
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Aging Neurosci. , 10 February 2023
Sec. Parkinson’s Disease and Aging-related Movement Disorders
Volume 15 - 2023 | https://doi.org/10.3389/fnagi.2023.1091919
 Jingru Ren1†
Jingru Ren1† Gaiyan Zhou1
Gaiyan Zhou1 Yajie Wang1Ronggui Zhang1
Yajie Wang1Ronggui Zhang1 Zhiying Guo1Hao Zhou1
Zhiying Guo1Hao Zhou1 Huifen Zheng2
Huifen Zheng2 Yu Sun3Changyan Ma4
Yu Sun3Changyan Ma4 Ming Lu5
Ming Lu5 Weiguo Liu1*
Weiguo Liu1*Objective: Variants in the glucocerebrosidase (GBA) gene are the most common and significant risk factor for Parkinson’s disease (PD). However, the impact of GBA variants on PD disease progression in the Chinese population remains unclear. This study aimed to explore the significance of GBA status on motor and cognitive impairment in a longitudinal cohort of Chinese patients with PD.
Methods: The entire GBA gene was screened by long-range polymerase chain reaction (LR-PCR) and next generation sequencing (NGS). A total of 43 GBA-related PD (GBA-PD) and 246 non-GBA-mutated PD (NM-PD) patients with complete clinical data at baseline and at least one follow-up were recruited for this study. The associations of GBA genotype with rate of motor and cognitive decline, as measured by Unified PD Rating Scale (UPDRS) motor and Montreal Cognitive Assessment (MoCA), were assessed by linear mixed-effect models.
Results: The estimated (standard error, SE) UPDRS motor [2.25 (0.38) points/year] and MoCA [−0.53 (0.11) points/year] progression rates in the GBA-PD group were significantly faster than those in the NM-PD group [1.35 (0.19); −0.29 (0.04) points/year; respectively]. In addition, the GBA-PD group showed significantly faster estimated (SE) bradykinesia [1.04 (0.18) points/year], axial impairment [0.38 (0.07) points/year], and visuospatial/executive [−0.15 (0.03) points/year] progression rates than the NM-PD group [0.62 (0.10); 0.17 (0.04); −0.07 (0.01) points/year; respectively].
Conclusion: GBA-PD is associated with faster motor and cognitive decline, specifically greater disability in terms of bradykinesia, axial impairment, and visuospatial/executive function. Better understanding of GBA-PD progression may help predict prognosis and improve clinical trial design.
Parkinson’s disease (PD) is a complex chronic neurodegenerative condition (Bloem et al., 2021). Due to insufficient understanding of its pathogenesis, no disease-modifying therapies are currently available (Lang and Espay, 2018). With the relentless progression of neurodegeneration in PD, motor and cognitive impairments greatly affect patients’ quality of life and cause a huge socioeconomic burden (Feigin et al., 2019). Given that motor and cognitive deterioration rates vary considerably among PD patients (Armstrong and Okun, 2020), identifying the genes that modulate disease progression is potentially important for improving trial design—especially in neuroprotective therapies—and developing personalized treatment approaches.
One of the most effective ways to identify genetic modifiers of PD progression is to examine established PD susceptibility genes (Bandres-Ciga et al., 2020). Heterozygous variants in the glucocerebrosidase (GBA; OMIM 606463) gene, which encodes the lysosomal enzyme β-glucocerebrosidase (GCase), have been identified as the most common and significant genetic risk factor for PD worldwide (Sidransky et al., 2009). The frequency of GBA variants is ethnically heterogeneous, estimated to be 10–31% in Ashkenazi Jewish (AJ), 3–12% in non-AJ North Americans, and 2–11% in Chinese populations (Neumann et al., 2009; Mao et al., 2010; Gan-Or et al., 2015; Li et al., 2020; Ren et al., 2022). Notably, a highly homologous pseudogene (GBAP1), located 16 kb downstream, shares 96% sequence identity with the functional GBA gene, making GBA sequencing more challenging (Horowitz et al., 1989; Hruska et al., 2008).
Clinically, GBA-related PD (GBA-PD) is characterized by an earlier age of onset, more severe motor and non-motor symptoms (specifically cognitive decline, rapid eye movement sleep disorder, depression, and autonomic dysfunction), and lower survival rates compared with non-GBA-mutated PD (NM-PD) (Brockmann et al., 2015; Malek et al., 2018; Stoker et al., 2020; Zhao et al., 2020). Importantly, GBA variant severity underlies the phenotypic spectrum of PD (Petrucci et al., 2020; Ren et al., 2022). Several studies have shown that GBA status is an independent risk factor for cognitive decline in PD and that GBA genotype severity is linked to the rate of cognitive impairment progression (Cilia et al., 2016; Liu et al., 2016; Lunde et al., 2018; Stoker et al., 2020; Szwedo et al., 2022). However, the distinct pattern of progression in cognitive impairment in GBA-PD patients remains unclear. Although PD is predominantly a movement disorder, few studies have examined the impact of GBA status on progression of motor decline (Davis M. Y. et al., 2016; Ortega et al., 2021). In a recent study of Northern European PD patients, GBA variants were associated with more aggressive motor impairment (Maple-Grødem et al., 2021).
To the best of our knowledge, the influence of GBA variants on motor and cognitive impairment has not been previously investigated in the Chinese PD population. To address this gap, we studied a longitudinal cohort of Chinese PD patients to comprehensively explore the role of GBA status in the progression of motor and cognitive decline, especially patterns of motor and cognitive progression.
This prospective comprehensive study of a large cohort of Chinese patients with GBA-PD or NM-PD was conducted at the Department of Neurology of the Affiliated Brain Hospital of Nanjing Medical University from 2012 to 2022. The inclusion criteria were: (1) a diagnosis of PD by a movement disorder specialist according to the United Kingdom PD Society Brain Bank clinical diagnostic criteria (Gibb and Lees, 1988), regardless of the presence of a family history of PD; and (2) received genetic testing for GBA variants. The exclusion criteria were: (1) diagnosed with atypical or secondary parkinsonism disorders; (2) a history of serious chronic diseases, such as kidney or heart failure; and (3) brain magnetic resonance imaging (MRI) scans revealing clinically significant lesions. Patients who met these criteria were invited to participate in in-depth annual follow-up at our institution if available.
