doi: 10.3389/fnmol.2017.00035
Fromm, S. A., Kamenz, J., Nøldeke, E. R., Neu, A., Zocher, G., and Sprangers, R. (2014). In vitro reconstitution of a cellular phase-transition process that involves the mRNA decapping machinery. Angew. Chem. Int. Ed. Eng. 53, 7354–7359. doi: 10.1002/anie.201402885
Ganassi, M., Mateju, D., Bigi, I., Mediani, L., Poser, I., Lee, H. O., et al. (2016). A surveillance function of the HSPB8-BAG3-HSP70 chaperone complex ensures stress granule integrity and dynamism. Mol. Cell 63, 796–810. doi: 10.1016/j.molcel.2016.07.021
Gopal, P. P., Nirschl, J. J., Klinman, E., and Holzbaur, E. L. (2017). Amyotrophic lateral sclerosis-linked mutations increase the viscosity of liquid-like TDP-43 RNP granules in neurons. Proc. Natl. Acad. Sci. U. S. A. 114, E2466–e2475. doi: 10.1073/pnas.1614462114
Gøtz, J., Halliday, G., and Nisbet, R. M. (2019). Molecular pathogenesis of the Tauopathies. Annu. Rev. Pathol. 14, 239–261. doi: 10.1146/annurev-pathmechdis-012418-012936
Gwon, Y., Maxwell, B. A., Kolaitis, R. M., Zhang, P., Kim, H. J., and Taylor, J. P. (2021). Ubiquitination of G3BP1 mediates stress granule disassembly in a context-specific manner. Science 372:eabf6548. doi: 10.1126/science.abf6548
Hastings, R. L., and Boeynaems, S. (2021). Designer condensates: A toolkit for the biomolecular architect. J. Mol. Biol. 433:166837. doi: 10.1016/j.jmb.2021.166837
Hernãndez-Vega, A., Braun, M., Scharrel, L., Jahnel, M., Wegmann, S., Hyman, B. T., et al. (2017). Local nucleation of microtubule bundles through tubulin concentration into a condensed tau phase. Cell Rep. 20, 2304–2312. doi: 10.1016/j.celrep.2017.08.042
Hipp, M. S., Kasturi, P., and Hartl, F. U. (2019). The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 20, 421–435. doi: 10.1038/s41580-019-0101-y
Hoffmann, C., Sansevrino, R., Morabito, G., Logan, C., Vabulas, R. M., Ulusoy, A., et al. (2021). Synapsin condensates recruit alpha-Synuclein. J. Mol. Biol. 433:166961. doi: 10.1016/j.jmb.2021.166961
Hyman, A. A., Weber, C. A., and Jþlicher, F. (2014). Liquid-liquid phase separation in biology. Annu. Rev. Cell Dev. Biol. 30, 39–58. doi: 10.1146/annurev-cellbio-100913-013325
Jain, A., and Vale, R. D. (2017). RNA phase transitions in repeat expansion disorders. Nature 546, 243–247. doi: 10.1038/nature22386
Jiang, L., Ash, P. E. A., Maziuk, B. F., Ballance, H. I., Boudeau, S., Abdullatif, A. A., et al. (2019). TIA1 regulates the generation and response to toxic tau oligomers. Acta Neuropathol. 137, 259–277. doi: 10.1007/s00401-018-1937-5
Kanaan, N. M., Hamel, C., Grabinski, T., and Combs, B. (2020). Liquid-liquid phase separation induces pathogenic tau conformations in vitro. Nat. Commun. 11:2809. doi: 10.1038/s41467-020-16580-3
Kasthuri, N., Hayworth, K. J., Berger, D. R., Schalek, R. L., Conchello, J. A., Knowles-Barley, S., et al. (2015). Saturated reconstruction of a volume of neocortex. Cells 162, 648–661. doi: 10.1016/j.cell.2015.06.054
Kim, G., Lee, S. E., Jeong, S., Lee, J., Park, D., and Chang, S. (2021). Multivalent electrostatic pi-cation interaction between synaptophysin and synapsin is responsible for the coacervation. Mol. Brain 14:137. doi: 10.1186/s13041-021-00846-y
Kramer, M. L., and Schulz-Schaeffer, W. J. (2007). Presynaptic alpha-synuclein aggregates, not Lewy bodies, cause neurodegeneration in dementia with Lewy bodies. J. Neurosci. 27, 1405–1410. doi: 10.1523/JNEUROSCI.4564-06.2007
Kwon, I., Xiang, S., Kato, M., Wu, L., Theodoropoulos, P., Wang, T., et al. (2014). Poly-dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science 345, 1139–1145.
