
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Aging Neurosci. , 01 February 2023
Sec. Parkinson’s Disease and Aging-related Movement Disorders
Volume 15 - 2023 | https://doi.org/10.3389/fnagi.2023.1067954
 Arianna Manini1,2
Arianna Manini1,2 Valeria Casiraghi1,3
Valeria Casiraghi1,3 Alberto Brusati1,4Alessio Maranzano1,2
Alberto Brusati1,4Alessio Maranzano1,2 Francesco Gentile1,2Eleonora Colombo1Ruggero Bonetti1,2
Francesco Gentile1,2Eleonora Colombo1Ruggero Bonetti1,2 Silvia Peverelli1Sabrina Invernizzi1
Silvia Peverelli1Sabrina Invernizzi1 Davide Gentilini4,5Stefano Messina1
Davide Gentilini4,5Stefano Messina1 Federico Verde1,6
Federico Verde1,6 Barbara Poletti1Isabella Fogh7
Barbara Poletti1Isabella Fogh7 Claudia Morelli1
Claudia Morelli1 Vincenzo Silani1,6
Vincenzo Silani1,6 Antonia Ratti1,3†
Antonia Ratti1,3† Nicola Ticozzi1,6*†
Nicola Ticozzi1,6*†Background: The UNC13A gene is an established susceptibility locus for amyotrophic lateral sclerosis (ALS) and a determinant of shorter survival after disease onset, with up to 33.0 months difference in life expectancy for carriers of the rs12608932 risk genotype. However, its overall effect on other clinical features and ALS phenotypic variability is controversial.
Methods: Genotype data of the UNC13A rs12608932 SNP (A–major allele; C–minor allele) was obtained from a cohort of 972 ALS patients. Demographic and clinical variables were collected, including cognitive and behavioral profiles, evaluated through the Edinburgh Cognitive and Behavioral ALS Screen (ECAS) – Italian version and the Frontal Behavioral Inventory (FBI); upper and lower motor neuron involvement, assessed by the Penn Upper Motor Neuron Score (PUMNS) and the Lower Motor Neuron Score (LMNS)/Medical Research Council (MRC) scores, respectively; the ALS Functional Rating Scale Revised (ALSFRS-R) score at evaluation and progression rate; age and site of onset; survival. The comparison between the three rs12608932 genotypes (AA, AC, and CC) was performed using the additive, dominant, and recessive genetic models.
Results: The rs12608932 minor allele frequency was 0.31 in our ALS cohort, in comparison to 0.33–0.41 reported in other Caucasian ALS populations. Carriers of at least one minor C allele (AC + CC genotypes) had a shorter median survival than patients with the wild-type AA genotype (−11.7 months, p = 0.013), even after adjusting for age and site of onset, C9orf72 mutational status and gender. Patients harboring at least one major A allele (AA + AC genotypes) and particularly those with the wild-type AA genotype showed a significantly higher PUMNS compared to CC carriers (p = 0.015 and padj = 0.037, respectively), thus indicating a more severe upper motor neuron involvement. Our analysis did not detect significant associations with all the other clinical parameters considered.
Conclusion: Overall, our findings confirm the role of UNC13A as a determinant of survival in ALS patients and show the association of this locus also with upper motor neuron involvement.
Amyotrophic lateral sclerosis (ALS) is a rapidly progressive motor neuron disease (MND), which usually occurs in the sixth or seventh decades, and leads to death within 3–5 years from symptoms onset (Talbott et al., 2016). Over the last years, ALS was found to be part of a spectrum disorder defined as MND-frontotemporal dementia (FTD) continuum (Burrell et al., 2016). Indeed, 5–10% of ALS patients are diagnosed with FTD and up to 50% of ALS patients develop cognitive decline or behavioral dysfunction, albeit not fulfilling the diagnostic criteria for FTD (Burrell et al., 2016). Similarly, a significant proportion of FTD patients shows signs of motor neuron involvement.
While most ALS cases are sporadic (sporadic ALS; SALS), approximately 10% of patients show a family history (familial ALS; FALS) with mutations in more than 30 genes identified so far (Goutman et al., 2022). ALS-associated causative genes account for 60–80% FALS and 10% SALS cases (Renton et al., 2014). However, the most recurring genetic defects are found in only four genes, and are represented by hexanucleotide repeat expansions in C9orf72, followed by mutations in SOD1, TARDBP, and FUS.
In line with a multifactorial view of ALS genetic architecture in sporadic cases, large international efforts have been conducted to identify susceptibility loci by genome-wide association studies (GWAS; Renton et al., 2014). Historically, in 2009 van Es and colleagues first identified a significant association peak, mapping at chromosome 19p13.3, within the intron 20–21 of the Unc-13 Homolog A (UNC13A) gene (van Es et al., 2009). Only one common single nucleotide polymorphism (SNP), namely rs12608932, reached genome-wide significance (p = 2.53 × 10−14) in a two-stage combined analysis for a total of 4,855 ALS patients and 14,953 control subjects (van Es et al., 2009). Subsequent replication studies and colocalization analyses across neurodegenerative diseases have not only confirmed the association of this locus with ALS susceptibility (van Rheenen et al., 2016), but also revealed that UNC13A is a shared susceptibility locus for ALS and FTD (van Rheenen et al., 2016; Karch et al., 2018), especially in presence of TDP-43 proteinopathy (Diekstra et al., 2014).
Patients homozygous for the minor C allele of the UNC13A rs12608932 SNP show a reduction of survival of 5.0–33.0 months compared to the other genotypes (Diekstra et al., 2012; Chiò et al., 2013; Vidal-Taboada et al., 2015; van Rheenen et al., 2016; Yang et al., 2019; Tan et al., 2020). Furthermore, the minor allele has been associated, although not consistently, with higher age at onset, more frequent bulbar onset and reduced forced vital capacity (Diekstra et al., 2012; The ALSGEN Consortium, 2013; Chen et al., 2014; Vidal-Taboada et al., 2015; Tan et al., 2020). Only a limited number of studies have investigated the relationship of UNC13A with behavioral and cognitive features of ALS patients, highlighting an association of the minor C allele with imaging, neuropsychological and pathological markers of FTD in ALS, as well as with higher rates of reported disinhibition and behavioral impairment (Placek et al., 2019; Tan et al., 2020).
In this scenario, we combined genotype data of rs12608932 and a broad variety of clinical variables, including age and site of onset, survival, upper (UMN) and lower motor neuron (LMN) signs, functional status, disease progression, cognitive dysfunction and behavioral symptoms within a large cohort of 972 Italian ALS patients, to explore the contribution of the UNC13A locus to ALS phenotypic variability.
A total of 972 patients, affected by ALS and other motor neuron diseases (primary lateral sclerosis, PLS and progressive muscular atrophy, PMA) according to the El Escorial revised criteria (Brooks et al., 2000), were enrolled at IRCCS Istituto Auxologico Italiano between 2013 and 2022. The Ethics Committee of IRCCS Istituto Auxologico Italiano approved the study (2021_05_18). Written informed consent for employing pseudo-anonymized clinical data for research purposes was obtained from all patients at the time of evaluation. The study was conducted in accordance with the principles of the Declaration of Helsinki.