A total of 737 unrelated PD patients were recruited from January 2012 to June 2020. Of these 79 (10.7%) were confirmed to have GBA variants and the remaining 658 (89.3%) had no GBA variants. Among the 79 patients with GBA variants, 43 were followed up at least once with complete clinical data at baseline (GBA-PD group). Of the 658 patients without GBA variants, 246 were followed up at least once with complete clinical data at baseline (NM-PD group) (Supplementary Figure 1).
This study was approved by the Medical Ethics Committee of the Affiliated Brain Hospital of Nanjing Medical University. All participants provided written informed consent prior to entering the study.
Standardized procedures to collect detailed demographic and clinical data were conducted through face-to-face interviews with all PD patients. The levodopa equivalent daily dose (LEDD) was calculated in accordance with an established method (Tomlinson et al., 2010). The Unified PD Rating Scale (UPDRS) part II, part III, and modified Hoehn–Yahr (H–Y) stage were used to assess activities of daily living (ADL), motor impairment, and disease severity, respectively. The severity of specific motor characteristics in PD patients was determined by summing the following relevant UPDRS III items: tremor (items 20 and 21), rigidity (item 22), bradykinesia (items 23–26 and 31), and axial impairment (item 27–30). Cognitive ability was measured with the Mini-Mental State Examination (MMSE) and Montreal Cognitive Assessment (MoCA). The MoCA comprises 30 items divided into 7 domains, namely, visuospatial/executive (5 points), naming (3 points), attention (6 points), language (3 points), abstraction (2 points), delayed recall (5 points), and orientation (6 points). Total MoCA scores were adjusted for education by adding one point for individuals with ≤12 years of education (Nasreddine et al., 2005).
The entire GBA gene was screened using the long-range polymerase chain reaction (LR-PCR) protocol to avoid nearby GBAP1 contamination followed by next generation sequencing (NGS), as previously described (Ren et al., 2022). All identified variants were confirmed by repeating amplification and resequencing. We adopted the traditional nomenclature for GBA alleles, referring to the processed protein and excluding the 39-residue signal peptide. Similar to previous studies (Liu et al., 2016; Petrucci et al., 2020; Ren et al., 2022), GBA variants were divided into five types: mild [causing Gaucher disease (GD) type 1], severe (causing GD type 2 or 3), risk (associated with risk factors of PD, but meaningless for GD), complex (≥2 variants, with no confirmation of cis distribution), and variants of unknown significance (VUS). Pathogenicity predictions of the identified non-synonymous GBA variants were assessed with 22 computational methods, including 9 function-prediction methods (FATHMM, fitCons, LRT, MutationAssessor, MutationTaster, PolyPhen2-HVAR, PROVEAN, SIFT, and VEST3), 4 conservation methods (GERP++, phastCons, phyloP, and SiPhy), and 9 ensemble methods (CADD, DANN, Eigen, FATHMM-MKL, GenoCanyon, M-CAP, MetaLR, MetaSVM, and REVEL) (Supplementary Table 1). Scores for the 22 methods were downloaded directly from the dbNSFP database v3. The cutoffs used to distinguish deleterious non-synonymous variants were adopted from a previous study (Li et al., 2018).
Statistical analyses were performed using IBM SPSS Statistics version 27.0 or Stata version 17.0. Baseline between-group differences were compared using linear regression analysis for continuous variables and logistic regression analysis for binary variables, adjusting for the confounders listed in the Table 1. Linear mixed-effects models were used to examine the relationship between GBA genotype and longitudinal motor and cognitive measures assessed as repeat UPDRS motor and MoCA scores. As linear mixed-effects models can accommodate unbalanced data due to missing data points, dropout, unequal visit