Lasker, K., Boeynaems, S., Lam, V., Scholl, D., Stainton, E., Briner, A., et al. (2022). The material properties of a bacterial-derived biomolecular condensate tune biological function in natural and synthetic systems. Nat. Commun. 13:5643.
Lautenschlæger, J. (2022). Protein phase separation hotspots at the presynapse. Open Biol. 12:210334. doi: 10.1098/rsob.210334
Lee, H. G., Casadesus, G., Zhu, X., Castellani, R. J., Mcshea, A., Perry, G., et al. (2009). Cell cycle re-entry mediated neurodegeneration and its treatment role in the pathogenesis of Alzheimer's disease. Neurochem. Int. 54, 84–88.
Lee, K. H., Zhang, P., Kim, H. J., Mitrea, D. M., Sarkar, M., Freibaum, B. D., et al. (2016). C9orf72 dipeptide repeats impair the assembly, dynamics, and function of membrane-less organelles. Cells 167, 774–788.e17. doi: 10.1016/j.cell.2016.10.002
Lester, E., Ooi, F. K., Bakkar, N., Ayers, J., Woerman, A. L., Wheeler, J., et al. (2021). Tau aggregates are RNA-protein assemblies that mislocalize multiple nuclear speckle components. Neuron 109, 1675–1691.e9.
Li, P., Banjade, S., Cheng, H. C., Kim, S., Chen, B., Guo, L., et al. (2012). Phase transitions in the assembly of multivalent signalling proteins. Nature 483, 336–340. doi: 10.1038/nature10879
Liang, M., Jin, G., Xie, X., Zhang, W., Li, K., Niu, F., et al. (2021). Oligomerized liprin-α promotes phase separation of ELKS for compartmentalization of presynaptic active zone proteins. Cell Rep. 34:108901. doi: 10.1016/j.celrep.2021.108901
Liao, Y. C., Fernandopulle, M. S., Wang, G., Choi, H., Hao, L., Drerup, C. M., et al. (2019). RNA granules hitchhike on lysosomes for long-distance transport, using Annexin A11 as a molecular tether. Cells 179, 147–164.e20. doi: 10.1016/j.cell.2019.08.050
Lin, Y., Mori, E., Kato, M., Xiang, S., Wu, L., Kwon, I., et al. (2016). Toxic PR poly-dipeptides encoded by the C9orf72 repeat expansion target LC domain polymers. Cells 167, 789–802.e12. doi: 10.1016/j.cell.2016.10.003
Ling, J. P., Pletnikova, O., Troncoso, J. C., and Wong, P. C. (2015). TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science 349, 650–655.
Ling, S. C., Polymenidou, M., and Cleveland, D. W. (2013). Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79, 416–438. doi: 10.1016/j.neuron.2013.07.033
Liu, G., Peng, J., Liao, Z., Locascio, J. J., Corvol, J. C., Zhu, F., et al. (2021). Genome-wide survival study identifies a novel synaptic locus and polygenic score for cognitive progression in Parkinson's disease. Nat. Genet. 53, 787–793.
