
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Aging Neurosci. , 06 May 2022
Sec. Parkinson’s Disease and Aging-related Movement Disorders
Volume 14 - 2022 | https://doi.org/10.3389/fnagi.2022.897688
 Shun Yoshida1,2
Shun Yoshida1,2 Takafumi Hasegawa1*
Takafumi Hasegawa1*
Retromer is a highly integrated multimeric protein complex that mediates retrograde cargo sorting from endosomal compartments. In concert with its accessory proteins, the retromer drives packaged cargoes to tubular and vesicular structures, thereby transferring them to the trans-Golgi network or to the plasma membrane. In addition to the endosomal trafficking, the retromer machinery participates in mitochondrial dynamics and autophagic processes and thus contributes to cellular homeostasis. The retromer components and their associated molecules are expressed in different types of cells including neurons and glial cells, and accumulating evidence from genetic and biochemical studies suggests that retromer dysfunction is profoundly involved in the pathogenesis of neurodegenerative diseases including Alzheimer’s Disease and Parkinson’s disease. Moreover, targeting retromer components could alleviate the neurodegenerative process, suggesting that the retromer complex may serve as a promising therapeutic target. In this review, we will provide the latest insight into the regulatory mechanisms of retromer and discuss how its dysfunction influences the pathological process leading to neurodegeneration.
Membrane trafficking is an evolutionarily conserved cellular process by which proteins and other macromolecules reach their destinations without crossing a membrane. Multiple lines of evidence have revealed that the defects in membrane trafficking are profoundly involved in the pathogenesis of neurodegenerative diseases (Hasegawa et al., 2017a). In particular, much interest has been focused on retromer because recent genetic and biological studies have underscored the significance of the retromer sorting machinery in the pathogenesis of Alzheimer’s Disease (AD) and Parkinson’s disease (PD) (Zhang et al., 2018). Retromer is considered a master regulator of retrograde cargo trafficking, e.g., transport from early endosomes (EEs) to the trans-Golgi network (TGN) and the plasma membrane (Seaman, 2021). On the other hand, retromer participates in the mitochondrial dynamics and the autophagic system, which are key processes in the maintenance of neuronal homeostasis (Cui et al., 2018). Moreover, pharmacological chaperones that stabilize retromer function successfully prevent neurodegeneration in cellular and animal models, suggesting that retromer is a promising target for disease-modifying therapy (Seaman, 2021). In this review, we will summarize the molecular basis of retromer function and discuss its pleiotropic roles in the causation and prevention of neurodegeneration.
The term “retromer” was first used to describe an essential protein complex for the transport of vacuolar protein sorting 10p (Vps10p), a transmembrane receptor, from endosomes to the TGN in Saccharomyces cerevisiae (Seaman et al., 1998). Structurally, the retromer complex comprises five distinct proteins, namely Vps26p, Vps29p, Vps35p, Vps5p, and Vps17p.
In mammals, retromer usually comprises a VPS26/VPS29/VPS35 heterotrimer complex because it lacks robust interaction with sorting nexin 1 (SNX1), a mammalian homolog of Vps5p (Seaman, 2021). In cooperation with the VPS26/VPS29/VPS35 trimeric structure, SNX orchestrates endosomal cargo sorting (Figure 1). Similarly, as their yeast counterpart Vps5p, SNX1 and SNX2 in mammals carry a Bim/Amphiphysin/Rvs (BAR) domain that drives membrane curvature and tubulation on the endosomal membrane (Carlton et al., 2005). Although the affinity of SNX1/2 for retromer is rather weak and transient, these SNX synergistically function in cargo retrieval from endosomes to the TGN (Bujny et al., 2007; Rojas et al., 2007). In addition, SNX5 and SNX6, which form heterodimers with SNX1/2, can directly interact with retromer cargoes such as cation-independent mannose-6-phosphate receptor (CI-MPR) (Wassmer et al., 2009; Simonetti et al., 2017; Yong et al., 2020). Likewise, SNX27 binds retromer subunit VPS26, and these proteins cooperatively drive the cargo transport from endosomes to the plasma membrane (Steinberg et al., 2013; Gallon et al., 2014).
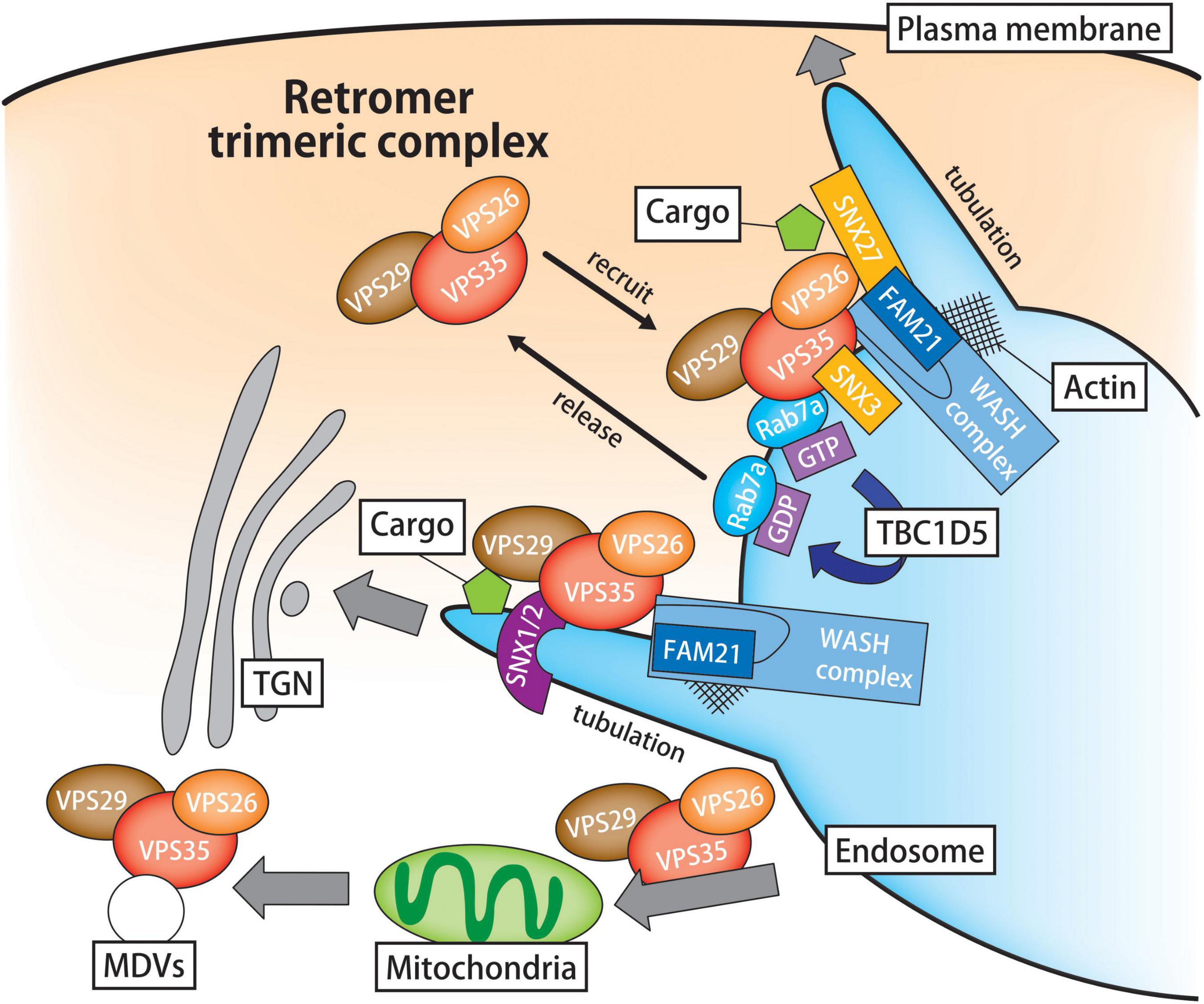
Figure 1. Schematic illustration of the retromer-mediated sorting pathway. Retromer is a hetero-trimeric protein complex composed of VPS26, VPS29, and VPS35. In cooperation with its associated proteins, the retromer sorting machine plays a primary role in the retrograde cargo trafficking from the endosomes to the TGN or plasma membrane. In the first step of retromer-mediated cargo transport, the retromer core is recruited to the endosomal surface under the control of Rab7a and SNX3. Because the endosomal recruitment of retromer complex largely depends on Rab7 activity, the inactivation of Rab7a by TBC1D5 promotes the release of retromer from endosomes. Lipid membrane deformation is crucial for intracellular trafficking and organelle remodeling. In the endosomal microdomain, the WASH complex modulates the process of endosomal tubulation through the activation of actin nucleation and polymerization. In addition to endosomal transport, retromer participates in the mitochondria dynamics through the cargo trafficking from the endosomes to the mitochondria and MDV-mediated trafficking. VPS, vacuolar protein sorting; TGN, trans-Golgi network; SNX, sorting nexin; TBC1D5, TBC1 domain family member 5; WASH, Wiskott-Aldrich Syndrome protein and scar homolog.
In the first step of retromer-dependent cargo transport, the retromer core is recruited to the endosomal surface under the control of Rab7a and SNX3 (Rojas et al., 2008; Seaman et al., 2009). Interestingly, the inactivation of Rab7a by TBC1 domain family member 5 (TBC1D5), a Rab7a GTPase-activating protein (GAP), promotes the release of retromer from endosomes (Seaman et al., 2009; Ye et al., 2020). Meanwhile, SNX3 binds to the endosomal membrane, thereby initiating retromer-mediated retrograde transport irrespective of SNX1/2 and SNX5/6 (Strochlic et al., 2007; Harterink et al., 2011). In the endosomal microdomain, the Wiskott-Aldrich syndrome protein and scar homolog (WASH) complex modulates the process of endosomal tubulation (Linardopoulou et al., 2007; Gomez and Billadeau, 2009) (Figure 1). The WASH molecular machinery is a macromolecular protein complex composed of WASH1, FAM21, strumpellin- and WASH-interacting protein, strumpellin and coiled-coil domain-containing protein 53. VPS35 interacts with the unstructured C-terminal tail of FAM21 and recruits FAM21 to endosomes, whereas the FAM21-WASH interaction occurs through its N-terminus, thereby regulating actin polymerization (Harbour et al., 2012; Hao et al., 2013). Additionally, FAM21 interacts with SNX27, directing SNX27-retromer cargoes to the plasma membrane (Temkin et al., 2011).
Apart from the endosomal cargo sorting, the retromer is involved in mitochondrial dynamics and the autophagic system (Figure 1). An unbiased molecular screening revealed that both VPS35 and VPS26 bind the mitochondrial-anchored protein ligase (MAPL) (Braschi et al., 2010). The recruitment of VPS35 to mitochondria regulates the transport of MAPL to peroxisomes via mitochondrial-derived vesicles (MDVs). Moreover, VPS35 participates in the recycling of dynamin-like protein 1 (DLP1), a mitochondrial fission protein (Wang W. et al., 2016). In that sense, retromer may control mitochondrial dynamics through cargo-protein trafficking. Another line of evidence suggested a regulatory role for retromer in autophagy lysosomal pathway. Actually, proteomics analysis of autophagosome composition in MCF7 cells identified VPS35 as an autophagosome-associated protein (Dengjel et al., 2012). When retromer is depleted, Atg9 aberrantly remains in EEs and interferes with subsequent autophagosome formation (Ravussin et al., 2021). In addition, retromer participates in mitophagy by regulating Rab7 activity with TBC1D5 (Jimenez-Orgaz et al., 2018). Cumulatively, these findings provide a scientific basis for the fundamental role of retromer in the maintenance of cellular homeostasis and stress tolerance.