intervals, and staggered first visit times and account for the correlation between repeated measures across individuals, they are often used to analyze the trajectory of longitudinal markers. Disease duration was used as the time scale considering its correlation with motor and cognitive function. A participant-specific random effect was used to explain repeated-measures correlations within the same participant. Time, GBA genotype, and their interaction were included as fixed effects. Motor and cognitive progression was modeled adjusting for sex, age at baseline, and LEDD or education. All models had random intercepts of the participants’ IDs as well as a random slope of time. Predictive margins of motor and cognitive decline were plotted using margin and marginsplot in Stata. Findings with two-tailed P-values < 0.05 were considered statistically significant.
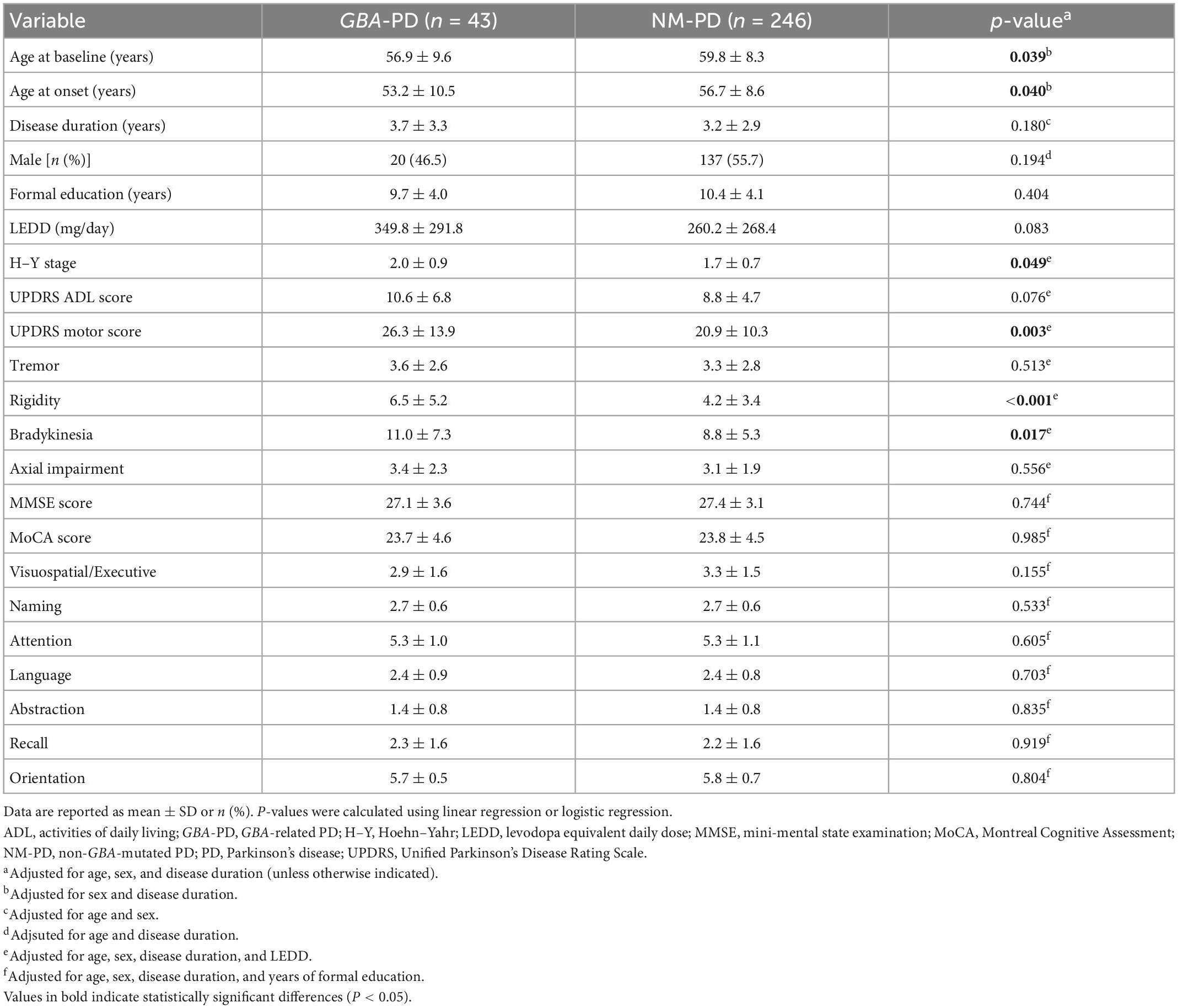
Table 1. Baseline demographic and clinical features of GBA-related PD (GBA-PD) and non-GBA mutated PD (NM-PD) patients.
A total of 31 distinct variants were identified in the 43 GBA-PD patients (Supplementary Table 2). Of these 43 cases, the variant severity classes were 3 mild, 16 severe, 1 risk, 3 complex, and 20 unknown. The in silico pathogenicity predictions of the identified non-synonymous GBA variants are presented in Supplementary Table 1. Only D399H and W393C were predicted to be deleterious by all 22 computational methods. Notably, R163Q, a common GBA variant in the Chinese population, was predicted to be tolerable by 14 computational methods in the in silico analyses.
Table 1 presents the baseline demographic and clinical profiles of the GBA-PD and NM-PD groups. A total of 289 patients (43 carriers and 246 non-carriers) were included in this analysis. The mean follow-up time was 2.6 years in the GBA-PD group and 3.5 years in the NM-PD group. Consistent with our previous report (Ren et al., 2022), PD patients with and without GBA mutations were similar in terms of disease duration, sex, formal education, LEDD, ADL, and cognitive impairment. Compared with NM-PD patients, GBA-PD patients had a younger baseline age, earlier age of onset, greater modified H–Y stages, and more severe UPDRS motor scores, adjusting for potential confounding factors. In addition, GBA-PD patients had more severe rigidity and bradykinesia scores than NM-PD patients. However, PD patients with and without GBA mutations did not differ in terms of severity of tremor or axial impairment.
Linear mixed-effects models were used to examine the association of GBA genotype with the rate of change in UPDRS motor scores and subscores, adjusting for sex, age at baseline, and LEDD at visit (Figure 1 and Table 2). The estimated (standard error, SE) progression rate of UPDRS motor score in the GBA-PD group [2.25 (0.38) points/year; P < 0.001] was significantly faster than that in the NM-PD group [1.35 (0.19) points/year; P < 0.001] [difference, 0.90 (0.37) points/year; P = 0.017] (Supplementary Figure 2). For GBA-PD, this remained significant after excluding VUS [difference, 1.64 (0.45) points/year; P < 0.001]. In addition, the progression rate of UPDRS motor score did not differ between the non-severe GBA-PD and NM-PD groups [difference, 0.73 (0.44) points/year; P = 0.099], whereas the estimated (SE) progression rate of UPDRS motor score in the severe GBA-PD group [2.65 (0.61) points/year; P < 0.001] was significantly faster than that in the NM-PD group [1.25 (0.18) points/year; P < 0.001] [difference, 1.40 (0.60) points/year; P = 0.020]. The magnitude of the impact on motor impairment in the severe GBA-PD group was greater than that in the overall GBA-PD group.
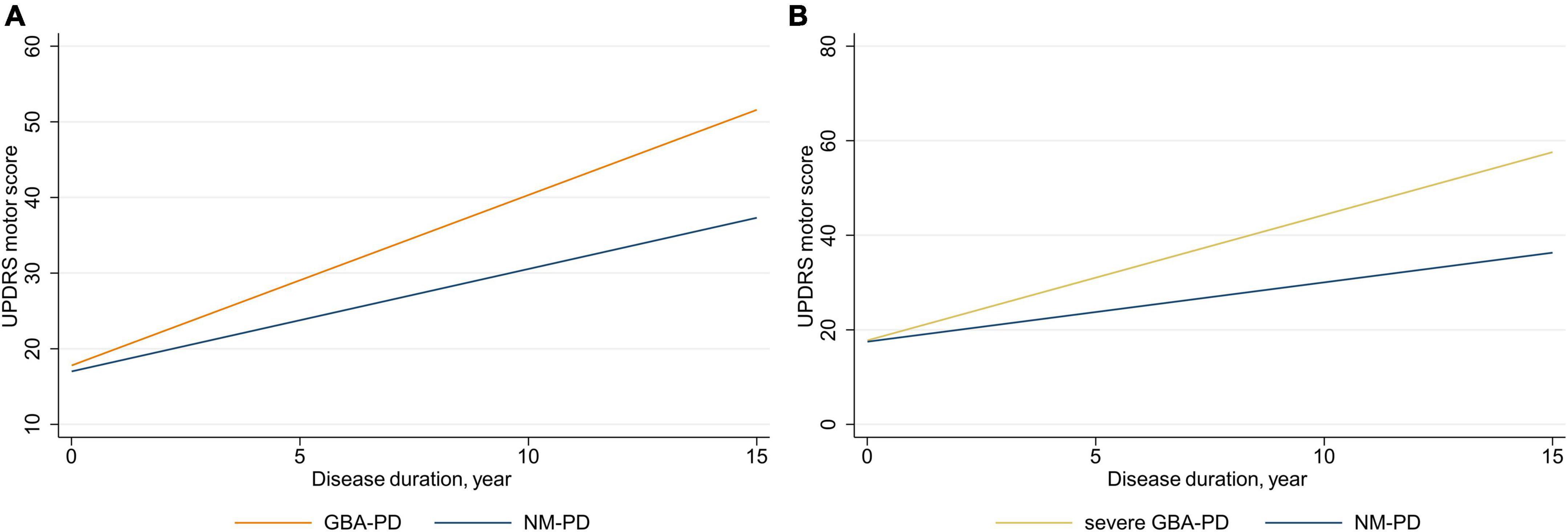
Figure 1. Longitudinal trajectories of mean Unified Parkinson’s Disease Rating Scale (UPDRS) motor scores. (A) GBA-PD vs. NM-PD; (B) severe GBA-PD vs. NM-PD. GBA-PD, GBA-related PD; NM-PD, non-GBA-mutated PD; PD, Parkinson’s disease.

