 Germaine Hiu-Fai Chan
Germaine Hiu-Fai Chan
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Aging Neurosci., 10 June 2022
Sec. Parkinson’s Disease and Aging-related Movement Disorders
Volume 14 - 2022 | https://doi.org/10.3389/fnagi.2022.895430
This article is part of the Research TopicGenetic and Molecular Diversity in Parkinson's DiseaseView all 9 articles
Parkinson’s disease (PD) is a common neurodegenerative disease. At present, 5–10% of PD patients are found to have monogenic form of the disease. Each genetic mutation has its own unique clinical features and disease trajectory. It is unclear if the genetic background can affect the outcome of device-aided therapies in these patients. In general, monogenic PD patients have satisfactory motor outcome after receiving invasive therapies. However, their long-term outcome can vary with their genetic mutations. It appears that patients with leucine-rich repeat kinase-2 (LRRK2) and PRKN mutations tended to have good outcome following deep brain stimulation (DBS) surgery. However, those with Glucocerebrosidase (GBA) mutation were found to have poorer cognitive performance, especially after undergoing subthalamic nucleus DBS surgery. In this review, we will provide an overview of the outcomes of device-aided therapies in PD patients with different genetic mutations.
Parkinson’s disease (PD) is the second commonest neurodegenerative disease affecting both motor and non-motor domains (Kalia and Lang, 2015). At present, no treatment is available to stop or slow down disease progression. However, currently available therapies can offer symptomatic relief to the patients (Kalia and Lang, 2015). In general, with the use of oral dopaminergic treatment, their symptoms can be controlled for a few years after symptom onset before developing motor and non-motor complications (Aquino and Fox, 2015; Kalia and Lang, 2015).
Device-aided therapies including deep brain stimulation (DBS), continuous apomorphine subcutaneous infusion (CASI), and levodopa/carbidopa intestinal gel infusion (LCIG) have been used in the management of advanced PD when oral pharmacological treatments are no longer sufficient to control the symptoms or when the patients cannot tolerate the drugs. Nevertheless, not all the PD patients are suitable candidates for these treatments. The inclusion and exclusion criteria of device-aided therapies of advanced parkinsonism are summarized in Table 1 (Defer et al., 1999; Bronstein et al., 2011; Fernandez and Odin, 2011; Volkmann et al., 2013; Worth, 2013; Odin et al., 2015; Trenkwalder et al., 2015; Moro and Lang, 2016; Munhoz et al., 2016; Timpka et al., 2017; Williams et al., 2017; Burack et al., 2018).
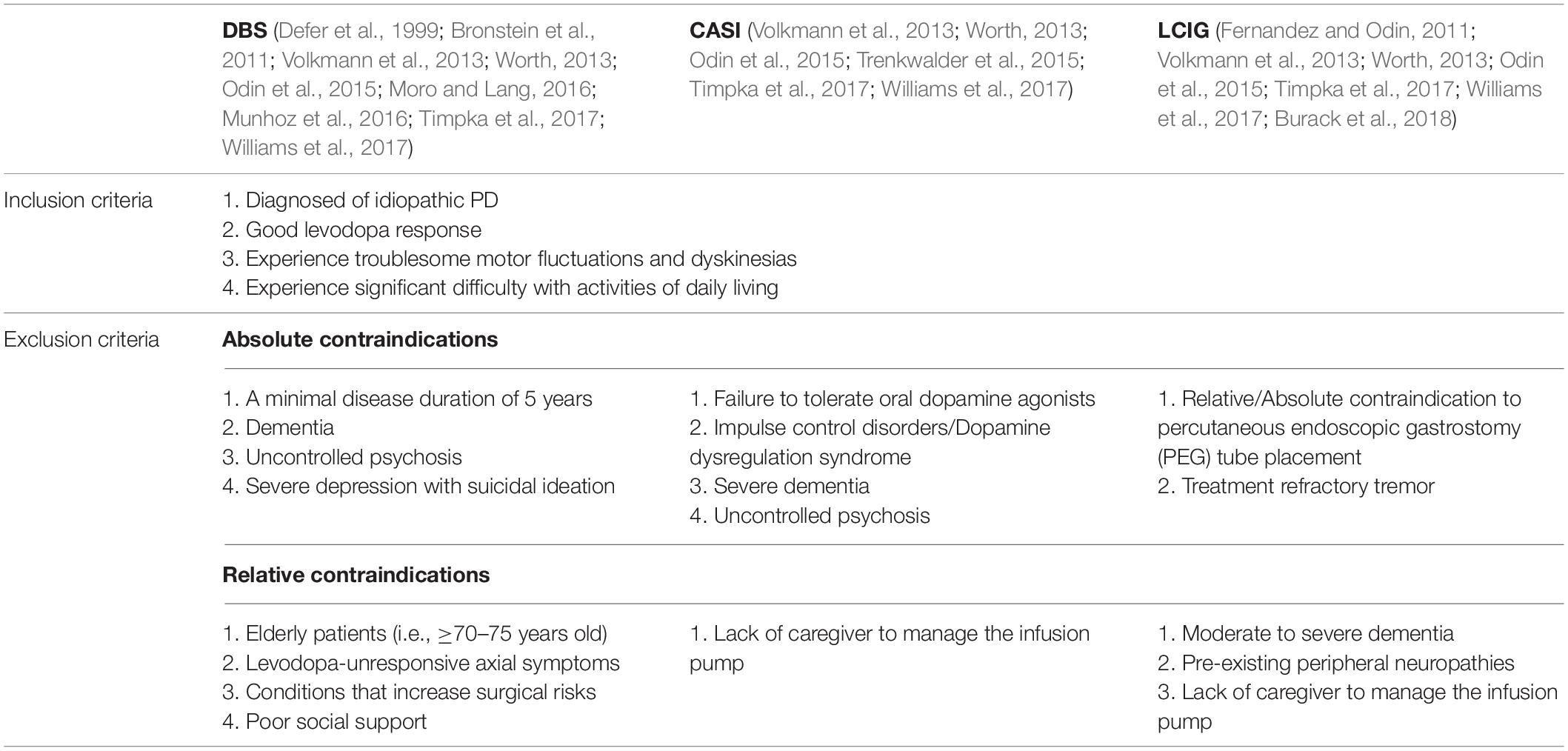
Table 1. Inclusion and exclusion criteria for device-aided therapies of advanced PD.
