 Bing-Nan Su
Bing-Nan Su Ren-Juan Shen
Ren-Juan Shen Zhuo-Lin Liu1,2,3
Zhuo-Lin Liu1,2,3 Yang Li
Yang Li Zi-Bing Jin
Zi-Bing Jin
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Aging Neurosci. , 10 August 2022
Sec. Cellular and Molecular Mechanisms of Brain-aging
Volume 14 - 2022 | https://doi.org/10.3389/fnagi.2022.948279
This article is part of the Research Topic Rare and Common Neurodegenerative Retinal Diseases: From Molecular Mechanisms to the Identification of Novel Mutation-independent Therapeutic Strategies View all 3 articles
Purpose: Mutation in the USH2A gene is the most common cause of inherited retinal dystrophy (IRD), including non-syndromic retinitis pigmentosa (RP) and Usher syndrome II (USH2). Gene editing and therapy targeting USH2A, especially the hotspot region, would benefit a large proportion of IRD patients. In this study, we comprehensively analyzed the genetic spectrum of the USH2A gene, aiming to identify global hot spot mutations in USH2A-related IRDs and differences in hot spot regions across continents.
Materials and methods: A retrospective USH2A-related IRD study was conducted, including our IRD cohort, and reported USH2A studies worldwide.
Results: A total of 3,972 mutated USH2A alleles of approximately 1,935 patients were collected from 33 cohort studies worldwide, containing 102 alleles of 51 patients in our IRD cohort. Mutations in exon 13 were the most common, reaching 18.4% globally and a higher frequency of 22% in America, 19.2% in Europe, and a lower 12% in East Asia. Pathogenic mutations that affected 10 of the 72 exons of USH2A, exon 2, exon 13, exon 41–43, exon 50, exon 54, exon 57, exon 61, and exon 63 in total were responsible for half of global USH2A mutant alleles. With base editors including adenine base editor (ABE), cytidine base editor (CBE), and glycosylase base editor (GBE), 76.3% of single nucleotide variations (SNVs) and 58% of all mutations in USH2A are correctable. Meantime, four novel pathogenic mutations were revealed in our IRD cohort, p. (Val1130Cysfs*72), p. (Ala2139fs*14), p. (Gly4139Arg), and p. (Val4166Cysfs*7).
Conclusion: In this study, we revealed four novel mutations, expanding the spectrum of USH2A mutations, and importantly presented global hotspot exons and mutations of USH2A as well as the proportion of SNVs that can be restored by different base editors, providing a perspective for exploring high-efficiency and broader-reaching gene editing and gene therapies.
Inherited retinal dystrophies (IRDs), affecting approximately one in 1,000 people worldwide, is a group of genetically and clinically heterogeneous disorders that severely destroy visual function. For many years, patients with IRDs had severe visual impairment and psychological burdens because lacking effective treatments. Retinitis pigmentosa (RP) is the most common form of IRD, affecting about 435,000 patients in China and almost two million worldwide (Berger et al., 2010).
Biallelic variants in the USH2A gene cause either Usher syndrome type 2 (USH2) or non-syndromic RP. In both conditions, the retinal phenotype includes progressive night blindness and constriction of the visual field, followed by central vision loss. In addition, USH2 is typically associated with congenital moderate to severe sensorineural hearing impairment (Weston et al., 2000; Hartong et al., 2006; Huang et al., 2015).
The USH2A gene was mapped to 1q41, spanning 800.05 kb with 72 exons, and encodes a transmembrane protein of 5202 amino acids named usherin, which contains laminin EGF motifs, a transmembrane domain, and repeats of fibronectin type III motifs. The usherin protein is predominantly expressed in the photoreceptor of the retina and the hair cells in the cochlea, and could be found in the basement membranes of many other tissues (Kimberling et al., 1995; Weston et al., 2000; van Wijk et al., 2004; Liu et al., 2007).
Many studies have shown that mutations in USH2A are responsible for at least 7% of non-syndromic RP and 57–67.7% of USH2. So far, more than 1,000 pathogenic or likely pathogenic variants have been reported in HGMD1 and ClinVar.2 Two ancestral variants, c.2299delG and c.2276G > T, are the most frequent in Europe and America (Carss et al., 2017; Stone et al., 2017). Two variants, c.2802T > G and c.8559-2A > G, are the most commonly reported in the Chinese cohort (Huang et al., 2015; Gao et al., 2021). The c.2299delG, c.2276G > T, and c.2802T > G are all located in exon 13, making exon 13 a hotspot region of USH2A mutations.
Research in gene editing and gene therapy of ocular disease has made rapid progress. Luxturna has been recently approved as a gene augmentation therapy drug for patients with RPE65-associated retinal dystrophy. Antisense oligonucleotide (AON) has also made impressive progress in treating IRDs. Three AON drugs have been used in clinical trials to treat IRDs (Slijkerman et al., 2016; Dulla et al., 2021). Base editing is one of the most recent and remarkable advances in gene editing. Base editors convert one base into another directly, without creating a double-strand DNA break (DSB), thereby minimizing the generation of excess undesired DSB-associated by-products. Up to now, three classes of DNA base editors have been established: ABE (converting A-T base pair to G-C base pair), CBE (converting C-G base pair to T-A base pair), and GBE (converting C-G base pair to G-C base pair) (Rees and Liu, 2018; Porto et al., 2020). SNVs make up most of the human pathogenic mutations, indicating that base editing will have great potential for treating a variety of genetic disorders (Goodwin et al., 2016; Landrum et al., 2016; Rees and Liu, 2018). Gene therapies and gene editing targeting USH2A, especially the hotspot mutant region, would benefit many patients with IRDs. In this study, we comprehensively analyzed the genetic spectrum of the USH2A gene, aiming to identify global hotspot mutant exons and variants in USH2A-related IRDs and differences in hotspot regions across continents. Furthermore, we predicted the proportion of USH2A mutations that could be corrected by base editing based on the mutation collection we integrated.
The study was conducted in compliance with the tenets of the Declaration of Helsinki. Institutional Ethics approval and informed consent was obtained from the newly recruited patients in this study. Clinical and genetic diagnoses of patients in our IRD cohort of nearly 1,500 probands were reassessed. A medical history along with ophthalmic examinations was re-evaluated, including the initial symptoms, onset age, and best-corrected visual acuity (BCVA), fundus photography, optical coherence tomography (OCT), and full-field electroretinography (ERG). Pathogenicity of each variant was reassessed and categorized as pathogenic, likely pathogenic, uncertain significance, benign and likely benign according to ACMG/AMP classification guidelines (Richards et al., 2015). Variants categorized as benign or likely benign were excluded. Seventeen patients diagnosed with Usher syndrome and 34 patients with RP who were verified to harbor biallelic pathogenic mutations were enrolled. Informed consent was obtained from all study participants.
Global reported variants were collected through a comprehensive literature review. Keywords “USH2A”, “Usher”, “inherited retinal dystrophy”, and “retinitis pigmentosa” were used to search for genetic studies with a large sample size in the last 5–10 years. Genetic variation data were collected when available.
