 Xingxing Chen
Xingxing Chen Enhao He1
Enhao He1 Jiang Xu
Jiang Xu- 1Brain Science and Advanced Technology Institute, Hubei Province Key Laboratory of Occupational Hazard Identification and Control, School of Medicine, Wuhan University of Science and Technology, Wuhan, Hubei, China
- 2Geriatric Hospital Affiliated to Wuhan University of Science and Technology, Wuhan, Hubei, China
- 3Hubei Key Laboratory of Nerve Injury and Functional Reconstruction, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei, China
Huntingtin-associated protein 1 (HAP1), the first identified HTT-binding partner, is highly expressed in the central nervous system, and has been found to associated with neurological diseases. Mounting evidence suggests that HAP1 functions as a component of cargo-motor molecules to bind various proteins and participates in intracellular trafficking. It is known that the failure of intracellular transport is a key contributor to the progression of neurodegenerative disorders (NDs) including Alzheimer’s disease (AD), Huntington’s disease (HD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), spinal and bulbar muscular atrophy (SBMA) and spinocerebellar ataxia (SCA). The link between HAP1 and various NDs is supported by growing evidence. This review aims to provide a comprehensive overview of the intracellular trafficking function of HAP1 and its involvement in NDs.
Introduction
Neurodegenerative diseases (NDs) including Alzheimer’s disease (AD), Huntington’s chorea (HD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), spinal and bulbar muscular atrophy (SBMA) and different types of spinocerebellar ataxia (SCA) share important pathological features characterized by the age-dependent and slowly progressive degeneration of selectively vulnerable nerve cells. NDs are caused by the accumulation of misfolded proteins, though they exhibit heterogeneous clinical and pathological phenotypes (Dugger and Dickson, 2017). While the pathogenic mechanisms of NDs are poorly understood, many lines of evidence point to a role of disrupted neuronal trafficking during the development of these diseases (Suzuki et al., 2006; Schreij et al., 2016; Burk and Pasterkamp, 2019; Singh and Muqit, 2020; Blackstone et al., 2021; Liu et al., 2021). The defects in intracellular trafficking typically result in mislocalization of proteins and the accumulation of undegraded proteins in neurons.
Huntingtin-associated protein 1 (HAP1) was first identified in yeast two-hybrid screens to interact with huntingtin (HTT), a protein known to cause HD when there is increased polyglutamine (polyQ) expansion in HTT (Li et al., 1995). Unlike HTT that is ubiquitously expressed in the brain and peripheral tissues, HAP1 is mainly enriched in neurons (Li et al., 1995; Gutekunst et al., 1998), suggesting that HAP1 may be associated with selective neurodegeneration in HD. In the rodent animals, HAP1 is more abundant in the limbic and hypothalamic regions (Fujinaga et al., 2004). However, our recent study indicates that HAP1 is ubiquitously expressed in different regions in the monkey and human brains, such as cortex, striatum (putamen and caudate), hippocampus and hypothalamus, indicating that HAP1 is expressed in a species-dependent manner (Chen et al., 2022). It has been reported that HAP1-immunoreactive products are exhibited as cytoplasmic puncta or “stigmoid bodies” (STBs), a special cellular structure ranged from 0.5 to 3 μm in diameter (Gutekunst et al., 1998; Li et al., 1998a). Although HAP1 immuno-positive puncta are also seen in the monkey brain, most of the puncta were often small (< 1 μm in diameter) (Chen et al., 2022). STBs appear to be formed by intracellular microtubule-dependent multiple fusion of small HAP1 inclusions (Fujinaga et al., 2009). HAP1 in the rat consists of two isoforms (HAP1A and HAP1B) that differ in their C-terminal sequence (Li et al., 1995). Human HAP1 has a major form (molecular weight is about 75kD) that is very similar to rat Hap1A (Li et al., 1998b). Increasing evidence suggests that HAP1 is a component of cargo-motor molecules that binds various proteins and participates in intracellular trafficking (Table 1). Abnormal interaction of mutant proteins with HAP1 affects the intracellular transport of selected molecules, which can critically contribute to the pathogenesis of neurodegeneration.
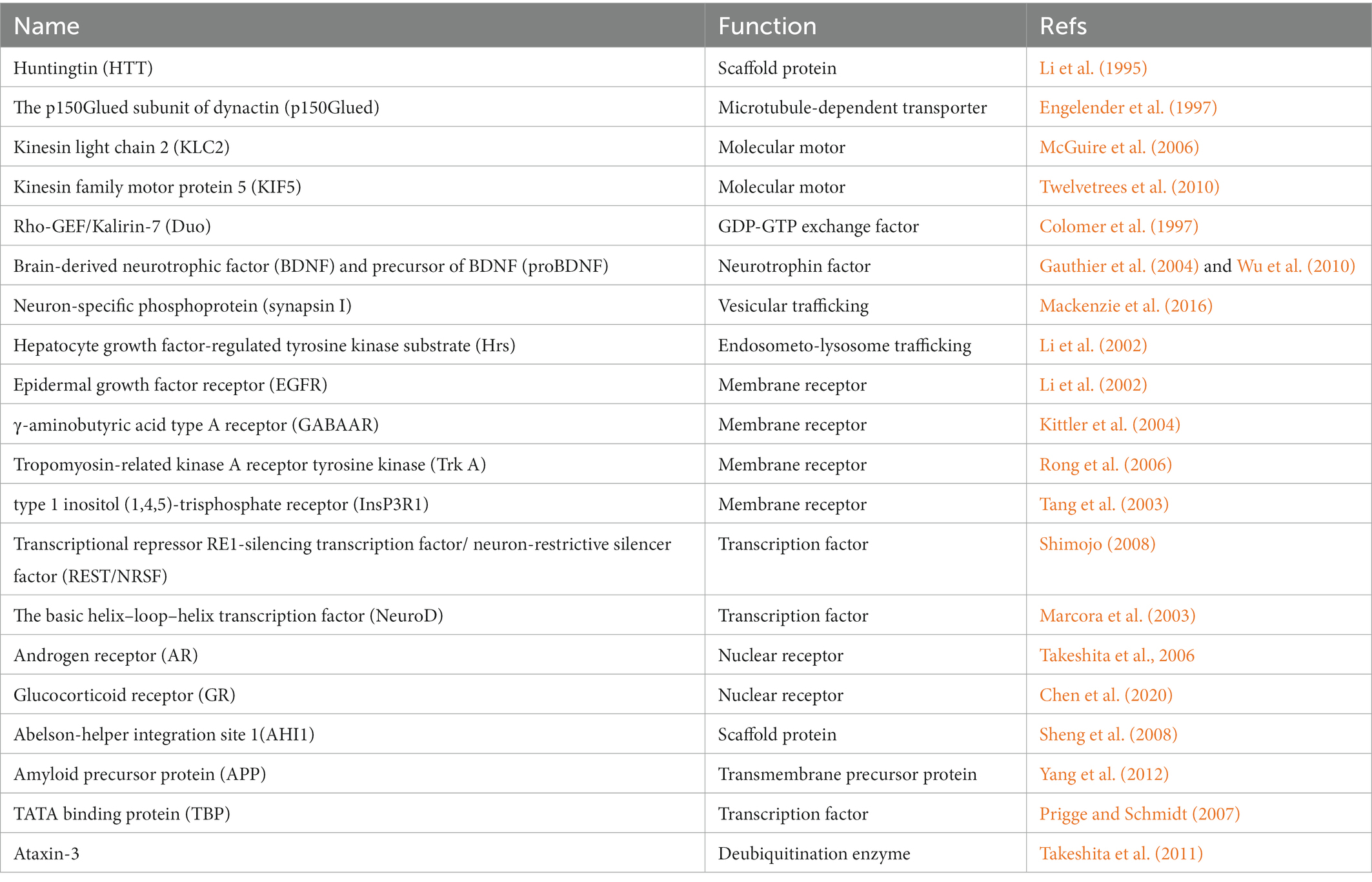
Table 1. HAP1-interacting proteins.