To date, approximately 5 to 10% of PD patients are found to have monogenic form of the disease, with either an autosomal dominant or autosomal recessive pattern of inheritance (Lesage and Brice, 2009). Different types of genetic parkinsonism have their own distinct clinical features (both motor and non-motor symptoms), disease trajectories and treatment outcomes (Hardy et al., 2009; Niemann and Jankovic, 2019). Unlike dystonia (Andrews et al., 2010; Tisch and Kumar, 2021), the current view on the association between genetic mutation and treatment response to DBS surgery remains equivocal in PD. Besides, genetics testing is not routinely included in the pre-procedural workup of device-aided therapies in advanced PD. It is unclear if the genetic background of PD patients will affect their treatment outcomes.
Therefore, the objective of this minireview is to provide an overview of the outcomes of device-aided therapies in patients with genetic parkinsonism and to provide insight on the need to include genetic testing when choosing device-aided therapies for advanced PD patients.
Twenty-three studies with keywords including “DBS,” “apomorphine,” “levodopa carbidopa intestinal gel,” “PD,” “genetic,” and genes including leucine-rich repeat kinase-2 (LRRK2), SNCA, PRKN, PINK1, and Glucocerebrosidase (GBA) were identified from PubMed search. The results are summarized in Table 2.

Table 2. Summary of treatment outcomes of device-aided therapies in patients with monogenic parkinsonism.
Mutations in the LRRK2 gene are the commonest mutations in sporadic and familial PD. Patients with LRRK2 parkinsonism share clinical features and disease courses which resemble closely to those with idiopathic PD. They often manifest as late-onset PD with typical clinical features of asymmetrical, tremor-dominant parkinsonism with bradykinesia and rigidity. Dystonia, especially painful off-period foot dystonia, is more common in LRRK2 mutation carriers after starting dopaminergic treatment. They tend to have a lower rate of cognitive impairment and hyposmia (Healy et al., 2008; Tolosa et al., 2020). Sleep complaints are frequently seen. Unlike the idiopathic PD patients, their sleep dysfunction is characterized by frequent sleep onset insomnia with less prominent REM sleep behavioral disorders (Pont-Sunyer et al., 2015). Most of these patients have excellent response to dopaminergic treatment (Healy et al., 2008). Even though they have a similar rate of dyskinesia as the non-carriers, it takes a longer period for them to develop dyskinesia after starting levodopa (Healy et al., 2008).
Among the PD patients with LRRK2 mutations, they were more likely to be referred for surgery because of severe dyskinesia (Leaver et al., 2021). They often responded well to DBS surgery. Schüpbach et al. (2007) showed that subthalamic nucleus (STN) DBS is beneficial in LRRK2-PD patients. Artusi et al. (2019) described that motor function improved by 46% in LRRK2 mutation carriers after surgery. Nonetheless, the treatment outcome can vary with the gene mutation in LRRK2-PD patients. Carriers of LRRK2 G2019S gene mutation, the commonest gene mutation in LRRK2-PD patients, were found to have excellent response to STN DBS and their treatment outcome was not inferior to that of the non-carriers (Greenbaum et al., 2013; Sayad et al., 2016). Besides, sustained benefit was seen in two patients with LRRK2 G2019S mutation 9 years after implantation of STN neurostimulator (Schüpbach et al., 2007). Good surgical outcome was also reported in carriers of LRRK2-G2385R gene mutations, which is prevalent in the Hans Chinese population (Chen et al., 2019). On the other hand, PD patients with R1441G mutation seemed to have unsatisfactory outcome from DBS surgery (Gómez-Esteban et al., 2008).
The majority of LRRK2 carriers had a stable cognitive performance after surgery (Schüpbach et al., 2007; Artusi et al., 2019). However, a patient with T2031S mutation developed hallucination and behavioral disorder with addiction to levodopa 5 years post-operatively (Artusi et al., 2019).
Although most of the studies were conducted in patients undergoing STN DBS surgery Leaver et al. (2021), revealed that both STN and globus pallidus internus (GPi) were effective surgical targets in controlling motor symptoms. However, there were three patients developing depression following STN DBS surgery while none of the patients with GPi DBS implanted experienced mood dysfunction (Leaver et al., 2021).