Alzheimer’s Disease (AD) is the most common cause of progressive dementia among older populations. The histopathological signature of AD is the deposit of extracellular senile plaques and intracellular neurofibrillary tangles, which are composed mainly of aggregated amyloid-β (Aβ) and phosphorylated tau, respectively (DeTure and Dickson, 2019). The synergistic neurotoxicity of these two proteins in AD has been extensively studied, and growing evidence suggests that the retromer sorting pathway exerts a substantial impact on the generation of AD pathology through Aβ production and tau accumulation (Zhang et al., 2018). Microarray analysis using the entorhinal cortex and the dentate gyrus from the autopsied brain tissue of patients with AD demonstrated that the expression of the retromer subunits VPS35 and VPS26 is markedly reduced at both the mRNA and protein levels. This finding is further corroborated by experiments in a cultured cellular model revealing that VPS35 silencing leads to a significant increase of endogenous Aβ level (Small et al., 2005). Like AD, the expression level of VPS35 is significantly decreased in the brains of patients with distinct primary tauopathies such as progressive supranuclear palsy (PSP) and Pick’s disease, and downregulation of VPS35 results in the exacerbation of motor and learning impairments and accumulation of pathological tau in a relevant mouse model (Vagnozzi et al., 2019). Moreover, a gene-association study between AD and single nucleotide polymorphisms (SNPs) in 15 retromer-related genes revealed a positive association for several retromer-associated genes (e.g., SNX3, RAB7A, KIAA1033, and SNX1) (Vardarajan et al., 2012). Furthermore, copy number variation analysis and whole exome sequencing in sporadic early-onset AD identified a de novo deleterious variant (L625P) in VPS35 in a French cohort (Rovelet-Lecrux et al., 2015). The pathogenic role of retromer in AD is also supported by studies using different animal models. Human amyloid precursor protein (APP) transgenic (Tg) mice (Tg2576 and J20) exhibit a progressive decrease in the expression levels of VPS35, VPS26, and CI-MPR (Chu and Praticò, 2017; Tammineni et al., 2017). In Macaca fascicularis, an age-dependent decline in the endosomal sorting machinery including the retromer is closely related to the intracellular accumulation of APP and Aβ (Kimura et al., 2009, 2016). Lifestyle-related diseases, such as hypertension and diabetes, are known as major risk factors for AD, and interestingly, a mouse model of type 2 diabetes revealed hippocampus-specific retromer deficiency similarly as observed in an APP Tg mouse model (Morabito et al., 2014).
A plethora of evidence suggests that the endosomal sorting machinery including the retromer has a great impact on the biogenesis and transport of Aβ peptides in healthy and diseased brains (Willén et al., 2017; Kimura and Yanagisawa, 2018). Cellular and animal model studies demonstrated that retromer deficiency facilitates the buildup of toxic Aβ oligomers in the endosomal compartments, resulting in abnormal endosomal enlargement and subsequent neuronal cell death (Muhammad et al., 2008; Wen et al., 2011; Bhalla et al., 2012; Ansell-Schultz et al., 2018). In the amyloidogenic pathway, Aβ synthesis is initiated through the proteolytic cleavage of APP by β-secretase [β-APP-cleaving enzyme-1 (BACE1)] on the plasma membrane, TGN, and EEs, followed by the transport to the multivesicular bodies (MVBs) (Rajendran et al., 2006; Burgos et al., 2010; Willén et al., 2017). BACE1 produces the N-terminal fragment of APP called soluble peptide APPβ, and the C-terminal fragment of APP named β-CTF (also known as C99). Subsequently, the Aβ peptide is generated as a fragment, in which β-CTF is cleaved by γ-secretase within endosomes. Importantly, the retromer complex contributes to the retrograde transport of APP, BACE1, γ-secretase, and related proteins from the endosomes, and thus, its malfunction causes the aberrant endosomal retention of these molecules, leading to the overproduction of Aβ (Wen et al., 2011; Bhalla et al., 2012; Choy et al., 2012; Cuartero et al., 2012; Kanatsu et al., 2018).
The retrograde trafficking of APP is mediated by the Vps10 domain-containing proteins SorLA and sortilin-related Vps10 domain-containing receptor 1 (SorCS1) through their interactions with the retromer complex (Pallesen and Vaegter, 2012) (Figure 2). Intriguingly, the SORL1 gene, a gene encoding SorLA which is abundantly expressed in the central nervous system, is associated with both late- and early-onset forms of AD (Rogaeva et al., 2007; Pottier et al., 2012). In addition, the protein expression of SorLA is significantly lower in brain tissue and cerebrospinal fluid (CSF) from patients with sporadic AD compared to controls, (Scherzer et al., 2004; Ma et al., 2009). Intriguingly the reduced expression of SorLA in the brains begins even in the prodromal phase of AD, and low SorLA expression is correlated with cognitive function (Sager et al., 2007). In agreement with these findings, overexpression of SorLA decreased the expression level of APP and Aβ in cellular and mouse models, whereas loss of SorLA increased the Aβ load (Andersen et al., 2005; Offe et al., 2006; Schmidt et al., 2007; Dodson et al., 2008). Mechanistically, SorLA co-localizes with APP in EEs, and transports APP to the TGN in association with the retromer complex (Andersen et al., 2005; Fjorback et al., 2012). Collectively, these results suggest that the lack of interaction between APP and SorLA perturbs the retrograde trafficking of APP and SorLA from EEs to the TGN or plasma membrane, leading to aberrant endosomal retention of APP and Aβ in the AD brain.
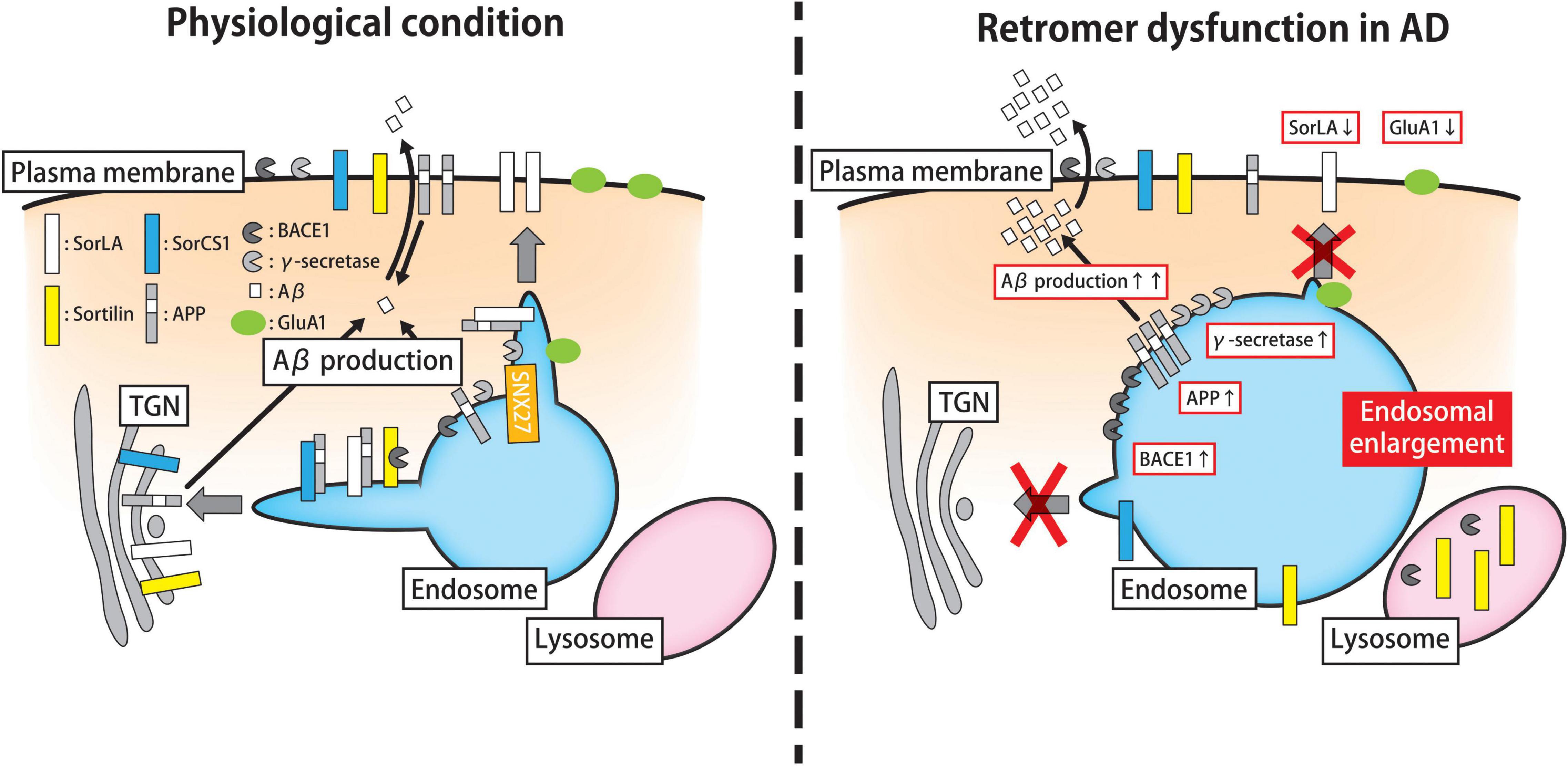
Figure 2. Roles of the retromer machinery in the trafficking and metabolism of AD-related proteins. Endosomal trafficking plays a key role in the processing of APP and the biogenesis of Aβ peptides. Under physiological conditions (left panel), the Aβ peptide is synthesized through the proteolytic cleavage of APP by BACE1 (also known as β-secretase) and γ-secretase on the plasma membrane, early endosomes, and TGN. Retromer complex contributes to the retrograde transport of APP, BACE1, γ-secretase, and related proteins (e.g., SorLA and SorCS1) from endosomes. The SorLA-mediated transport of APP to the plasma membrane requires the support of SNX27. In addition to the endosomal pathway, the retromer might participate in surface recycling of the AMPAR subunit GluA1, thereby modulating synaptic plasticity. In AD brains, in which retromer function is compromised (right panel), APP, BACE1, γ-secretase, and associated proteins accumulate in early endosomes, thereby increasing Aβ production with hypertrophic changes in endosomal compartments. Besides the amyloidogenic pathway, retromer malfunction perturbs the cell surface recycling of GluA1, which may influence on synaptic plasticity. APP, amyloid precursor protein; Aβ, amyloid-β; BACE1, β-APP-cleaving enzyme-1; TGN, trans-Golgi network; SNX sorting nexin; AMPAR, α-amino-3-hydroxy-5-methyl-4-isoxazole propionate receptor; AD, Alzheimer’s Disease.
Notably, the SorLA-mediated retrieval of APP to the plasma membrane is regulated by SNX27; thus, the depletion of SNX27 leads to the accumulation of SorLA and APP in EEs, thereby promoting Aβ production (Huang et al., 2016). Indeed, the coding variants of SORL1 identified in the familial and sporadic forms of AD bind APP less well, and HEK293 cells overexpressing mutant SORL1 displayed increased Aβ secretion in culture medium (Vardarajan et al., 2015). Similarly, SorLA-deficient human induced pluripotent stem cell (iPSC)-derived neurons specifically exhibit the abnormal enlargement of EEs with APP accumulation, which mimics affected neurons in AD brains (Cataldo et al., 2000; Knupp et al., 2020). Moreover, genetic cohort studies demonstrated that variants in SORCS1, a gene encoding another Vps10 family protein, are significantly associated with AD as well as type 1 and type 2 diabetes mellitus (Goodarzi et al., 2007; Paterson et al., 2010; Reitz et al., 2011). Similarly as SorLA protein, overexpression of SORCS1 in HEK293 cells transfected with mutant APP reduces Aβ secretion into the culture medium. Conversely, SORCS1 silencing significantly increases Aβ secretion together with the decline of VPS35 level in the mice brain (Lane et al., 2010; Reitz et al., 2011). Regarding the mode of action, it is likely that SORCS1 does not directly modulate the endocytic uptake of APP, but rather, it regulates the exit of APP and/or CTFs out of EEs, resulting in increased APP translocation to the TGN as well as decreased Aβ secretion (Lane et al., 2010, 2013).
The scission of APP by BACE1 is putatively the rate-limiting step in Aβ synthesis. As a type 1 transmembrane aspartic protease, BACE1 activity is highest in acidic compartments including the endosomal compartments and TGN, making it plausible that the regulation of the post-Golgi transport of BACE1 plays an important role in the processing of APP and Aβ genesis (Sun and Zhang, 2017). In neuronal cells, BACE1 is transported from EEs to the TGN through retromer-mediated transport, and thus, the loss of retromer function promotes the retention of BACE1 in endosomes, resulting in increased binding of APP to BACE1 and consequently, Aβ production (Wen et al., 2011; Cuartero et al., 2012) (Figure 2). During this process, sortilin, a Vps10 domain-containing protein, cooperatively regulates the trafficking of BACE1. Specifically, sortilin on the cell surface is taken up by cells via adaptor protein 1-dependent endocytosis, which is followed by transport to the TGN with BACE1 (Canuel et al., 2008; Finan et al., 2011). The expression level of sortilin is correlated with Aβ production and is markedly elevated in the brain tissue of patients with AD and retromer-deficient mice (Kim et al., 2010; Finan et al., 2011). Supporting this result, the N-terminal fragments of two BACE1 substrates, namely APP-like 1 and close homolog of L1, are substantially increased in the CSF of forebrain-specific Vps35 KO mice and patients in the prodromal stage of AD (Simoes et al., 2020). Altogether, these findings strongly suggest that the subcellular trafficking of BACE1 and the amyloidogenic APP processing pathway largely depend on the retromer function.