Patients in our cohort underwent targeted exome sequencing (TES) or whole-exome sequencing (WES) were recruited. SNP array was used to detect the copy number variation (CNV) and real-time quantitative PCR was performed to confirm CNV. Suspected SNVs and small indels were verified by Sanger sequencing, as was the co-segregation analysis of available family members.
Multiple databases were used to annotate and evaluate the variants, and the pathogenicity of missense variants was predicted in silico by Mutation Taster, PolyPhen-2, and SIFT. The Human Gene Mutation Database and ClinVar database were screened for previously reported variants. Classification of variants was based on the ACMG guidelines (Richards et al., 2015).
In total, 51 patients in our IRD cohort were classified into three categories based on different combinations of mutation types. Bilateral BCVA of patients was taken into analysis. To eliminate the inter-correlation of both eyes, linear mixed-effects model was used to explore the association between mutations and other variables (e.g., BCVA). Both Mean ± SD and median (interquartile range) were used for basic statistical description. Analysis was done using SAS 9.4. The significance level was set to be 0.05 and was adjusted according to Bonferroni criteria for multiple comparisons.
Fifty-one patients, including 17 USH and 34 RP, were included in the study after reassessment and identifying pathogenic USH2A biallelic mutations. All patients complained of night blindness and constriction of the visual field. Patients diagnosed with USH suffered different degrees of hearing loss. 64% of patients had onset of night blindness after 10 years old, consistent with previous reports that patients with USH2A mutations usually develop RP in the second decade of their life or later.
All patients’ fundus showed typical RP presentations, including waxy pallor disk, attenuated retinal vasculature, and RPE degeneration. However, we also found a unique characteristic feature of RPE degeneration exhibited by USH2A mutated patients. In our cohort, patients were divided into three sub-groups, NBP (no bone-spicule), SBP (scattered bone spicule), and EBP (extensive bone-spicule) according to whether there was bone-spicule pigmentation in the posterior pole of the fundus (Figure 1). NBP was present in 62% of patients, and an additional 17% had sporadic bone-spicule deposits along the vascular arcade, which was defined as SBP (Figure 1D). Moreover, they all had the same presentation of RPE depigmentation, which manifests as small pale patches (Figure 1B).

Figure 1. Clinical manifestations of USH2A-related inherited retinal dystrophy (IRD) patients. (A–C) Fundus photos present no bone-spicule pigmentation (NBP), scattered bone spicule (SBP), and extensive bone-spicule (EBP) in the posterior pole of the fundus. The black arrowheads indicate the bone-spicule pigmentation, white arrowheads indicated the RPE depigmentation presenting as small pale patches. (D,E) Distribution of USH2A-related IRDs patients presenting as NBP, SBP, and EBP. The X-axis of E represents the disease duration of <10, 11–20, 21–30, and >30 years.
Damage to photoreceptor and RPE function in patients with RP typically progresses from the periphery to the center. Interestingly, this NBP and SBP presentation did not strongly correlate with disease duration. However, among the four groups of different disease duration, the proportion of patients presenting NBP was the highest (Figure 1E).
A total of 102 mutant alleles and 50 variants in USH2A were detected in our IRD cohort (Figure 2). Information of all USH2A variants in our IRD cohort was listed in Supplementary Table 1. Among the 102 alleles, 62 were missense mutations, and 40 were null mutations consisting of six stop-gain mutations, ten frameshift mutations, three splicing mutations, and a copy number variation. Four novel mutations, p. (Val1130Cysfs*72), p. (Ala2139fs*14), p. (Gly4139Arg), and p. (Val4166Cysfs*7) were newly reported, all of which were absent in the global healthy control database (Tables 1, 2). Despite valine at residue 1130 and 4166 being less conserved (Figure 3C), all three frameshift mutations will result in protein truncation, indicating strong evidence of pathogenicity. A patient diagnosed with typical non-syndromic RP harbored the novel missense mutation c.12415G > C, p. (Gly4139Arg). The p. (Gly4139Arg) mutation was located in an evolutionary conserved domain called fibronectin type III (FN3) and was predicted to be damaging or intolerant by multiple in silico predictive algorithms (Figure 2 and Table 2). A three-dimensional protein model of usherin was constructed using PyMOL to visualize the predicted impact of amino acid change at position 4139 (Figures 3A,B). As the model shows, the change from glycine to arginine would result in the formation of a new hydrogen bond between amino acid residues 4139 and 4065, which may affect the folding and stability of the protein. Co-segregation analysis in available family members showed the mutation segregated within the pedigree. Therefore, the p. (Gly4139Arg) mutation was defined as a likely pathogenic mutation according to the guideline of ACMG. The detailed clinical and genetic information of patients who harbored novel mutations was listed in Tables 1, 2.
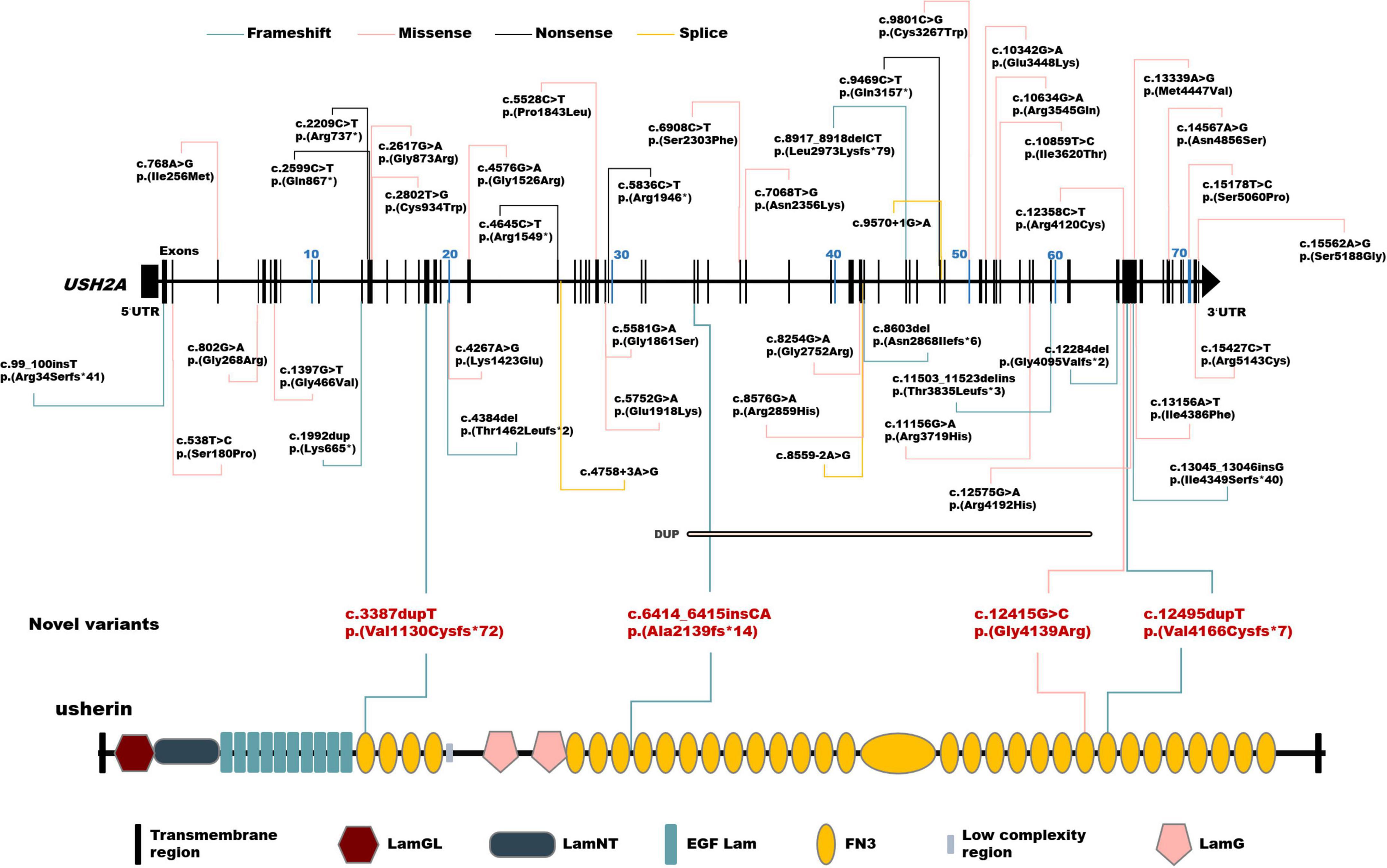
Figure 2. Schematic distribution of the USH2A variants identified in this study. Upper, schematic presentation of the USH2A gene and the distribution of variants identified in our inherited retinal dystrophy (IRD) cohort. Novel variants are highlighted in red. Lower, usherin domains encoded by USH2A. LamGL, laminin G-like domain; LamNT, laminin N-terminal domain; EGF_Lam, laminin-type epidermal growth factor-like domain; FN3, fibronectin type III domain; LamG, laminin G domain.