Reduction in dopaminergic medication dosage was reported in the carriers of LRRK2 mutation, especially in those who underwent STN DBS surgery (Artusi et al., 2019; Leaver et al., 2021).
Furthermore, as LRRK2 mutation carriers are known to be associated with sleep dysfunction (Pont-Sunyer et al., 2015), they may not be good candidates for CASI. As for LCIG therapy, they appeared to be good candidates for this treatment because of good response to levodopa (Healy et al., 2008). Nonetheless, no studies are available for the use of infusion therapy in this group of patients.
SNCA mutation was the first gene discovered to cause autosomal-dominant PD. The disease often starts in the fourth or fifth decades. However, one-third of SNCA-PD patients has an early age at onset (i.e., 20–40 years). Atypical features, such as anterocollis/retrocollis, pyramidal signs and alien limb syndrome, can be seen in the SNCA mutation carriers. Moreover, cognitive decline is found in 70% of patients. They often respond well to dopaminergic treatments (Trinh et al., 2018).
The overall treatment outcome of DBS surgery in SNCA-PD patients is satisfactory but it may differ with the type of gene mutation. Shimo et al. (2014) reported that a PD patient with SNCA duplication benefited from STN DBS surgery. Following the surgery, the motor fluctuation improved without developing hallucinations or dementia. Youn et al. (2022) showed that motor fluctuation improved in three PD patients with SNCA duplication after STN DBS surgery. Sustained benefit was observed for up to 10 years post-operatively. There was a significant reduction in the mean levodopa equivalent daily dose (LEDD). Non-motor function remained stable. However, in the patient with SNCA missense mutation (c.158C.A [p.A53E]), although there was motor benefit from the surgery, the patient suffered from natural progression of the disease. She became wheelchair bound and demented 3.5 years after implantation of neurostimulator (Youn et al., 2022).
So far, no case reports are available for the use of CASI and LCIG treatments in SNCA mutation carriers. Infusion pump therapies may be considered in these patients who respond to dopaminergic treatment well and do not suffer from severe dementia.
PRKN mutation is frequently seen in patients with early onset autosomal recessive PD and juvenile PD. The disease usually starts in the third and fourth decades. Typical clinical manifestations at disease onset include limb bradykinesia and tremor, as well as symmetrical foot dystonia (Lücking et al., 2000). Besides, these patients tend to develop depressed mood without executive dysfunction or cognitive impairment (Song et al., 2020). In general, they have a good response to levodopa and a slower disease progression. Dyskinesia is common at a later stage of disease (Lücking et al., 2000).
In contrast, there were case reports of atypical presentations of PRKN mutation carriers. Klein et al. (2000) described an Italian family with PRKN deletions presenting as adult-onset tremor-dominant parkinsonism. Gao et al. (2020) reported a Han Chinese family with PRKN mutation suffering from symmetrical resting tremor of lower limbs without rigidity.
In general, the outcome of STN DBS in this group of patients was favorable. Studies reported that motor function improved by 46 to 84% after surgery (Capecci et al., 2004; Lohmann et al., 2008; Kim et al., 2014; Artusi et al., 2019; de Oliveira et al., 2019). Disabling dyskinesia was found to improve or disappear with STN neurostimulation (Capecci et al., 2004; Kim et al., 2014; Artusi et al., 2019). Sustained improvements in motor function was reported 2 to 5 years post-operatively (Lücking et al., 2000). Nonetheless, postural stability and gait was worse in PRKN carriers than in non-carriers after receiving STN DBS (Kim et al., 2014).
Besides, significant decline in cognitive function was not seen in these patients who received STN DBS surgery (Artusi et al., 2019). Medication reduction was reported (Capecci et al., 2004; Lohmann et al., 2008; Kim et al., 2014; Artusi et al., 2019; de Oliveira et al., 2019). Artusi et al. (2019) revealed that the LEDD was reduced by 61% in PRKN mutation carriers after surgery.
Limited data is available for PRKN mutation carriers undergoing pallidal stimulation. A United Kingdom group reported limited motor benefit in this group of patients (21% improvement with GPi DBS vs. 31% improvement with STN DBS). However, motor complications, in terms of UPDRS-IV scores, improved by 70%. In addition, there was in an increment in the dopaminergic medication dosage after undergoing GPi DBS surgery (Angeli et al., 2013).
Furthermore, the type of gene mutation in PRKN mutation carriers may have an impact on the response to DBS surgery. A French group indicated that as compared with the non-carriers and single heterozygous mutation carriers, there was a significant reduction in dopaminergic medication dosage in patients with homozygous or compound heterozygous mutations after surgery (Lohmann et al., 2008). Yet, little information is available concerning the association between surgical outcome and genetic background of PRKN-PD patients and it is premature to conclude if those with 2 PRKN mutations tend to have better treatment response.
On the other hand, CASI therapy may be beneficial in the PRKN mutation carriers, especially when severe cognitive impairment is known to be rare in this group of patients. Lim et al. (2021) reported that a 47-year-old Malaysian woman with PD who carried homozygous PRKN mutation (p.Cys441Arg, c.1321T>C, and exon 12) showing excellent response from CASI treatment.
As for LCIG therapy, there is a lack of literature concerning its use in PRKN mutation carriers. However, this group seldom has marked cognitive impairment or peripheral neuropathies and so they may consider this treatment when motor complications occur.