In addition to the amyloidogenic pathway, the retromer machinery is likely to modulate the subcellular trafficking of cargo molecules related to AD pathogenesis. One example is triggering receptor expressed on myeloid cells 2 (TREM2), a risk gene for AD and an important regulator of microglial functions (Guerreiro et al., 2013; Jonsson et al., 2013; Lee et al., 2018). TREM2 is a transmembrane receptor of the immunoglobulin superfamily that is mainly expressed in monocytes, macrophages, dendritic cells, and microglia, and it undergoes shutting between the plasma membrane and endosomal compartments in association with retromer (Yin et al., 2016). Particularly, the loss of retromer components perturbs plasma membrane-resident TREM2 but increases its lysosomal translocation for degradation, which impairs the microglial activation and phagocytic clearance of Aβ (Lucin et al., 2013; Yin et al., 2016). Consistent with this finding, R47H TREM2, an AD-associated mutant, disrupts the binding to VPS35, and it is destined for lysosomal degradation (Yin et al., 2016). The importance of retromer function in the microglial clearance of Aβ is further supported by a recent study showing that microglia-specific Vps35 conditional KO 5XFAD mice showed impaired microglial uptake of Aβ and disease-associated microglia development in the brains, resulting in the exacerbation of Aβ-related pathology and cognitive decline (Ren et al., 2022).
Other evidence demonstrated the putative role of retromer in the alteration of synaptic plasticity in AD (Figure 2). In hippocampal neurons, VPS35 deficiency impairs the surface recycling of α-amino-3-hydroxy-5-methyl-4-isoxazole propionate receptor (AMPAR) subunit GluA1 during long-term potentiation (LTP), resulting in dendritic spine deficit (Tian et al., 2015; Temkin et al., 2017). In addition, Vps26B, a brain-enriched paralog of Vps26 in mammals, potentiates the activity-dependent retrograde trafficking of GluA1 during LTP (Bugarcic et al., 2011; Simoes et al., 2021). It is interesting that silencing of Vps26B, but not Vps26A, in mice significantly decreases SorLA levels in the plasma membrane, which is accompanied by increased Aβ and tau accumulation in brain tissue and CSF (Simoes et al., 2021).
Given the multifaceted roles of retromer in the subcellular trafficking of AD-related proteins, one can imagine that the genetic engineering or pharmacological stabilization of retromer components may have a potentially beneficial effect on the neurodegenerative process of AD. For example, intracerebral AAV-mediated gene transfer of VPS35 in triple Tg (3xTg) mice {i.e., a human mutant presenilin 1 [M146V] knockin (KI), mutant APP [KM670/671NL] and tau [P301L] transgene} ameliorates cognitive dysfunction, which is associated with significant decreases in Aβ deposition and phosphorylated tau levels (Li et al., 2020a). Moreover, overexpression of VPS35 in cultured cellular models increases the expression of cathepsin D (CTSD), a lysosomal aspartic protease, thereby promoting the autophagic clearance of pathological tau aggregates (Vagnozzi et al., 2019). In addition to retromer gene transduction, retromer stabilization by the chemical chaperones R33 and R55 can mitigate AD-related pathology. Specifically, both R33 and R55 stabilize the retromer complex by binding the interface between VPS35 and VPS29, thereby preventing their degradation (Mecozzi et al., 2014). In the aforementioned 3xTg mice, R33 successfully prevented memory deficit along with reducing the intracerebral Aβ burden and phosphorylated tau levels (Li et al., 2020b). Likewise, in human iPSC-derived neurons from patients with AD, both R33 and R55 reduced tau phosphorylation in an APP-independent manner (Young et al., 2018). Finally, a recent cellular and animal model study demonstrated that the administration of R33 ameliorated the retention of APP in EE and increased the level of phosphorylated tau under high glucose condition (Chae et al., 2022). Taken together, these results open up a new therapeutic avenue for targeting retromer in AD. Future studies are required to further evaluate the efficacy of retromer-modulating drugs in different types of cellular and animal models.
Parkinson’s disease (PD), the second most common neurodegenerative disease, is clinically characterized by a progressive movement disability and a variety of non-motor symptoms. The neuropathological hallmarks of PD are the preferential loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) and the appearance of cytoplasmic inclusions called Lewy bodies (LBs), which are mainly composed of hyperphosphorylated, aggregated α-synuclein (α-syn) (Baba et al., 1998).
After the discovery of the missense mutations in the VPS35 gene in a late-onset, dominantly inherited familial form of PD (PARK17), the retromer function in the pathogenesis of PD has been highlighted (Vilariño-Güell et al., 2011; Zimprich et al., 2011). The exogenous induction of PD-related leucine-rich repeat kinase 2 (LRRK2, PARK8) and Rab7L1 also impairs the retromer-mediated transport of MPR with abnormal lysosomal swelling. Somewhat surprisingly, the expression of wild-type (WT) VPS35 (WTVPS35) can rescue the phenotypes induced by LRRK2 or RAB7L1 variants both in vitro and in vivo, suggesting that these three genes might operate in a common cellular pathway (MacLeod et al., 2013). Moreover, in the brain tissue from LRRK2 mutation carriers, the insoluble form of VPS35 is prominently increased probably because of retromer or lysosomal dysfunction (Zhao et al., 2018). Although the mechanisms by which VPS35 and LRRK2 synergistically participate in the pathogenesis of PD remains unclear, several possibilities have been postulated. The pathogenic D620N VPS35 (D620NVPS35) mutant enhanced LRRK2-mediated Rab10 phosphorylation in cellular and mouse models. Conversely, an in vivo study using a fly model revealed that Drosophila vps35 (dvps35) and LRRK2 cooperatively modulate synaptic vesicle endocytosis through the endosomal pathway (Inoshita et al., 2017; Mir et al., 2018). Several lines of evidence also suggest a molecular interaction between VPS35 and PARKIN (PARK2), the most common cause of autosomal recessive young-onset parkinsonism. In a Drosophila model, vps35 genetically interacted with PARKIN but not with PINK1 (PTEN-induced putative kinase 1), and notably, vps35 overexpression rescued several parkin-mutant phenotypes (Malik et al., 2015). As an E3 ubiquitin ligase, parkin directly interacts with VPS35 through its RING1 domain, thereby modulating retromer function through VPS35 ubiquitination (Williams et al., 2018). Additionally, as a vesicle-associated protein, α-syn can influence retromer-mediated sorting by interfering with the interaction between SNX3 and PI(3)P (Patel et al., 2018; Kobayashi et al., 2019). Another interesting finding was that the loss of iPLA2-VIA, a Drosophila homolog of PLA2G6 (PARK14), inhibits the retromer-mediated transport of sphingolipids from endosomes to the TGN, resulting in the lysosomal dysfunction due to ceramide overload in the lysosomes (Lin et al., 2018). Intriguingly, similar results were observed upon loss of vps26 or vps35 or overexpression of α-syn in this fly model, indicating that these defects might be common in the pathogenesis of PD. In addition to genetic models mimicking familial forms of PD, VPS35 overexpression may have a protective effect on toxin-induced models such as rotenone-induced Drosophila PD model (Linhart et al., 2014; Dhungel et al., 2015; Williams et al., 2018). Taken together, these findings indicate that the retromer sorting machinery may configure a common biological pathway involved in PD.
Although the molecular mechanism underlying neuronal loss in PD remains unclear, the critical roles of endosomes and their associated trafficking process in the pathophysiology of PD have emerged (Hasegawa et al., 2011, 2017a,b; Konno et al., 2012; Sugeno et al., 2014; Yoshida and Hasegawa, 2022). In particular, the discovery of VPS35 as a responsible gene for PARK17 has attracted great attention because this finding revealed a causal relationship between the retromer machinery and PD (Vilariño-Güell et al., 2011; Zimprich et al., 2011). Among the VPS35 mutations so far identified, the D620N missense mutation in the C-terminus of VPS35 has been consistently reported in unrelated PD families from different ethnicities (Sassone et al., 2021). Based on these results, genetically engineered animal models harboring D620NVPS35 have been created, and they have variable phenotypes. In a viral-mediated gene transfer rat model, the expression of human D620NVPS35 in the SNpc resulted in prominent dopaminergic neuron loss with axonal pathology, whereas D620NVPS35 Tg aged mice generated via Rosa26-based transgenesis did not exhibit apparent motor impairment or neurodegeneration (Tsika et al., 2014; Vanan et al., 2020). On the contrary, Tg flies expressing human D620NVPS35 or P316SVPS35 displayed a detrimental phenotype including dopaminergic neuron loss, locomotor dysfunction, a shortened lifespan, and susceptibility toward PD-linked environmental toxins (Wang et al., 2014). There are several conflicting research findings about D620NVPS35 KI mice; however, some of them exhibit levodopa-responsive motor impairment with dopaminergic neuron loss in the SNpc (Chen et al., 2019; Chiu et al., 2020; Niu et al., 2021). Notably, D620NVPS35 KI mice display phosphorylated tau accumulation and tangle-like pathology instead of LB pathology (Ishizu et al., 2016; Chen et al., 2019; Chiu et al., 2020), which may have mimicked the autopsy findings in the Japanese PARK17 patient carrying VPS35 mutation displaying “pure nigral” degeneration without LB pathology in the brain (Bono et al., 2020).
The mechanisms by which mutant VPS35 induces the PD-related pathology remain uncertain; however, several possibilities have been proposed: (i) toxic α-syn accumulation attributable to lysosomal dysfunction, (ii) synaptic dysfunction, and (iii) impaired mitochondrial dynamics and mitophagy (Figure 3). Retromer plays a key role in the lysosomal sorting of CTSD, a major lysosomal hydrolase in α-syn degradation (Sevlever et al., 2008). Namely, upon arrival in the Golgi apparatus, newly synthesized lysosomal enzymes including CTSD are modified with mannose 6-phosphate residues, which are recognized by CI-MPR in the TGN. CTSD is translocated to endosomes and released for further sorting to lysosomes. Retromer retrieves the unoccupied CI-MPR from endosomes to the TGN, where they participate in further cycles of CTSD sorting. Hence, retromer malfunction decreases the levels of the active form of CTSD in lysosomes and thus leads to abnormal α-syn accumulation (Follett et al., 2014; Miura et al., 2014). Through comparative stable isotope labeling by amino acids in cell culture (SILAC)-based analysis, the major defect of D620NVPS35 is attributed to its insufficient interaction with the actin-nucleating WASH complex, which results in perturbation of endosome-to-TGN trafficking (McGough et al., 2014). Likewise, exogenous expression of D620NVPS35 in HeLa cells can rescue lysosomal proteolytic defect and altered autophagic flux caused by the silencing of endogenous VPS35; however, this mutant fails to support the retrieval of CI-MPR from endosomes to the TGN (Cui et al., 2021). Similarly as D620NVPS35, R524WVPS35 and A320VVPS35 can also interfere with retrograde cargo sorting in the endosome-to-TGN pathway (Follett et al., 2016; Wu et al., 2020).
