 Qian Zeng1Hongxu Pan1Yuwen Zhao1
Qian Zeng1Hongxu Pan1Yuwen Zhao1 Yige Wang1
Yige Wang1 Qian Xu1
Qian Xu1 Jieqiong Tan2Xinxiang Yan1
Jieqiong Tan2Xinxiang Yan1 Jinchen Li3,4,5
Jinchen Li3,4,5 Beisha Tang1,2,3,4
Beisha Tang1,2,3,4 Jifeng Guo1,2,3,4,6,7*
Jifeng Guo1,2,3,4,6,7*- 1Department of Neurology, Xiangya Hospital, Central South University, Changsha, China
- 2Key Laboratory of Hunan Province in Neurodegenerative Disorders, Central South University, Changsha, China
- 3Bioinformatics Center & National Clinical Research Center for Geriatric Disorders, Xiangya Hospital, Central South University, Changsha, China
- 4Center for Medical Genetics & Hunan Key Laboratory of Medical Genetics, School of Life Sciences, Central South University, Changsha, China
- 5Department of Geriatrics, Xiangya Hospital, Central South University, Changsha, China
- 6Hunan International Scientific and Technological Cooperation Base of Neurodegenerative and Neurogenetic Diseases, Changsha, China
- 7Engineering Research Center of Hunan Province in Cognitive Impairment Disorders, Central South University, Changsha, China
Objective: Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) is a hereditary cerebral small vessel disease caused by mutations in the NOTCH3 gene. Previous studies have established a link between NOTCH3 variants and Parkinson’s disease (PD) in terms of neuropathology and clinical characteristics. In this study, we aimed to explore the role of NOTCH3 gene in PD in a large Chinese cohort.
Methods: A total of 1,917 patients with early-onset or familial PD and 1,652 matched controls were included. All variants were divided into common or rare types by minor allele frequency (MAF) at a threshold of 0.01 (MAF > 0.01 into common variants and others into rare variants). Common variants were subjected to single-variant tests by PLINK, then gene-based analyses were used for rare variants with the optimized sequence kernel association test (SKAT-O). For genotype–phenotype correlation assessment, regression models were conducted to compare clinical features between the studied groups.
Results: Three common variants (rs1044006, rs1043997, and rs1043994) showed a nominal protective effect against PD. However, none of these SNPs survived Bonferroni correction. The results in the validation cohort revealed a significant but opposite association between these variants and PD. The gene-based analyses of rare variants showed no significant associations of NOTCH3 with PD. Although we did not find significant associations in the following genotype–phenotype analysis, the higher clinical scores of motor symptoms in NOTCH3-variant carriers were of interest.
Conclusion: Our results indicated that NOTCH3 gene may not play an important role in the early-onset or familial PD of Chinese population.
Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disease. While the etiology of PD is not fully understood, environmental factors, aging, and genetic factors are often reported as triggers. A recent study has reported an overall frequency of known PD gene mutations of 1.4% in a large United Kingdom population-based cohort (Tan et al., 2019). Interestingly, marked heritability can be observed in some PD families without known genetic factors. Using genome-wide complex trait analysis for PD, it is estimated that only 27% of PD heritability is accounted for (Keller et al., 2012). Apart from known PD genes, it is worth exploring unknown genetic factors contributing to PD.
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), the most common hereditary cerebral small vessel disease (CSVD), is caused by pathogenic mutations in NOTCH3 located on chromosome 19p13 (Joutel et al., 1996). It is clinically characterized by recurrent strokes, progressive subcortical dementia, migraine and psychiatric conditions (Dichgans et al., 1998). The NOTCH3 gene encodes a single-pass transmembrane protein in smooth muscle cells and pericytes (Joutel et al., 2000). The extracellular domain of the protein contains 34 epidermal growth factor (EGF)-like repeats. Each EGF-like repeat (EGFr) domain comprises six conserved cysteine residues that form three disulfide bonds. Virtually all CADASIL mutations lead to the loss or addition of a cysteine residue and therefore to an odd number of cysteine residues within a given EGFr domain (Joutel et al., 1997).
As far as PD is concerned, the inclusions of aggregated misfolded a-synuclein, also known as Lewy bodies, play a significant role in the PD neurodegenerative process (Marogianni et al., 2020). CADASIL shares some similarities with PD in the pathology of proteinopathies. Several studies on CADASIL have exhibited dramatic abnormal protein accumulation in the vascular media of penetrating arteries (Monet-Lepretre et al., 2013; Zhang et al., 2015). Protein accumulation includes the conformational change of NOTCH3 may result from vascular redox changes (Zhang et al., 2014). Recent work has implicated that overlapping molecular mechanisms may drive PD and CADASIL (Young et al., 2020). For example, dopamine is known to play a role in the formation of multimerization of synuclein (Planchard et al., 2014; Mor et al., 2019). Dopamine and norepinephrine can also form conjugates with the NOTCH3 N-terminal fragment, which facilitate multimerization of the protein (Young et al., 2020). In 2003, Van Gerpen et al. reported a case of parkinsonism diagnosed as CADASIL for the first time (Van Gerpen et al., 2003). A later study described a patient diagnosed as CADASIL by skin biopsy, presenting with parkinsonism (Wegner et al., 2007). More and more studies have confirmed that the parkinsonian phenotype may represent one of the CADASIL manifestations (Valenti et al., 2011; Ragno et al., 2013; Erro et al., 2014; Guo et al., 2021). A recent analysis on the magnetic resonance imaging of 139 PD patients demonstrated that rare NOTCH3 variants might be involved in the neuropathology associated with idiopathic PD (Ramirez et al., 2020). However, the contribution of the NOTCH3 variants to the risk and clinical characteristics of PD has not been systematically described.