Lyon, A. S., Peeples, W. B., and Rosen, M. K. (2021). A framework for understanding the functions of biomolecular condensates across scales. Nat. Rev. Mol. Cell Biol. 22, 215–235. doi: 10.1038/s41580-020-00303-z
Mackenzie, I. R., Nicholson, A. M., Sarkar, M., Messing, J., Purice, M. D., Pottier, C., et al. (2017). TIA1 mutations in amyotrophic lateral sclerosis and frontotemporal dementia promote phase separation and Alter stress granule dynamics. Neuron 95, 808–816.e9. doi: 10.1016/j.neuron.2017.07.025
Magalhåes, P., and Lashuel, H. A. (2022). Opportunities and challenges of alpha-synuclein as a potential biomarker for Parkinson's disease and other synucleinopathies. NPJ Parkinsons Dis. 8:93.
Mathieu, C., Pappu, R. V., and Taylor, J. P. (2020). Beyond aggregation: pathological phase transitions in neurodegenerative disease. Science 370, 56–60.
Mcdonald, N. A., Fetter, R. D., and Shen, K. (2020). Assembly of synaptic active zones requires phase separation of scaffold molecules. Nature 588, 454–458.
Milovanovic, D., Wu, Y., Bian, X., and De Camilli, P. (2018). A liquid phase of synapsin and lipid vesicles. Science 361, 604–607. doi: 10.1126/science.aat5671
Mitrea, D. M., Cika, J. A., Guy, C. S., Ban, D., Banerjee, P. R., Stanley, C. B., et al. (2016). Nucleophosmin integrates within the nucleolus via multi-modal interactions with proteins displaying R-rich linear motifs and rRNA. elife, 5:e13571. doi: 10.7554/eLife.13571
Molliex, A., Temirov, J., Lee, J., Coughlin, M., Kanagaraj, A. P., Kim, H. J., et al. (2015). Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cells 163, 123–133. doi: 10.1016/j.cell.2015.09.015
Mori, K., Arzberger, T., Græsser, F. A., Gijselinck, I., May, S., Rentzsch, K., et al. (2013a). Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. 126, 881–893.
Mori, K., Weng, S. M., Arzberger, T., May, S., Rentzsch, K., Kremmer, E., et al. (2013b). The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339, 1335–1338.
Murakami, T., Qamar, S., Lin, J. Q., Schierle, G. S., Rees, E., Miyashita, A., et al. (2015). ALS/FTD mutation-induced phase transition of FUS liquid droplets and reversible hydrogels into irreversible hydrogels impairs RNP granule function. Neuron 88, 678–690. doi: 10.1016/j.neuron.2015.10.030
Nahm, M., Lim, S. M., Kim, Y. E., Park, J., Noh, M. Y., Lee, S., et al. (2020). ANXA11 mutations in ALS cause dysregulation of calcium homeostasis and stress granule dynamics. Sci. Transl. Med. 12:566. doi: 10.1126/scitranslmed.aax3993
Nedelsky, N. B., and Taylor, J. P. (2019). Bridging biophysics and neurology: aberrant phase transitions in neurodegenerative disease. Nat. Rev. Neurol. 15, 272–286. doi: 10.1038/s41582-019-0157-5
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. doi: 10.1126/science.1134108
Nott, T. J., Petsalaki, E., Farber, P., Jervis, D., Fussner, E., Plochowietz, A., et al. (2015). Phase transition of a disordered nuage protein generates environmentally responsive membraneless organelles. Mol. Cell 57, 936–947. doi: 10.1016/j.molcel.2015.01.013
Pak, C. W., Kosno, M., Holehouse, A. S., Padrick, S. B., Mittal, A., Ali, R., et al. (2016). Sequence determinants of intracellular phase separation by complex Coacervation of a disordered protein. Mol. Cell 63, 72–85. doi: 10.1016/j.molcel.2016.05.042
Park, D., Wu, Y., Lee, S. E., Kim, G., Jeong, S., Milovanovic, D., et al. (2021). Cooperative function of synaptophysin and synapsin in the generation of synaptic vesicle-like clusters in non-neuronal cells. Nat. Commun. 12:263. doi: 10.1038/s41467-020-20462-z
Patel, A., Lee, H. O., Jawerth, L., Maharana, S., Jahnel, M., Hein, M. Y., et al. (2015). A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cells 162, 1066–1077. doi: 10.1016/j.cell.2015.07.047
Pechstein, A., Tomilin, N., Fredrich, K., Vorontsova, O., Sopova, E., Evergren, E., et al. (2020). Vesicle clustering in a living synapse depends on a Synapsin region that mediates phase separation. Cell Rep. 30, 2594–2602.e3. doi: 10.1016/j.celrep.2020.01.092
Peskett, T. R., Rau, F., Odriscoll, J., Patani, R., Lowe, A. R., and Saibil, H. R. (2018). A liquid to solid phase transition underlying pathological huntingtin Exon1 aggregation. Mol. Cell 70, 588–601.e6.