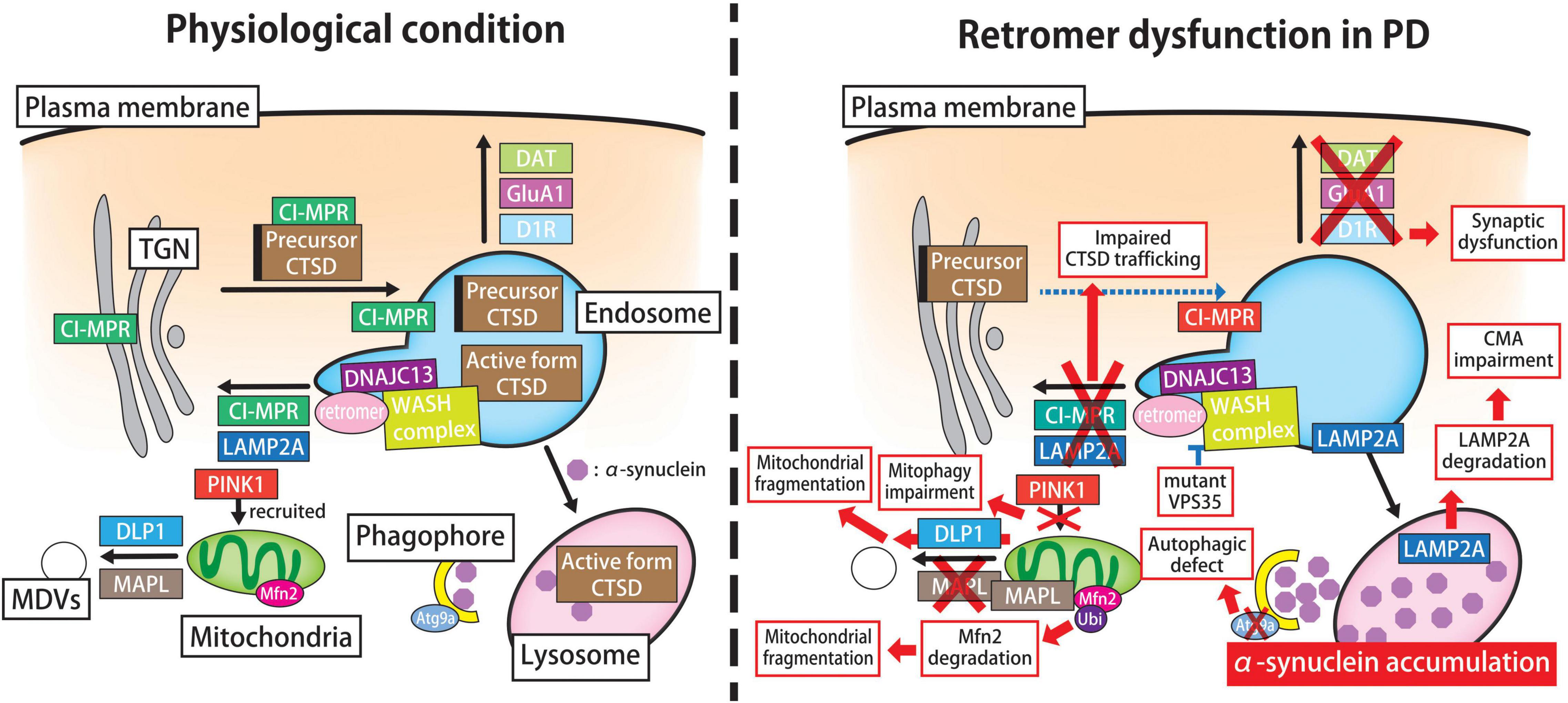
Figure 3. Retromer-mediated pathogenic pathway in PD. In physiological states (left panel), the retromer participates in the activation and CI-MPR-mediated sorting of CTSD, a major lysosomal hydrolase for α-syn degradation. The clearance of neurotoxic α-syn species largely depends on the autophagy-lysosomal pathway, especially macroautophagy and CMA. Retromer modulates the retrograde transport of LAMP2A, a receptor for CMA, from endosomes to the TGN, thereby preventing its degradation in lysosomes. In addition, retromer regulates the surface recycling of GluA1, D1R and DAT and thus influences synaptic function. Furthermore, retromer might participate in the autophagic machinery and mitochondrial dynamics, which are key processes in the maintenance of neuronal homeostasis. In the PD brain (right panel), retromer function is disturbed by aging, environmental toxin exposure, or genetic alterations. The familial PD-linked mutant VPS35 disrupts the molecular interaction between VPS35 and the WASH complex, which hampers retromer-mediated cargo sorting to the TGN. The perturbation of endosome-TGN trafficking hampers CTSD trafficking, thereby leading to abnormal α-syn accumulation in lysosomes. Likewise, retromer failure impairs the endosome-to-TGN retrieval of LAMP2A and accelerates its degradation in lysosomes, which compromises the CMA-mediated degradation of cytotoxic α-syn. Moreover, mutant VPS35 causes autophagic failure, which is partly explained by the mislocalization of autophagy protein ATG9A. Additionally, retromer failure increases the expression level of GluA1 and D1R on the plasma membrane, and thus induces synaptic dysfunction. In addition, mutant VPS35 has detrimental effects on mitochondrial fission-fusion dynamics and PINK1-PARKIN-mediated mitophagy, which results in mitochondrial dysfunction and fragmentation. The PD-linked DNAJC13 mutation influences on the trafficking of multiple cargoes possibly due to the alteration of the membrane dynamics via retromer-related WASH complex and SNX1. CI-MPR, cation-independent mannose-6-phosphate receptor; CTSD, cathepsin D; CMA, chaperone-mediated autophagy; LAMP2A, lysosomal-associated membrane protein 2A; TGN, trans-Golgi network; D1R, dopamine D1 receptor; DAT, dopamine transporter; PD, Parkinson’s disease; VPS, vacuolar protein sorting; WASH, Wiskott-Aldrich Syndrome protein and scar homolog; ATG, autophagy-related; PINK1, PTEN-induced putative kinase 1; SNX sorting nexin; DNAJC13, DnaJ heat shock protein family (Hsp40) member C13.
In PD and other synucleinopathies, one of the major concerns is the mode of α-syn clearance. Although some researchers have emphasized the importance of the ubiquitin-proteasome system for α-syn degradation, numerous studies have suggested that its degradation largely depends on the autophagy lysosomal pathway, especially macroautophagy and chaperone-mediated autophagy (CMA) (Cuervo et al., 2004; Oshima et al., 2016). Because retromer function is closely involved in the maintenance of autophagy-mediated proteostasis (Figure 3), it is easy to assume that retromer malfunction could influence the clearance of toxic α-syn and subsequent neurodegeneration. Indeed, PD-linked D620NVPS35 impairs WASH complex recruitment to the endosomes and thus causes autophagic failure, which is partly explained by the mislocalization of the autophagy protein ATG9A (Zavodszky et al., 2014). Furthermore, dopaminergic neurons expressing D620NVPS35 and neurons in D620NVPS35 KI mice exhibit impaired endosome-to-Golgi retrieval of LAMP2A, thereby accelerating LAMP2A degradation in lysosomes (Tang et al., 2015a; Niu et al., 2021). Collectively, these results suggest that retromer malfunction impairs the cellular clearance of α-syn via the autophagy-lysosome pathway, thereby accelerating neurodegeneration possibly due to the accumulation of toxic, misfolded α-syn species.
Growing evidence suggests that retromer can influence mammalian nervous system development and synaptic neurotransmission in healthy and diseased brains (Brodin and Shupliakov, 2018). Although limited evidence is available, several studies claim that the disorder in retromer function by PD-related VPS35 mutations may affect synaptic function (Figure 3). In mouse primary cortical neurons, the presence of D620NVPS35 was less frequently present in dendritic spines than WTVPS35, and D620NVPS35 tended to form clusters with FAM21 in EEs (Munsie et al., 2015; Kadgien et al., 2021). In the synaptic nerve terminal, VPS35 participates in the cell surface recycling of GluA1, dopamine D1 receptor (D1R), and dopamine transporter (DAT), and thus, D620NVPS35 might increase the surface expression of these receptors, thereby producing chronic stress in neuronal circuits (Munsie et al., 2015; Wang C. et al., 2016; Wu et al., 2017; Kadgien et al., 2021).
Mitochondria have long been recognized as a key component in the pathogenesis of PD (Hasegawa et al., 2006; Bose and Beal, 2016). Another interesting idea is that the VPS35 pathogenic mutation may have a detrimental effect on mitochondrial fission-fusion dynamics and mitophagy (Figure 3). Indeed, aberrant mitochondrial fragmentation and impaired mitophagy have been observed in fibroblasts from patients bearing D620NVPS35 (Wang W. et al., 2016; Hanss et al., 2021). The underlying mechanism of mitochondrial fragmentation induced by VPS35 deficiency is supposed to be aberrant trafficking of MAPL (also known as mitochondrial E3 ubiquitin ligase-1) and dynamin-like protein 1 (DLP1). More specifically, D620NVPS35 impairs the trafficking of MAPL from mitochondria to MDVs, and the overloaded MAPL in mitochondria ubiquitinates mitofusin-2 (MFN2), thereby promoting mitochondrial fragmentation (Tang et al., 2015b). Alternatively, D620NVPS35 enhances the VPS35-DLP1 interaction and increases the turnover of the DLP1 complex in mitochondria, which induces neurodegeneration by increasing the rate of mitochondrial fission (Wang W. et al., 2016). It is widely accepted that both PINK1 and PARKIN participate in the quality control pathway to sense damaged mitochondria and target them for degradation through mitophagy (Tanaka, 2020). A recent study using SH-SY5Y cells carrying the D620NVPS35 demonstrated that mutant VPS35 impairs the PINK1-PARKIN-mediated mitophagy through impaired PINK1 recruitment to mitochondria, suggesting a converging pathophysiological cascade among VPS35, PINK1, and PARKIN in PD (Ma et al., 2021).
Another important player in the endosomal cargo sorting is a DNAJC13 [DnaJ heat shock protein family (Hsp40) member C13], which associates with SNX1 and has been linked to an autosomal-dominant, late-onset familial form of PD (PARK21) (McGough and Cullen, 2013; Freeman et al., 2014; Vilariño-Güell et al., 2014; Gustavsson et al., 2015). The neuropathological feature in DNAJC13 N855S (N855SDNAJC13) mutation carriers is the presence of the brainstem or transitional type of LB pathology (Appel-Cresswell et al., 2014; Vilariño-Güell et al., 2014). DNAJC13 is a human homolog of receptor-mediated endocytosis 8 (RME-8) in nematodes and is ubiquitously expressed including the nervous system (Fujibayashi et al., 2008). Structurally, it includes four conserved IWN repeats, which are characterized by seven invariant residues, including isoleucine, tryptophan, and asparagine, and a Hsc70-binding J-domain (Hasegawa et al., 2017b). In a fly model, N855SDNAJC13 exacerbated α-syn-mediated motor dysfunction, a rough eye phenotype, and the loss of dopaminergic neurons, which recapitulates the clinicopathological features of PD (Yoshida et al., 2018). In concert with SNX1 and FAM21, DNAJC13 is recruited to EEs, where it participates in multidirectional endosomal sorting including the retrieval of CI-MPR (Hasegawa et al., 2017b). It remains unclear whether the PD-linked DNJAC13 mutant could directly impede retromer function; however, the expression of N855SDNAJC13 in cultured cells alters the membrane dynamics of retromer-related SNX1 and influences the trafficking of multiple cargoes, e.g., the transport of epidermal growth factor receptor (EGFR) to the lysosomes, the recycling of transferrin receptor (TfR) to the cell surface, notch receptor recycling, and the transport of ATG9A to the phagophores (Gomez-Lamarca et al., 2015; Yoshida et al., 2018; Follett et al., 2019; Besemer et al., 2021). Further work is required to precisely identify the pathophysiological role of DNAJC13 in the neurodegenerative process leading to PD.
Although there is only limited evidence available at present, the retromer sorting system may also contribute to the etiopathogenesis of less common neurodegenerative diseases such as amyotrophic lateral sclerosis (ALS) and Huntington’s disease (HD). Amyotrophic lateral sclerosis is a fatal neurodegenerative disease clinically characterized by the selective loss of both upper and lower motor neurons. Although most ALS cases are sporadic, approximately 10% of cases are familial (FALS) and predominantly associated with Mendelian-inherited mutations in genes including Cu/Zn superoxide dismutase (SOD1) and C9ORF72. Notably, the iPSC-derived motor neurons from patients with FALS carrying C9ORF72 hexanucleotide repeat expansion or G93A SOD1 mutation exhibit retromer deficiency, and retromer stabilization by chemical chaperone attenuated the locomotive activity and motor neuron loss in G93A SOD1 Tg mice (Aoki et al., 2017; Muzio et al., 2020). Likewise, the neuron-specific deletion of VPS35 results in the selective loss of ventral horn motor neurons with the formation of p62-positive inclusions in the spinal cord, mimicking the neuropathological features of sporadic ALS (Sargent et al., 2021).
Huntington’s disease (HD) is a progressive, dominantly inherited neurodegenerative disorder clinically manifesting as involuntary movement and cognitive and psychiatric impairment. The cardinal genetic defect in HD is the abnormally elongated polyglutamine repeat expansion in the huntingtin (HTT), and striatal medium spiny neurons (MSNs) are known as the most vulnerable cells in HD. In MSNs, SorCS2 interacts with VPS35, thereby regulating the surface trafficking of the NR2A subunit of N-methyl-D-aspartate (NMDA) receptor. Intriguingly, SorCS2 selectively interacts with mutant huntingtin but not WT huntingtin, and it is mislocalized to perinuclear clusters in the striatal neurons of patients with HD and model mice, indicating that retromer affects the pathogenesis of HD by modulating SorCS2-mediated NR2A trafficking in MSNs (Ma et al., 2017).