The pace of genetic discovery in complex diseases has accelerated exponentially for decades. In the last dozen years, many studies have been working on the genetic risk of PD. Multiple new technologies, such as next-generation sequencing (NGS) and single-cell RNA sequencing, have emerged to help with understanding the genetic architecture of disease. Whole-exome sequencing (WES) is a powerful tool to detect genes causative of multifactorial diseases, including PD. Therefore, to evaluate the association between NOTCH3 and PD, we performed WES in a Chinese cohort and explored the clinical features between variant carriers and non-carriers.
Materials and methods
Study subjects
PD patients with age at onset (AAO) no more than 50 years or with a family history of PD were recruited from Xiangya Hospital and other cooperating centers of Parkinson’s Disease & Movement Disorders Multicenter Database and Collaborative Network in China (PD-MDCNC, http://pd-mdcnc.com). The diagnoses were confirmed by experienced movement disorder specialists according to the United Kingdom Parkinson’s Disease Society Brain Bank criteria (Hughes et al., 1992) or Movement Disorders Society (MDS) clinical diagnostic criteria (Postuma et al., 2015). Patients underwent a comprehensive neurological assessment. Demographic and clinical data, including the age, AAO, disease duration, as well as motor and non-motor manifestations, were retrieved from the PD-MDCNC database. Controls of Chinese ancestry enrolled from communities had no neurological or psychiatric system diseases. Written informed consents were obtained from all participants, and this study was approved by the Ethics Committee of Xiangya Hospital (Central South University). DNA of PD patients was isolated from peripheral blood leukocytes.
Whole-exome sequencing analysis
As described in our previous study (Zeng et al., 2022), WES was performed and quality control was conducted. The Agilent SureSelect Human All Exon V6 Kit was used for exome capture. Paired-end sequencing was generated by the Illumina X10 platform with an average coverage of 123 times. The Burrows-Wheeler Aligner (BWA) was used to align sequence reads to the human reference genome (hg19; Li and Durbin, 2009). PCR duplicates were removed with Picard tools.1 Filtered alignments were further processed to improve the alignment quality, including base quality-score recalibration (BQSR) and local realignment around insertions/deletions (indels) using the Genome Analysis Toolkit (GATK; McKenna et al., 2010). ANNOVAR (Yang and Wang, 2015) and VarCards (Li et al., 2018a) were used to annotate gene regions (RefSeq, hg19), amino acid alterations, functional consequences, and allele frequencies in the East Asian population [the Genome Aggregation Database (gnomAD), Exome Aggregation Consortium (ExAC) database] of genetic variation from exome sequencing data. ReVe (Li et al., 2018b) was used to predict the effects of amino acid changes on protein functional status and the threshold was set to 0.7.
PLINK 1.90 (Christopher Chang/Grail Inc; Chang et al., 2015) was used for quality control. To ensure analysis of high-fidelity variants, variants were filtered to exclude those with allele depth < 5, a read depth < 10, or genotype quality score < 20. We also removed variants with a missing rate >0.1, or variants deviated significantly (p < 0.0001) from Hardy–Weinberg equilibrium. Individuals with discordant sex information, a missing genotype rate >0.1, or heterozygosity more than three standard deviations from the mean were excluded. Related individuals were detected by calculating identity by descent (IBD), and those with cryptic relatedness were removed (IBD > 0.15). Principal component analysis (PCA) was performed to check for genetic ancestry and only unrelated subjects of Chinese descent were retained. Besides, subjects with pathogenic/likely pathogenic variants of PD disease-causing genes were excluded from the analysis (Zhao et al., 2020).
Statistical analysis
We selected variants in the coding regions of NOTCH3 based on the DNA reference sequence NM_000435 (hg19, chr19:15,271,473-15,311,716) for further analysis. All variants were divided into common or rare types by minor allele frequency (MAF). Rare variants were further categorized into synonymous, missense, loss-of-function (stop gain/loss, frameshift, and splicing mutations), damaging missense (ReVe > 0.7), and previously reported to be associated with CADASIL. We performed logistic regression for common variants (MAF > 0.01) using PLINK with case/control status as an outcome. To further explore the association between NOTCH3 common variants and PD, we conducted a validation study using data from the Parkinson’s Disease DNA Variant browser2 (Kim et al., 2021). This public platform integrates sequencing data from multiple sources, and the vast majority of data are from European ancestry. Difference in the allele frequency was compared by Fisher’s exact tests.
The optimized sequence kernel association test (SKAT-O) method (Lee et al., 2012) in R software (v3.6.3) was used to analyze variants with MAF < 0.01. Age at enrollment, sex and the first five principal components of ancestry were used as covariates. Besides, variants within the EGFr domain were exclusively analyzed. Clinical characteristics were compared between two groups based on their genotype: patients with rare variants which had been previously reported and patients negative for these variants. Linear and logistic regression analyses were performed for genotype–phenotype correlations with adjustment for age at enrollment, disease duration, and sex. Value of p < 0.05 was defined as statistically significant after Bonferroni correction.
