
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Aging Neurosci. , 30 June 2022
Sec. Cellular and Molecular Mechanisms of Brain-aging
Volume 14 - 2022 | https://doi.org/10.3389/fnagi.2022.878958
This article is part of the Research Topic Rare and Common Neurodegenerative Retinal Diseases: From Molecular Mechanisms to the Identification of Novel Mutation-independent Therapeutic Strategies View all 3 articles
 Daniela Intartaglia1†
Daniela Intartaglia1† Giuliana Giamundo1,2†
Giuliana Giamundo1,2† Federica Naso1Edoardo Nusco1Simona Di Giulio1
Federica Naso1Edoardo Nusco1Simona Di Giulio1 Francesco Giuseppe Salierno1
Francesco Giuseppe Salierno1 Elena Polishchuk1
Elena Polishchuk1 Ivan Conte1,2*
Ivan Conte1,2*Autophagy is a critical metabolic process that acts as a major self-digestion and recycling pathway contributing to maintain cellular homeostasis. An emerging field of research supports the therapeutic modulation of autophagy for treating human neurodegenerative disorders, in which toxic aggregates are accumulated in neurons. Our previous study identified Ezrin protein as an inhibitor of autophagy and lysosomal functions in the retina; thus, in turn, identifying it as a potential pharmacological target for increasing retinal cell clearance to treat inherited retinal dystrophies in which misfolded proteins have accumulated. This study aimed to verify the therapeutic inhibition of Ezrin to induce clearance of toxic aggregates in a mouse model for a dominant form of retinitis pigmentosa (i.e., RHOP23H/+). We found that daily inhibition of Ezrin significantly decreased the accumulation of misfolded RHOP23H aggregates. Remarkably, induction of autophagy, by a drug-mediated pulsatile inhibition of Ezrin, promoted the lysosomal clearance of disease-linked RHOP23H aggregates. This was accompanied with a reduction of endoplasmic reticulum (ER)-stress, robust decrease of photoreceptors' cell death, amelioration in both retinal morphology and function culminating in a better preservation of vision. Our study opens new perspectives for a pulsatile pharmacological induction of autophagy as a mutation-independent therapy paving the way toward a more effective therapeutic strategy to treat these devastating retinal disorders due to an accumulation of intracellular toxic aggregates.
Retinitis pigmentosa (RP) is a heterogeneous group of inherited retinal disorders (IRDs), affecting ~1.5 million people worldwide (Berson, 1996; Rattner et al., 1999; Hartong et al., 2006; Athanasiou et al., 2013). These conditions display genetic and clinical heterogeneity, with variable time of onset of the disease from childhood to middle age (Gu et al., 1999). Patients with this disorder typically develop night blindness and progressive loss of peripheral vision due to rod photoreceptor cell death. Secondary cone photoreceptor cell death causes legal blindness at later stages of RP. RP phenotypes can be generated by autosomal dominant, recessive or X-linked recessive modes, and ~60 genes have been implicated in RP disease (Farrar et al., 2002; Daiger et al., 2007). Although more than 60 RP genes have been identified to date, we are still far from an efficient therapy. At present, the recent success of clinical trials is providing sound evidence for the use of gene-based complementation therapeutic strategies to treat genetic recessive traits (Illing et al., 2002; Saliba et al., 2002; Mendes et al., 2005; Mendes and Cheetham, 2008; Mitter et al., 2012; Rodriguez-Muela et al., 2013; Mohlin et al., 2014). Among these studies, the successful reversal of blindness in individuals with Leber Congenital Amaurosis (LCA), a congenital form of IRD, represents a milestone result for treatment of genetic diseases and has been encouraging to the entire field of gene therapy (Mendes et al., 2005; Mitter et al., 2012; Chen et al., 2013; Rodriguez-Muela et al., 2013; Mohlin et al., 2014). Autosomal dominant retinitis pigmentosa (adRP) accounts for ~20% of all RP cases and is characterized by significant allelic and non-allelic heterogeneity. The difficulties posed by dominant genetic traits are owed to the nature of the disease. Typically, the dominant mutations affecting one allele cause complex sequels of events eventually leading to cell dysfunction and death through partial loss-of-function, gain-of-function, dominant negative, or toxic effects (Daiger et al., 2014). If one considers mutation-specific treatments, in the case of rhodopsin causing adRP, the theoretical number of therapeutics necessary to be designed and developed would be more than 200, compared to recessive genetic diseases in which one single correct copy of a gene can in principle cure different loss of function mutations. In addition, clinical studies for such numerous therapeutic protocols appear unrealistic and unfeasible. The recent advances in adRP have pointed out to the role of mistrafficking and accumulation of mutated and unfolded proteins in impairing normal cellular function and inducing toxicity in photoreceptor (PR) cells. Mutations in the Rhodopsin (RHO) gene, affecting the amino acidic sequence of the rod-specific protein rhodopsin, are responsible primarily for adRP and account for 30–40% of this form of RP. One of the most common causes of adRP is due to a missense mutation in the RHO gene, which results in the substitution of histidine for proline at amino acid residue 23 (RHOP23H) (Liu et al., 1996; Saliba et al., 2002). Mutated RHOP23H results highly aggregation-prone within the ER leading to cellular stress and degradation by the ubiquitin–proteasome system (UPS) in response to the activation of unfolded protein response (UPR) (Illing et al., 2002; Saliba et al., 2002; Mendes et al., 2005). Importantly, autophagy is activated by ER-stress and contributes to the clearance of cellular debris in neurodegenerative disorders (Mendes and Cheetham, 2008; Mohlin et al., 2014). More importantly, autophagy has been shown to be an important housekeeping in the retina and specific deletion of the essential autophagy gene Atg5 in rods induced retinal degeneration (Mitter et al., 2012; Chen et al., 2013; Rodriguez-Muela et al., 2013). The recent data implies autophagy as a complementary pathway for degradation of misfolded proteins in photoreceptor cells (Kunchithapautham and Rohrer, 2007). Moreover, current findings demonstrated an increase of autophagy flux in the inner segments of RHOP23H/+ rod photoreceptor cells, at onset and progression of retinal disease (Qiu et al., 2019), suggesting that it may be involved in the clearance of RHOP23H protein in early stage of the disease or participating in the rod cell death at later stage. Interesting, valproic acid-mediated induction of autophagy in a RHOP23H/+ Xenopus laevis model demonstrated a protective effect of autophagy against retinal degeneration (Wen et al., 2019). On the other hand, increase of autophagy at PN14, corresponding to post symptom-onset stage, in which photoreceptors' death is already evident, worsen retinal degeneration in the RHOP23H/+ mice (Qiu et al., 2019), whereas inhibition of autophagy ameliorates retinal degeneration. These findings support the dual function of autophagy, suggesting a transition from a pro-survival to pro-apoptosis function in response to irreversible RHOP23H accumulation and cellular stress. However, the protective role of the autophagy pathway in the photoreceptor cells is still largely unknown. In our previous research, we demonstrated that miR-211/Ezrin axis controls lysosomal biogenesis and function at the beginning of light–dark transitions in the retinal pigment epithelium (RPE)/PR cells and plays an important role in the activation of cell clearance. In line with these findings, pharmacological inhibition of Ezrin activity induced a daily pulse of autophagy in both RPE/PR cells rescuing imbalance of autophagy and retinal degeneration in miR-211−/− mice (Naso et al., 2020). Importantly, this pharmacological therapy was safe and well-tolerated. Thus, the aim of this work was to assess the induction of selective autophagy via Ezrin inhibition as pharmacological therapy to rescue PRs' cell death from RHOP23H accumulation and cellular stress at the onset of molecular and clinical symptoms of retinal degeneration; thus, in turn, blocking the transition of cell fate from survival to death. Our results show that a daily pulse of selective autophagy pathway in the RHOP23H/+ mice plays a protective role in promoting cell survival by reducing RHOP23H accumulation and ER-stress, opening a new frontier for potential clinical application of autophagy activators as therapeutic compounds useful to counteract the onset and progression of retinal degeneration.
The P23H mouse line (RHOP23H/+) employed in this study was generated as previously described (Sakami et al., 2011). All studies on animals were conducted in strict accordance with the institutional guidelines for animal research and approved by the Italian Ministry of Health; Department of Public Health, Animal Health, Nutrition and Food Safety in accordance with the law on animal experimentation (article 31; D.L. 26/2014; protocol number: 0016304-21/07/2020-DGSAF-MDS-P). RHOP23H/+ mice were maintained on the C57Bl/6J background. In all experiments, we used as controls aged-matched littermates of RHOP23H/+ mice.
Drug treatments were performed by once-daily intraperitoneal injection of NSC668394, at a dose of 0.226 mg/kg, as previously described (Celik et al., 2016).
Scotopic and photopic electrophysiological recordings were performed as described (Barbato et al., 2017). A National Instruments amplifier with a xenon Ganzfeld stimulator (CSO, Costruzione Strumenti Oftalmici, Florence, Italy) was used to record mice. Briefly, mice were dark-adapted for at least 3 h. Animals were anesthetized and positioned in a stereotaxic apparatus under dim red light. Their pupils were dilated with a drop of 0.5% tropicamide (Visufarma, Rome, Italy), and body temperature was maintained at 37.5°C. The electrophysiological signals were recorded through gold-plate electrodes inserted under the lower eyelids in contact with the cornea. The electrodes in each eye were referred to a needle electrode inserted subcutaneously at the level of the corresponding frontal region. The different electrodes were connected to a two-channel amplifier. For ERG analysis in dark-adapted conditions (scotopic), eyes were stimulated with light flashes. Five different light intensity stimuli were used ranging from 1 × 10−4 to 20 cd·s/m2. Amplitudes of a- and b-waves were plotted as a function of increasing light intensity. After the completion of responses obtained in scotopic conditions, the recording session continued with the purpose of dissecting the cone pathway through the photopic ERG. Photopic cone responses were isolated in light conditions with a constant background illumination of 50 cd·s/m2, with 10 flashes, and a light intensity of 20 cd·s/m2.