Table 2. Association between GBA carrier status and annual change in Unified Parkinson’s Disease Rating Scale (UPDRS) motor scores and subscores using linear mixed-effects models.
Differences in motor decline were driven primarily by changes in bradykinesia and axial impairment scores. The estimated (SE) progression rate of bradykinesia score in the GBA-PD group [1.04 (0.18) points/year; P < 0.001] was significantly faster than that in the NM-PD group [0.62 (0.10) points/year; P < 0.001] [difference, 0.41 (0.18) points/year; P = 0.024]. Furthermore, the estimated (SE) progression rate of bradykinesia score in the severe GBA-PD group [1.17 (0.30) points/year; P < 0.001] was significantly faster than that in the NM-PD group [0.56 (0.09) points/year; P < 0.001] [difference, 0.62 (0.29) points/year; P = 0.033]. Similarly, the estimated (SE) progression rate of axial impairment score in the GBA-PD group [0.38 (0.07) points/year; P < 0.001] was significantly faster than that in the NM-PD group [0.17 (0.04) points/year; P < 0.001; difference, 0.20 (0.07) points/year; P = 0.005], and the estimated (SE) progression rate of axial impairment score in the severe GBA-PD group [0.42 (0.13) points/year; P = 0.001] was significantly faster than that in the NM-PD group [0.16 (0.04) points/year; P < 0.001] [difference, 0.26 (0.12) points/year; P = 0.033].
Next, we used linear mixed-effects models to explore the association of GBA genotype with the rate of annual decline in MoCA scores and subscores adjusted for sex, age at baseline, and education (Figure 2 and Table 3). The estimated (SE) progression rate of MoCA score in the GBA-PD group [−0.53 (0.11) points/year; P < 0.001] was significantly faster than that in the NM-PD group [−0.29 (0.04) points/year; P < 0.001] [difference, −0.25 (0.12) points/year; P = 0.035]. For GBA-PD, this remained significant after excluding VUS [difference, −0.40 (0.15) points/year; P = 0.009]. Furthermore, the progression rate of MoCA motor score did not differ between the non-severe GBA-PD and NM-PD groups [difference, −0.20 (0.14) points/year; P = 0.147], whereas the estimated (SE) progression rate of MoCA score in the severe GBA-PD group [−0.68 (0.19) points/year; P < 0.001] was significantly faster than that in the NM-PD group [−0.29 (0.04) points/year; P < 0.001] [difference, −0.40 (0.20) points/year; P = 0.046]. The magnitude of the impact on cognitive impairment in the severe GBA-PD group was greater than that in the entire GBA-PD group.
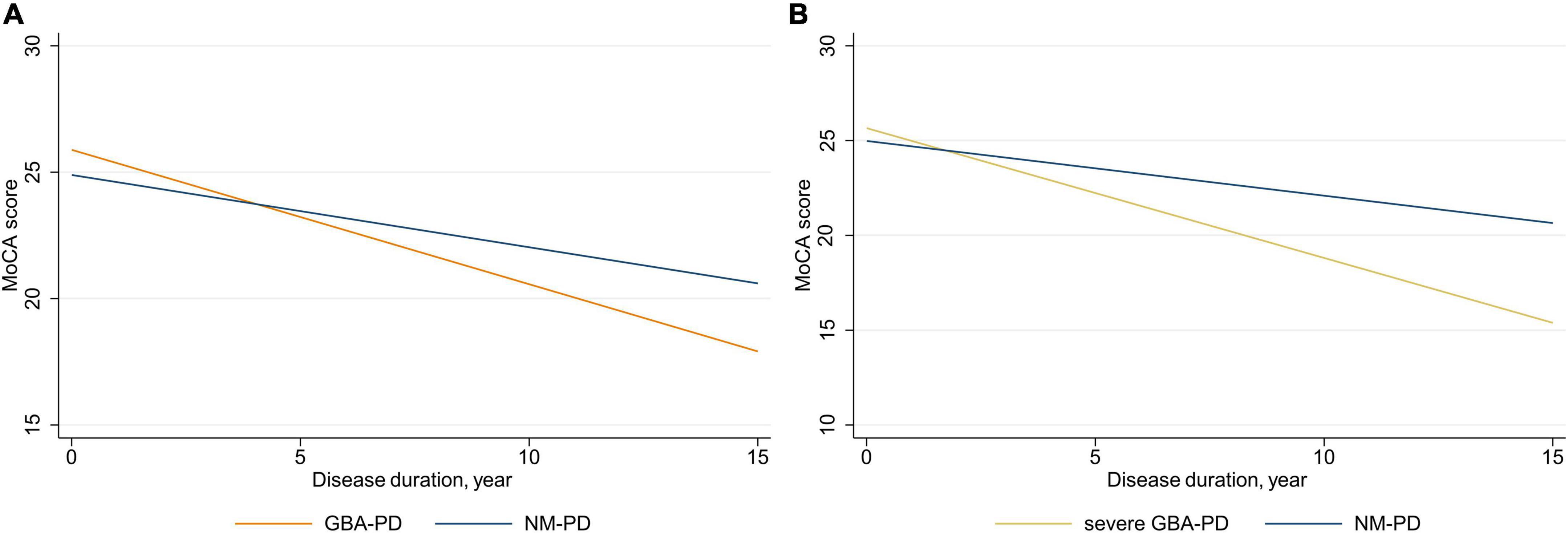
Figure 2. Longitudinal trajectories of mean Montreal Cognitive Assessment (MoCA) scores. (A) GBA-PD vs. NM-PD; (B) severe GBA-PD vs. NM-PD. GBA-PD, GBA-related PD; NM-PD, non-GBA-mutated PD; PD, Parkinson’s disease.