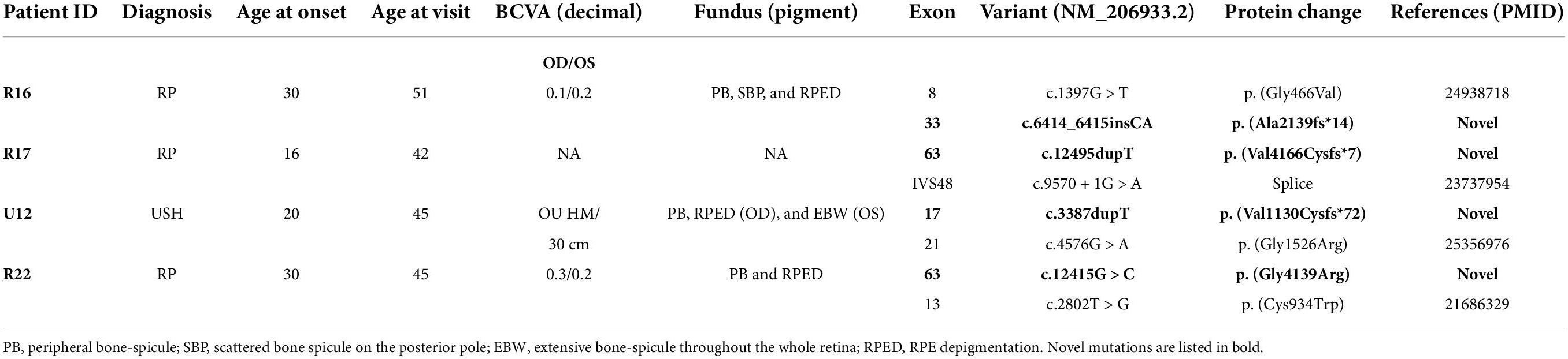
Table 1. USH2A novel variants and clinical manifestation of these patients.

Table 2. Pathogenicity of USH2A novel variants found in our cohort.
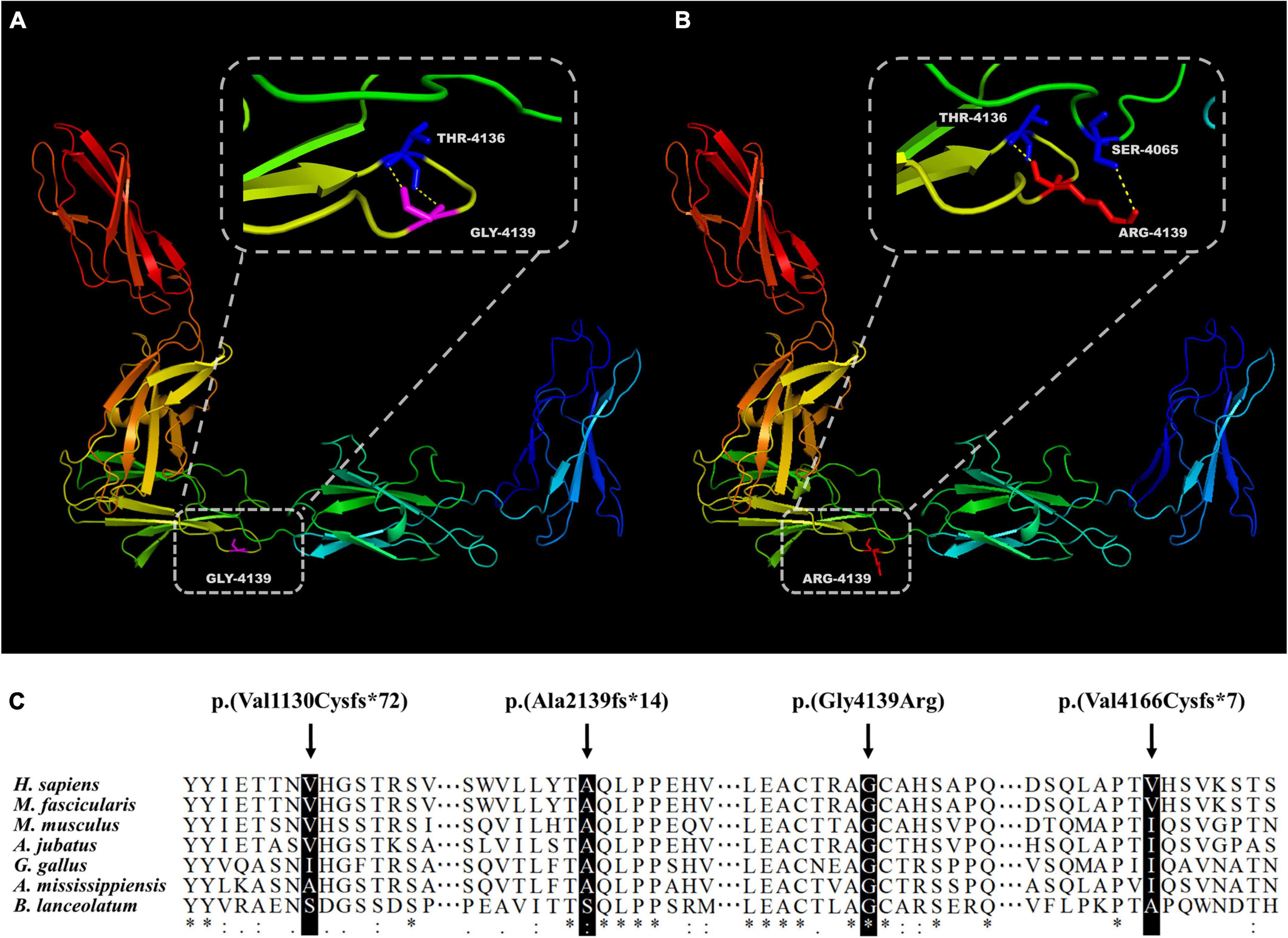
Figure 3. Evaluation of novel mutations in USH2A. Presentation of five adjacent FN3 domains and residue 4139 in wildtype (A) and mutant (B) usherin. (C) Alignment of the usherin protein sequence of residues 1130, 2139, 4139, and 4166 to its orthologous protein sequences in different species. The asterisk “*” indicates positions with a single and fully conserved residue, colon “:” indicates highly conserved positions, and “.” indicates positions with a weak conservation.