We recorded the following demographic and clinical variables: gender, age at onset and at diagnosis, site of onset, clinical phenotype, ALS Functional Rating Scale – Revised version (ALSFRS-R) score at evaluation (Cedarbaum et al., 1999), and progression rate [calculated using the formula (48 – ALSFRS-R score)/disease duration at evaluation expressed in months].
The burden of UMN signs was explored with the Penn Upper Motor Neuron Score (PUMNS; Quinn et al., 2020), while involvement of LMN was measured with a modified version of the LMN score (LMNS; Devine et al., 2016), and the Medical Research Council (MRC) muscle scale, as previously described (Maranzano et al., 2022).
We assessed the prevalence of cognitive impairment by administering the Edinburgh Cognitive and Behavioural ALS Screen (ECAS) – Italian version to a subset of 254 patients (Poletti et al., 2016). The ECAS total score is composed of subdomains investigating the ALS-specific cognitive decline (language, verbal fluency, and executive functions; ALS-specific score) and others which explore memory and visuospatial functions (ALS non-specific score). Based on the performance at ECAS and according to the Strong revised criteria, patients were classified as cognitively normal (ALScn), behaviorally impaired (ALSbi), cognitively impaired (ALSci), or both cognitively and behaviorally impaired (ALScbi; Strong et al., 2017).
In a subset of 203 patients, the presence of behavioral symptoms was explored using the ECAS Carer Interview and the Frontal Behavioral Inventory (FBI; Alberici et al., 2007). In addition to the total ECAS Carer Interview score (range 0–10), we recorded also the number of behavioral symptoms for each patient (disinhibition, apathy/inertia, loss of sympathy/empathy, perseverative/stereotyped/compulsive/ritualistic behavior and hyperorality/altered food preferences; range 0–5). As regards FBI, from 0 to 3 points can be attributed to each of 24 items (total score 0–72), which investigate both negative (FBI-A) and positive/disinhibited behaviors (FBI-B).
We employed the diagnostic criteria for behavioral variant FTD (bvFTD) and primary progressive aphasia (PPA) to define the presence of ALS/FTD (Gorno-Tempini et al., 2011; Rascovsky et al., 2011).
All patients included in the study were screened for the hexanucleotide repeat expansion in C9orf72 and for mutations in SOD1 (all 5 exons), TARDBP (exon 6) and FUS (exons 5, 6, 13, 14 and 15), as previously described (Corrado et al., 2009, 2010; Ratti et al., 2022).
For 865 ALS patients, UNC13A rs12608932 genotyping was performed with the Illumina SNP arrays as previously described (Manini et al., 2022). For the remaining 107 patients, allele-specific polymerase chain reaction (PCR) was performed using 2 primers to amplify the whole region (forward: GGGGCAGCTTACATCATCCAT; reverse: GGATGTATAGGCAGATGGACA) and 2 allele-specific primers (reference: CCACCCATCAATTTATCCAA; alternative: ACAGACGAAAAATGGATGGG). Amplicons were then resolved on 3% agarose gel to discriminate the presence of a 273-bp band (allele C) and a 178-bp band (allele A).
Statistical analyses were performed with the IBM Statistical Package for the Social Sciences (SPSS) version 26. Values were reported as medians and interquartile range (IQR) for not normally distributed quantitative variables, or frequencies (%) for categorical variables. We compared variables across the three rs12608932 genotypes by employing three different genetic models: additive (CC vs. AC vs. AA), dominant [(CC + CA) vs. AA], and recessive [CC vs. (AA + AC)], where A and C are the major and minor alleles, respectively. Chi-square or Fisher exact tests were used to compare categorical variables, as appropriate. The non-parametric Kruskal–Wallis one-way analysis of variance (ANOVA) was run to compare quantitative variables between genotypes, since their distribution was not similar for all groups, as assessed by visual inspection of boxplots, and normality assumptions for parametric tests were not met according to the Kolmogorov–Smirnov normality test. When possible, post hoc analysis was performed to compare subgroups, including pairwise comparisons using the z-test of two proportions and the (Dunn 1961) procedure with a Bonferroni correction for multiple testing. The impact of the rs12608932 SNP on survival was assessed by the univariate Kaplan–Meier survival analysis with a log-rank comparison test and multivariate Cox regression model analysis, using gender, age at onset, and site of onset as covariates. Censoring was applied for patients alive at last follow-up. p values <0.05 were considered statistically significant. In the Kaplan–Meier survival analysis, log-rank pairwise comparisons were run to determine which genotypes had different survival distributions under the additive model. A Bonferroni correction was made with statistical significance accepted at the p < 0.0167 level. Pairwise deletion was used to handle missing data.
We genotyped the UNC13A rs12608932 SNP in a cohort of 972 Italian ALS patients, with a prevalence of males (n = 616, 63.4%) over females (n = 356, 36.6%; Table 1). Only 44 patients (4.8%) had a positive family history for ALS (FALS), while most cases were sporadic (SALS). The C9orf72 repeat expansion was present in 31 patients (7 FALS, 24 SALS), whereas 10 had a mutation in TARDBP (4 FALS, 6 SALS), 3 in FUS (all SALS) and 1 in SOD1 (FALS). We were able to retrieve the age at onset of 887 patients (median 61.6 years, IQR 52.7–69.6), the survival after disease onset of 886 (median 25.9 months, IQR 14.0–45.7), and the site of onset of 903, which was bulbar in 224 cases (24.8%) and spinal in the remaining 679 (75.2%). The median ALSFRS-R score, available for 516 patients, was 40 (IQR 35–43). Median PUMNS, recorded for 767 patients, was 9 (IQR 4–15). Amongst the 765 patients for whom the LMNS was available, median score was 2 (IQR 2–6). The median MRC total score, collected for 597 patients, was 54 (IQR 47–58). ECAS and FBI were performed in 254 and 203 patients, respectively. Concerning the ECAS, the median total score was 105 (IQR 90–114), the median ALS-specific score was 78 (IQR 67–85), and the median ALS non-specific score was 27 (IQR 24–30). Based on the performance at ECAS and according to the Strong revised criteria, 112 out of 254 patients (44.1%) could be classified as ALScn, 45 (17.7%) as ALSbi, 66 (26.0%) as ALSci, and 32 (12.6%) as ALScbi. As regards FBI, the median total score was 2 (IQR 0–5), with a median A score of 1 (IQR 0–4), and a median B score of 0 (IQR 0–2). According to the Rascovsky and Gorno-Tempini criteria for bvFTD and PPA, respectively, 26 (2.7%) of our patients were affected by ALS/FTD. All the demographic and clinical features of the cohort are reported in Table 1.