HeLa cells were obtained from American Type Culture Collection (ATCC) and were cultured in Dulbecco's Modified Eagle Medium (DMEM) (Gibco) supplemented with 10% (v/v) FBS and 5% penicillin-streptomycin. Cell lines were maintained at 37°C, 5% CO2 in a humidified incubator according to the guidelines provided by the vendors. To analyze rhodopsin degradation, cells were plated for 24 h and then treated with 200-nM Bafilomycin A1 (Baf) (Sigma–Aldrich, B1793) for 3 h, 100-μg/ml cycloheximide (CHX) (Sigma–Aldrich, C4859), 100-mM Bortezomib (BTZ) (Sigma–Aldrich, 5043140001) and 10 μM of NSC668394 or DMSO as previously reported (Bulut et al., 2012).
Cells were transfected at 70–80% confluence with 2.5 μg of pCS2-RHO P23H for 48 h. For plasmid transfection Lipofectamine 2000 (Invitrogen, 12566014) was used, following the manufacturer's protocol.
Mouse eyes were enucleated and the RPE was separated from the retina. Cells were collected after transfections or treatments to extract total protein. Both mice and cell samples were lysed by using RIPA buffer (150-mM sodium chloride, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 50-mM Tris, pH 8.0) with inhibitors cocktail (Thermo Fischer Scientific, 78420). The concentration of total protein was determined by Bradford analysis and quantified by using a NanoDrop ND-8000 spectrophotometer (NanoDrop Technologies). Proteins were fractionated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and transferred to PVDF membranes (EMD Millipore, IPVH00010), then blocked in Tween 0.1%-Tris-buffered saline containing 5% non-fat milk or 5% bovine serum albumin (Sigma–Aldrich, 9048-46-8) for 1 h at room temperature and subsequently incubated overnight at 4°C with primary antibodies. For the Western blot analysis, the following antibodies were used: Mouse anti-Lamp1 (1:500, Sigma–Aldrich, L1418), mouse anti-Ezrin (1:1000, Novex, 357300), rabbit anti-phospho-Ezrin (Thr567) (1:700, Sigma-Aldrich, PA5-37763), rabbit anti-SQSTM1/p62 (1:1000, Sigma–Aldrich, P0067), rabbit anti-LC3 (1:1,000, Novus LC3B/MAP1LC3B), mouse anti-GAPDH (1:1000, Santa Cruz, SC-32233), rabbit anti-Perk (1:1,000, Cell Signaling, 3192), rabbit anti-BiP (1:1,000, Cell Signaling, 3177), rabbit anti-Xbp1s (1:1,000, Cell Signaling, 12782), mouse anti-Chop (1:1,000, Cell Signaling, 2895), mouse anti-RHO (1:500, Santa Cruz, sc-57432). After washing with 1% TBS, the membranes were incubated for 1 h at room temperature with the following secondary antibodies: Goat anti-rabbit IgG antibody, HPR conjugate and goat anti-mouse IgG antibody, HPR conjugate (1:10,000 EMD Millipore, 12–348; 12–349). Western blot detection was done with a GE detector (GE Healthcare Life Sciences) and quantified using ImageJ software.
Mouse eyes were fixed overnight in 4% paraformaldehyde in PBS at 4°C and then cryopreserved by treatment, first with 15% and then with 30% sucrose in phosphate-buffered saline and embedded in OCT. Twenty-micrometer cryosections were collected on slides (Superfrost Plus; Fisher Scientific, Pittsburgh, PA). Cells were fixed with 4% formaldehyde (Sigma–Aldrich) for 10 min at room temperature followed by washing with 1% PBS. After the fixation, the cells were permeated with blocking buffer (0.5% BSA, 0.005% saponin, 0.02% NaN3) for 1 h at room temperature. The following primary antibodies were used: rabbit anti-LAMP1 (1:400, abcam, ab24170), mouse anti-RHO (1:100, abcam, ab5417), rabbit anti-c-Arrestin (1:500, EMD Millipore, AB15282), rabbit anti-GFAP (1:400, Dako GA52461-2), rabbit anti-Iba-1 (1:1,000, Wako 019-19741). All incubations were performed overnight at 4°C. After washing with 1% PBS, slides were incubated with the following secondary antibodies: Alexa 594 goat anti-rabbit/mouse/rat (1:1,000, Invitrogen A-11037 rabbit, A-11032 mouse) or Alexa 488 goat anti-rabbit/mouse/rat (1:1,000, Invitrogen A-11008 rabbit, A-11001 mouse) and DAPI (1:500, Vector Laboratories H-1200) for 1 h at room temperature; then, the slides were washed with 1% PBS and mounted with PBS/glycerol and imaged with a Zeiss LSM700 microscope. All samples were imaged, and images were identically processed.
The number of apoptotic cells was analyzed by TdT-mediated dUTP nick end labeling (TUNEL), using the in situ Cell Death Detection Kit, Fluorescein (Roche 11684795910) following the manufacturer's directions. Twelve-micrometer cryosections were collected on slides and subjected to TUNEL assay. To consider the presence of unspecific results, some retina sections were incubated with the reaction mix without TUNEL reaction enzyme. Sections were observed with a Leica DM-5500 microscope and then confocal images were acquired using the LSM700 Zeiss Confocal Microscopy system. The number of TUNEL-positive cells was evaluated in the dorsal and ventral part of the retina by manual counts with a Leica DM-6000 microscope, with the objective Leica ∞/0.17/D, HCX PL FLUOTAR, 40X/0.75 that has an area of 0.31 mm2.
Measurements of ONL thickness and OS length were performed in the superior and inferior retina, located equidistant from the optic nerve head (ONH). Retina sections were analyzed by using a Leica DM-6000 microscope, with the objective Leica ∞/0.17/D, HCX PL FLUOTAR, 40X/0.75 that has an area of 0.31 mm2.
The number of normal cones/area was manually estimated in the superior and inferior retina, located equidistant from the ONH by using a Leica DM-6000 microscope, with the objective Leica ∞/0.17/D, HCX PL FLUOTAR, 40X/0.75 that has an area of 0.31 mm2. We matched the number of cone segments and dendrites clearly demarcated by the cone arrestin immunological staining.
Fluorescent images of dorsal and ventral region of mice retina were captured at 20X magnification using LSM700 Zeiss Confocal Microscopy system, converted to gray-scale and normalized to background staining, using ImageJ. Quantification of GFAP+ reactivity was measured as mean values to define fluorescence signal intensity (IntDen/Area) and as the area occupied by fluorescent-labeling in each region of interest.
To quantify the numbers of microglial cells, the number of Iba-1-positive cells was evaluated in the dorsal and ventral part of the retina by manual counts in each considered retinal layer: GCL, IPL, OPL, and ONL. These counts were pooled to obtain a mean number of microglial cells per layer, per retinal region and per animal group.
Hela cells were fixed with a mixture of 4% paraformaldehyde (PFA) and 0.05% glutaraldehyde (GA) for 10 min at room temperature, then washed with 4% PFA once to remove the residual GA and fixed again with 4% PFA for 30 min at room temperature. Next, the cells were incubated with a blocking/permeabilizing mixture (0.5% BSA, 0.1% saponin, 50-mM NH4Cl) for 30 min and subsequently with the primary monoclonal antibody anti-RHO, diluted 1:100 in blocking/permeabilizing solution. The following day, the cells were washed and incubated with the secondary antibody, the anti-mouse Fab fragment coupled to 1.4-nm gold particles (diluted 1:50 in blocking/permeabilizing solution) for 2 h at room temperature. The cells were then post-fixed as described in Polishchuk and Polishchuk (2019). After dehydration, the specimens were embedded in epoxy resin and polymerized at 60°C for 72 h. Thin 60-nm sections were cut on a Leica EM UC7 microtome. The EM images were acquired from thin sections using a FEI Tecnai-12 electron microscope equipped with a VELETTA CCD digital camera (FEI, Eindhoven, the Netherlands).
Total RNA was extracted from murine eyes using the miRNeasy Kit (QIAGEN 217004) according to the manufacturer's instructions. Ribonucleic acid was quantified using a NanoDrop ND-8000 spectrophotometer (NanoDrop Technologies). The cDNAs were generated using the QuantiTect Reverse Transcription Kit (Qiagen) for the qRT-PCR analysis. The qRT-PCR reactions were performed with nested primers (Table 1) and carried out with the Roche Light Cycler 480 system. The qRT-PCR reaction was performed using cDNA (200–500 ng), 10 μl of the SYBR Green Master Mix (ROCHE), and 400-nM primers, in a total volume of 20 μl. The PCR conditions for all the genes were as follows: Preheating, 95°C for 60 s; cycling, 45 cycles of 95°C for 10 s, 60°C for 10 s, and 72°C for 15 s. Quantified results were expressed in terms of cycle threshold (Ct). The Ct values were averaged for each triplicate. The HPRT gene was used as the endogenous control for the experiments. Differences between the mean Ct values of the tested genes and those of the reference gene were calculated as DCtgene = Ctgene – Ctreference. Relative expression was analyzed as 2-DCt. Relative fold changes in expression levels were determined as 2-DDCt.

Table 1. Primers used in qRT-PCR.
All data are expressed as mean ± standard error unless indicated otherwise. The number of samples in each experiment is provided in appropriate methods description or figure legends. The statistical analysis was carried out at least from three independent experiments for each genotype and experimental conditions. In all experiments, the significance of differences between groups was evaluated and made using t-test. The p-values of significance are indicated where appropriate in the figure legends.