We classified the 50 different mutations detected in our IRD cohort into two categories: missense, and null (including nonsense, frameshift, splice, and copy number variation). The 51 patients were then divided into three groups based on different combinations of mutation types for two alleles: USH2Anull/null (n = 10), USH2Amissense/missense (n = 21), and USH2Anull/missense (n = 20; Supplementary Table 3). Compared with patients with two null mutations, those patients who harbored two or one missense mutation were more likely to develop non-syndromic RP than USH (with an adjusted P-value of 0.040 and 0.019, respectively). There was no statistically significant difference between USH2Amissense/missense and USH2Anull/missense (Supplementary Table 4). Meanwhile, the age at onset of both patients with biallelic null mutations and patients with biallelic missense mutations was younger than that of patients with one missense and one null mutation (with an adjusted P-value of 0.045 and 0.029, respectively) (Figure 4 and Supplementary Table 5). There was no statistical difference in BCVA and disease progression among the three groups.
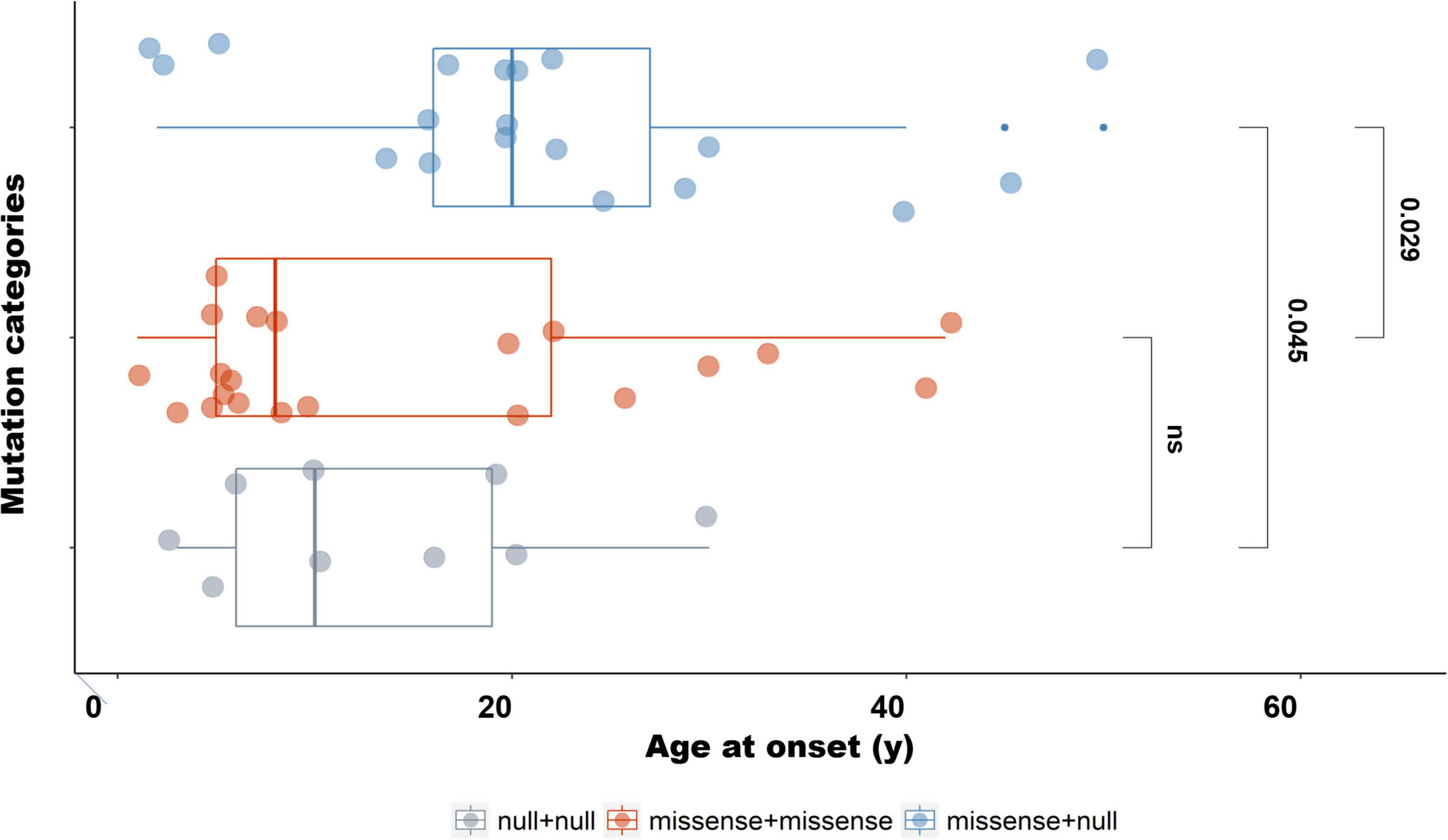
Figure 4. Age at onset of patients with different mutation types. The age at onset of patients with biallelic missense mutations was significantly younger than that of patients harboring one missense with one null mutation (Padjusted = 0.029). The age at onset of patients with one missense and one null mutation was significantly older than that of patients with biallelic null mutations (Padjusted = 0.045).
A total of 3870 mutant USH2A alleles of approximately 1,884 patients were collected from 33 cohort studies with large sample sizes conducted in the United Kingdom, Spain, Germany, Italy, United States, Japan, Korea, China, etc. One hundred and two alleles of 51 patients detected in our IRD cohort were calculated together, making the sample size of 1,935 patients with 3972 mutant alleles (Supplementary Table 2).
Overall, the frequency of mutations detected in exon 13 was the highest, reaching 18.4%. The other two exons in the top3 were exon 63 with a frequency of 10% and exon 61 with 5.5%. Variants affected or detected in the following ten exons, including exon 2, exon 13, exon 41–43, exon 50, exon 54, exon 57, exon 61, and exon 63, accounting for more than half of the cohort. Global mutation hotspots are displayed in Figure 5A. Exon 13 harbored three of the top five hotspot variants: c.2299del, c.2802T > G, and c.2276G > T. The frameshift mutation c.2299del, p. (Glu767Serfs*21), which leads to the formation of truncated usherin protein, accounted for the highest frequency of 7.7% of USH2A mutations worldwide. Both missense mutations c.2802T > G, p. (Cys934Trp) and c.2276G > T, p. (Cys759Phe) were located in regions that encode the functional and evolutionary conserved EGF-Lam domain and were predicted to be detrimental to the correct folding of usherin (Xu et al., 2011; Reurink et al., 2022). Unexpectedly, no mutations occurred in exon 63 with a frequency higher than 1%, while the pathologic variants accounted for 3.5% of all the mutant alleles, ranking second.