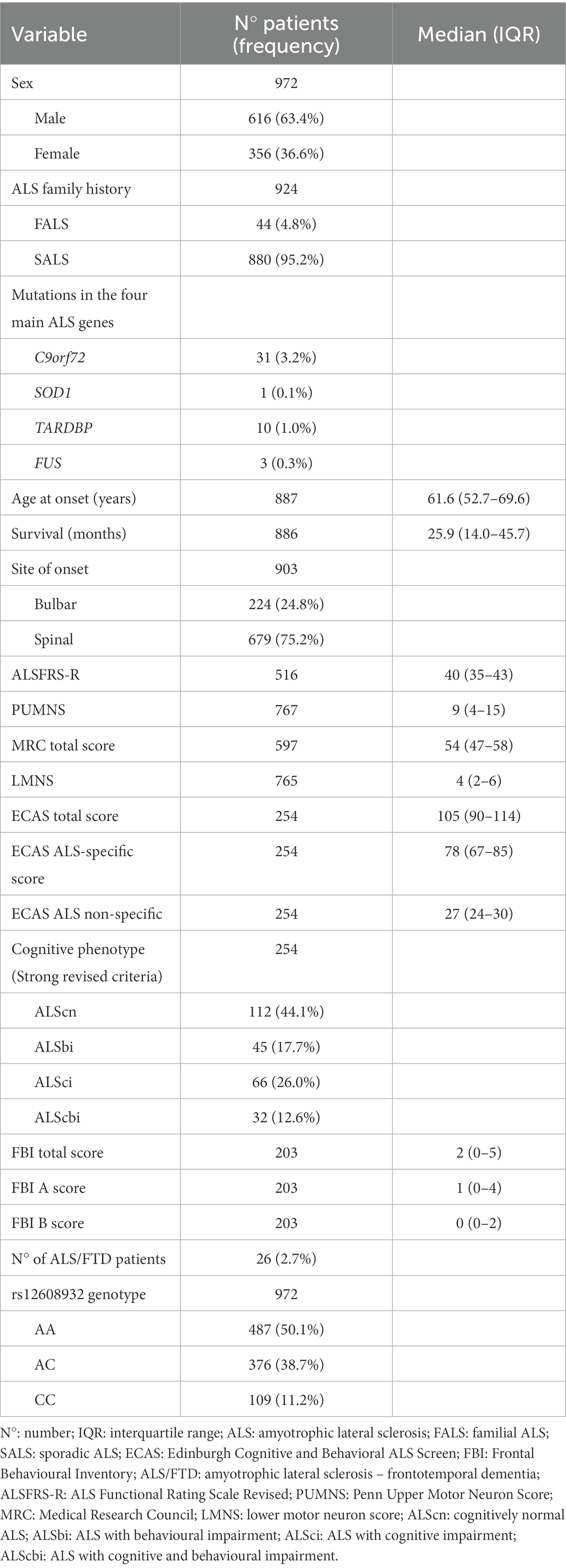
Table 1. Demographic and clinical features and genotype data of the ALS cohort.
The minor allele frequency (MAF) of the rs12608932 SNP C within our cohort was 0.31, in line with the MAF value reported in control subjects of Caucasian origin (0.29–0.36) and slightly lower compared to non-Caucasian ALS populations (0.33–0.41; Yang et al., 2019). The prevalence of the different genotypes was 50.1% (n = 487) for AA, 38.7% (n = 376) for AC, and 11.2% (n = 109) for CC (Table 1).
We found an association between rs12608932 and median survival after disease onset under the additive and dominant models. According to the additive model, we found that the median survival was 55.4 (95% CI, 47.9–63.0) in AA, 43.5 (36.1–50.7) in AC and 56.0 months (26.7–85.3) in CC patients (p = 0.025; Table 2; Figure 1A). Post hoc analysis revealed a statistically significant difference in survival distributions between the AC vs. AA carriers [χ2(1) = 7.407, p = 0.006], but not between the CC vs. AA carriers [χ2(1) = 0.335, p = 0.563], nor between the AC vs. CC carriers [χ2(1) = 0.906, p = 0.341]. The percentage of censored cases was similar between subjects harboring the AA and CC genotypes (60.9 and 61.5%, respectively), but quite lower in the AC carriers (51.9%).
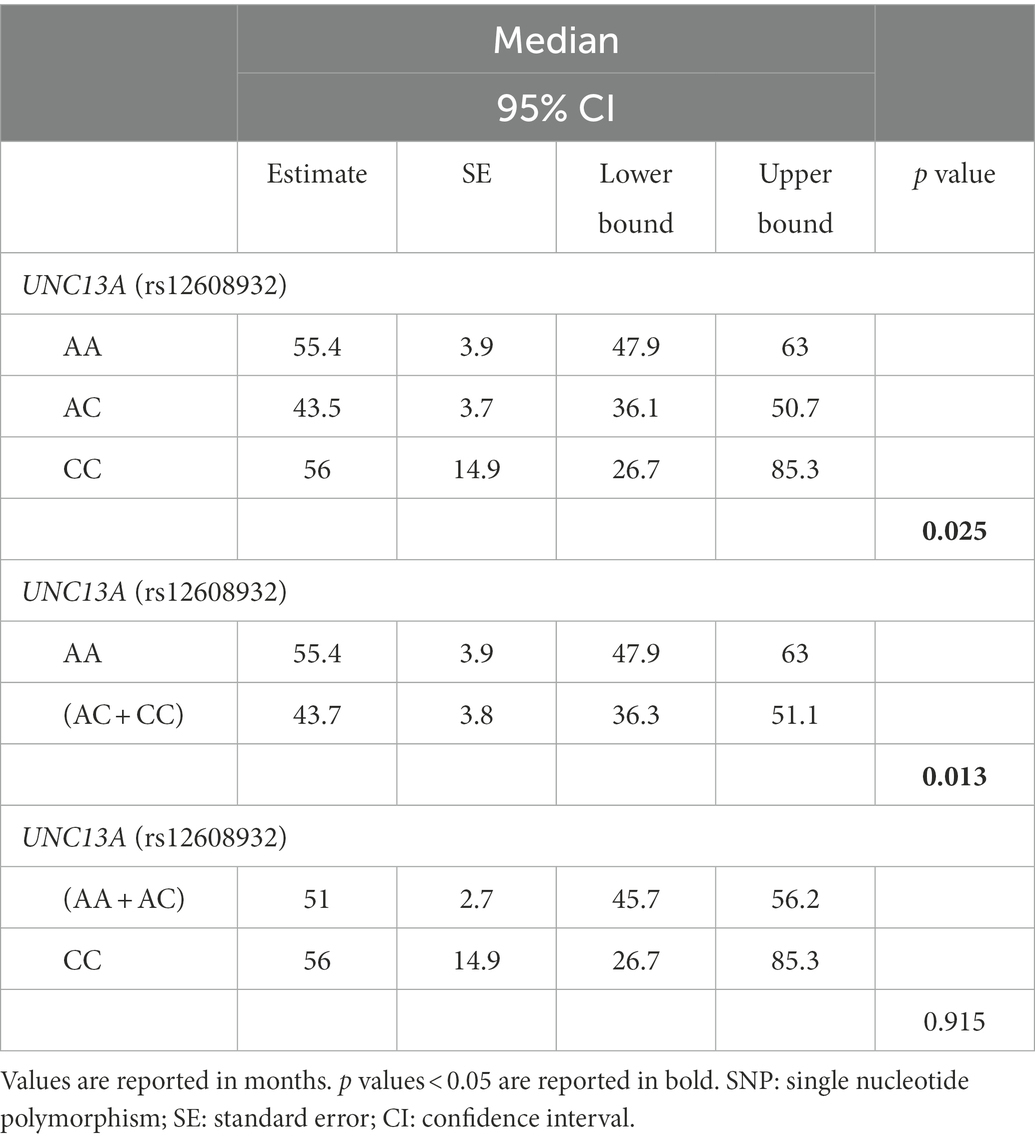
Table 2. Comparison of median survival time, and associated statistics, between carriers of the [AA vs. AC vs. CC] genotypes (additive model), [AA vs. (AC + CC)] genotypes (dominant model), and [(AA + AC) vs. CC] genotypes (recessive model) of the UNC13A rs12608932 SNP.