PINK1 mutation is an important genetic cause of early onset PD. Their clinical manifestations are indistinguishable from that of other early onset PD patients. They usually have a good response to levodopa and a slow disease progression. Some patients may develop dementia at a later stage of the disease (Kasten et al., 2018).
Moro et al. (2008) showed that bilateral STN DBS was beneficial in a patient with PINK1 mutation. Balestrino et al. (2021) reported that a patient with homozygous PINK1 mutation [619C>T-p. (Arg207*)] developed motor fluctuation and subsequently received STN DBS surgery 13 years after disease onset. Transient improvement was noted after surgery. However, 1 year after the surgery, her condition worsened with freezing of gait and dyskinesia (Balestrino et al., 2021). Borellini et al. (2017, 2021) described successful short-term outcome of bilateral pallidal stimulation in a Filipino woman with homozygous L347P PINK1 mutation who suffered from painful dystonia of lower limbs and progressive gait dysfunction. Nevertheless, despite optimal medical treatment and programming, her condition deteriorated due to worsening of dystonia, and she had to walk with aids 4 years after surgery (Borellini et al., 2017, 2021).
So far, no data is available for CASI and LCIG treatments in this group of mutation carriers.
Glucocerebrosidase mutations are the commonest genetic risk factor for the development of PD. A multicenter study conducted in 16 centers worldwide demonstrated a strong association between GBA mutation and PD, especially in the Ashkenazi Jewish population (Sidransky et al., 2009). The clinical features of GBA-PD patients are similar to that of idiopathic PD patients (Winder-Rhodes et al., 2013). Nonetheless, GBA mutation carriers usually present at a younger age (Wang et al., 2014). They tend to have more symmetrical parkinsonian features (Sidransky et al., 2009). They have a more rapid motor progression with a greater impact on postural stability and gait (Brockmann et al., 2015; Davis et al., 2016). Levodopa induced dyskinesia are frequently seen (Lesage et al., 2011). In addition, non-motor symptoms are more common and severe in GBA-PD patients. They include dementia, visual hallucination, REM sleep behavioral disorders, and autonomic dysfunction (Brockmann et al., 2011; Jesús et al., 2016). The progression of cognitive decline is faster in GBA mutation carriers (Brockmann et al., 2015; Davis et al., 2016). Moreover, they tend to require a higher dosage of dopaminergic medications (Brockmann et al., 2015).
The disease trajectory can be affected by the type of GBA variants. Jesús et al. (2016) indicated that deleterious GBA variants (including L444P mutation) were associated with a more rapid progression to dementia and visual hallucination. On the contrary, benign GBA variants (including E326K mutation) tended to develop dyskinesia (Jesús et al., 2016).
As a whole, motor outcome was satisfactory in GBA mutation carriers undergoing STN DBS surgery (Artusi et al., 2019). The UPDRS motor score improved by 49% in these patients after surgery (Artusi et al., 2019). However, post-operative cognitive impairment was found to be more common and severe in this group of patients (Lythe et al., 2017; Artusi et al., 2019; Mangone et al., 2020). Pal et al. (2022) revealed that the combined effects of GBA mutation and STN DBS may increase the risk of cognitive decline (Pal et al., 2022). Also, non-motor symptoms (with cardiovascular symptoms as an exception) were found to be more prevalent and severe after surgery (Lythe et al., 2017). Besides, the quality of life worsened significantly in GBA mutation carriers after receiving DBS surgery, with greatest deterioration in domains such as mobility, activities of daily living, cognition, and communication (Lythe et al., 2017). Furthermore, it appeared that the severity of the type of GBA mutation was not linked to a difference in the rate of cognitive decline after operation (Pal et al., 2022).
In contrast, Angeli et al. (2013) reported 2 GBA-PD patients receiving GPi DBS surgery. Their motor benefit was not as robust as that of those with STN DBS implanted (22% improvement with GPi DBS vs. 40% improvement with STN DBS). Yet, the motor complications (especially dyskinesia) markedly improved after surgery. They were able to achieve a greater amount of medication reduction too (Angeli et al., 2013). In addition, this group showed satisfactory outcome in a GBA-PD patient undergoing DBS surgery of unilateral ventral intermediate nucleus of the thalamus (Angeli et al., 2013).
So far, there were no studies concerning the use of infusion therapy in GBA mutation carriers. Anyway, as cognitive impairment is frequently seen in these patients, they may not be good candidates for CASI and LCIG therapies.
This article offered an overview of the outcomes of device-aided therapies in patients with advanced PD with different genetic mutations. At present, the majority of studies were conducted in patients receiving DBS surgery. Data concerning other therapeutic options such as infusion therapies and lesioning surgeries are lacking.
In general, the short-term motor outcome of most patients with genetic parkinsonism is satisfactory. Their motor function and motor fluctuation improved markedly post-op. However, in patients with certain genetic mutations, their condition may deteriorate with time due to cognitive impairment and levodopa-resistant axial features, even when the pre-operative workup indicated that they were suitable candidates for DBS surgery. For instance, in GBA-PD patients, it has been reported that the long-term outcome was limited by the development of dementia after implantation of STN neurostimulators. In the PINK1 mutation carriers, despite transient therapeutic benefit after surgery, patients were found to develop axial symptoms and dyskinesia, which barely responded to medical treatments and titration of neurostimulator settings. To make things more complicated, variants within genetic mutation matter and LRRK2 mutation is a good example. In general, carriers of LRRK2 mutation are known to respond to DBS surgery very well. Yet, some Spanish LRRK2-PD patients with R1441G mutation were found to have suboptimal response from DBS surgery.