In this review, we summarized the functional roles of the retromer as an endosomal trafficking regulator and its implication in the pathogenesis of neurodegenerative disorders, including AD, PD, ALS, and HD. In particular, after the discovery of missense mutations in VPS35, a core component of retromer, in autosomal dominant forms of PD, the role of retromer-mediated endosomal sorting came into the limelight in the etiopathogenesis of PD. However, several questions remain to be clarified (e.g., why neurons are selectively vulnerable to the retromer dysfunction; why do mutations in the same retromer-associated gene result in multiple phenotypes and different disorders; are disease-associated protein aggregates an indicator of retromer malfunction, a driver of retromer impairment, or both). Currently, most researchers postulate that defects in the retromer machinery affect the proteostasis and cellular burden of cytotoxic proteins including α-syn and Aβ; however, the causal relationship between these protein aggregates and neuronal cell loss is unclear, especially in PD because patients with PARK2 typically display pure nigral degeneration without LBs, and LB pathology may not be present in patients with dominantly inherited familial PD with LRRK2 and VPS35 mutations (Hayashi et al., 2000; Hasegawa et al., 2009; Bono et al., 2020). In that sense, dissecting the molecular mechanisms responsible for changes in retromer-mediated synaptic neurotransmission and mitochondrial dynamics may help to clarify the pathophysiological cascades of neurodegenerative disorders. Of course, other scenarios that we might not anticipate are also possible. Although stabilization of the retromer complex by pharmacological chaperones can direct disease-causing proteins away from a pathogenic pathway and mitigate neurodegeneration both in vivo and in vitro, we must continue to decipher the mechanism by which the distinct retromer components and their associated proteins cooperatively function in endosomal sorting and the change of cellular circumstances after machineries are perturbed. Further investigation will uncover the underlying molecular mechanisms of retromer-mediated neurodegeneration and provide a crucial insight into the development of disease-modifying therapy.
SY and TH wrote the manuscript. TH provided expertise and edited the manuscript. Both authors contributed to the article and approved the submitted version.
This work was supported in part by a Grant-in-Aid for Scientific Research (C) [grant number 20K07896] and a Grant-in-Aid for Young Scientists [grant number 19K16998] from the Ministry of Education, Culture, Sports, Science and Technology (MEXT); and a Grant-in-aid for the Strategic Research Program for Brain Sciences from the Japan Agency for Medical Research and Development (AMED); Biogen Research Grants.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Andersen, O. M., Reiche, J., Schmidt, V., Gotthardt, M., Spoelgen, R., Behlke, J., et al. (2005). Neuronal sorting protein-related receptor sorLA/LR11 regulates processing of the amyloid precursor protein. Proc. Natl. Acad. Sci. U.S.A. 102, 13461–13466. doi: 10.1073/pnas.0503689102
Ansell-Schultz, A., Reyes, J. F., Samuelsson, M., and Hallbeck, M. (2018). Reduced retromer function results in the accumulation of amyloid-beta oligomers. Mol. Cell. Neurosci. 93, 18–26. doi: 10.1016/j.mcn.2018.09.003
Aoki, Y., Manzano, R., Lee, Y., Dafinca, R., Aoki, M., Douglas, A. G. L., et al. (2017). C9orf72 and RAB7L1 regulate vesicle trafficking in amyotrophic lateral sclerosis and frontotemporal dementia. Brain 140, 887–897. doi: 10.1093/brain/awx024
Appel-Cresswell, S., Rajput, A. H., Sossi, V., Thompson, C., Silva, V., McKenzie, J., et al. (2014). Clinical, positron emission tomography, and pathological studies of DNAJC13 p.N855S Parkinsonism. Mov. Disord. 29, 1684–1687. doi: 10.1002/mds.26019
Baba, M., Nakajo, S., Tu, P. H., Tomita, T., Nakaya, K., Lee, V. M., et al. (1998). Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am. J. Pathol. 152, 879–884.
Besemer, A. S., Maus, J., Ax, M. D. A., Stein, A., Vo, S., Freese, C., et al. (2021). Receptor-mediated endocytosis 8 (RME-8)/DNAJC13 is a novel positive modulator of autophagy and stabilizes cellular protein homeostasis. Cell. Mol. Life Sci. CMLS 78, 645–660. doi: 10.1007/s00018-020-03521-y
Bhalla, A., Vetanovetz, C. P., Morel, E., Chamoun, Z., Di Paolo, G., and Small, S. A. (2012). The location and trafficking routes of the neuronal retromer and its role in amyloid precursor protein transport. Neurobiol. Dis. 47, 126–134. doi: 10.1016/j.nbd.2012.03.030
Bono, K., Hara-Miyauchi, C., Sumi, S., Oka, H., Iguchi, Y., and Okano, H. J. (2020). Endosomal dysfunction in iPSC-derived neural cells from Parkinson’s disease patients with VPS35 D620N. Mol. Brain 13:137. doi: 10.1186/s13041-020-00675-5
Bose, A., and Beal, M. F. (2016). Mitochondrial dysfunction in Parkinson’s disease. J. Neurochem. 139(Suppl. 1), 216–231. doi: 10.1111/jnc.13731
Braschi, E., Goyon, V., Zunino, R., Mohanty, A., Xu, L., and McBride, H. M. (2010). Vps35 mediates vesicle transport between the mitochondria and peroxisomes. Curr. Biol. CB 20, 1310–1315. doi: 10.1016/j.cub.2010.05.066
Brodin, L., and Shupliakov, O. (2018). Retromer in synaptic function and pathology. Front. Synaptic Neurosci. 10:37. doi: 10.3389/fnsyn.2018.00037
Bugarcic, A., Zhe, Y., Kerr, M. C., Griffin, J., Collins, B. M., and Teasdale, R. D. (2011). Vps26A and Vps26B subunits define distinct retromer complexes. Traffic (Copenhagen, Denmark) 12, 1759–1773. doi: 10.1111/j.1600-0854.2011.01284.x
Bujny, M. V., Popoff, V., Johannes, L., and Cullen, P. J. (2007). The retromer component sorting nexin-1 is required for efficient retrograde transport of Shiga toxin from early endosome to the trans Golgi network. J. Cell Sci. 120(Pt. 12), 2010–2021. doi: 10.1242/jcs.003111
Burgos, P. V., Mardones, G. A., Rojas, A. L., daSilva, L. L., Prabhu, Y., Hurley, J. H., et al. (2010). Sorting of the Alzheimer’s disease amyloid precursor protein mediated by the AP-4 complex. Dev. Cell 18, 425–436. doi: 10.1016/j.devcel.2010.01.015
Canuel, M., Lefrancois, S., Zeng, J., and Morales, C. R. (2008). AP-1 and retromer play opposite roles in the trafficking of sortilin between the Golgi apparatus and the lysosomes. Biochem. Biophys. Res. Commun. 366, 724–730. doi: 10.1016/j.bbrc.2007.12.015
Carlton, J. G., Bujny, M. V., Peter, B. J., Oorschot, V. M., Rutherford, A., Arkell, R. S., et al. (2005). Sorting nexin-2 is associated with tubular elements of the early endosome, but is not essential for retromer-mediated endosome-to-TGN transport. J. Cell Sci. 118(Pt. 19), 4527–4539. doi: 10.1242/jcs.02568
Cataldo, A. M., Peterhoff, C. M., Troncoso, J. C., Gomez-Isla, T., Hyman, B. T., and Nixon, R. A. (2000). Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer’s disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am. J. Pathol. 157, 277–286. doi: 10.1016/s0002-9440(10)64538-5
Chae, C. W., Choi, G. E., Jung, Y. H., Lim, J. R., Cho, J. H., Yoon, J. H., et al. (2022). High glucose-mediated VPS26a down-regulation dysregulates neuronal amyloid precursor protein processing and tau phosphorylation. Br. J. Pharmacol. doi: 10.1111/bph.15836 [Epub ahead of print].
Chen, X., Kordich, J. K., Williams, E. T., Levine, N., Cole-Strauss, A., Marshall, L., et al. (2019). Parkinson’s disease-linked D620N VPS35 knockin mice manifest tau neuropathology and dopaminergic neurodegeneration. Proc. Natl. Acad. Sci. U.S.A. 116, 5765–5774. doi: 10.1073/pnas.1814909116
Chiu, C. C., Weng, Y. H., Huang, Y. Z., Chen, R. S., Liu, Y. C., Yeh, T. H., et al. (2020). (D620N) VPS35 causes the impairment of Wnt/β-catenin signaling cascade and mitochondrial dysfunction in a PARK17 knockin mouse model. Cell Death Dis. 11:1018. doi: 10.1038/s41419-020-03228-9
Choy, R. W., Cheng, Z., and Schekman, R. (2012). Amyloid precursor protein (APP) traffics from the cell surface via endosomes for amyloid β (Aβ) production in the trans-Golgi network. Proc. Natl. Acad. Sci. U.S.A. 109, E2077–E2082. doi: 10.1073/pnas.1208635109
Chu, J., and Praticò, D. (2017). The retromer complex system in a transgenic mouse model of AD: influence of age. Neurobiol. Aging 52, 32–38. doi: 10.1016/j.neurobiolaging.2016.12.025
Cuartero, Y., Mellado, M., Capell, A., Alvarez-Dolado, M., and Verges, M. (2012). Retromer regulates postendocytic sorting of β-secretase in polarized Madin-Darby canine kidney cells. Traffic (Copenhagen, Denmark) 13, 1393–1410. doi: 10.1111/j.1600-0854.2012.01392.x
Cuervo, A. M., Stefanis, L., Fredenburg, R., Lansbury, P. T., and Sulzer, D. (2004). Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science (New York, N.Y.) 305, 1292–1295. doi: 10.1126/science.1101738
Cui, Y., Yang, Z., and Teasdale, R. D. (2018). The functional roles of retromer in Parkinson’s disease. FEBS Lett. 592, 1096–1112. doi: 10.1002/1873-3468.12931
Cui, Y., Yang, Z., Flores-Rodriguez, N., Follett, J., Ariotti, N., Wall, A. A., et al. (2021). Formation of retromer transport carriers is disrupted by the Parkinson disease-linked Vps35 D620N variant. Traffic (Copenhagen, Denmark) 22, 123–136. doi: 10.1111/tra.12779
Dengjel, J., Høyer-Hansen, M., Nielsen, M. O., Eisenberg, T., Harder, L. M., Schandorff, S., et al. (2012). Identification of autophagosome-associated proteins and regulators by quantitative proteomic analysis and genetic screens. Mol. Cell. Proteomics 11:M111.014035. doi: 10.1074/mcp.M111.014035
DeTure, M. A., and Dickson, D. W. (2019). The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 14:32. doi: 10.1186/s13024-019-0333-5
Dhungel, N., Eleuteri, S., Li, L. B., Kramer, N. J., Chartron, J. W., Spencer, B., et al. (2015). Parkinson’s disease genes VPS35 and EIF4G1 interact genetically and converge on α-synuclein. Neuron 85, 76–87. doi: 10.1016/j.neuron.2014.11.027
Dodson, S. E., Andersen, O. M., Karmali, V., Fritz, J. J., Cheng, D., Peng, J., et al. (2008). Loss of LR11/SORLA enhances early pathology in a mouse model of amyloidosis: evidence for a proximal role in Alzheimer’s disease. J. Neurosci. 28, 12877–12886. doi: 10.1523/jneurosci.4582-08.2008
Finan, G. M., Okada, H., and Kim, T. W. (2011). BACE1 retrograde trafficking is uniquely regulated by the cytoplasmic domain of sortilin. J. Biol. Chem. 286, 12602–12616. doi: 10.1074/jbc.M110.170217
