 Shan Huang1,2,3,4
Shan Huang1,2,3,4 Jun Wang3,4Dong-Yu Fan3,4,5Tong Luo3,4Yanli Li1Yun-Feng Tu3,4,6Ying-Ying Shen3,4Gui-Hua Zeng3,4Dong-Wan Chen3,4
Jun Wang3,4Dong-Yu Fan3,4,5Tong Luo3,4Yanli Li1Yun-Feng Tu3,4,6Ying-Ying Shen3,4Gui-Hua Zeng3,4Dong-Wan Chen3,4 Ye-Ran Wang3,4Li-Yong Chen7
Ye-Ran Wang3,4Li-Yong Chen7 Yan-Jiang Wang3,4,8,9*
Yan-Jiang Wang3,4,8,9* Junhong Guo1*
Junhong Guo1*- 1Department of Neurology, First Affiliated Hospital, Shanxi Medical University, Taiyuan, China
- 2First Clinical Medical College, Shanxi Medical University, Taiyuan, China
- 3Department of Neurology, Daping Hospital, Third Military Medical University, Chongqing, China
- 4Chongqing Key Laboratory of Ageing and Brain Diseases, Chongqing, China
- 5Shigatse Branch, Xinqiao Hospital, Third Military Medical University, Shigatse, China
- 6Department of Biomedical Engineering, Chongqing University, Chongqing, China
- 7Department of Anaesthesiology, Daping Hospital, Third Military Medical University, Chongqing, China
- 8State Key Laboratory of Trauma, Burn and Combined Injury, Institute of Surgery Research, Daping Hospital, Third Military Medical University, Chongqing, China
- 9Center for Excellence in Brain Science and Intelligence Technology, Chinese Academy of Sciences (CAS), Shanghai, China
Background: Cognitive impairment (CI) has become a worldwide health problem. The relationship between CI and uric acid (UA) is contradictory.
Objective: We included participants with a full spectrum of CI, from cognitively unimpaired (CU) to dementia, from the Chongqing Ageing & Dementia Study (CADS).
Methods: First, we identified the relationships between serum UA (sUA) and cognitive function in different stages of CI. Second, we analyzed these relationships among different stages and types of CI. Finally, we explored the association between sUA and amyloid/tangle/neurodegeneration (ATN) biomarkers.
Results: We recruited 427 participants from the CADS, including 382 participants with mini-mental state examination (MMSE) evaluation. The levels of sUA were positively correlated with MMSE scores (p < 0.001), and the correlation was prominent in the course of dementia and in the type of Alzheimer’s disease (AD). The levels of UA had a positive correlation with plasma amyloid-β 42 (Aβ42) (p = 0.004). Higher levels of sUA weakened the correlation of MMSE scores with CSF ATN biomarkers and the correlation of CSF Aβ42 with tau.
Conclusion: UA is positively correlated with cognitive function, especially in the advanced stage of AD. The probable neuroprotective effects of sUA mainly act on Aβ42 and the downstream pathological cascade.
Introduction
Dementia is currently the seventh leading cause of death among all diseases and one of the major causes of disability and dependency among older people. It is estimated that more than 55 million people live with dementia, and the number could rise to 78 million by 2030 and to 139 million by 2050 ([WHO], 2021b). Alzheimer’s disease (AD) is the most common type of age-related dementia, accounting for 60-70% of cases ([WHO], 2021a). However, the pathogenesis is not fully understood, and there is a lack of early diagnosis and effective therapy (Jia et al., 2020).
Oxidative damage is increasingly aggravated during aging and is an important pathological process of cognitive impairment (CI) and AD; in turn, oxidative damage can accelerate aging and AD development. The imbalance between the production of oxidative stress reactive oxygen species (ROS) and reactive nitrogen species (RNS), and antioxidant can contribute to the pathology of AD (Butterfield and Halliwell, 2019). For examples, oxidative damage could lead to the accumulation of amyloid-abeta (Aβ), neurofibrillary tangles, synaptic damage and microglia response (Cheignon et al., 2018; Buccellato et al., 2021).
Uric acid (UA), the final metabolite of purine, is regulated by diet intake, metabolism and renal excretion. UA is a natural antioxidant in the human body that scavenges oxygen free radicals. For examples, it can improve the removal of ROS, RNS, peroxyl radicals and hydrogen radicals and reduce the pathological process associated with oxidative stress in circulation, such as autoxidation of hemoglobin, lipid peroxidation and peroxide production by macrophage (Becker et al., 1991; Sautin and Johnson, 2008).
It has been reported that UA can prevent aging and age-related diseases (Becker et al., 1991; Sautin and Johnson, 2008). Thus, the relationship between UA and dementia has gained increasing attention. Some studies have suggested that serum UA (sUA) is a protective factor of cognition and is decreased in AD patients compared with cognitively unimpaired (CU) individuals (Polidori and Mecocci, 2002; Kim et al., 2006; Cankurtaran et al., 2013; Al-khateeb et al., 2015; Tana et al., 2018; Boccardi et al., 2021). Prospective studies have suggested that higher baseline sUA levels are associated with a lower risk of developing dementia and mild cognitive impairment (MCI) (Wang et al., 2017; Scheepers et al., 2019; Chen et al., 2021). However, UA could also be prooxidative and proinflammatory under certain conditions (So and Thorens, 2010; Kimura et al., 2021). Other studies have suggested that sUA may aggravate amyloidosis burden in the brain and have found higher sUA levels in participants with preclinical AD or mild-moderate AD (Zuliani et al., 2018; Li et al., 2021). We investigated the underlying reasons for the contradictory results and found that participants in these studies were in different disease stages, including preclinical, MCI, and dementia. In addition, the subtypes of dementia were different in these studies.
To investigate whether sUA functions and changes differently in different stages and types of CI, we observed the dynamic changes in sUA levels during the whole progression of AD and the relationship of sUA with different types of dementia. Furthermore, we analyzed the relationship of sUA and amyloid/tangle/neurodegeneration (ATN) biomarkers in a Chinese cohort.
Materials and methods
The inclusion and exclusion criteria of participants
This study was based on data from the Chongqing Ageing & Dementia Study (CADS). CADS is an on- going cohort study based on the southwest Chinese population and from Daping Hospital, aimed to explore the evolution mechanisms of ageing to AD and identify biomarkers of diagnosis and intervention for AD. Participants were recruited from CADS between January 2012 and January 2021. Participants were included with clinical evaluations, neuropsychological evaluations (mini-mental state examination (MMSE) evaluations, clinical dementia rating (CDR) scores etc.), and serum renal function biomarkers (including sUA) data. Participants were excluded for the following reasons: concomitant neurological disorders, including symptomatic stroke, intracranial infection, multiple sclerosis, Parkinson’s disease, epilepsy, metabolic encephalopathy, hydrocephalus etc.; psychiatric disorders, including schizophrenia, depression etc.; severe systematic disorders, including tumors, severe liver insufficiency, severe renal insufficiency and severe infection; refusal of blood sampling or lumbar puncture. This study was approved by the Institutional Review Board of Daping Hospital, and all participants and their caregivers provided informed consent.
Cerebrospinal fluid sampling and testing
Participants received lumbar puncture after a fast following the standard procedure, and cerebrospinal fluid (CSF) samples were collected in polypropylene tubes and centrifuged at 2,000 g and 4°C for 10 min (Blennow et al., 2010). These processed CSF samples were stored frozen at −80°C until used. The levels of CSF amyloid-β 40 (Aβ40), amyloid-β 42 (Aβ42), total tau (T-tau) and phosphorylated tau-181 (P-tau181) were analyzed using commercially available enzyme-linked immunosorbent assay (ELISA) kits (INNOTEST, Fujirebio Europe, Ghent, Belgium) (Carrillo et al., 2013). CSF neurofilament light chain (NFL) was analyzed using a single molecule array (SIMOA) onboard an automated SIMOA HD-1 analyzer (Quanterix United States). Preanalytical process and quality control strictly followed protocols from the Alzheimer’s Association guidelines, and our laboratory is one participant of the quality control (QC) program (code Lab129)(Mattsson et al., 2011, 2013; Hansson et al., 2021). Informed consent was obtained before sampling CSF, and the interval among CSF sampling and clinical evaluation was shorter than two weeks.