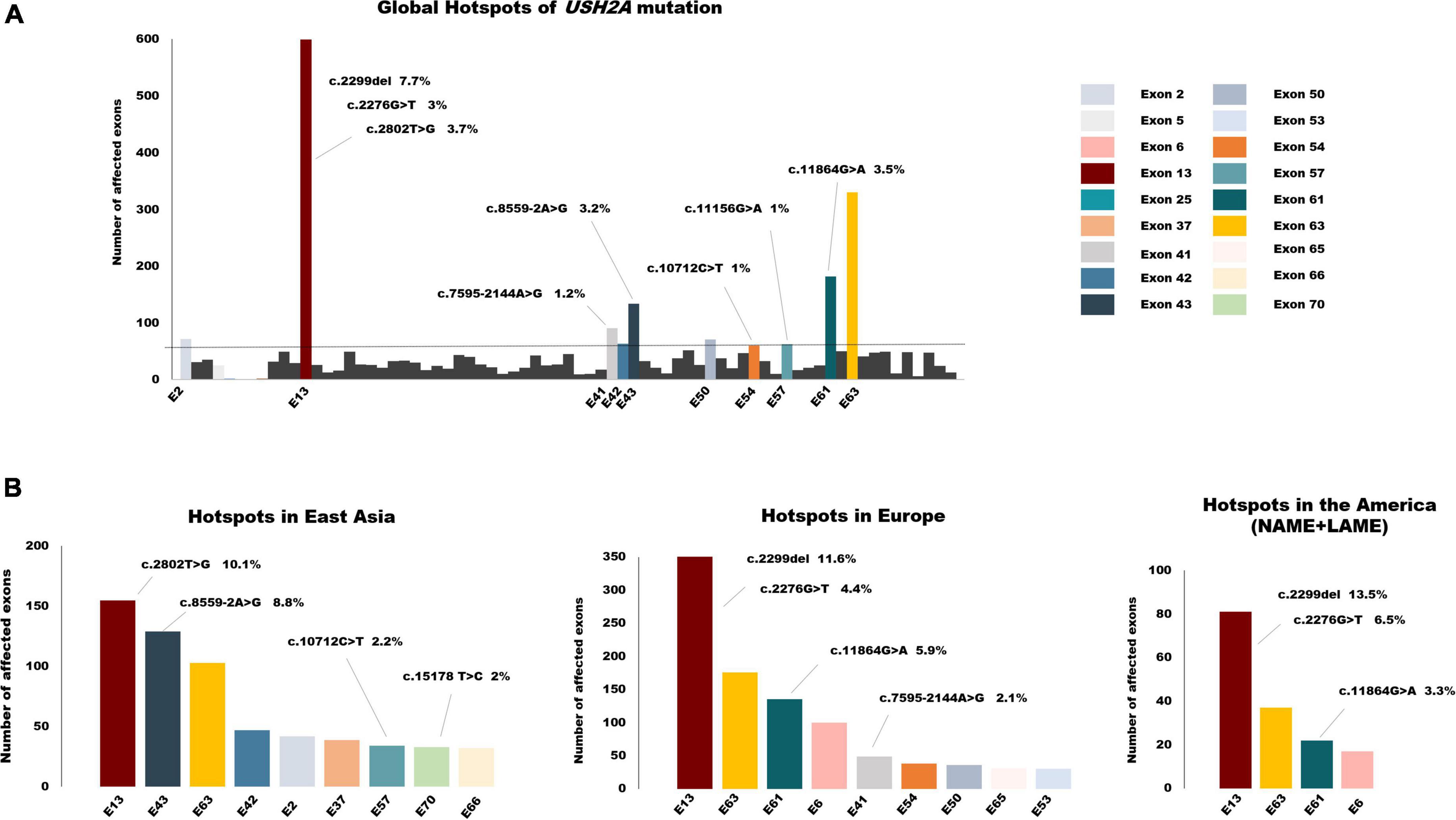
Figure 5. The global spectrum of USH2A variants. (A) Global hotspots of USH2A mutation. Exons colored are the top 10 hotspot mutant exons which accounted for more than half of all the alleles in total. (B) Hotspot exons and mutations of USH2A in different continents. Mutant alleles with a frequency of >1% were listed above the relevant exons.
Considering the small sample size of South Asia and Africa, we mainly focused on the mutation distribution in East Asia, Europe, and America (Figure 5B). As the mutation distributions in Latin America (LAME) and North America (NAME) were nearly identical, the data for these two regions were combined. There are some similarities among the three continents in the distribution of USH2A pathogenic variants. First, exon 13 was still the most commonly affected exon, with the highest frequency of 22% in America, 19.2% in Europe, and 12% in East Asia. Second, mutations that affected exon 63 frequently occurred in all three continents, with a nearly same frequency of around 10%. However, the mutation spectrum in different regions still varies, which may be due to the disparity in ethnicity. The splicing variant c.8559-2A > G, which has been demonstrated to result in skipping of exon 43 (Nakanishi et al., 2010), was common in the Asia cohort, especially in East Asia. Only one allele was reported in a study conducted in Germany. This splicing variant accounted for 8.8% of patients diagnosed with USH2A-related IRDs in East Asia, making exon 43 the second common mutant exon. Similarly, a deep intronic variant c.7595-2144A > G, leading to the insertion of an out-of-frame pseudo-exon (PE) (Vaché et al., 2012), was detected only in Caucasians and was one of the most frequent mutations in the Europe cohort.
Of the total 3972 mutant alleles listed in this study, missense variants comprised the highest proportion (46%). In contrast, null frameshift and nonsense variants, as well as splice site variants, accounted for 19, 17, and 10%, respectively (Figure 6A). In addition to causing missense mutations, point mutations can also lead to nonsense, splice site, and synonymous mutations. Therefore, as shown in Figure 6B, SNVs accounted for 76% of pathogenic USH2A variants, which is consistent with the consensus that SNVs are the most common mutation type in most human diseases (Goodwin et al., 2016; Landrum et al., 2016). 58% of total variants, which is 76.3% of SNVs, can be corrected according to the editing principle of ABE, CBE, and GBE. 23.7% of SNVs that involve alteration between purines and pyrimidines cannot be restored by these three base editors (Figure 6C). The proportion of SNVs that can be corrected by base editors was shown in Figure 6D. ABE made up the majority (52%), followed by CBE (29%) and GBE (19%). Notably, for a particular set of SNVs, the mutant nucleotide cannot be repaired according to the original editing principle of ABE (converting A-T base pair to G-C base pair), CBE (converting C-G base pair to T-A base pair), and GBE (converting G-C base pair to C-G base pair), but the amino acid encoded can be restored by changing another base within the same codon, thereby increasing the proportion of editable mutations. Excitingly, a hotspot mutation of USH2A in East Asia: c.2802T > G, p. (Cys934Trp) could be corrected by this strategy. Detailed information can be found in Supplementary Table 2.