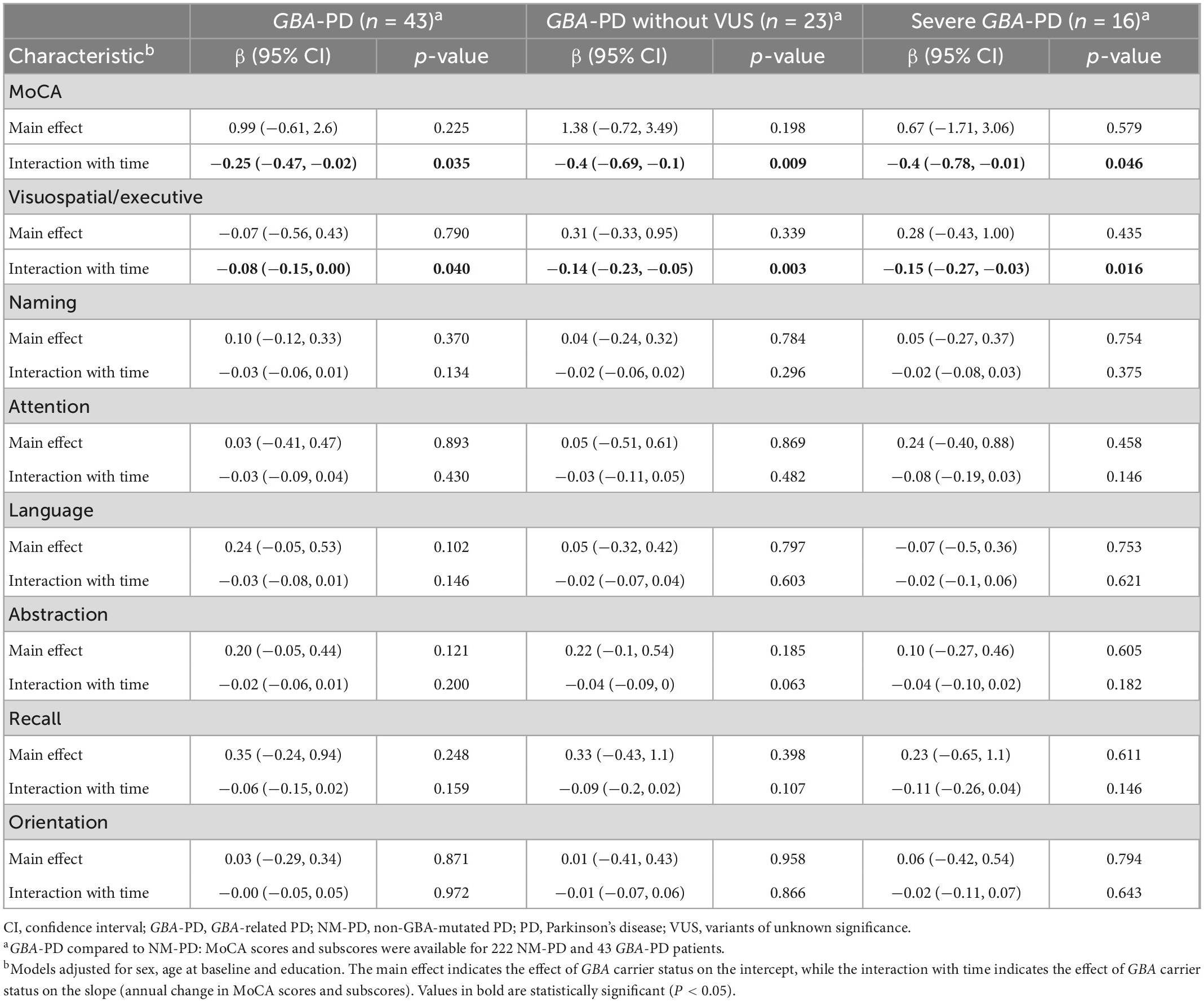
Table 3. Association between GBA carrier status and annual change in Montreal Cognitive Assessment (MoCA) scores and subscores using linear mixed-effects models.
Differences in cognitive decline were driven primarily by changes in visuospatial/executive score. The estimated (SE) progression rate of visuospatial/executive score in the GBA-PD group [−0.15 (0.03) points/year; P < 0.001] was significantly faster than that in the NM-PD group [−0.07 (0.01) points/year; P < 0.001] [difference, −0.08 (0.04) points/year; P = 0.040]. Further, the estimated (SE) progression rate of visuospatial/executive score in the severe GBA-PD group [−0.22 (0.06) points/year; P < 0.001] was significantly faster than that in the NM-PD group [−0.07 (0.01) points/year; P < 0.001] [difference, −0.15 (0.06) points/year; P = 0.016].
To our knowledge, this is the first paper to systematically assess the association of GBA genotype with long-term motor and cognitive decline in a longitudinal Chinese PD cohort. The results showed that both motor and cognitive impairment progressed more rapidly in the GBA-PD patients, especially the severe subgroup, compared to the NM-PD patients. Furthermore, examination of changes in specific motor and cognitive domains revealed that between-group differences in progression were primarily attributable to bradykinesia, axial impairment, and visuospatial/executive functioning.