Results
Characteristics of variants identified by WES
All samples underwent WES and Supplementary Table 1 presented the results of the sequencing. After quality control, 1,917 patients with early-onset or familial PD and 1,652 controls were available for analysis. Brief descriptions of included samples were given in Supplementary Table 2. Among our cohort, seven common variants and 207 rare variants in NOTCH3 gene were detected. A total of 124 nonsynonymous variants were located at coding regions (two splicing and 40 damaging missense variants). We identified 102 rare variants in the EGFr domain of NOTCH3 in our samples, of which 24 were predicted to be damaging by ReVe. As shown in Figure 1, these rare variants were enriched in EGFr 4, EGFr 9 and EGFr 14. In addition, four variants had been reported to be associated with CADASIL, and three of them were in the EGFr domain. Further detailed information regarding the prioritized rare variants (splicing, damaging missense or previously reported) was shown in Supplementary Table 3.
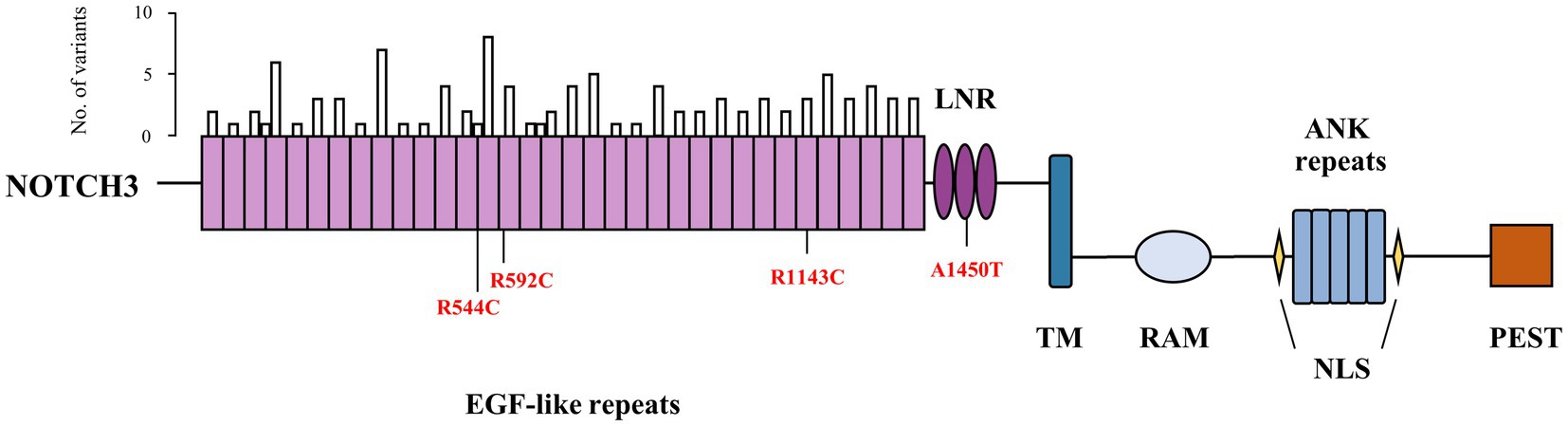
Figure 1. Schematic diagram of domain structure and rare variants of NOTCH3. The bar above represented number of rare variants within epidermal growth factor-like repeat domain. Previously reported variants were depicted as red. EGF, epidermal growth factor; LNR, Lin12-Notch repeats; TM, transmembrane domain; RAM, RBP-jκ-associated molecule domain; ANK, ankyrin; NLS, nuclear localization sequence; PEST, domain rich in proline, glutamate, serine and threonine.
Common and rare variants association tests
Among seven common variants identified, the single nucleotide polymorphisms (SNPs) rs1044006, rs1043997 and rs1043994 showed a nominal protective effect against PD (Table 1). However, none of these SNPs survived Bonferroni correction (threshold of significance = 7.14 × 10−3 for seven variants). In the validation study, we found significant differences in the MAF of three SNPs between PD cases and controls when considered the whole data cohorts (Supplementary Table 4). Of note, the association direction was opposite from our study, that the altered allele was higher in PD patients in the validation cohort. In analyses restricted to each cohort separately, only rs1044006 was associated with a higher odds of PD in the International Parkinson’s Disease Genomics Consortium (IPDGC) genome-wide association study (GWAS) Cohort, and the association direction of three SNPs varied in different cohorts.