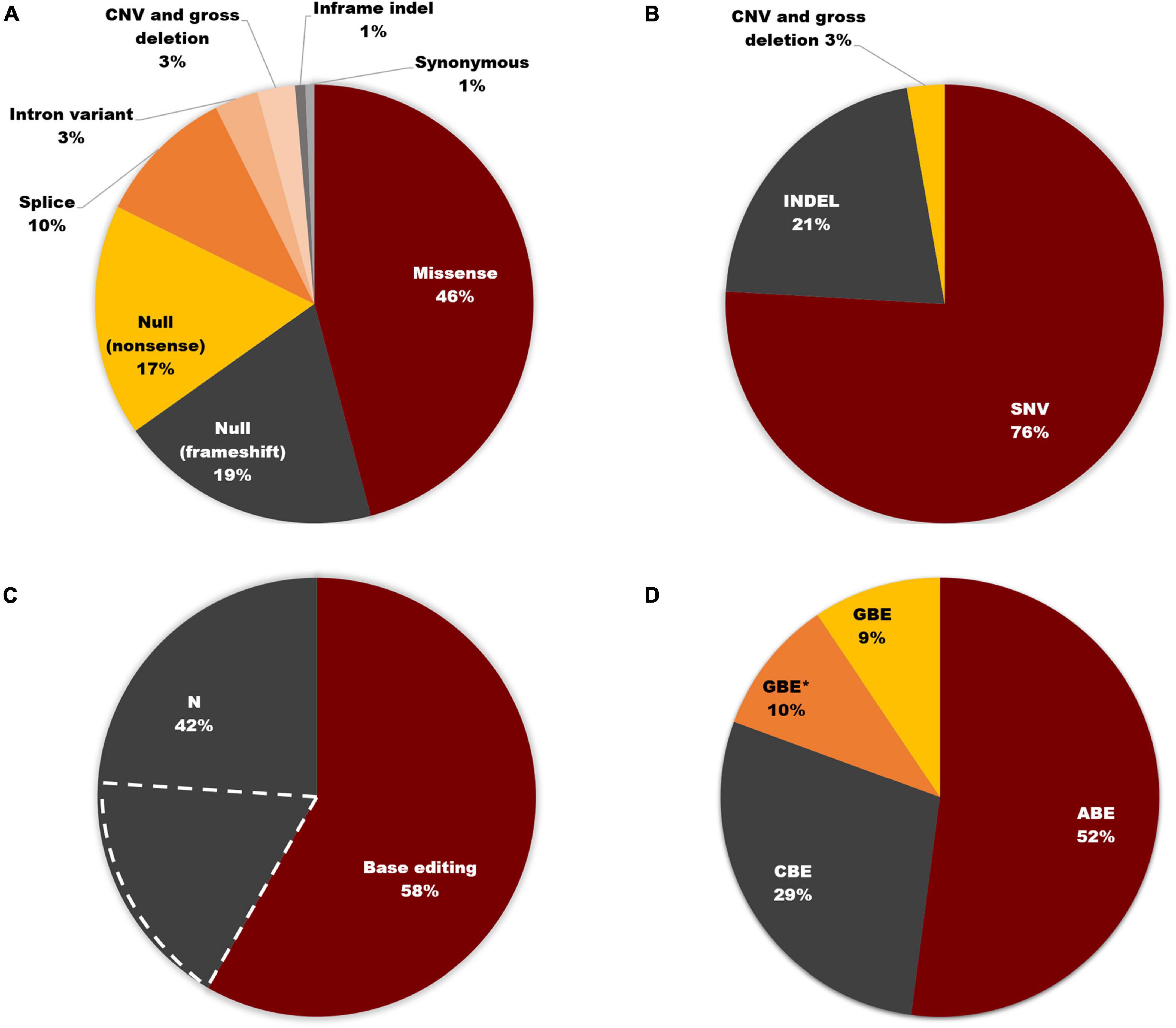
Figure 6. Base editing on USH2A. (A) Distribution of USH2A mutation types (n = 3,972 alleles). (B) SNVs account for 76% of all USH2A variants. (C) 58% of total variants can be corrected according to the editing principle of ABE, CBE, and GBE. The region marked by dashed white lines represents the SNVs that cannot be restored by three base editors. (D) Proportion of SNVs that can be corrected by different base editors. GBE with an asterisk (GBE*) represents SNVs in which the mutant nucleotide cannot be corrected through the original editing principle of ABE, CBE, and GBE, but the amino acid encoded will be restored by changing another base within the same codon using GBE.
Tremendous work has been done to seek therapeutic breakthroughs in hereditary retinal diseases (Chen et al., 2013; Mandai et al., 2017; Echigoya et al., 2018; Jin et al., 2019; Pendse et al., 2019; Zhang et al., 2021; Zhao et al., 2021). Significant progress has been achieved in research on cell replacement therapy, AON-based gene therapy, gene editing, etc. Among them, base editing has been promising to restore the vision in IRD patients. In the present study, we specifically described the possible application of three established base editors: ABE, CBE, and GBE on USH2A-related IRDs. Meanwhile, we predicted and delineated the proportion of USH2A mutations that can be corrected by base editing based on the global genetic landscape of USH2A that we depicted. SNV is the largest class of known human pathogenic mutations (Goodwin et al., 2016; Landrum et al., 2016). Thereby the correction of single point mutations will conceivably play an essential role in the future treatment of genetic disorders.
Among the USH2A SNVs, 18% of variants remained uncorrectable with these three base editors, and for all USH2A mutations, there are still 42% cannot be restored. The good news is that a new gene editing method termed “prime editing (PE)” has been developed. It is theoretically possible that PE can mediate indels and all SNVs (Anzalone et al., 2019), and a growing number of studies are underway to assess the editing efficacy, safety, and off-target concerns (Crunkhorn, 2021; Jiang et al., 2022).
Another promising approach for the future treatment of USH2A-related IRDs is exon skipping. Mutations in exon 13, including the recurrent mutations c.2299del, c.2276G > T, and c.2802T > G, are estimated to be responsible for approximately 18% of patients with USH2A-related IRDs around the world. Using antisense phosphorodiamidate morpholino oligomers (PMOs) to target exon 13 of zebrafish ush2a, Dulla et al. (2021) demonstrated that skipping of exon 13 using AON enables the synthesis of a shorter but functional USH2A protein, with the function of photoreceptor cells and retina restored.
Since mutations in USH2A are among the most common causes of non-syndromic RP and Usher syndromes, therapies targeting the hotspot mutant regions of USH2A would benefit a large proportion of IRD patients. In this large-scale retrospective study, we comprehensively reviewed nearly 4000 USH2A mutations from 33 genetic studies worldwide and presented the global mutation hotspot. Pathogenic mutations that affected ten of the 72 exons of USH2A were responsible for half of global USH2A mutant alleles. Thus, the rescue of dysfunction resulting from mutations on these exons may resolve more than half of USH2A-associated IRDs patients worldwide. Meanwhile, distribution and hotspot mutations of USH2A vary widely among different ethnic groups. For example, mutations in affected exon 43 were distinctly common in East Asia and the variant c.7595-2144A > G may be a unique mutation in Caucasians.