To assess whether pharmacological induction of autophagy exerts a protective effect when delivered in a fully differentiated retina before the molecular and clinical signs of PRs degeneration, we daily administered the Ezrin's inhibitor “NSC668394” in RHOP23H/+ mice from PN6 to PN60 using intraperitoneal injection of NSC668394, at a dose of 0.226 mg/kg as previously described (Celik et al., 2016). The starting point of intervention better represents the pre-symptomatic stage of the disease in human patients. Following the daily injection from PN6 onwards, we recorded an improved ERG responses at PN30 and PN60 in NSC668394-treated eyes vs. controls (Figure 1). Importantly, we recorded an amelioration in both scotopic a-wave and b-wave amplitudes at PN30 (Figures 1A,C), which persisted until PN60 (Figures 1D,F). Moreover, photopic ERG records, in the presence of background light, appeared higher amplitudes in NSC668394-treated animals compared to control (Figures 1C,F). Specifically, the a-wave amplitude of the RHOP23H/+ treated mice showed a trend of increase both at PN30 and PN60, albeit statistical significance was only observed at PN60 (Figures 1A,D). Notably, a significant increase in the b-wave amplitude was observed at both PN30 and PN60 (Figures 1B,E). Thus, we next sought to determine whether the pharmacological NSC668394 treatment was indeed related to rescue of morphological and molecular detrimental cascade underlying PRs' ER-stress and cell death. Importantly, daily injections of NSC668394 to RHOP23H/+ mice over two consecutive months (beginning at PN6) statistically ameliorated both outer nuclear layer (ONL) thickness and density in both dorsal and ventral portions of retina of the injected eyes, as detected by immunofluorescence assays (Figures 2A–H). First, we performed a morphometric analysis in PN18 retinas of NSC668394- and DMSO-treated RHOP23H/+ mice when the first evidence of alteration of the retinal structure appears (Supplementary Figures S1, S2). Importantly, NSC668394-treated RHOP23H/+ mice showed a relevant increase of the thickness of the ONL compared to DMSO-treated mice both in the dorsal and ventral retinal areas (Supplementary Figures S1A,G). Consistent with this finding, treatment of RHOP23H/+ mice with NSC668394 resulted in a marked preservation of thickness of rod outer segments (OS), detected by immunostaining with anti-Rhodopsin antibody (Supplementary Figures S1A,C,H). Moreover, we measured a complete rescue of cone morphology in NSC668394-treated RHOP23H/+ that were like those recorded in DMSO-wild-type (WT) mice, as demonstrated by quantifying cones with cone segments (CS) and dendrites clearly labeled by immunostaining with anti-c-Arrestin antibody (Supplementary Figures S1D–F,I). Most importantly, we obtained similar beneficial effects also from NSC668394-treated RHOP23H/+ animals injected at PN6 and analyzed at PN60 (Figure 2), when retinal degeneration is advanced. Treatment of RHOP23H/+ mice with NSC668394 led to an increased ONL thickness and density in both dorsal and ventral retina, as measured by DAPI staining and compared with DMSO-treated control mice (Figures 2A,H). Consistent with photoreceptor function assessments, significant recovery was also detected on the thickness of rod OS (Figures 2A–C,I), cones' morphology in NSC668394-treated RHOP23H/+ that were like those recorded in DMSO-WT mice (Figures 2D,F,J). Besides, we sought to determine whether the pharmacological NSC668394 administration had an impact on PRs viability. Notably, treatment with NSC668394 resulted in a significant decreased number of TUNEL-positive photoreceptor cells compared to DMSO-treated mice, supporting a reduction in PRs' cell death (Figure 3, Supplementary Figure S2). This outcome was significant in Ezrin inhibited- RHOP23H/+ mice at PN18 (Supplementary Figure S2), but it became even more consistent in treated retina at PN60 (Figures 3A,D). Next, we investigated possible inflammatory effects of Ezrin inhibition in the RHOP23H/+ retinae. For this purpose, we performed immunofluorescence staining for glial fibrillary acidic protein (GFAP) and ionized calcium-binding adapter molecule 1 (Iba-1) of RHOP23H/+ mice at PN60 (Figures 3E,M). Firstly, to measure activation of retinal gliosis, we quantified the fluorescent integrated density (IntDen) and the area of GFAP immunoreactivity in dorsal and ventral region of the retina. In RHOP23H/+ mice GFAP immunoreactivity was significantly higher than in the age-matched WT control and it extended to the ONL, indicating Müller cells activation and retinal gliosis. Remarkably, NSC668394 treatment rescued gliosis as demonstrated by reduction of area of the GFAP immunoreactivity and IntDen (Figures 3E,I). In support of beneficial effect of the NSC668394 administration in RHOP23H/+ mice, we also observed a reduction of the number of microglia cells in the RHOP23H/+ retinal cryosections. We quantified the number of microglia cells by Iba-1 immunostaining, hallmark of activated microglial cells and retinal inflammatory response. Under DMSO treatment, RHOP23H/+ mice showed over twice as many Iba-1-positive cells compared to WT retinal cryosections, confirming that the RHOP23H accumulation in rods induced cell death and secondarily strong activation of retinal microglia, which was localized in the ONL, inner nuclear layer (INL) and ganglion cell layer (GCL). Consistent with protective effects, NSC668394 decreased Iba1-positive cell number in NSC668394-RHOP23H/+ treated animals compared to DMSO controls (Figures 3J,M). This result supports that the protective effect of NSC668394 in RHOP23H/+ mice may be mediated by the clearance of unfolded RHOP23H aggregates via targeting Ezrin activity and induction of autophagy pathways.
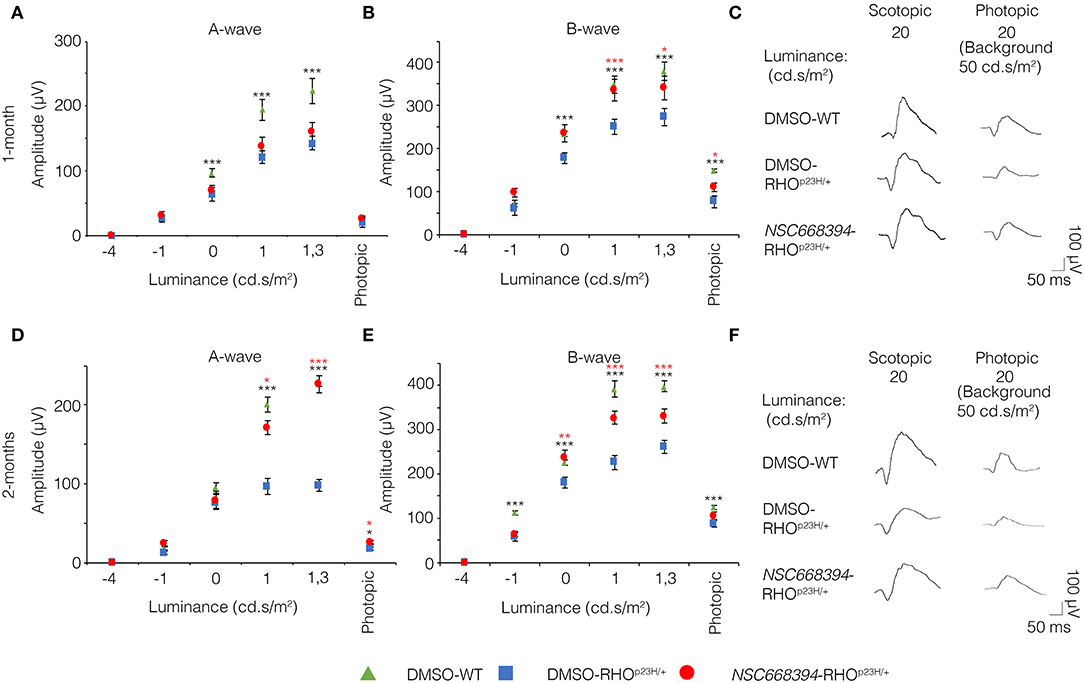
Figure 1. NSC668394 RHOP23H/+ mice treatment results in progressive functional rescue of photoreceptors deficiency. (A,B) Representative ERG (a- and b-wave), plotted as a function of stimulus intensity, from 1-month old DMSO-treated WT (green triangle), DMSO-treated RHOP23H/+ (blue square), and NSC668394-treated RHOP23H/+ (red circle) mice. Error bars represent SEM. ***p ≤ 0.005, *p ≤ 0.05 t-test (DMSO-RHOP23H/+ vs. DMSO-WT; NSC668394-RHOP23H/+ vs. DMSO-RHOP23H/+). (C) Representative ERG (scotopic and photopic responses) traces of DMSO-treated WT, DMSO-treated RHOP23H/+, and NSC668394-treated RHOP23H/+ mice at 1-month of age. Scotopic indicates ashes of 20.0 cd·s/m2 light intensity; photopic indicates ashes of 20.0 cd·s/m2 light intensity on a constant background illumination of 50 cd·s/m2. (D,E) Representative ERG (a- and b-wave), plotted as a function of stimulus intensity, from 2 months-old DMSO-treated WT (green triangle), DMSO-treated RHOP23H/+ (blue square) and NSC668394-treated RHOP23H/+ (red circle) mice. Error bars represent SEM. ***p ≤ 0.005, **p ≤ 0.01, *p ≤ 0.05 t-test (DMSO-RHOP23H/+ vs. DMSO-WT; NSC668394-RHOP23H/+ vs. DMSO-RHOP23H/+). (F) Representative ERG (scotopic and photopic responses) traces of DMSO-treated WT, DMSO-treated RHOP23H/+ and NSC668394-treated RHOP23H/+ mice at 2 months of age. Scotopic indicates ashes of 20.0 cd·s/m2 light intensity; photopic indicates ashes of 20.0 cd·s/m2 light intensity on a constant background illumination of 50 cd·s/m2.