Fjorback, A. W., Seaman, M., Gustafsen, C., Mehmedbasic, A., Gokool, S., Wu, C., et al. (2012). Retromer binds the FANSHY sorting motif in SorLA to regulate amyloid precursor protein sorting and processing. J. Neurosci. 32, 1467–1480. doi: 10.1523/jneurosci.2272-11.2012
Follett, J., Bugarcic, A., Yang, Z., Ariotti, N., Norwood, S. J., Collins, B. M., et al. (2016). Parkinson disease-linked Vps35 R524W mutation impairs the endosomal association of retromer and induces α-synuclein aggregation. J. Biol. Chem. 291, 18283–18298. doi: 10.1074/jbc.M115.703157
Follett, J., Fox, J. D., Gustavsson, E. K., Kadgien, C., Munsie, L. N., Cao, L. P., et al. (2019). DNAJC13 p.Asn855Ser, implicated in familial parkinsonism, alters membrane dynamics of sorting nexin 1. Neurosci. Lett. 706, 114–122. doi: 10.1016/j.neulet.2019.04.043
Follett, J., Norwood, S. J., Hamilton, N. A., Mohan, M., Kovtun, O., Tay, S., et al. (2014). The Vps35 D620N mutation linked to Parkinson’s disease disrupts the cargo sorting function of retromer. Traffic (Copenhagen, Denmark) 15, 230–244. doi: 10.1111/tra.12136
Freeman, C. L., Hesketh, G., and Seaman, M. N. (2014). RME-8 coordinates the activity of the WASH complex with the function of the retromer SNX dimer to control endosomal tubulation. J. Cell Sci. 127(Pt. 9), 2053–2070. doi: 10.1242/jcs.144659
Fujibayashi, A., Taguchi, T., Misaki, R., Ohtani, M., Dohmae, N., Takio, K., et al. (2008). Human RME-8 is involved in membrane trafficking through early endosomes. Cell Struct. Funct. 33, 35–50. doi: 10.1247/csf.07045
Gallon, M., Clairfeuille, T., Steinberg, F., Mas, C., Ghai, R., Sessions, R. B., et al. (2014). A unique PDZ domain and arrestin-like fold interaction reveals mechanistic details of endocytic recycling by SNX27-retromer. Proc. Natl. Acad. Sci. U.S.A. 111, E3604–E3613. doi: 10.1073/pnas.1410552111
Gomez, T. S., and Billadeau, D. D. (2009). A FAM21-containing WASH complex regulates retromer-dependent sorting. Dev. Cell 17, 699–711. doi: 10.1016/j.devcel.2009.09.009
Gomez-Lamarca, M. J., Snowdon, L. A., Seib, E., Klein, T., and Bray, S. J. (2015). Rme-8 depletion perturbs Notch recycling and predisposes to pathogenic signaling. J. Cell Biol. 210, 303–318. doi: 10.1083/jcb.201411001
Goodarzi, M. O., Lehman, D. M., Taylor, K. D., Guo, X., Cui, J., Quiñones, M. J., et al. (2007). SORCS1: a novel human type 2 diabetes susceptibility gene suggested by the mouse. Diabetes 56, 1922–1929. doi: 10.2337/db06-1677
Guerreiro, R., Wojtas, A., Bras, J., Carrasquillo, M., Rogaeva, E., Majounie, E., et al. (2013). TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 368, 117–127. doi: 10.1056/NEJMoa1211851
Gustavsson, E. K., Trinh, J., Guella, I., Vilariño-Güell, C., Appel-Cresswell, S., Stoessl, A. J., et al. (2015). DNAJC13 genetic variants in parkinsonism. Mov. Disord. 30, 273–278. doi: 10.1002/mds.26064
Hanss, Z., Larsen, S. B., Antony, P., Mencke, P., Massart, F., Jarazo, J., et al. (2021). Mitochondrial and clearance impairment in p.D620N VPS35 patient-derived neurons. Mov. Disord. 36, 704–715. doi: 10.1002/mds.28365
Hao, Y. H., Doyle, J. M., Ramanathan, S., Gomez, T. S., Jia, D., Xu, M., et al. (2013). Regulation of WASH-dependent actin polymerization and protein trafficking by ubiquitination. Cell 152, 1051–1064. doi: 10.1016/j.cell.2013.01.051
Harbour, M. E., Breusegem, S. Y., and Seaman, M. N. (2012). Recruitment of the endosomal WASH complex is mediated by the extended ‘tail’ of Fam21 binding to the retromer protein Vps35. Biochem. J. 442, 209–220. doi: 10.1042/bj20111761
Harterink, M., Port, F., Lorenowicz, M. J., McGough, I. J., Silhankova, M., Betist, M. C., et al. (2011). A SNX3-dependent retromer pathway mediates retrograde transport of the Wnt sorting receptor Wntless and is required for Wnt secretion. Nat. Cell Biol. 13, 914–923. doi: 10.1038/ncb2281
Hasegawa, K., Stoessl, A. J., Yokoyama, T., Kowa, H., Wszolek, Z. K., and Yagishita, S. (2009). Familial parkinsonism: study of original Sagamihara PARK8 (I2020T) kindred with variable clinicopathologic outcomes. Parkinsonism Relat. Disord. 15, 300–306. doi: 10.1016/j.parkreldis.2008.07.010
Hasegawa, T., Konno, M., Baba, T., Sugeno, N., Kikuchi, A., Kobayashi, M., et al. (2011). The AAA-ATPase VPS4 regulates extracellular secretion and lysosomal targeting of α-synuclein. PLoS One 6:e29460. doi: 10.1371/journal.pone.0029460
Hasegawa, T., Matsuzaki-Kobayashi, M., Takeda, A., Sugeno, N., Kikuchi, A., Furukawa, K., et al. (2006). Alpha-synuclein facilitates the toxicity of oxidized catechol metabolites: implications for selective neurodegeneration in Parkinson’s disease. FEBS Lett. 580, 2147–2152. doi: 10.1016/j.febslet.2006.03.018
Hasegawa, T., Sugeno, N., Kikuchi, A., Baba, T., and Aoki, M. (2017a). Membrane trafficking illuminates a path to Parkinson’s disease. Tohoku J. Exp. Med. 242, 63–76. doi: 10.1620/tjem.242.63
Hasegawa, T., Yoshida, S., Sugeno, N., Kobayashi, J., and Aoki, M. (2017b). DnaJ/Hsp40 family and Parkinson’s disease. Front. Neurosci. 11:743. doi: 10.3389/fnins.2017.00743
Hayashi, S., Wakabayashi, K., Ishikawa, A., Nagai, H., Saito, M., Maruyama, M., et al. (2000). An autopsy case of autosomal-recessive juvenile parkinsonism with a homozygous exon 4 deletion in the parkin gene. Mov. Disord. 15, 884–888.
Huang, T. Y., Zhao, Y., Li, X., Wang, X., Tseng, I. C., Thompson, R., et al. (2016). SNX27 and SORLA interact to reduce amyloidogenic subcellular distribution and processing of amyloid precursor protein. J. Neurosci. 36, 7996–8011. doi: 10.1523/jneurosci.0206-16.2016
Inoshita, T., Arano, T., Hosaka, Y., Meng, H., Umezaki, Y., Kosugi, S., et al. (2017). Vps35 in cooperation with LRRK2 regulates synaptic vesicle endocytosis through the endosomal pathway in Drosophila. Hum. Mol. Genet. 26, 2933–2948. doi: 10.1093/hmg/ddx179
Ishizu, N., Yui, D., Hebisawa, A., Aizawa, H., Cui, W., Fujita, Y., et al. (2016). Impaired striatal dopamine release in homozygous Vps35 D620N knock-in mice. Hum. Mol. Genet. 25, 4507–4517. doi: 10.1093/hmg/ddw279
Jimenez-Orgaz, A., Kvainickas, A., Nägele, H., Denner, J., Eimer, S., Dengjel, J., et al. (2018). Control of RAB7 activity and localization through the retromer-TBC1D5 complex enables RAB7-dependent mitophagy. EMBO J. 37, 235–254. doi: 10.15252/embj.201797128
Jonsson, T., Stefansson, H., Steinberg, S., Jonsdottir, I., Jonsson, P. V., Snaedal, J., et al. (2013). Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 368, 107–116. doi: 10.1056/NEJMoa1211103
Kadgien, C. A., Kamesh, A., and Milnerwood, A. J. (2021). Endosomal traffic and glutamate synapse activity are increased in VPS35 D620N mutant knock-in mouse neurons, and resistant to LRRK2 kinase inhibition. Mol. Brain 14:143. doi: 10.1186/s13041-021-00848-w
Kanatsu, K., Hori, Y., Ebinuma, I., Chiu, Y. W., and Tomita, T. (2018). Retrograde transport of γ-secretase from endosomes to the trans-Golgi network regulates Aβ42 production. J. Neurochem. 147, 110–123. doi: 10.1111/jnc.14477
Kim, E., Lee, Y., Lee, H. J., Kim, J. S., Song, B. S., Huh, J. W., et al. (2010). Implication of mouse Vps26b-Vps29-Vps35 retromer complex in sortilin trafficking. Biochem. Biophys. Res. Commun. 403, 167–171. doi: 10.1016/j.bbrc.2010.10.121
Kimura, N., and Yanagisawa, K. (2018). Traffic jam hypothesis: relationship between endocytic dysfunction and Alzheimer’s disease. Neurochem. Int. 119, 35–41. doi: 10.1016/j.neuint.2017.07.002
Kimura, N., Inoue, M., Okabayashi, S., Ono, F., and Negishi, T. (2009). Dynein dysfunction induces endocytic pathology accompanied by an increase in Rab GTPases: a potential mechanism underlying age-dependent endocytic dysfunction. J. Biol. Chem. 284, 31291–31302. doi: 10.1074/jbc.M109.012625
Kimura, N., Samura, E., Suzuki, K., Okabayashi, S., Shimozawa, N., and Yasutomi, Y. (2016). Dynein dysfunction reproduces age-dependent retromer deficiency: concomitant disruption of retrograde trafficking is required for alteration in β-amyloid precursor protein metabolism. Am. J. Pathol. 186, 1952–1966. doi: 10.1016/j.ajpath.2016.03.006
Knupp, A., Mishra, S., Martinez, R., Braggin, J. E., Szabo, M., Kinoshita, C., et al. (2020). Depletion of the AD risk gene SORL1 selectively impairs neuronal endosomal traffic independent of amyloidogenic APP processing. Cell Rep. 31:107719. doi: 10.1016/j.celrep.2020.107719
Kobayashi, J., Hasegawa, T., Sugeno, N., Yoshida, S., Akiyama, T., Fujimori, K., et al. (2019). Extracellular α-synuclein enters dopaminergic cells by modulating flotillin-1-assisted dopamine transporter endocytosis. FASEB J. 33, 10240–10256. doi: 10.1096/fj.201802051R
Konno, M., Hasegawa, T., Baba, T., Miura, E., Sugeno, N., Kikuchi, A., et al. (2012). Suppression of dynamin GTPase decreases α-synuclein uptake by neuronal and oligodendroglial cells: a potent therapeutic target for synucleinopathy. Mol. Neurodegener. 7:38. doi: 10.1186/1750-1326-7-38
Lane, R. F., Raines, S. M., Steele, J. W., Ehrlich, M. E., Lah, J. A., Small, S. A., et al. (2010). Diabetes-associated SorCS1 regulates Alzheimer’s amyloid-beta metabolism: evidence for involvement of SorL1 and the retromer complex. J. Neurosci. 30, 13110–13115. doi: 10.1523/jneurosci.3872-10.2010
Lane, R. F., Steele, J. W., Cai, D., Ehrlich, M. E., Attie, A. D., and Gandy, S. (2013). Protein sorting motifs in the cytoplasmic tail of SorCS1 control generation of Alzheimer’s amyloid-β peptide. J. Neurosci. 33, 7099–7107. doi: 10.1523/jneurosci.5270-12.2013
Lee, C. Y. D., Daggett, A., Gu, X., Jiang, L. L., Langfelder, P., Li, X., et al. (2018). Elevated TREM2 gene dosage reprograms microglia responsivity and ameliorates pathological phenotypes in Alzheimer’s disease models. Neuron 97, 1032–1048.e1035. doi: 10.1016/j.neuron.2018.02.002
Li, J. G., Chiu, J., and Praticò, D. (2020a). Full recovery of the Alzheimer’s disease phenotype by gain of function of vacuolar protein sorting 35. Mol. Psychiatry 25, 2630–2640. doi: 10.1038/s41380-019-0364-x
Li, J. G., Chiu, J., Ramanjulu, M., Blass, B. E., and Praticò, D. (2020b). A pharmacological chaperone improves memory by reducing Aβ and tau neuropathology in a mouse model with plaques and tangles. Mol. Neurodegener. 15:1. doi: 10.1186/s13024-019-0350-4