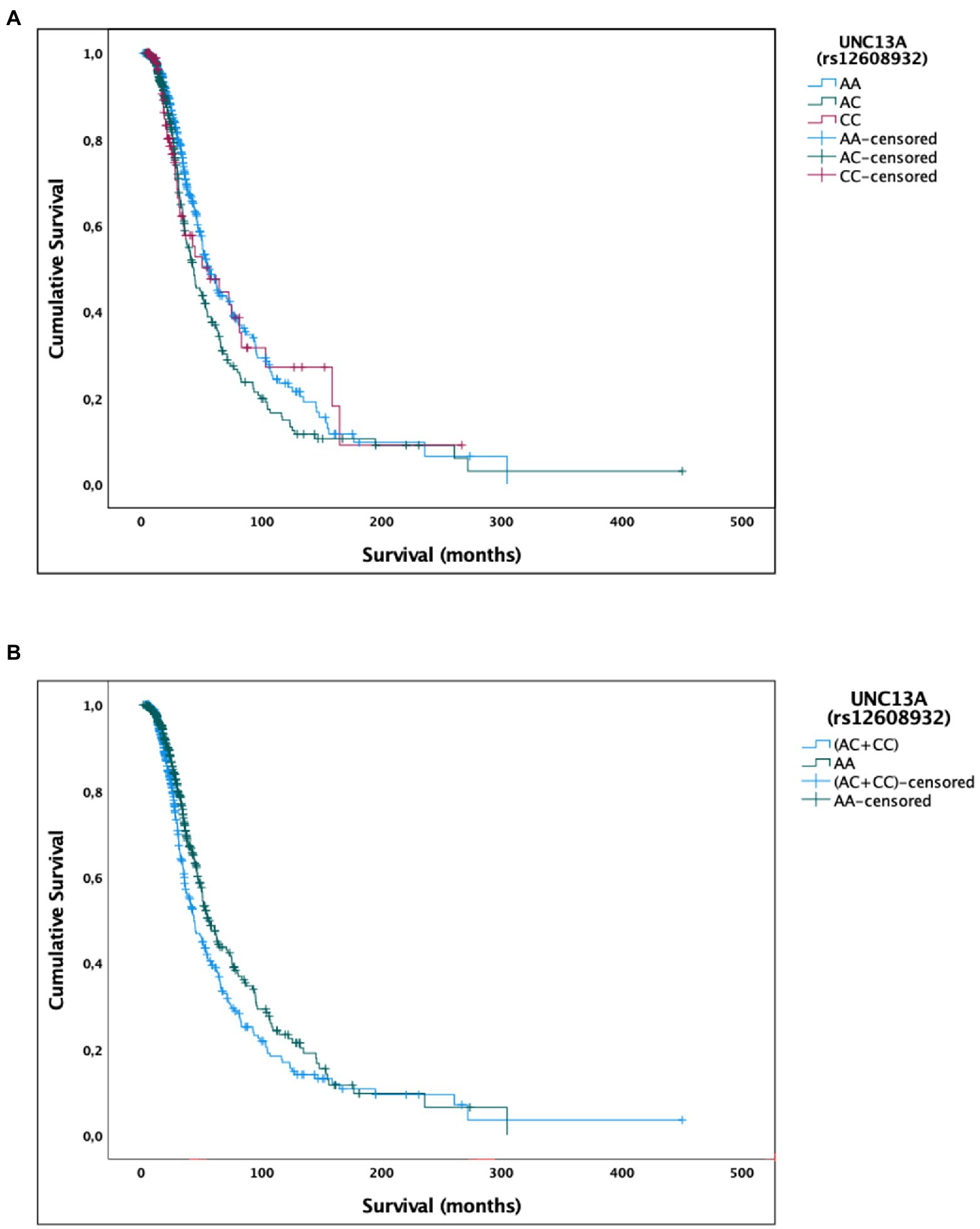
Figure 1. Univariate Kaplan–Meier survival analysis for the UNC13A rs12608932 SNP. (A) Additive model. (B) Dominant model.
As regards the dominant model, the median survival of patients harboring the CC and AC genotypes (43.7 months, 95% CI, 36.3–51.1) was significantly reduced compared to the AA carriers [55.4 months, 95% CI, 47.9–63.0 months; χ2(2) = 6.233, p = 0.013; Table 2; Figure 1B]. A similar percentage of censored cases was present in AA carriers (60.9%) and (CC + AC) genotypes (54.0%).
No statistically significant difference in median survival time was instead detected when considering the recessive model [CC vs. (AA + AC) carriers; Table 2].
A multivariate Cox regression model, including the additive model for the rs12608932 SNP, age at onset, C9orf72 mutational status, gender, and site of onset, significantly predicted survival [χ2(5) = 101.786, p < 0.001]. Among covariates, only the rs12608932 genotypes under the additive model [B = 0.219, Exp(B) = 1.245, 95% CI = 1.069–1.450, p = 0.005], the age at onset [B = 0.043, Exp(B) = 1.044, 95% CI = 1.034–1.054, p < 0.001], and the presence of a pathological repeat expansion in C9orf72 [B = −0.810, Exp(B) = 0.445, 95% CI = 0.277–0.716, p < 0.001] correlated with survival. Similarly, a Cox regression model, including the same covariates, but replacing the additive model for rs12608932 with the dominant one, significantly predicted survival [χ2(5) = 104.918, p < 0.001]. Again, the only covariates significantly associated to survival were the rs12608932 genotypes under the dominant model [B = 0.335, Exp(B) = 1.398, 95% CI = 1.135–1.721, p = 0.002], the age at onset [B = 0.042, Exp(B) = 1.043, 95% CI = 1.034–1.053, p < 0.001], and the presence of a pathological repeat expansion in C9orf72 [B = −0.784, Exp(B) = 0.457, 95% CI = 0.284–0.735, p = 0.001]. Regression coefficients and standard errors are reported in Supplementary Table 1.
We then tested possible associations with different clinical variables and scales. Among them, rs12608932 was significantly associated with the PUMNS under the additive [median value 10 (IQR 4–16) in AA vs. 9 (IQR 4–16) in AC vs. 8 (2–13) in CC, p = 0.044] and recessive models [median value 8 (2–13) in CC vs. 9 (IQR 4–16) in (AA + AC), p = 0.015; Supplementary Table 2; Figure 2]. A post hoc analysis showed that the PUMNS was significantly lower in CC carriers compared to AA carriers (p = 0.012, padjusted = 0.037; Figure 2A), whereas no significant difference subsisted between the AC and AA genotypes (p = 0.579, padjusted = 1.000) nor between the CC and AC genotypes after correction for multiple tests (p = 0.036, padjusted = 0.108).