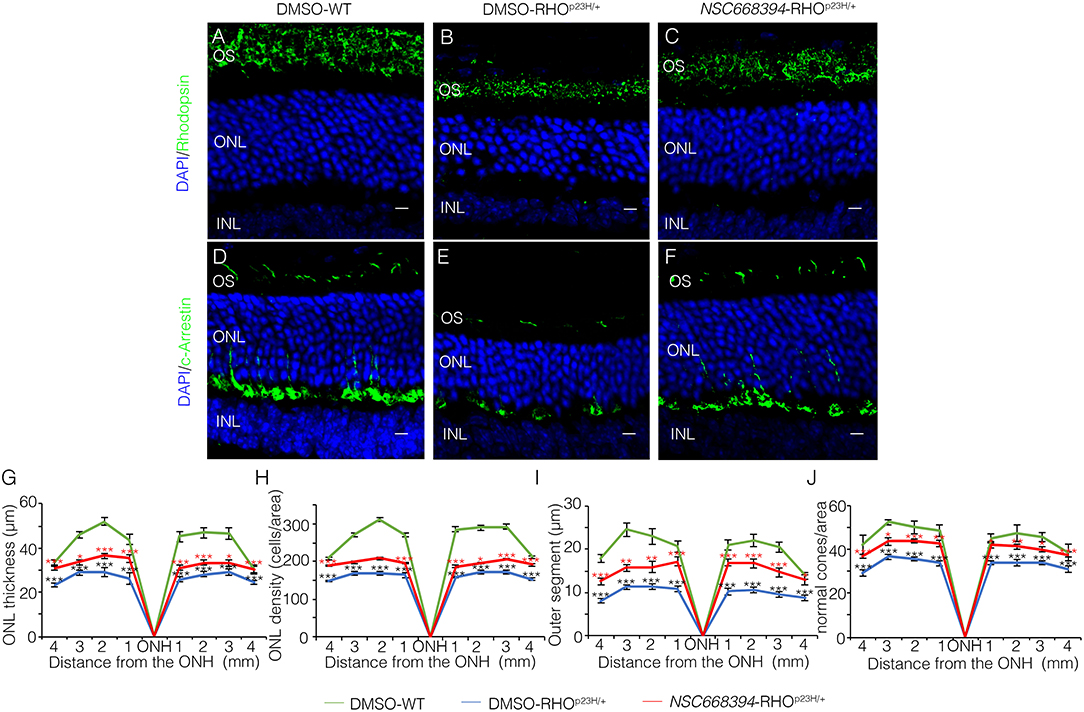
Figure 2. Two-months-old NSC668394-treated RHOP23H/+ mice show retinal morphology recovery. Representative images of retina cryosection immunostained with anti-Rhodopsin (A–C) and anti-c-Arrestin (D,F) antibodies from DMSO-treated WT (A,D), DMSO-treated RHOP23H/+ (B,E) and NSC668394-treated RHOP23H/+ (C,F) mice at PN60. Nuclei are counterstained with DAPI (blue). At least n = 5 mice per group. Scale bar 10 μm. OS, outer segment; ONL, outer nuclear layer; INL, inner nuclear layer. (G) Graphs show the measure of ONL thickness from the retina of DMSO-treated WT (green line), DMSO-treated RHOP23H/+ (blue line), and NSC668394-treated RHOP23H/+ (red line) mice at 2 months of age. (H) Graphs show measure of ONL density (cells/area) from the retina of DMSO-treated WT (green line), DMSO-treated RHOP23H/+ (blue line), and NSC668394-treated RHOP23H/+ (red line) mice at 2 months of age. (I) Graphs show the reduction of OS length from 2 months of age onward from each analyzed retina region of DMSO-treated WT (green line), DMSO-treated RHOP23H/+ (blue line) and NSC668394-treated RHOP23H/+ (red line) mice. (J) Graphs show normal cone morphology (cones/area) from the retina of DMSO-treated WT (green line), DMSO-treated RHOP23H/+ (blue line), and NSC668394-treated RHOP23H/+ (red line) mice at 2 months of age. Error bars represent SEM. ***p ≤ 0.005, **p ≤ 0.01, *p ≤ 0.05 t-test. (DMSO-RHOP23H/+ vs. DMSO-WT; NSC668394-RHOP23H/+ vs. DMSO-RHOP23H/+).
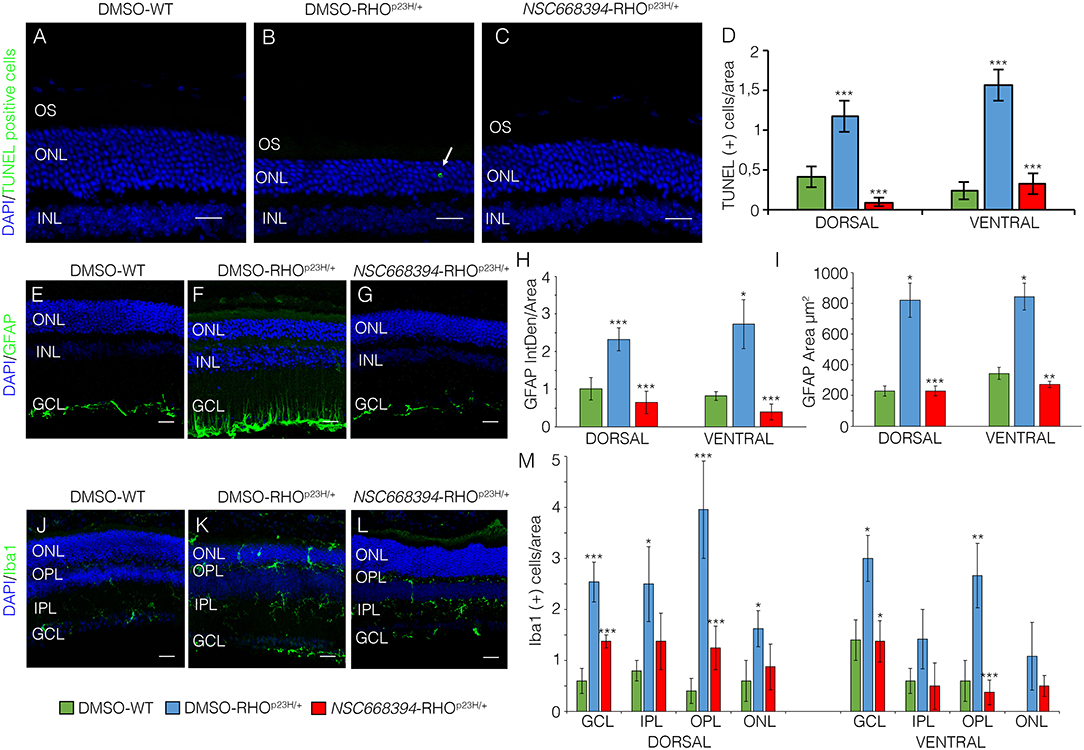
Figure 3. The NSC668394-treated RHOP23H/+ mice display a reduction in retinal cell death, microglia and inflammation. Representative images of DMSO-treated WT (A), DMSO-treated RHOP23H/+ (B) and NSC668394-treated RHOP23H/+ (C) retinas of 2 months of age stained with TUNEL-fluorescein. Positive cells are indicated with white arrows. Nuclei are counterstained with DAPI (blue). At least n = 4 mice per group. Scale bar 20 μm. (D) Graph shows the number of TUNEL positive cells from the dorsal and ventral retina of DMSO-treated WT, DMSO-treated RHOP23H/+ and NSC668394-treated RHOP23H/+ mice at 2 months of age. Error bars represent SEM. Representative images of DMSO-treated WT (E), DMSO-treated RHOP23H/+ (F) and NSC668394-treated RHOP23H/+ (G) retinas of 2 months of age stained with GFAP. Nuclei are counterstained with DAPI (blue). At least n = 3 mice per group. Scale bar 20 μm. (H) Bar graph is the quantitative analysis of optical density for GFAP fluorescence from the dorsal and ventral retina of DMSO-treated WT, DMSO-treated RHOP23H/+ and NSC668394-treated RHOP23H/+ mice at 2 months of age. Error bars represent SEM. (I) Bar graph shows quantification of GFAP-immunoreactive area in the dorsal and ventral retina of DMSO-treated WT, DMSO-treated RHOP23H/+, and NSC668394-treated RHOP23H/+ mice at 2 months of age. Error bars represent SEM. Representative images of DMSO-treated WT (J), DMSO-treated RHOP23H/+ (K), and NSC668394-treated RHOP23H/+ (L) retinas of 2 months of age stained with Iba-1. Nuclei are counterstained with DAPI (blue). At least n = 3 mice per group. Scale bar 20 μm. (M) Bar graph represents the number of Iba-1 positive (Iba-1+) cells in the dorsal and ventral retinal layers of DMSO-treated WT, DMSO-treated RHOP23H/+ and NSC668394-treated RHOP23H/+ mice at 2 months of age. Error bars represent SEM. OS, outer segment; ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer. ***p ≤ 0.005, **p ≤ 0.01, *p ≤ 0.05 t-test. (DMSO-RHOP23H/+ vs. DMSO-WT; NSC668394-RHOP23H/+ vs. DMSO-RHOP23H/+).
The recent discoveries have proven that lysosomes may reduce misfolded protein accumulation via activation of autophagy pathway (Mendes and Cheetham, 2008; Sakami et al., 2011). We sought to assess whether the protective effect of NSC668394 is mediated by activation of lysosomal clearance of RHOP23H, we analyzed NSC668394 administration on Hela cells overexpressing RHOP23H (HeLaRHO−P23H). Interestingly, culturing HeLaRHO−P23H cells in the presence of NSC668394 led to a significant decrease of RHOP23H in both insoluble and soluble fraction, as demonstrated by Western blot analyses, suggesting that there was a major change in rhodopsin degradation (Figure 4A). To further support that NSC668394 treatment induced lysosomal clearance of RHOP23H, we assessed lysosomal cargo degradation on Ezrin-inhibited Hela RHOP23H cells followed by a treatment with Bafilomycin A1 (Baf), a known inhibitor of vacuolar-type H(+)-ATPase that promotes alkalinization of the lysosomal lumen and inhibition of lysosomal cargo degradation, leading to an accumulation of undigested material within the lumen of already fused autolysosomes, and prevents new autophagosomes and lysosomes fusion (Yoshimori et al., 1991). As expected, treatment with Baf on HelaRHO−P23H cells showed a slight increase of RHOP23H accumulation in the lumen of autolysosomes (LAMP1-positive organelles) compared with DMSO-treated cells (Figures 4B–C”). Treatment with NSC668394 on HelaRHO−P23H cells increased the number of autolysosomes structures and showed a moderate increase of RHOP23H accumulation in the lumen of autolysosomes (LAMP1-positive organelles) compared with DMSO-treated cells (Figures 4B–B”,D–D”). Notably, administration of Baf after 3 h of NSC668394 treatment (Baf/NSC668394-treated HelaRHO−P23H) induced a high increase of autolysosomes structures containing RHOP23H in the lumen, in accordance with a block of cargo degradation in already increased number of autolysosomes (Figures 4E–E”). These data were also confirmed in the 661W photoreceptor cell line (Supplementary Figure S3) (Wheway et al., 2019), supporting the notion that NSC668394 treatment increases RHOP23H degradation via autophagy pathway. Furthermore, to better investigate on rhodopsin sub-cellular localization, we performed an IEM analysis (Figure 5) and carried out morphometric issues. Hela RHOP23H cells showed an increase of autolysosome-like structures, compared to normal levels, presumably due to the presence of the misfolded protein. Interestingly, the number and the volume of the autolysosome-like structures were even more increased in Baf/NSC668394-treated HelaRHO−P23H cells (Figures 5A–D), confirming the treatment effectiveness. Most importantly, we found a significant increase in the number of rhodopsin-positive autolysosome-like structures in Baf/NSC668394-treated HelaRHO−P23H cells (Figures 5A–E). Subsequently, we tested whether the reduction of RHOP23H protein was also due to a block of protein synthesis or increase of proteasome activity. Remarkably, administration of NSC668394 on HelaRHO−P23H in the presence of cycloheximide (CHX) treatment, a well-known inhibitor of protein synthesis, further showed a faster degradation of RHOP23H protein in NSC668394-treated cells compared with DMSO-treated control cells (Supplementary Figure S4A). Taken together, these data indicate that reduction of RHOP23H protein in NSC668394-treated HelaRHO−P23H is not due to an altered protein synthesis. To avoid the possibility that NSC668394 might induce proteasome activity, we provide insights into how NSC668394 regulates RHOP23H clearance. To this end, NSC668394-treated HeLaRHO−P23H cells were cultured in presence of the Bortezomib (BTZ), a proteasome inhibitor. Notably, following NSC668394 treatment, a significant decrease of RHOP23H was also detected upon BTZ conditions, as demonstrated by Western blot analysis (Supplementary Figure S4B). On the contrary, the levels of RHOP23H protein were markedly increased in DMSO-treated HeLaRHO−P23H cells upon BTZ conditions (Supplementary Figure S4B). Taken together, these data indicate that pharmacological NSC668394 treatment specifically results in activation of lysosomal RHOP23H degradation.