Huntingtin-associated protein 1-associated intracellular trafficking
As a cytosolic protein in neurons, HAP1 lacks conserved transmembrane domains and nuclear localization signals. It contains a coiled-coil domain in the middle region and multiple N-myristoylation sites which are found in a variety of proteins associated with membrane proteins and involved in intracellular trafficking (Li and Li, 2005), Consistently, the electron microscopy results show that HAP1 is distributed in microtubules and membranous organelles (Gutekunst et al., 1998). Intracellular anterograde and retrograde transport of membrane organelles requires the molecular motors kinesin and dynein. The motor protein dynein, which is involved in retrograde transport, is required to drive vesicles along microtubules with the assistance of microtubule transporter dynactin. HAP1 might be involved in intracellular trafficking via its interaction with p150Glued that is the largest subunit of dynactin and mediates the binding between microtubules and dynein (Engelender et al., 1997; Tempes et al., 2020). HAP1 interacts with p150Glued to form a stable complex that may mediate the microtubule-dependent retrograde of membranous organelles (Engelender et al., 1997). HAP1 also participates in anterograde transport in association with kinesin, which consists of two heavy chains (KHC) and two light chains (KLC) for moving along microtubules with the energy generated by ATP hydrolysis. There is an interaction between HAP1 and kinesin light chain 2 (KLC2), and deletion of HAP1 results in significant suppression of kinesin-dependent amyloid precursor protein (APP) transport in neurons (McGuire et al., 2006). Further, the association of HAP1 with p150Glued and KLC can be reduced by the phosphorylation of the C-terminus of HAP1A, and increased when HAP1A is dephosphorylated (Rong et al., 2006). In addition, HAP1 associates with Rac1 guanine nucleotide exchange factor Kalirin-7/Duo that has characteristics of membrane cytoskeletal proteins (Colomer et al., 1997). The interactions of HAP1 with these cytoskeletal proteins allow HAP1 to act as a scaffold protein to participate in intracellular trafficking, which is supported by increasing evidence.
First, HAP1 is reportedly associated with synaptic vesicles (Gutekunst et al., 1998). Studies have shown an important role of HAP1 in the vesicular transport of brain-derived neurotrophic factor (BDNF) and proBDNF, a precursor of BDNF (Gauthier et al., 2004; Wu et al., 2010; Yang et al., 2011). BDNF is a member of the neurotrophin family regulating neuronal development and plasticity. It is synthesized as proBDNF that is cleaved in the Golgi or immature secretory vesicles to form mature BDNF. BDNF is transported anterogradely in neurons and released from the nerve terminal in an activity-dependent manner. HTT, HAP1, and p150Glued form a complex that is involved in vesicular transport of BDNF along microtubules (Gauthier et al., 2004). BDNF transport is blocked when HAP1 level is reduced by small-interfering RNA (siRNA). HAP1 also participates in axonal transport and activity-dependent release of proBDNF by interacting with the BDNF prodomain (Wu et al., 2010). HAP1 and proBDNF form a complex with sortilin to be engaged in Golgi-endosome transport, preventing proBDNF degradation and modulating its targeting to endosomes, microtubules, and neuritis (Yang et al., 2011). The regulation of microtubule-dependent vesicles transport by HAP1 may involve synapsin I, a neuron-specific phosphoprotein that is associated with synaptic vesicles during neurotransmitter release to regulate synaptic plasticity and neuronal development (Cesca et al., 2010; Mackenzie et al., 2016). These findings suggest a direction for investigation of how HAP1 regulates synaptic function.
Second, HAP1 appears to be important for the stability and recycling of membrane receptors. HAP1 was reported to interact with hepatocyte growth factor-regulated tyrosine kinase substrate (Hrs), a key protein for endosometo-lysosome trafficking of membrane receptors (Li et al., 2002). Moreover, HAP1 is able to stabilize the internalized membrane receptors, such as epidermal growth factor receptor (EGFR), γ-aminobutyric acid type A receptor (GABAAR, the major inhibitory receptor in the brain) and tropomyosin-related kinase A receptor tyrosine kinase (TrkA), a nerve growth factor (NGF) receptor required for neurite outgrowth (Li et al., 2002; Kittler et al., 2004; Rong et al., 2006). When the interaction of HAP1 with microtubule-dependent transporters is suppressed, these membrane receptors cannot be efficiently transported to the appropriate cellular sites and are instead targeted to the lysosomes for degradation, resulting in a decrease of their levels and defective neural activity. HAP1 also binds to other membrane receptors, such as the type 1 inositol (1, 4, 5)-trisphosphate receptor (InsP3R1), an intracellular Ca2+ channel localized in the endoplasmic reticulum. The InsP3R1 carboxy terminus binds HAP1A and HTT to form the InsP3R1/HAP1A/HTT ternary complex (Tang et al., 2003). HAP1 can affect InsP3R1-mediated Ca2+ release by regulating the binding of InsP3R1 and HTT (Tang et al., 2004; Arteaga-Bracho et al., 2016). All these findings provide an insight into the regulation of neuronal function by HAP1 through its effect on membrane receptors.
Third, HAP1 is involved in the nuclear translocation of transcription factors. HAP1, HTT, p150Glued, the transcriptional repressor RE1-silencing transcription factor/neuron-restrictive silencer factor (REST/NRSF), and the REST/NRSF-interacting LIM domain protein (RILP) were found to form a complex involved in the translocation of REST/NRSF into the nucleus, a process in which HAP1 controls REST/NRSF cellular localization in neurons (Shimojo, 2008; Zhu et al., 2019). HAP1 acts as a scaffold protein to provide binding sites for two or more components of a signaling pathway so they can be tightly linked together (Smith and Scott, 2002; Marcora et al., 2003). For example, HTT interacts with NeuroD, a basic helix–loop–helix transcription factor important for neuronal development and survival via HAP1. HAP1 and HTT facilitate activation of NeuroD by mixed-lineage kinase 2 (MLK2) (Marcora et al., 2003). NeuroD activation is correlated with its translocation from the cytoplasm to the nucleus (Petersen et al., 2002). In addition, HAP1 appears to associate with nuclear translocation with steroid hormone receptors that are ligand-dependent transcription factors and include androgen receptor (AR) and glucocorticoid receptor (GR) (Fujinaga et al., 2011). In line with this, it has been shown that HAP1 interacts with AR (Takeshita et al., 2006). Our recent findings also suggest that HAP1 interacts with GR and can stabilize the level of GR, indicating that HAP1 may be involved in the regulation of stress in the central nervous system (Chen et al., 2020). Consistent with this finding, the Abelson-helper integration site 1(AHI1), which forms a stable protein complex with HAP1 (Sheng et al., 2008), also regulates the nuclear translocation of GR (Wang et al., 2021).