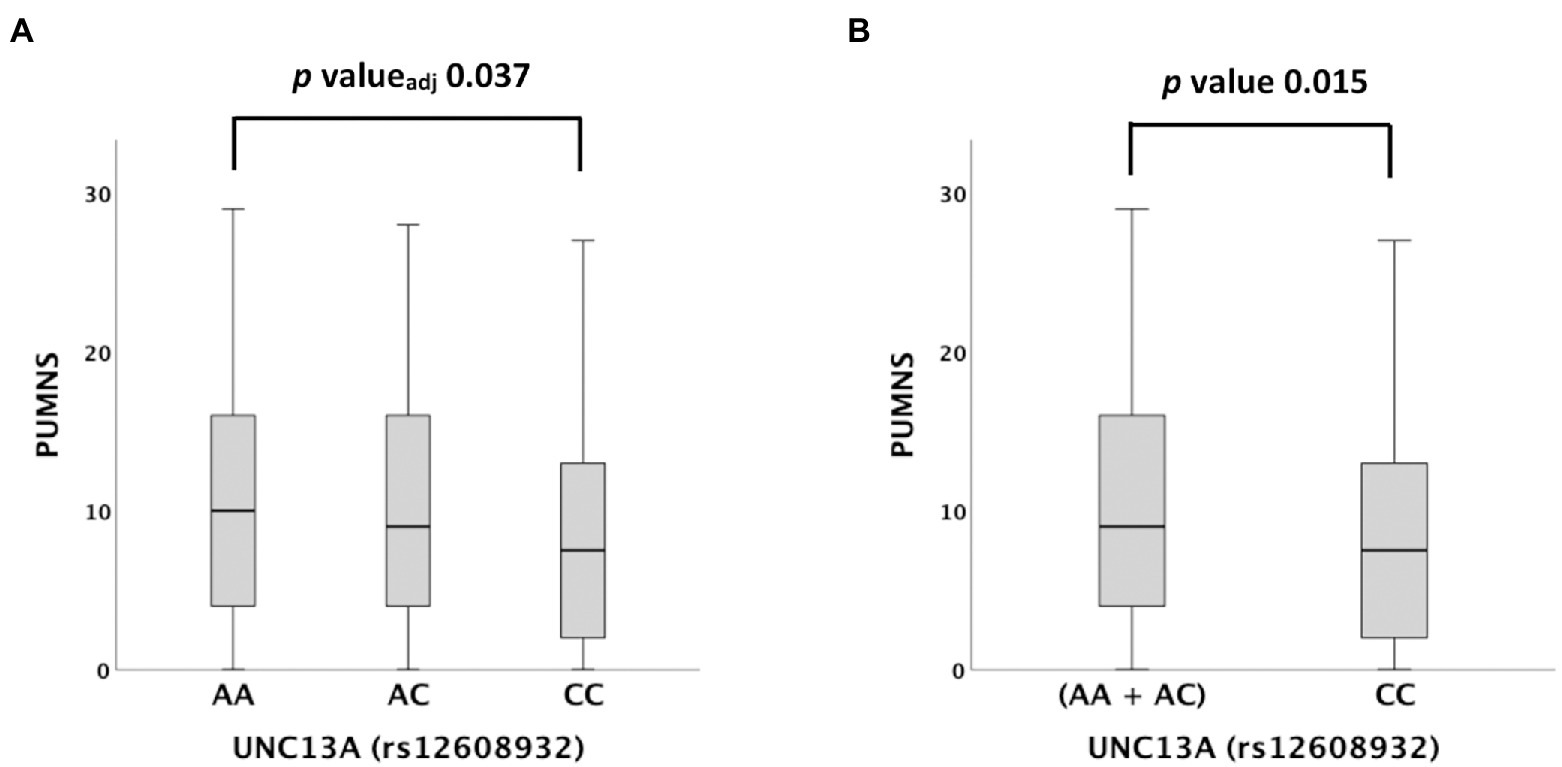
Figure 2. Distribution of PUMNS amongst UNC13A rs12608932 genotypes according to the Kruskal–Wallis one-way analysis of variance for independent samples. (A) Additive model. (B) Recessive model. For each group, the bold line shows the median, the gray box includes the middle 50% of the data and whiskers show the minimum and maximum values. Empty circles represent outliers.
No significant association was found between rs12608932 genotypes and several clinical features and parameters, including age or site of onset; ALSFRS-R and progression rate; LMNS and MRC total score; ECAS scores (total, ALS-specific and ALS non-specific); FBI scores (total, A and B); ECAS carer interview total score and number of symptoms recorded; distribution of the four ALS types according to the Strong criteria (ALScn, ALSci, ALSbi and ALScbi); prevalence of cognitively impaired vs. unimpaired patients [i.e., (ALSci + ALScbi) vs. (ALScn + ALSbi)] and of behaviorally impaired vs. unimpaired patients [i.e., (ALSbi + ALScbi) vs. (ALScn + ALSci)]; presence of ALS/FTD.
UNC13A protein is widely expressed in both the central and the peripheral nervous system and is localized at the presynaptic membrane, where it controls the release of hormones, peptides, and neurotransmitters, including glutamate (Aravamudan et al., 1999; Augustin et al., 1999). UNC13A is involved in the priming of vesicles at the presynaptic membrane, which precedes their exocytotic fusion and content release in the synaptic cleft (Zikich et al., 2008). Mice lacking Unc13a (Munc13-1) show impaired glutamatergic neurotransmission and structurally altered neuromuscular junctions (Varoqueaux et al., 2005).
The rs12608932 SNP in UNC13A gene is in linkage disequilibrium (LD) with other two SNPs, rs12973192 and rs56041637 (a CATC-repeat insertion), all mapping within intron 20–21 (Ma et al., 2022). Recently, a cryptic exon containing a premature stop codon and whose inclusion is prevented by TDP-43 splicing activity was identified in this intron (Brown et al., 2022; Ma et al., 2022). In motor and cortical-like excitatory neurons derived from induced pluripotent stem cells (iPSCs), the depletion of nuclear TDP-43 led to this cryptic exon inclusion, and, consequently, to a reduced UNC13A protein synthesis due to mRNA nonsense-mediated decay (Brown et al., 2022; Ma et al., 2022). Also in brain samples of patients affected by frontotemporal lobar degeneration with TDP-43 proteinopathy (FTLD-TDP) the inclusion of the UNC13A cryptic exon was observed (Ma et al., 2022) together with reduced UNC13A protein level (Brown et al., 2022). Furthermore, the combination of RNA-seq, SNP genotyping and real-time PCR data from post-mortem brain tissues of FTD and ALS patients revealed an association between the top risk variants of UNC13A and increased expression of the cryptic exon-containing transcripts (Brown et al., 2022; Ma et al., 2022). Also, in HEK293T cells knocked-down for TDP-43, the three UNC13A SNPs (rs12608932, rs56041637 and mainly rs12973192) correlated with an increased inclusion of UNC13A cryptic exon due to a decreased TDP-43 binding affinity to the SNP-containing RNA sequence (Ma et al., 2022). This effect was rescued by the expression of TDP-43, thus suggesting a mechanistic link between UNC13A risk alleles and TDP-43 splicing activity in ALS/FTD pathophysiology (Ma et al., 2022).