Although researchers from different countries have made a great effort to find genotype-phenotype correlations for USH2A mutations, no clear correlations have been established. It has been observed in a number of studies that patients with USH2 typically have an earlier onset and a faster clinical course of retinal degeneration than patients with non-syndromic RP. Protein-truncating variation has been demonstrated to correlate with earlier onset of vision loss (Blanco-Kelly et al., 2015; Lenassi et al., 2015; Pierrache et al., 2016). Furthermore, patients harboring biallelic truncating variants tended to exhibit more severe hearing loss than those with a single truncating variant or two non-truncating variants (Steele-Stallard et al., 2013). However, findings on genotype-phenotype correlation usually vary among studies of different scope and methodology. In our 51-patient cohort study, no statistical difference was found across three groups of different genotypes in disease progression and severity. Nevertheless, we found that patients harboring one or biallelic missense mutations were more likely to develop RP, which is similar to previous findings (Bork et al., 2001; Jiang et al., 2015; Inaba et al., 2020; Zhu et al., 2021). Meanwhile, the age at onset of both patients with biallelic null mutations and patients with both missense mutations was younger than that of patients with one missense and one null mutation, which is somewhat different from the findings of previous studies (Bork et al., 2001; Gao et al., 2021), indicating high heterogeneity of USH2A-related disorders. Patients with double null mutations presented a trend of reaching the state of low vision or legal blindness earlier than those who harbored one or no truncated mutations, but the differences were not statistically significant. Larger sample sizes and precise clinical follow-up information are needed. Clinical data of these large global samples need to be collected to search for a straightforward genotype-phenotype correlation.
In brief, we conducted a retrospective study of USH2A-related IRDs and provided a global mutation spectrum and hotspot in different regions, highlighting the critical therapeutic targets for the development of base editing treatments. Meanwhile, we revealed four novel mutations, expanding the spectrum of USH2A mutations, and reported a unique presentation of RPE depigmentation as patchy pale areas with no bone spicule on the posterior pole, increasing awareness of clinical manifestations of USH2A-related IRDs. Our findings provide a comprehensive perspective for exploring high-efficiency and broader-reaching gene editing and gene therapy.
The original contributions presented in this study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by Medical Ethics Committee of Beijing Tongren Hospital. The patients/participants provided their written informed consent to participate in this study.
Z-BJ designed the study, recruited the patients, recorded clinical data, and revised the manuscript. R-JS and Z-LL performed the experiments, ophthalmic examinations, and genetic analysis. B-NS and R-JS reviewed the literature and performed the analysis. B-NS wrote the manuscript. YL performed data analysis and revised the manuscript. All authors contributed to the article and approved the submitted version.
This study was supported by the National Natural Science Foundation of China (82125007) and National Key R&D Program of China (2017YFA0105300).
We would like to thank all the patients and family members for their participation in this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnagi.2022.948279/full#supplementary-material
Anzalone, A. V., Randolph, P. B., Davis, J. R., Sousa, A. A., Koblan, L. W., Levy, J. M., et al. (2019). Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157.
Berger, W., Kloeckener-Gruissem, B., and Neidhardt, J. (2010). The molecular basis of human retinal and vitreoretinal diseases. Prog Retin. Eye Res. 29, 335–375.
Blanco-Kelly, F., Jaijo, T., Aller, E., Avila-Fernandez, A., López-Molina, M. I., Giménez, A., et al. (2015). Clinical aspects of usher syndrome and the USH2A gene in a cohort of 433 patients. JAMA Ophthalmol. 133:157. doi: 10.1001/jamaophthalmol.2014.4498
Bork, J. M., Peters, L. M., Riazuddin, S., Bernstein, S. L., Ahmed, Z. M., Ness, S. L., et al. (2001). Usher syndrome 1D and nonsyndromic autosomal recessive deafness DFNB12 are caused by allelic mutations of the novel cadherin-like gene CDH23. Am. J. Hum. Genet. 68, 26–37. doi: 10.1086/316954
Carss, K. J., Arno, G., Erwood, M., Stephens, J., Sanchis-Juan, A., Hull, S., et al. (2017). Comprehensive rare variant analysis via whole-genome sequencing to determine the molecular pathology of inherited retinal disease. Am. J. Hum. Genet. 100, 75–90.
Chen, G., Shi, X., Sun, C., Li, M., Zhou, Q., Zhang, C., et al. (2013). VEGF-mediated proliferation of human adipose tissue-derived stem cells. PLoS One 8:e73673. doi: 10.1371/journal.pone.0073673
Crunkhorn, S. (2021). Prime editing rescues mice from genetic disorders. Nat. Rev. Drug Discov. 20:740.
Dulla, K., Slijkerman, R., van Diepen, H. C., Albert, S., Dona, M., Beumer, W., et al. (2021). Antisense oligonucleotide-based treatment of retinitis pigmentosa caused by USH2A exon 13 mutations. Mol. Ther. 29, 2441–2455. doi: 10.1016/j.ymthe.2021.04.024
Echigoya, Y., Lim, K. R. Q., Nakamura, A., and Yokota, T. (2018). Multiple exon skipping in the Duchenne muscular dystrophy hot spots: Prospects and challenges. J. Pers. Med. 8:41. doi: 10.3390/jpm8040041
Gao, F. J., Wang, D. D., Chen, F., Sun, H. X., Hu, F. Y., Xu, P., et al. (2021). Prevalence and genetic-phenotypic characteristics of patients with USH2A mutations in a large cohort of Chinese patients with inherited retinal disease. Br. J. Ophthalmol. 105, 87–92. doi: 10.1136/bjophthalmol-2020-315878
Goodwin, S., McPherson, J. D., and McCombie, W. R. (2016). Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 17, 333–351. doi: 10.1038/nrg.2016.49
Hartong, D. T., Berson, E. L., and Dryja, T. P. (2006). Retinitis pigmentosa. Lancet 368, 1795–1809.
Huang, X. F., Huang, F., Wu, K. C., Wu, J., Chen, J., Pang, C. P., et al. (2015). Genotype-phenotype correlation and mutation spectrum in a large cohort of patients with inherited retinal dystrophy revealed by next-generation sequencing. Genet. Med. 17, 271–278. doi: 10.1038/gim.2014.138
Inaba, A., Maeda, A., Yoshida, A., Kawai, K., Hirami, Y., Kurimoto, Y., et al. (2020). Truncating variants contribute to hearing loss and severe retinopathy in USH2A-associated retinitis pigmentosa in Japanese patients. Int. J. Mol. Sci. 21:7817. doi: 10.3390/ijms21217817
Jiang, L., Liang, X., Li, Y., Wang, J., Zaneveld, J. E., Wang, H., et al. (2015). Comprehensive molecular diagnosis of 67 Chinese Usher syndrome probands: High rate of ethnicity specific mutations in Chinese USH patients. Orphanet J. Rare Dis. 10:110. doi: 10.1186/s13023-015-0329-3
Jiang, T., Zhang, X. O., Weng, Z., and Xue, W. (2022). Deletion and replacement of long genomic sequences using prime editing. Nat. Biotechnol. 40, 227–234. doi: 10.1038/s41587-021-01026-y
Jin, Z. B., Gao, M. L., Deng, W. L., Wu, K. C., Sugita, S., Mandai, M., et al. (2019). Stemming retinal regeneration with pluripotent stem cells. Prog. Retin. Eye Res. 69, 38–56.