Prokopenko, D., Morgan, S. L., Mullin, K., Hofmann, O., Chapman, B., Kirchner, R., et al. (2021). Whole-genome sequencing reveals new Alzheimer's disease-associated rare variants in loci related to synaptic function and neuronal development. Alzheimers Dement. 17, 1509–1527.
Ratovitski, T., Jiang, M., Omeally, R. N., Rauniyar, P., Chighladze, E., Faragõ, A., et al. (2022). Interaction of huntingtin with PRMTs and its subsequent arginine methylation affects HTT solubility, phase transition behavior and neuronal toxicity. Hum. Mol. Genet. 31, 1651–1672.
Ray, S., Singh, N., Kumar, R., Patel, K., Pandey, S., Datta, D., et al. (2020). α-Synuclein aggregation nucleates through liquid-liquid phase separation. Nat. Chem. 12, 705–716.
Renton, A. E., Majounie, E., Waite, A., Simõn-Sãnchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268.
Reshetniak, S., and Rizzoli, S. O. (2021). The vesicle cluster as a major organizer of synaptic composition in the short-term and long-term. Curr. Opin. Cell Biol. 71, 63–68. doi: 10.1016/j.ceb.2021.02.007
Roychaudhuri, R., Yang, M., Hoshi, M. M., and Teplow, D. B. (2009). Amyloid beta-protein assembly and Alzheimer disease. J. Biol. Chem. 284, 4749–4753.
Saberi, S., Stauffer, J. E., Jiang, J., Garcia, S. D., Taylor, A. E., Schulte, D., et al. (2018). Sense-encoded poly-GR dipeptide repeat proteins correlate to neurodegeneration and uniquely co-localize with TDP-43 in dendrites of repeat-expanded C9orf72 amyotrophic lateral sclerosis. Acta Neuropathol. 135, 459–474. doi: 10.1007/s00401-017-1793-8
Savastano, A., Flores, D., Kadavath, H., Biernat, J., Mandelkow, E., and Zweckstetter, M. (2021). Disease-associated tau phosphorylation hinders tubulin assembly within tau condensates. Angew. Chem. Int. Ed. Eng. 60, 726–730.
Sawner, A. S., Ray, S., Yadav, P., Mukherjee, S., Panigrahi, R., Poudyal, M., et al. (2021). Modulating α-Synuclein liquid-liquid phase separation. Biochemistry 60, 3676–3696.
Schuster, B. S., Dignon, G. L., Tang, W. S., Kelley, F. M., Ranganath, A. K., Jahnke, C. N., et al. (2020). Identifying sequence perturbations to an intrinsically disordered protein that determine its phase-separation behavior. Proc. Natl. Acad. Sci. U. S. A. 117, 11421–11431. doi: 10.1073/pnas.2000223117
Shin, Y., and Brangwynne, C. P. (2017). Liquid phase condensation in cell physiology and disease. Science 357: eaaf4382. doi: 10.1126/science.aaf4382
Siahaan, V., Krattenmacher, J., Hyman, A. A., Diez, S., Hernãndez-Vega, A., Lansky, Z., et al. (2019). Kinetically distinct phases of tau on microtubules regulate kinesin motors and severing enzymes. Nat. Cell Biol. 21, 1086–1092.