Blood sampling and testing
Blood samples were collected for the analysis of AD core biomarkers and basic tests, including renal function biomarkers. Fasting blood was collected between 07:00 a.m. and 09:00 a.m. A portion of blood samples was centrifuged within an hour of collection, and ethylenediaminetetraacetic acid plasma was aliquoted in 0.5-ml polypropylene tubes and stored at −80°C until the measurements of plasma AD core biomarkers (Aβ40, Aβ42, and T-tau) by a SIMOA Human Neurology 3-PlexA assay kit (Quanterix, United States) onboard the automated SIMOA HD-1 analyzer (Quanterix United States). Another portion of blood samples was used to measure basic tests, including apolipoprotein E (APOE) genotypes and sUA. APOE genotypes (rs429358 and rs7412) were determined by the polymerase chain reaction restriction fragment length polymorphism method. The sUA was measured on a Beckman AU-5800 instrument (Beckman Coulter, Inc., United States) by the uricase-peroxidase method (Reportable range: 89-1785 μmol/L; Sensitivity: the absorbance change value range was between 0.05 and 0.13 with a concentration of 314 μmol/L; Repeatability: the coefficient of variation of quality control was <5.0%; Inter-batch variability: the relative range of quality control between batches was <10.0%), and the biological reference intervals were 208.0-428.0 μmol/L for males and 155.0-357.0 μmol/L for females. Informed consent was obtained before sampling blood, and the interval among blood sampling and clinical evaluation was shorter than two weeks.
Cognitive assessment and Alzheimer’s disease diagnosis
Cognitive function was evaluated by adjusted MMSE scores and CDR scores. CU is defined as scores ≥28 for middle school education and above, scores ≥24 for primary school education and scores ≥21 for illiteracy (Folstein et al., 1975); MCI is defined as scores 25-27 for middle school education and above, scores 21-23 for primary school education and scores 18-20 for illiteracy, and MCI corresponds to the prodromal stage of dementia (Petersen et al., 1999; Winblad et al., 2004); dementia is defined as scores ≤24 for middle school education and above, scores ≤20 for primary school education and scores ≤17 for illiteracy (Folstein et al., 1975). CDR scores are used to evaluate the severity of dementia, and mild dementia is defined as scores = 1; moderate dementia is defined as scores = 2; and severe dementia is defined as scores = 3 (Morris, 1993).
Clinical diagnosis of AD was made according to the recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease in 2011 (McKhann et al., 2011; Jack et al., 2012). In CADS, CSF Aβ42 ≤ 933 pg/mL (A +), CSF phosphorylated tau181 (P-tau181) > 48.7 pg/mL (T +), and CSF total tau (T-tau) >313 pg/mL (N +) were used to define abnormal amyloid, tangle levels or neurodegeneration in the brain.
Statistical analysis
Normally distributed continuous variables are presented as the mean (standard deviation, SD), and non-normally distributed continuous variables are presented as the median (interquartile range, IQR). Categorical variables are presented as frequencies (percentages). Student’s t-test and analysis of variance were used to compare normally distributed continuous data across groups, and the Mann–Whitney U test and Kruskal–Wallis test were used to compare non-normally distributed continuous data. Regression analysis was used to identify the independent influencing factors. Analysis of covariates or multivariable model by regression analysis was used for adjusting the covariates of sUA after t-test or Mann–Whitney U test. The chi-squared test was used for categorical data. Pearson’s (normal distribution) or Spearman’s (non-normal distribution) analysis was used to evaluate correlations, and partial correlation analysis was used to adjust the covariates of sUA. Hierarchical regression analysis were used to evaluate the moderating effect. All hypothesis testing was two-sided, and p<0.05 was considered statistically significant. All statistical analyses were performed using R programming language (version 4.1.1) and SPSS software (version 26.0, Inc., Chicago, United States).
Results
Demographic characteristics and uric acid-related covariables
We recruited 427 participants from the CADS, including 382 participants with MMSE evaluation (Table 1). We screened UA-related covariables and found that age (p = 0.027), sex (p < 0.001), APOE gene (p = 0.007) and glomerular filtration rate (GFR) estimated by the Chronic Kidney Disease Epidemiology Collaboration creatinine equation (CKD-EPI eGFRCr) (p = 0.002) were related to sUA (Figure 1). Regression analysis suggested that the four covariables were independent influencing factors of sUA. In our cohorts, sUA had no correlation with hypertension, diabetes, dyslipidaemia or coronary heart disease (CHD).
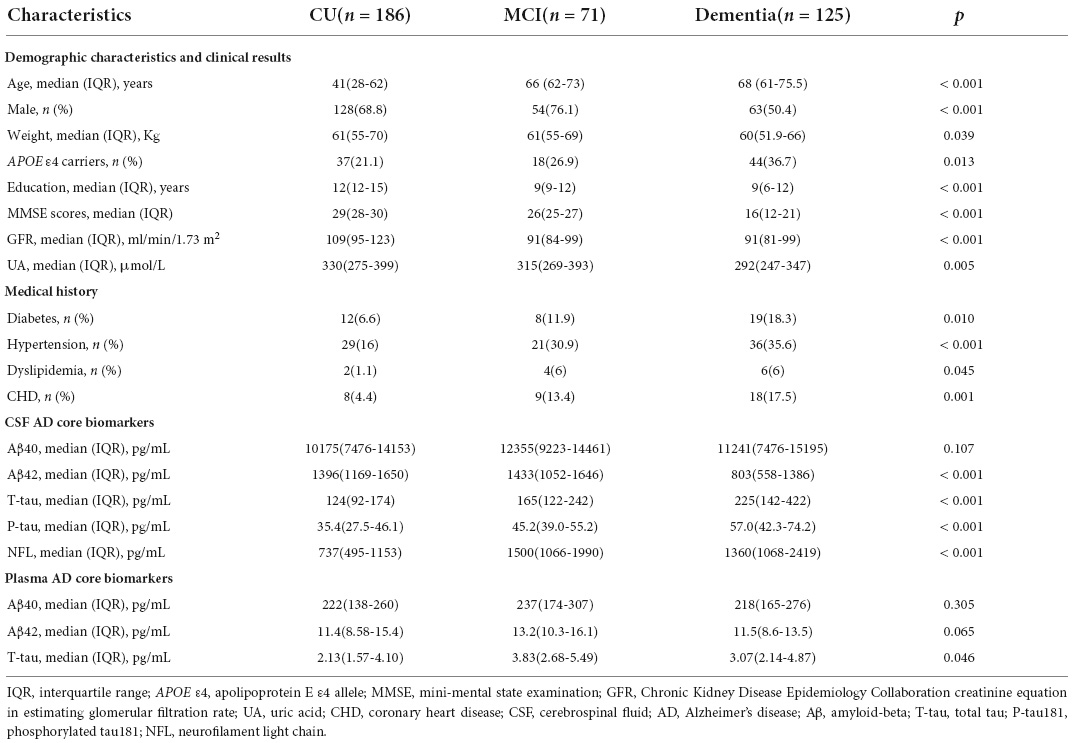
Table 1. Characteristics of the cohorts from CADS.