Lin, G., Lee, P. T., Chen, K., Mao, D., Tan, K. L., Zuo, Z., et al. (2018). Phospholipase PLA2G6, a parkinsonism-associated gene, affects Vps26 and Vps35, retromer function, and ceramide levels, similar to α-synuclein gain. Cell Metab. 28, 605–618.e606. doi: 10.1016/j.cmet.2018.05.019
Linardopoulou, E. V., Parghi, S. S., Friedman, C., Osborn, G. E., Parkhurst, S. M., and Trask, B. J. (2007). Human subtelomeric WASH genes encode a new subclass of the WASP family. PLoS Genet. 3:e237. doi: 10.1371/journal.pgen.0030237
Linhart, R., Wong, S. A., Cao, J., Tran, M., Huynh, A., Ardrey, C., et al. (2014). Vacuolar protein sorting 35 (Vps35) rescues locomotor deficits and shortened lifespan in Drosophila expressing a Parkinson’s disease mutant of Leucine-Rich Repeat Kinase 2 (LRRK2). Mol. Neurodegener. 9:23. doi: 10.1186/1750-1326-9-23
Lucin, K. M., O’Brien, C. E., Bieri, G., Czirr, E., Mosher, K. I., Abbey, R. J., et al. (2013). Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer’s disease. Neuron 79, 873–886. doi: 10.1016/j.neuron.2013.06.046
Ma, K. Y., Fokkens, M. R., Reggiori, F., Mari, M., and Verbeek, D. S. (2021). Parkinson’s disease-associated VPS35 mutant reduces mitochondrial membrane potential and impairs PINK1/Parkin-mediated mitophagy. Transl. Neurodegener. 10:19. doi: 10.1186/s40035-021-00243-4
Ma, Q. L., Galasko, D. R., Ringman, J. M., Vinters, H. V., Edland, S. D., Pomakian, J., et al. (2009). Reduction of SorLA/LR11, a sorting protein limiting beta-amyloid production, in Alzheimer disease cerebrospinal fluid. Arch. Neurol. 66, 448–457. doi: 10.1001/archneurol.2009.22
Ma, Q., Yang, J., Milner, T. A., Vonsattel, J. G., Palko, M. E., Tessarollo, L., et al. (2017). SorCS2-mediated NR2A trafficking regulates motor deficits in Huntington’s disease. JCI Insight 2:e88995. doi: 10.1172/jci.insight.88995
MacLeod, D. A., Rhinn, H., Kuwahara, T., Zolin, A., Di Paolo, G., McCabe, B. D., et al. (2013). RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson’s disease risk. Neuron 77, 425–439. doi: 10.1016/j.neuron.2012.11.033
Malik, B. R., Godena, V. K., and Whitworth, A. J. (2015). VPS35 pathogenic mutations confer no dominant toxicity but partial loss of function in Drosophila and genetically interact with parkin. Hum. Mol. Genet. 24, 6106–6117. doi: 10.1093/hmg/ddv322
McGough, I. J., and Cullen, P. J. (2013). Clathrin is not required for SNX-BAR-retromer-mediated carrier formation. J. Cell Sci. 126(Pt. 1), 45–52. doi: 10.1242/jcs.112904
McGough, I. J., Steinberg, F., Jia, D., Barbuti, P. A., McMillan, K. J., Heesom, K. J., et al. (2014). Retromer binding to FAM21 and the WASH complex is perturbed by the Parkinson disease-linked VPS35(D620N) mutation. Curr. Biol. CB 24, 1670–1676. doi: 10.1016/j.cub.2014.06.024
Mecozzi, V. J., Berman, D. E., Simoes, S., Vetanovetz, C., Awal, M. R., Patel, V. M., et al. (2014). Pharmacological chaperones stabilize retromer to limit APP processing. Nat. Chem. Biol. 10, 443–449. doi: 10.1038/nchembio.1508
Mir, R., Tonelli, F., Lis, P., Macartney, T., Polinski, N. K., Martinez, T. N., et al. (2018). The Parkinson’s disease VPS35[D620N] mutation enhances LRRK2-mediated Rab protein phosphorylation in mouse and human. Biochem. J. 475, 1861–1883. doi: 10.1042/bcj20180248
Miura, E., Hasegawa, T., Konno, M., Suzuki, M., Sugeno, N., Fujikake, N., et al. (2014). VPS35 dysfunction impairs lysosomal degradation of α-synuclein and exacerbates neurotoxicity in a Drosophila model of Parkinson’s disease. Neurobiol. Dis. 71, 1–13. doi: 10.1016/j.nbd.2014.07.014
Morabito, M. V., Berman, D. E., Schneider, R. T., Zhang, Y., Leibel, R. L., and Small, S. A. (2014). Hyperleucinemia causes hippocampal retromer deficiency linking diabetes to Alzheimer’s disease. Neurobiol. Dis. 65, 188–192. doi: 10.1016/j.nbd.2013.12.017
Muhammad, A., Flores, I., Zhang, H., Yu, R., Staniszewski, A., Planel, E., et al. (2008). Retromer deficiency observed in Alzheimer’s disease causes hippocampal dysfunction, neurodegeneration, and Abeta accumulation. Proc. Natl. Acad. Sci. U.S.A. 105, 7327–7332. doi: 10.1073/pnas.0802545105
Munsie, L. N., Milnerwood, A. J., Seibler, P., Beccano-Kelly, D. A., Tatarnikov, I., Khinda, J., et al. (2015). Retromer-dependent neurotransmitter receptor trafficking to synapses is altered by the Parkinson’s disease VPS35 mutation p.D620N. Hum. Mol. Genet. 24, 1691–1703. doi: 10.1093/hmg/ddu582
Muzio, L., Sirtori, R., Gornati, D., Eleuteri, S., Fossaghi, A., Brancaccio, D., et al. (2020). Retromer stabilization results in neuroprotection in a model of Amyotrophic Lateral Sclerosis. Nat. Commun. 11:3848. doi: 10.1038/s41467-020-17524-7
Niu, M., Zhao, F., Bondelid, K., Siedlak, S. L., Torres, S., Fujioka, H., et al. (2021). VPS35 D620N knockin mice recapitulate cardinal features of Parkinson’s disease. Aging Cell 20:e13347. doi: 10.1111/acel.13347
Offe, K., Dodson, S. E., Shoemaker, J. T., Fritz, J. J., Gearing, M., Levey, A. I., et al. (2006). The lipoprotein receptor LR11 regulates amyloid beta production and amyloid precursor protein traffic in endosomal compartments. J. Neurosci. 26, 1596–1603. doi: 10.1523/jneurosci.4946-05.2006
Oshima, R., Hasegawa, T., Tamai, K., Sugeno, N., Yoshida, S., Kobayashi, J., et al. (2016). ESCRT-0 dysfunction compromises autophagic degradation of protein aggregates and facilitates ER stress-mediated neurodegeneration via apoptotic and necroptotic pathways. Sci. Rep. 6:24997. doi: 10.1038/srep24997
Pallesen, L. T., and Vaegter, C. B. (2012). Sortilin and SorLA regulate neuronal sorting of trophic and dementia-linked proteins. Mol. Neurobiol. 45, 379–387. doi: 10.1007/s12035-012-8236-2
Patel, D., Xu, C., Nagarajan, S., Liu, Z., Hemphill, W. O., Shi, R., et al. (2018). Alpha-synuclein inhibits Snx3-retromer-mediated retrograde recycling of iron transporters in S. cerevisiae and C. elegans models of Parkinson’s disease. Hum. Mol. Genet. 27, 1514–1532. doi: 10.1093/hmg/ddy059
Paterson, A. D., Waggott, D., Boright, A. P., Hosseini, S. M., Shen, E., Sylvestre, M. P., et al. (2010). A genome-wide association study identifies a novel major locus for glycemic control in type 1 diabetes, as measured by both A1C and glucose. Diabetes 59, 539–549. doi: 10.2337/db09-0653
Pottier, C., Hannequin, D., Coutant, S., Rovelet-Lecrux, A., Wallon, D., Rousseau, S., et al. (2012). High frequency of potentially pathogenic SORL1 mutations in autosomal dominant early-onset Alzheimer disease. Mol. Psychiatry 17, 875–879. doi: 10.1038/mp.2012.15
Rajendran, L., Honsho, M., Zahn, T. R., Keller, P., Geiger, K. D., Verkade, P., et al. (2006). Alzheimer’s disease beta-amyloid peptides are released in association with exosomes. Proc. Natl. Acad. Sci. U.S.A. 103, 11172–11177. doi: 10.1073/pnas.0603838103
Ravussin, A., Brech, A., Tooze, S. A., and Stenmark, H. (2021). The phosphatidylinositol 3-phosphate-binding protein SNX4 controls ATG9A recycling and autophagy. J. Cell Sci. 134:jcs250670. doi: 10.1242/jcs.250670
Reitz, C., Tokuhiro, S., Clark, L. N., Conrad, C., Vonsattel, J. P., Hazrati, L. N., et al. (2011). SORCS1 alters amyloid precursor protein processing and variants may increase Alzheimer’s disease risk. Ann. Neurol. 69, 47–64. doi: 10.1002/ana.22308
Ren, X., Yao, L., Wang, Y., Mei, L., and Xiong, W. C. (2022). Microglial VPS35 deficiency impairs Aβ phagocytosis and Aβ-induced disease-associated microglia, and enhances Aβ associated pathology. J. Neuroinflamm. 19:61. doi: 10.1186/s12974-022-02422-0
Rogaeva, E., Meng, Y., Lee, J. H., Gu, Y., Kawarai, T., Zou, F., et al. (2007). The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat. Genet. 39, 168–177. doi: 10.1038/ng1943
Rojas, R., Kametaka, S., Haft, C. R., and Bonifacino, J. S. (2007). Interchangeable but essential functions of SNX1 and SNX2 in the association of retromer with endosomes and the trafficking of mannose 6-phosphate receptors. Mol. Cell. Biol. 27, 1112–1124. doi: 10.1128/mcb.00156-06
Rojas, R., van Vlijmen, T., Mardones, G. A., Prabhu, Y., Rojas, A. L., Mohammed, S., et al. (2008). Regulation of retromer recruitment to endosomes by sequential action of Rab5 and Rab7. J. Cell Biol. 183, 513–526. doi: 10.1083/jcb.200804048
Rovelet-Lecrux, A., Charbonnier, C., Wallon, D., Nicolas, G., Seaman, M. N., Pottier, C., et al. (2015). De novo deleterious genetic variations target a biological network centered on Aβ peptide in early-onset Alzheimer disease. Mol. Psychiatry 20, 1046–1056. doi: 10.1038/mp.2015.100
Sager, K. L., Wuu, J., Leurgans, S. E., Rees, H. D., Gearing, M., Mufson, E. J., et al. (2007). Neuronal LR11/sorLA expression is reduced in mild cognitive impairment. Ann. Neurol. 62, 640–647. doi: 10.1002/ana.21190
Sargent, D., Cunningham, L. A., Dues, D. J., Ma, Y., Kordich, J. J., Mercado, G., et al. (2021). Neuronal VPS35 deletion induces spinal cord motor neuron degeneration and early post-natal lethality. Brain Commun. 3:fcab208. doi: 10.1093/braincomms/fcab208
Sassone, J., Reale, C., Dati, G., Regoni, M., Pellecchia, M. T., and Garavaglia, B. (2021). The role of VPS35 in the pathobiology of Parkinson’s disease. Cell. Mol. Neurobiol. 41, 199–227. doi: 10.1007/s10571-020-00849-8
Scherzer, C. R., Offe, K., Gearing, M., Rees, H. D., Fang, G., Heilman, C. J., et al. (2004). Loss of apolipoprotein E receptor LR11 in Alzheimer disease. Arch. Neurol. 61, 1200–1205. doi: 10.1001/archneur.61.8.1200
Schmidt, V., Sporbert, A., Rohe, M., Reimer, T., Rehm, A., Andersen, O. M., et al. (2007). SorLA/LR11 regulates processing of amyloid precursor protein via interaction with adaptors GGA and PACS-1. J. Biol. Chem. 282, 32956–32964. doi: 10.1074/jbc.M705073200
Seaman, M. N. J. (2021). The retromer complex: from genesis to revelations. Trends Biochem. Sci. 46, 608–620. doi: 10.1016/j.tibs.2020.12.009
Seaman, M. N., Harbour, M. E., Tattersall, D., Read, E., and Bright, N. (2009). Membrane recruitment of the cargo-selective retromer subcomplex is catalysed by the small GTPase Rab7 and inhibited by the Rab-GAP TBC1D5. J. Cell Sci. 122(Pt. 14), 2371–2382. doi: 10.1242/jcs.048686
Seaman, M. N., McCaffery, J. M., and Emr, S. D. (1998). A membrane coat complex essential for endosome-to-Golgi retrograde transport in yeast. J. Cell Biol. 142, 665–681. doi: 10.1083/jcb.142.3.665
Sevlever, D., Jiang, P., and Yen, S. H. (2008). Cathepsin D is the main lysosomal enzyme involved in the degradation of alpha-synuclein and generation of its carboxy-terminally truncated species. Biochemistry 47, 9678–9687. doi: 10.1021/bi800699v