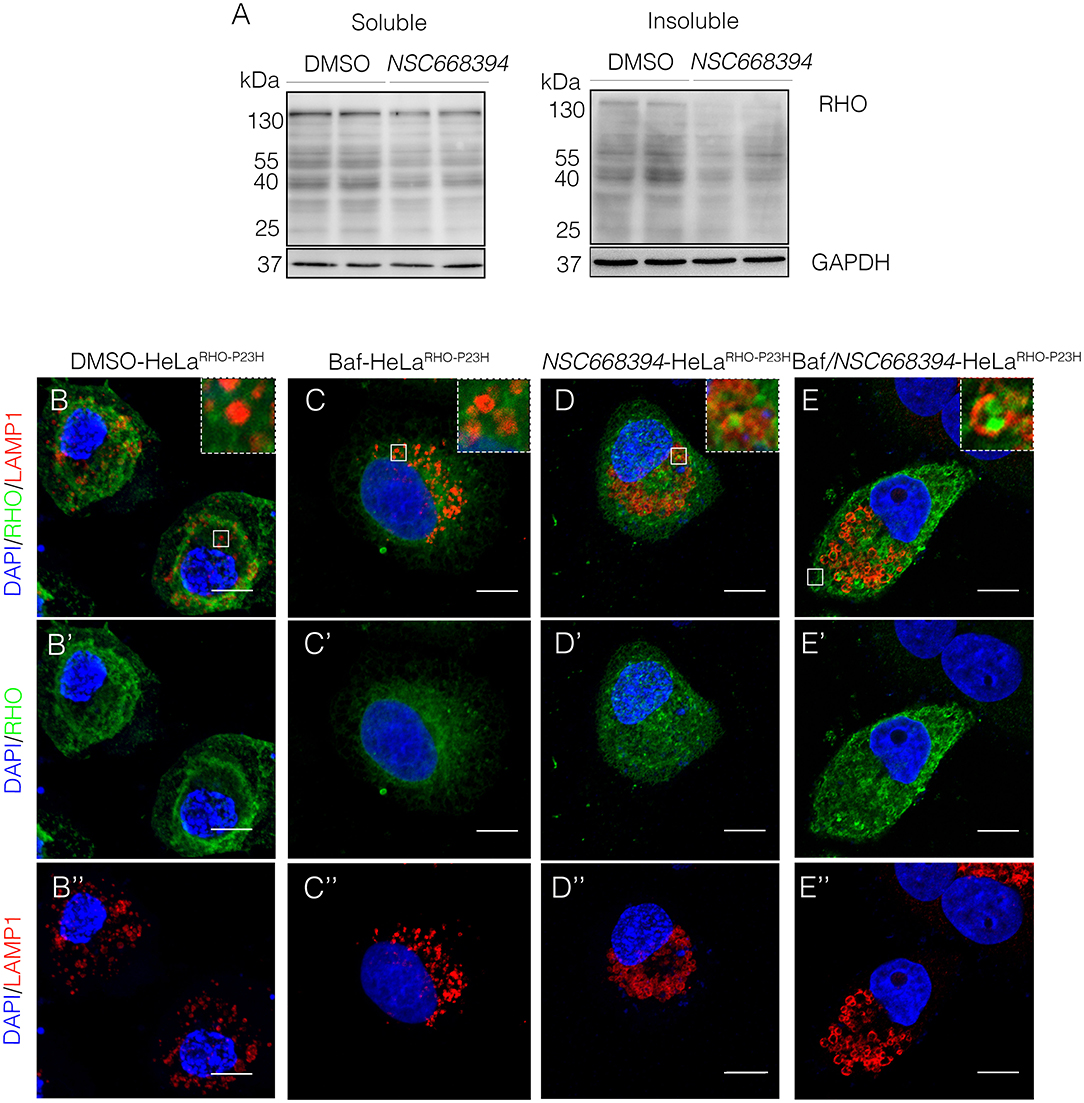
Figure 4. The NSC668394-treated HeLaRHO−P23H cells show an autophagy-related reduction of protein accumulation. (A) Representative Western blot analysis of RHO protein in the soluble and insoluble fraction of HeLaRHO−P23H cells treated with DMSO and NSC668394. Representative images of HeLaRHO−P23H cells immunostained with anti-RHO and anti-LAMP1 from DMSO-treated (B−B”), Baf-treated (C−C”), NSC668394-treated (D−D”), and Baf/NSC668394-treated (E−E”) HeLaRHO−P23H cells. Nuclei are counterstained with DAPI (blue). Scale bar 10 μm.
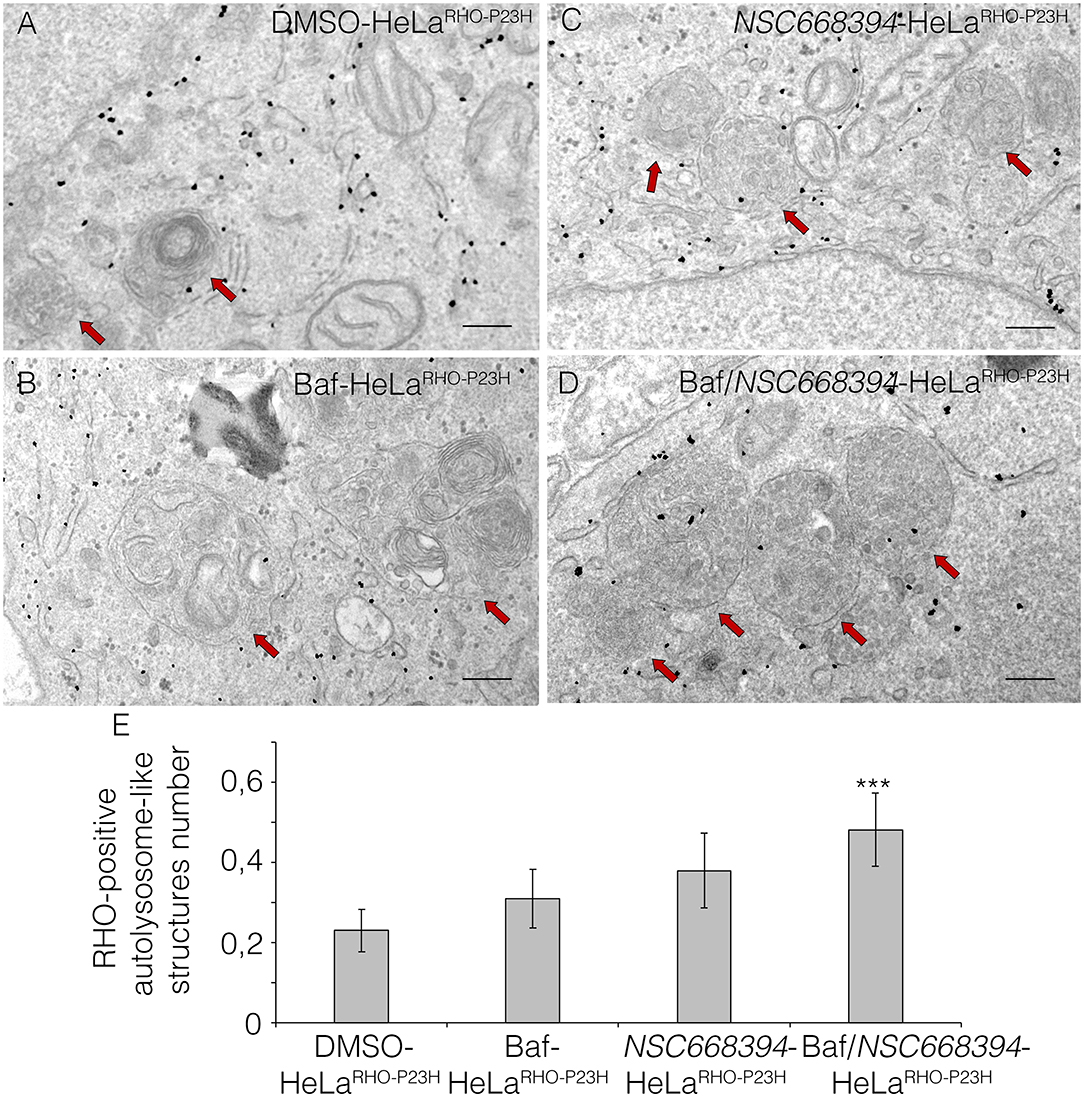
Figure 5. Ezrin inhibition induces RHO disposal via lysosomal activity. Immuno-electron microscopy (IEM) analysis of HeLaRHO−P23H cells treated with DMSO (A), Baf (B), NSC668394 (C), and Baf/NSC668394 (D). Scale bar 250 nm. The RHO-positive autolysosome-like structures are indicated with red arrows. (E) The blot shows the quantification of the number of RHO-positive autolysosome-like structures. Bar graphs represent mean values ± SEM of independent experiments. ***p ≤ 0.005 t-test (Baf/NSC66834-HeLaRHO−P23H vs. DMSO-HeLaRHO−P23H).