The role of HAP1 in HD
HD is a monogenetic neurodegenerative disorder, with autosomal dominant inheritance, and displays unrelenting progressive motor, psychiatric and cognitive symptoms that are caused by severe neuronal loss in the striatum, hippocampus, and cerebral neocortex (Dom et al., 1976; Hedreen and Folstein, 1995; Murphy et al., 2000). The expansion of CAG trinucleotide repeat in the causative HTT gene on chromosome 4 leads to a prolonged polyQ repeat of variable length in the N-terminal region of HTT (Walker, 2007; McColgan and Tabrizi, 2018), which causes HTT to aggregate and accumulate in the nucleus and cytoplasm (DiFiglia et al., 1997; Gutekunst et al., 1999). Although HTT is expressed widely in the central nervous system (Li et al., 1993; Sharp et al., 1995; Bhide et al., 1996), mutant HTT (mHTT) with the expanded polyQ repeat causes selective neurodegeneration in the brain (Vonsattel et al., 1985; Wang et al., 2014; Arteaga-Bracho et al., 2016). HAP1 interacts with polyQ-expanded HTT more tightly in a polyQ length dependent manner (Li et al., 1995). In particular, our recent research shows that HAP1 is correlatively expressed with HTT in monkey and human brains (Chen et al., 2022), suggesting that abnormal protein–protein interactions may be an important contributor to the selective neuronal pathogenesis of HD (Figure 1).
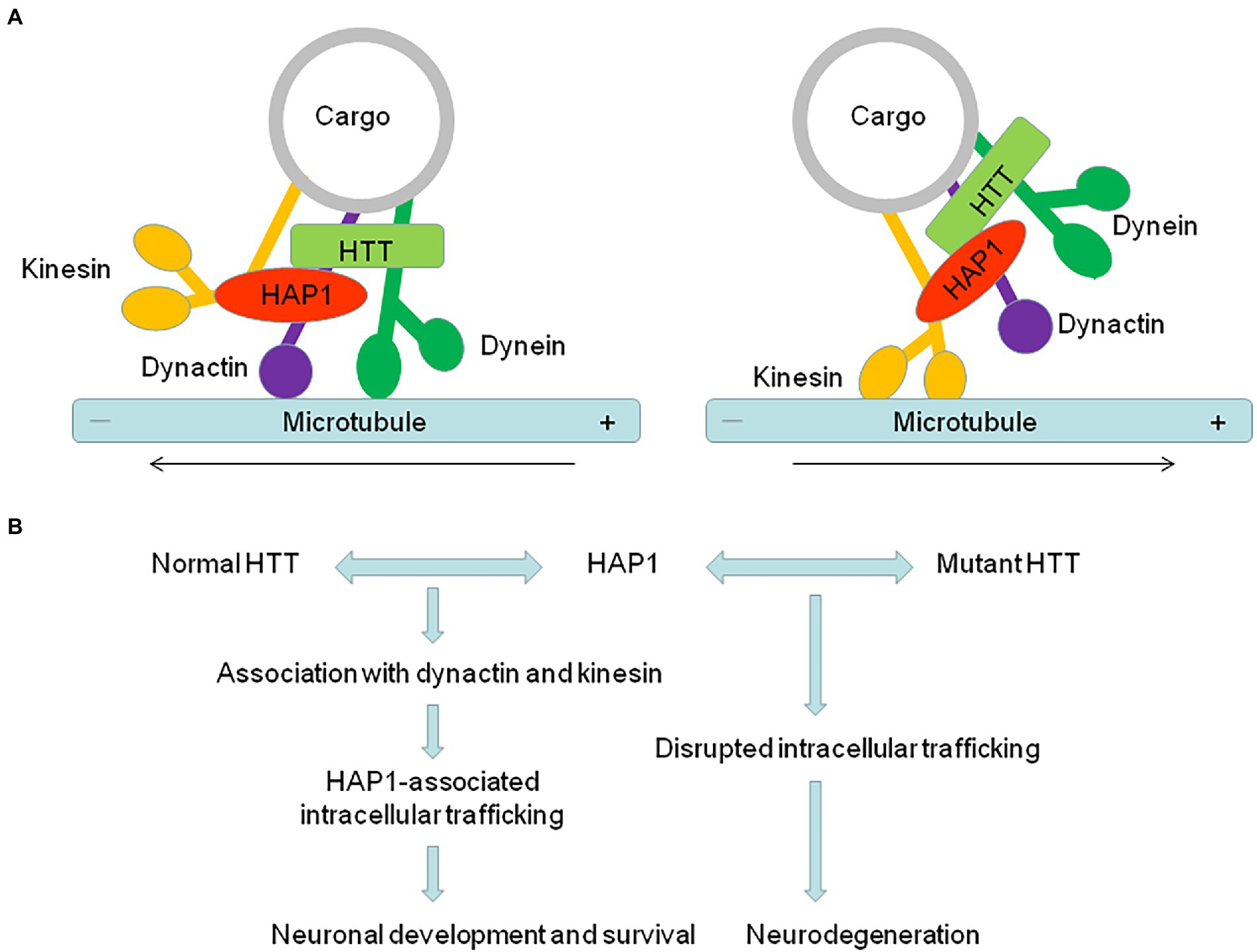
Figure 1. The abnormal interaction of HAP1 with mutant HTT is an important contributor to the pathogenesis of neurodegeneration in HD. (A) HAP1 is involved in intracellular trafficking via its interaction with dynactin and kinesin. The dynactin complex promotes retrograde trafficking, while kinesin promotes anterograde trafficking. (B) HAP1-dependent intracellular trafficking important for neuronal functions is affected by the abnormal interaction of HAP1 with mutant HTT, therefore resulting in neurodegeneration in HD.