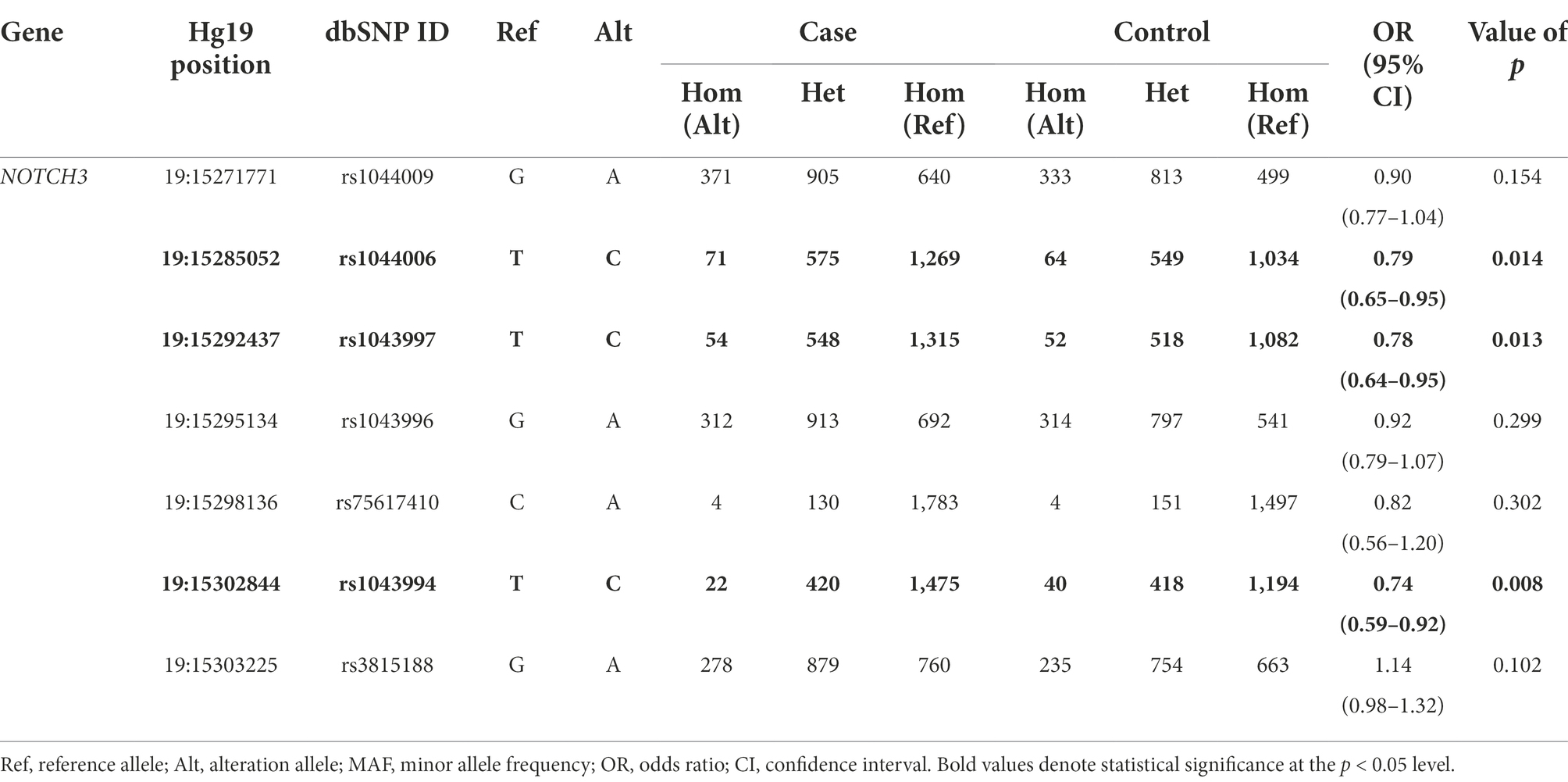
Table 1. Association analysis of common variants in the coding region.
To evaluate the effects of rare variants, stratified association analyses were performed, as shown in Table 2. Of the rare variants in NOTCH3 gene, no significant difference in variant burden was found between cases and controls. Further stratification by variant properties showed similar results. In analyses restricted to variants that had been previously reported, the SKAT-O analysis revealed no significant associations between these variants and PD status (0.6% vs. 0.5%, p = 0.669). Considering more than 95% of CADASIL mutations are missense mutations altering the number of cysteine residues within the EGFr (Abramycheva et al., 2015), we further compared the frequencies of variants lying within the EGFr domain, and Table 2 showed a similar distribution between the two groups.
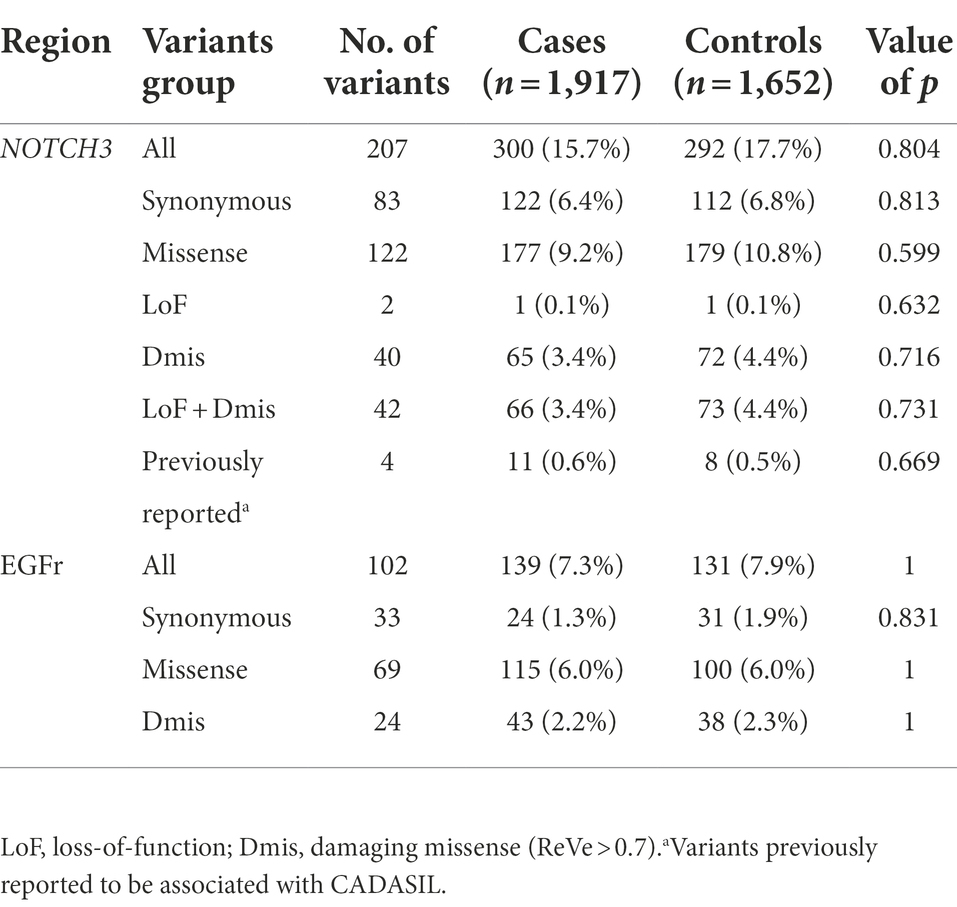
Table 2. Gene-based association results of rare variants.