Simoes, S., Guo, J., Buitrago, L., Qureshi, Y. H., Feng, X., Kothiya, M., et al. (2021). Alzheimer’s vulnerable brain region relies on a distinct retromer core dedicated to endosomal recycling. Cell Rep. 37:110182. doi: 10.1016/j.celrep.2021.110182
Simoes, S., Neufeld, J. L., Triana-Baltzer, G., Moughadam, S., Chen, E. I., Kothiya, M., et al. (2020). Tau and other proteins found in Alzheimer’s disease spinal fluid are linked to retromer-mediated endosomal traffic in mice and humans. Sci. Transl. Med. 12:eaba6334. doi: 10.1126/scitranslmed.aba6334
Simonetti, B., Danson, C. M., Heesom, K. J., and Cullen, P. J. (2017). Sequence-dependent cargo recognition by SNX-BARs mediates retromer-independent transport of CI-MPR. J. Cell Biol. 216, 3695–3712. doi: 10.1083/jcb.201703015
Small, S. A., Kent, K., Pierce, A., Leung, C., Kang, M. S., Okada, H., et al. (2005). Model-guided microarray implicates the retromer complex in Alzheimer’s disease. Ann. Neurol. 58, 909–919. doi: 10.1002/ana.20667
Steinberg, F., Gallon, M., Winfield, M., Thomas, E. C., Bell, A. J., Heesom, K. J., et al. (2013). A global analysis of SNX27-retromer assembly and cargo specificity reveals a function in glucose and metal ion transport. Nat. Cell Biol. 15, 461–471. doi: 10.1038/ncb2721
Strochlic, T. I., Setty, T. G., Sitaram, A., and Burd, C. G. (2007). Grd19/Snx3p functions as a cargo-specific adapter for retromer-dependent endocytic recycling. J. Cell Biol. 177, 115–125. doi: 10.1083/jcb.200609161
Sugeno, N., Hasegawa, T., Tanaka, N., Fukuda, M., Wakabayashi, K., Oshima, R., et al. (2014). Lys-63-linked ubiquitination by E3 ubiquitin ligase Nedd4-1 facilitates endosomal sequestration of internalized α-synuclein. J. Biol. Chem. 289, 18137–18151. doi: 10.1074/jbc.M113.529461
Sun, M., and Zhang, H. (2017). Par3 and aPKC regulate BACE1 endosome-to-TGN trafficking through PACS1. Neurobiol. Aging 60, 129–140. doi: 10.1016/j.neurobiolaging.2017.08.024
Tammineni, P., Jeong, Y. Y., Feng, T., Aikal, D., and Cai, Q. (2017). Impaired axonal retrograde trafficking of the retromer complex augments lysosomal deficits in Alzheimer’s disease neurons. Hum. Mol. Genet. 26, 4352–4366. doi: 10.1093/hmg/ddx321
Tanaka, K. (2020). The PINK1-Parkin axis: an overview. Neurosci. Res. 159, 9–15. doi: 10.1016/j.neures.2020.01.006
Tang, F. L., Erion, J. R., Tian, Y., Liu, W., Yin, D. M., Ye, J., et al. (2015a). VPS35 in dopamine neurons is required for endosome-to-Golgi retrieval of Lamp2a, a receptor of chaperone-mediated autophagy that is critical for α-synuclein degradation and prevention of pathogenesis of Parkinson’s disease. J. Neurosci. 35, 10613–10628. doi: 10.1523/jneurosci.0042-15.2015
Tang, F. L., Liu, W., Hu, J. X., Erion, J. R., Ye, J., Mei, L., et al. (2015b). VPS35 deficiency or mutation causes dopaminergic neuronal loss by impairing mitochondrial fusion and function. Cell Rep. 12, 1631–1643. doi: 10.1016/j.celrep.2015.08.001
Temkin, P., Lauffer, B., Jäger, S., Cimermancic, P., Krogan, N. J., and von Zastrow, M. (2011). SNX27 mediates retromer tubule entry and endosome-to-plasma membrane trafficking of signalling receptors. Nat. Cell Biol. 13, 715–721. doi: 10.1038/ncb2252
Temkin, P., Morishita, W., Goswami, D., Arendt, K., Chen, L., and Malenka, R. (2017). The retromer supports AMPA receptor trafficking during LTP. Neuron 94, 74–82.e75. doi: 10.1016/j.neuron.2017.03.020
Tian, Y., Tang, F. L., Sun, X., Wen, L., Mei, L., Tang, B. S., et al. (2015). VPS35-deficiency results in an impaired AMPA receptor trafficking and decreased dendritic spine maturation. Mol. Brain 8:70. doi: 10.1186/s13041-015-0156-4
Tsika, E., Glauser, L., Moser, R., Fiser, A., Daniel, G., Sheerin, U. M., et al. (2014). Parkinson’s disease-linked mutations in VPS35 induce dopaminergic neurodegeneration. Hum. Mol. Genet. 23, 4621–4638. doi: 10.1093/hmg/ddu178
Vagnozzi, A. N., Li, J. G., Chiu, J., Razmpour, R., Warfield, R., Ramirez, S. H., et al. (2019). VPS35 regulates tau phosphorylation and neuropathology in tauopathy. Mol. Psychiatry 26, 6992–7005.
Vanan, S., Zeng, X., Chia, S. Y., Varnäs, K., Jiang, M., Zhang, K., et al. (2020). Altered striatal dopamine levels in Parkinson’s disease VPS35 D620N mutant transgenic aged mice. Mol. Brain 13:164. doi: 10.1186/s13041-020-00704-3
Vardarajan, B. N., Bruesegem, S. Y., Harbour, M. E., Inzelberg, R., Friedland, R., St George-Hyslop, P., et al. (2012). Identification of Alzheimer disease-associated variants in genes that regulate retromer function. Neurobiol. Aging 33, 2231.e2215–2231.e2230. doi: 10.1016/j.neurobiolaging.2012.04.020
Vardarajan, B. N., Zhang, Y., Lee, J. H., Cheng, R., Bohm, C., Ghani, M., et al. (2015). Coding mutations in SORL1 and Alzheimer disease. Ann. Neurol. 77, 215–227. doi: 10.1002/ana.24305
Vilariño-Güell, C., Rajput, A., Milnerwood, A. J., Shah, B., Szu-Tu, C., Trinh, J., et al. (2014). DNAJC13 mutations in Parkinson disease. Hum. Mol. Genet. 23, 1794–1801. doi: 10.1093/hmg/ddt570
Vilariño-Güell, C., Wider, C., Ross, O. A., Dachsel, J. C., Kachergus, J. M., Lincoln, S. J., et al. (2011). VPS35 mutations in Parkinson disease. Am. J. Hum. Genet. 89, 162–167. doi: 10.1016/j.ajhg.2011.06.001
Wang, C., Niu, M., Zhou, Z., Zheng, X., Zhang, L., Tian, Y., et al. (2016). VPS35 regulates cell surface recycling and signaling of dopamine receptor D1. Neurobiol. Aging 46, 22–31. doi: 10.1016/j.neurobiolaging.2016.05.016
Wang, H. S., Toh, J., Ho, P., Tio, M., Zhao, Y., and Tan, E. K. (2014). In vivo evidence of pathogenicity of VPS35 mutations in the Drosophila. Mol. Brain 7:73. doi: 10.1186/s13041-014-0073-y
Wang, W., Wang, X., Fujioka, H., Hoppel, C., Whone, A. L., Caldwell, M. A., et al. (2016). Parkinson’s disease-associated mutant VPS35 causes mitochondrial dysfunction by recycling DLP1 complexes. Nat. Med. 22, 54–63. doi: 10.1038/nm.3983
Wassmer, T., Attar, N., Harterink, M., van Weering, J. R., Traer, C. J., Oakley, J., et al. (2009). The retromer coat complex coordinates endosomal sorting and dynein-mediated transport, with carrier recognition by the trans-Golgi network. Dev. Cell 17, 110–122. doi: 10.1016/j.devcel.2009.04.016
Wen, L., Tang, F. L., Hong, Y., Luo, S. W., Wang, C. L., He, W., et al. (2011). VPS35 haploinsufficiency increases Alzheimer’s disease neuropathology. J. Cell Biol. 195, 765–779. doi: 10.1083/jcb.201105109
Willén, K., Edgar, J. R., Hasegawa, T., Tanaka, N., Futter, C. E., and Gouras, G. K. (2017). Aβ accumulation causes MVB enlargement and is modelled by dominant negative VPS4A. Mol. Neurodegener. 12:61. doi: 10.1186/s13024-017-0203-y
Williams, E. T., Glauser, L., Tsika, E., Jiang, H., Islam, S., and Moore, D. J. (2018). Parkin mediates the ubiquitination of VPS35 and modulates retromer-dependent endosomal sorting. Hum. Mol. Genet. 27, 3189–3205. doi: 10.1093/hmg/ddy224
Wu, S., Fagan, R. R., Uttamapinant, C., Lifshitz, L. M., Fogarty, K. E., Ting, A. Y., et al. (2017). The dopamine transporter recycles via a retromer-dependent postendocytic mechanism: tracking studies using a novel fluorophore-coupling approach. J. Neurosci. 37, 9438–9452. doi: 10.1523/jneurosci.3885-16.2017
Wu, Y. R., Lin, C. H., Chao, C. Y., Chang, C. W., Chen, C. M., and Lee-Chen, G. J. (2020). Rare VPS35 A320V variant in taiwanese Parkinson’s disease indicates disrupted CI-MPR sorting and impaired mitochondrial morphology. Brain Sci. 10:783. doi: 10.3390/brainsci10110783
Ye, H., Ojelade, S. A., Li-Kroeger, D., Zuo, Z., Wang, L., Li, Y., et al. (2020). Retromer subunit, VPS29, regulates synaptic transmission and is required for endolysosomal function in the aging brain. eLife 9:e51977. doi: 10.7554/eLife.51977
Yin, J., Liu, X., He, Q., Zhou, L., Yuan, Z., and Zhao, S. (2016). Vps35-dependent recycling of Trem2 regulates microglial function. Traffic (Copenhagen, Denmark) 17, 1286–1296. doi: 10.1111/tra.12451
Yong, X., Zhao, L., Deng, W., Sun, H., Zhou, X., Mao, L., et al. (2020). Mechanism of cargo recognition by retromer-linked SNX-BAR proteins. PLoS Biol. 18:e3000631. doi: 10.1371/journal.pbio.3000631
Yoshida, S., and Hasegawa, T. (2022). Deciphering the prion-like behavior of pathogenic protein aggregates in neurodegenerative diseases. Neurochem. Int. 155:105307. doi: 10.1016/j.neuint.2022.105307
Yoshida, S., Hasegawa, T., Suzuki, M., Sugeno, N., Kobayashi, J., Ueyama, M., et al. (2018). Parkinson’s disease-linked DNAJC13 mutation aggravates alpha-synuclein-induced neurotoxicity through perturbation of endosomal trafficking. Hum. Mol. Genet. 27, 823–836. doi: 10.1093/hmg/ddy003
Young, J. E., Fong, L. K., Frankowski, H., Petsko, G. A., Small, S. A., and Goldstein, L. S. B. (2018). Stabilizing the retromer complex in a human stem cell model of Alzheimer’s disease reduces TAU phosphorylation independently of amyloid precursor protein. Stem Cell Rep. 10, 1046–1058. doi: 10.1016/j.stemcr.2018.01.031
Zavodszky, E., Seaman, M. N., Moreau, K., Jimenez-Sanchez, M., Breusegem, S. Y., Harbour, M. E., et al. (2014). Mutation in VPS35 associated with Parkinson’s disease impairs WASH complex association and inhibits autophagy. Nat. Commun. 5:3828. doi: 10.1038/ncomms4828
Zhang, H., Huang, T., Hong, Y., Yang, W., Zhang, X., Luo, H., et al. (2018). The retromer complex and sorting nexins in neurodegenerative diseases. Front. Aging Neurosci. 10:79. doi: 10.3389/fnagi.2018.00079
Zhao, Y., Perera, G., Takahashi-Fujigasaki, J., Mash, D. C., Vonsattel, J. P. G., Uchino, A., et al. (2018). Reduced LRRK2 in association with retromer dysfunction in post-mortem brain tissue from LRRK2 mutation carriers. Brain 141, 486–495. doi: 10.1093/brain/awx344
Keywords: retromer, membrane trafficking, neurodegeneration, Alzheimer’s Disease, Parkinson’s disease
Citation: Yoshida S and Hasegawa T (2022) Beware of Misdelivery: Multifaceted Role of Retromer Transport in Neurodegenerative Diseases. Front. Aging Neurosci. 14:897688. doi: 10.3389/fnagi.2022.897688
Received: 16 March 2022; Accepted: 19 April 2022;
Published: 06 May 2022.
Edited by:
Anamitra Ghosh, Wave Life Sciences Ltd., United StatesReviewed by:
Ashok Iyaswamy, Hong Kong Baptist University, Hong Kong SAR, ChinaCopyright © 2022 Yoshida and Hasegawa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Takafumi Hasegawa, dGhhc2VnYXdhQG1lZC50b2hva3UuYWMuanA=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.