HAP1 and HTT are associated with trafficking proteins to participate in intracellular transport. Since HAP1 binds mHTT more tightly than normal HTT, HAP1-dependent transport of various vesicles or receptors could be affected by the abnormal interaction of HAP1 with mHTT. For example, mHTT can dissociate the HTT/HAP1/p150Glued complex from microtubules, together with the weakened interaction between HAP1 and proBDNF in HD, affecting the transport and release of BDNF (Gauthier et al., 2004; Wu et al., 2010). Decreased BDNF transport results in the loss of neurotrophic support and increased neuronal toxicity. Consistent with that, mHTT reduces the association of HAP1 with p150Glued and KLC and thereby decreases the intracellular level of TrkA, whose internalization and trafficking are required for neurite outgrowth (Rong et al., 2006). In HD mouse model, the transport of GABAAR, which mediates inhibitory postsynaptic currents (IPSCs), and α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor, which mediates excitatory postsynaptic currents (EPSCs), is also impaired. HAP1 interacts with the kinesin family motor protein 5 (KIF5), and this interaction-mediated anterograde transport of GABAARs and GluR2-containing AMPARs along microtubules in dendrites is disrupted by mHTT, leading to disrupted excitatory/inhibitory balance in HD (Twelvetrees et al., 2010; Mandal et al., 2011; Yuen et al., 2012). Moreover, mHTT alters the InsP3R1/HAP1A/HTT complex and subsequently sensitizes InsP3R1 activation to facilitate Ca2+ release in medium spiny striatal neurons (MSN) (Tang et al., 2003), providing a molecular mechanism to explain the alterations of intracellular Ca2+ signaling that are believed to play a significant role in the pathogenesis of HD (Kolobkova et al., 2017). HAP1 also seems to be involved in other neuropathological changes in HD. Disruption of autophagy has been implicated in HD, in which both soluble and aggregated polyQ-HTT are cleared by autophagy (Ravikumar et al., 2002; Qin et al., 2003). HAP1, as a dynein-activating adaptor, drives autophagosomal transport by binding the dynein–dynactin complex via canonical and noncanonical interactions (Cason and Carman, 2021). The regulation of autophagosome dynamics by HAP1 is disrupted by mHTT, leading to defective cargo degradation and the accumulation of polyQ-HTT observed in the neurons in HD (Wong and Holzbaur, 2014). These different lines of evidence indicate that mHTT affects various kinds of HAP1-dependent intracellular trafficking important for neuronal functions, therefore playing an important role in neurodegeneration.
HAP1 together with other HTT interacting proteins may be involved in the neuronal degeneration in the striatum, a region that is most affected in HD (Vonsattel et al., 1985; Vonsattel and DiFiglia, 1998). The small G protein Rhes, an E3 ligase for attachment of small ubiquitin-like modifier, is enriched in the striatum and binds selectively to mHTT and enhances sumoylation of mHTT (Subramaniam and Snyder, 2011). Sumoylation decreases the formation of mHTT aggregates and promotes cell death as sumoylated mHTT is more toxic (Subramaniam and Snyder, 2011; Soares et al., 2022). HAP1 deficiency in adult HD knock-in (KI) mouse brains caused more Rhes to bind mHTT and increased the level of sumoylated mHTT, leading to neuronal loss in the striatum (Liu et al., 2020). Neuronal loss induced by HAP1 deletion appears to require the presence of mHTT, consistent with our recent observation that deletion of HAP1 exacerbated neurotoxicity of mHTT in the organotypic brain slices of adult monkeys (Chen et al., 2022). However, how the interaction of mHTT and HAP1 mediates neuronal dysfunction in the primate brains needs to be further studied.
The role of HAP1 in AD
Alzheimer’s disease is the most common form of neurodegenerative dementia worldwide, and its prevalence continues to grow with increase in the aged population worldwide. The initial stages of AD are characterized by deficits in the ability to encode and store new memories, followed by progressive cognitive and behavioral changes that occur at the later stages (Soria Lopez et al., 2019). The main pathological changes in AD include extracellular beta-amyloid (Aβ) plaques accumulation and intracellular neurofibrillary tangles, leading to synaptic damage or loss and neurodegeneration (Bloom, 2014). It is known that neurons rely on microtubule-dependent transport machinery for survival and development. Failure of axonal transport has been proposed as a key contributor in the progression of neurodegenerative disorders such as AD (De Vos et al., 2008; Vicario-Orri et al., 2015). A number of studies suggest that axonal swellings consisting of accumulated abnormal amounts of microtubule-associated proteins, organelles, and vesicles are the important early pathological features in mouse AD models and human AD (Stokin et al., 2005).
Reductions in microtubule-dependent transport may increase Aβ levels and amyloid plaques deposition in AD. The Aβ peptides are proteolytic fragments of the type I transmembrane receptor-like APP protein that plays a role in promoting neurite outgrowth (Sosa et al., 2017). APP is axonally transported, endocytosed and sorted to different cellular compartments where APP is cleaved by several secretases to generate Aβ (Tang, 2009). The kinesin-dependent APP axonal transport is affected by HAP1 in neurons, and lack of HAP1 decreases the transport of APP vesicles from the cell body to neuritis (McGuire et al., 2006). HAP1 and APP have been found to be highly colocalized to interact with each other in mice and human brain (Yang et al., 2012). HAP1 regulates APP trafficking to the non-amyloidogenic pathway and reduces Aβ level in neurons (Yang et al., 2012). HAP1’s close partner AHI1 also participates in APP trafficking and processing to rescue AD pathology (Ting et al., 2019). Recent study suggests that transport of endosomes and vesicles containing APP and Trks is inhibited in AD mouse models (Lazarov et al., 2007). Deficit of APP trafficking affects neurotrophic signaling as APP interacts with the NGF receptors Trks and mediate neuronal survival and differentiation (Zhang et al., 2013). In line with this, HAP1 is important for the stability and sorting of Trks (Rong et al., 2006; Xiang et al., 2014), and HAP1 reduction selectively affects survival and growth of postnatal mice (Xiang et al., 2014). In conclusion, APP-related transport mechanism is a potential therapeutic strategy for AD, in which HAP1 may play an important role.
The role of HAP1 in other NDs
HAP1 participates in the neuropathology of certain subtypes of SCAs, a group of autosomal dominantly inherited neurodegenerative disorders, that share the characteristic of progressive ataxia resulting from degeneration of cerebellum and its connections. There are over 40 SCAs, including common SCAs (SCA1, SCA2, and SCA3/Machado-Joseph, SCA6, SCA7, and SCA17) and dentatorubral-pallidoluysian atrophy (DRPLA), all of which are caused by an expansion of a CAG trinucleotide repeat in the distinct disease genes (Soong and Morrison, 2018). HAP1 has been reported to associate with TATA binding protein (TBP) and ataxin-3, which are the causative agents of polyQ-expansion-dependent neuropathology of SCA17 and SCA3, respectively (Prigge and Schmidt, 2007; Takeshita et al., 2011). In SCA17 patients, overaccumulation TBP assembles nuclear aggregates, resulting in neuronal loss (van Roon-Mom et al., 2005). HAP1 binds specifically to the conserved C-terminal domain of TBP to prevent nuclear localization of excessive TBP by sequestering a subset of TBP into STBs (Prigge and Schmidt, 2007). Analogously, HAP1 interacts with ataxin-3 intracellularly through its N-terminus Josephin domain and modifies its pathophysiological involvement in SCA3 (Takeshita et al., 2011).
HAP1 also seems to be involved in the neuropathology of SBMA, a neurodegenerative and neuromuscular genetic disease characterized by progressive proximal (bulbar and limb) muscle atrophy, weakness and fasciculations, that is caused by the expansion of a polyQ-encoding CAG tract in the AR gene (Takeshita et al., 2006; Sengupta et al., 2022). HAP1 can interact with AR through its ligand-binding domain in a polyQ-length-dependent manner and protect cells from polyQ-expanded AR-induced apoptosis by sequestering polyQ-expanded AR in STBs (Takeshita et al., 2006). HAP1/STB critically involves in pathogenesis of SBMA as an important intrinsic neuroprotectant determining the threshold for cellular vulnerability to apoptosis.