At present, genetic testing is not regularly included in the panel of pre-procedural workup of device-aided therapies of advanced PD. Nonetheless, genetic mutation may affect the long-term effects of these invasive therapies. Even though pre-procedural workup results indicate that patients of certain genetic mutations are good candidates of device-aided therapies, the presence of genetic mutations can influence treatment outcome in the long run. Therefore, it may be reasonable to include genetic testing in advanced PD patients when considering invasive therapies.
Nevertheless, currently available data is not sufficient to recommend or refute the use of device-aided therapies in monogenic parkinsonism because there are many genes responsible for genetic parkinsonism (both causative and risk factor gene) and the phenotypes of different mutations vary greatly. Moreover, the number of patients with monogenic PD in each center is very low and so it is difficult to conduct large-scale trials of invasive therapies in these patients.
This review had a few limitations. Most of the available literature comprises retrospective, anecdotal and short-term studies. Secondly, most of the studies focused on the motor outcome following device-aided therapies. The impact of these treatments on non-motor issues, especially cognitive performance, was often neglected. Thirdly, little information is available concerning the use of infusion therapies and lesioning surgery in patients with monogenic PD. It is uncertain if these patients can have a different outcome with invasive therapies other than DBS surgery.
In conclusion, monogenic PD patients usually have satisfactory motor outcome after receiving invasive therapies. However, their genetic background can affect the long-term outcome of device-aided therapies due to development of axial symptoms and cognitive impairment. In general, patients with LRRK2 and PRKN mutations tended to enjoy good surgical outcome following DBS surgery. Yet, carriers of GBA mutation, especially those with STN DBS implanted, were found to have poorer cognitive performance. Therefore, it may be useful to incorporate genetic data in the management of advanced PD patients. Long term multicenter studies are required in the future in order to draw conclusions of predicting outcomes of invasive therapies based on the genetic information.
GC was responsible for designing the study and writing the manuscript.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Andrews, C., Aviles-Olmos, I., Hariz, M., and Foltynie, T. (2010). Which patients with dystonia benefit from deep brain stimulation? A metaregression of individual patient outcomes. J. Neurol. Neurosurg. Psychiatry 81, 1383–1389. doi: 10.1136/jnnp.2010.207993
Angeli, A., Mencacci, N. E., Duran, R., Aviles-Olmos, I., Kefalopoulou, Z., Candelario, J., et al. (2013). Genotype and phenotype in Parkinson’s disease: lessons in heterogeneity from deep brain stimulation. Mov. Disord. 28, 1370–1375. doi: 10.1002/mds.25535
Aquino, C. C., and Fox, S. H. (2015). Clinical spectrum of levodopa-induced complications. Mov. Disord. 30, 80–89. doi: 10.1002/mds.26125
Artusi, C. A., Dwivedi, A. K., Romagnolo, A., Pal, G., Kauffman, M., Mata, I., et al. (2019). Association of subthalamic deep brain stimulation with motor, functional, and pharmacologic outcomes in patients with monogenic parkinson disease: a systematic review and meta-analysis. JAMA Netw. Open 2:e187800. doi: 10.1001/jamanetworkopen.2018.7800
Balestrino, R., Ledda, C., Romagnolo, A., Bozzali, M., Giulietti, G., Montanaro, E., et al. (2021). Motor and non-motor outcomes of subthalamic deep brain stimulation in a case of juvenile PARK-PINK1. Brain Stimul. 14, 725–727. doi: 10.1016/j.brs.2021.04.002
Borellini, L., Cogiamanian, F., and Ardolino, G. (2021). Globus pallidus internus deep brain stimulation in PINK-1 related Parkinson’s disease: an update. Parkinsonism Relat. Disord. 82, 104–105. doi: 10.1016/j.parkreldis.2020.11.023
Borellini, L., Cogiamanian, F., Carrabba, G., Locatelli, M., Rampini, P., Di Fonzo, A., et al. (2017). Globus pallidus internus deep brain stimulation in PINK-1 related Parkinson’s disease: a case report. Parkinsonism Relat. Disord. 38, 93–94. doi: 10.1016/j.parkreldis.2017.02.018
Brockmann, K., Srulijes, K., Hauser, A. K., Schulte, C., Csoti, I., Gasser, T., et al. (2011). GBA-associated PD presents with nonmotor characteristics. Neurology 77, 276–280. doi: 10.1212/WNL.0b013e318225ab77