Clinical features of PD patients with the NOTCH3 variants
Previous studies have discovered that some CADASIL patients can present with parkinsonism (Wegner et al., 2007; Ragno et al., 2013). In our cohort, four variants had been previously reported to be associated with CADASIL. A total of 11 PD patients carrying these variants were included in our cohort. Patients that harbored at least one previously reported variant were referred to as NOTCH3-variant carriers, whereas those who did not were referred to as NOTCH3-variant non-carriers. To better understand the clinical characteristics of NOTCH3-variant carriers, PD patients carrying rare variants which had been previously reported were compared with non-carriers. Descriptions and results for the comparisons of the clinical features across the two groups of PD patients were included in Table 3. The sex, AAO and disease duration at evaluation of the two groups showed no statistical differences. Although the differences were not significant, we still found possible impacts of previously reported variants in NOTCH3 on clinical severity. The results demonstrated more prevalent motor symptoms in NOTCH3-variant carriers, especially more severe in stiffness (p = 0.278), bradykinesia (p = 0.547) and postural instability (p = 0.119). With regard to dopaminergic complications, we found that dyskinesias (27.3% vs. 15.0%; OR = 2.57, p = 0.194) were more common in NOTCH3-variant carriers. In terms of non-motor features, it seemed that depression, autonomic symptom dysfunction and fatigue were more frequently observed in NOTCH3-variant carriers. Notably, the scores of Mini-mental state examination (MMSE) did not differ between groups.
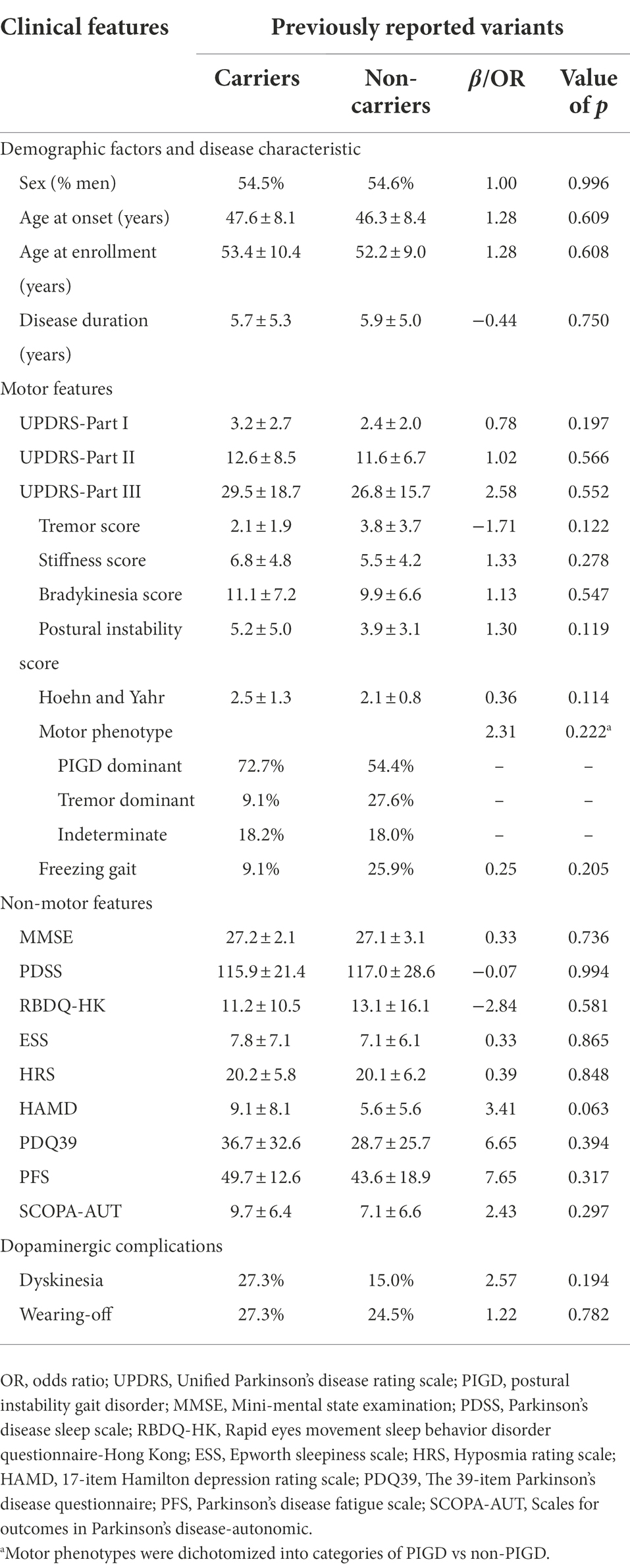
Table 3. Comparison of clinical features in carriers and non-carriers of NOTCH3 variants.