UPDRS motor score, which is an operational standard for assessing symptom severity, has the advantage of good intra- and inter-rater reliability, making it one of the most used primary outcomes in observational studies and clinical trials. However, surprisingly few studies have used UPDRS motor score to evaluate progression of motor impairment. In our study, GBA-PD patients showed faster motor decline, as measured by rate of change in UPDRS motor score, consistent with recent longitudinal findings (Davis M. Y. et al., 2016; Maple-Grødem et al., 2021; Ortega et al., 2021). Furthermore, we found that severe GBA-PD patients experienced more rapid motor progression than overall GBA-PD patients, compared to NM-PD patients, implying that motor impairment progression is associated with GBA variant severity. In contrast to our findings, (Davis A. A. et al., 2016) found no significant association between either variant (N370S or K26R) and annual change in UPDRS motor score in the “on” or “off” state in PD patients from two cohorts. These inconsistent findings could relate to the fact that the GBA-PD group in our study included all GBA variants, rather than analysis of individual GBA variants. Considering the low frequency of individual GBA variants, greater collaboration is required to study individual GBA variants. In the largest genome-wide association study to date involving 12 cohorts, T369M was associated with faster progression of motor impairment severity to H–Y stage 3 (Iwaki et al., 2019). Alternatively, as done in this study, screening the entire GBA gene and classifying GBA severity, in the absence of GBAP1 interference, could clarify whether GBA status affects motor decline progression in PD patients.
Numerous studies have reported an association between GBA variants and more rapid progression of cognitive symptoms, as assessed using the MMSE, in PD patients (Cilia et al., 2016; Liu et al., 2016; Lunde et al., 2018; Stoker et al., 2020; Szwedo et al., 2022). However, there is a paucity of longitudinal studies using the MoCA to evaluate cognitive features in GBA-PD patients (Ortega et al., 2021). It is well known that the MoCA is more sensitive than the MMSE for detecting mild, domain-specific changes in global cognitive performance in PD (Hoops et al., 2009; Skorvanek et al., 2018). Thus, our finding that GBA-PD patients experienced faster cognitive decline, as represented by annual change in MoCA, provides further evidence that GBA is a major risk factor for cognitive impairment. We also observed that severe GBA-PD patients had faster cognitive progression than entire GBA-PD patients, relative to NM-PD patients. Although previous studies have used different classification methods for GBA variants, it has been consistently demonstrated that the progression of cognitive impairment increases with GBA variant severity (Cilia et al., 2016; Liu et al., 2016; Lunde et al., 2018; Szwedo et al., 2022). Several studies have reported no link between GBA risk variants and cognitive impairment, which may be attributed to the small sample sizes or combining E326K and T369M variants with synonymous or intronic variants, which dilutes the effect of risk variants (Winder-Rhodes et al., 2013; Jesús et al., 2016). Recent longitudinal studies have demonstrated that cognitive impairment progresses more rapidly in risk GBA-PD patients (Davis M. Y. et al., 2016; Lunde et al., 2018; Straniero et al., 2020; Szwedo et al., 2022). Therefore, both severe and risk GBA variants appear to modify cognitive progression in PD and could help explain clinical heterogeneity among GBA-PD patients.
Since GBA-PD is associated with faster progression of motor decline and global cognitive impairment, we further investigated whether and to what extent GBA status was related to changes in specific motor features and cognitive domains. We found that progression rates for subscores related to bradykinesia, axial impairment, and visuospatial/executive function differed significantly between groups. Regarding specific motor symptoms, a similar study reported that GBA variants were significantly associated with worsening of bradykinesia and axial impairment (Maple-Grødem et al., 2021), which is comparable to our results. It is well known that cognitive impairment is a clinical signature of GBA-PD. Bradykinesia and rigidity impairment are recognized as independent risk factors for cognitive impairment in PD (Aarsland et al., 2021), which explains why GBA-PD patients show rapid progression in these areas. Regarding specific cognitive domains, our results are consistent with those of a recent 10-year longitudinal study of 10 GBA-PD and 20 NM-PD patients reporting that GBA-PD patients had greater deterioration in visuospatial function compared with NM-PD patients (Leocadi et al., 2022). In a previous multicenter cross-sectional study, GBA-PD patients showed a unique cognitive pattern of significant impairment of working memory/executive function and visuospatial abilities (Mata et al., 2016). Our data provide an important missing piece of the puzzle, demonstrating for the first time in a large longitudinal study that cognitive progression in GBA-PD is specifically manifested as visuospatial/executive function impairments.
This study has several strengths, including (1) establishing the first GBA-PD longitudinal cohort in a Chinese population; (2) demonstrating that the pattern of rapid motor and cognitive progression in GBA-PD is characterized by severely impaired bradykinesia, axial impairment, and visuospatial/executive function; and (3) screening the entire GBA gene without GBAP1 interference for more accurate variant-specific analysis. However, there are also several limitations that should be considered. (1) An insufficient sample size for the genotype group precluded assessment of mild or risk GBA variants. Larger longitudinal studies are thus needed to determine whether motor or cognitive progression rates are affected by mild or risk GBA variations. (2) All PD patients in this study underwent GBA gene testing. However, some PD patients may have common PD-related genes, such as LRRK2, PRKN, APOE, and PICALM, which may have influenced the results. Therefore, it is necessary to exclude common PD-related genes. (3) Lastly, we used MoCA for cognitive screening and did not perform detailed neuropsychological tests of each cognitive domain, as suggested by the Movement Disorder Society (Litvan et al., 2012).
This is the first longitudinal follow-up study to comprehensively assess motor and cognitive progression in Chinese GBA-PD patients. We found that GBA-PD is associated with faster motor and cognitive decline, which was specifically characterized by greater impairment in bradykinesia, axial impairment, and visuospatial/executive function. These findings have important clinical implications for understanding the role of GBA variants on patterns of motor and cognitive decline in the natural course of PD, which could help clinicians more accurately predict prognosis and design clinical trials for potential disease-modifying therapies.
The original contributions presented in this study are included in this article/Supplementary material, further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by the Ethics Committee of the Affiliated Brain Hospital of Nanjing Medical University. The patients/participants provided their written informed consent to participate in this study.
WL organized the project and critically revised the manuscript. JR organized the project, drafted the preliminary manuscript, and performed statistical analysis. GZ, YW, and RZ collected data and performed statistical analysis. ZG, HaZ, HuZ, and YS critiqued the statistical analysis and interpreted the data. CM and ML critically revised the manuscript. All authors contributed to the article and approved the submitted manuscript.
This study was funded by the National Key Research and Development Program of China, Grant/Award Numbers: 2017YFC1310302 and 2016YFC1306600; National Natural Science Foundation of China, Grant/Award Number: 81571348; and Science and Technology Program of Jiangsu Province, Grant/Award Number: BE2019611.