The role of UNC13A as a disease modifier in ALS has been broadly explored in the last decade. The rs12608932 minor allele C, the risk allele responsible for increased disease susceptibility (van Es et al., 2009; Diekstra et al., 2012; The ALSGEN Consortium, 2013; Vidal-Taboada et al., 2015; Yang et al., 2019), has been associated also with reduced survival (Diekstra et al., 2012; Chiò et al., 2013; Vidal-Taboada et al., 2015; van Rheenen et al., 2016; Yang et al., 2019; Tan et al., 2020; Ma et al., 2022), which was significantly increased by the treatment with lithium carbonate within three randomized clinical trials (van Eijk et al., 2017). The impact of the rs12608932 SNP on survival after ALS, but not FTD, onset was confirmed also in carriers of pathogenic repeat expansions in the C9orf72 gene (van Blitterswijk et al., 2014). In our work, we have confirmed the association of the C minor allele with reduced survival of ALS patients. Indeed, the median survival of patients harboring at least one copy of the minor allele of rs12608932 is almost 1 year shorter than homozygotes for the major one. This difference remained significant even after adjusting for possible confounding factors, including age at onset, C9orf72 mutational status, gender, and site of onset. However, when we ran log-rank pairwise comparisons in the Kaplan–Meier survival analysis to determine which genotypes had different survival distributions under the additive model, we found a significant difference in survival distributions only between the AC vs. AA carriers, but not between the CC vs. AA carriers. Since the 95% CI and the standard error of median survival of CC carriers, who were a minority (11.2%), were far higher than in the other genotypes, it is possible that the results of the post hoc analysis might have been influenced by the elevated rate of variability of estimated survival within this group of patients.
An association between the rs12608932 risk allele C and higher age at onset, more frequent bulbar onset and reduced forced vital capacity at diagnosis has been reported in ALS (The ALSGEN Consortium, 2013; Tan et al., 2020), albeit inconsistently (Diekstra et al., 2012; Chen et al., 2014; Vidal-Taboada et al., 2015). According to the ALSFRS-R, patients who were homozygous for the major allele (AA) showed slower progression of symptoms compared to the other genotypes (Vidal-Taboada et al., 2015). In subsequent works, however, either the homozygotes for the major allele (AA) showed the lowest ALSFRS-R compared to the other rs12608932 genotypes (Placek et al., 2019), or no association between the ALSFRS-R and the rs12608932 was found at all (Tan et al., 2020), so that the definite association with this clinical parameter is not clear. Unlike previous works, however, we detected no association of the minor allele C with higher age at onset, bulbar onset and ALSFRS-R, as well as with the presence of clinical LMN signs assessed by LMNS and MRC total score.
Conversely, we found a previously undescribed association of rs12608932 with UMN involvement. Specifically, we showed that patients who are homozygous for the minor allele (CC genotype) have a significantly reduced burden of UMN signs compared to the carriers of the two other possible genotypes, especially compared to those homozygous for the major allele (AA genotype), thus suggesting a “protective” role of the minor C allele against UMN involvement. Noteworthy, the presence of the minor allele, which is associated with reduced survival, does not correlate with the UMN involvement. This finding is in line with current literature, according to which the UMN scores are scarcely associated with survival (Devine et al., 2016).
In fact, the assessment of UMN involvement in ALS might be tricky, due to the simultaneous coexistence of muscle atrophy and LMN signs (Huynh et al., 2016). While the investigation of LMN degeneration is strongly supported by neurophysiological findings, evaluation of UMN involvement mostly relies on clinical clues. In this scenario, the PUMNS has proved to be an efficient, easy and reliable tool to quantify the burden of UMN deterioration in ALS (Quinn et al., 2020). In addition to that, different neurophysiological and imaging biomarkers of corticomotoneuronal pathology have been developed in the last decades, including the increased susceptibility skewness of the precentral cortex through quantitative susceptibility mapping algorithms, which showed a significant correlation with PUMNS (Contarino et al., 2020). Therefore, future studies should extend our genotype–phenotype analysis to additional variables derived from an extensive electrophysiological and neuroimaging assessment, which might take into account early features of motor neuron degeneration.
To date, few works have instead assessed the role of UNC13A risk allele on modulating behavioral and cognitive profiles in ALS (Placek et al., 2019; Tan et al., 2020). The rs12608932 minor allele has been associated with imaging, neuropsychological and pathological markers of FTD in ALS, including: (i) significantly reduced cortical thickness in dorsal/ventromedial prefrontal, and anterior/middle temporal areas (Placek et al., 2019; Tan et al., 2020); (ii) lower scores at the reverse digit span test, which mainly evaluates working memory, and at the ECAS, especially concerning the language subdomain (Placek et al., 2019; Tan et al., 2020); (iii) higher rates of behavioral disturbances (Tan et al., 2020); (iv) widespread TDP-43 pathology in middle frontal/temporal and motor cortex (Placek et al., 2019). Here, in our ALS cohort, we did not replicate previous findings regarding the correlation between rs12608932 and the performance at ECAS (total, ALS-specific and ALS non-specific scores). Furthermore, Tan and colleagues demonstrated a higher prevalence of patients classified as ALS-bi and as ALS-FTD, as well as an higher frequency of disinhibition in carriers of the minor allele C (Tan et al., 2020). Within our cohort, instead, we did not detect a different distribution of patients classified according to the Strong revised criteria or a different prevalence of ALS/FTD patients amongst the rs12608932 genotypes under any of the genetic models considered. Further, no significant differences in behavioral symptoms, recorded through the ECAS carer interview and the FBI, emerged.
We recognize that our work has some limitations, including the fact that employing pairwise deletions to handle missing data might have resulted in biased estimates. However, we must point out that the proportion of missing data within our cohort was relatively low. In addition, the ECAS, ECAS Carer Interview and FBI scales were administered only to a minority of patients.
Overall, our findings confirm that UNC13A plays a key role in influencing survival after ALS onset and, for the first time, reveal a strong correlation of this locus with UMN involvement. The identification of disease modifiers in ALS is pivotal. In this scenario, an extensive phenotyping of patients, achieved by the simultaneous employment of clinical scales and neuroimaging, neurophysiological and laboratory findings, properly correlated with genotype data, might be helpful in the definition of ALS subgroups, which should guide the follow-up of patients and, hopefully, future clinical trials design.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: www.zenodo.org, 10.5281/zenodo.7128674.
The studies involving human participants were reviewed and approved by IRCCS Istituto Auxologico Italiano. The patients/participants provided their written informed consent to participate in this study.
AMan, AR, AB, and NT: conceptualization. AMan, VC, AB, DG, SP, SI, and NT: methodology. AMan, AB, AMar, and NT: formal analysis. AMan, AMar, SM, FV, BP, CM, and NT: investigation. AR, IF, VS, and NT: resources. AMan, AB, AMar, FG, EC, and NT: data curation. AMan: writing – original draft preparation. AMan, AR, and NT: writing – review and editing. AR, VS, and NT: supervision. AR and NT: project administration. VS, AR, and NT: funding acquisition. All authors contributed to the article and approved the submitted version.