Siegert, A., Rankovic, M., Favretto, F., Ukmar-Godec, T., Strohæker, T., Becker, S., et al. (2021). Interplay between tau and α-synuclein liquid-liquid phase separation. Protein Sci. 30, 1326–1336. doi: 10.1002/pro.4025
Smith, B. N., Topp, S. D., Fallini, C., Shibata, H., Chen, H. J., Troakes, C., et al. (2017). Mutations in the vesicular trafficking protein annexin A11 are associated with amyotrophic lateral sclerosis. Sci. Transl. Med. 9:eaad9157. doi: 10.1126/scitranslmed.aad9157
Sønmez, A., Mustafa, R., Ryll, S. T., Tuorto, F., Wacheul, L., Ponti, D., et al. (2021). Nucleolar stress controls mutant Huntington toxicity and monitors Huntington's disease progression. Cell Death Dis. 12:1139. doi: 10.1038/s41419-021-04432-x
Soto, C., and Pritzkow, S. (2018). Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 21, 1332–1340. doi: 10.1038/s41593-018-0235-9
Stein, J. L., Hua, X., Morra, J. H., Lee, S., Hibar, D. P., Ho, A. J., et al. (2010). Genome-wide analysis reveals novel genes influencing temporal lobe structure with relevance to neurodegeneration in Alzheimer's disease. NeuroImage 51, 542–554. doi: 10.1016/j.neuroimage.2010.02.068
Su, X., Ditlev, J. A., Hui, E., Xing, W., Banjade, S., Okrut, J., et al. (2016). Phase separation of signaling molecules promotes T cell receptor signal transduction. Science 352, 595–599.
Tan, R., Lam, A. J., Tan, T., Han, J., Nowakowski, D. W., Vershinin, M., et al. (2019). Microtubules gate tau condensation to spatially regulate microtubule functions. Nat. Cell Biol. 21, 1078–1085. doi: 10.1038/s41556-019-0375-5
Tollervey, J. R., Curk, T., Rogelj, B., Briese, M., Cereda, M., Kayikci, M., et al. (2011). Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Neurosci. 14, 452–458.
Turakhiya, A., Meyer, S. R., Marincola, G., Bøhm, S., Vanselow, J. T., Schlosser, A., et al. (2018). ZFAND1 recruits p97 and the 26S proteasome to promote the clearance of Arsenite-induced stress granules. Mol. Cell 70, 906–919.e7. doi: 10.1016/j.molcel.2018.04.021
Ukmar-Godec, T., Hutten, S., Grieshop, M. P., Rezaei-Ghaleh, N., Cima-Omori, M. S., Biernat, J., et al. (2019). Lysine/RNA-interactions drive and regulate biomolecular condensation. Nat. Commun. 10:2909. doi: 10.1038/s41467-019-10792-y
Uversky, V. N. (2002). Natively unfolded proteins: a point where biology waits for physics. Protein Sci. 11, 739–756. doi: 10.1110/ps.4210102
Vanderweyde, T., Apicco, D. J., Youmans-Kidder, K., Ash, P. E. A., Cook, C., Rocha, L. D. A., et al. (2016). Interaction of tau with the RNA-binding protein TIA1 regulates tau pathophysiology and toxicity. Cell Rep. 15, 1455–1466.