We thank all patients who actively participated in this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnagi.2023.1091919/full#supplementary-material
Aarsland, D., Batzu, L., Halliday, G. M., Geurtsen, G. J., Ballard, C., Ray Chaudhuri, K., et al. (2021). Parkinson disease-associated cognitive impairment. Nat. Rev. Dis. Primers 7:47. doi: 10.1038/s41572-021-00280-3
Armstrong, M. J., and Okun, M. S. (2020). Diagnosis and treatment of Parkinson disease: A review. JAMA 323, 548–560. doi: 10.1001/jama.2019.22360
Bandres-Ciga, S., Diez-Fairen, M., Kim, J. J., and Singleton, A. B. (2020). Genetics of Parkinson’s disease: An introspection of its journey towards precision medicine. Neurobiol. Dis. 137:104782. doi: 10.1016/j.nbd.2020.104782
Bloem, B. R., Okun, M. S., and Klein, C. (2021). Parkinson’s disease. Lancet 397, 2284–2303. doi: 10.1016/s0140-6736(21)00218-x
Brockmann, K., Srulijes, K., Pflederer, S., Hauser, A. K., Schulte, C., Maetzler, W., et al. (2015). GBA-associated Parkinson’s disease: Reduced survival and more rapid progression in a prospective longitudinal study. Mov. Disord. 30, 407–411. doi: 10.1002/mds.26071
Cilia, R., Tunesi, S., Marotta, G., Cereda, E., Siri, C., Tesei, S., et al. (2016). Survival and dementia in GBA-associated Parkinson’s disease: The mutation matters. Ann. Neurol. 80, 662–673. doi: 10.1002/ana.24777
Davis, A. A., Andruska, K. M., Benitez, B. A., Racette, B. A., Perlmutter, J. S., and Cruchaga, C. (2016). Variants in GBA, SNCA, and MAPT influence Parkinson disease risk, age at onset, and progression. Neurobiol. Aging 37, 209.e1–209.e7. doi: 10.1016/j.neurobiolaging.2015.09.014
Davis, M. Y., Johnson, C. O., Leverenz, J. B., Weintraub, D., Trojanowski, J. Q., Chen-Plotkin, A., et al. (2016). Association of GBA mutations and the E326K polymorphism with motor and cognitive progression in Parkinson disease. JAMA Neurol. 73, 1217–1224. doi: 10.1001/jamaneurol.2016.2245
Feigin, V. L., Nichols, E., Alam, T., Bannick, M. S., Beghi, E., Blake, N., et al. (2019). Global, regional, and national burden of neurological disorders, 1990-2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 18, 459–480. doi: 10.1016/s1474-4422(18)30499-x
Gan-Or, Z., Amshalom, I., Kilarski, L. L., Bar-Shira, A., Gana-Weisz, M., Mirelman, A., et al. (2015). Differential effects of severe vs mild GBA mutations on Parkinson disease. Neurology 84, 880–887. doi: 10.1212/wnl.0000000000001315
Gibb, W. R., and Lees, A. J. (1988). The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 51, 745–752. doi: 10.1136/jnnp.51.6.745
Hoops, S., Nazem, S., Siderowf, A. D., Duda, J. E., Xie, S. X., Stern, M. B., et al. (2009). Validity of the MoCA and MMSE in the detection of MCI and dementia in Parkinson disease. Neurology 73, 1738–1745. doi: 10.1212/WNL.0b013e3181c34b47
Horowitz, M., Wilder, S., Horowitz, Z., Reiner, O., Gelbart, T., and Beutler, E. (1989). The human glucocerebrosidase gene and pseudogene: Structure and evolution. Genomics 4, 87–96. doi: 10.1016/0888-7543(89)90319-4
Hruska, K. S., LaMarca, M. E., Scott, C. R., and Sidransky, E. (2008). Gaucher disease: Mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum. Mutat. 29, 567–583. doi: 10.1002/humu.20676
Iwaki, H., Blauwendraat, C., Leonard, H. L., Kim, J. J., Liu, G., Maple-Grødem, J., et al. (2019). Genomewide association study of Parkinson’s disease clinical biomarkers in 12 longitudinal patients’ cohorts. Mov. Disord. 34, 1839–1850. doi: 10.1002/mds.27845
Jesús, S., Huertas, I., Bernal-Bernal, I., Bonilla-Toribio, M., Cáceres-Redondo, M. T., Vargas-González, L., et al. (2016). GBA variants influence motor and non-motor features of Parkinson’s disease. PLoS One 11:e0167749. doi: 10.1371/journal.pone.0167749
Lang, A. E., and Espay, A. J. (2018). Disease modification in Parkinson’s disease: Current approaches, challenges, and future considerations. Mov. Disord. 33, 660–677. doi: 10.1002/mds.27360
Leocadi, M., Canu, E., Donzuso, G., Stojkovic, T., Basaia, S., Kresojević, N., et al. (2022). Longitudinal clinical, cognitive, and neuroanatomical changes over 5 years in GBA-positive Parkinson’s disease patients. J. Neurol. 269, 1485–1500. doi: 10.1007/s00415-021-10713-4
Li, J., Zhao, T., Zhang, Y., Zhang, K., Shi, L., Chen, Y., et al. (2018). Performance evaluation of pathogenicity-computation methods for missense variants. Nucleic Acids Res. 46, 7793–7804. doi: 10.1093/nar/gky678
Li, N., Wang, L., Zhang, J., Tan, E. K., Li, J., Peng, J., et al. (2020). Whole-exome sequencing in early-onset Parkinson’s disease among ethnic Chinese. Neurobiol. Aging 90, 150.e5–150.e11. doi: 10.1016/j.neurobiolaging.2019.12.023
Litvan, I., Goldman, J. G., Tröster, A. I., Schmand, B. A., Weintraub, D., Petersen, R. C., et al. (2012). Diagnostic criteria for mild cognitive impairment in Parkinson’s disease: Movement disorder society task force guidelines. Mov. Disord. 27, 349–356. doi: 10.1002/mds.24893
Liu, G., Boot, B., Locascio, J. J., Jansen, I. E., Winder-Rhodes, S., Eberly, S., et al. (2016). Specifically neuropathic Gaucher’s mutations accelerate cognitive decline in Parkinson’s. Ann. Neurol. 80, 674–685. doi: 10.1002/ana.24781
Lunde, K. A., Chung, J., Dalen, I., Pedersen, K. F., Linder, J., Domellöf, M. E., et al. (2018). Association of glucocerebrosidase polymorphisms and mutations with dementia in incident Parkinson’s disease. Alzheimers Dement. 14, 1293–1301. doi: 10.1016/j.jalz.2018.04.006
Malek, N., Weil, R. S., Bresner, C., Lawton, M. A., Grosset, K. A., Tan, M., et al. (2018). Features of GBA-associated Parkinson’s disease at presentation in the UK Tracking Parkinson’s study. J. Neurol. Neurosurg. Psychiatry 89, 702–709. doi: 10.1136/jnnp-2017-317348
Mao, X. Y., Burgunder, J. M., Zhang, Z. J., An, X. K., Zhang, J. H., Yang, Y., et al. (2010). Association between GBA L444P mutation and sporadic Parkinson’s disease from Mainland China. Neurosci. Lett. 469, 256–259. doi: 10.1016/j.neulet.2009.12.007
Maple-Grødem, J., Dalen, I., Tysnes, O. B., Macleod, A. D., Forsgren, L., Counsell, C. E., et al. (2021). Association of GBA genotype with motor and functional decline in patients with newly diagnosed Parkinson disease. Neurology 96, e1036–e1044.