The mutation P23H rhodopsin was reported to activate ubiquitin/proteasome system (Chapple et al., 2001) and trigger the ER-stress response, which, in turn, lead to cell death (Griciuc et al., 2010). We therefore hypothesized that if NSC668394 directly induces the clearance of RHOP23H aggregates in the photoreceptor cells, the clearance of RHOP23H misfolded protein should reduce ER-stress response in vivo. Notably, NSC668394 treatment reduced the levels of Perk, Xbp1, and Chop proteins, commonly used as UPR markers, compared with vehicle-treated RHOP23H/+ mice at PN18 (Figure 6A). Consistent with this observation, we detected a significant increase in Bip protein level in NSC668394-RHOP23H/+ retina, as analyzed by Western blot analysis (Figure 6A). In addition, we observed reduced ER-stress and UPR markers, as demonstrated by BiP, Chop, and Xbp1 gene expression levels in NSC668394-RHOP23H/+ compared with DMSO-WT and DMSO-RHOP23H/+ mice. Notably, BiP expression level was increased following NSC668394 treatment, suggesting a possible BiP-mediated protective effect. Accordingly, Chop and Xbp1 gene expression levels resulted significatively reduced in NSC668394-RHOP23H/+ mice compared with DMSO-RHOP23H/+ mice at PN18. Consistently, the expression levels of both Atf4 and Atf6 genes in NSC668394-RHOP23H/+ mice resulted similar to DMSO-WT mice, whereas DMSO-RHOP23H/+ mice showed higher Atf4 and Atf6 expression levels compared to DMSO-WT mice (Figure 6C). Taken together, these results strongly support that pharmacological NSC663894 administration reduces both UPR and ER-stress pathways. Interestingly, RHOP23H/+ mice showed impairment of autophagy pathway as a consequence of RHOP23H-mediated ER-stress at the onset and during the progression of retinal degeneration in mice (Sizova et al., 2014; Qiu et al., 2019; Kakavand et al., 2020). We therefore hypothesized that if NSC668394 treatment reduces ER-stress should re-establish and normalize autophagy pathway.
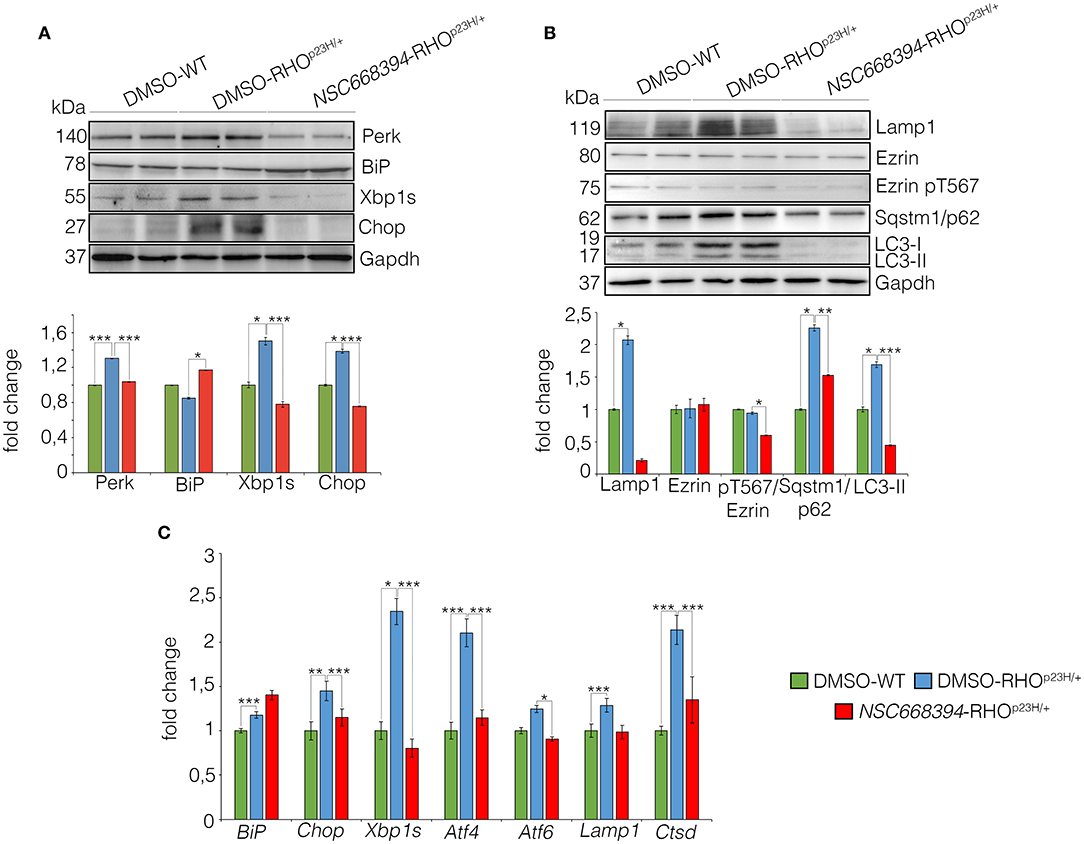
Figure 6. The NSC668394 reduces ER-stress and autophagy in RHOP23H/+ mice. (A) Representative Western blot analysis of Perk, BiP, Xbp1s and Chop proteins from DMSO-treated WT, DMSO-treated and NSC668394-treated RHOP23H/+ mice at PN18. The plot shows the quantification of these proteins normalized to the Gapdh loading control. Bar graphs represent mean values ± SEM of independent experiments (at least n = 3 mice). (B) Representative Western blot analysis of Lamp1, Ezrin, Ezrin pT567, Sqstm1/p62, and LC3 proteins from retina of DMSO treated WT, DMSO-treated RHOP23H/+ and NSC668394-treated RHOP23H/+ mice at PN18. The plot shows the quantification of these proteins normalized to the Gapdh loading control. Bar graphs represent mean values ± SEM of independent experiments (at least n = 3 mice). (C) qRT-PCR assay for RPE of DMSO-treated WT, DMSO-treated RHOP23H/+, and NSC668394-treated RHOP23H/+ mice at PN18. The plot shows the expression level of Bip, Chop, Xbp1s, Atf4, Atf6, Lamp1 and Ctsd normalized to the Hprt control. Bar graphs represent means values ± SEM of independent experiments (n = 3 mice). ***p ≤ 0.005, **p ≤ 0.01, *p ≤ 0.05 t-test (DMSO-RHOP23H/+ vs. DMSO-WT; NSC668394-RHOP23H/+ vs. DMSO-RHOP23H/+).
As expected, daily injection of NSC668394 over 2 consecutive weeks reduced the active pT567-Ezrin protein level, without affecting Ezrin total protein level, and was efficient in rescuing imbalance of autophagy pathway in the retina of RHOP23H/+ mice when compared with vehicle-injected (DMSO) control animals (Figure 6B). Western blot analysis showed a normalization of Lamp1, SQSTM1/p62, and LC3-II protein levels, consistent with a rescue of autophagy pathway (Figure 6B). Interestingly, we also observed a reduction of LC3I. Accordingly, this was due to an increase of autophagic flux, leading to a rapid conversion of LC3-I in LC3-II, which is rapidly degraded.
Accordingly, retinas from NSC668394-RHOP23H/+ treated mice exhibited Lamp1 and Ctsd expression levels similar to those detected in the retinas from DMSO-WT mice, supporting a normalization of autophagy pathway. Opposite results were observed in the retinas from RHOP23H/+ mice treated with DMSO, which exhibited increase of both Lamp1 and Ctsd expression levels compared to DMSO-WT mice (Figure 6C), in line with previous results (Sizova et al., 2014). The ability of NSC668394to regulate differently autophagic flux was consistent with autophagic flux on RHOP23H/+ mice, compared with WT mice, that showed a rapid increase at PN6-PN12, followed by a reduction PN18-PN30 of autophagy, as showed by Western blot analysis (Supplementary Figure S4C).
Altogether, these data show that NSC668394-mediated activation of cell clearance exerts a protective effect in IRD model and may represent an innovative strategy to treat retinal dystrophies due to accumulation of misfolded protein.
Recently, autophagy inducers have gained increasing attention owing to its role in facilitating the degradation of insoluble protein aggregates protecting against their toxicity in neurodegenerative disorders in the central nervous system. However, there is little direct information on the forces driving autophagy pathway in retinal degeneration and how autophagy responds to misfolded protein accumulation in IRDs representing key questions that have been poorly addressed. Notably, translation of biological knowledge into pre-clinical and clinical studies for IRD remains challenging because there is a lack of autophagy inducers compounds specifically targeting autophagy pathway in the retina, which is finely modulated in PRs and RPE by light intensity. Recently, we demonstrated that daily induction of lysosomal biogenesis and function in RPE/PRs is post-transcriptionally regulated by a gene network employing the light-responsive miR-211, and its target gene, namely, Ezrin (Naso et al., 2020). Importantly, we demonstrated that pharmacological activation of lysosomal biogenesis and function, through NSC668394-mediated inhibition of Ezrin, rescued both lipofuscin accumulation and PRs degeneration in miR-211−/− mice, pointing to a new lysosomal-based therapeutic intervention to treat retinal degeneration (Naso et al., 2020). Notably, Ezrin has also been validated as a key element influencing the critical steps of rhodopsin biogenesis, suggesting its role in controlling rhodopsin-laden membrane delivery to the rod outer segments (Deretic et al., 2004), but no role for Ezrin in clearance of rhodopsin has been reported thus far. The P23H mutation in rhodopsin is one of the most common causes of adRP, in which misfolded RHOP23H is retained in the ER (Gorbatyuk et al., 2010). This event generates ER-stress that, in turn, activates autophagy pathway causing a secondary proteasome insufficiency and activation of cell death pathways. Therefore, a precocious remotion of RHOP23H accumulation would alleviate ER-stress and consequently would prevent photoreceptors' cell death. Here, we hypothesized that early pharmacological inhibition of Ezrin would promote a pulsatile increase of autophagy for reducing misfolded RHOP23H accumulation at the onset of its retention in the ER. This pharmacological treatment would prevent ER-stress and photoreceptors' cell death and rescue vision. Therefore, we investigated the systemic administration of NSC668394 compound in RHOP23H/+ mice, in which misfolded rhodopsin protein, harboring the proline23-to-histidine mutation, accumulates in the ER.