Vazquez, L. E., Chen, H. J., Sokolova, I., Knuesel, I., and Kennedy, M. B. (2004). SynGAP regulates spine formation. J. Neurosci. 24, 8862–8872. doi: 10.1523/JNEUROSCI.3213-04.2004
Wang, J., Choi, J. M., Holehouse, A. S., Lee, H. O., Zhang, X., Jahnel, M., et al. (2018). A molecular grammar governing the driving forces for phase separation of prion-like RNA binding proteins. Cells 174, 688–699.e16. doi: 10.1016/j.cell.2018.06.006
Wang, B., Maxwell, B. A., Joo, J. H., Gwon, Y., Messing, J., Mishra, A., et al. (2019). ULK1 and ULK2 regulate stress granule disassembly through phosphorylation and activation of VCP/p97. Mol. Cell 74, 742–757.e8. doi: 10.1016/j.molcel.2019.03.027
Wegmann, S., Eftekharzadeh, B., Tepper, K., Zoltowska, K. M., Bennett, R. E., Dujardin, S., et al. (2018). Tau protein liquid-liquid phase separation can initiate tau aggregation. EMBO J. 37:e98049. doi: 10.15252/embj.201798049
White, M. R., Mitrea, D. M., Zhang, P., Stanley, C. B., Cassidy, D. E., Nourse, A., et al. (2019). C9orf72 poly(PR) dipeptide repeats disturb biomolecular phase separation and disrupt nucleolar function. Mol. Cell 74, 713–728.e6. doi: 10.1016/j.molcel.2019.03.019
Wong, C. E., Jin, L. W., Chu, Y. P., Wei, W. Y., Ho, P. C., and Tsai, K. J. (2021). TDP-43 proteinopathy impairs mRNP granule mediated postsynaptic translation and mRNA metabolism. Theranostics 11, 330–345.
Workman, R. J., and Pettitt, B. M. (2021). Thermodynamic compensation in peptides following liquid-liquid phase separation. J. Phys. Chem. B 125, 6431–6439.
Wright, P. E., and Dyson, H. J. (2015). Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 16, 18–29. doi: 10.1038/nrm3920
Wu, X., Cai, Q., Shen, Z., Chen, X., Zeng, M., Du, S., et al. (2019). RIM and RIM-BP form presynaptic active-zone-like condensates via phase separation. Mol. Cell 73, 971–984.e5. doi: 10.1016/j.molcel.2018.12.007
Zeng, M., Chen, X., Guan, D., Xu, J., Wu, H., Tong, P., et al. (2018). Reconstituted postsynaptic density as a molecular platform for understanding synapse formation and plasticity. Cells 174, 1172–1187.e16. doi: 10.1016/j.cell.2018.06.047
Zeng, M., Shang, Y., Araki, Y., Guo, T., Huganir, R. L., and Zhang, M. (2016). Phase transition in postsynaptic densities underlies formation of synaptic complexes and synaptic plasticity. Cells 166, 1163–1175.e12.
Zhang, P., Fan, B., Yang, P., Temirov, J., Messing, J., Kim, H. J., et al. (2019). Chronic optogenetic induction of stress granules is cytotoxic and reveals the evolution of ALS-FTD pathology. elife 8:e39578. doi: 10.7554/eLife.39578
Zhang, X., Lin, Y., Eschmann, N. A., Zhou, H., Rauch, J. N., Hernandez, I., et al. (2017). RNA stores tau reversibly in complex coacervates. PLoS Biol. 15:e2002183. doi: 10.1371/journal.pbio.2002183
Zhang, X., Vigers, M., Mccarty, J., Rauch, J. N., Fredrickson, G. H., Wilson, M. Z., et al. (2020). The proline-rich domain promotes tau liquid-liquid phase separation in cells. J. Cell Biol. 219, 11. doi: 10.1083/jcb.202006054
Zhang, Y., Wu, K. M., Yang, L., Dong, Q., and Yu, J. T. (2022). Tauopathies: new perspectives and challenges. Mol. Neurodegener. 17:28.
Keywords: biomolecular condensates, liquid–liquid phase separation, phase transition, protein aggregation, neurodegenerative disease, synapse, RNA transport granule
Citation: Nam J and Gwon Y (2023) Neuronal biomolecular condensates and their implications in neurodegenerative diseases. Front. Aging Neurosci. 15:1145420. doi: 10.3389/fnagi.2023.1145420
Edited by:
Tae In Kam, School of Medicine, Johns Hopkins University, United StatesReviewed by:
Nam Chul Kim, University of Minnesota Twin Cities, United StatesSiranjeevi Nagaraj, Université libre de Bruxelles, Belgium
Copyright © 2023 Nam and Gwon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Youngdae Gwon, eWd3b25Ac2trdS5lZHU=