Discussion
The NOTCH3 gene encodes a single-pass transmembrane protein belonging to an evolutionarily conserved NOTCH receptor family. In 1993, the data obtained from two French CADASIL families allowed the gene locus of the disease to be assigned to chromosome 19q12 (Tournier-Lasserve et al., 1993). In our study, we applied WES to a cohort of 1,917 PD patients and 1,652 controls. Genetic and phenotypic data were analyzed with regression analyses and the SKAT-O method. Our results indicated that NOTCH3 gene may not play an important role in PD of Chinese population.
Among the common variants that were detected in our cohort, rs1044006, rs1043997, and rs1043994 exhibited suggestive associations with PD. Contrary to what we had supposed, these SNPs exerted a protective effect against PD. Furthermore, our study detected significant association results in the opposite direction in an integrated European population cohort, which can be attributed to the ethnic difference in the MAF of these SNP. But the association direction of each SNP varied when considering the genomic data sets separately. Thus, our results may be a chance finding and needs to be replicated in future studies. For rare variants, we explored the possible association of PD in different variants groups. Consistent with the findings by Dilliott et al. (2021), we did not detect a significant enrichment of rare nonsynonymous variants among PD patients when compared to the controls. To date, more than 300 NOTCH3 mutations have been reported, and it was shown that the vast majority of the pathogenic mutations for CADASIL were clustered in EGFr 1–6 (Chabriat et al., 2020). Rare variants identified in our cohort enriched in EGFr 4, EGFr 9 and EGFr 14, and gene-based burden tests restricted to the EGFr domain also did not achieve a value of p <0.05. Typically, mutations in the NOTCH3 gene are associated with CADASIL, a monogenic subtype of CSVD. In our cohort, it is unclear whether the control participants had any history of CSVD without obvious symptoms or had CSVD imaging characteristics, which could lead to false negative results.
A previous study found that parkinsonism may not be a rare manifestation in CADASIL. These patients with the R1006C mutation of NOTCH3 had higher frequencies of akinesia, rigidity and postural instability, as well as a poor response to levodopa (Ragno et al., 2013). Recently, evidence has shown that the NOTCH3 rare variants may significantly contribute to the increased white matter hyperintensities burden of PD patients, which in turn may negatively influence cognition (Ramirez et al., 2020). Although we did not find significant associations in the following genotype–phenotype analysis, the higher clinical scores of motor symptoms in NOTCH3-variant carriers were of interest, providing a possibility of the association with NOTCH3 variants and motor signs in PD. We found stiffness, bradykinesia and postural instability were seen more commonly in PD patients with variants that had been previously reported, whereas rest tremor was always absent. Although literature data which used the Montreal Cognitive Assessment (MoCA) to assess global cognition supported an association between the presence of NOTCH3 and cognitive impairment (Ramirez et al., 2020), we did not detect significant differences in cognitive capabilities between NOTCH3-variant carriers and non-carriers based on the MMSE scores.
However, this result might have occurred due to the small number of NOTCH3-variant carriers. Hence, it calls for a validation study that will have access to a cohort with a larger size of NOTCH3-variant carriers, thus increasing the power of detection. In addition, the MMSE is the most widely used cognitive screening measure for general cognitive evaluation (Anderson, 2019). Considering its variation in sensitivity and specificity, MMSE might be too insensitive to detect subtle differences between two groups.
The main strength of our study is the large sample size of Chinese origin in addition to the use of uniform clinical evaluation methods by PD-MDCNC database. However, some additional limitations need to be considered. First, the majority of non-motor symptoms were evaluated based on the patients’ responses to a questionnaire. More powerful and objective tests were needed to enhance the reliability of non-motor features. On the other hand, due to its retrospective design, the phenotypic assessment had some missing data. For future studies, missing data should be complemented to facilitate an optimal assessment. Third, because of a lack of magnetic resonance imaging, we failed to validate the relationship between white matter lesions and PD in NOTCH3-positive patients, which might play a role in the pathogenesis of cognitive impairment. Meanwhile, false negative results may be produced due to the absence of magnetic resonance imaging in the control participants. Finally, WES method failed to identify copy number variations and non-coding variants, so it is necessary for extensive analysis to explore the link between variants within these regions and PD.
Taken together, we comprehensively investigated the NOTCH3 gene in a large cohort of Chinese PD patients and healthy controls. Although no evidence for significant associations between NOTCH3 variants and PD was noted, replications in different ethnic cohorts and further functional studies are needed.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://db.cngb.org/cnsa/, CNP0002638.
Ethics statement
The studies involving human participants were reviewed and approved by the Ethics Committee of Xiangya Hospital (Central South University). The patients/participants provided their written informed consent to participate in this study.
Author contributions
QZ carried out genetic analysis and wrote the manuscript. HP, YZ, and YW processed genomic data. QX, JT, XY, and JL helped the study design and data interpretation. BT and JG critically revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the Central Public-Interest Scientific Institution Basal Research Fund of Chinese Academy of Medical Sciences (grant nos. 2018-12M-HL-025 and 2019-RC-HL-025); National Natural Science Foundation of China (grant nos. 81974202, 81873785, 82071439, and U20A20355); Technology Major Project of Hunan Provincial Science and Technology Department (grant no. 2021SK1010); Hunan Province Innovative Construction Project Science (grant no. 2019SK2335); Innovative team program from Department of Science & Technology of Hunan Province (grant no. 2019RS1010); Innovation-driven Team Project from Central South University (grant no. 2020CX016); and National Key Research and Development Program of China (grant no. 2021YFC2501204).