Brockmann, K., Srulijes, K., Pflederer, S., Hauser, A. K., Schulte, C., Maetzler, W., et al. (2015). GBA-associated Parkinson’s disease: reduced survival and more rapid progression in a prospective longitudinal study. Mov. Disord. 30, 407–411. doi: 10.1002/mds.26071
Bronstein, J. M., Tagliati, M., Alterman, R. L., Lozano, A. M., Volkmann, J., Stefani, A., et al. (2011). Deep brain stimulation for Parkinson disease: an expert consensus and review of key issues. Arch. Neurol. 68:165. doi: 10.1001/archneurol.2010.260
Burack, M., Aldred, J., Zadikoff, C., Vanagunas, A., Klos, K., Bilir, B., et al. (2018). Implementing levodopa-carbidopa intestinal gel for parkinson disease: insights from US practitioners. Mov. Disord. Clin. Pract. 5, 383–393. doi: 10.1002/mdc3.12630
Capecci, M., Passamonti, L., Annesi, F., Annesi, G., Bellesi, M., Candiano, I. C., et al. (2004). Chronic bilateral subthalamic deep brain stimulation in a patient with homozygous deletion in the parkin gene. Mov. Disord. 19, 1450–1452. doi: 10.1002/mds.20250
Chen, S., Liu, H., Wu, Q. Q., Xu, S. J., Li, W. G., Chen, T., et al. (2019). Effect of LRRK2 G2385R variant on subthalamic deep brain stimulation efficacy in parkinson’s disease in a han chinese population. Front. Neurol. 10:1231. doi: 10.3389/fneur.2019.01231
Davis, M. Y., Johnson, C. O., Leverenz, J. B., Weintraub, D., Trojanowski, J. Q., Chen-Plotkin, A., et al. (2016). Association of GBA mutations and the E326K polymorphism with motor and cognitive progression in parkinson disease. JAMA Neurol. 73, 1217–1224. doi: 10.1001/jamaneurol.2016.2245
de Oliveira, L. M., Barbosa, E. R., Aquino, C. C., Munhoz, R. P., Fasano, A., and Cury, R. G. (2019). Deep brain stimulation in patients with mutations in parkinson’s disease-related genes: a systematic review. Mov. Disord. Clin. Pract. 6, 359–368. doi: 10.1002/mdc3.12795
Defer, G. L., Widner, H., Marié, R. M., Rémy, P., and Levivier, M. (1999). Core assessment program for surgical interventional therapies in Parkinson’s disease (CAPSIT-PD). Mov. Disord. 14, 572–584. doi: 10.1002/1531-8257(199907)14:4<572::aid-mds1005<3.0.co;2-c
Fernandez, H. H., and Odin, P. (2011). Levodopa-carbidopa intestinal gel for treatment of advanced Parkinson’s disease. Curr. Med. Res. Opin. 27, 907–919. doi: 10.1185/03007995.2011.560146
Gao, C., Huang, T., Chen, R., Yuan, Z., Tian, Y., and Zhang, Y. A. (2020). Han chinese family with early-onset parkinson’s disease carrying novel frameshift mutation and compound heterozygous mutation of PRKN appearing incompatible with MDS clinical diagnostic criteria. Front. Neurol. 11:582323. doi: 10.3389/fneur.2020.582323
Gómez-Esteban, J. C., Lezcano, E., Zarranz, J. J., González, C., Bilbao, G., Lambarri, I., et al. (2008). Outcome of bilateral deep brain subthalamic stimulation in patients carrying the R1441G mutation in the LRRK2 dardarin gene. Neurosurgery 62, 857–862.
Greenbaum, L., Israeli-Korn, S. D., Cohen, O. S., Elincx-Benizri, S., Yahalom, G., Kozlova, E., et al. (2013). The LRRK2 G2019S mutation status does not affect the outcome of subthalamic stimulation in patients with Parkinson’s disease. Parkinsonism Relat. Disord. 19, 1053–1056. doi: 10.1016/j.parkreldis.2013.07.005
Hardy, J., Lewis, P., Revesz, T., Lees, A., and Paisan-Ruiz, C. (2009). The genetics of Parkinson’s syndromes: a critical review. Curr. Opin. Genet. Dev. 19, 254–265. doi: 10.1016/j.gde.2009.03.008
Healy, D. G., Falchi, M., O’Sullivan, S. S., Bonifati, V., Durr, A., Bressman, S., et al. (2008). International LRRK2 Consortium. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol. 7, 583–590. doi: 10.1016/S1474-4422(08)70117-0
Jesús, S., Huertas, I., Bernal-Bernal, I., Bonilla-Toribio, M., Cáceres-Redondo, M. T., Vargas-González, L., et al. (2016). GBA variants influence motor and non-motor features of parkinson’s disease. PLoS One 11:e0167749. doi: 10.1371/journal.pone.0167749
Kalia, L. V., and Lang, A. E. (2015). Parkinson’s disease. Lancet 386, 896–912. doi: 10.1016/S0140-6736(14)61393-3
Kasten, M., Hartmann, C., Hampf, J., Schaake, S., Westenberger, A., Vollstedt, E. J., et al. (2018). Genotype-phenotype relations for the parkinson’s disease genes parkin, PINK1, DJ1: MDSGene systematic review. Mov. Disord. 33, 730–741. doi: 10.1002/mds.27352
Kim, H. J., Yun, J. Y., Kim, Y. E., Lee, J. Y., Kim, H. J., Kim, J. Y., et al. (2014). Parkin mutation and deep brain stimulation outcome. J. Clin. Neurosci. 21, 107–110. doi: 10.1016/j.jocn.2013.03.026
Klein, C., Pramstaller, P. P., Kis, B., Page, C. C., Kann, M., Leung, J., et al. (2000). Parkin deletions in a family with adult-onset, tremor-dominant parkinsonism: expanding the phenotype. Ann. Neurol. 48, 65–71.