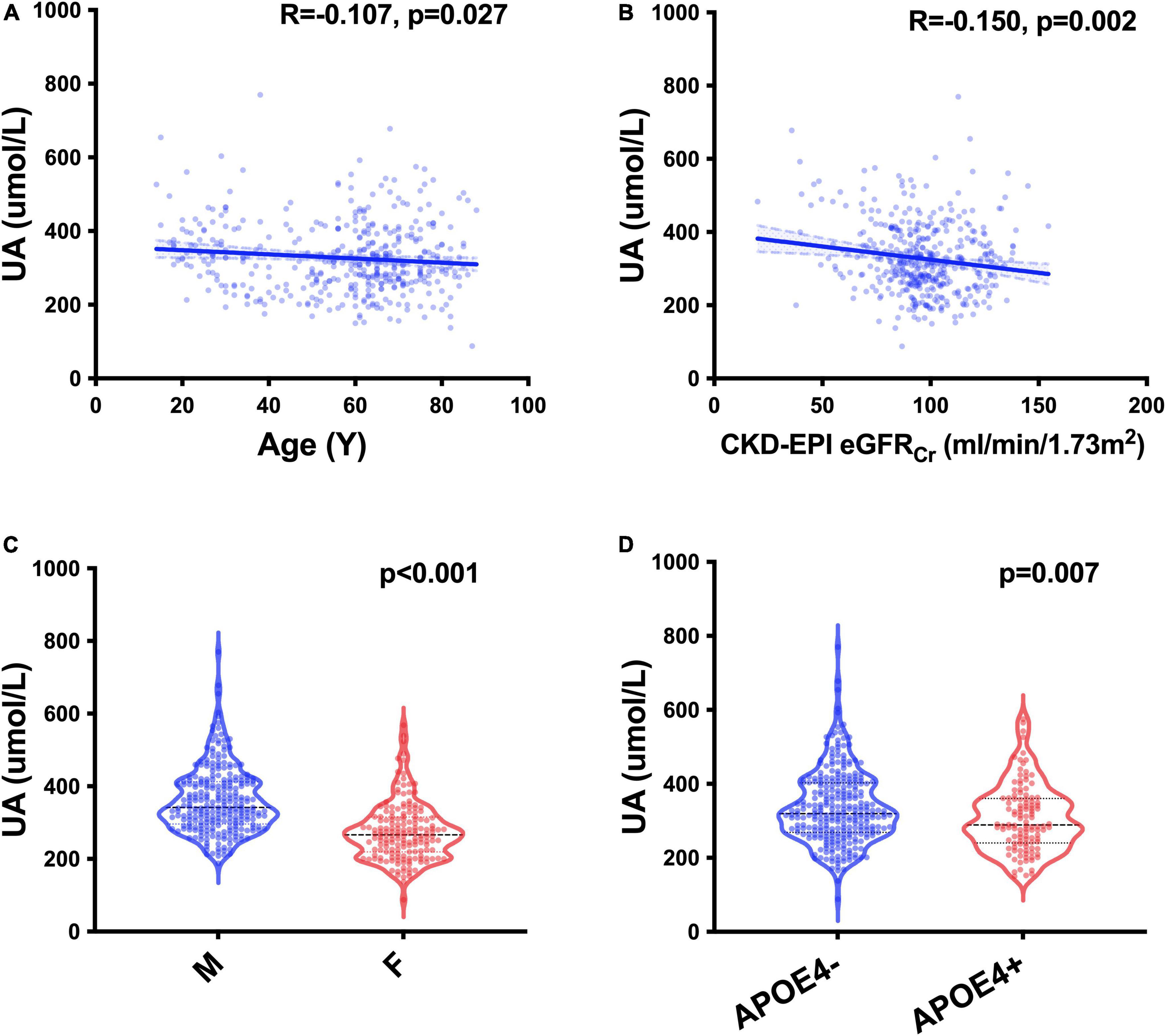
Figure 1. UA-related factors. Spearman’s correlation analysis between the levels of sUA and age (A) and CKD-EPI eGFRCr (B); Mann–Whitney U test of the levels of sUA grouped by sex (C) and the status of APOE ε4 carriers (D).
The relationships of uric acid and cognitive function
To investigate the relationship between UA and cognition, we first analyzed the correlation between the levels of sUA and MMSE scores, and there was a positive correlation between them (R = 0.228, p < 0.001) (Figure 2A). The correlation remained statistically significant after adjustment (p = 0.039). The levels of sUA were stratified into 4 subgroups from the lowest to the highest levels, and subgroups 1-4 corresponded to quartiles 1–4 (Q1 - Q4) of the sUA levels (male: (0, 296), [296, 341.9), [314.9, 412.7), [412.7, ∞) μmol/L; female: (0, 219.4), [219.4, 266.7), [266.7, 315.2), [315.2, ∞) μmol/L). We compared the proportion of CI phases among the subgroups of sUA from Q1 to Q4, and the proportion of dementia and MCI in Q1 (the lowest levels of sUA) was significantly higher than that in Q2 (p = 0.007), Q3 (p = 0.029) and Q4 (p = 0.029) (Figure 2B).
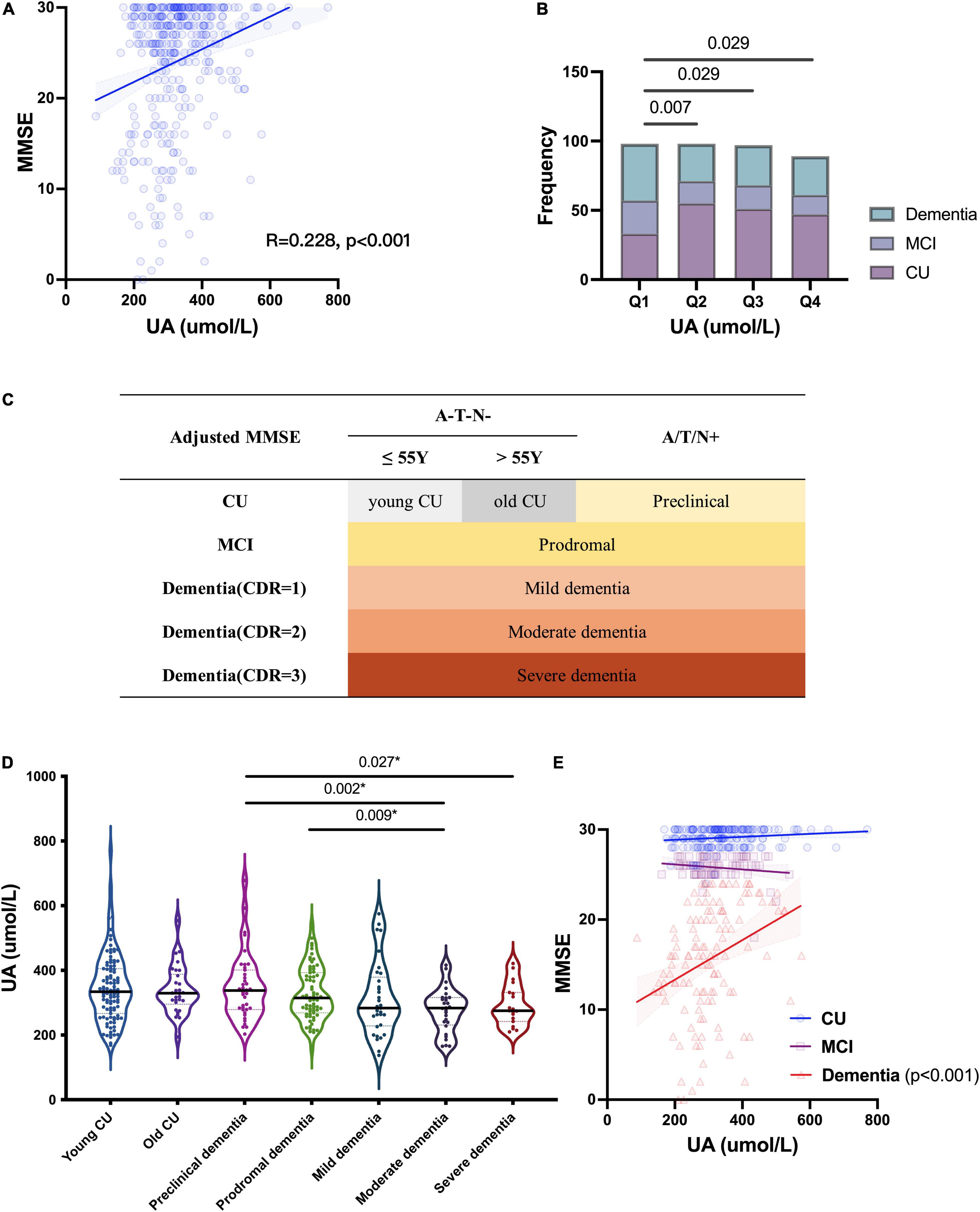
Figure 2. Relationship of UA and cognitive function. (A) Spearman’s correlation analysis between the levels of sUA and MMSE scores. (B) Proportion of CI phrases in different levels of sUA (Q1 - Q4: the levels of sUA were stratified into 4 subgroups from the lowest to the highest levels as quartiles 1–4). (C) Staging as MMSE, CDR and cut-offs of ATN biomarkers; A/T/N + means positive on at least one ATN biomarkers. (D) Kruskal–Wallis test for the levels of sUA among the different cognitive stages. The p value, two-tailed Mann–Whitney U test between two subgroups; (E) Spearman’s correlation analysis between the levels of sUA and MMSE scores in the stages CU, MCI and dementia. The p value, two-tailed chi-squared test.