Mata, I. F., Leverenz, J. B., Weintraub, D., Trojanowski, J. Q., Chen-Plotkin, A., Van Deerlin, V. M., et al. (2016). GBA Variants are associated with a distinct pattern of cognitive deficits in Parkinson’s disease. Mov. Disord. 31, 95–102. doi: 10.1002/mds.26359
Nasreddine, Z. S., Phillips, N. A., Bédirian, V., Charbonneau, S., Whitehead, V., Collin, I., et al. (2005). The montreal cognitive assessment, MoCA: A brief screening tool for mild cognitive impairment. J. Am. Geriatr. Soc. 53, 695–699.
Neumann, J., Bras, J., Deas, E., O’Sullivan, S. S., Parkkinen, L., Lachmann, R. H., et al. (2009). Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain 132(Pt 7), 1783–1794. doi: 10.1093/brain/awp044
Ortega, R. A., Wang, C., Raymond, D., Bryant, N., Scherzer, C. R., Thaler, A., et al. (2021). Association of dual LRRK2 G2019S and GBA variations with Parkinson disease progression. JAMA Netw. Open 4:e215845. doi: 10.1001/jamanetworkopen.2021.5845
Petrucci, S., Ginevrino, M., Trezzi, I., Monfrini, E., Ricciardi, L., Albanese, A., et al. (2020). GBA-related Parkinson’s disease: Dissection of genotype-phenotype correlates in a large italian cohort. Mov. Disord. 35, 2106–2111. doi: 10.1002/mds.28195
Ren, J., Zhang, R., Pan, C., Xu, J., Sun, H., Hua, P., et al. (2022). Prevalence and genotype-phenotype correlations of GBA-related Parkinson disease in a large Chinese cohort. Eur. J. Neurol. 29, 1017–1024. doi: 10.1111/ene.15230
Sidransky, E., Nalls, M. A., Aasly, J. O., Aharon-Peretz, J., Annesi, G., Barbosa, E. R., et al. (2009). Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med. 361, 1651–1661. doi: 10.1056/NEJMoa0901281
Skorvanek, M., Goldman, J. G., Jahanshahi, M., Marras, C., Rektorova, I., Schmand, B., et al. (2018). Global scales for cognitive screening in Parkinson’s disease: Critique and recommendations. Mov. Disord. 33, 208–218. doi: 10.1002/mds.27233
Stoker, T. B., Camacho, M., Winder-Rhodes, S., Liu, G., Scherzer, C. R., Foltynie, T., et al. (2020). Impact of GBA1 variants on long-term clinical progression and mortality in incident Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 91, 695–702. doi: 10.1136/jnnp-2020-322857
Straniero, L., Asselta, R., Bonvegna, S., Rimoldi, V., Melistaccio, G., Soldà, G., et al. (2020). The SPID-GBA study: Sex distribution, penetrance, incidence, and dementia in GBA-PD. Neurol. Genet. 6:e523. doi: 10.1212/nxg.0000000000000523
Szwedo, A. A., Dalen, I., Pedersen, K. F., Camacho, M., Bäckström, D., Forsgren, L., et al. (2022). GBA and APOE impact cognitive decline in Parkinson’s disease: A 10-year population-based study. Mov. Disord. 37, 1016–1027. doi: 10.1002/mds.28932
Tomlinson, C. L., Stowe, R., Patel, S., Rick, C., Gray, R., and Clarke, C. E. (2010). Systematic review of levodopa dose equivalency reporting in Parkinson’s disease. Mov. Disord. 25, 2649–2653. doi: 10.1002/mds.23429
Winder-Rhodes, S. E., Evans, J. R., Ban, M., Mason, S. L., Williams-Gray, C. H., Foltynie, T., et al. (2013). Glucocerebrosidase mutations influence the natural history of Parkinson’s disease in a community-based incident cohort. Brain 136(Pt 2), 392–399. doi: 10.1093/brain/aws318
Keywords: cognitive decline, GBA, longitudinal, motor impairment, Parkinson’s disease
Citation: Ren J, Zhou G, Wang Y, Zhang R, Guo Z, Zhou H, Zheng H, Sun Y, Ma C, Lu M and Liu W (2023) Association of GBA genotype with motor and cognitive decline in Chinese Parkinson’s disease patients. Front. Aging Neurosci. 15:1091919. doi: 10.3389/fnagi.2023.1091919
Received: 07 November 2022; Accepted: 23 January 2023;
Published: 10 February 2023.
Edited by:
Luca Marsili, University of Cincinnati, United StatesReviewed by:
Simona Petrucci, Sapienza University of Rome, ItalyCopyright © 2023 Ren, Zhou, Wang, Zhang, Guo, Zhou, Zheng, Sun, Ma, Lu and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weiguo Liu,  d2dsaXVuYmhAc2luYS5jb20=
d2dsaXVuYmhAc2luYS5jb20=
†ORCID: Jingru Ren, orcid.org/0000-0002-9186-6621
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.