The role of HAP1 in PD and ALS has been less reported. PD, the second most common degenerative disease of the central nervous system, is characterized neuropathologically by loss of dopaminergic neurons and formation of α-synuclein (α-Syn)-containing Lewy bodies in the substantia nigra, manifesting as reduced facilitation of voluntary movements (Spillantini et al., 1997; Damier et al., 1999). ALS, also known as motor neuron disease, is primarily characterized by progressive loss of motor neurons in the brain and the spinal cord, which leads to muscle weakness and eventual paralysis (Hardiman et al., 2017). Accumulation of misfolded proteins, such as α-Syn in PD, TAR DNA-binding protein 43 (TDP-43) in ALS, is also the main event triggering pathological abnormalities responsible for PD and ALS (Soto and Pritzkow, 2018). It would be interesting to investigate whether HAP1 also affects the intracellular trafficking of these proteins to participate in the neuropathology of PD and ALS.
In addition to central nervous system (CNS), HAP1 is also present in the enteric nervous system (ENS) (Tarif and Islam, 2021; Tarif et al., 2022) that is embedded in the wall of the gastrointestinal tract and contains polarized neural circuits responsible for controlling a wide variety of gastrointestinal functions (Furness et al., 2014). The ENS, sharing many features with the brain, can act as a potential portal for pathogenesis of NDs and is also a neurodegenerative target (Chalazonitis and Rao, 2018). HAP1 is abundantly expressed in excitatory and inhibitory motor neurons of myenteric plexuses, and secretomotor and vasodilator neurons of submucosal plexuses (Tarif and Islam, 2021; Tarif et al., 2022). The fact suggests that HAP1-associated intracellular trafficking plays an important role in these neurons of ENS. It is intriguing to consider that the dysfunction of HAP1 in ENS could be a potential risk factor for certain NDs.
Conclusion
The interactions of HAP1 with cytoskeletal proteins allow HAP1 to participate in the intracellular trafficking of various proteins, stability and recycling of membrane receptors, nuclear translocation of transcription factors and nuclear receptors. Thus, acting as a scaffold to link microtubule transporters with different cargos, HAP1 may stabilize protein complexes that are required for protein trafficking to the appropriate cellular sites. It could also function as an adaptor for two or more components of a signaling pathway. HAP1 interacts with a number of proteins that are involved in NDs, such as HTT, TBP, ataxin-3, and AR. Abnormal interactions of mutant proteins with HAP1 affect the intracellular transport of selected molecules, which is an important contributor to the pathogenesis of NDs. For example, HAP1-dependent intracellular trafficking could be affected by the abnormal interaction of HAP1 with mHTT, as HAP1 binds mHTT more tightly than normal HTT in HD. On the other hand, many major human NDs, including AD, HD, PD, ALS, SBMA, and SCA, display axonal pathologies including abnormal accumulations of proteins and organelles. Such pathologies highlight damage to axonal transport, a key contributor in the progression of NDs. Given the function of HAP1 in intracellular transport that is vitally important for neuronal function and survival, dysfunction of HAP1 may cause axonal transport defect that can account for a number of NDs. Thus, improving or restoring the function of HAP1 could be a potential therapeutic strategy for treating NDs.
Author contributions
XC and JX wrote the manuscript. EH, CS, and YZ edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
Our research work is supported by grants from Natural Science Foundation of Hubei Province (2022CFB216), Key Research Project of Ministry of Science and Technology of China (2022ZD021160) and National Natural Science Foundation of China (82071272).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Arteaga-Bracho, E. E., Gulinello, M., Winchester, M. L., Pichamoorthy, N., Petronglo, J. R., Zambrano, A. D., et al. (2016). Postnatal and adult consequences of loss of huntingtin during development: Implications for Huntington's disease. Neurobiol. Dis. 96, 144–155. doi: 10.1016/j.nbd.2016.09.006
Bhide, P. G., Day, M., Sapp, E., Schwarz, C., Sheth, A., Kim, J., et al. (1996). Expression of normal and mutant huntingtin in the developing brain. J. Neurosci. 16, 5523–5535. doi: 10.1523/jneurosci.16-17-05523.1996
Blackstone, C., Elwood, F., Plun-Favreau, H., and Lewis, P. A. (2021). Vesicle trafficking and pathways to neurodegeneration. Mol. Neurodegener. 16, 1–4. doi: 10.1186/s13024-021-00480-1
Bloom, G. S. (2014). Amyloid-β and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 71, 505–508. doi: 10.1001/jamaneurol.2013.5847
Burk, K., and Pasterkamp, R. J. (2019). Disrupted neuronal trafficking in amyotrophic lateral sclerosis. Acta Neuropathol. 137, 859–877. doi: 10.1007/s00401-019-01964-7
Cason, S. E., and Carman, P. J. (2021). Sequential dynein effectors regulate axonal autophagosome motility in a maturation-dependent pathway. J. Cell Biol. 220:e202010179. doi: 10.1083/jcb.202010179
Cesca, F., Baldelli, P., Valtorta, F., and Benfenati, F. (2010). The synapsins: key actors of synapse function and plasticity. Prog. Neurobiol. 91, 313–348. doi: 10.1016/j.pneurobio.2010.04.006
Chalazonitis, A., and Rao, M. (2018). Enteric nervous system manifestations of neurodegenerative disease. Brain Res. 1693, 207–213. doi: 10.1016/j.brainres.2018.01.011
Chen, X., Sun, Y., Chen, L., Chen, X. S., Pan, M., Zhang, Y., et al. (2022). Differential expression and roles of Huntingtin and Huntingtin-associated protein 1 in the mouse and primate brains. Cell. Mol. Life Sci. 79, 1–20. doi: 10.1007/s00018-022-04577-8
Chen, X., Xin, N., Pan, Y., Zhu, L., Yin, P., Liu, Q., et al. (2020). Huntingtin-associated protein 1 in mouse hypothalamus stabilizes glucocorticoid receptor in stress response. Front. Cell. Neurosci. 14:125. doi: 10.3389/fncel.2020.00125
Colomer, V., Engelender, S., Sharp, A. H., Duan, K., Cooper, J. K., Lanahan, A., et al. (1997). Huntingtin-associated protein 1 (HAP1) binds to a Trio-like polypeptide, with a rac1 guanine nucleotide exchange factor domain. Hum. Mol. Genet. 6, 1519–1525. doi: 10.1093/hmg/6.9.1519
Damier, P., Hirsch, E. C., Agid, Y., and Graybiel, A. M. (1999). The substantia nigra of the human brain. II. Patterns of loss of dopamine-containing neurons in Parkinson's disease. Brain 122, 1437–1448. doi: 10.1093/brain/122.8.1437