This work was financially supported by the Italian Ministry of Health (Ricerca Corrente to IRCCS Istituto Auxologico Italiano).
The authors would thank all the patients who participated in this study.
VS received compensation for consulting services and/or speaking activities from AveXis, Cytokinetics, Italfarmaco, Liquidweb Srl, Novartis Pharma AG, and Zambon. He is on the Editorial Board of Amyotrophic Lateral Sclerosis and Frontotemporal Degeneration, European Neurology, American Journal of Neurodegenerative Diseases, Frontiers in Neurology, and Exploration of Neuroprotective Therapy. FV and CM are Review Editor of Frontiers in Aging Neuroscience. BP received compensation for consulting services and/or speaking activities from Liquidweb S.r.l. She is Associate Editor for Frontiers in Neuroscience. NT received compensation for consulting services from Amylyx Pharmaceuticals and Zambon Biotech SA. He is Associate Editor for Frontiers in Aging Neuroscience.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnagi.2023.1067954/full#supplementary-material
Alberici, A., Geroldi, C., Cotelli, M., Adorni, A., Calabria, M., Rossi, G., et al. (2007). The frontal behavioural inventory (Italian version) differentiates frontotemporal lobar degeneration variants from Alzheimer’s disease. Neurol. Sci. 28, 80–86. doi: 10.1007/s10072-007-0791-3
Aravamudan, B., Fergestad, T., Davis, W. S., Rodesch, C. K., and Broadie, K. (1999). Drosophila Unc-13 is essential for synaptic transmission. Nat. Neurosci. 2, 965–971. doi: 10.1038/14764
Augustin, I., Rosenmund, C., Südhof, T. C., and Brose, N. (1999). Munc13-1 is essential for fusion competence of glutamatergic synaptic vesicles. Nature 400, 457–461. doi: 10.1038/22768
Brooks, B. R., Miller, R. G., Swash, M., and Munsat, T. L. (2000). El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 1, 293–299. doi: 10.1080/146608200300079536
Brown, A.-L., Wilkins, O. G., Keuss, M. J., Hill, S. E., Zanovello, M., Lee, W. C., et al. (2022). TDP-43 loss and ALS-risk SNPs drive mis-splicing and depletion of UNC13A. Nature 603, 131–137. doi: 10.1038/s41586-022-04436-3
Burrell, J. R., Halliday, G. M., Kril, J. J., Ittner, L. M., Götz, J., Kiernan, M. C., et al. (2016). The frontotemporal dementia-motor neuron disease continuum. Lancet 388, 919–931. doi: 10.1016/S0140-6736(16)00737-6
Cedarbaum, J. M., Stambler, N., Malta, E., Fuller, C., Hilt, D., Thurmond, B., et al. (1999). The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. J. Neurol. Sci. 169, 13–21. doi: 10.1016/S0022-510X(99)00210-5
Chen, X., Huang, R., Chen, Y., Zheng, Z., Chen, K., Song, W., et al. (2014). Association analysis of four candidate genetic variants with sporadic amyotrophic lateral sclerosis in a Chinese population. Neurol. Sci. 35, 1089–1095. doi: 10.1007/s10072-014-1656-1
Chiò, A., Mora, G., Restagno, G., Brunetti, M., Ossola, I., Barberis, M., et al. (2013). UNC13A influences survival in Italian amyotrophic lateral sclerosis patients: a population-based study. Neurobiol. Aging 34, 357.e1–357.e5. doi: 10.1016/j.neurobiolaging.2012.07.016
Contarino, V. E., Conte, G., Morelli, C., Trogu, F., Scola, E., Calloni, S. F., et al. (2020). Toward a marker of upper motor neuron impairment in amyotrophic lateral sclerosis: a fully automatic investigation of the magnetic susceptibility in the precentral cortex. Eur. J. Radiol. 124:108815. doi: 10.1016/j.ejrad.2020.108815
Corrado, L., Del Bo, R., Castellotti, B., Ratti, A., Cereda, C., Penco, S., et al. (2010). Mutations of FUS gene in sporadic amyotrophic lateral sclerosis. J. Med. Genet. 47, 190–194. doi: 10.1136/jmg.2009.071027
Corrado, L., Ratti, A., Gellera, C., Buratti, E., Castellotti, B., Carlomagno, Y., et al. (2009). High frequency of TARDBP gene mutations in Italian patients with amyotrophic lateral sclerosis. Hum. Mutat. 30, 688–694. doi: 10.1002/humu.20950
Devine, M. S., Ballard, E., O’Rourke, P., Kiernan, M. C., Mccombe, P. A., and Henderson, R. D. (2016). Targeted assessment of lower motor neuron burden is associated with survival in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 17, 184–190. doi: 10.3109/21678421.2015.1125502
Diekstra, F. P., Van Deerlin, V. M., van Swieten, J. C., Al-Chalabi, A., Ludolph, A. C., Weishaupt, J. H., et al. (2014). C9orf72 and UNC13A are shared risk loci for amyotrophic lateral sclerosis and frontotemporal dementia: a genome-wide meta-analysis. Ann. Neurol. 76, 120–133. doi: 10.1002/ana.24198
Diekstra, F. P., van Vught, P. W. J., van Rheenen, W., Koppers, M., Pasterkamp, R. J., van Es, M. A., et al. (2012). UNC13A is a modifier of survival in amyotrophic lateral sclerosis. Neurobiol. Aging 33, 630.e3–630.e8. doi: 10.1016/j.neurobiolaging.2011.10.029
Dunn, O. J. (1961). Multiple Comparisons among Means. J. Am. Stat. Assoc. 56, 52–64. doi: 10.1080/01621459.1961.10482090
Gorno-Tempini, M. L., Hillis, A. E., Weintraub, S., Kertesz, A., Mendez, M., Cappa, S. F., et al. (2011). Classification of primary progressive aphasia and its variants. Neurology 76, 1006–1014. doi: 10.1212/WNL.0b013e31821103e6
Goutman, S. A., Hardiman, O., Al-Chalabi, A., Chiò, A., Savelieff, M. G., Kiernan, M. C., et al. (2022). Emerging insights into the complex genetics and pathophysiology of amyotrophic lateral sclerosis. Lancet Neurol. 21, 465–479. doi: 10.1016/S1474-4422(21)00414-2
Huynh, W., Simon, N. G., Grosskreutz, J., Turner, M. R., Vucic, S., and Kiernan, M. C. (2016). Assessment of the upper motor neuron in amyotrophic lateral sclerosis. Clin. Neurophysiol. 127, 2643–2660. doi: 10.1016/j.clinph.2016.04.025
Karch, C. M., Wen, N., Fan, C. C., Yokoyama, J. S., Kouri, N., Ross, O. A., et al. (2018). Selective genetic overlap between amyotrophic lateral sclerosis and diseases of the frontotemporal dementia spectrum. JAMA Neurol. 75, 860–875. doi: 10.1001/jamaneurol.2018.0372