Uric acid in different stages of cognitive impairment
Then, we presented sUA in the different stages of CI (Figure 2C). There is a dynamic decline in sUA levels with the progression of CI (Figure 2D). The levels of sUA were lower in moderate dementia than in preclinical dementia (p = 0.002) and MCI (p = 0.009) and lower in severe dementia than in preclinical dementia (p = 0.027) (Figure 2D). These differences were still statistically significant after adjustment (p = 0.003, 0.026, 0.029, respectively). We further verified the correlation between UA and cognition in different cognitive stages, and the positive correlation was more significant in dementia (R = 0.382, p < 0.001) than in the two earlier stages (Figure 2E).
Uric acid in different types of cognitive impairment
We divided participants into different types of CI: CU contained Aβ– young CU and Aβ– old CU; AD contained Aβ+ CU (preclinical AD), Aβ+ MCI (prodromal AD) and Aβ+ dementia; Non-AD CI contained Aβ– MCI and Aβ– dementia. MMSE scores were positively correlated with sUA in AD (R = 0.398, p = 0.002) but not in Aβ– old CU (R = 0.292, p = 0.118) or non-AD CI (R = 0.095, p = 0.334), and the positive correlation was still significant after adjustment (p = 0.013) (Figure 3A). The levels of sUA were lower in the Aβ+ dementia subgroup than in the Aβ– dementia subgroup (p = 0.007) (Figure 3B). Throughout the process of AD, we found that sUA levels first increased and then decreased, and the peak was in the stage of MCI (Figure 3C). The levels of sUA were lower in Aβ+ moderate dementia than in old CU (p < 0.001), Aβ+ CU (p = 0.018) and Aβ+ MCI (p = 0.003). These differences were still statistically significant after adjustment (p = 0.014, 0.018, 0.023, respectively).
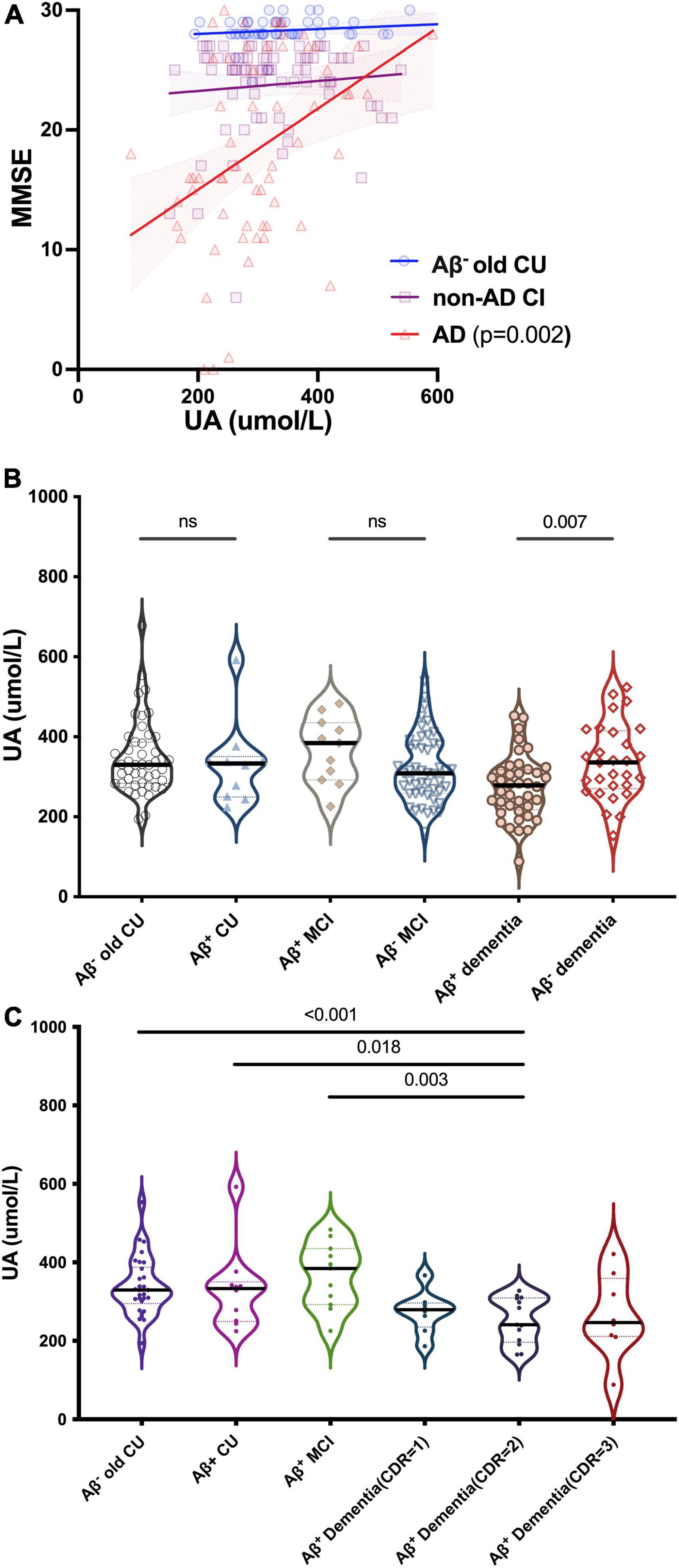
Figure 3. Relationships between UA and cognition in CU and different types of CI. (A) Spearman’s correlation analysis between the levels of sUA and MMSE scores in old CU, non-AD CI and AD. (B) Kruskal–Wallis test for the levels of sUA between cognitive subgroups. (C) Dynamic changes of sUA along for the process of AD. The p value, two-tailed Spearman’s correlation (A) or Mann–Whitney U test (B,C). ns, not significant.