Leaver, K., Viser, A., Kopell, B. H., Ortega, R. A., Miravite, J., Okun, M. S., et al. (2021). Clinical profiles and outcomes of deep brain stimulation in G2019S LRRK2 Parkinson disease. J. Neurosurg. 1–8. doi: 10.3171/2021.7.JNS21190 [Epub ahead of print].
Lesage, S., and Brice, A. (2009). Parkinson’s disease: from monogenic forms to genetic susceptibility factors. Hum. Mol. Genet. 18, R48–R59. doi: 10.1093/hmg/ddp012
Lesage, S., Anheim, M., Condroyer, C., Pollak, P., Durif, F., Dupuits, C., et al. (2011). French parkinson’s disease genetics study group. Large-scale screening of the Gaucher’s disease-related glucocerebrosidase gene in Europeans with Parkinson’s disease. Hum. Mol. Genet. 20, 202–210. doi: 10.1093/hmg/ddq454
Lim, S. Y., Ahmad-Annuar, A., Lohmann, K., Tan, A. H., Tay, Y. W., Lim, J. L., et al. (2021). Clinical phenotype of Parkinson’s disease with a homozygous PRKN p.Cys441Arg mutation. Neurol. Asia 26, 161–166.
Lohmann, E., Welter, M. L., Fraix, V., Krack, P., Lesage, S., Laine, S., et al. (2008). French parkinson’s disease genetics study group. Are parkin patients particularly suited for deep-brain stimulation? Mov. Disord. 23, 740–743. doi: 10.1002/mds.21903
Lücking, C. B., Dürr, A., Bonifati, V., Vaughan, J., De Michele, G., Gasser, T., et al. (2000). French parkinson’s disease genetics study group; european consortium on genetic susceptibility in parkinson’s disease. association between early-onset parkinson’s disease and mutations in the parkin gene. N. Engl. J. Med. 342, 1560–1567. doi: 10.1056/NEJM200005253422103
Lythe, V., Athauda, D., Foley, J., Mencacci, N. E., Jahanshahi, M., Cipolotti, L., et al. (2017). GBA-Associated parkinson’s disease: progression in a deep brain stimulation cohort. J. Parkinsons Dis. 7, 635–644. doi: 10.3233/JPD-171172
Mangone, G., Bekadar, S., Cormier-Dequaire, F., Tahiri, K., Welaratne, A., Czernecki, V., et al. (2020). Early cognitive decline after bilateral subthalamic deep brain stimulation in Parkinson’s disease patients with GBA mutations. Parkinsonism Relat. Disord. 76, 56–62. doi: 10.1016/j.parkreldis.2020.04.002
Moro, E., and Lang, A. E. (2016). Criteria for deep-brain stimulation in Parkinson’s disease: review and analysis. Expert Rev. Neurother. 6, 1695–1705. doi: 10.1586/14737175.6.11.1695
Moro, E., Volkmann, J., König, I. R., Winkler, S., Hiller, A., Hassin-Baer, S., et al. (2008). Bilateral subthalamic stimulation in Parkin and PINK1 parkinsonism. Neurology 70, 1186–1191. doi: 10.1212/01.wnl.0000307748.11216.03
Munhoz, R. P., Picillo, M., Fox, S. H., Bruno, V., Panisset, M., Honey, C. R., et al. (2016). Eligibility criteria for deep brain stimulation in parkinson’s disease, tremor, and dystonia. Can. J. Neurol. Sci. 43, 462–471. doi: 10.1017/cjn.2016.35
Niemann, N., and Jankovic, J. (2019). Juvenile parkinsonism: differential diagnosis, genetics, and treatment. Parkinsonism Relat. Disord. 67, 74–89. doi: 10.1016/j.parkreldis.2019.06.025
Odin, P., Ray Chaudhuri, K., Slevin, J. T., Volkmann, J., Dietrichs, E., Martinez-Martin, P., et al. (2015). Collective physician perspectives on non-oral medication approaches for the management of clinically relevant unresolved issues in Parkinson’s disease: consensus from an international survey and discussion program. Parkinsonism Relat. Disord. 21, 1133–1144. doi: 10.1016/j.parkreldis.2015.07.020
Pal, G., Mangone, G., Hill, E. J., Ouyang, B., Liu, Y., Lythe, V., et al. (2022). Parkinson disease and subthalamic nucleus deep brain stimulation: cognitive effects in GBA mutation carriers. Ann. Neurol. 91, 424–435. doi: 10.1002/ana.26302
Pont-Sunyer, C., Iranzo, A., Gaig, C., Fernández-Arcos, A., Vilas, D., Valldeoriola, F., et al. (2015). Sleep disorders in parkinsonian and nonparkinsonian LRRK2 mutation carriers. PLoS One 10:e0132368. doi: 10.1371/journal.pone.0132368
Sayad, M., Zouambia, M., Chaouch, M., Ferrat, F., Nebbal, M., Bendini, M., et al. (2016). Greater improvement in LRRK2 G2019S patients undergoing Subthalamic Nucleus Deep Brain Stimulation compared to non-mutation carriers. BMC Neurosci. 17:6. doi: 10.1186/s12868-016-0240-4
Schüpbach, M., Lohmann, E., Anheim, M., Lesage, S., Czernecki, V., Yaici, S., et al. (2007). Subthalamic nucleus stimulation is efficacious in patients with Parkinsonism and LRRK2 mutations. Mov. Disord. 22, 119–122. doi: 10.1002/mds.21178