Ma, X. R., Prudencio, M., Koike, Y., Vatsavayai, S. C., Kim, G., Harbinski, F., et al. (2022). TDP-43 represses cryptic exon inclusion in the FTD–ALS gene UNC13A. Nature 603, 124–130. doi: 10.1038/s41586-022-04424-7
Manini, A., Ratti, A., Brusati, A., Maranzano, A., Fogh, I., Peverelli, S., et al. (2022). TMEM106B acts as a modifier of cognitive and motor functions in amyotrophic lateral sclerosis. Int. J. Mol. Sci. 23:9276. doi: 10.3390/ijms23169276
Maranzano, A., Poletti, B., Solca, F., Torre, S., Colombo, E., Faré, M., et al. (2022). Upper motor neuron dysfunction is associated with the presence of behavioural impairment in patients with amyotrophic lateral sclerosis. Eur. J. Neurol. 29, 1402–1409. doi: 10.1111/ene.15243
Placek, K., Baer, G. M., Elman, L., McCluskey, L., Hennessy, L., Ferraro, P. M., et al. (2019). UNC13A polymorphism contributes to frontotemporal disease in sporadic amyotrophic lateral sclerosis. Neurobiol. Aging 73, 190–199. doi: 10.1016/j.neurobiolaging.2018.09.031
Poletti, B., Solca, F., Carelli, L., Madotto, F., Lafronza, A., Faini, A., et al. (2016). The validation of the Italian Edinburgh cognitive and behavioural ALS screen (ECAS). Amyotroph. Lateral Scler. Front. Degener. 17, 489–498. doi: 10.1080/21678421.2016.1183679
Quinn, C., Edmundson, C., Dahodwala, N., and Elman, L. (2020). Reliable and efficient scale to assess upper motor neuron disease burden in amyotrophic lateral sclerosis. Muscle Nerve 61, 508–511. doi: 10.1002/mus.26764
Rascovsky, K., Hodges, J. R., Knopman, D., Mendez, M. F., Kramer, J. H., Neuhaus, J., et al. (2011). Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 134, 2456–2477. doi: 10.1093/brain/awr179
Ratti, A., Peverelli, S., D’Adda, E., Colombrita, C., Gennuso, M., Prelle, A., et al. (2022). Genetic and epigenetic disease modifiers in an Italian C9orf72 family expressing ALS, FTD or PD clinical phenotypes. Amyotroph. Lateral Scler. Frontotemporal Degener. 23, 292–298. doi: 10.1080/21678421.2021.1962355
Renton, A. E., Chiò, A., and Traynor, B. J. (2014). State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 17, 17–23. doi: 10.1038/nn.3584
Strong, M. J., Abrahams, S., Goldstein, L. H., Woolley, S., Mclaughlin, P., Snowden, J., et al. (2017). Amyotrophic lateral sclerosis - frontotemporal spectrum disorder (ALS-FTSD): revised diagnostic criteria. Amyotroph. Lateral Scler. Front. Degener. 18, 153–174. doi: 10.1080/21678421.2016.1267768
Talbott, E. O., Malek, A. M., and Lacomis, D. (2016). The epidemiology of amyotrophic lateral sclerosis. Handb. Clin. Neurol. 138, 225–238. doi: 10.1016/B978-0-12-802973-2.00013-6
Tan, H. H. G., Westeneng, H., Burgh, H. K., Es, M. A., Bakker, L. A., Veenhuijzen, K., et al. (2020). The distinct traits of the UNC13A polymorphism in amyotrophic lateral sclerosis. Ann. Neurol. 88, 796–806. doi: 10.1002/ana.25841
The ALSGEN Consortium (2013). Age of onset of amyotrophic lateral sclerosis is modulated by a locus on 1p34.1. Neurobiol. Aging 34, 357.e7–357.e19. doi: 10.1016/j.neurobiolaging.2012.07.017
van Blitterswijk, M., Mullen, B., Wojtas, A., Heckman, M. G., Diehl, N. N., Baker, M. C., et al. (2014). Genetic modifiers in carriers of repeat expansions in the C9ORF72 gene. Mol. Neurodegener. 9:38. doi: 10.1186/1750-1326-9-38
van Eijk, R. P. A., Jones, A. R., Sproviero, W., Shatunov, A., Shaw, P. J., Leigh, P. N., et al. (2017). Meta-analysis of pharmacogenetic interactions in amyotrophic lateral sclerosis clinical trials. Neurology 89, 1915–1922. doi: 10.1212/WNL.0000000000004606
van Es, M. A., Veldink, J. H., Saris, C. G. J., Blauw, H. M., van Vught, P. W. J., Birve, A., et al. (2009). Genome-wide association study identifies 19p13.3 (UNC13A) and 9p21.2 as susceptibility loci for sporadic amyotrophic lateral sclerosis. Nat. Genet. 41, 1083–1087. doi: 10.1038/ng.442
van Rheenen, W., Shatunov, A., Dekker, A. M., McLaughlin, R. L., Diekstra, F. P., Pulit, S. L., et al. (2016). Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat. Genet. 48, 1043–1048. doi: 10.1038/ng.3622
Varoqueaux, F., Sons, M. S., Plomp, J. J., and Brose, N. (2005). Aberrant morphology and residual transmitter release at the Munc13-deficient mouse neuromuscular synapse. Mol. Cell. Biol. 25, 5973–5984. doi: 10.1128/MCB.25.14.5973-5984.2005
Vidal-Taboada, J. M., Lopez-Lopez, A., Salvado, M., Lorenzo, L., Garcia, C., Mahy, N., et al. (2015). UNC13A confers risk for sporadic ALS and influences survival in a Spanish cohort. J. Neurol. 262, 2285–2292. doi: 10.1007/s00415-015-7843-z
Yang, B., Jiang, H., Wang, F., Li, S., Wu, C., Bao, J., et al. (2019). UNC13A variant rs12608932 is associated with increased risk of amyotrophic lateral sclerosis and reduced patient survival: a meta-analysis. Neurol. Sci. 40, 2293–2302. doi: 10.1007/s10072-019-03951-y
Keywords: amyotrophic lateral sclerosis, UNC13A, alleles, genotype, motor neurons, behavioral symptoms
Citation: Manini A, Casiraghi V, Brusati A, Maranzano A, Gentile F, Colombo E, Bonetti R, Peverelli S, Invernizzi S, Gentilini D, Messina S, Verde F, Poletti B, Fogh I, Morelli C, Silani V, Ratti A and Ticozzi N (2023) Association of the risk factor UNC13A with survival and upper motor neuron involvement in amyotrophic lateral sclerosis. Front. Aging Neurosci. 15:1067954. doi: 10.3389/fnagi.2023.1067954
Edited by:
Michael Swash, Queen Mary University of London, United KingdomReviewed by:
Emanuele Buratti, International Centre for Genetic Engineering and Biotechnology, ItalyCopyright © 2023 Manini, Casiraghi, Brusati, Maranzano, Gentile, Colombo, Bonetti, Peverelli, Invernizzi, Gentilini, Messina, Verde, Poletti, Fogh, Morelli, Silani, Ratti and Ticozzi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicola Ticozzi, ✉ bi50aWNvenppQGF1eG9sb2dpY28uaXQ=
†These authors share co-senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.