Several studies have reported that the level of autophagy proteins resulted to be decreased in RHOP23H/+ rat retina, before the pathological signs of retinal degeneration, suggesting that an initial decline of autophagy pathway may contribute to the onset of the disease. On the other hand, the recent findings demonstrated that prolonged activation of autophagy participates in the deficiency of proteasome activity and activation of apoptosis at the onset of RHOP23H/+ pathological signs (Qiu et al., 2019). Therefore, the regulatory mechanisms controlling autophagy and its crosstalk among survival/cell death pathways need to be further investigated. We demonstrated that daily NSC668394 treatment before the onset of the pathological accumulation of RHOP23H may induce autophagy as a protective mechanism for eliminating and reducing misfolded RHOP23H in the photoreceptor cells. Our data showed that administration of NSC668394 compound rescues retinal phenotype (Figure 2, Supplementary Figure S1) and establishes normal ER-stress (Figure 6) preserving photoreceptors homeostasis and function, ultimately recovering visual function in RHOP23H/+ mice (Figure 1). The increased ERG responses (Figure 1) are mainly due to a clearance of RHOP23H accumulation. This mechanism is corroborated by the reduction of both UPR and ER-stress pathways (Figure 6), accompanied by the decrease of TUNEL staining (Figure 3, Supplementary Figure S2) and a visible increase in ONL thickness in the retinas of NSC668394-treated RHOP23H/+ mice, at the analyzed stages (Figure 2, Supplementary Figure S1). In support of this mechanism, RHOP23H aggregates were degraded within lysosomes upon NSC668394 stimulation, in vitro, as detected by IEM analysis (Figure 5). The recent studies on therapeutic molecules for neurodegeneration has put forward several new candidates, including autophagy inducers in the neuronal cells (Silva et al., 2020). Thus, the observation that an increase in an autophagy-mediated clearance of aggregates in retinal cells could be not surprising, but there is still a lack of evidence for its efficacy. To our knowledge, our work is one of the first demonstration of pharmacological upregulation of autophagy may rescues phenotypes in IRD.
These findings support the notion that precocious intervention on RHOP23H accumulation from PN6 onward, when pathological signs of disease are not evident in RHOP23H/+ retina, is highly effective and capable of halting the progression of retinal degeneration, preserving visual function. Thus, with the implication of the use of autophagy inducers to rescue multiple disease mechanisms common to genetically different pathologies, this study highlights that the time span within which potential treatments may positively modify the onset and progression of a disease need a more accurate attention.
In a preclinical trial study, the pharmacological therapy would ideally be applied in earlier disease stage, when pathological signs of altered molecular networks are not yet evident and detected. This may provide the best protective effect. This is particularly relevant for blindness condition, in which the detrimental molecular cascade leads to irreversible cell death of photoreceptor cells, and no useful vision remains to be rescued. Consistently, AAV-miR-211 delivery, a post-transcriptional repressor of Ezrin, was recently shown to preserve retinal function when injected before the onset of first pathological signs. Therapeutic rescue was associated with a reduction of apoptosis and amelioration of photoreceptor morphology in RHOP347S mouse, suggesting that induction of autophagy can represent a mutation-independent therapeutic strategy to block disease onset and progression (Karali et al., 2020). In support of this notion, enhanced lysosomal activity by NSC668394 also plays crucial role in lipofuscin degradation representing an attractive therapeutic target to treat Age-related macular degeneration and other related diseases (Naso et al., 2020).
Therefore, understanding the time span within which potential treatments may positive modifier and how the therapeutic use of autophagy inducers may be applied in the retina is still fundamental to move the field forward and provide clinical studies for evaluating the therapeutic efficacy of autophagy inducers in treating retinal diseases, in which toxic misfolded proteins accumulate.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
All studies on animals were conducted in strict accordance with the institutional guidelines for animal research and approved by Italian Ministry of Health; Department of Public Health, Animal Health, Nutrition and Food Safety in accordance with the law on animal experimentation (article 31; D.L. 26/2014; protocol number: 0016304-21/07/2020-DGSAF-MDS-P).
DI, GG, FN, SDG, FGS, EN, and EP performed the experiments and analyzed the data. DI and GG contributed to the experimental design, implementation, and interpretation. EN and FGS contributed to the technical part of the work. DI, GG, and IC conceived the experiments, analyzed the data, and wrote the manuscript. All authors read and approved the final manuscript.
Work in the Conte group was supported by grants from the Million Dollar Bike Ride Grant Program MDBR-21-103-CHM, International Retinal Research Foundation, Italian Telethon Foundation TMICCBX16TT and MIUR FISR2020IP_03551, and Sanfilippo Children's Foundations and National MPS Society.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We are grateful to Phoebe Ashley-Norman for critical reading of the manuscript and Eugenio Del Prete for statistical analysis support.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnagi.2022.878958/full#supplementary-material
Supplementary Figure S1| The NSC668394-treated RHOP23H/+ mice at PN18 show rescue of retinal phenotype. Representative images of retina cryosection immunostained with anti-Rhodopsin (A,C) and anti-c-Arrestin (D,F) antibodies from DMSO-treated WT (A,D), DMSO-treated RHOP23H/+ (B,E) and NSC668394-treated RHOP23H/+ (C,F) mice at PN18. Nuclei are counterstained with DAPI (blue). At least n = 3 mice per group. Scale bar 10 μm. OS, outer segment; ONL, outer nuclear layer; INL, inner nuclear layer. (G) Graphs show the measure of ONL thickness from the retina of DMSO-treated WT (green line), DMSO-treated RHOP23H/+ (blue line) and NSC668394-treated RHOP23H/+ (red line) mice at PN18. (H) Graphs show the reduction of OS length from PN18 onward from each analyzed retina region of DMSO-treated WT (green line), DMSO-treated RHOP23H/+ (blue line) and NSC668394-treated RHOP23H/+ (red line) mice. (I) Graphs show normal cone morphology (cones/area) from the retina of DMSO-treated WT (green line), DMSO-treated RHOP23H/+ (blue line) and NSC668394-treated RHOP23H/+ (red line) mice at PN18. Error bars represent SEM. ***p ≤ 0.005, *p ≤ 0.05 t-test (DMSO-RHOP23H/+ vs. DMSO-WT; NSC668394-RHOP23H/+ vs. DMSO-RHOP23H/+).
Supplementary Figure S2| The NSC668394 treatment reduces TUNEL positive cells in PN18 RHOP23H/+ mice retina. Representative images of DMSO-treated WT (A), DMSO-treated RHOP23H/+ (B) and NSC668394-treated RHOP23H/+ (C) retinas of PN18 mice stained with TUNEL-fluorescein. Positive cells are indicated with white arrows. Nuclei are counterstained with DAPI (blue). At least n = 3 mice per group. Scale bar 20 μm. ONL, outer nuclear layer; INL, inner nuclear layer; GCL, ganglion cell layer. (D) Graph shows the number of TUNEL positive cells from the dorsal and ventral retina of DMSO-treated WT, DMSO-treated RHOP23H/+ and NSC668394-treated RHOP23H/+ mice at PN18. Error bars represent SEM. ***p ≤ 0.005, **p ≤ 0.01 t-test (DMSO-RHOP23H/+ vs. DMSO-WT; NSC668394-RHOP23H/+ vs. DMSO-RHOP23H/+).
Supplementary Figure S3| NSC668394-treated 661WRHO−P23H cells show lysosomal clearance of RHOP23H. Representative images of 661WRHO−P23H cells immunostained with anti-RHO and anti-LAMP1 from DMSO-treated (A,A”); Baf-treated (B,B”); NSC668394-treated (C,C”); and Baf/NSC66834-treated (D,D”) HeLaRHO−P23H cells. Nuclei are counterstained with DAPI (blue). Scale bar 10 μm.
Supplementary Figure S4| (A) Representative Western blot analysis of RHO protein from 0, 2, 4, and 6 h of CHX in DMSO and NSC668394 conditions. The graph shows the quantification of RHO protein normalized to the GAPDH loading control. Bar graphs represent mean values ± SEM of independent experiments. (B) Representative Western blot analysis of RHO protein from 0 and 24 h of BTZ in DMSO and NSC668394 conditions. The plot shows the quantification of RHO protein normalized to the GAPDH loading control. Bar graphs represent mean values ± SEM of independent experiments. ***p ≤ 0.005, **p ≤ 0.01, *p ≤ 0.05 t-test. (NSC668394-HeLaRHO−P23H vs. DMSO-HeLaRHO−P23H). (C) Representative Western blot analysis of Lamp1, Sqstm1/p62 and LC3 proteins from retina of WT and RHOP23H/+ mice at PN6, PN12, PN18, and PN30. The plot shows the quantification of these proteins normalized to the Gapdh loading control. Bar Graphs represent means values ± SEM of independent experiments (n = 3 mice). ***p ≤ 0.005, **p ≤ 0.01, *p ≤ 0.05 t-test (RHOP23H/+ vs. WT).