Association between uric acid and ATN biomarkers
We analyzed the correlations between the levels of sUA and AD biomarkers in CSF and plasma. The levels of sUA had positive correlations with the levels of CSF Aβ42 (R = 0.133, p = 0.017), plasma Aβ42 (R = 0.211, p = 0.004) and plasma Aβ40 (R = 0.170, p = 0.019) (Figures 4A,G,H). There were no significant correlations between the levels of sUA and CSF Aβ40 (p = 0.082), CSF Aβ42/Aβ40 (p = 0.256), CSF P-tau181 (p = 0.082), CSF T-tau (p = 0.364), CSF NFL (p = 0.986) and plasma T-tau (p = 0.532) (Figures 4B-F,I). The difference in the correlation between plasma Aβ42 and sUA was still statistically significant after adjustment (p = 0.019). We also compared the levels of sUA grouping by the cut-offs of the ATN biomarkers. The levels of sUA were lower in A + than A− (p = 0.014) and in N + than N− (p = 0.030), and there were no significant differences in T + and T− (p = 0.058) (Figure 4J).
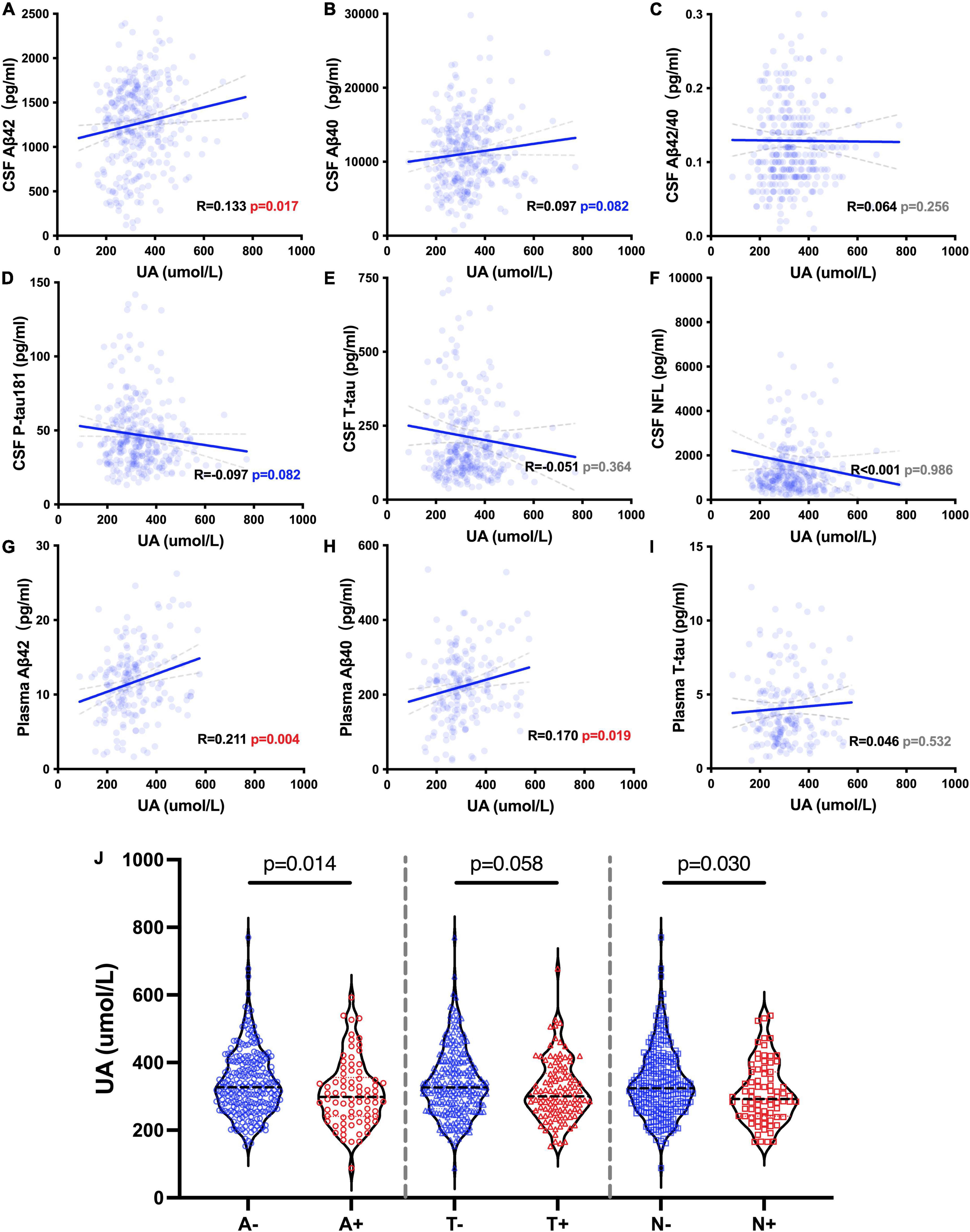
Figure 4. UA-related ATN biomarkers. (A–I) Spearman’s correlation analysis between the levels of sUA and ATN biomarkers. (J) Mann–Whitney U test between groups according to the cut-offs of CSF ATN biomarkers, including A– and A +, T– and T + and N– and N +.
We also explored whether UA moderated cognitive function by interacting with ATN biomarkers. The levels of sUA were stratified into 4 subgroups from the lowest to the highest levels as described above (Q1 – Q4). The analyses showed that high levels of sUA alleviated the association of CSF Aβ42 with MMSE scores (ΔR2 = 0.041, p < 0.001) and CSF P-tau181 with MMSE scores (ΔR2 = 0.024, p = 0.001) (Figures 5A,B), and sUA levels had no significant moderating effect on the association of CSF T-tau with MMSE scores (ΔR2 = 0.002, p = 0.381). The hyperphosphorylation of tau is the main cascade under Aβ42, so we explored whether sUA played a role in this key pathologic process of AD (Hardy and Higgins, 1992; Hardy and Selkoe, 2002). The analyses showed that high levels of sUA alleviated the association of CSF Aβ42 with CSF P-tau181 (ΔR2 = 0.037, p = 0.001) and CSF T-tau (ΔR2 = 0.044, p < 0.001) (Figure 5C, D).