Acknowledgments
The authors thank all participants for their contribution to this research. We are grateful for resources from the Bioinformatics Center of Xiangya Hospital and High Performance Computing Center of Central South University.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnagi.2022.995330/full#supplementary-material
Footnotes
References
Abramycheva, N., Stepanova, M., Kalashnikova, L., Zakharova, M., Maximova, M., Tanashyan, M., et al. (2015). New mutations in the Notch3 gene in patients with cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy (CADASIL). J. Neurol. Sci. 349, 196–201. doi: 10.1016/j.jns.2015.01.018
Anderson, N. D. (2019). State of the science on mild cognitive impairment (MCI). CNS Spectr. 24, 78–87. doi: 10.1017/S1092852918001347
Chabriat, H., Joutel, A., Tournier-Lasserve, E., and Bousser, M. G. (2020). CADASIL: yesterday, today, tomorrow. Eur. J. Neurol. 27, 1588–1595. doi: 10.1111/ene.14293
Chang, C. C., Chow, C. C., Tellier, L. C., Vattikuti, S., Purcell, S. M., and Lee, J. J. (2015). Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4:7. doi: 10.1186/s13742-015-0047-8
Dichgans, M., Mayer, M., Uttner, I., Bruning, R., Muller-Hocker, J., Rungger, G., et al. (1998). The phenotypic spectrum of CADASIL: clinical findings in 102 cases. Ann. Neurol. 44, 731–739. doi: 10.1002/ana.410440506
Dilliott, A. A., Abdelhady, A., Sunderland, K. M., Farhan, S. M. K., Abrahao, A., Binns, M. A., et al. (2021). Contribution of rare variant associations to neurodegenerative disease presentation. NPJ Genom. Med. 6:80. doi: 10.1038/s41525-021-00243-3
Erro, R., Lees, A. J., Moccia, M., Picillo, M., Penco, S., Mosca, L., et al. (2014). Progressive parkinsonism, balance difficulties, and supranuclear gaze palsy. JAMA Neurol. 71, 104–107. doi: 10.1001/jamaneurol.2013.5149
Guo, W., Xu, B., Sun, H., Ma, J., Mei, S., Zeng, J., et al. (2021). Case report: progressive asymmetric parkinsonism secondary to CADASIL without dementia. Front. Neurol. 12:760164. doi: 10.3389/fneur.2021.760164
Hughes, A. J., Daniel, S. E., Kilford, L., and Lees, A. J. (1992). Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J. Neurol. Neurosurg. Psychiatry 55, 181–184. doi: 10.1136/jnnp.55.3.181
Joutel, A., Andreux, F., Gaulis, S., Domenga, V., Cecillon, M., Battail, N., et al. (2000). The ectodomain of the Notch3 receptor accumulates within the cerebrovasculature of CADASIL patients. J. Clin. Invest. 105, 597–605. doi: 10.1172/JCI8047
Joutel, A., Corpechot, C., Ducros, A., Vahedi, K., Chabriat, H., Mouton, P., et al. (1996). Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature 383, 707–710. doi: 10.1038/383707a0
Joutel, A., Vahedi, K., Corpechot, C., Troesch, A., Chabriat, H., Vayssiere, C., et al. (1997). Strong clustering and stereotyped nature of Notch3 mutations in CADASIL patients. Lancet 350, 1511–1515. doi: 10.1016/S0140-6736(97)08083-5
Keller, M. F., Saad, M., Bras, J., Bettella, F., Nicolaou, N., Simon-Sanchez, J., et al. (2012). Using genome-wide complex trait analysis to quantify ‘missing heritability’ in Parkinson’s disease. Hum. Mol. Genet. 21, 4996–5009. doi: 10.1093/hmg/dds335
Kim, J. J., Makarious, M. B., Bandres-Ciga, S., Gibbs, J. R., Ding, J., Hernandez, D. G., et al. (2021). The Parkinson’s disease DNA variant browser. Mov. Disord. 36, 1250–1258. doi: 10.1002/mds.28488
Lee, S., Emond, M. J., Bamshad, M. J., Barnes, K. C., Rieder, M. J., Nickerson, D. A., et al. (2012). Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am. J. Hum. Genet. 91, 224–237. doi: 10.1016/j.ajhg.2012.06.007
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, J., Shi, L., Zhang, K., Zhang, Y., Hu, S., Zhao, T., et al. (2018a). Var cards: an integrated genetic and clinical database for coding variants in the human genome. Nucleic Acids Res. 46, D1039–D1048. doi: 10.1093/nar/gkx1039
Li, J., Zhao, T., Zhang, Y., Zhang, K., Shi, L., Chen, Y., et al. (2018b). Performance evaluation of pathogenicity-computation methods for missense variants. Nucleic Acids Res. 46, 7793–7804. doi: 10.1093/nar/gky678
Marogianni, C., Sokratous, M., Dardiotis, E., Hadjigeorgiou, G. M., Bogdanos, D., and Xiromerisiou, G. (2020). Neurodegeneration and inflammation-an interesting interplay in Parkinson’s disease. Int. J. Mol. Sci. 21:8421. doi: 10.3390/ijms21228421
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