Athanasiou, D., Aguila, M., Bevilacqua, D., Novoselov, S. S., Parfitt, D. A., and Cheetham, M. E. (2013). The cell stress machinery and retinal degeneration. FEBS Lett. 587, 2008–2017. doi: 10.1016/j.febslet.2013.05.020
Barbato, S., Marrocco, E., Intartaglia, D., Pizzo, M., Asteriti, S., Naso, F., et al. (2017). MiR-211 is essential for adult cone photoreceptor maintenance and visual function. Sci. Rep. 7, 17004. doi: 10.1038/s41598-017-17331-z
Berson, E. L.. (1996). Retinitis pigmentosa: unfolding its mystery. Proc Natl Acad Sci U. S. A. 93, 4526–4528. doi: 10.1073/pnas.93.10.4526
Bulut, G., Hong, S. H., Chen, K., Beauchamp, E. M., Rahim, S., Kosturko, G. W., et al. (2012). Small molecule inhibitors of ezrin inhibit the invasive phenotype of osteosarcoma cells. Oncogene 31, 269–281. doi: 10.1038/onc.2011.245
Celik, H., Bulut, G., Han, J., Graham, G. T., Minas, T. Z., Conn, E. J., et al. (2016). Ezrin inhibition up-regulates stress response gene expression. J. Biol. Chem. 291, 13257–13270. doi: 10.1074/jbc.M116.718189
Chapple, J. P., Grayson, C., Hardcastle, A. J., Saliba, R. S., van der Spuy, J., and Cheetham, M. E. (2001). Unfolding retinal dystrophies: a role for molecular chaperones? Trends Mol. Med. 7, 414–421. doi: 10.1016/S1471-4914(01)02103-7
Chen, Y., Sawada, O., Kohno, H., Le, Y. Z., Subauste, C., Maeda, T., et al. (2013). Autophagy protects the retina from light-induced degeneration. The Journal of biological chemistry 288, 7506–7518. doi: 10.1074/jbc.M112.439935
Daiger, S. P., Bowne, S. J., and Sullivan, L. S. (2007). Perspective on genes and mutations causing retinitis pigmentosa. Arch. Ophthalmol. 125, 151–158. doi: 10.1001/archopht.125.2.151
Daiger, S. P., Bowne, S. J., and Sullivan, L. S. (2014). Genes and mutations causing autosomal dominant retinitis pigmentosa. Cold Spring Harb. Perspect. Med. 5, a017129. doi: 10.1101/cshperspect.a017129
Deretic, D., Traverso, V., Parkins, N., Jackson, F., Rodriguez de Turco, E. B., and Ransom, N. (2004). Phosphoinositides, ezrin/moesin, and rac1 regulate fusion of rhodopsin transport carriers in retinal photoreceptors. Mol. Biol. Cell 15, 359–370. doi: 10.1091/mbc.e03-04-0203
Farrar, G. J., Kenna, P. F., and Humphries, P. (2002). On the genetics of retinitis pigmentosa and on mutation-independent approaches to therapeutic intervention. EMBO J. 21, 857–864. doi: 10.1093/emboj/21.5.857
Gorbatyuk, M. S., Knox, T., LaVail, M. M., Gorbatyuk, O. S., Noorwez, S. M., Hauswirth, W. W., et al. (2010). Restoration of visual function in P23H rhodopsin transgenic rats by gene delivery of BiP/Grp78. Proc. Natl. Acad. Sci. U. S. A. 107, 5961–5966. doi: 10.1073/pnas.0911991107
Griciuc, A., Aron, L., Piccoli, G., and Ueffing, M. (2010). Clearance of Rhodopsin(P23H) aggregates requires the ERAD effector VCP. Biochim. Biophys. Acta 1803, 424–434. doi: 10.1016/j.bbamcr.2010.01.008
Gu, S., Kumaramanickavel, G., Srikumari, C. R., Denton, M. J., and Gal, A. (1999). Autosomal recessive retinitis pigmentosa locus RP28 maps between D2S1337 and D2S286 on chromosome 2p11-p15 in an Indian family. J Med Genet. 36, 705–707.
Hartong, D. T., Berson, E. L., and Dryja, T. P. (2006). Retinitis pigmentosa. Lancet 368, 1795–1809. doi: 10.1016/S0140-6736(06)69740-7
Illing, M. E., Rajan, R. S., Bence, N. F., and Kopito, R. R. (2002). A rhodopsin mutant linked to autosomal dominant retinitis pigmentosa is prone to aggregate and interacts with the ubiquitin proteasome system. J. Biol. Chem. 277, 34150–34160. doi: 10.1074/jbc.M204955200
Kakavand, K., Jobling, A. I., Greferath, U., Vessey, K. A., de Iongh, R. U., and Fletcher, E. L. (2020). Photoreceptor degeneration in Pro23His transgenic rats (Line 3) involves autophagic and necroptotic mechanisms. Front. Neurosci. 14, 581579. doi: 10.3389/fnins.2020.581579
Karali, M., Guadagnino, I., Marrocco, E., De Cegli, R., Carissimo, A., Pizzo, M., et al. (2020). AAV-miR-204 protects from retinal degeneration by attenuation of microglia activation and photoreceptor cell death. Mol. Ther. Nucleic Acids 19, 144–156. doi: 10.1016/j.omtn.2019.11.005
Kunchithapautham, K., and Rohrer, B. (2007). Autophagy is one of the multiple mechanisms active in photoreceptor degeneration. Autophagy 3, 65–66. doi: 10.4161/auto.3431
Liu, X., Garriga, P., and Khorana, H. G. (1996). Structure and function in rhodopsin: correct folding and misfolding in two point mutants in the intradiscal domain of rhodopsin identified in retinitis pigmentosa. Proc. Natl. Acad. Sci. U. S. A. 93, 4554–4559. doi: 10.1073/pnas.93.10.4554
Mendes, H. F., and Cheetham, M. E. (2008). Pharmacological manipulation of gain-of-function and dominant-negative mechanisms in rhodopsin retinitis pigmentosa. Hum. Mol. Genet. 17, 3043–3054. doi: 10.1093/hmg/ddn202
Mendes, H. F., van der Spuy, J., Chapple, J. P., and Cheetham, M. E. (2005). Mechanisms of cell death in rhodopsin retinitis pigmentosa: implications for therapy. Trends Mol. Med. 11, 177–185. doi: 10.1016/j.molmed.2005.02.007
Mitter, S. K., Rao, H. V., Qi, X., Cai, J., Sugrue, A., Dunn, W. A. Jr., et al. (2012). Autophagy in the retina: a potential role in age-related macular degeneration. Adv. Exp. Med. Biol. 723, 83–90. doi: 10.1007/978-1-4614-0631-0_12
Mohlin, C., Taylor, L., Ghosh, F., and Johansson, K. (2014). Autophagy and ER-stress contribute to photoreceptor degeneration in cultured adult porcine retina. Brain Res. 1585, 167–183. doi: 10.1016/j.brainres.2014.08.055
Naso, F., Intartaglia, D., Falanga, D., Soldati, C., Polishchuk, E., Giamundo, G., et al. (2020). Light-responsive microRNA miR-211 targets Ezrin to modulate lysosomal biogenesis and retinal cell clearance. EMBO J. 39, e102468. doi: 10.15252/embj.2019102468
Polishchuk, E. V., and Polishchuk, R. S. (2019). Pre-embedding labeling for subcellular detection of molecules with electron microscopy. Tissue Cell 57, 103–110. doi: 10.1016/j.tice.2018.11.002
Qiu, Y., Yao, J., Jia, L., Thompson, D. A., and Zacks, D. N. (2019). Shifting the balance of autophagy and proteasome activation reduces proteotoxic cell death: a novel therapeutic approach for restoring photoreceptor homeostasis. Cell Death Dis. 10, 547. doi: 10.1038/s41419-019-1780-1
Rattner, A., Sun, H., and Nathans, J. (1999). Molecular genetics of human retinal disease. Annu. Rev. Genet. 33, 89–131. doi: 10.1146/annurev.genet.33.1.89
Rodriguez-Muela, N., Koga, H., Garcia-Ledo, L., de la Villa, P., de la Rosa, E. J., Cuervo, A. M., et al. (2013). Balance between autophagic pathways preserves retinal homeostasis. Aging Cell 12, 478–488. doi: 10.1111/acel.12072
Sakami, S., Maeda, T., Bereta, G., Okano, K., Golczak, M., Sumaroka, A., et al. (2011). Probing mechanisms of photoreceptor degeneration in a new mouse model of the common form of autosomal dominant retinitis pigmentosa due to P23H opsin mutations. J. Biol. Chem. 286, 10551–10567. doi: 10.1074/jbc.M110.209759
Saliba, R. S., Munro, P. M., Luthert, P. J., and Cheetham, M. E. (2002). The cellular fate of mutant rhodopsin: quality control, degradation and aggresome formation. J. Cell Sci. 115, 2907–2918. doi: 10.1242/jcs.115.14.2907
Silva, M. C., Nandi, G. A., Tentarelli, S., Gurrell, I. K., Jamier, T., Lucente, D., et al. (2020). Prolonged tau clearance and stress vulnerability rescue by pharmacological activation of autophagy in tauopathy neurons. Nat. Commun. 11, 3258. doi: 10.1038/s41467-020-16984-1
Sizova, O. S., Shinde, V. M., Lenox, A. R., and Gorbatyuk, M. S. (2014). Modulation of cellular signaling pathways in P23H rhodopsin photoreceptors. Cell Signal 26, 665–672. doi: 10.1016/j.cellsig.2013.12.008
Wen, R. H., Stanar, P., Tam, B., and Moritz, O. L. (2019). Autophagy in Xenopus laevis rod photoreceptors is independently regulated by phototransduction and misfolded RHO(P23H). Autophagy 15, 1970–1989. doi: 10.1080/15548627.2019.1596487
Wheway, G., Nazlamova, L., Turner, D., and Cross, S. (2019). 661W photoreceptor cell line as a cell model for studying retinal ciliopathies. Front. Genet. 10, 308. doi: 10.3389/fgene.2019.00308
Keywords: Ezrin, RHOP23H/+, autophagy, retinal degeneration, ER-stress
Citation: Intartaglia D, Giamundo G, Naso F, Nusco E, Di Giulio S, Salierno FG, Polishchuk E and Conte I (2022) Induction of Autophagy Promotes Clearance of RHOP23H Aggregates and Protects From Retinal Degeneration. Front. Aging Neurosci. 14:878958. doi: 10.3389/fnagi.2022.878958
Received: 18 February 2022; Accepted: 19 May 2022;
Published: 30 June 2022.
Edited by:
Sabrina Carrella, Stazione Zoologica Anton Dohrn Napoli, ItalyReviewed by:
Serena Mirra, University of Barcelona, SpainCopyright © 2022 Intartaglia, Giamundo, Naso, Nusco, Di Giulio, Salierno, Polishchuk and Conte. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ivan Conte, Y29udGVAdGlnZW0uaXQ=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.