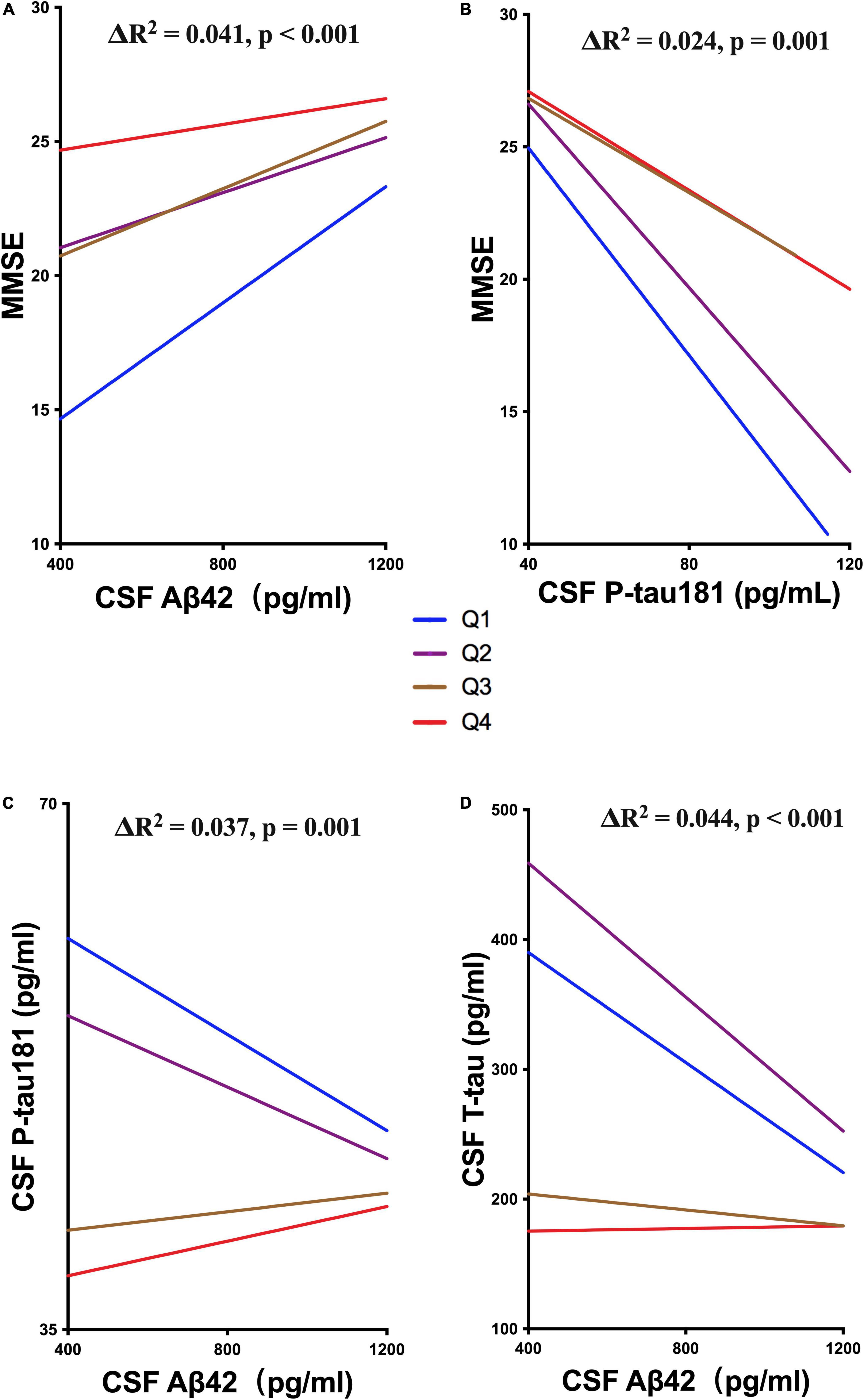
Figure 5. Moderating effect of UA and ATN biomarkers. (A,B) Hierarchical regression analysis of the moderating effect of the sUA levels in the association of CSF Aβ42 and P-tau181 with MMSE scores. (C,D) Hierarchical regression analysis of the moderating effect of the sUA levels in the association of CSF Aβ42 with P-tau181 and T-tau.
Discussion
UA is a result of purine metabolism, and it could be neuroprotective as a powerful antioxidant (mainly in blood) or neurotoxic as a prooxidant (mainly intracellular) or an inflammatory factor (Ames et al., 1981; Becker et al., 1991; So and Thorens, 2010; Kimura et al., 2021). As the results of our study, we tend to look at UA as a neuroprotective factor. Oxidative stress plays an important role in the pathological process of dementia, and UA has probable neuroprotective effects as a powerful antioxidant (Ames et al., 1981; Proctor, 2008). At the cellular or animal level, UA was found to protect hippocampal neurons from oxidative stress, and it can block nitrotyrosine formation in neurons and endothelial cells and maintain the integrity of the BBB (Hooper et al., 1998; Yu et al., 1998). Clinical studies have suggested that lower levels of UA could be a risk factor in multiple nervous system diseases, including AD (Schlesinger and Schlesinger, 2008; Peng et al., 2012; Abraham and Drory, 2014; Moccia et al., 2015; Scheepers et al., 2019; Guan et al., 2021). In AD, pathological biomarkers in the ATNX framework are associated with oxidative stress (Kaplow et al., 2020). UA could reduce the Aβ-induced tyrosine nitration of synaptophysin and the nitration and hyperphosphorylation of tau as a peroxynitrite scavenger (Tran et al., 2003; Zhang et al., 2006), and high levels of sUA could alleviate the effect of CSF Aβ42 and tau on cognitive decline (Ye et al., 2016).
UA could present different levels in different stages of CI. Taking AD as an example, first, several cross-sectional studies have observed higher levels of sUA in CI individuals than in CU individuals in preclinical AD or prodromal AD (Zuliani et al., 2018; Li et al., 2021). Second, longitudinal studies with sUA levels to predict the incidence of AD should pay attention to the stage of AD at baseline (Latourte et al., 2018). These results may not mean that sUA was a risk factor for AD, but there could be a compensatory increase in sUA levels for antioxidative effects confronting the early pathology in AD. In that light, high levels of sUA in the early phase of AD could be a biomarker of a high risk of dementia, which could suggest an attempted protective response. In addition, sUA could play different roles in different types of CI. Hyperuricaemia and gout could be linked to multiple comorbidities, including stroke, hypertension and diabetes, and these comorbidities are risk factors for dementia (Richette et al., 2020; Choi et al., 2022). The effect of UA on cognitive function primarily depends on the type of CI, and high sUA levels could lead to vascular dementia (VaD) but not AD (Cervellati et al., 2014; Latourte et al., 2018).
In our study, age, sex, APOE gene and GFR were independent influencing factors of sUA. These relationships suggested relatively low metabolism or insufficient nutrition in aged and female individuals. APOE is a metabolism-related gene, and the status of carrying APOE ε4 could be an interacting factor of UA. sUA excreted almost in the kidney, and renal dysfunction with a low GFR could lead to high levels of sUA.
There is a full spectrum of CI from CU to dementia, and we could take a panoramic view of the change in UA during the process of CI. We found a positive correlation between sUA levels and MMSE scores in AD patients, and the levels of sUA were lower in AD patients than in CU and non-AD patients, especially in the phase of dementia. The proportion of dementia and MCI was higher in group with lower levels of sUA. Additional analyses of UA and ATN biomarkers suggested the positive correlation of sUA with Aβ42, and sUA could alleviate the malignant effects of ATN biomarkers on cognitive function and Aβ42 on tau. The first increasing and then falling trend of sUA levels could be due to the early compensatory antioxidative function for Aβ42 and the later decompensation and self-exhaustion for anti-Aβ42 and the Aβ-tau pathological process or other amplifying vicious cascade in the later stage of AD. Therefore, sUA could be a protective factor of CI, and this effect predominantly acted on dementia stage and AD.
In the future, we should derive the exact causal relations between sUA and CI. Mendelian randomization (MR) studies could investigate the causal relationship of AD with UA, and more research based on familial AD cohorts is needed. There was a small quantity of data on hyperuricaemia, gout and hypouricaemia in our cohort. According to the present studies, high sUA levels were still protective not only in AD but also in VaD (Hong et al., 2015; Lu et al., 2016). Therefore, we should be committed to the management of UA in patients with AD and keep them in the status of relatively high levels of sUA based on the risk of cardiovascular diseases under control (Euser et al., 2009). According to the neuroprotective effects of UA, inosine, the precursor of urate, has been used in neurodegeneration disease as urate elevation therapy, and it could also be used in AD with hypouricaemia (Schwarzschild et al., 2021).
Conclusion
UA is positively correlated with cognitive function and could be a protective factor of AD, especially in the advanced stage. Throughout the process of AD, sUA levels first increased and then decreased, and the peak was in the stage of MCI. The probable neuroprotective effects of sUA mainly act on Aβ42 and the downstream pathological cascade.
Data availability statement
The original contributions presented in the study are included in this article/supplementary material, further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving human participants were reviewed and approved by Institutional Review Board of Daping Hospital. Written informed consent to participate in this study was provided by the participants or their legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
JG, Y-JW, and SH contributed to the study design, data interpretation, and writing of the report. JW, TL, YL, D-YF, Y-FT, Y-YS, G-HZ, D-WC, Y-RW, L-YC, SH, Y-JW, and JG contributed to the data collection, data analysis, data interpretation, and revision of the manuscript. All authors had full access to study data for interpretation and drafting of the report.