Monet-Lepretre, M., Haddad, I., Baron-Menguy, C., Fouillot-Panchal, M., Riani, M., Domenga-Denier, V., et al. (2013). Abnormal recruitment of extracellular matrix proteins by excess Notch3 ECD: a new pathomechanism in CADASIL. Brain 136, 1830–1845. doi: 10.1093/brain/awt092
Mor, D. E., Daniels, M. J., and Ischiropoulos, H. (2019). The usual suspects, dopamine and alpha-synuclein, conspire to cause neurodegeneration. Mov. Disord. 34, 167–179. doi: 10.1002/mds.27607
Planchard, M. S., Exley, S. E., Morgan, S. E., and Rangachari, V. (2014). Dopamine-induced alpha-synuclein oligomers show self-and cross-propagation properties. Protein Sci. 23, 1369–1379. doi: 10.1002/pro.2521
Postuma, R. B., Berg, D., Stern, M., Poewe, W., Olanow, C. W., Oertel, W., et al. (2015). MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 30, 1591–1601. doi: 10.1002/mds.26424
Ragno, M., Berbellini, A., Cacchio, G., Manca, A., Di Marzio, F., Pianese, L., et al. (2013). Parkinsonism is a late, not rare, feature of CADASIL: a study on Italian patients carrying the R1006C mutation. Stroke 44, 1147–1149. doi: 10.1161/STROKEAHA.111.000458
Ramirez, J., Dilliott, A. A., Binns, M. A., Breen, D. P., Evans, E. C., Beaton, D., et al. (2020). Parkinson’s disease, NOTCH3 genetic variants, and white matter Hyperintensities. Mov. Disord. 35, 2090–2095. doi: 10.1002/mds.28171
Tan, M. M. X., Malek, N., Lawton, M. A., Hubbard, L., Pittman, A. M., Joseph, T., et al. (2019). Genetic analysis of Mendelian mutations in a large UK population-based Parkinson’s disease study. Brain 142, 2828–2844. doi: 10.1093/brain/awz191
Tournier-Lasserve, E., Joutel, A., Melki, J., Weissenbach, J., Lathrop, G. M., Chabriat, H., et al. (1993). Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy maps to chromosome 19q12. Nat. Genet. 3, 256–259. doi: 10.1038/ng0393-256
Valenti, R., Bianchi, S., Pescini, F., D’Eramo, C., Inzitari, D., Dotti, M. T., et al. (2011). First report of a pathogenic mutation on exon 24 of the NOTCH3 gene in a CADASIL family. J. Neurol. 258, 1632–1636. doi: 10.1007/s00415-011-5983-3
Van Gerpen, J. A., Ahlskog, J. E., and Petty, G. W. (2003). Progressive supranuclear palsy phenotype secondary to CADASIL. Parkinsonism Relat. Disord. 9, 367–369. doi: 10.1016/s1353-8020(02)00146-3
Wegner, F., Strecker, K., Schwarz, J., Wagner, A., Heinritz, W., Sommerer, F., et al. (2007). Vascular parkinsonism in a CADASIL case with an intact nigrostriatal dopaminergic system. J. Neurol. 254, 1743–1745. doi: 10.1007/s00415-007-0529-4
Yang, H., and Wang, K. (2015). Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat. Protoc. 10, 1556–1566. doi: 10.1038/nprot.2015.105
Young, K. Z., Cartee, N. M. P., Ivanova, M. I., and Wang, M. M. (2020). Thiol-mediated and catecholamine-enhanced multimerization of a cerebrovascular disease enriched fragment of NOTCH3. Exp. Neurol. 328:113261. doi: 10.1016/j.expneurol.2020.113261
Zeng, Q., Pan, H., Zhao, Y., Wang, Y., Xu, Q., Tan, J., et al. (2022). Evaluation of common and rare variants of Alzheimer’s disease-causal genes in Parkinson’s disease. Parkinsonism Relat. Disord. 97, 8–14. doi: 10.1016/j.parkreldis.2022.02.016
Zhang, X., Lee, S. J., Young, K. Z., Josephson, D. A., Geschwind, M. D., and Wang, M. M. (2014). Latent NOTCH3 epitopes unmasked in CADASIL and regulated by protein redox state. Brain Res. 1583, 230–236. doi: 10.1016/j.brainres.2014.08.018
Zhang, X., Lee, S. J., Young, M. F., and Wang, M. M. (2015). The small leucine-rich proteoglycan BGN accumulates in CADASIL and binds to NOTCH3. Transl. Stroke Res. 6, 148–155. doi: 10.1007/s12975-014-0379-1
Keywords: CADASIL, whole-exome sequencing, common variants, NOTCH3, Parkinson’s disease
Citation: Zeng Q, Pan H, Zhao Y, Wang Y, Xu Q, Tan J, Yan X, Li J, Tang B and Guo J (2022) Association between NOTCH3 gene and Parkinson’s disease based on whole-exome sequencing. Front. Aging Neurosci. 14:995330. doi: 10.3389/fnagi.2022.995330
Edited by:
Sabina-Capellari, University of Bologna, ItalyReviewed by:
Neven Maksemous, Queensland University of Technology, AustraliaAleksandra Pavlovic, University of Belgrade, Serbia
Paul Dunn, Bond University, Australia
Copyright © 2022 Zeng, Pan, Zhao, Wang, Xu, Tan, Yan, Li, Tang and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jifeng Guo, Z3VvamlmZW5nMjAwM0AxNjMuY29t