De Vos, K. J., Grierson, A. J., Ackerley, S., and Miller, C. C. (2008). Role of axonal transport in neurodegenerative diseases. Annu. Rev. Neurosci. 31, 151–173. doi: 10.1146/annurev.neuro.31.061307.090711
DiFiglia, M., Sapp, E., Chase, K. O., Davies, S. W., Bates, G. P., Vonsattel, J. P., et al. (1997). Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277, 1990–1993. doi: 10.1126/science.277.5334.1990
Dom, R., Malfroid, M., and Baro, F. (1976). Neuropathology of Huntington's chorea. Studies of the ventrobasal complex of the thalamus. Neurology 26, 64–68. doi: 10.1212/wnl.26.1.64
Dugger, B. N., and Dickson, D. W. (2017). Pathology of neurodegenerative diseases. Cold Spring Harb. Perspect. Biol. 9:a028035. doi: 10.1101/cshperspect.a028035
Engelender, S., Sharp, A. H., Colomer, V., Tokito, M. K., Lanahan, A., Worley, P., et al. (1997). Huntingtin-associated protein 1 (HAP1) interacts with the p150Glued subunit of dynactin. Hum. Mol. Genet. 6, 2205–2212. doi: 10.1093/hmg/6.13.2205
Fujinaga, R., Kawano, J., Matsuzaki, Y., Kamei, K., Yanai, A., Sheng, Z., et al. (2004). Neuroanatomical distribution of huntingtin-associated protein 1-mRNA in the male mouse brain. J. Comp. Neurol. 478, 88–109. doi: 10.1002/cne.20277
Fujinaga, R., Takeshita, Y., Uozumi, K., Yanai, A., Yoshioka, K., Kokubu, K., et al. (2009). Microtubule-dependent formation of the stigmoid body as a cytoplasmic inclusion distinct from pathological aggresomes. Histochem. Cell Biol. 132, 305–318. doi: 10.1007/s00418-009-0618-9
Fujinaga, R., Takeshita, Y., Yoshioka, K., Nakamura, H., Shinoda, S., Islam, M. N., et al. (2011). Intracellular colocalization of HAP1/STBs with steroid hormone receptors and its enhancement by a proteasome inhibitor. Exp. Cell Res. 317, 1689–1700. doi: 10.1016/j.yexcr.2011.05.004
Furness, J. B., Callaghan, B. P., Rivera, L. R., and Cho, H. J. (2014). The enteric nervous system and gastrointestinal innervation: integrated local and central control. Adv. Exp. Med. Biol. 817, 39–71. doi: 10.1007/978-1-4939-0897-4_3
Gauthier, L. R., Charrin, B. C., Borrell-Pagès, M., Dompierre, J. P., Rangone, H., Cordelières, F. P., et al. (2004). Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cells 118, 127–138. doi: 10.1016/j.cell.2004.06.018
Gutekunst, C. A., Li, S. H., Yi, H., Ferrante, R. J., Li, X. J., and Hersch, S. M. (1998). The cellular and subcellular localization of huntingtin-associated protein 1 (HAP1): comparison with huntingtin in rat and human. J. Neurosci. 18, 7674–7686. doi: 10.1523/jneurosci.18-19-07674.1998
Gutekunst, C. A., Li, S. H., Yi, H., Mulroy, J. S., Kuemmerle, S., Jones, R., et al. (1999). Nuclear and neuropil aggregates in Huntington's disease: relationship to neuropathology. J. Neurosci. 19, 2522–2534. doi: 10.1523/jneurosci.19-07-02522.1999
Hardiman, O., Al-Chalabi, A., Chio, A., Corr, E. M., Logroscino, G., Robberecht, W., et al. (2017). Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers. 3:17071. doi: 10.1038/nrdp.2017.71
Hedreen, J. C., and Folstein, S. E. (1995). Early loss of neostriatal striosome neurons in Huntington's disease. J. Neuropathol. Exp. Neurol. 54, 105–120. doi: 10.1097/00005072-199501000-00013
Kittler, J. T., Thomas, P., Tretter, V., Bogdanov, Y. D., Haucke, V., Smart, T. G., et al. (2004). Huntingtin-associated protein 1 regulates inhibitory synaptic transmission by modulating gamma-aminobutyric acid type A receptor membrane trafficking. Proc. Natl. Acad. Sci. U. S. A. 101, 12736–12741. doi: 10.1073/pnas.0401860101
Kolobkova, Y. A., Vigont, V. A., Shalygin, A. V., and Kaznacheyeva, E. V. (2017). Huntington's Disease: Calcium Dyshomeostasis and Pathology Models. Acta Nat. 9, 34–46. doi: 10.32607/20758251-2017-9-2-34-46
Lazarov, O., Morfini, G. A., Pigino, G., Gadadhar, A., Chen, X., Robinson, J., et al. (2007). Impairments in fast axonal transport and motor neuron deficits in transgenic mice expressing familial Alzheimer's disease-linked mutant presenilin 1. J. Neurosci. 27, 7011–7020. doi: 10.1523/jneurosci.4272-06.2007
Li, Y., Chin, L. S., Levey, A. I., and Li, L. (2002). Huntingtin-associated protein 1 interacts with hepatocyte growth factor-regulated tyrosine kinase substrate and functions in endosomal trafficking. J. Biol. Chem. 277, 28212–28221. doi: 10.1074/jbc.M111612200
Li, S. H., Gutekunst, C. A., Hersch, S. M., and Li, X. J. (1998a). Association of HAP1 isoforms with a unique cytoplasmic structure. J. Neurochem. 71, 2178–2185. doi: 10.1046/j.1471-4159.1998.71052178.x
Li, S. H., Hosseini, S. H., Gutekunst, C. A., Hersch, S. M., Ferrante, R. J., and Li, X. J. (1998b). A human HAP1 homologue. Cloning, expression, and interaction with huntingtin. J. Biol. Chem. 273, 19220–19227. doi: 10.1074/jbc.273.30.19220
Li, X. J., and Li, S. H. (2005). HAP1 and intracellular trafficking. Trends Pharmacol. Sci. 26, 1–3. doi: 10.1016/j.tips.2004.11.001
Li, X. J., Li, S. H., Sharp, A. H., Nucifora, F. C. Jr., Schilling, G., Lanahan, A., et al. (1995). A huntingtin-associated protein enriched in brain with implications for pathology. Nature 378, 398–402. doi: 10.1038/378398a0
Li, S. H., Schilling, G., Young, W. S. 3rd, Li, X. J., Margolis, R. L., Stine, O. C., et al. (1993). Huntington's disease gene (IT15) is widely expressed in human and rat tissues. Neuron 11, 985–993. doi: 10.1016/0896-6273(93)90127-d
Liu, Q., Cheng, S., Yang, H., Zhu, L., and Pan, Y. (2020). Loss of Hap1 selectively promotes striatal degeneration in Huntington disease mice. Proc. Natl. Acad. Sci. U. S. A. 117, 20265–20273. doi: 10.1073/pnas.2002283117
Liu, J., Huang, Y., Li, T., Jiang, Z., Zeng, L., and Hu, Z. (2021). The role of the Golgi apparatus in disease (Review). Int. J. Mol. Med. 47:38. doi: 10.3892/ijmm.2021.4871
Mackenzie, K. D., Lumsden, A. L., Guo, F., Duffield, M. D., Chataway, T., Lim, Y., et al. (2016). Huntingtin-associated protein-1 is a synapsin I-binding protein regulating synaptic vesicle exocytosis and synapsin I trafficking. J. Neurochem. 138, 710–721. doi: 10.1111/jnc.13703
Mandal, M., Wei, J., Zhong, P., Cheng, J., Duffney, L. J., Liu, W., et al. (2011). Impaired alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor trafficking and function by mutant huntingtin. J. Biol. Chem. 286, 33719–33728. doi: 10.1074/jbc.M111.236521
Marcora, E., Gowan, K., and Lee, J. E. (2003). Stimulation of NeuroD activity by huntingtin and huntingtin-associated proteins HAP1 and MLK2. Proc. Natl. Acad. Sci. U. S. A. 100, 9578–9583. doi: 10.1073/pnas.1133382100
McColgan, P., and Tabrizi, S. J. (2018). Huntington's disease: a clinical review. Eur. J. Neurol. 25, 24–34. doi: 10.1111/ene.13413