Funding
This study was supported by the National Natural Science Foundation of China (81930028) and the National Key R & D Program of China (2018YFA0109600).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abraham, A., and Drory, V. E. (2014). Influence of serum uric acid levels on prognosis and survival in amyotrophic lateral sclerosis: a meta-analysis. J. Neurol. 261, 1133–1138. doi: 10.1007/s00415-014-7331-x
Al-khateeb, E., Althaher, A., Al-khateeb, M., Al-Musawi, H., Azzouqah, O., Al-Shweiki, S., et al. (2015). Relation between uric acid and Alzheimer’s disease in elderly Jordanians. J. Alzheimer’s Dis. 44, 859–865. doi: 10.3233/JAD-142037
Ames, B. N., Cathcart, R., Schwiers, E., and Hochstein, P. (1981). Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: a hypothesis. Proc.Natl. Acad. Sci. U.S.A. 78, 6858–6862. doi: 10.1073/pnas.78.11.6858
Becker, B. F., Reinholz, N., Leipert, B., Raschke, P., Permanetter, B., and Gerlach, E. (1991). Role of uric acid as an endogenous radical scavenger and antioxidant. Chest 100:176S–181S.
Blennow, K., Hampel, H., Weiner, M., and Zetterberg, H. (2010). Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat. Rev. Neurol. 6, 131–144. doi: 10.1038/nrneurol.2010.4
Boccardi, V., Carino, S., Marinelli, E., Lapenna, M., Caironi, G., Bianco, A. R., et al. (2021). Uric acid and late-onset Alzheimer’s disease: results from the ReGAl 2.0 project. Aging Clin. Exp. Res. 33, 361–366. doi: 10.1007/s40520-020-01541-z
Buccellato, F. R., D’Anca, M., Fenoglio, C., Scarpini, E., and Galimberti, D. (2021). Role of Oxidative Damage in Alzheimer’s Disease and Neurodegeneration: From Pathogenic Mechanisms to Biomarker Discovery. Antioxidants 10:1353. doi: 10.3390/antiox10091353
Butterfield, D. A., and Halliwell, B. (2019). Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 20, 148–160. doi: 10.1038/s41583-019-0132-6
Cankurtaran, M., Yesil, Y., Kuyumcu, M. E., Oztürk, Z. A., Yavuz, B. B., Halil, M., et al. (2013). Altered levels of homocysteine and serum natural antioxidants links oxidative damage to Alzheimer’s disease. J. Alzheimer’s Dis. 33, 1051–1058. doi: 10.3233/JAD-2012-121630
Carrillo, M. C., Blennow, K., Soares, H., Lewczuk, P., Mattsson, N., Oberoi, P., et al. (2013). Global standardization measurement of cerebral spinal fluid for Alzheimer’s disease: an update from the Alzheimer’s Association Global Biomarkers Consortium. Alzheimers Dement. 9, 137–140. doi: 10.1016/j.jalz.2012.11.003
Cervellati, C., Romani, A., Seripa, D., Cremonini, E., Bosi, C., Magon, S., et al. (2014). Oxidative balance, homocysteine, and uric acid levels in older patients with Late Onset Alzheimer’s Disease or Vascular Dementia. J. Neurol. Sci. 337, 156–161. doi: 10.1016/j.jns.2013.11.041
Cheignon, C., Tomas, M., Bonnefont-Rousselot, D., Faller, P., Hureau, C., and Collin, F. (2018). Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 14, 450–464. doi: 10.1016/j.redox.2017.10.014
Chen, C., Li, X., Lv, Y., Yin, Z., Zhao, F., Liu, Y., et al. (2021). High Blood Uric Acid Is Associated With Reduced Risks of Mild Cognitive Impairment Among Older Adults in China: A 9-Year Prospective Cohort Study. Front. aging Neurosci. 13:747686. doi: 10.3389/fnagi.2021.747686
Choi, H. K., McCormick, N., and Yokose, C. (2022). Excess comorbidities in gout: the causal paradigm and pleiotropic approaches to care. Nat. Rev. Rheumatol. 18, 97–111. doi: 10.1038/s41584-021-00725-9
Euser, S. M., Hofman, A., Westendorp, R. G. J., and Breteler, M. M. B. (2009). Serum uric acid and cognitive function and dementia. Brain 132, 377–382. doi: 10.1093/brain/awn316
Folstein, M. F., Folstein, S. E., and McHugh, P. R. (1975). Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J. Psychiatric Res. 12, 189–198.
Guan, H., Geng, Z., Yuan, W., and Chang, B. (2021). Association of Serum Uric Acid Levels in Meige’s Syndrome. Front. Neurosci. 15:755056. doi: 10.3389/fnins.2021.755056
Hansson, O., Batrla, R., Brix, B., Carrillo, M. C., Corradini, V., Edelmayer, R. M., et al. (2021). The Alzheimer’s Association international guidelines for handling of cerebrospinal fluid for routine clinical measurements of amyloid β and tau. Alzheimer’s Dement. 17, 1575–1582. doi: 10.1002/alz.12316
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356.
Hardy, J. A., and Higgins, G. A. (1992). Alzheimer’s disease: the amyloid cascade hypothesis. Science 256, 184–185.
Hong, J.-Y., Lan, T.-Y., Tang, G.-J., Tang, C.-H., Chen, T.-J., and Lin, H.-Y. (2015). Gout and the risk of dementia: a nationwide population-based cohort study. Arthritis Res. Ther. 17:139. doi: 10.1186/s13075-015-0642-1
Hooper, D. C., Spitsin, S., Kean, R. B., Champion, J. M., Dickson, G. M., Chaudhry, I., et al. (1998). Uric acid, a natural scavenger of peroxynitrite, in experimental allergic encephalomyelitis and multiple sclerosis. Proc.Natl. Acad. Sci. U.S.A. 95, 675–680.
Jack, C. R., Knopman, D. S., Weigand, S. D., Wiste, H. J., Vemuri, P., Lowe, V., et al. (2012). An operational approach to National Institute on Aging-Alzheimer’s Association criteria for preclinical Alzheimer disease. Ann. Neurol. 71, 765–775. doi: 10.1002/ana.22628
Jia, L., Quan, M., Fu, Y., Zhao, T., Li, Y., Wei, C., et al. (2020). Dementia in China: epidemiology, clinical management, and research advances. Lancet Neurol. 19, 81–92. doi: 10.1016/S1474-4422(19)30290-X
Kaplow, J., Vandijck, M., Gray, J., Kanekiyo, M., Huyck, E., Traynham, C. J., et al. (2020). Concordance of Lumipulse cerebrospinal fluid t-tau/Aβ42 ratio with amyloid PET status. Alzheimer’s Dement. 16, 144–152. doi: 10.1002/alz.12000
Kim, T.-S., Pae, C.-U., Yoon, S.-J., Jang, W.-Y., Lee, N. J., Kim, J.-J., et al. (2006). Decreased plasma antioxidants in patients with Alzheimer’s disease. Int. J. Geriatric. Psychiatry 21, 344–348.