Shimo, Y., Natori, S., Oyama, G., Nakajima, M., Ishii, H., Arai, H., et al. (2014). Subthalamic deep brain stimulation for a Parkinson’s disease patient with duplication of SNCA. Neuromodulation 17, 102–103. doi: 10.1111/ner.12034
Sidransky, E., Nalls, M. A., Aasly, J. O., Aharon-Peretz, J., Annesi, G., Barbosa, E. R., et al. (2009). Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med. 361, 1651–1661. doi: 10.1056/NEJMoa0901281
Song, J., Shen, B., Yang, Y. J., Liu, F. T., Zhao, J., Tang, Y. L., et al. (2020). Non-motor symptoms in parkinson’s disease patients with parkin mutations: more depression and less executive dysfunction. J. Mol. Neurosci. 70, 246–253. doi: 10.1007/s12031-019-01444-3
Timpka, J., Nitu, B., Datieva, V., Odin, P., and Antonini, A. (2017). Device-Aided treatment strategies in advanced Parkinson’s Disease. Int. Rev. Neurobiol. 132, 453–474. doi: 10.1016/bs.irn.2017.03.001
Tisch, S., and Kumar, K. R. (2021). Pallidal deep brain stimulation for monogenic dystonia: the effect of gene on outcome. Front. Neurol. 11:630391. doi: 10.3389/fneur.2020.630391
Tolosa, E., Vila, M., Klein, C., and Rascol, O. (2020). LRRK2 in Parkinson disease: challenges of clinical trials. Nat. Rev. Neurol. 16, 97–107. doi: 10.1038/s41582-019-0301-2
Trenkwalder, C., Chaudhuri, K. R., García Ruiz, P. J., LeWitt, P., Katzenschlager, R., Sixel-Döring, F., et al. (2015). Expert consensus group report on the use of apomorphine in the treatment of Parkinson’s disease - Clinical practice recommendations. Parkinsonism Relat. Disord. 21, 1023–1030. doi: 10.1016/j.parkreldis.2015.06.012
Trinh, J., Zeldenrust, F. M. J., Huang, J., Kasten, M., Schaake, S., Petkovic, S., et al. (2018). Genotype-phenotype relations for the Parkinson’s disease genes SNCA, LRRK2, VPS35: MDSGene systematic review. Mov. Disord. 33, 1857–1870. doi: 10.1002/mds.27527
Volkmann, J., Albanese, A., Antonini, A., Chaudhuri, K. R., Clarke, C. E., de Bie, R. M., et al. (2013). Selecting deep brain stimulation or infusion therapies in advanced Parkinson’s disease: an evidence-based review. J. Neurol. 260, 2701–2714. doi: 10.1007/s00415-012-6798-6
Wang, C., Cai, Y., Gu, Z., Ma, J., Zheng, Z., Tang, B. S., et al. (2014). Chinese parkinson study group. clinical profiles of parkinson’s disease associated with common leucine-rich repeat kinase 2 and glucocerebrosidase genetic variants in Chinese individuals. Neurobiol. Aging 35, e1–.e6. doi: 10.1016/j.neurobiolaging.2013.08.012
Weiss, D., Brockmann, K., Srulijes, K., Meisner, C., Klotz, R., Reinbold, S., et al. (2012). Long-term follow-up of subthalamic nucleus stimulation in glucocerebrosidase-associated Parkinson’s disease. J. Neurol. 259, 1970–1972. doi: 10.1007/s00415-012-6469-7
Williams, D. R., Evans, A. H., Fung, V. S. C., Hayes, M., Iansek, R., Kimber, T., et al. (2017). Practical approaches to commencing device-assisted therapies for Parkinson disease in Australia. Intern. Med. J. 47, 1107–1113. doi: 10.1111/imj.13398
Winder-Rhodes, S. E., Evans, J. R., Ban, M., Mason, S. L., Williams-Gray, C. H., Foltynie, T., et al. (2013). Glucocerebrosidase mutations influence the natural history of Parkinson’s disease in a community-based incident cohort. Brain. 136(Pt 2) 392–399. doi: 10.1093/brain/aws318
Worth, P. F. (2013). When the going gets tough: how to select patients with Parkinson’s disease for advanced therapies. Pract. Neurol. 13, 140–152. doi: 10.1136/practneurol-2012-000463
Keywords: Parkinson’s disease, genetics, deep brain stimulation, apomorphine, levodopa/carbidopa intestinal gel
Citation: Chan GHF (2022) The Role of Genetic Data in Selecting Device-Aided Therapies in Patients With Advanced Parkinson’s Disease: A Mini-Review. Front. Aging Neurosci. 14:895430. doi: 10.3389/fnagi.2022.895430
Received: 13 March 2022; Accepted: 16 May 2022;
Published: 10 June 2022.
Edited by:
Siti Aishah Sulaiman, National University of Malaysia, MalaysiaReviewed by:
Dejan Georgiev, University Medical Centre, Ljubljana, SloveniaCopyright © 2022 Chan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Germaine Hiu-Fai Chan, Q2hmODYyQGhhLm9yZy5oaw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.