McGuire, J. R., Rong, J., Li, S. H., and Li, X. J. (2006). Interaction of Huntingtin-associated protein-1 with kinesin light chain: implications in intracellular trafficking in neurons. J. Biol. Chem. 281, 3552–3559. doi: 10.1074/jbc.M509806200
Murphy, K. P., Carter, R. J., Lione, L. A., Mangiarini, L., Mahal, A., Bates, G. P., et al. (2000). Abnormal synaptic plasticity and impaired spatial cognition in mice transgenic for exon 1 of the human Huntington's disease mutation. J. Neurosci. 20, 5115–5123. doi: 10.1523/jneurosci.20-13-05115.2000
Petersen, H. V., Jensen, J. N., Stein, R., and Serup, P. (2002). Glucose induced MAPK signalling influences NeuroD1-mediated activation and nuclear localization. FEBS Lett. 528, 241–245. doi: 10.1016/s0014-5793(02)03318-5
Prigge, J. R., and Schmidt, E. E. (2007). HAP1 can sequester a subset of TBP in cytoplasmic inclusions via specific interaction with the conserved TBP(CORE). BMC Mol. Biol. 8:76. doi: 10.1186/1471-2199-8-76
Qin, Z. H., Wang, Y., Kegel, K. B., Kazantsev, A., Apostol, B. L., Thompson, L. M., et al. (2003). Autophagy regulates the processing of amino terminal huntingtin fragments. Hum. Mol. Genet. 12, 3231–3244. doi: 10.1093/hmg/ddg346
Ravikumar, B., Duden, R., and Rubinsztein, D. C. (2002). Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 11, 1107–1117. doi: 10.1093/hmg/11.9.1107
Rong, J., McGuire, J. R., Fang, Z. H., Sheng, G., Shin, J. Y., Li, S. H., et al. (2006). Regulation of intracellular trafficking of huntingtin-associated protein-1 is critical for TrkA protein levels and neurite outgrowth. J. Neurosci. 26, 6019–6030. doi: 10.1523/jneurosci.1251-06.2006
Schreij, A. M., Fon, E. A., and McPherson, P. S. (2016). Endocytic membrane trafficking and neurodegenerative disease. Cell. Mol. Life Sci. 73, 1529–1545. doi: 10.1007/s00018-015-2105-x
Sengupta, M., Pluciennik, A., and Merry, D. E. (2022). The role of ubiquitination in spinal and bulbar muscular atrophy. Front. Mol. Neurosci. 15:1020143. doi: 10.3389/fnmol.2022.1020143
Sharp, A. H., Loev, S. J., Schilling, G., Li, S. H., Li, X. J., Bao, J., et al. (1995). Widespread expression of Huntington's disease gene (IT15) protein product. Neuron 14, 1065–1074. doi: 10.1016/0896-6273(95)90345-3
Sheng, G., Xu, X., Lin, Y. F., Wang, C. E., Rong, J., Cheng, D., et al. (2008). Huntingtin-associated protein 1 interacts with Ahi1 to regulate cerebellar and brainstem development in mice. J. Clin. Invest. 118, 2785–2795. doi: 10.1172/jci35339
Shimojo, M. (2008). Huntingtin regulates RE1-silencing transcription factor/neuron-restrictive silencer factor (REST/NRSF) nuclear trafficking indirectly through a complex with REST/NRSF-interacting LIM domain protein (RILP) and dynactin p150 Glued. J. Biol. Chem. 283, 34880–34886. doi: 10.1074/jbc.M804183200
Singh, P. K., and Muqit, M. M. K. (2020). Parkinson's: a disease of aberrant vesicle trafficking. Annu. Rev. Cell Dev. Biol. 36, 237–264. doi: 10.1146/annurev-cellbio-100818-125512
Smith, F. D., and Scott, J. D. (2002). Signaling complexes: junctions on the intracellular information super highway. Curr. Biol. 12, R32–R40. doi: 10.1016/s0960-9822(01)00646-7
Soares, E. S., Prediger, R. D., Brocardo, P. S., and Cimarosti, H. I. (2022). SUMO-modifying Huntington’s disease. IBRO Neurosci. Rep. 12, 203–209. doi: 10.1016/j.ibneur.2022.03.002
Soong, B. W., and Morrison, P. J. (2018). Spinocerebellar ataxias. Handb. Clin. Neurol. 155, 143–174. doi: 10.1016/b978-0-444-64189-2.00010-x
Soria Lopez, J. A., González, H. M., and Léger, G. C. (2019). Alzheimer's disease. Handb. Clin. Neurol. 167, 231–255. doi: 10.1016/b978-0-12-804766-8.00013-3
Sosa, L. J., Cáceres, A., Dupraz, S., Oksdath, M., Quiroga, S., and Lorenzo, A. (2017). The physiological role of the amyloid precursor protein as an adhesion molecule in the developing nervous system. J. Neurochem. 143, 11–29. doi: 10.1111/jnc.14122
Soto, C., and Pritzkow, S. (2018). Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 21, 1332–1340. doi: 10.1038/s41593-018-0235-9
Spillantini, M. G., Schmidt, M. L., Lee, V. M., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997). Alpha-synuclein in Lewy bodies. Nature 388, 839–840. doi: 10.1038/42166
Stokin, G. B., Lillo, C., Falzone, T. L., Brusch, R. G., Rockenstein, E., Mount, S. L., et al. (2005). Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science 307, 1282–1288. doi: 10.1126/science.1105681
Subramaniam, S., and Snyder, S. H. (2011). Huntington's disease is a disorder of the corpus striatum: focus on Rhes (Ras homologue enriched in the striatum). Neuropharmacology 60, 1187–1192. doi: 10.1016/j.neuropharm.2010.10.025
Suzuki, T., Araki, Y., Yamamoto, T., and Nakaya, T. (2006). Trafficking of Alzheimer's disease-related membrane proteins and its participation in disease pathogenesis. J. Biochem. 139, 949–955. doi: 10.1093/jb/mvj121
Takeshita, Y., Fujinaga, R., Kokubu, K., Islam, M. N., Jahan, M. R., Yanai, A., et al. (2011). Interaction of ataxin-3 with huntingtin-associated protein 1 through Josephin domain. Neuroreport 22, 232–238. doi: 10.1097/WNR.0b013e32834505f4
Takeshita, Y., Fujinaga, R., Zhao, C., Yanai, A., and Shinoda, K. (2006). Huntingtin-associated protein 1 (HAP1) interacts with androgen receptor (AR) and suppresses SBMA-mutant-AR-induced apoptosis. Hum. Mol. Genet. 15, 2298–2312. doi: 10.1093/hmg/ddl156
Tang, B. L. (2009). Neuronal protein trafficking associated with Alzheimer disease: from APP and BACE1 to glutamate receptors. Cell Adhes. Migr. 3, 118–128. doi: 10.4161/cam.3.1.7254
Tang, T. S., Tu, H., Chan, E. Y., Maximov, A., Wang, Z., Wellington, C. L., et al. (2003). Huntingtin and huntingtin-associated protein 1 influence neuronal calcium signaling mediated by inositol-(1,4,5) triphosphate receptor type 1. Neuron 39, 227–239. doi: 10.1016/s0896-6273(03)00366-0
Tang, T. S., Tu, H., Orban, P. C., Chan, E. Y., Hayden, M. R., and Bezprozvanny, I. (2004). HAP1 facilitates effects of mutant huntingtin on inositol 1,4,5-trisphosphate-induced Ca release in primary culture of striatal medium spiny neurons. Eur. J. Neurosci. 20, 1779–1787. doi: 10.1111/j.1460-9568.2004.03633.x
Tarif, A. M. M., and Islam, M. N. (2021). Immunohistochemical expression and neurochemical phenotypes of huntingtin-associated protein 1 in the myenteric plexus of mouse gastrointestinal tract. Cell Tissue Res. 386, 533–558. doi: 10.1007/s00441-021-03542-4
Tarif, A. M. M., Islam, M. N., Jahan, M. R., Afrin, M., Meher, M. M., Nozaki, K., et al. (2022). Neurochemical phenotypes of huntingtin-associated protein 1 in reference to secretomotor and vasodilator neurons in the submucosal plexuses of rodent small intestine. Neurosci. Res. doi: 10.1016/j.neures.2022.12.023 (in press).