Kimberling, W. J., Weston, M. D., Moller, C., van Aarem, A., Cremers, C. W., Sumegi, J., et al. (1995). Gene mapping of Usher syndrome type IIA: Localization of the gene to a 2.1-cM segment on chromosome 1q41. Am. J. Hum. Genet. 56, 216–223.
Landrum, M. J., Lee, J. M., Benson, M., Brown, G., Chao, C., Chitipiralla, S., et al. (2016). ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 44, D862–D868.
Lenassi, E., Vincent, A., Li, Z., Saihan, Z., Coffey, A. J., Steele-Stallard, H. B., et al. (2015). A detailed clinical and molecular survey of subjects with nonsyndromic USH2A retinopathy reveals an allelic hierarchy of disease-causing variants. Eur. J. Hum. Genet. 23, 1318–1327. doi: 10.1038/ejhg.2014.283
Liu, X., Bulgakov, O. V., Darrow, K. N., Pawlyk, B., Adamian, M., Liberman, M. C., et al. (2007). Usherin is required for maintenance of retinal photoreceptors and normal development of cochlear hair cells. Proc. Natl. Acad. Sci. U.S.A. 104, 4413–4418. doi: 10.1073/pnas.0610950104
Mandai, M., Watanabe, A., Kurimoto, Y., Hirami, Y., Morinaga, C., Daimon, T., et al. (2017). Autologous induced stem-cell-derived retinal cells for macular degeneration. N. Engl. J. Med. 376, 1038–1046.
Nakanishi, H., Ohtsubo, M., Iwasaki, S., Hotta, Y., Mizuta, K., Mineta, H., et al. (2010). Hair roots as an mRNA source for mutation analysis of Usher syndrome-causing genes. J. Hum. Genet. 55, 701–703. doi: 10.1038/jhg.2010.83
Pendse, N. D., Lamas, V., Pawlyk, B. S., Maeder, M. L., Chen, Z. Y., Pierce, E. A., et al. (2019). In vivo assessment of potential therapeutic approaches for USH2A-associated diseases. Adv. Exp. Med. Biol. 1185, 91–96. doi: 10.1007/978-3-030-27378-1_15
Pierrache, L. H. M., Hartel, B. P., van Wijk, E., Meester-Smoor, M. A., Cremers, F. P., de Baere, E., et al. (2016). Visual prognosis in USH2A-associated retinitis pigmentosa is worse for patients with Usher syndrome type IIa than for those with nonsyndromic retinitis pigmentosa. Ophthalmology 123, 1151–1160. doi: 10.1016/j.ophtha.2016.01.021
Porto, E. M., Komor, A. C., Slaymaker, I. M., and Yeo, G. W. (2020). Base editing: Advances and therapeutic opportunities. Nat. Rev. Drug Discov. 19, 839–859.
Rees, H. A., and Liu, D. R. (2018). Publisher correction: Base editing: precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 19:801. doi: 10.1038/s41576-018-0068-0
Reurink, J., de Vrieze, E., Li, C. H. Z., van Berkel, E., Broekman, S., Aben, M., et al. (2022). Scrutinizing pathogenicity of the USH2A c.2276 G > T; p.(Cys759Phe) variant. NPJ Genom. Med. 7:37. doi: 10.1038/s41525-022-00306-z
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17, 405–424.
Slijkerman, R. W., Vaché, C., Dona, M., García-García, G., Claustres, M., Hetterschijt, L., et al. (2016). Antisense oligonucleotide-based splice correction for USH2A-associated retinal degeneration caused by a frequent deep-intronic mutation. Mol. Ther. Nucleic Acids 5:e381. doi: 10.1038/mtna.2016.89
Steele-Stallard, H. B., Le Quesne Stabej, P., Lenassi, E., Luxon, L. M., Claustres, M., Roux, A. F., et al. (2013). Screening for duplications, deletions and a common intronic mutation detects 35% of second mutations in patients with USH2A monoallelic mutations on Sanger sequencing. Orphanet J. Rare Dis. 8:122. doi: 10.1186/1750-1172-8-122
Stone, E. M., Andorf, J. L., Whitmore, S. S., DeLuca, A. P., Giacalone, J. C., Streb, L. M., et al. (2017). Clinically focused molecular investigation of 1000 consecutive families with inherited retinal disease. Ophthalmology 124, 1314–1331. doi: 10.1016/j.ophtha.2017.04.008
Vaché, C., Besnard, T., le Berre, P., García-García, G., Baux, D., Larrieu, L., et al. (2012). Usher syndrome type 2 caused by activation of an USH2A pseudoexon: Implications for diagnosis and therapy. Hum. Mutat. 33, 104–108. doi: 10.1002/humu.21634
van Wijk, E., Pennings, R. J. E., te Brinke, H., Claassen, A., Yntema, H. G., Hoefsloot, L. H., et al. (2004). Identification of 51 novel exons of the Usher syndrome type 2A (USH2A) gene that encode multiple conserved functional domains and that are mutated in patients with usher syndrome type II. Am. J. Hum. Genet. 74, 738–744. doi: 10.1086/383096
Weston, M. D., Eudy, J. D., Fujita, S., Yao, S., Usami, S., Cremers, C., et al. (2000). Genomic structure and identification of novel mutations in Usherin, the gene responsible for Usher syndrome type IIa. Am. J. Hum. Genet. 66, 1199–1210. doi: 10.1086/302855
Xu, W., Dai, H., Lu, T., Zhang, X., Dong, B., and Li, Y. (2011). Seven novel mutations in the long isoform of the USH2A gene in Chinese families with nonsyndromic retinitis pigmentosa and Usher syndrome Type II. Mol. Vis. 17, 1537–1552.
Zhang, H., Su, B., Jiao, L., Xu, Z. H., Zhang, C. J., Nie, J., et al. (2021). Transplantation of GMP-grade human iPSC-derived retinal pigment epithelial cells in rodent model: The first pre-clinical study for safety and efficacy in China. Ann. Transl. Med. 9:245. doi: 10.21037/atm-20-4707
Zhao, D., Li, J., Li, S., Xin, X., Hu, M., Price, M. A., et al. (2021). Glycosylase base editors enable C-to-A and C-to-G base changes. Nat. Biotechnol. 39, 35–40.
Keywords: USH2A, mutation hotspot, Usher syndrome, retinitis pigmentosa, gene editing, inherited retinal dystrophy
Citation: Su B-N, Shen R-J, Liu Z-L, Li Y and Jin Z-B (2022) Global spectrum of USH2A mutation in inherited retinal dystrophies: Prompt message for development of base editing therapy. Front. Aging Neurosci. 14:948279. doi: 10.3389/fnagi.2022.948279
Received: 19 May 2022; Accepted: 07 July 2022;
Published: 10 August 2022.
Edited by:
Enrico Maria Surace, University of Naples Federico II, ItalyReviewed by:
Danna Chen, Changsha Medical University, ChinaCopyright © 2022 Su, Shen, Liu, Li and Jin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zi-Bing Jin, amluemliaW5nQGZveG1haWwuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.