Kimura, Y., Tsukui, D., and Kono, H. (2021). Uric Acid in Inflammation and the Pathogenesis of Atherosclerosis. Int. J. Mol. Sci. 22, 12394. doi: 10.3390/ijms222212394
Latourte, A., Soumaré, A., Bardin, T., Perez-Ruiz, F., Debette, S., and Richette, P. (2018). Uric acid and incident dementia over 12 years of follow-up: a population-based cohort study. Ann. Rheumatic Dis. 77, 328–335. doi: 10.1136/annrheumdis-2016-210767
Li, L.-L., Ma, Y.-H., Bi, Y.-L., Sun, F.-R., Hu, H., Hou, X.-H., et al. (2021). Serum Uric Acid May Aggravate Alzheimer’s Disease Risk by Affecting Amyloidosis in Cognitively Intact Older Adults: The CABLE Study. J. Alzheimer’s Dis. 81, 389–401. doi: 10.3233/JAD-201192
Lu, N., Dubreuil, M., Zhang, Y., Neogi, T., Rai, S. K., Ascherio, A., et al. (2016). Gout and the risk of Alzheimer’s disease: a population-based, BMI-matched cohort study. Ann. Rheumatic Dis. 75, 547–551. doi: 10.1136/annrheumdis-2014-206917
Mattsson, N., Andreasson, U., Persson, S., Arai, H., Batish, S. D., Bernardini, S., et al. (2011). The Alzheimer’s Association external quality control program for cerebrospinal fluid biomarkers. Alzheimers Dement. 7:386–395.e6. doi: 10.1016/j.jalz.2011.05.2243
Mattsson, N., Andreasson, U., Persson, S., Carrillo, M. C., Collins, S., Chalbot, S., et al. (2013). CSF biomarker variability in the Alzheimer’s Association quality control program. Alzheimers Dement 9, 251–261. doi: 10.1016/j.jalz.2013.01.010
McKhann, G. M., Knopman, D. S., Chertkow, H., Hyman, B. T., Jack, C. R., Kawas, C. H., et al. (2011). The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 7, 263–269. doi: 10.1016/j.jalz.2011.03.005
Moccia, M., Lanzillo, R., Costabile, T., Russo, C., Carotenuto, A., Sasso, G., et al. (2015). Uric acid in relapsing-remitting multiple sclerosis: a 2-year longitudinal study. J. Neurol. 262, 961–967. doi: 10.1007/s00415-015-7666-y
Morris, J. C. (1993). The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 43, 2412–2414. doi: 10.1212/wnl.43.11.2412-a
Peng, F., Yang, Y., Liu, J., Jiang, Y., Zhu, C., Deng, X., et al. (2012). Low antioxidant status of serum uric acid, bilirubin and albumin in patients with neuromyelitis optica. Eur. J. Neurol. 19, 277–283. doi: 10.1111/j.1468-1331.2011.03488.x
Petersen, R. C., Smith, G. E., Waring, S. C., Ivnik, R. J., Tangalos, E. G., and Kokmen, E. (1999). Mild cognitive impairment: clinical characterization and outcome. Archives Neurol. 56, 303–308.
Polidori, M. C., and Mecocci, P. (2002). Plasma susceptibility to free radical-induced antioxidant consumption and lipid peroxidation is increased in very old subjects with Alzheimer disease. J. Alzheimer’s Dis. 4, 517–522. doi: 10.3233/jad-2002-4608
Proctor, P. H. (2008). Uric acid and neuroprotection. Stroke 39, e126. doi: 10.1161/STROKEAHA.108.524462
Richette, P., Doherty, M., Pascual, E., Barskova, V., Becce, F., Castaneda, J., et al. (2020). 2018 updated European League Against Rheumatism evidence-based recommendations for the diagnosis of gout. Ann. Rheumatic Dis. 79, 31–38. doi: 10.1136/annrheumdis-2019-215315
Sautin, Y. Y., and Johnson, R. J. (2008). Uric acid: the oxidant-antioxidant paradox. Nucleosides Nucleotides Nucleic Acids 27, 608–619. doi: 10.1080/15257770802138558
Scheepers, L. E. J. M., Jacobsson, L. T. H., Kern, S., Johansson, L., Dehlin, M., and Skoog, I. (2019). Urate and risk of Alzheimer’s disease and vascular dementia: a population-based study. Alzheimer’s Dement. 15, 754–763. doi: 10.1016/j.jalz.2019.01.014
Schlesinger, I., and Schlesinger, N. (2008). Uric acid in Parkinson’s disease. Mov. Disord. 23, 1653–1657. doi: 10.1002/mds.22139
Schwarzschild, M. A., Ascherio, A., Casaceli, C., Curhan, G. C., Fitzgerald, R., Kamp, C., et al. (2021). Effect of Urate-Elevating Inosine on Early Parkinson Disease Progression: The SURE-PD3 Randomized Clinical Trial. JAMA 326, 926–939. doi: 10.1001/jama.2021.10207
So, A., and Thorens, B. (2010). Uric acid transport and disease. J. Clin. Investig. 120, 1791–1799. doi: 10.1172/JCI42344
Tana, C., Ticinesi, A., Prati, B., Nouvenne, A., and Meschi, T. (2018). Uric Acid and Cognitive Function in Older Individuals. Nutrients 10:975. doi: 10.3390/nu10080975
Tran, M. H., Yamada, K., Nakajima, A., Mizuno, M., He, J., Kamei, H., et al. (2003). Tyrosine nitration of a synaptic protein synaptophysin contributes to amyloid beta-peptide-induced cholinergic dysfunction. Mol. Psychiatry 8, 407–412. doi: 10.1038/sj.mp.4001240
Wang, T., Wu, Y., Sun, Y., Zhai, L., and Zhang, D. (2017). A Prospective Study on the Association between Uric Acid and Cognitive Function among Middle-Aged and Older Chinese. J. Alzheimer’s Dis. 58, 79–86. doi: 10.3233/JAD-161243
Winblad, B., Palmer, K., Kivipelto, M., Jelic, V., Fratiglioni, L., Wahlund, L. O., et al. (2004). Mild cognitive impairment–beyond controversies, towards a consensus: report of the International Working Group on Mild Cognitive Impairment. J. Int. Med. 256, 240–246. doi: 10.1111/j.1365-2796.2004.01380.x
Ye, B. S., Lee, W. W., Ham, J. H., Lee, J. J., Lee, P. H., and Sohn, Y. H. (2016). Does serum uric acid act as a modulator of cerebrospinal fluid Alzheimer’s disease biomarker related cognitive decline? Eur. J. Neurol. 23, 948–957. doi: 10.1111/ene.12969
Yu, Z. F., Bruce-Keller, A. J., Goodman, Y., and Mattson, M. P. (1998). Uric acid protects neurons against excitotoxic and metabolic insults in cell culture, and against focal ischemic brain injury in vivo. J. Neurosci. Res. 53, 613–625. doi: 10.1002/(SICI)1097-4547(19980901)53:5<613::AID-JNR11>3.0.CO;2-1
Zhang, Y.-J., Xu, Y.-F., Liu, Y.-H., Yin, J., Li, H.-L., Wang, Q., et al. (2006). Peroxynitrite induces Alzheimer-like tau modifications and accumulation in rat brain and its underlying mechanisms. FASEB J 20, 1431–1442. doi: 10.1096/fj.05-5223com
Keywords: Alzheimer’s disease, uric acid, Aβ42, tau, cognitive impairment
Citation: Huang S, Wang J, Fan D-Y, Luo T, Li Y, Tu Y-F, Shen Y-Y, Zeng G-H, Chen D-W, Wang Y-R, Chen L-Y, Wang Y-J and Guo J (2022) The association of serum uric acid with cognitive impairment and ATN biomarkers. Front. Aging Neurosci. 14:943380. doi: 10.3389/fnagi.2022.943380
Received: 13 May 2022; Accepted: 29 June 2022;
Published: 18 July 2022.
Edited by:
Anupom Borah, Assam University, IndiaReviewed by:
Muhammed Khairujjaman Mazumder, Dhemaji College, IndiaManas Das, Gauhati University, India
Copyright © 2022 Huang, Wang, Fan, Luo, Li, Tu, Shen, Zeng, Chen, Wang, Chen, Wang and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan-Jiang Wang, eWFuamlhbmdfd2FuZ0B0bW11LmVkdS5jbg==; Junhong Guo, bmV1cm9ndW9AMTYzLmNvbQ==