Tempes, A., Weslawski, J., Brzozowska, A., and Jaworski, J. (2020). Role of dynein-dynactin complex, kinesins, motor adaptors, and their phosphorylation in dendritogenesis. J. Neurochem 155, 10–28. doi: 10.1111/jnc.15010
Ting, L. L., Lu, H. T., Yen, S. F., Ngo, T. H., Tu, F. Y., Tsai, I. S., et al. (2019). Expression of AHI1 rescues amyloidogenic pathology in Alzheimer's disease model cells. Mol. Neurobiol. 56, 7572–7582. doi: 10.1007/s12035-019-1587-1
Twelvetrees, A. E., Yuen, E. Y., Arancibia-Carcamo, I. L., MacAskill, A. F., Rostaing, P., Lumb, M. J., et al. (2010). Delivery of GABAARs to synapses is mediated by HAP1-KIF5 and disrupted by mutant huntingtin. Neuron 65, 53–65. doi: 10.1016/j.neuron.2009.12.007
van Roon-Mom, W. M., Reid, S. J., Faull, R. L., and Snell, R. G. (2005). TATA-binding protein in neurodegenerative disease. Neuroscience 133, 863–872. doi: 10.1016/j.neuroscience.2005.03.024
Vicario-Orri, E., Opazo, C. M., and Muñoz, F. J. (2015). The pathophysiology of axonal transport in Alzheimer's disease. J. Alzheimers Dis. 43, 1097–1113. doi: 10.3233/jad-141080
Vonsattel, J. P., and DiFiglia, M. (1998). Huntington disease. J. Neuropathol. Exp. Neurol. 57, 369–384. doi: 10.1097/00005072-199805000-00001
Vonsattel, J. P., Myers, R. H., Stevens, T. J., Ferrante, R. J., Bird, E. D., and Richardson, E. P. Jr. (1985). Neuropathological classification of Huntington's disease. J. Neuropathol. Exp. Neurol. 44, 559–577. doi: 10.1097/00005072-198511000-00003
Wang, N., Gray, M., Lu, X. H., Cantle, J. P., Holley, S. M., Greiner, E., et al. (2014). Neuronal targets for reducing mutant huntingtin expression to ameliorate disease in a mouse model of Huntington's disease. Nat. Med. 20, 536–541. doi: 10.1038/nm.3514
Wang, B., Xin, N., Qian, X., Zhai, L., Miao, Z., and Yang, Y. (2021). Ahi1 regulates the nuclear translocation of glucocorticoid receptor to modulate stress response. Transl. Psychiatry 11:188. doi: 10.1038/s41398-021-01305-x
Wong, Y. C., and Holzbaur, E. L. (2014). The regulation of autophagosome dynamics by huntingtin and HAP1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. J. Neurosci. 34, 1293–1305. doi: 10.1523/jneurosci.1870-13.2014
Wu, L. L., Fan, Y., Li, S., Li, X. J., and Zhou, X. F. (2010). Huntingtin-associated protein-1 interacts with pro-brain-derived neurotrophic factor and mediates its transport and release. J. Biol. Chem. 285, 5614–5623. doi: 10.1074/jbc.M109.073197
Xiang, J., Yang, H., Zhao, T., Sun, M., Xu, X., Zhou, X. F., et al. (2014). Huntingtin-associated protein 1 regulates postnatal neurogenesis and neurotrophin receptor sorting. J. Clin. Invest. 124, 85–98. doi: 10.1172/jci69206
Yang, M., Lim, Y., Li, X., Zhong, J. H., and Zhou, X. F. (2011). Precursor of brain-derived neurotrophic factor (proBDNF) forms a complex with Huntingtin-associated protein-1 (HAP1) and sortilin that modulates proBDNF trafficking, degradation, and processing. J. Biol. Chem. 286, 16272–16284. doi: 10.1074/jbc.M110.195347
Yang, G. Z., Yang, M., Lim, Y., Lu, J. J., Wang, T. H., Qi, J. G., et al. (2012). Huntingtin associated protein 1 regulates trafficking of the amyloid precursor protein and modulates amyloid beta levels in neurons. J. Neurochem. 122, 1010–1022. doi: 10.1111/j.1471-4159.2012.07845.x
Yuen, E. Y., Wei, J., Zhong, P., and Yan, Z. (2012). Disrupted GABAAR trafficking and synaptic inhibition in a mouse model of Huntington's disease. Neurobiol. Dis. 46, 497–502. doi: 10.1016/j.nbd.2012.02.015
Zhang, Y. W., Chen, Y., Liu, Y., Zhao, Y., Liao, F. F., and Xu, H. (2013). APP regulates NGF receptor trafficking and NGF-mediated neuronal differentiation and survival. PLoS One 8:e80571. doi: 10.1371/journal.pone.0080571
Keywords: Huntingtin-associated protein 1, neurodegenerative diseases, intracellular trafficking, aging, selective neurodegeneration
Citation: Chen X, He E, Su C, Zeng Y and Xu J (2023) Huntingtin-associated protein 1-associated intracellular trafficking in neurodegenerative diseases. Front. Aging Neurosci. 15:1100395. doi: 10.3389/fnagi.2023.1100395
Edited by:
Pablo Helguera, Instituto Ferreyra INIMEC CONICET, ArgentinaReviewed by:
Md Nabiul Islam, Yamaguchi University School of Medicine, JapanNobuyuki Kimura, Okayama University of Science, Japan
Copyright © 2023 Chen, He, Su, Zeng and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xingxing Chen, ✉ Y2hlbnhpbmd4aW5nODBAYWxpeXVuLmNvbQ==; Jiang Xu, ✉ eHVqaWFuZ0BodXN0LmVkdS5jbg==